 Oriana Kreutzfeld
Oriana Kreutzfeld Josephine Grützke
Josephine Grützke Alyssa Ingmundson
Alyssa Ingmundson Katja Müller
Katja Müller Kai Matuschewski
Kai Matuschewski
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 08 December 2021
Sec. Human and Medical Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.742153
This article is part of the Research Topic Genetics of Apicomplexans and Apicomplexan-Related Parasitic Diseases View all 13 articles
Host cell remodeling is critical for successful Plasmodium replication inside erythrocytes and achieved by targeted export of parasite-encoded proteins. In contrast, during liver infection the malarial parasite appears to avoid protein export, perhaps to limit exposure of parasite antigens by infected liver cells. HSP101, the force-generating ATPase of the protein translocon of exported proteins (PTEX) is the only component that is switched off during early liver infection. Here, we generated transgenic Plasmodium berghei parasite lines that restore liver stage expression of HSP101. HSP101 expression in infected hepatocytes was achieved by swapping the endogenous promoter with the ptex150 promoter and by inserting an additional copy under the control of the elongation one alpha (ef1α) promoter. Both promoters drive constitutive and, hence, also pre-erythrocytic expression. Transgenic parasites were able to complete the life cycle, but failed to export PEXEL-proteins in early liver stages. Our results suggest that PTEX-dependent early liver stage export cannot be restored by addition of HSP101, indicative of alternative export complexes or other functions of the PTEX core complex during liver infection.
Host cell remodeling is critical for successful Plasmodium replication inside erythrocytes and achieved by targeted export of parasite-encoded proteins. Therefore, the parasite exports a large set of proteins to remodel the host erythrocyte, which is particularly important for plasma membrane fluidity and adhesive properties (Jonsdottir et al., 2021). The repertoire of exported proteins, termed exportome, is estimated to be approximately 10% of the Plasmodium proteome, and a quarter of the exported proteins are likely essential for parasite survival during blood stage development (Maier et al., 2008; Spielmann and Gilberger, 2015). Exported proteins have three major destinations in host erythrocytes, i) parasite-generated membranous structures, e.g. Maurer’s clefts (Mundwiler-Pachlatko and Beck, 2013) and caveolae-vesicle complexes (Akinyi et al., 2012), ii) the erythrocyte membrane cytoskeleton (Warncke and Beck, 2019), and iii) the erythrocyte plasma membrane (Wahlgren et al., 2017).
Export of proteins across two apposed membranes, the parasite plasma membrane and the parasitophorous vacuole membrane (PVM), requires a tightly coordinated translocation process. Similar to the classic secretory pathway, Plasmodium exported proteins enter the endoplasmic reticulum (ER) and are eventually secreted into the PV, although the underlying mechanism is still inadequately understood (Matthews et al., 2019a). The signature export label for this destination is a small host cell targeting (HT) sequence or Plasmodium export element (PEXEL), comprising the amino acid residues RxLxE/Q/D located downstream of the ER signal sequence (Hiller et al., 2004; Marti et al., 2004; Mesén-Ramírez et al., 2016). A different class of exported proteins lack this motif, so-called PEXEL/HT negative exported proteins (PNEPs), but share the downstream translocation pathway (Grüring et al., 2012; Heiber et al., 2013). Both PEXEL and PNEP proteins are believed to be loaded into secretory vesicles to be transported and released into the PV. Once in the PV, proteins need to translocate the PVM to reach the erythrocyte cytoplasm, a process mediated by the Plasmodium translocon of exported proteins (PTEX) (de Koning-Ward et al., 2009).
Studies in human and rodent Plasmodium species revealed that the PTEX complex consists of three core proteins, EXP2, HSP101, PTEX150, which are flanked by two auxiliary proteins, PTEX88 and TRX2 (de Koning-Ward et al., 2009; Matthews et al., 2013; Matz et al., 2013), as well as the GPI-anchored surface protein P113 (Elsworth et al., 2016). Additional identified auxiliary proteins include the Export Protein Interacting complex, which comprises of the parasitophorous vacuole protein 1 (PV1), PV2 and EXP3 (Batinovic et al., 2017). EXP2 plays a central role as a membrane-spanning component, but simultaneously acts as a nutrient channel in a PTEX-independent manner (Garten et al., 2018). HSP101 is considered to be the force-generating motor protein and a member of the ClpB-type AAA+-ATPase family (Beck et al., 2014; Matthews et al., 2019b). HSP101 contains a cargo-binding pore loop at the amino-terminal end and is most likely responsible for protein unfolding and active translocation through the complex. Although there is little evidence for a distinct functional role of PTEX150, numerous studies consistently confirmed that PTEX150 appears to play a structural role linking EXP2 and HSP101 (Bullen et al., 2012; Elsworth et al., 2014; Elsworth et al., 2016; Garten et al., 2018). Experimental genetics studies of the three core components in human and rodent Plasmodium species corroborated the importance of these proteins for parasite blood stage survival, and loss-of-function mutants failed to propagate during blood infection (Matthews et al., 2013; Matz et al., 2013; Beck et al., 2014; Elsworth et al., 2014). The role of PTEX88 in protein translocation is less well understood. Knockout of PTEX88 in a murine malaria model reduced experimental cerebral malaria and cytoadherence (Matthews et al., 2013; Matz et al., 2013; Matz et al., 2015; Chisholm et al., 2016). It, thus, might play a role in translocation of specific proteins important for parasite virulence.
Although host cell remodeling and protein export are remarkably expanded in asexual blood stages, they occur both in gametocytes and during liver stage development (Ingmundson et al., 2014). This observation is further corroborated by the presence of syntenic genes in closely related Hepatocystis parasites, which only replicate in the liver and lack blood schizogony (Ejotre et al., 2021), yet harbor orthologous genes for EXP2 (HEP_00110700), HSP101 (HEP_00056400), PTEX150 (HEP_ 00473300) and PTEX88 (HEP_004505500) (Aunin et al., 2020). One hypothesis is that during liver infection the malarial parasite appears to curtail protein export. As metabolically active cells and in contrast to red blood cells, hepatocytes permit the developing parasite to exploit the host cell, likely limiting the need for intensive remodeling and, thereby, directing energy towards population expansion (Prudencio et al., 2006; Silvie et al., 2008a).
Moreover, hepatocytes harbor an active MHCI pathway with the potential to present peptide fragments of intracellular parasitic antigens to the host’s immune cells (Chakravarty et al., 2007). To avoid recognition by the host, the parasite might keep protein translocation during early to mid liver stage development to a minimum. To date, only peptides of sporozoite origin could be identified as targets of CD8+ T cells (Bongfen et al., 2007; Hafalla et al., 2013; Müller et al., 2017). Recently, a liver stage specific peptide, Kb-17, was identified, but its contribution to protective immune responses remains to be determined (Pichugin et al., 2018).
To date, three proteins were reported to translocate during liver stage growth, namely circumsporozoite protein (CSP) (Singh et al., 2007), liver-specific protein 2 (LISP2) (Orito et al., 2013), and sporozoite- and liver stage-expressed tryptophan rich protein (SLTRiP) (Jaijyan et al., 2015). The extent of CSP export in infected hepatocytes remains highly controversial (Singh et al., 2007; Cockburn et al., 2011), partly because CSP might be already released into the host’s cytoplasm during sporozoite cell traversal and upon hepatocyte entry. Immunofluorescence assays with LISP2 antibodies demonstrated onset of export 24 h after infection, yet parasites harboring a C-terminal mCherry tagged LISP2 failed to export the tagged protein to the hepatocyte cytoplasm (Orito, et al., 2013). SLTRiP export in liver stages was detected as early as 12 h after infection, with 80% of infected host cells showing export of SLTRiP (Jaijyan et al., 2015). A central question is whether a potential liver stage exportome is translocated by similar or distinct mechanisms as compared to translocation during blood infection.
The notion of very restricted protein export during the first schizogony in infected hepatocytes is supported by additional experimental genetics evidence. Fusion of the PEXEL motif of Plasmodium berghei CSP to the model antigen ovalbumin and expression under the control of the UIS4 promoter failed to induce export in infections with transgenic parasites, although CD8+ T cell proliferation was increased (Montagna et al., 2014). Moreover, during liver stage maturation HSP101 is not expressed until 48 h after infection, whereas all other PTEX components are present (Matz et al., 2015), offering a plausible explanation for restricted protein export in these stages. Investigation of KAHRPL-GFP PEXEL dependent export in liver stages showed absence of such export despite the presence of EXP2 and PTEX150. Further experiments with HA-tagged HSP101 parasites confirmed the absence of HSP101 in liver stages, an indicator for lack of PEXEL dependent export in liver stages (Kalanon et al., 2016). A valid hypothesis, experimentally addressed in the present study, is that the Plasmodium PTEX ATPase HSP101 might be a limiting factor for liver stage protein export. Alternatively, the protein unfolding and translocation functions of HSP101 might be substituted by a distinct, yet unidentified factor.
An important implication is whether increasing antigen export in early liver stages would result in increased immune recognition of Plasmodium-infected cells, by presentation of parasite-derived epitopes via MHCI. We, therefore, sought to define liver stage export in HSP101 over-expressing parasites and to study these parasite lines for potential enhanced CD8+ T cell responses and protection in cell-based immunization.
To this end, we designed P. berghei lines that express HSP101 (PBANKA_0931200) during liver stage maturation. This gain-of-function approach is expected to allow PTEX reconstitution, since the other major PTEX components, PTEX150 and EXP2, and the auxiliary factor PTEX88 are expressed during liver stage development (Matz et al., 2015). Reconstitution of the PTEX export machinery might facilitate protein export in early liver stages and, thus, might promote antigen presentation and recognition of parasites by the host immune system.
To compare PEXEL-dependent export in P. berghei blood and liver stages we generated several reporter lines, which encode mCherry fusion proteins that contain predicted CSP-PEXEL and IBIS-PEXEL motifs at their amino-terminal end (Figure 1A; Supplementary Figure S1). Export of the fluorescent reporter proteins into red blood cells was analyzed by live immunofluorescence imaging. The first set of transgenic parasite lines expressed the PEXEL-containing reporter proteins under the control of the IBIS1 promoter, which is active in several life cycle stages, including in liver and blood stages. Reporter proteins that included additional amino acids after the RxLxQ/E/D motif, CSP1-70-mCherry and IBIS1-118-mCherry, were exported to the infected erythrocytes (Figure 1A). When a truncated version of an IBIS1 fusion protein, IBIS11-90-mCherry, was expressed under the control of the IBIS1 promoter, the red fluorescent signal was confined to the parasite and displayed an enrichment at the PV. A similar confinement to the parasite and a stronger signal at the PV was detected for transgenic parasites that expressed CSP1-70-mCherry under the control of the strong and ubiquitous HSP70 promoter (Figure 1A), indicative that excessive protein may congest the PTEX translocon.
