 Jiaxin Xue
Jiaxin Xue Jin Han
Jin Han Xiaopeng Zhao
Xiaopeng Zhao Li Zhen1
Li Zhen1 Shanshan Mei
Shanshan Mei Zhiyang Hu
Zhiyang Hu Xiuzhen Li
Xiuzhen Li- 1Prenatal Diagnosis Center, Guangzhou Women and Children’s Medical Center, Guangzhou, China
- 2Department of Obstetrics and Gynecology, Guangzhou Medical University, Guangzhou, China
- 3Division of Neonatology, Guangzhou Women and Children’s Medical Center, Guangzhou, China
- 4Division of Obstetrics, Guangzhou Women and Children’s Medical Center, Guangzhou, China
- 5Shenzhen People’s Hospital, Shenzhen, China
- 6Division of Endocrinology, Guangzhou Women and Children’s Medical Center, Guangzhou, China
Transaldolase (TALDO) deficiency is a rare autosomal recessive disorder caused by variants in the TALDO1 gene that commonly results in multisystem dysfunction. Herein, we reported compound heterozygous variants in a Chinese prenatal case with TALDO deficiency using whole-exome sequencing (WES) for trios and Sanger sequencing. The heterozygous variants were located on the TALDO1 gene: NM_006755.2:c.574C > T(Chr11:g.763456C > T), a missense variant in exon 5 paternally inherited; NM_006755.2:c.462-2A > G(Chr11:g.763342A > G), a splicing aberration in intron 4 maternally inherited. The qualitative analysis of urinary polyols in neonatal urine indicated that xylitol + arabitol and ribitol in the proband’s urine were significantly increased. These findings expand the variation spectrum of the TALDO1 gene, provide solid evidence for the counseling of the family in regard to future pregnancies, strongly support the application of WES in prenatal diagnosis, and further prove that effective postpartum treatments could improve prognosis.
Introduction
Transaldolase (TALDO) deficiency (OMIM 606003), a rare metabolic congenital defect of the pentose phosphate pathway (PPP), is caused by homozygous or compound heterozygous variants of the TALDO1 gene (Wamelink et al., 2008) located on chromosome 11p15. Its main clinical manifestations usually appear in the neonatal period, while they are relatively rare in the antenatal period. The typical symptoms include coagulopathy, thrombocytopenia, liver dysfunction, hepatosplenomegaly, hepatic fibrosis, hemolytic anemia, generalized edema, dysmorphic features, and renal dysfunction that rarely occurs. Prolonged activated partial thromboplastin time (APTT), prothrombin time (PT) and thrombin time (TT), low cholesterol, high alkaline phosphatase (AKP), as well as elevated total bilirubin (TBIL), direct bilirubin (DBIL), total bile acid (TBA), and β2-microglobulin (β2-MG), can indicate liver and renal dysfunction in some reported cases (Valayannopoulos et al., 2006).
The PPP has two main functions: 1) It provides reduced nicotine adenine dinucleotide phosphate (the cofactor of redox reaction for organism biosynthesis), and 2) it offers ribose-5-phosphate to the nucleic acid. The PPP is divided into oxidative (nonreversible) and nonoxidative (reversible) enzymatic reactions/parts. Moreover, TALDO is the second enzyme of the nonoxidative part tightly linking the PPP and glycolysis pathway (Verhoeven et al., 2005).
To date, approximately 39 cases diagnosed with TALDO deficiency have been reported, but the incidence is unclear (Verhoeven et al., 2001; Eyaid et al., 2013; Rodan and Berry, 2017; Halabi et al., 2019; Lee-Barber et al., 2019; Williams et al., 2019; Lafci et al., 2021) (Table 3). Yet, the pathophysiology leading to TALDO deficiency remains unclear due to the low number of reported cases. TALDO deficiency can also have high variability in clinical manifestations and outcomes, even within the same family (Tylki-Szymanska et al., 2009; Leduc et al., 2014). Herein, we reported a novel compound heterozygous variant in a Chinese prenatal case with multiorgan dysfunction confirmed as TALDO deficiency by prenatal molecular diagnosis.
Materials and Methods
Ethics Approval
After receiving written informed consent from both of the parents, WES (trio analysis of the proband, mother, and father) was carried out. Our study was approved by the Ethics Committee of Guangzhou Women and Children’s Medical Center and Guangzhou Medical University, and it conformed with the ethical standards of experiments on human subjects.
