
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 22 October 2021
Sec. Genomic Assay Technology
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.739054
 Jagadheshwar Balan1
Jagadheshwar Balan1 Garrett Jenkinson1
Garrett Jenkinson1 Asha Nair1
Asha Nair1 Neiladri Saha1
Neiladri Saha1 Tejaswi Koganti1Jesse Voss2Christopher Zysk3Emily G. Barr Fritcher4Christian A. Ross5Caterina Giannini4Aditya Raghunathan4Benjamin R. Kipp4Robert Jenkins2Cris Ida2
Tejaswi Koganti1Jesse Voss2Christopher Zysk3Emily G. Barr Fritcher4Christian A. Ross5Caterina Giannini4Aditya Raghunathan4Benjamin R. Kipp4Robert Jenkins2Cris Ida2 Kevin C. Halling2Patrick R. Blackburn6
Kevin C. Halling2Patrick R. Blackburn6 Surendra Dasari1
Surendra Dasari1 Gavin R. Oliver1
Gavin R. Oliver1 Eric W. Klee1*
Eric W. Klee1*Detecting gene fusions involving driver oncogenes is pivotal in clinical diagnosis and treatment of cancer patients. Recent developments in next-generation sequencing (NGS) technologies have enabled improved assays for bioinformatics-based gene fusions detection. In clinical applications, where a small number of fusions are clinically actionable, targeted polymerase chain reaction (PCR)-based NGS chemistries, such as the QIAseq RNAscan assay, aim to improve accuracy compared to standard RNA sequencing. Existing informatics methods for gene fusion detection in NGS-based RNA sequencing assays traditionally use a transcriptome-based spliced alignment approach or a de-novo assembly approach. Transcriptome-based spliced alignment methods face challenges with short read mapping yielding low quality alignments. De-novo assembly-based methods yield longer contigs from short reads that can be more sensitive for genomic rearrangements, but face performance and scalability challenges. Consequently, there exists a need for a method to efficiently and accurately detect fusions in targeted PCR-based NGS chemistries. We describe SeekFusion, a highly accurate and computationally efficient pipeline enabling identification of gene fusions from PCR-based NGS chemistries. Utilizing biological samples processed with the QIAseq RNAscan assay and in-silico simulated data we demonstrate that SeekFusion gene fusion detection accuracy outperforms popular existing methods such as STAR-Fusion, TOPHAT-Fusion and JAFFA-hybrid. We also present results from 4,484 patient samples tested for neurological tumors and sarcoma, encompassing details on some novel fusions identified.
Gene fusions are potentially pathogenic events that result from genomic structural rearrangements including inversions, translocations, and interstitial deletions. Gene fusions are frequently observed in cancer, and while their prevalence varies by tumor type they have been estimated to account for 20% of all cancer morbidity (Mitelman et al., 2007). Identification of gene fusions in a tumor can help guide therapeutic decision-making since fused protein products can represent targets of small molecule inhibitors or other novel treatments. Examples include the treatment of recurrent PTPRZ1-MET fusion-positive glioblastoma using the MET kinase inhibitor crizotinib, KIAA1549-BRAF fusion driven pediatric pilocytic astrocytoma treatment using the MEK inhibitor trametinib, and ALK-EML4 fusion-positive lung cancer using tyrosine kinase inhibitor lorlatinib (Bender et al., 2016; Jain et al., 2017; Shaw et al., 2019).
RNA sequencing (RNA-Seq) assays for gene fusion detection provide improvements in throughput, sensitivity and specificity over traditional DNA and protein-based approaches like fluorescence in situ hybridization (FISH) and immunohistochemistry (IHC) (Wang et al., 2009; Abel et al., 2014; Moskalev et al., 2014; Pekar-Zlotin et al., 2015; Haynes et al., 2019). RNA-based strategies to detect gene fusions use transcriptome-wide sequencing with ribosomal RNA-depleted, fractionated messenger RNA or target specific genes of interest using polymerase chain reaction (PCR) based amplicon RNA-Seq, or bait hybridization (capture and ligation) using assays such as QIAGEN’s QIAseq RNAscan panel or Illumina’s SureSelect RNA capture (Wang et al., 2009; Blomquist et al., 2013; Drilon et al., 2015). The PCR-based amplicon RNA-Seq methods offer inexpensive and highly accurate sequencing of fusion transcripts, simultaneously assessing dozens to thousands of targets (Blomquist et al., 2013) and enabling detection of lowly expressed fusions at a high sequencing depth. While PCR-based approaches have traditionally been limited due to stochastic errors that propagate to all PCR cycles (Thilly, 1993), the addition of unique molecular indexes (UMI) in ligation adapters has recently been used to alleviate the impact of such errors (Kivioja et al., 2012), and can improve the sensitivity of gene fusion detection assays. QIAseq NGS assay panels have been demonstrated to provide robust correlation with RT-PCR and low PCR-bias (Wong et al., 2019), and so was the chemistry used in this study (see Methods, Library Preparation and Sequencing section).