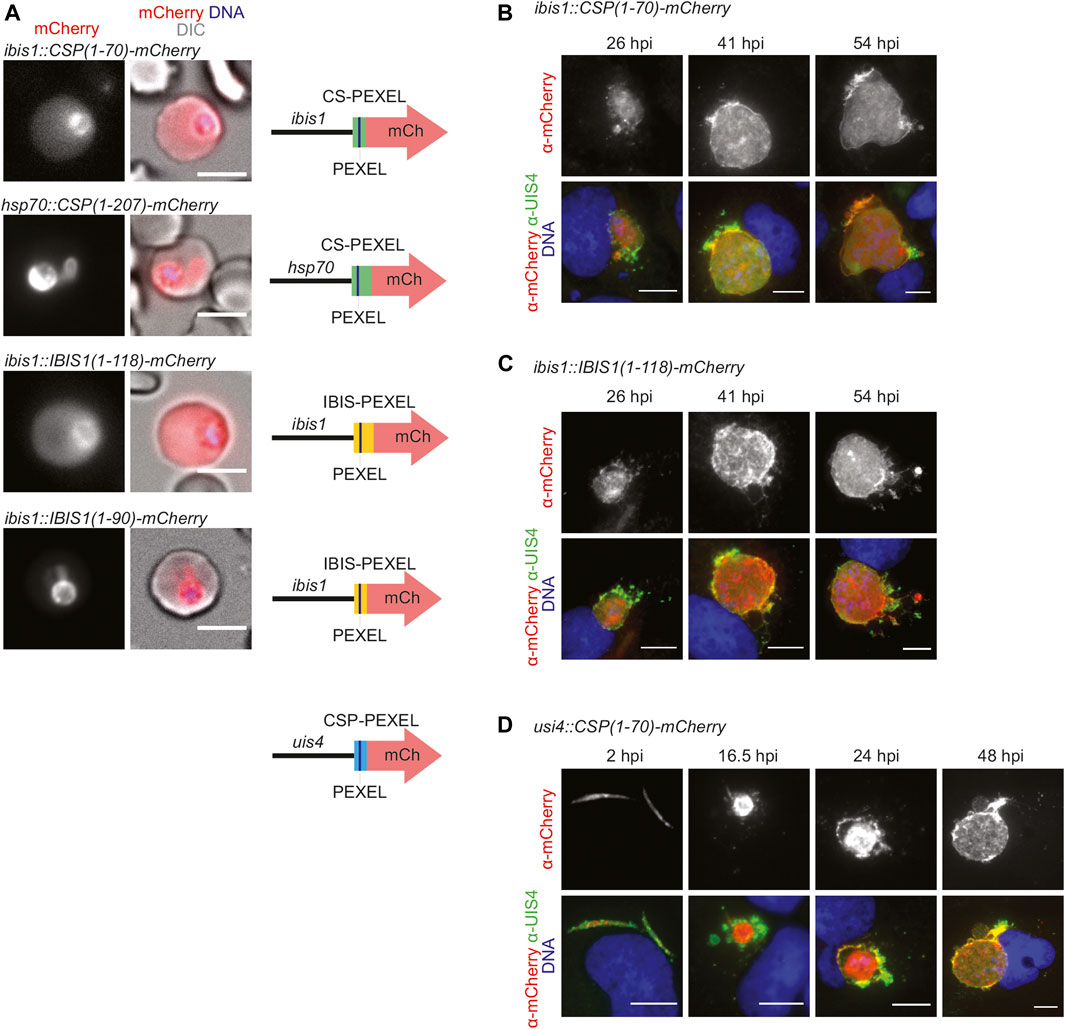
FIGURE 1. PEXEL-dependent export of mCherry reporter protein occurs during blood but not liver infection. (A) Live fluorescence imaging of parasite-infected erythrocytes. Shown are the mCherry signal (left), a merge with DIC images (center), and graphic images of the reporter constructs (right). Depicted are promoters, PEXEL regions (green, yellow, blue), the PEXEL motif (line), and the mCherry open reading frame (red arrow). Top row, erythrocyte infected with a transgenic CSP1-70-mCherry parasite under the control of the IBIS1 promoter. Upper center row, erythrocyte infected with a transgenic CSP1-70-mCherry parasite under the control of the hsp70 promoter. Lower center row, erythrocyte infected with a transgenic IBIS1-118-mCherry parasite under the control of the IBIS1 promoter. Bottom row, erythrocyte infected with a transgenic IBIS1-90-mCherry parasite under the control of the IBIS1 promoter. Bar, 5 μm. (B–D) Immunofluorescent staining of Huh7 cells infected with the parasite lines expressing either (B) CSP1-70-mCherry, (C) IBIS111-118-mCherry, or (D) CSP-PEXEL-mCherry under the control of the promoter indicated and at the indicated time points after sporozoite infections. Shown are α-mCherry signals (top row) and merge images (bottom row) of α-mCherry (red), α-UIS4 (green), and Hoechst 33,342 (blue) stain. Bar, 10 µm.
We wanted to determine the fate of the two fusion proteins, CSP1-70-mCherry and IBIS1-118-mCherry, that were exported during blood infection, in liver stages. When hepatoma cells were infected with the lines described above that express these proteins from the IBIS1 promoter, mCherry remained confined within the liver-stage PVM (Figures 1B,C). Because IBIS1 is not expressed until the mid-liver stage (Ingmundson et al., 2012), we generated an additional transgenic line in which CSP1-70-mCherry is under the control of the UIS4 promoter to examine the localization of the reporter early in the pre-erythrocytic stage (Silvie et al., 2014). Fluorescent imaging of hepatoma cells infected with these transgenic sporozoites revealed expression of mCherry as early as 2 h after infection and throughout liver stage maturation up until 48 h later, but the signal remained within the membranes of the parasitophorous vacuole and associated tubovesicular network (Figure 1D).
Together, PEXEL-dependent export of reporter constructs was readily detected in blood stages, but liver stage export was not observed.
To test whether PEXEL-dependent protein export in P. berghei liver stages is limited by the absence of the PTEX component HSP101, we next generated transgenic parasite lines that express HSP101 using two complementary strategies, i) by insertion of an additional HSP101 copy under the control of a constitutively active promoter, and ii) by swapping the endogenous promoter with a constitutive promoter. Both strategies are predicted to allow full reconstitution of a functional PTEX in liver stages, since PTEX150, EXP2, and PTEX88 are expressed during liver stage maturation (Matz et al., 2015).
Plasmids for P. berghei transfection were assembled in the pBAT and the pBART-SIL6 vector (Kooij et al., 2012). These targeting vectors harbor the drug-selectable hDHFR-yFcu cassette for positive/negative selection and a GFP expression cassette. The latter is used to acquire isogenic lines by fluorescence-activated cell sorting (FACS) and for live imaging of the parasite cytoplasm during the entire parasite life cycle. The pBART-SIL6 vector also contains two homologous regions to integrate into a silent region of chromosome 6 (SIL6) (Supplementary Figure S2).
We generated two targeting plasmids, which express an additional copy P. berghei HSP101 (PbANKA_0931200) under the control of the constitutive promoter of elongation factor 1α (ef1α, PBANKA_113330) either as a fusion protein with a triple Myc tag only (ef1α:HSP101-myc) or an additional mCherry tag (ef1α:HSP101-mCherry) (Supplementary Figure S3A). Upon transfection and positive selection transgenic parasites were readily obtained and labeled ef1α:HSP101-myc or -mCherry, respectively. As a complementary approach, we replaced the endogenous promoter with the promoter of PTEX150 (PBANKA_1008500), which is expressed during liver stage development (Matz et al., 2015) and is expected to permit the appropriate temporal expression of HSP101 (Supplementary Figure S3C). The corresponding parasite line, which was selected after transfection was labeled HSP101:5′ptex150 to denote the promoter swap and retainment of HSP101 at the original locus. For all three parasite lines the desired integration was confirmed by diagnostic PCR, demonstrating 5′ and 3′ integration and absence of WT parasites (Supplementary Figures S3B,D). Successful generation of the two ef1α:HSP101 parasite lines and HSP101:5′ptex150 parasites already indicated that additional expression from a second copy or exchange of the endogenous HSP101 does not interfere with parasite blood stage development. HSP101:5′ptex150 parasites harbor the endogenous HSP101 copy under the ptex150 promoter. As correct PTEX assembly is vital for parasite survival (Matthews et al., 2013; Matz et al., 2013; Beck et al., 2014; Elsworth et al., 2014), the ability to generate and grow HSP101:5′ptex150 parasites indicated that correct PTEX assembly was achieved with HSP101 expressed under the ptex150 promoter. Hence, we could characterize life cycle progression and analyze protein export during parasite maturation in the liver.
We next examined life cycle progression of the transgenic parasite lines. ef1α:HSP101-mCherry blood stages developed normally and displayed an mCherry signal consistent with PV localization (Supplementary Figures S4A,B), indicative of proper localization of the HSP101-mCherry fusion protein. Female Anopheles stephensi mosquitoes were allowed to feed on mice infected with ef1α:HSP101-mCherry parasites. Successful development in the mosquito was determined on day 10 for oocysts, day 14 for midgut sporozoites and day 17 for salivary gland sporozoite formation and results were compared to P. berghei WT parasites (bergreen). ef1α:HSP101-mCherry-infected mosquitoes showed normal oocyst and sporozoite development, and HSP101-mCherry was moderately expressed in ef1α:HSP101-mCherry oocysts and sporozoites (Supplementary Figure S4C). Midgut infectivity and sporozoite numbers in freshly dissected salivary glands of all three transgenic lines were similar to WT (Supplementary Table S1).
Overall, transgenic parasites underwent normal life cycle progression enabling analysis of liver stage development and protein export.
We first examined whether ef1α:HSP101-mCherry and HSP101:5′ptex150 sporozoites are able to undergo pre-erythrocytic development and establish a patent blood infection. Groups of C57BL/6 mice were infected intravenously with 10,000 of WT, ef1α:HSP101-mCherry or HSP101:5′ptex150 sporozoites and monitored for the time to occurrence of the first blood stage parasite, termed pre-patency, and parasite growth dynamics (Figure 2). We noticed a slight delay of approximately 1 day in pre-patency in mice infected with ef1α:HSP101-mCherry parasites in comparison to WT (Figure 2A). The delayed onset of blood infection was clearly visible in the analysis of blood stage propagation (Figure 2C).