Case Presentation
A 33-year-old pregnant woman, gravida 2, para 1, was referred to our hospital at 34 weeks because of ultrasonic abnormalities. Fetal middle cerebral artery peak systolic velocity (MCA-PSV) kept increasing from 24 gestational weeks, reaching 93.97 cm/s [>1.5 MoM (Multiples of the Median)] at 33 gestational weeks. Additional anomalies included a slightly high echo of the right lobe of the liver, cardiomegaly with the cardiothoracic ratio of 0.61, a small amount of pericardial effusion, and placental thickness of 46 mm.
Similar manifestations, including cardiac enlargement, hepatosplenomegaly, placental thickness, elevated MCA-PSV, high umbilical artery resistance, and intrauterine growth restriction (IUGR), were observed during the first pregnancy (II:1). Following fetal distress, at 36 weeks, a baby boy was born by cesarean section weighing 1,860 g (<10th). His Apgar scores were normal (9′-10′-10′), while the neonatal peripheral blood test detected that hemoglobin (HGB) and platelet (PLT) were low. Repeated examinations of coagulation showed extended APTT, PT, and TT. Brain ultrasound suggested a head injury with subependymal hemorrhage. Therefore, II:1 received human immunoglobulin and blood transfusion to prevent infection and improve blood coagulation. The neonate did not recover and consequently died of disseminated intravascular coagulation (DIC), a low-birth-weight, and hypoproteinemia at 18 days. Clinical findings of the two affected fetuses (II:1 and II:2) are summarized in Tables 1, 2.
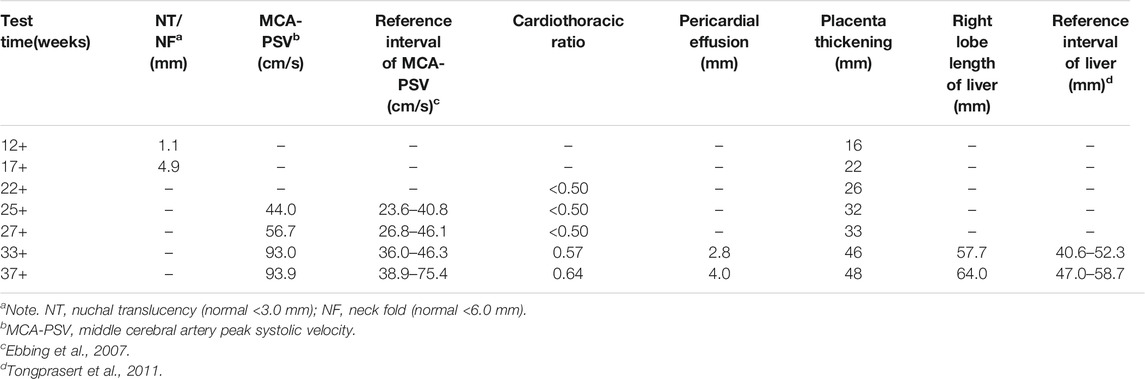
TABLE 1. Ultrasound findings of II:2 at different gestational weeks.
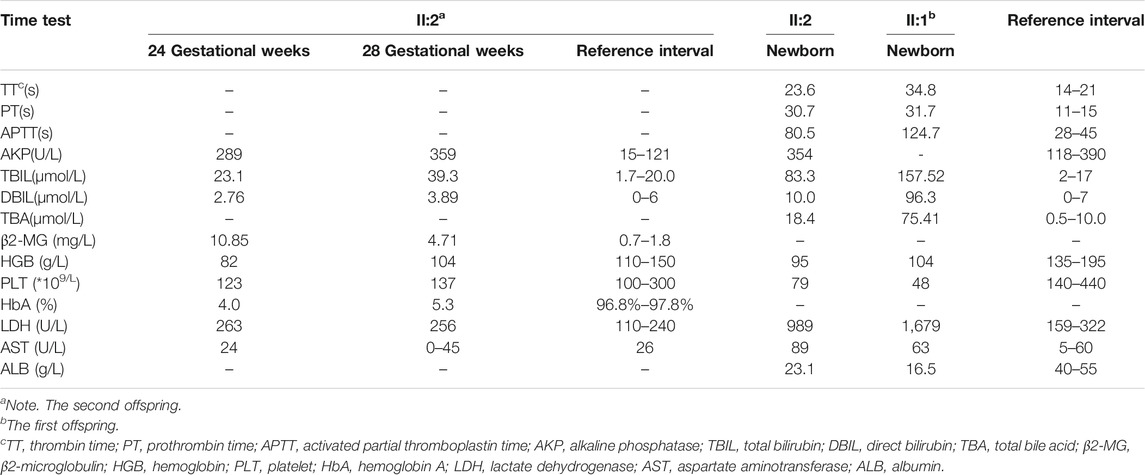
TABLE 2. The laboratory results of II:2 as determined directly in 24+ and 28+ weeks through the umbilical cord blood tests and after birth between II:1 and II:2.