Several bioinformatics tools are available for detecting gene fusions that use wide variety of approaches. Brian et al. and Trung et al. have each outlined benchmarking of 15 gene fusion identification tools in their studies and detailed the methods and performance (Vu et al., 2018; Haas et al., 2019). One common approach uses transcriptome-based spliced alignment. This method is reliant on the accurate mapping of short reads to the transcriptome, which can be challenging due to genome repetitiveness, sequence homology, incomplete transcriptome annotations, and novel patient sequences not well-represented by the reference genome. Matteo et al. elaborate on limitations of several gene fusion identification tools using short read alignment technologies and report key factors such as read length, quality scores and number of reads supporting each fusion call (Carrara et al., 2013a). De-novo assembly-based approaches yield longer contigs from short reads and address some of the limitations of short-read alignment but are computationally intensive and often not scalable to large scale clinical testing (Carrara et al., 2013b; Davidson et al., 2015; Haas et al., 2019). Hybrid approaches such as the method implemented in JAFFA (Davidson et al., 2015) combine the approaches described above, but so far, no hybrid method achieves a balance of scalability and accuracy.
Herein we describe SeekFusion (https://hub.docker.com/repository/docker/jagadhesh89/seekfusion), a time-efficient pipeline that leverages de-novo assembly and alignment based approaches to accurately identify gene fusions utilizing PCR-UMI-based amplicon RNA-Seq. SeekFusion performs rapid alignment to gene sequences, then groups and filters aligned reads for de-novo assembly. Assembled contigs are realigned to a reference genome and fusion genes are identified, annotated and reported in a VCF format. Using verified clinical cases and synthetic controls, we demonstrate that SeekFusion outperforms existing pipelines by balancing high analytical sensitivity and specificity with computational efficiency. The algorithm is written using Python and bash and the functions are wrapped in WDL and processed using Cromwell (Voss, 2017) for ease of deployment by end-users in their own compute environments.
The study (Figure 1) involves developing a bioinformatics method for neurological cancer and sarcoma cancer clinical NGS assays aimed at targeting gene fusions. Towards development of the assay, few positive samples, negative samples and in-silico samples were selected, sequenced and analyzed across multiple fusion callers. The fusion callers were benchmarked carefully, results were summarized and compared to orthogonal assay results to identify the winning method. SeekFusion, an internally developed pipeline is highly optimized and accurate compared to the other methods that included STAR-Fusion, JAFFA and Tophat-Fusion. SeekFusion was then used towards verification of the developed assay, the assays were then implemented clinically for gene fusion identification post New York State (NYS) approval for the laboratory developed tests.