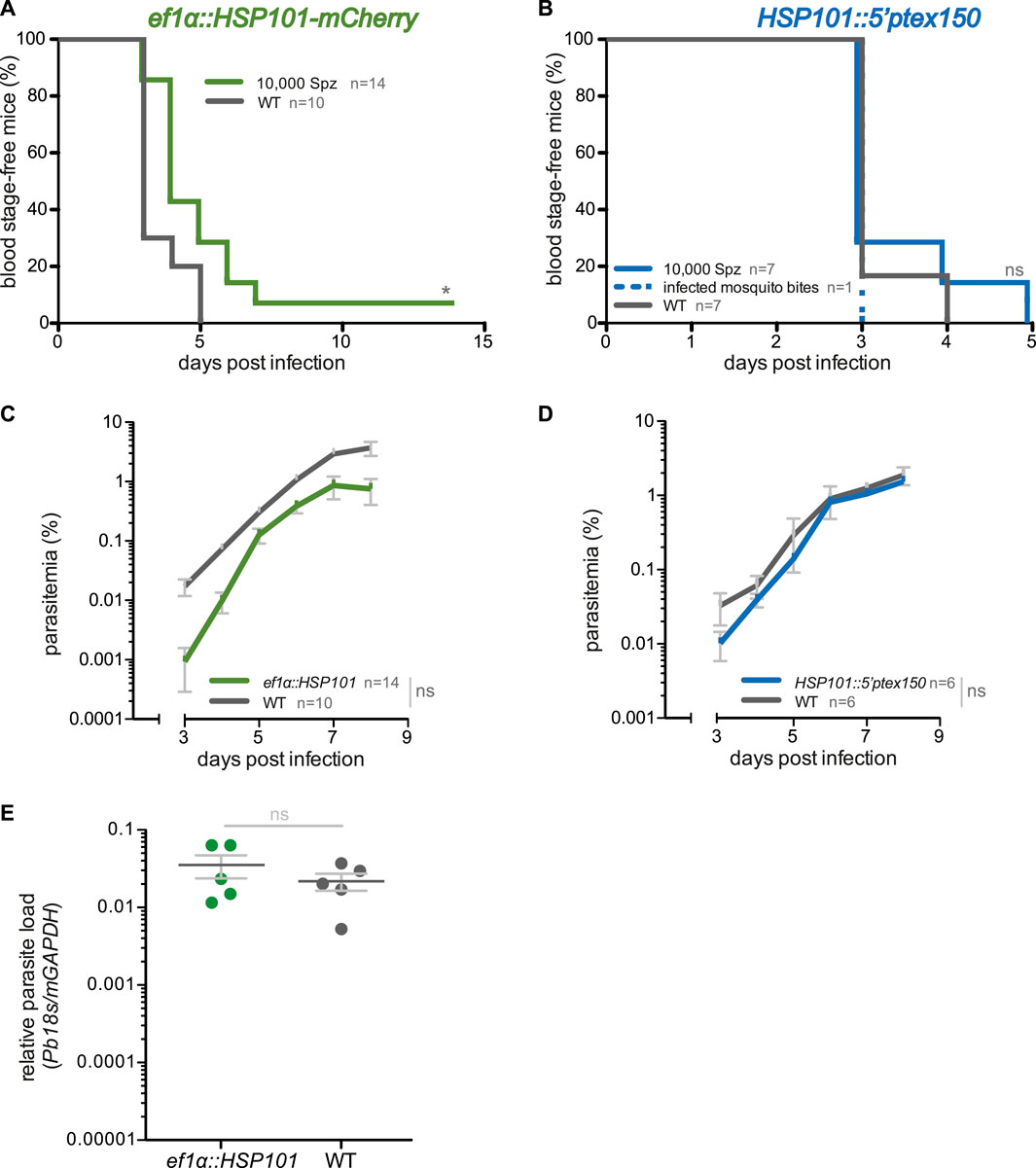
FIGURE 2. Blood infections induced by ef1α:HSP101-mCherry and hsp101:5′ptex150 sporozoites. (A,B) Shown are Kaplan-Meier analyses of blood infection over time after intravenous injection of 10,000 sporozoites isolated from (A) ef1α:HSP101-mcherry or (B) hsp101:5′ptex150-infected Anopheles stephensi mosquitoes. Exposure to 15 hsp101:5′ptex150-infected mosquitoes is included in (B). (C,D) Blood stage growth in (C) ef1α:HSP101-mCherry or (D) hsp101:5′ptex150-infected mice. (E) qPCR analysis of relative parasite liver load 48 h after sporozoite infection. Shown are mean values (±SD). Blood infection was monitored daily by microscopic examination of Giemsa-stained blood films. *, p < 0.05; n. s., non-significant (Log rank test for Kaplan-Meier analysis and slope analysis; Mann-Whitney-U test for relative parasite liver load).
We independently confirmed this finding by intravenous injection of the second transgenic line, ef1α:HSP101-myc, which also displayed a 1 day delay in patency (Supplementary Figure S5). But exposure to 15 ef1α:HSP101-myc-infected mosquitoes resulted in a normal prepatent period of 3 days (Supplementary Figure S5). This finding prompted us to quantify the parasite RNA levels in livers infected with mature liver stages, i.e. 48 h after sporozoite inoculation (Figure 2E). In this analysis, no significant differences were detectable between ef1α:HSP101-mCherry and WT-infected mice, indicating that the observed delay is not due to slower liver stage maturation. Remarkably, and in contrast to ef1α:HSP101-mCherry infections, there was no difference in HSP101:5′ptex150 sporozoites parasite growth as compared to WT (Figures 2B,D).
We infected hepatoma cells with freshly dissected salivary gland sporozoites and monitored liver stage size, morphology, HSP101 expression, and numbers of infected cells (Figure 3). The sizes of liver stages were similar for all transgenic lines and WT parasites. In ef1α:HSP101-mCherry parasites successful expression of HSP101 could be detected, using the red mCherry and myc tags as indicator. HSP101-mCherry localized to the liver stage PVM (Figure 3A). This localization was independently confirmed in hepatoma cells infected with ef1α:HSP101-myc sporozoites (Figure 3B). For HSP101:5′ptex150 parasites growth was monitored by immunofluorescence analysis (Figure 3C). All three transgenic lines showed similar growth rates compared to WT (Figure 3).
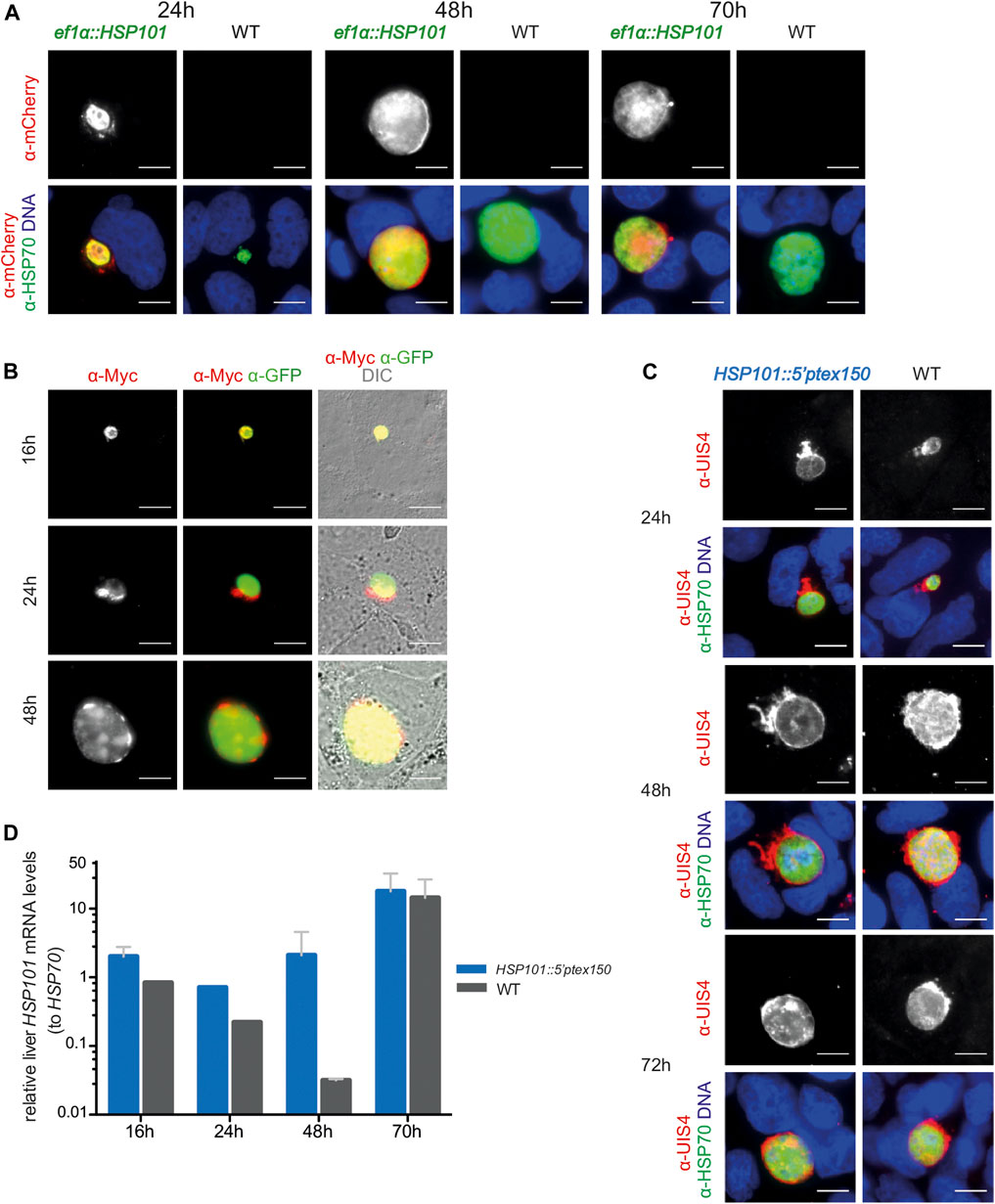
FIGURE 3. Ectopic expression of HSP101 in liver stages results in PV localization. (A) Localization of HSP101-mCherry in ef1α:HSP101-mCherry-infected hepatoma cells at 24, 48, and 72 h after infection. Shown are immunofluorescent images with α-mCherry antibody (top row) and merge images (bottom row) displaying the signals of α-mCherry (red), α-HSP70 (green), and Hoechst 33,342 (blue). Bars, 10 µm. (B) Localization of HSP101-myc in ef1α:HSP101-myc-infected hepatoma cells at 16, 24, 48 h after infection. Shown are immunofluorescent images with an α-myc antibody (left column), α-myc (red) and α-GFP (green) antibodies (center column), and merge images (right column) displaying the signals of α-myc (red), α-GFP (green), and DIC. Bars, 10 µm. (C) Liver stage development of hsp101:5′ptex150 parasites at 24, 48, and 72 h after infection. Shown are immunofluorescent images with an α-UIS4 antibody (top), and merge images (bottom) displaying the signals of α-UIS4 (red), α-HSP70 (green), and Hoechst 33,342 (blue). Bars, 10 µm. (D) Steady state HSP101 mRNA levels in hsp101:5′ptex150 liver stages. Shown are relative mRNA levels determined qRT-PCR from total RNA of hsp101:5′ptex150 or WT-infected hepatoma cells at indicated time points after infection. ∆∆CT values of HSP101 mRNA values were normalized to HSP70. Depicted are mean values (±SD) (n = 2, except for 24h, n = 1).
In contrast to ef1α:HSP101-mCherry parasites, HSP101 expression in HSP101:5′ptex150 could not be verified by immunofluorescence, since the endogenous open reading frame was retained. As a proxy for expression, we determined steady-state HSP101 mRNA levels (Figure 3D). As intended, HSP101 mRNA levels were significantly elevated at 24 and 48 h in HSP101:5′ptex150 liver stages.
In conclusion, gain of HSP101 expression resulted in successful liver stage development and HSP101 localization to the PVM in infected liver cells. While onset of blood infection was delayed in ef1α:HSP101-mCherry parasites, HSP101:5′ptex150 infections developed parasite infections indistinguishable from WT.