To assess the risk of recurrence, cordocentesis was performed for genetic diagnosis, including karyotype analysis and chromosomal microarray analysis (CMA), to clarify the potential cause of the disease two times in another hospital, but the results were negative. Consequently, WES was performed on the proband and his healthy parents (Figures 1A,B) to search for potential variants. The detailed examinations during pregnancy are listed in Table 1.
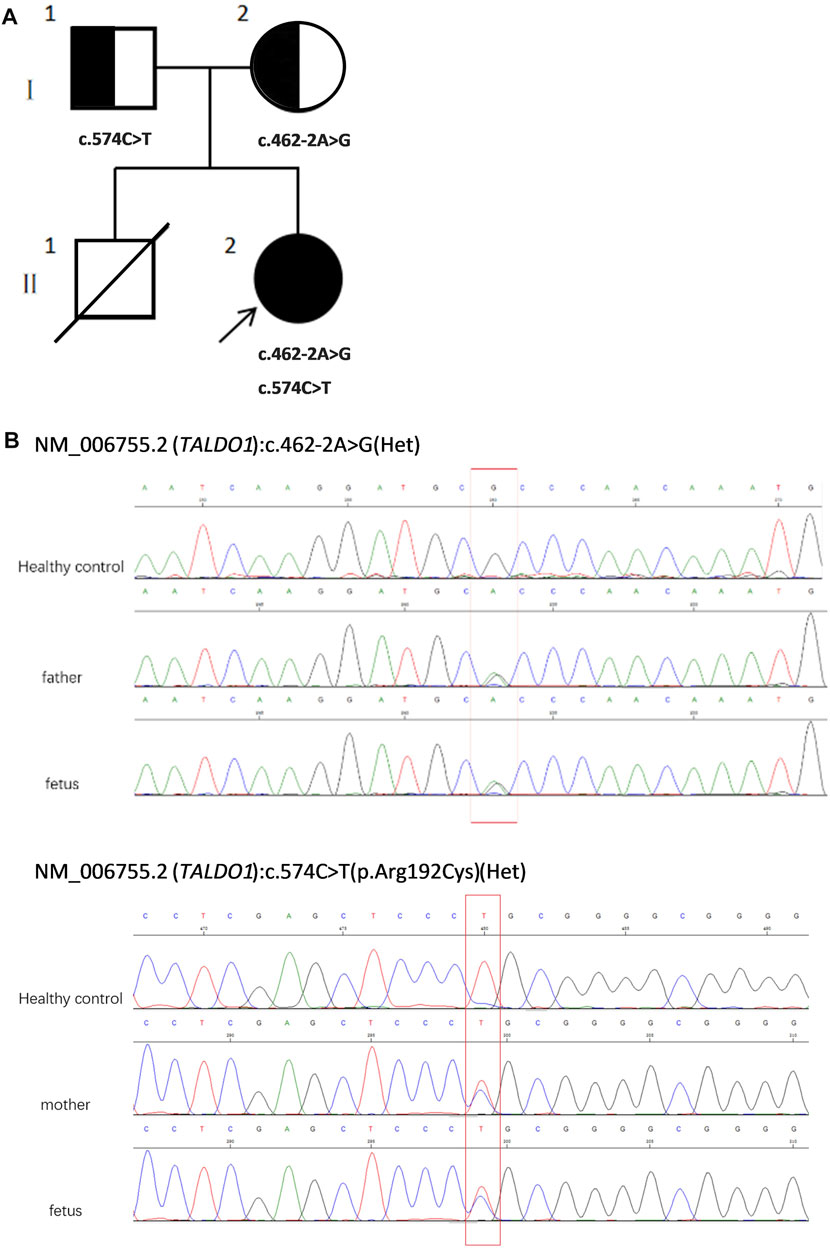
FIGURE 1. (A) Pedigree of the family. (B) Sequencing of TALDO1 gene (reference cDNA sequence, NM_006755.2) revealed two heterozygous variations, resulting in A to G splicing at nucleotide position 462–2 (c.462-2A > G) and C to T substitution at nucleotide position 574 [c.574C > T(p.Arg192Cyrs)]. Het, heterozygous.