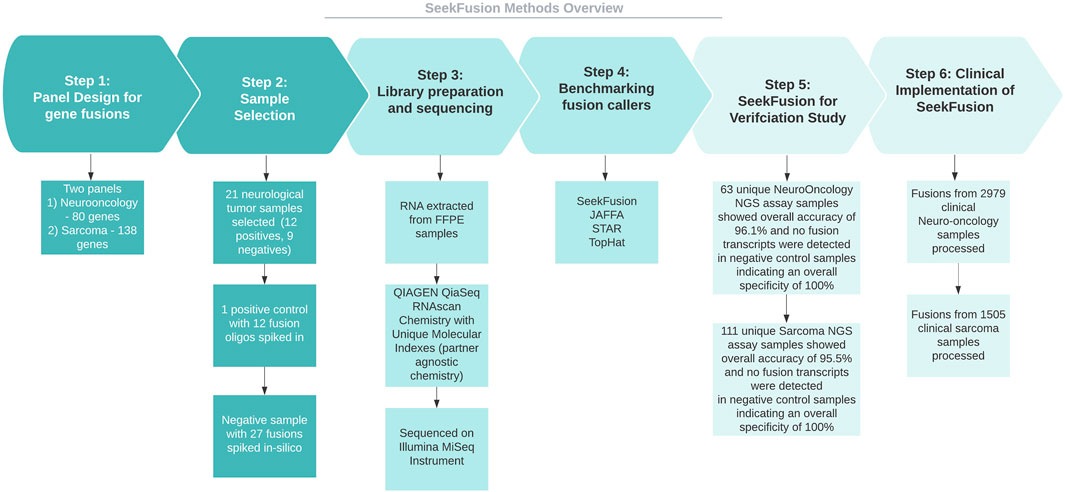
FIGURE 1. Methods overview. The study involves developing Neuorlogical oncology and Sarcoma clinical NGS assays aimed at targeting gene fusions. Towards development of the assay, few positive samples, negative samples and in-silico samples were selected, sequenced and analyzed across multiple fusion callers. The fusion calls were examined to identify the winning method, and the winning method was SeekFusion, which was internally developed and optimized for the assay. The method was then used towards verification of the assay for NYS approvals and was deployed clinically for gene fusion identification.
A neurological oncology panel was designed to target fusions in 80 genes (Supplementary Table S2) utilizing the Qiagen QIAseq RNAscan Custom Panel (Blessing et al., 2019). Targeted rearrangements were selected based on association with a variety of adult and pediatric central nervous system (CNS) tumors and potential utility in the differential diagnosis of these tumors or in the differentiation of molecularly defined tumor subtypes (e.g., ependymoma RELA fusion-positive) (Kim et al., 2017). A sarcoma assay was designed to diagnose specific soft tissue and bone tumors (sarcoma) based on the observed gene fusion in 138 genes (Supplementary Table S3). Targeted rearrangements were selected based on associations with a variety of sarcoma types such as rhabdomyosarcoma, synovial sarcoma, and Ewing’s sarcoma. Both neurological oncology and sarcoma assays’ chemistry is designed to target a list of genes known to be involved in rearrangements using gene specific primers but uses a universal primer on the partner gene, making it a partner-agnostic chemistry capable of novel gene fusion identification. Specimen requirements and types for the assays have been detailed in Supplementary Methods section.
Twenty-one neurological tumor samples were obtained following IRB-approved protocols. Of the 21 samples, 12 were fusion positive and 9 were fusion negative (Table 1). In addition to these clinical samples of known gene fusion status, we included a positive control, created from fusion-negative samples to which 13 gene fusion oligonucleotides were spiked in. Finally, a normal control sample was sequenced, and 27 unique fusions were added in-silico as positive controls. In total, benchmarking was performed using 52 known positive gene fusions. All samples were processed using the 80-gene QIAseq RNAscan neurological oncology NGS panel.

TABLE 1. List of samples for benchmarking.
Gene fusions detected while benchmarking was confirmed using OncoScan FFPE Assay Kit (Thermo Fisher Scientific, Waltham, MA) chromosomal microarray (CMA) and reverse transcription polymerase chain reaction (RT-PCR) with or without subsequent direct Sanger sequencing as previously described (Gliem and Aypar, 2017).
RNA was extracted from macrodissected, unstained slides using the QIAamp miRNeasy FFPE kit and nucleic acid was quantitated with the NanoDrop 2.0 system (Blessing et al., 2019). Library preparation was performed with the QIAseq RNAscan chemistry using 200 ng of RNA per manufacturer’s protocol recommendations. Final libraries were quantitated using a Qubit fluorometer, with 8 equimolar samples pooled and sequenced on an Illumina MiSeq instrument. Raw sequencing data was de-multiplexed into FASTQ files and processed through the SeekFusion pipeline.
SeekFusion was designed in Cromwell/WDL to enable standalone or server mode workflow execution. Server mode performs tasks in parallel for every gene targeted (Figure 2). The pipeline is configurable for each module, and parameters can be fine-tuned through the profile settings file for the pipeline. Details of the individual steps comprising the pipeline are described in the following sections.