To determine whether ef1α:HSP101-mCherry or HSP101:5′ptex150 parasites display export of PEXEL-containing proteins during early liver stage development, we performed genetic crosses of the three transgenic parasite lines with four parasite lines that express reporter proteins (Table 1). Three of the lines express reporter proteins that were successfully exported during blood infection, namely uis4:CSP1-70-mCherry, uis4:IBIS1-118-mCherry, and ibis1:IBIS1-118-mCherry (Figure 1), and one line, uis4:CSP1-70-OVA, expresses the model antigen ovalbumin fused to the predicted CSP PEXEL motif (Montagna et al., 2014), which is also predicted to be exported in the blood stage. Genetic crosses were obtained by feeding Anopheles stephensi mosquitoes on mice, which were double infected with either ef1α:HSP101-myc or HSP101:5′ptex150 parasites and the export reporter line (Supplementary Figure S6). Sporozoites were then isolated for hepatoma cell infection.

TABLE 1. Export ability of reporter lines in transgenic HSP101-expressing liver stages.
After sexual recombination in the brief phase of the diploid/tetraploid zygote of the otherwise haploid parasite life cycle a quarter of the mixed sporozoite population carries both desired genetic modification, since they are coded on different chromosomes. They can be directly visualized by fluorescent imaging, with GFP only in HSP101 expressing parasites and mCherry or ovalbumin in the reporter constructs. We evaluated protein export in liver stages by both, live imaging and after antibody staining. As exemplified for one cross, ef1α:HSP101-myc and UIS4:CSP1-70-mCherry, the tagged protein stayed confined to the PV at all time points during liver stage maturation, similar to the reporter line in a WT background (Figure 4A). We systematically determined protein export in all nine crosses and noted similar observations of no indication for export into the host hepatocyte. We independently tested the distribution of CSP (Singh, et al., 2007; Cockburn, et al., 2011) by antibody staining. Even in the presence of HSP101 we could not capture CSP signal outside the area of the ef1α:HSP101-myc parasite (Figure 4B).
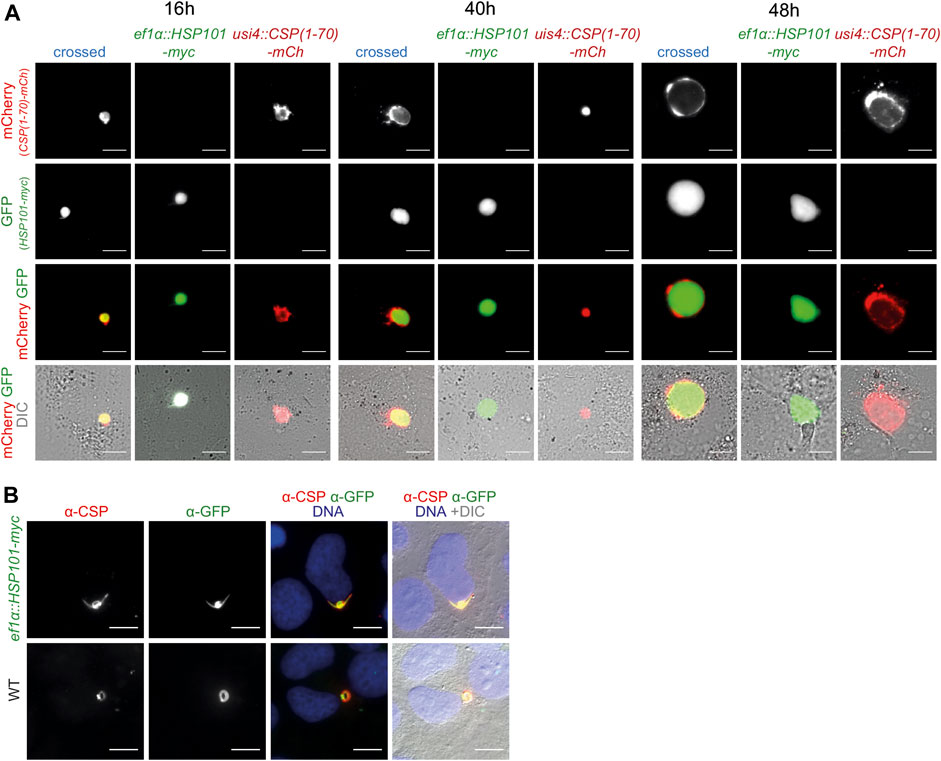
FIGURE 4. Ecctopic expression of HSP101 in liver stages results in PV localization. (A) Live imaging of hepatoma cells infected with sporozoites from a genetic cross of ef1α:HSP101-myc and UIS4:CSP-PEXEL-mCherry (crossed, blue), and the two individual strains, ef1α:HSP101-myc (green), UIS4:CSP-PEXEL-mCherry (red). Shown are the PEXEL model protein fused to mCherry (top row, red), cytoplasmic GFP of the ef1α:HSP101-myc parasite (upper center row, green), the merge of both signals (lower center row), and a merge of both fluorescent signals with DIC images (bottom row). Note that the mCherry-tagged model antigen remains confined to the PV in HSP101-expressing parasites. Bars, 10 µm. (B) Immunofluorescence analysis of CSP in ef1α:HSP101-myc (top row) and WT (bottom row) parasites, 4 h after infection. Shown are the signals of α-CSP (left) and α-GFP (centre left), the merge of both signals together with Hoechst 33,342 (blue; centre right), and a merge of all fluorescent signals with DIC images (right). Bars, 10 µm.
Thus, protein export could not be induced in ef1α:HSP101-myc or HSP101:5′ptex150 parasites, despite a range of PEXEL sequences and assessment of different time points during liver stage maturation. Accordingly, HSP101 is not a limiting factor for PEXEL-dependent protein export during this life cycle stage.
We finally wanted to explore whether gain of HSP101 function modulated vaccine-induced protection. One hypothesis is that a larger repertoire of yet unidentified parasite antigens might increase immunogenicity during liver stage development, and processing of exported parasite proteins might be an important contributing factor. To test this notion, protective efficacy of immunization with live attenuated ef1α:HSP101-mCherry or HSP101:5′ptex150 parasites was compared to WT immunizations (Figure 5). Groups of C57BL/6 mice received a prime/boost vaccination scheme of 10,000 sporozoites, followed by azithromycin treatment to elicit late liver stage arrest (Friesen et al., 2010). Challenge was done 3 months later by intravenous injection of 10,000 WT sporozoites (Figure 5A). This protocol has consistently shown incomplete sterile protection permitting evaluation of superior or inferior vaccination schemes (Friesen and Matuschewski, 2011). ef1α:HSP101-mCherry and WT sporozoite -immunized mice displayed substantial sterile protection, with three and four out of five mice, respectively, blood infection-free (Figure 5B). In contrast, only two out of 10 HSP101:5′ptex150 -immunized mice were protected in comparison to seven out of 10 in WT-immunized mice (Figure 5C). Protection in HSP101:5′ptex150 -immunized mice under azithromycin cover vanished completely when challenged at a late time point, 260 days after immunization (Figure 5D).
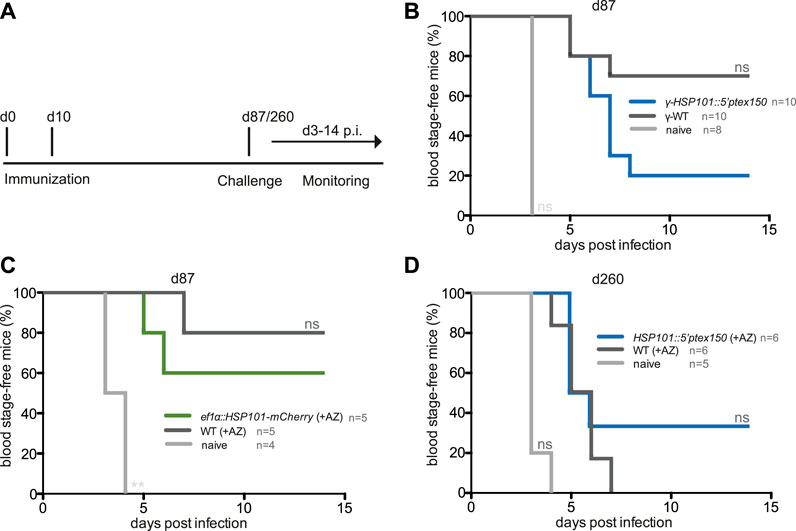
FIGURE 5. Inferior protection in mice vaccinated with attenuated ef1α:HSP101-mCherry or HSP101:5′ptex150 sporozoites. (A) Overview of immunization and challenge protocol. C57bl/6 mice were immunized twice at a 10-days interval with 10,000 intravenously injected (i.v.) sporozoites. Challenge infections with 10,000 i. v. WT sporozoites was done at indicated time points. Monitoring of blood infection was done daily by microscopic examination of Giemsa-stained blood films. (B–D) Kaplan-Meier analysis of blood infection over time after intravenous sporozoite injection. (B) Immunizations with irradiated HSP101:5′ptex150 (blue) and WT (black) sporozoites. (C) Immunizations with azithromycin-arrested ef1α:HSP101-mCherry (green) and WT (black) sporozoites. (B,C) Challenge infections were done on day 87 (77 days after the booster immunization). (D) Immunizations with azithromycin-arrested HSP101:5′ptex150 (blue) and WT (black) sporozoites. Challenge infections were done on day 260 (250 days after the booster immunization). Naive mice (grey) served as infection controls in all experiments. n. s, non-significant (Log rank (Mantel-Cox) test).
Collectively, immunizations with live ef1α:HSP101-mCherry or HSP101:5′ptex150 sporozoites followed by attenuation with azithromycin does not lead to superior protection, but instead results in reduced vaccine efficacy.
In this study, we systematically analyzed PEXEL-dependent protein export during liver stage maturation and determined whether HSP101 is the limiting factor for the apparent paucity of secreted proteins. We generated and investigated transgenic Plasmodium berghei parasite lines that express fluorescent reporter proteins or HSP101 in liver stages, and, by genetic crosses, both simultaneously. We could show that reporter proteins, which are exported to the erythrocyte cytoplasm, fail to cross the membranes of the parasitophorous vacuole and tubovesicular network during liver stage development. For instance, P. berghei IBIS1 contains a conventional PEXEL motif that mediates export in Plasmodium falciparum blood stages (Petersen et al., 2015), but this is apparently not efficiently recognized for protein export in liver stages. Accordingly, any protein export occurring in the liver stages is not analogous to protein export in the blood infection stage.
We also analyzed an unconventional PEXEL sequence, of the sporozoite surface antigen CSP. However, when mCherry was linked to this motif the corresponding fusion protein remained confined to the PV of developing liver stages, although it was efficiently targeted to host erythrocytes during blood infection. Our data corroborate the notion that CSP is constantly shed during sporozoite transmigration, but not actively exported after conversion to liver stage development (Cockburn, et al., 2011), and refute the suggestion that CSP is exported to the host nucleus to reprogram the infected hepatocyte (Singh et al., 2007). As of now, LISP2 and SLTriP remain the only liver stage proteins reported to translocate during mid-liver stage development. Carboxy-terminal mCherry tagging of LISP2 leads to accumulation of mCherry in the PV, and it was suggested that carboxy-terminal processing of LISP2 is required for export (Orito, et al., 2013). Signatures of exported but PEXEL-negative proteins remain poorly understood (Spielmann and Gilberger 2010), and in perspective, discovery of liver stage-specific export signals might help to define the liver stage exportome.