Metabolite Analyses
Urine xylitol + arabitol and ribitol were measured using gas chromatography-mass spectrometry (GC-MS). Urine sample preparation was based on urease pretreatment methods. Samples were standardized to 0.25 mg creatinine. Derivatization was performed with 100 μl bis-(trimethylsilyl) trifluoracetamide + 1% trimethylchlorosilane and was allowed to react at 60°C for 10 min. The metabolites were chromatographically analyzed as trimethylsilyl compounds.
Whole-Exome Sequencing
Genomic DNA was randomly fragmented and purified using the magnetic particle method. WES was performed on an IIIumina HiSeq 2,500 sequencer (Illumina, San Diego, CA, United States) for a minimal of 10.14 Gb read-depth per case. Sequencing reads after quality control were aligned to the human reference genome by BWA (hg19). Nucleotide changes of aligned reads were reviewed using NextGENe software (Version 2.4.1.2) (SofGenetics, State College, PA, United States). Novel variants were filtered against the 1,000 Genomes database (http://www.1000genomes.org/), dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi), and the Genome Aggregation database (gnomad.broadinstitute.org). Databases, including ClinVar (version: #372716), OMIM (version: #602063.0005), ClinGen (version: #CA5788214), and Human Gene Mutation database, were used. In addition, software (SIFT, Polyphen, MutationTaster, PROVEAN and REVEL) was used to predict the impact of missense variants. For the splicing variant, the in silico prediction tools were dbscSNV and MaxEntScan. Common variants (with high minor allele frequency in normal population; gnomAD) were eliminated. Finally, polymerase chain reaction (PCR) was performed to amplify the affected fragment of TALDO1 gene using specific primers, and the purified PCR products were applied to Sanger sequencing to affirm the variant(s).
Results
The umbilical cord blood samples of the fetus (II:2) in 24 and 28 gestational weeks in another hospital showed fetal anemia, thrombocytopenia, coagulation dysfunction, and elevated liver enzymes [lactate dehydrogenase (LDH) and β2-MG] (Table 2). Urine test for metabolic compounds using GC-MS showed an elevation of xylitol + arabitol at 170,388 mmol/mol creatinine (normal 0–1,151 mmol/mol creatinine) and ribitol at 193,301 mmol/mol creatinine (normal 0–886 mmol/mol creatinine) (Figure 2).
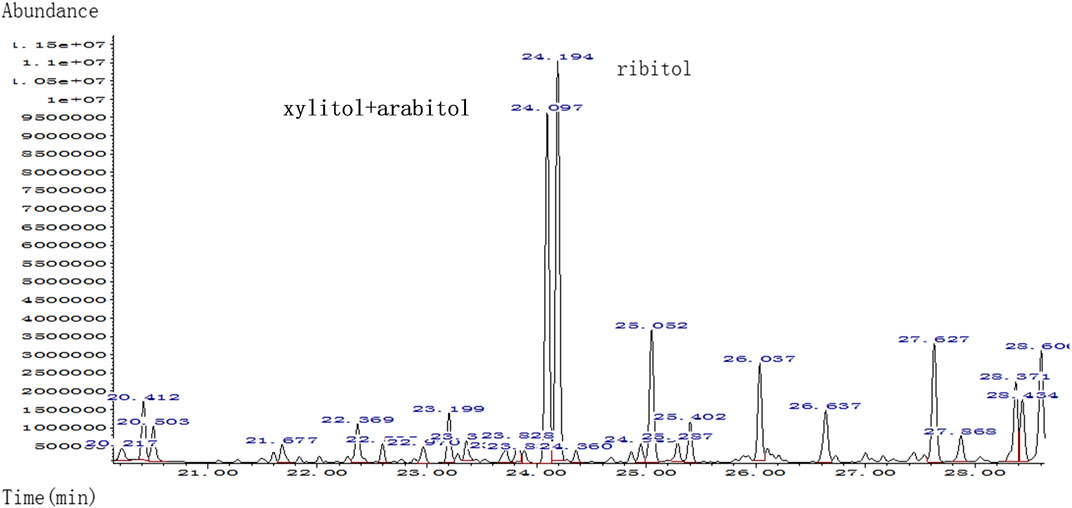
FIGURE 2. Quality analysis of polyols in urine sample of patient by urine gas chromatography-mass spectrometry (GC-MS); markedly elevated xylitol + arabitol and ribitol were detected.