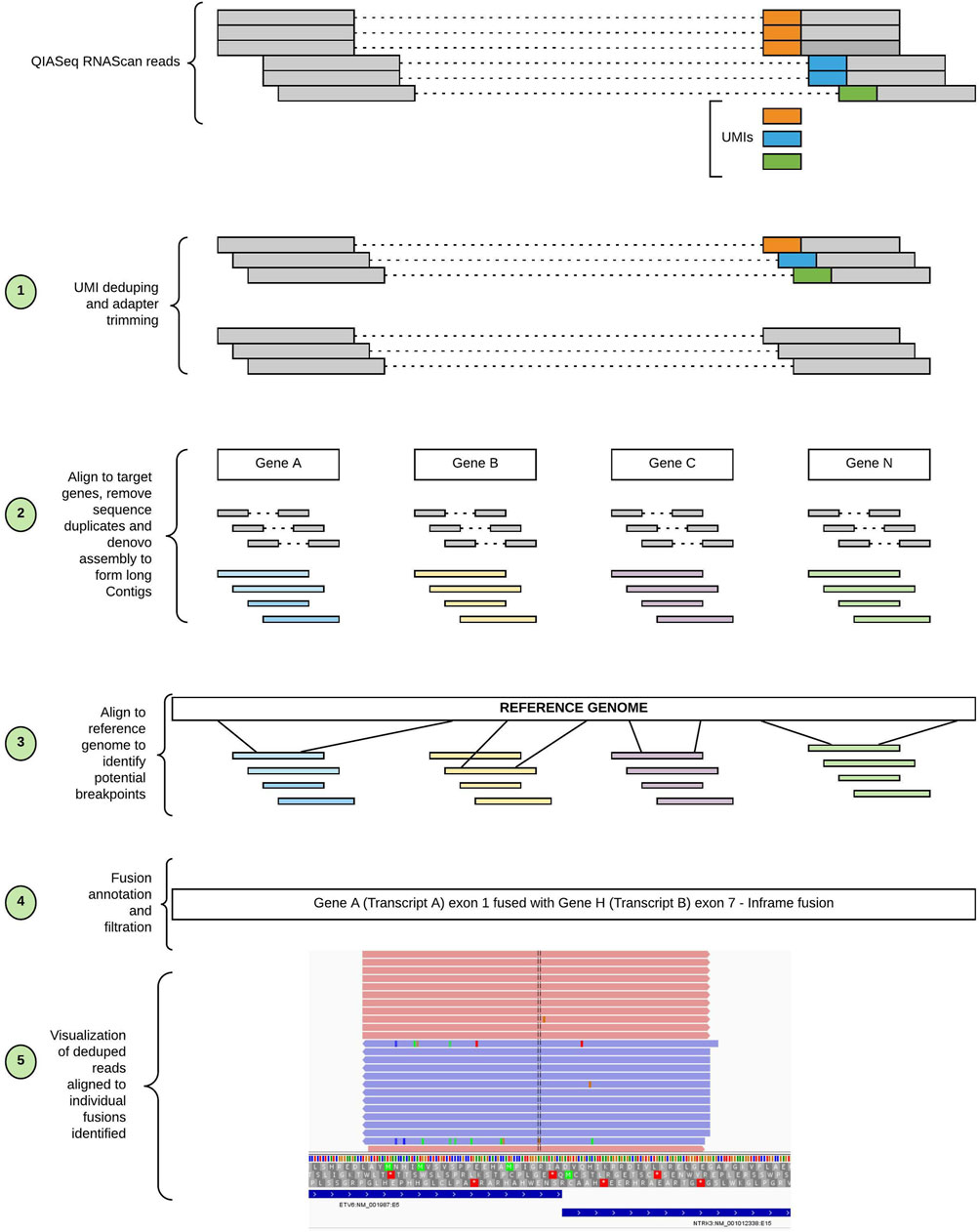
FIGURE 2. Workflow of SeekFusion pipeline. The pipeline begins with deduping the reads utilizing the UMIs; the deduped reads are then aligned to target genes. The aligned reads are pooled based on the genes and for each gene-based read pool a de-novo assembly is performed to construct longer contigs. The contigs are aligned to the reference genome to identify potential breakpoints. The potential breakpoints are annotated, and reads are aligned to the breakpoints to visualize the fusions.