In other pathogens, protein translocons fail to export larger, stably folded proteins, therefore limiting translocation. For instance, bacterial type III secretion systems fail to translocate stably folded fluorescent proteins (Akeda and Galan 2005). In this study, reporter constructs harbored a PEXEL linked to mCherry, which is a relatively packed and large (29 kDa) protein, potentially limiting export of the reporter constructs in the liver stage in spite of efficient export in the blood. However, the model antigen OVA can be translocated by bacterial type III secretion systems (Wiedig et al., 2005), and is also not detected outside the liver stage PV when fused to the region of CSP that drives protein translocation across the blood stage PVM. Tagging with fluorescent proteins does not appear to limit protein translocation in the blood, and export of the mCherry reporters we used in our study was consistently detected in asexual blood stages. Translocation of fluorescent proteins in the blood stage requires functional HSP101 for protein unfolding (Matthews et al., 2019b), so the lack of mCherry export in the liver in the absence of HSP101 is perhaps expected. However, we were able to demonstrate that expression of untagged HSP101 in the liver stage from the PTEX150 promoter still did not allow liver-stage parasites to export the mCherry reporter constructs. Clearly, more work is warranted to decipher protein export by liver stage Plasmodium. To this end, smaller tags that permit immunodetection might further minimize constraints by protein folding.
Plasmodium is an obligate intracellular pathogen and has to avoid immune recognition inside the only nucleated host cell, the hepatocyte, during the first population expansion phase in the liver. Parasite replication inside a PV is a powerful shield to minimize antigen exposure at the surface of the infected cell. The corresponding trade-offs are nutrient depletion and waste accumulation. During blood infection extensive erythrocyte remodeling poses no danger, since these terminally differentiated host cells are devoid of MHC class I -dependent antigen presentation. Previous studies have proposed a concept of regional PV export protein storage in asexual blood stages (Bullen, et al., 2012). These export areas are proposed to localize to specific bulges in the PV, which are defined by PTEX assembly, which in turn might be initiated by EXP2 localization determined by host cell factors (McMillan et al., 2013; Meibalan et al., 2015; de Koning-Ward et al., 2016). Proteins resistant to unfolding get trapped in loop-like PVM extensions in blood stages, and unblocking the unfolding capacity only partially recovers protein export, as cargo remains spatially segregated from the export machinery (Charnaud et al., 2018). Although our results indicate localization of cargo proteins in the PV, previous studies propose that proteins need to remain in defined export zones for efficient translocation, which might be determined by yet unknown proteins. PV-localized accessory proteins are involved in the export of proteins in blood stages (Elsworth et al., 2016; Batinovic et al., 2017). Whether this complex system relies on further accessory proteins, thereby raising the question whether these proteins are present in liver stages, remains to be determined.
Proper PTEX assembly plays a major role in effective protein export. Despite introducing HSP101 expression in liver stages to facilitate PTEX assembly, the mCherry signal of reporter constructs were confined to the parasite PV and not detectable in the hepatocyte cytoplasm. Thus, we conclude that transgenic parasites were not able to recreate the PEXEL-dependent export machinery in liver stages. Although expression of all core components was induced in the transgenic parasites, we were unable to confirm proper assembly of the complex in liver stages. Carboxy-terminal truncation of PTEX150, the linker between the pore spanning EXP2 and the AAA+ ATPase HSP101, led to decrease in quantity of other PTEX components (Elsworth et al., 2016), suggesting complex destabilization. In this study, HSP101 was carboxy-terminally tagged, possibly interfering with correct protein folding. Indeed, recent structural analysis of PTEX complex assembly revealed stable binding of the EXP2 protomer to the carboxy-terminal ends of the HSP101 protomer, thereby placing the HSP101 cargo tunnel directly on the top of the EXP2-PTEX150 pore (Ho, et al., 2018). This suggests that interfering with the HSP101 carboxy-terminus, and as a consequence, with proper protein folding may prevent PTEX assembly; incomplete or unstable complex assembly has been proposed to inhibit protein translocon activity (de Koning-Ward, et al., 2016). Tagging HSP101 with mCherry has been shown to disrupt its function (Matthews et al., 2019b). Specifically, C-terminal mCherry tagging has been shown to interfere with unfolding of soluble translocated proteins and to interfere with parasite growth in the blood. Our findings of reduced growth of the ef1α:HSP101-mCherry are consistent with these findings. A smaller tag was well tolerated in this study (Matthews et al., 2019b). We note that we also expressed not only a myc-tagged HSP101, but also a native HSP101 protein in our promoter swap approach. Since these parasites displayed similar paucity of protein export, we consider dysfunctional PTEX assembly due to their carboxy-terminal tags less likely.
In vitro infection of hepatoma cells with ef1α:HSP101-mCherry parasites revealed HSP101 expression. However, the mCherry tag signal in the PV was weak, questioning if reconstruction of the machinery is feasible. As HSP101:5′ptxe150 parasites lack a tagged version of HSP101, mRNA levels confirmed increased liver HSP101 transcription compared to WT parasites, but whether HSP101 localizes to the PV remains to be determined. While we cannot rule out the possibility of post-translational repression, we believe this is unlikely to be the case because the regulatory regions used are well characterized, and visualization of HSP101-mCherry in early liver stages was possible in ef1α:HSP101-mCherry parasites. In contrast, endogenous HSP101 is controlled at the transcriptional level in this stage; however, low mRNA levels were detectable in WT parasites, whereas HSP101 protein is absent in early liver stages (Matz et al., 2015), raising the questions whether endogenous HSP101 is post-transcriptionally repressed.
It remains to be determined if further, yet unidentified, parasite and host proteins can contribute to protein export in liver stages. The core components EXP2, PTEX150 and HSP101 are essential for PTEX assembly and blood stage parasite survival (de Koning-Ward et al., 2009; Matthews et al., 2013; Matz et al., 2013). Previous work established that decreased HSP101 levels blocked export and led to vesicular accumulation of proteins in blood stages (Beck et al., 2014; Elsworth et al., 2014). Ho et al. indicate that the stoichiometry of 6:7:7 for HSP101-PTEX150-EXP2 is a prerequisite for functional PTEX assembly (Ho et al., 2018). Presently, it remains unknown whether the ef1α promoter or the promoter swap with the ptex150 promoter yields sufficient protein. This can be experimentally tested by placing HSP101 under the control of a strong pre-erythrocytic promoter.
In vivo infections demonstrated half a day delay in pre-patency for ef1α:HSP101-mCherry and HSP101:5′ptex150. Similarly, subsequent blood stage growth was delayed and parasitemia remained lower, however slope analysis demonstrated no significant differences in growth rate. Thus, it can be postulated that intra-hepatic growth is slightly affected, whereas blood stage development remains unaffected by HSP101 overexpression. This notion is strengthened by parasite loads of infected mouse livers, which were slightly elevated at 48 h after infection in ef1α:HSP101-mCherry compared to WT -infected cohorts. As budding of merozoite-filled merosomes initiates as early as 48 h after infection in WT parasites (Sturm et al., 2006), slightly elevated parasite loads in ef1α:HSP101-mCherry infected mice could indicate a delay in maturation and budding of merosomes leading to the observed delay in pre-patency. Whether this is actually caused by HSP101 overexpression itself or subsequent up- or downregulation or obstruction of other liver stage proteins remains to be determined.
To address PEXEL-dependent export in liver stages it is of interest to determine PTEX assembly with the additional HSP101 copy in engineered parasites. Lack of P. berghei-specific PTEX component antibodies prevented confirmation of PTEX assembly with immunoprecipitation. However, despite the lack of PTEX assembly verification, we observed localization of HSP101 to the PV of the parasite in liver stages and could confirm elevated HSP101 mRNA levels in early liver stages. Additionally, PTEX assembly is vital for parasite survival in blood stages, therefore, a functional PTEX complex and correct HSP101 positioning in the complex is present in the promoter swap parasites HSP101:5′ptex. Localization of the tagged HSP101 copy in ef1a:HSP101-mCherry parasites was observed in the cytoplasm and a more prominent stain in the PV of the parasite. We note that the bulky mCherry tag could interfere with the correct localization of HSP101. This might affect PTEX assembly in parasites harboring an additional copy of HSP101.
Taking advantage of pathogen stage conversion for immunization strategies is one way to attack the parasite at its developmental bottleneck (Kreutzfeld et al., 2017). Studies have shown that superior immunity can be achieved by a developmental arrest shortly before liver-to-blood stage conversion, as a larger and broader array of antigens is presented compared to early arresting GAPs (Butler et al., 2011; Haussig et al., 2011). Therefore, we performed immunizations with azithromycin attenuated late arresting liver stages and challenged vaccinated animals to explore whether restoring HSP101 in liver stages can elicit superior immune responses. We, however, detected inferior protection, and whether this can be attributed to alteration of protein export during liver stage development needs to be explored further.
In conclusion, a functional PTEX complex, as seen in infected erythrocytes, cannot be reconstructed solely by ectopic HSP101. Our work also highlights that liver stage-host cell interactions remain relatively ill defined. The tight control of protein export during liver development likely contributes to poor recognition and cytolysis by the host immune system. Ultimately, identifying candidate liver stage-specific protein export signals, defining the contribution of PTEX in liver stage protein export, and characterizing the liver stage exportome by proteomic approaches are key to a better mechanistic understanding of immunity and vaccine development against Plasmodium pre-erythrocytic stages.
All animal work was conducted in accordance with the German “Tierschutzgesetz in der Fassung vom 18. Mai 2006 (BGBl. I S. 1207)”, which implements the directive 86/609/EEC from the European Union and the European Convention for the protection of vertebrate animals used for experimental and other scientific purposes. The ethics committee of MPI-IB and the Berlin state authorities (LAGeSo Reg# G0469/09 and G0294/15) approved the protocol.
P. berghei ANKA cl507 parasites that constitutively express GFP under the control of the EF1α promoter (Franke-Fayard et al., 2004) and P. berghei ANKA bergreen parasites that constitutively express GFP under the control of the HSP70 promoter (Matz et al., 2013) were used in our experiments. 6–8 weeks old female NMRI or C57BL/6 mice used in this study were either purchased from Charles River Laboratories, Janvier, or bred in-house. NMRI mice were used for transfection experiments, blood stage infections and transmission to Anopheles stephensi mosquitos. C57BL/6 mice were used for sporozoite infections and subsequent immunological studies.