WES revealed compound heterozygosity of variants in the TALDO1 gene in the proband: maternally inherited likely splicing aberration NM_006755.2(TALDO1):c.462-2A > G(Chr11:g.763342A > G), and paternally inherited missense variant NM_006755.2(TALDO1):c.574C > T(Chr11:g.763456C > T). Both parents were heterozygous carriers and phenotypically normal. Sanger sequencing of the patient and his family members further validated these results (Figures 1A,B). According to the ACMG standards, both variants were defined as likely pathogenic (c.462-2A > G: PVS1 + PM2, c.574C > T: PM2 + PM3 + PM5 + PP3). The two variants had a very low carrying rate in some databases: c.462-2A > G is not recorded in gnomAD, dbSNP, or 1,000 Genomes databases; the minor allele frequency of c.574C > T is .00001591 (4 heterozygotes) in gnomAD, and 0.000008243 (1 heterozygote) in ExAC. c.574C > T is a missense variant located in exon 5, which is also described on dbSNP (rs751425603) and reported as pathogenicity in ClinVar (variation ID: 381,759). c.462-2A > G is a splicing variant located in intron 4 and may lead to abnormal mRNA splicing that affects protein expression. The splicing variant c.462-2A > G in MaxEntScan score was from 10.76 in wild type to 2.824 in mutant type. This variant is predicted to cause a loss of function of the protein.
Outcome
The couple decided to continue the pregnancy after genetic counseling. A baby girl was born at 38 weeks, with a weight of 2,760 g. Apgar scores were normal (8′-8′-8′) after delivery. At birth, the baby had dysmorphic features (hirsutism, low hair implantation), mild pallor, and cutis laxa. She also presented low skin temperature, quick breath with groaning, thick breath sounds in both lungs with moist rales, and abdominal distention with the visible vascular network. The baby was hospitalized at the Neonatal Department in our hospital for 9 days (Figures 3A,B). She had hepatosplenomegaly and developed jaundice. A peripheral blood test showed HGB of 95 g/L (normal 110–150 g/L) with fragmented red cells on film, thrombocytopenia, and mild neutropenia, consistent with those in utero. Serum TBIL was 83.3 µmol/L (normal 2–17 µmol/L) with DBIL of 10.0 µmol/L (normal 0–7 µmol/L). LDH was also increased to 989 U/L (normal 159–322 U/L) together with marginally elevated transaminases, bile acids and alkaline phosphatase (ALP). Albumin was 23.1 g/L (normal 40–55 g/L) and PT was 31.7 s (normal 11–15 s). The infant received continuous ventilation for 9 days. Fresh frozen plasma and fibrinogen infusion were given to improve thrombocytopenia and coagulation. Blood glucose level was stable and was closely monitored. GC-MS indicated elevated urinary xylitol + arabitol and ribitol levels.
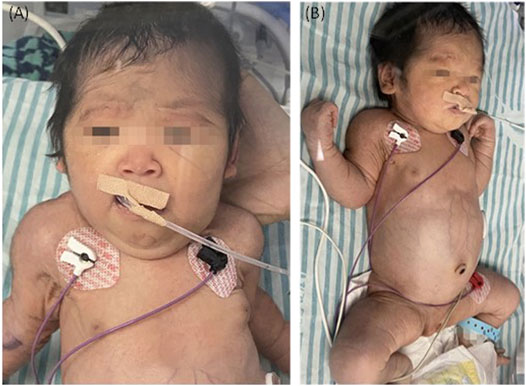
FIGURE 3. The TALDO-deficient dysmorphic feature-hirsutism (forehead), low hair implantation, mild pallor, and cuties laxa with visible vascular network of the patient. The abdomen was also grossly distended with dilated visible veins [(A) frontal view, (B) side view].
After her condition gradually improved, the patient was discharged from the hospital and was regularly followed up. At the age of 9 months, HGB was still slightly decreased (100 g/L), while red blood cell and PLT were both increased to 4.7 × 1012/L (normal 3.5–5.0 × 1012/L) and 517 × 109/L (normal 100–300 × 109/L), respectively. DBIL, TBIL, TBA, LDH, and AKP levels were normal, whereas aspartate aminotransferase (AST) mildly elevated to 84 U/L. The dysmorphic features and cutis laxa were not observed. Thus far, the child has shown normal physical and cognitive development.