PCR duplication is a byproduct of amplification steps in sequencing protocols that produces multiple copies of individual sequence fragments and artificially inflates their abundance. The QIAseq RNAscan chemistry utilizes UMIs that are random nucleotide sequences incorporated into sequence fragments prior to amplification to enable the identification of PCR duplicate reads originating from the same sequence fragment. Barring technical and sequencing errors, each of these duplicated reads will share identical nucleotide sequence. In reality, however, sequencing errors introduce differences in duplicate reads. The “deduping” module accounts for these errors and attempts to identify the PCR duplicates irrespective of sequencing error. It then builds a single consensus read from identified PCR duplicates and produces an updated Phred quality score for each base that accurately represents the summary evidence when estimating the probability of an erroneous base call. This approach produces higher confidence base calls that facilitate accuracy of downstream processing and improves the ability to discern true calls.
The deduping algorithm assumes a prior probability for the nucleotides at a particular position and uses Bayes theorem to update the probability of the base in a particular position of a UMI using conditional independence (See Supplementary Methods).
FASTQ files are used as input to the deduping module. The algorithm is written in C and based on a highly efficient and customized data structure. Efficient binary representation of k-mers (up to length 16) enables perfect hashing and rapid computations of hamming distances. The algorithms also make use of BGZF compressed FASTQ files (this is the same compression method used in bam files) which enables indexing and fast random read access within the compressed FASTQ files.
Fastp (Chen et al., 2018) was implemented with default settings to trim the adapters from reads produced by the QIAseq RNAscan chemistry, with adapter sequences provided in a FASTA file using the--adapter_fasta option.
De-novo assembly is a computationally intensive process that has the potential to overwhelm computational resources (Sze et al., 2017). To alleviate this potential bottleneck, a pre-processing step was formulated to reduce the number of identical or high similarity reads entering the de-novo assembly stage. A target reference database for read alignment was created using the full nucleotide sequence of the longest coding transcript for all genes targeted by the neuro oncology NGS panel, identified by HGNC (Yates et al., 2017) gene names. BWA-mem (Li and Durbin, 2009) from Sentieon (Sentieon, Mountain View, CA, United States) was used to enable rapid alignment to the gene-based reference database. BWA-mem can be substituted with the open source BWA-mem tool using profile settings for the pipeline. Default parameters for paired-end alignment were used. Best alignments were selected for each read and binned into gene-specific SAM files utilizing the SAMTOOLS (Li et al., 2009) view functionality with the aligned BAM file. A custom python module was developed to identify alignments sharing identical length and chromosomal start and end coordinates and filter the subsequent output to five copies or fewer. Reads passing this stage were output in FASTA format for de-novo assembly. Following alignment-based reduction, reads were assembled using the CAP3 (Huang and Madan, 1999) de-novo assembly tool with default settings (Supplementary Table S1). Assembly was performed individually on each gene-specific SAM file to improve the specificity of the de-novo assembly.
De-novo assembled contigs were aligned to the human reference genome (hg19) using BLAT (Kent, 2002). Those generating multiple non-contiguous alignments were retained as potential fusion candidates or splice-variants for downstream characterization. The parameters for BLAT are specified in Supplementary Table S1. Custom modules were developed to remove multiple contigs mapping to identical genomic regions from BLAT results, reducing redundancy in the putative gene fusion breakpoints.
Filtering modules were developed to reduce false positive calls occurring due to mononucleotide repeats, homology, and other recurrent artifacts. To filter the false positives due to mononucleotide repeats, the filtering script looks for repetitive regions using a Mononucleotide Repeat Ratio (MRR). The mononucleotide repeat ratio calculation is described in the Supplementary Methods.
For filtering highly homologous regions, SeekFusion flags calls that are recurrent in every sample due to homology/low complexity using a blocklist. The blocklist calls are presented in the Supplementary Data. Events involving a single transcript are filtered from the output by default. An inclusion feature is provided to enable detection of clinically relevant single transcript events, such as epidermal growth factor receptor variant III (EGFRvIII) in glioblastoma (An et al., 2018).
A custom script was developed to annotate identified contig breakpoints and provide genomic context. Annotations generated include the gene, exon and coding frame status of the fusion, i.e., if the fusion is likely protein-coding. One of the key annotations provided by the annotation module is if a fusion is In-frame, which implies that there was no frame shift in the 3′-gene, regardless of single amino acid mutation or insertion events at the fusion junction. Table 2 contains examples of fusion annotations generated by the pipeline.