To express an additional copy of PbHSP101 (PbANKA_0931200) under a constitutive promoter the expression cassette was inserted into a silent locus on chromosome 6, using the pBART-SIL6 vector (Kooij et al., 2012). This plasmid harbors a drug selectable hDHFR-yFcu cassette for positive/negative selection, a GFP under the PbHSP70 promoter for parasite life cycle analysis, an mCherry/triple c-Myc (3xMyc) tag sequence for protein tagging under the control of the 3′ region of PbPPPK-DHPS for mRNA stability, and two homologous sequences for stable integration into the silent locus on chromosome 6. The HSP101 open reading frame was amplified using primers HSP101for and HSP101rev (Supplementary Table S2). The fragment was inserted via XbaI and AgeI 5′ to the mCherry/3xMyc tag. For the plasmids EF1α:HSP101-mCherry and EF1α:HSP101-myc the 5′ region of EF1α (PbANKA_113330) was amplified using primers EF1for and EF1rev (Supplementary Table S2). The fragment was inserted via BssHII and XbaI 5′ next to the HSP101 open reading frame. mCherry was excised in the plasmids EF1α:HSP101-mCherry, and replaced by a linker sequence, generated with Linkfor und Linkrev (Supplementary Table S2), fusing the HSP101 open reading frame and the 3xMyc tag (Supplementary Table S2). For swap of the endogenous promoter with the 5′ region of PTEX150 a pBART-based plasmid containing the PTEX150 promoter linking HSP101 ORF was generated. In brief, the amino-terminal portion of the HSP101 ORF (primers HSP101_amino_F, HSP101_amino_R in Supplementary Table S2) was integrated under the control of PTEX150 promoter (primers PTEX150-5′-PrimerF, PTEX150-5′-PrimerR in Supplementary Table S2) integrated with SacII and HpaI. The 5′UTR of HSP101 amplified with HSP101-5′-F and HSP101-5′-R (Supplementary Table S2). The 5′UTR together with the HSP101 amino-terminus served as homologous recombination sites (Supplementary Figure S3).
The mCherry-based export reporters were constructed in the B3D+mCherry vector (Silvie et al., 2008b). The sequence encoding the first 70 amino acids of CSP was amplified with CSPfor and CSP70rev (Supplementary Table S2) and cloned into the XbaI and SpeI sites of B3D+mCherry, and the 5′ genomic regions directly upstream of UIS4, HSP70 and IBIS1 were amplified with UIS4-5′for and UIS4-5′rev, HSP70-5′for and HSP70-5′rev, and IBIS1-5′for and IBIS1-5′rev (Supplementary Table S2), respectively, and inserted into the SacII and NotI sites. Plasmids were linearized with BsmI, HpaI or PacI, respectively, prior to transfection. IBIS11-90 and IBIS11-118 were amplified together with the IBIS1 5′ region using primers IBIS1-5′for and IBIS190rev or IBIS1118rev (Supplementary Table S2), respectively and inserted into the SacII and SpeI sites of B3D + mCherry (Supplementary Figure S2). The IBIS1 plasmids were linearized with PacI prior to transfection.
In order to generate stable transgenic parasite lines, NMRI mice were infected with 100 μl thawed cryopreserved P.b. ANKA blood stage parasites peritoneal and parasites were grown until a parasitemia of ∼3–5%. Parasite culture and schizont isolation were done as described (Janse et al., 2006). Schizonts were transfected with 5–10 μg ApaLI linearized plasmids using the AMAXA Nucleofector device (program U33). Electroporated parasites were mixed with additional 50 μl transfection medium and intravenously injected into NMRI mice. The linearized ef1α:HSP101-mCherry and ef1α:HSP101-myc plasmids integrated in the silent locus in chromosome 6 (Supplementary Figure S3), and positive recombination events were selected with pyrimethamine (70 mg/L) in the drinking water, followed by FACS of GFP-positive iRBCs to generate isogenic transgenic parasite lines. The linearized hsp101:5′ptex150 plasmid integrated between the endogenous HSP101 5′UTR and ORF resulting in a new, constitutively active promoter for HSP101 expression (Supplementary Figure S3C). Recombinant hsp101:5′ptex150 were selected by pyrimethamine, and an isogenic line was established by flowcytometric (FACS) cloning as described (Kenthirapalan et al., 2012). To verify successful integration, transgenic parasites DNA was extracted from blood stage cultures, and genotyping was performed with oligonucleotides specific for 5′ and 3′ integration (Supplementary Table S2) as well as for the WT locus (Supplementary Table S2, Supplementary Figure S3B+D).
Parasite blood stages were visualized in immunofluorescence assays (IFA). Infected RBCs were retrieved by tail vein punctures of infected mice, mixed with 1x PBS and placed on a poly-l-lysine covered 12 mm ø cover slips in a 24-well plate to settle. Cells were fixed with 4% PFA/0.0075% fresh glutaraldehyde (GA) at RT. After three washing steps cells were permeabilized with 0.2% Trition X-100 (in PBS) for 20 min at RT. Cells were washed and blocked with 3% BSA, 0.2% Triton X-100 (in PBS) for 30 min at RT on a shaker. Primary antibody was added in blocking solution for 1 hour at RT, followed by three washing steps and incubation with fluorophore labeled secondary antibody and Hoechst 33,342 in blocking solution for 1 h at RT. Additional washing steps removed remaining secondary antibody before cells were mounted with Fluoromount G on a glass slide. Live imaging of iRBC was performed to access expression of mCherry and GFP as well as adequate growth of the parasites. iRBCs were placed on a glass slide and covered with a cover slip. Cells were analyzed with an Zeiss Axio Observer immunofluorescence microscope, and images were taken at 630x magnification.
WT GFP-expressing P. berghei ANKA and transgenic strains were maintained by continuous cycling in NMRI mice and female Anopheles stephensi mosquitoes (Vanderberg, 1975). Mosquitoes were kept at 28°C (non-infected) or 20°C (infected) at 80% humidity. 10–14 days after mosquito infection, midguts were isolated and oocyst development analyzed by fluorescent microscopy. Salivary gland sporozoites were isolated on day 17 post infection from mosquitos in DMEM medium containing 10% fetal calf serum (FCS).
Salivary gland sporozoites were visualized by immunofluorescence assay (IFA). Briefly, 10,000 salivary gland sporozoites suspended in 3% BSA-RPMI were added into each ring of a Medco glass slide pre-coated with 3% BSA-RPMI and incubated for 15 min at 37°C. Slides were fixed with 4% paraformaldehyde (PFA), permeabilized with 0.2% TritonX-100 and blocked with 3% BSA. Sporozoites were stained with chicken anti-GFP antibody, followed by anti-chicken Alexa Fluor 488-coupled antibody (Invitrogen, OR, United States), rat anti mCherry antibody followed by anti-rat Alexa Fluor 546-coupled antibody (Invitrogen, OR, United States) and the nuclear stain Hoechst 33,342 (1:5,000; Invitrogen, OR, United States). Slides were mounted with Fluoromount-G (SouthernBiotech) prior to analysis by fluorescence microscopy using a Zeiss Axio Observer immunofluorescence microscope, and images were taken at 630x magnification.
For analysis of pre-patency and blood stage growth after sporozoite infection, 10,000 WT or transgenic sporozoites were injected intravenously into to C57BL/6 mice (n = 5 each). Tail punctures for blood smears were performed daily from day 3 after infection onwards until day 14. Smears were stained with a 10% Giemsa solution and analyzed by microscopic examination for presence and percentage of blood stage parasites. For natural mosquito infections 15 infected mosquitoes were kept in a separate container allowing a targeted blood meal on one C57BL/6 mouse. Mice were anesthetized with ketamine/xylazine intraperitoneal (i.p.), and mosquitoes were allowed to feed for 15 min. Mosquitoes were analyzed for successful feeding by examining blood uptake in the mosquito midgut.
Groups of 5 C57BL/6 mice were infected with 10,000 WT or EF1α:HSP101 sporozoites each, and the livers were harvested 42 h later. Livers were homogenized in TriZol (Thermo Fischer), and total RNA was extracted. cDNA was generated by reverse transcription with the RETROScript Kit (Ambion). Quantitative real-time PCR (qPCR) analysis was done with primers specific for Pb18S rRNA and mouse GAPDH for normalization (Supplementary Table S2). Relative liver parasite levels were measured applying the ∆∆Ct method.
For crossing two parasite lines in the mosquito, female NMRI mice were infected intravenously (i.v.) with the parasite lines. On day 4, parasitemia was determined and five million parasites of each line were intravenously injected into one female NMRI mouse for subsequent mosquito infection. After confirmation of exflagellation of male gametes mosquitoes were allowed to take a blood meal on the infected mouse.
Cell lines were kept according to standard cell culture conditions for human hepatoma cell lines (Huh7). In brief, Huh7 cells were cultured in DMEM complete (DMEM, 10% FCS, 1% Penicillin-Streptomycin) at 37°C and 5% CO2. Nunc Lab-Tek II 8-well Chamber Slides (ThermoFischer, NY, United States) were seeded with 30,000 Huh7 cells per well 24 h before infection. 10,000 WT or transgenic salivary gland sporozoites suspended in 100 μl DMEM-complete were added, centrifuged and allowed to settle for 2 h at RT. Cells were washed 2 h later to remove non-invaded sporozoites, and then medium was changed daily. Growth was terminated at 2, 16, 24, 40, 48, 54 and 72 h after infection by fixation with 4% PFA for 10 min at RT. Infected cells were blocked with PBS/10%FCS and permeabilized with 0.2% Triton (Roth, Karlsruhe, Germany) for 1 h at 37 °C. Primary antibodies (α-GFP, α-USI4, α-mCherry, α-Myc, α-PbHSP70, and α-CSP) were added for 1 hour at 37 °C. After washing, cells were incubated with the respective fluorescently labeled secondary antibody (α-chicken Alexa Fluor 488, α-mouse Alexa Fluor 546, α-mouse Alexa Fluor 488, α-rat Alexa Fluor 546, α-rat Alexa Fluor 488, α-rabbit Alexa Fluor 546, α-rabbit Alexa Fluor 488; Invitrogen, OR, United States) and Hoechst 33,342 (Invitrogen, OR, United States) for 1 hour at 37 °C. Subsequently, cells were washed and mounted with Fluoromount G (SouthernBiotech) and imaged by fluorescence microscopy (Zeiss Axio Imager Z2) with 630x magnification.
For protein export analysis of infected Huh7 cells with crossed transgenic parasite lines, live imaging of parasites was performed. Infected cells were cultured in Ibidi slides, washed once with PBS and remained in PBS solution for the time of imaging. Live imaging was performed with a Zeiss Axio Observer immunofluorescence microscope with 1,000 magnification. Duration of analysis was kept at a minimum to ensure cell survival.
To determine expression of liver stage specific genes, relative mRNA levels were determined in WT and transgenic parasites. 300,000 Huh7 cells/well were plated in a 24-well plate. One day later, salivary gland sporozoites were isolated from infected mosquitoes and Huh7 cells were infected with 100,000 sporozoites/well. For parasite sedimentation plates were centrifuged for 10 min at 3,000 rpm, no brake, and incubated at 37 °C and 5% CO2. Two hours later, cells were washed once to remove extracellular sporozoites. After 16, 24, 48 or 72 h cells were trypsinized with 200 μl for 3 min at 37 °C, and carefully resuspended in DMEM and washed with additional DMEM. After 5 min centrifugation at 1200 rpm with brake, the pellet was resuspended in 500 μl TRIzol, and RNA was extracted for subsequent cDNA synthesis and qPCR analysis as described previously. qPCR analysis was performed with HSP101 and HSP70 primers (Supplementary Table S2).