Discussion
TALDO deficiency is a rare autosomal recessive error of the PPP caused by a variant in the TALDO1 gene (Williams et al., 2019). TALDO1 gene encodes TALDO implicated as a major modulator between the PPP and glycolysis in a reversible reaction. TALDO catalyzes the conversion of glyceraldehyde-3-phosphate and sedoheptulose-7-phosphate into fructose-6-phosphate and erythrose-4-phosphate, which are also considered targets for the treatments of this condition. In addition, its absence can result in the accumulation of intermediate products (e.g., sedoheptulose, erythritol, and ribitol) and eventually cause lesions of the patent. TALDO deficiency has been associated with a range of phenotypes, including intrauterine lethality together with fetal multimalformation syndrome and hydrops fetalis. The most common clinical manifestations in neonates are cirrhosis, liver failure, hepatosplenomegaly, anemia, thrombocytopenia, dysmorphia, congenital heart defects, and tubulopathy (Verhoeven et al., 2001; Verhoeven et al.,2005; Valayannopoulos et al., 2006; Wamelink et al., 2008; Tylki-Szymanska et al., 2009; Balasubramaniam et al., 2011; Eyaid et al., 2013). Yet, prenatal diagnosis is very challenging. Abnormal findings in the fetus are rare. Some of the common manifestations in the antenatal period are IUGR (Verhoeven et al., 2001; Valayannopoulos et al., 2006; Wamelink et al., 2008), oligohydramnios, fetal splenomegaly, fetal distress (Wamelink et al., 2008), and hyperechogenic bowel (Banne et al., 2015). Also, TALDO deficiency can be easily misdiagnosed with gestational alloimmune liver disease (GALD). GALD is the result of maternal alloimmune injury, which includes neonatal liver failure (coagulation disorders, ascites, and hypoalbuminemia), intrahepatic, and extrahepatic iron accumulation (hemosiderosis).
In this study, a family that had experienced neonatal death following IUGR, hepatosplenomegaly, anemia with thrombopenia, and abnormal coagulation tests in a previous pregnancy (II:1) and recurrent fetal anemia, hepatosplenomegaly in the second pregnancy (II:2), was recruited for WES. A prenatal diagnosis of the fetus confirmed heterozygous variants in the TALDO1 gene in II:2. Yet, prenatal findings were different between II:1 and II:2. Fetal MCA-PSV increased from 24 gestational weeks, which reflected fetal anemia in utero. Additional ultrasound anomalies identified at 33 gestational weeks included slightly high echo of the right lobe of the liver, cardiomegaly with increased cardiothoracic ratio, a small amount of pericardial effusion and placental thickness, all of which suggested a progressive development of fetal anemia. After birth, the postpartum symptoms were clearer and more obvious, including dysmorphic features, liver dysfunction and hemolytic anemia. This case is consistent with the range of phenotypes most commonly observed; however, fetal anemia, liver dysfunction, and coagulopathy are the main manifestations.
The accumulation of sugars and polyols [e.g., sedoheptulose-7-phosphate, ribose-5-phosphate, ribulose-5-phosphate, xylulose-5-phosphate, and C5-polyols (i.e., D-ribitol and D-arabitol)] are believed to be the cause of liver involvement in TALDO deficiency. Higher concentrations of the polyols xylitol + arabitol and ribitol in the urine of the proband could be relevant of the phenotypes in II.2, but could also be related to the younger age, since the polyol concentrations were higher in the neonatal period in other patients and accumulated less when they were older (Wamelink et al., 2008). Although from the same family, patient II:1 had IUGR, anemia, hepatosplenomegaly, DIC, a low-birth-weight, and secondary hemorrhage (subependymal hemorrhage), yet, even considering that molecular analysis was not performed for patient II:1, it was likely that these phenotypes were associated with TALDO deficiency.
To the best of our knowledge, this case is the first prenatal diagnosis of TALDO deficiency in a Chinese population (Verhoeven et al., 2001; Eyaid et al., 2013; Rodan and Berry, 2017; Lee-Barber et al., 2019; Williams et al., 2019; Halabi et al., 2019; Lafci et al., 2021). Both variants of this case were defined as likely pathogenic. One of the variants [c.574C > T p.(Arg192Cys)], reported as pathogenicity in ClinVar (Variation ID: 381,759), was previously reported in an Arab patient, suggesting a founder effect in Arab populations (Wamelink et al., 2008). The other is a novel splicing variant (c.462-2A > G), which is predicted to affect splicing while not exon skipping. The in-silico tools are dbscSNV and MaxEntScan. They all predict altering TALDO1 exon splicing. To date, there have been 13 variants reported to cause this condition worldwide (Table 3). Individuals with the same variant show different clinical manifestations.

TABLE 3. Summary of clinical manifestations in the current patients with TALDO deficiency.