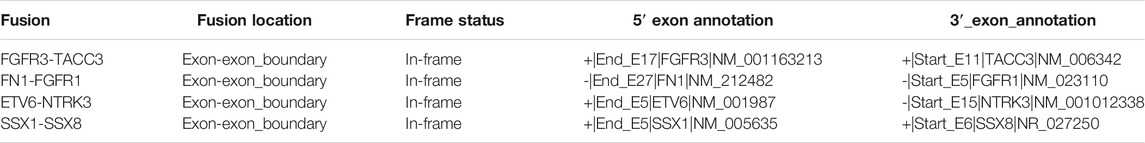
TABLE 2. Fusion annotation generated by the pipeline for documented fusions.
All reads originally passing the UMI-based deduping and adapter trimming steps were retrieved and aligned to the hg19 human reference transcriptome from Ensembl and Refseq using BWA-mem. Reads that produced perfect alignments to known gene transcripts were removed from further consideration as they likely represent normal transcriptional events. For each gene fusion candidate identified by the alignment of assembled contigs to the human genome, a chimeric construct of the purported fusion sequence was created (150 base pairs bidirectional beyond the putative breakpoint). Constructs annotated as occurring within a single known transcript were removed from further consideration, with the exception of select known aberrant single-gene events. The retained reads were aligned against the potentially chimeric gene fusion reference sequences using BWA-mem. Only reads spanning at least 10 bases beyond a breakpoint were considered as evidence of a potential fusion. A single supporting read was required as evidence of a fusion candidate and all candidates were output in a standardized variant call format (VCF) file. While minimal read support was required to facilitate downstream benchmarking, the supporting read threshold is user configurable. Finally, the fusion-construct aligned BAM file was used to create an Integrative Genomics Viewer (IGV) (Robinson et al., 2011) session to enable visual inspection of the evidence supporting each fusion candidate (Figure 3).
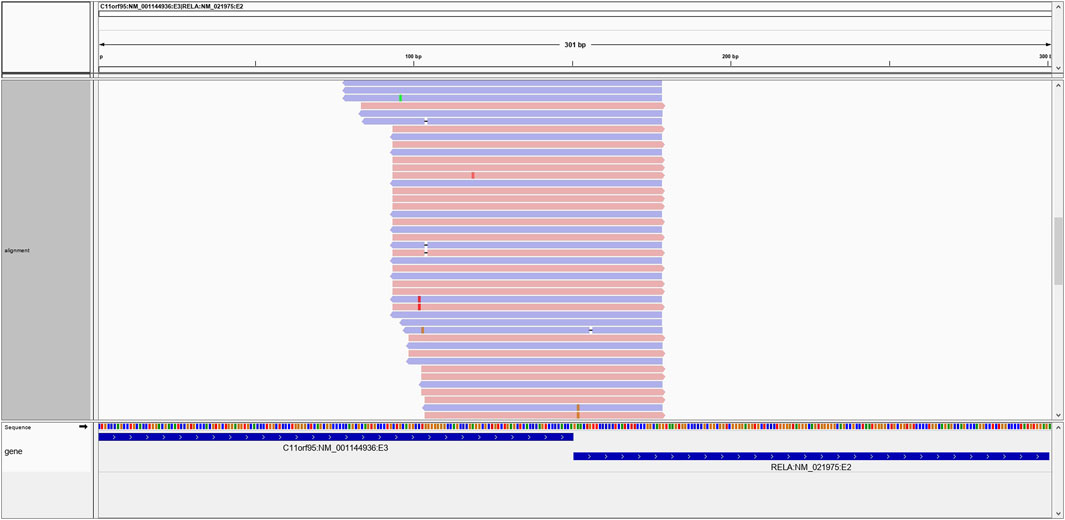
FIGURE 3. An example of the C11ORF95-RELA positive fusion on IGV. Multiple reads generate high-confidence alignments and span the breakpoint in support of the putative fusion transcript.
The pipeline is available in a Docker container (Docker, 2014) image and is available to download from https://hub.docker.com/repository/docker/jagadhesh89/seekfusion. The usage details are explained in the README file. In addition to the easy to use docker image, the source code is also made available via github (https://github.com/jagadhesh89/seekfusion).