To examine vaccine efficacy mice were immunized mice in a prime boost regimen (day 0 and day 7) with 10,000 sporozoites delivered i. v. Salivary gland sporozoites were attenuated either by a γ-irradiation dose of 12 × 104 cGy, or with azithromycin (AZ) cover (4.8 mg in 200 μl of sodium chloride solution per mouse) (Friesen et al., 2010). For challenge experiments, immunized and control mice were i.v. injected with 10,000 WT P. berghei ANKA sporozoites 87 or 260 days post immunization. All mice were checked for parasitemia by microscopical examination of Giemsa-stained blood smears starting at day 3 after challenge inoculation until day 14. Animals that remained parasitemia-free were continuously checked for parasitemia up till 45 days after the challenge.
Statistical significance was analyzed by non-parametrical Whitney-U test for non-normally distributed data (infected hepatoma cells) using GraphPad Prism V5.0c. Log rank (Mantel-Cox) test was applied for Kaplan-Meier analyses, and slope of parasite growth was analyzed using R (RStudio version 0.99.879).
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by The ethics committee of MPI-IB, Berlin.
OK, AI, KMü, and KMa designed the experiments; OK, KMü and JG performed experiments and analyzed data; OK and KMa wrote the paper; all authors commented and revised the manuscript.
This work was funded by the Deutsche Forschungsgemeinschaft through the research training group–PhD program 2046 “Parasite infections: From experimental models to natural systems” (project B1) and in part by the Max Planck Society.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Laura Golusda for her help with the mouse infections and Joachim Matz for expertise advice and help during the cloning process of the HSP101:5′ptex150 line. We acknowledge support by the German Research Foundation (DFG) and the Open Access Publication Fund of Humboldt-Universität zu Berlin.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.742153/full#supplementary-material
Akeda, Y., and Galán, J. E. (2005). Chaperone Release and Unfolding of Substrates in Type III Secretion. Nature 437, 911–915. doi:10.1038/nature03992
Akinyi, S., Hanssen, E., Meyer, E. V. S., Jiang, J., Korir, C. C., Singh, B., et al. (2012). A 95 kDa Protein of Plasmodium Vivax and P. cynomolgi Visualized by Three-Dimensional Tomography in the Caveola-Vesicle Complexes (Schüffner's Dots) of Infected Erythrocytes Is a Member of the PHIST Family. Mol. Microbiol. 84, 816–831. doi:10.1111/j.1365-2958.2012.08060.x
Aunin, E., Böhme, U., Sanderson, T., Simons, N. D., Goldberg, T. L., Ting, N., et al. (2020). Genomic and Transcriptomic Evidence for Descent from Plasmodium and Loss of Blood Schizogony in Hepatocystis Parasites from Naturally Infected Red colobus Monkeys. Plos Pathog. 16, e1008717. doi:10.1371/journal.ppat.1008717
Batinovic, S., McHugh, E., Chisholm, S. A., Matthews, K., Liu, B., Dumont, L., et al. (2017). An Exported Protein-Interacting Complex Involved in the Trafficking of Virulence Determinants in Plasmodium-Infected Erythrocytes. Nat. Commun. 8, 16044. doi:10.1038/ncomms16044
Beck, J. R., Muralidharan, V., Oksman, A., and Goldberg, D. E. (2014). PTEX Component HSP101 Mediates export of Diverse Malaria Effectors into Host Erythrocytes. Nature 511, 592–595. doi:10.1038/nature13574
Bongfen, S. E., Torgler, R., Romero, J. F., Renia, L., and Corradin, G. (2007). Plasmodium berghei-Infected Primary Hepatocytes Process and Present the Circumsporozoite Protein to Specific CD8+T Cells In Vitro. J. Immunol. 178, 7054–7063. doi:10.4049/jimmunol.178.11.7054
Bullen, H. E., Charnaud, S. C., Kalanon, M., Riglar, D. T., Dekiwadia, C., Kangwanrangsan, N., et al. (2012). Biosynthesis, Localization, and Macromolecular Arrangement of the Plasmodium falciparum Translocon of Exported Proteins (PTEX). J. Biol. Chem. 287, 7871–7884. doi:10.1074/jbc.M111.328591
Butler, N. S., Schmidt, N. W., Vaughan, A. M., Aly, A. S., Kappe, S. H. I., and Harty, J. T. (2011). Superior Antimalarial Immunity after Vaccination with Late Liver Stage-Arresting Genetically Attenuated Parasites. Cell Host & Microbe 9, 451–462. doi:10.1016/j.chom.2011.05.008
Chakravarty, S., Cockburn, I. A., Kuk, S., Overstreet, M. G., Sacci, J. B., and Zavala, F. (2007). CD8+ T Lymphocytes Protective against Malaria Liver Stages Are Primed in Skin-Draining Lymph Nodes. Nat. Med. 13, 1035–1041. doi:10.1038/nm1628
Charnaud, S. C., Jonsdottir, T. K., Sanders, P. R., Bullen, H. E., Dickerman, B. K., Kouskousis, B., et al. (2018). Spatial Organization of Protein export in Malaria Parasite Blood Stages. Traffic 19, 605–623. doi:10.1111/tra.12577
Chisholm, S. A., McHugh, E., Lundie, R., Dixon, M. W. A., Ghosh, S., O’Keefe, M., et al. (2016). Contrasting Inducible Knockdown of the Auxiliary PTEX Component PTEX88 in P. falciparum and P. berghei Unmasks a Role in Parasite Virulence. PLoS ONE 11, e0149296. doi:10.1371/journal.pone.0149296
Cockburn, I. A., Tse, S.-W., Radtke, A. J., Srinivasan, P., Chen, Y.-C., Sinnis, P., et al. (2011). Dendritic Cells and Hepatocytes Use Distinct Pathways to Process Protective Antigen from Plasmodium In Vivo. Plos Pathog. 7, e1001318. doi:10.1371/journal.ppat.1001318
de Koning-Ward, T. F., Dixon, M. W. A., Tilley, L., and Gilson, P. R. (2016). Plasmodium Species: Master Renovators of Their Host Cells. Nat. Rev. Microbiol. 14, 494–507. doi:10.1038/nrmicro.2016.79
de Koning-Ward, T. F., Gilson, P. R., Boddey, J. A., Rug, M., Smith, B. J., Papenfuss, A. T., et al. (2009). A Newly Discovered Protein export Machine in Malaria Parasites. Nature 459, 945–949. doi:10.1038/nature08104
Ejotre, I., Reeder, D. M., Matuschewski, K., and Schaer, J. (2021). Hepatocystis. Trends Parasitol. 37, 456–457. doi:10.1016/j.pt.2020.07.015
Elsworth, B., Matthews, K., Nie, C. Q., Kalanon, M., Charnaud, S. C., Sanders, P. R., et al. (2014). PTEX Is an Essential Nexus for Protein export in Malaria Parasites. Nature 511, 587–591. doi:10.1038/nature13555
Elsworth, B., Sanders, P. R., Nebl, T., Batinovic, S., Kalanon, M., Nie, C. Q., et al. (2016). Proteomic Analysis Reveals Novel Proteins Associated with the Plasmodium protein Exporter PTEX and a Loss of Complex Stability upon Truncation of the Core PTEX Component, PTEX150. Cell Microbiol. 18, 1551–1569. doi:10.1111/cmi.12596
Franke-Fayard, B., Trueman, H., Ramesar, J., Mendoza, J., van der Keur, M., van der Linden, R., et al. (2004). A Plasmodium berghei Reference Line that Constitutively Expresses GFP at a High Level throughout the Complete Life Cycle. Mol. Biochem. Parasitol. 137, 23–33. doi:10.1016/j.molbiopara.2004.04.007
Friesen, J., and Matuschewski, K. (2011). Comparative Efficacy of Pre-erythrocytic Whole Organism Vaccine Strategies against the Malaria Parasite. Vaccine 29, 7002–7008. doi:10.1016/j.vaccine.2011.07.034
Friesen, J., Silvie, O., Putrianti, E. D., Hafalla, J. C. R., Matuschewski, K., and Borrmann, S. (2010). Natural Immunization against Malaria: Causal Prophylaxis with Antibiotics. Sci. Transl. Med. 2, ra49. doi:10.1126/scitranslmed.3001058
Garten, M., Nasamu, A. S., Niles, J. C., Zimmerberg, J., Goldberg, D. E., and Beck, J. R. (2018). EXP2 Is a Nutrient-Permeable Channel in the Vacuolar Membrane of Plasmodium and Is Essential for Protein export via PTEX. Nat. Microbiol. 3, 1090–1098. doi:10.1038/s41564-018-0222-7
Grüring, C., Heiber, A., Kruse, F., Flemming, S., Franci, G., Colombo, S. F., et al. (2012). Uncovering Common Principles in Protein export of Malaria Parasites. Cell Host & Microbe 12, 717–729. doi:10.1016/j.chom.2012.09.010
Hafalla, J. C. R., Bauza, K., Friesen, J., Gonzalez-Aseguinolaza, G., Hill, A. V. S., and Matuschewski, K. (2013). Identification of Targets of CD8+ T Cell Responses to Malaria Liver Stages by Genome-wide Epitope Profiling. Plos Pathog. 9, e1003303. doi:10.1371/journal.ppat.1003303
Haussig, J. M., Matuschewski, K., and Kooij, T. W. A. (2011). Inactivation of a Plasmodium Apicoplast Protein Attenuates Formation of Liver Merozoites. Mol. Microbiol. 81, 1511–1525. doi:10.1111/j.1365-2958.2011.07787.x
Heiber, A., Kruse, F., Pick, C., Grüring, C., Flemming, S., Oberli, A., et al. (2013). Identification of New PNEPs Indicates a Substantial Non-PEXEL Exportome and Underpins Common Features in Plasmodium falciparum Protein Export. Plos Pathog. 9, e1003546. doi:10.1371/journal.ppat.1003546
Hiller, N. L., Bhattacharjee, S., van Ooij, C., Liolios, K., Harrison, T., Lopez-Estraño, C., et al. (2004). A Host-Targeting Signal in Virulence Proteins Reveals a Secretome in Malarial Infection. Science 306, 1934–1937. doi:10.1126/science.1102737
Ho, C.-M., Beck, J. R., Lai, M., Cui, Y., Goldberg, D. E., Egea, P. F., et al. (2018). Malaria Parasite Translocon Structure and Mechanism of Effector export. Nature 561, 70–75. doi:10.1038/s41586-018-0469-4
Ingmundson, A., Alano, P., Matuschewski, K., and Silvestrini, F. (2014). Feeling at home from Arrival to Departure: Protein export and Host Cell Remodelling during Plasmodium liver Stage and Gametocyte Maturation. Cell. Microbiol. 16, 324–333. doi:10.1111/cmi.12251