The prenatal diagnosis of TALDO deficiency remains a challenge and is usually confirmed by gene analysis. Thus far, there is still no effective treatment for TALDO deficiency. Yet, early and accurate prenatal diagnosis can lead to a better outcome and can provide better aid for prenatal management, including fetal surveillance strategy and appropriate postpartum treatment, as was the case in the present study. In particular, a higher frequency of fetal surveillance with targeted ultrasound can help identify early signs of clinical manifestations (e.g., elevated MCA-PSV, cardiomegaly and placental thickness), which are important prognostic indicators. Most important of all, it is inseparable from the joint efforts of multi-disciplinary team. Currently, there is only one gene known to cause TALDO deficiency. Further studies are warranted to comprehensively characterize the genetic contributions.
In conclusion, our data suggests that TALDO deficiency is a pleiotropic disorder that should be considered when investigating a prenatal case with unexplained hepatosplenomegaly or fetal anemia. Although no specific treatment is currently available, targeted molecular analysis of the TALDO1 gene in amniotic fluid or chorionic villi can be valuable in helping those suffering families to make informed reproductive choices.
Data Availability Statement
TThe original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by The ethics committee of Guangzhou Women and Children’s Medical Center and Guangzhou Medical University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This study was supported (or partially supported) by the Science and Technology Department of Guangdong Province (2016A020218003), Guangzhou Science Technology and Innovation Commission (201607010341), and Guangzhou Women and Children’s Medical Center (GWCMC2020-6-007).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed nor endorsed by the publisher.
Acknowledgments
The authors kindly acknowledge the participation of the family members and the staff members in the Prenatal Diagnosis Center of Guangzhou Women and Children’s Medical Center and Shenzhen People’s Hospital, Guangdong Province, China. The authors would also like to thank the Genome Aggregation database (gnomAD) for providing genome variant data.
References
Al-Shamsi, A. M., Ben-Salem, S., Hertecant, J., and Al-Jasmi, F. (2015). Transaldolase Deficiency Caused by the Homozygous p.R192C Mutation of the TALDO1 Gene in Four Emirati Patients With Considerable Phenotypic Variability. Eur. J. Pediatr. 174 (5), 661–668. doi:10.1007/s00431-014-2449-5
Balasubramaniam, S., Wamelink, M. M., Ngu, L.-H., Talib, A., Salomons, G. S., Jakobs, C., et al. (2011). Novel Heterozygous Mutations in TALDO1 Gene Causing Transaldolase Deficiency and Early Infantile Liver Failure. J. Pediatr. Gastroenterol. Nutr. 52, 113–116. doi:10.1097/MPG.0b013e3181f50388
Banne, E., Meiner, V., Shaag, A., Katz-Brull, R., Gamliel, A., and Korman, S. (2015). Transaldolase Deficiency: A New Case Expands the Phenotypic Spectrum. JIMD Rep. 26, 31–36. doi:10.1007/8904_2015_474
Ebbing, C., Rasmussen, S., and Kiserud, T. (2007). Middle Cerebral Artery Blood Flow Velocities and Pulsatility Index and the Cerebroplacental Pulsatility Ratio: Longitudinal Reference Ranges and Terms for Serial Measurements. Ultrasound. Obstet. Gynecol. 30 (3), 287–296. doi:10.1002/uog.4088
Eyaid, W., Al Harbi, T., Anazi, S., Wamelink, M. M. C., Jakobs, C., Al Salammah, M., et al. (2013). Transaldolase Deficiency: Report of 12 New Cases and Further Delineation of the Phenotype. J. Inherit. Metab. Dis. 36, 997–1004. doi:10.1007/s10545-012-9577-8
Fung, C. W., Siu, S., and Mak, C. (2007). A Rare Cause of Hepatosplenomegaly-Transaldolase Deficiency. J. Inherit. Metab. Dis. 30 (Supplement 1), 62.
Halabi, M., Alqoaer, K., and Asaad, Z. (2019). Severe Infantile Transaldolase Deficiency. Ajpr, 1–5. doi:10.9734/ajpr/2019/v2i430112
Jassim, N., Alghaihab, M., Saleh, S. A., Alfadhel, M., Wamelink, M. M., and Eyaid, W. (2014). Pulmonary Manifestations in a Patient With Transaldolase Deficiency. JIMD Rep. 12, 47–50. doi:10.1007/8904_2013_243
Lafci, N. G., Colak, F. K., Sahin, G., Sakar, M., Çetinkaya, S., and Savas-Erdeve, S. (2021). Hypergonadotrophic Hypogonadism in a Patient with Transaldolase Deficiency: Novel Mutation in the Pentose Phosphate Pathway. Hormones 20, 581–585. doi:10.1007/s42000-020-00252-4
Leduc, C. A., Crouch, E. E., Wilson, A., Lefkowitch, J., Wamelink, M. M. C., Jakobs, C., et al. (2013). Novel Association of Early Onset Hepatocellular Carcinoma with Transaldolase Deficiency. JIMD Rep. 12, 121–127. doi:10.1007/8904_2013_254
Lee-Barber, J., English, T. E., Britton, J. F., Sobreira, N., Goldstein, J., Valle, D., et al. (2019). Apparent Acetaminophen Toxicity in a Patient with Transaldolase Deficiency. JIMD Rep. 44, 9–15. doi:10.1007/8904_2018_116
Rodan, L. H., and Berry, G. T. (2016). N-Acetylcysteine Therapy in an Infant with Transaldolase Deficiency Is Well Tolerated and Associated with Normalization of Alpha Fetoprotein Levels. JIMD Rep. 31, 73–77. doi:10.1007/8904_2016_555
Tongprasert, F., Srisupundit, K., Luewan, S., and Tongsong, T. (2011). Normal Length of the Fetal Liver From 14 to 40 Weeks of Gestational Age. J. Clin. Ultrasound. 39 (2), 74–77. doi:10.1002/jcu.20756
Tylki-Szymańska, A., Stradomska, T. J., Wamelink, M. M. C., Salomons, G. S., Taybert, J., Pawłowska, J., et al. (2009). Transaldolase Deficiency in Two New Patients with a Relative Mild Phenotype. Mol. Genet. Metab. 97, 15–17. doi:10.1016/j.ymgme.2009.01.016
Valayannopoulos, V., Verhoeven, N. M., Mention, K., Salomons, G. S., Sommelet, D., Gonzales, M., et al. (2006). Transaldolase Deficiency: a New Cause of Hydrops Fetalis and Neonatal Multi-Organ Disease. J. Pediatr. 149, 713–717. doi:10.1016/j.jpeds.2006.08.016
Verhoeven, N. M., Huck, J. H. J., Roos, B., Struys, E. A., Salomons, G. S., Douwes, A. C., et al. (2001). Transaldolase Deficiency: Liver Cirrhosis Associated with a New Inborn Error in the Pentose Phosphate Pathway. Am. J. Hum. Genet. 68, 1086–1092. doi:10.1086/320108
Verhoeven, N. M., Wallot, M., Huck, J. H. J., Dirsch, O., Ballauf, A., Neudorf, U., et al. (2005). A Newborn with Severe Liver Failure, Cardiomyopathy and Transaldolase Deficiency. J. Inherit. Metab. Dis. 28, 169–179. doi:10.1007/s10545-005-5261-6
Wamelink, M. M., Struys, E. A., Salomons, G. S., Fowler, D., Jakobs, C., and Clayton, P. T. (2008). Transaldolase Deficiency in a Two-Year-Old Boy with Cirrhosis. Mol. Genet. Metab. 94, 255–258. doi:10.1016/j.ymgme.2008.01.011
Williams, M., Valayannopoulos, V., Altassan, R., Chung, W. K., Heijboer, A. C., Keng, W. T., et al. (2019). Clinical, Biochemical, and Molecular Overview of Transaldolase Deficiency and Evaluation of the Endocrine Function: Update of 34 Patients. Jrnl Inher Metab. Disea. 42, 147–158. doi:10.1002/jimd.12036
Keywords: Transaldolase deficiency, pentose phosphate pathway, TALDO1, prenatal diagnosis, whole-exome sequencing (WES)
Citation: Xue J, Han J, Zhao X, Zhen L, Mei S, Hu Z and Li X (2022) Prenatal Diagnosis of Fetus With Transaldolase Deficiency Identifies Compound Heterozygous Variants: A Case Report. Front. Genet. 12:752272. doi: 10.3389/fgene.2021.752272
Received: 02 August 2021; Accepted: 14 December 2021;
Published: 04 February 2022.
Edited by:
Natália Duarte Linhares, Genuity Science, IrelandReviewed by:
Vassili Valayannopoulos, Ultragenyx Pharmaceutical, United StatesMirjam Wamelink, Amsterdam University Medical Center, Netherlands
Copyright © 2022 Xue, Han, Zhao, Zhen, Mei, Hu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin Han, aGFuamluMTEyM0AxNjMuY29t