Ingmundson, A., Nahar, C., Brinkmann, V., Lehmann, M. J., and Matuschewski, K. (2012). The Exported Plasmodium berghei Protein IBIS1 Delineates Membranous Structures in Infected Red Blood Cells. Mol. Microbiol. 83, 1229–1243. doi:10.1111/j.1365-2958.2012.08004.x
Jaijyan, D. K., Singh, H., and Singh, A. P. (2015). A Sporozoite- and Liver Stage-Expressed Tryptophan-Rich Protein Plays an Auxiliary Role in Plasmodium Liver Stage Development and Is a Potential Vaccine Candidate. J. Biol. Chem. 290, 19496–19511. doi:10.1074/jbc.M114.588129
Janse, C. J., Franke-Fayard, B., Mair, G. R., Ramesar, J., ThielEngelmann, C. S., Engelmann, S., et al. (2006). High Efficiency Transfection of Plasmodium berghei Facilitates Novel Selection Procedures. Mol. Biochem. Parasitol. 145, 60–70. doi:10.1016/j.molbiopara.2005.09.007
Jonsdottir, T. K., Gabriela, M., Crabb, B. S., F. de Koning-Ward, T., and Gilson, P. R. (2021). Defining the Essential Exportome of the Malaria Parasite. Trends Parasitol. 37, 664–675. doi:10.1016/j.pt.2021.04.009
Kalanon, M., Bargieri, D., Sturm, A., Matthews, K., Ghosh, S., Goodman, C. D., et al. (2016). The Plasmodium Translocon of Exported Proteins Component EXP2 Is Critical for Establishing a Patent Malaria Infection in Mice. Cell Microbiol. 18, 399–412. doi:10.1111/cmi.12520
Kenthirapalan, S., Waters, A. P., Matuschewski, K., and Kooij, T. W. A. (2012). Flow Cytometry-Assisted Rapid Isolation of Recombinant Plasmodium berghei Parasites Exemplified by Functional Analysis of Aquaglyceroporin. Int. J. Parasitol. 42, 1185–1192. doi:10.1016/j.ijpara.2012.10.006
Kooij, T. W. A., Rauch, M. M., and Matuschewski, K. (2012). Expansion of Experimental Genetics Approaches for Plasmodium berghei with Versatile Transfection Vectors. Mol. Biochem. Parasitol. 185, 19–26. doi:10.1016/j.molbiopara.2012.06.001
Kreutzfeld, O., Müller, K., and Matuschewski, K. (2017). Engineering of Genetically Arrested Parasites (GAPs) for a Precision Malaria Vaccine. Front. Cel. Infect. Microbiol. 7, 198. doi:10.3389/fcimb.2017.00198
Maier, A. G., Rug, M., O'Neill, M. T., Brown, M., Chakravorty, S., Szestak, T., et al. (2008). Exported Proteins Required for Virulence and Rigidity of Plasmodium falciparum-infected Human Erythrocytes. Cell 134, 48–61. doi:10.1016/j.cell.2008.04.051
Marti, M., Good, R. T., Rug, M., Knuepfer, E., and Cowman, A. F. (2004). Targeting Malaria Virulence and Remodeling Proteins to the Host Erythrocyte. Science 306, 1930–1933. doi:10.1126/science.1102452
Matthews, K., Kalanon, M., Chisholm, S. A., Sturm, A., Goodman, C. D., Dixon, M. W. A., et al. (2013). The Plasmodium translocon of Exported Proteins (PTEX) Component Thioredoxin-2 Is Important for Maintaining normal Blood-Stage Growth. Mol. Microbiol. 89, 1167–1186. doi:10.1111/mmi.12334
Matthews, K. M., Kalanon, M., and de Koning-Ward, T. F. (2019b). Uncoupling the Threading and Unfoldase Actions of Plasmodium HSP101 Reveals Differences in Export between Soluble and Insoluble Proteins. mBio 10, e01106–19. doi:10.1128/mBio.01106-19
Matthews, K. M., Pitman, E. L., and Koning‐Ward, T. F. (2019a). Illuminating How Malaria Parasites export Proteins into Host Erythrocytes. Cell Microbiol. 21, e13009. doi:10.1111/cmi.13009
Matz, J. M., Goosmann, C., Brinkmann, V., Grützke, J., Ingmundson, A., Matuschewski, K., et al. (2015). The Plasmodium berghei Translocon of Exported Proteins Reveals Spatiotemporal Dynamics of Tubular Extensions. Sci. Rep. 5, 12532. doi:10.1038/srep12532
Matz, J. M., Matuschewski, K., and Kooij, T. W. A. (2013). Two Putative Protein export Regulators Promote Plasmodium Blood Stage Development In Vivo. Mol. Biochem. Parasitol. 191, 44–52. doi:10.1016/j.molbiopara.2013.09.003
McMillan, P. J., Millet, C., Batinovic, S., Maiorca, M., Hanssen, E., Kenny, S., et al. (2013). Spatial and Temporal Mapping of the PfEMP1 export Pathway in Plasmodium falciparum. Cel. Microbiol. 15, 1401–1418. doi:10.1111/cmi.12125
Meibalan, E., Comunale, M. A., Lopez, A. M., Bergman, L. W., Mehta, A., Vaidya, A. B., et al. (2015). Host Erythrocyte Environment Influences the Localization of Exported Protein 2, an Essential Component of the Plasmodium Translocon. Eukaryot. Cell 14, 371–384. doi:10.1128/EC.00228-14
Mesén-Ramírez, P., Reinsch, F., Blancke Soares, A., Bergmann, B., Ullrich, A.-K., Tenzer, S., et al. (2016). Stable Translocation Intermediates Jam Global Protein export in Plasmodium falciparum Parasites and Link the PTEX Component EXP2 with Translocation Activity. Plos Pathog. 12, e1005618. doi:10.1371/journal.ppat.1005618
Montagna, G. N., Beigier-Bompadre, M., Becker, M., Kroczek, R. A., Kaufmann, S. H. E., and Matuschewski, K. (2014). Antigen export during Liver Infection of the Malaria Parasite Augments Protective Immunity. mBio 5, e01321–14. doi:10.1128/mBio.01321-14
Müller, K., Gibbins, M. P., Matuschewski, K., and Hafalla, J. C. R. (2017). Evidence of Cross-Stage CD8+ T Cell Epitopes in Malaria Pre-erythrocytic and Blood Stage Infections. Parasite Immunol. 39, e12434. doi:10.1111/pim.12434
Mundwiler-Pachlatko, E., and Beck, H.-P. (2013). Maurer's Clefts, the enigma of Plasmodium falciparum. Proc. Natl. Acad. Sci. 110, 19987–19994. doi:10.1073/pnas.1309247110
Orito, Y., Ishino, T., Iwanaga, S., Kaneko, I., Kato, T., Menard, R., et al. (2013). Liver-specific Protein 2: a Plasmodium Protein Exported to the Hepatocyte Cytoplasm and Required for Merozoite Formation. Mol. Microbiol. 87, 66–79. doi:10.1111/mmi.12083
Petersen, W., Matuschewski, K., and Ingmundson, A. (2015). Trafficking of the Signature Protein of Intra-erythrocytic Plasmodium berghei-Induced Structures, IBIS1, to P. falciparum Maurer's Clefts. Mol. Biochem. Parasitol. 200, 25–29. doi:10.1016/j.molbiopara.2015.04.005
Pichugin, A., Zarling, S., Perazzo, L., Duffy, P. E., Ploegh, H. L., and Krzych, U. (2018). Identification of a Novel CD8 T Cell Epitope Derived from Plasmodium berghei Protective Liver-Stage Antigen. Front. Immunol. 9, 91. doi:10.3389/fimmu.2018.00091
Prudêncio, M., Rodriguez, A., and Mota, M. M. (2006). The Silent Path to Thousands of Merozoites: the Plasmodium Liver Stage. Nat. Rev. Microbiol. 4, 849–856. doi:10.1038/nrmicro1529
Silvie, O., Briquet, S., Müller, K., Manzoni, G., and Matuschewski, K. (2014). Post-transcriptional Silencing of UIS4 in Plasmodium berghei sporozoites Is Important for Host Switch. Mol. Microbiol. 91, 1200–1213. doi:10.1111/mmi.12528
Silvie, O., Mota, M. M., Matuschewski, K., and Prudêncio, M. (2008a). Interactions of the Malaria Parasite and its Mammalian Host. Curr. Opin. Microbiol. 11, 352–359. doi:10.1016/j.mib.2008.06.005
Silvie, O., Goetz, K., and Matuschewski, K. (2008b). A Sporozoite Asparagine-Rich Protein Controls Initiation of Plasmodium Liver Stage Development. PLoS Pathog. 4, e1000086. doi:10.1371/journal.ppat.1000086
Singh, A. P., Buscaglia, C. A., Wang, Q., Levay, A., Nussenzweig, D. R., Walker, J. R., et al. (2007). Plasmodium Circumsporozoite Protein Promotes the Development of the Liver Stages of the Parasite. Cell 131, 492–504. doi:10.1016/j.cell.2007.09.013
Spielmann, T., and Gilberger, T.-W. (2010). Protein export in Malaria Parasites: Do Multiple export Motifs Add up to Multiple export Pathways? Trends Parasitol. 26, 6–10. doi:10.1016/j.pt.2009.10.001
Sturm, A., Amino, R., van de Sand, C., Regen, T., Retzlaff, S., Rennenberg, A., et al. (2006). Manipulation of Host Hepatocytes by the Malaria Parasite for Delivery into Liver Sinusoids. Science 313, 1287–1290. doi:10.1126/science.1129720
Vanderberg, J. P. (1975). Development of Infectivity by the Plasmodium berghei Sporozoite. J. Parasitol. 61, 43–50. doi:10.2307/3279102
Wahlgren, M., Goel, S., and Akhouri, R. R. (2017). Variant Surface Antigens of Plasmodium falciparum and Their Roles in Severe Malaria. Nat. Rev. Microbiol. 15, 479–491. doi:10.1038/nrmicro.2017.47
Warncke, J. D., and Beck, H.-P. (2019). Host Cytoskeleton Remodeling throughout the Blood Stages of Plasmodium falciparum. Microbiol. Mol. Biol. Rev. 83, e00013–19. doi:10.1128/MMBR.00013-19
Keywords: malaria, plasmodium, protein export, pre-erythrocytic stage, PEXEL motif, PTEX translocon, ATPase, heat shock protein
Citation: Kreutzfeld O, Grützke J, Ingmundson A, Müller K and Matuschewski K (2021) Absence of PEXEL-Dependent Protein Export in Plasmodium Liver Stages Cannot Be Restored by Gain of the HSP101 Protein Translocon ATPase. Front. Genet. 12:742153. doi: 10.3389/fgene.2021.742153
Received: 15 July 2021; Accepted: 18 October 2021;
Published: 08 December 2021.
Edited by:
Jun-Hu Chen, National Institute of Parasitic Diseases, ChinaReviewed by:
Tania F. De Koning-Ward, Deakin University, AustraliaCopyright © 2021 Kreutzfeld, Grützke, Ingmundson, Müller and Matuschewski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kai Matuschewski, S2FpLk1hdHVzY2hld3NraUBodS1iZXJsaW4uZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.