Benchmarking was performed for SeekFusion, JAFFA-hybrid (Davidson et al., 2015), STAR-Fusion (Haas et al., 2019) and TOPHAT-Fusion (Kim and Salzberg, 2011), mirroring benchmarking analyses performed by the STAR-Fusion study (Haas et al., 2019). Selected samples were processed through the four pipelines for benchmarking in two separate analyses. In Analysis 1, FASTQs were trimmed with FASTP (Chen et al., 2018), subsequently deduped using UMIs, and supplied as inputs to the pipelines. In Analysis 2, the raw FASTQs were provided as pipeline inputs for the 12 gene fusion positive cases, the 9 gene fusion negative cases, and the positive controls. Analysis 2 was used to assess tool performance with raw sample FASTQs, without deduping, however the adapters and UMIs were trimmed to prune the reads for specific tools (see Supplementary Section S2.5). In Analysis 2, JAFFA-Hybrid posed a scalability challenge; the pipeline ran for over 24 h per sample before failure due to memory requirements. Due to the scalability issue, JAFFA-Hybrid was excluded from Analysis 2. However for the analysis 2, we benchmarked JAFFA-Direct to circumvent the scalability issues of JAFFA-hybrid.
Accuracy was measured based on putative gene fusion calls using the following equation at varying levels of fusion read support thresholds.
For in-silico generated fusions, supporting reads are a known quantity. Mean Absolute Percentage Error (de Myttenaere et al., 2016) (MAPE) and Symmetric Mean Absolute Percentage Error (Tofallis, 2015) (SMAPE) values were calculated as a measure of known supporting reads detected by the four algorithms. A Wilcoxon test was performed on the SMAPE values between SeekFusion and other tools. Key considerations on determining the winning tool from benchmarking involved high accuracy, high true positive rate, ease of installation, ease of usage in a clinical setting and turnaround time for the pipeline.
The neurological oncology assay was developed, verified, validated, New York (NY) State approved and implemented clinically using the SeekFusion algorithm. In the verification study, 63 total unique samples were processed (59 CNS tumor samples, 1 CNS tumor sample spiked with 13 known synthetic oligonucleotides and 3 brain gliosis samples). The NGS assay results were confirmed by RT-PCR and CMA tests. The overall accuracy of the NGS assay was 96.1% and no fusion transcripts were detected in negative control samples indicating an overall specificity of 100%. Following the clinical go-live of the assay, 2,979 clinical patient samples ordered for neurological oncology testing and were processed using the SeekFusion algorithm. A sarcoma assay was developed, verified, validated and approved for clinical testing by NY state. The assay uses the SeekFusion algorithm and evaluates 138 gene targets for presence of somatic gene fusions and common BCOR internal tandem duplications (ITDs) using the same QIAseq RNAscan chemistry. In the verification study, this next-generation sequencing (NGS) assay was performed in 111 sarcoma formalin-fixed, paraffin-embedded (FFPE) and cytology samples (86 fusion positive and 25 fusion negative). The NGS assay results were confirmed by RT-PCR and FISH tests. The overall accuracy of the NGS assay was 95.5%. No targeted gene fusions were detected in 20 negative control samples (100% specificity), A total of 1,505 clinical cases were processed using the SeekFusion algorithm. An IRB request was submitted and approved to share the clinically reported gene fusions.
The accuracy of SeekFusion was highest among the benchmarked tools, followed by JAFFA-hybrid, STAR-Fusion and TOPHAT-Fusion (Figure 4A). True positive rate was measured as a surrogate of clinical utility and SeekFusion consistently performed best among the tools at all levels of read support cut-off (Figure 4B). Read support cut-off is commonly applied to fusion detection algorithms for reducing false positive rate and improving accuracy (Liu et al., 2016). Read cut-offs for calling a fusion were assessed for all tools at levels of 1, 2, 3, 4, 5, 10, 20, 30, 40, 50, 60, 70, 80, 90 and 100 supporting reads, and accuracy and true-positive rates were calculated. Intuitively if an algorithm consistently reports gene fusions with fewer reads supporting the event, a higher read cut-off will result in a lower accuracy and true positive rate. JAFFA hybrid was ranked second, followed by STAR-Fusion; however, both JAFFA and STAR-Fusion produced higher numbers of calls beyond the known fusion events. These calls were closely examined and are documented in Supplementary File S1. Manual curation was performed, and the extra calls were classified as false positives; all these calls were due to homology, low complexity, or identified with very low frequency (Supplementary Table S5). Tophat Fusion performed the worst, with a high rate of false negatives. SeekFusion was the only tool observed to be 100% concordant with the expected positive gene fusions whereas STAR-Fusion, JAFFA-Hybrid and TOPHAT-Fusion detected 83, 77 and 1% respectively.
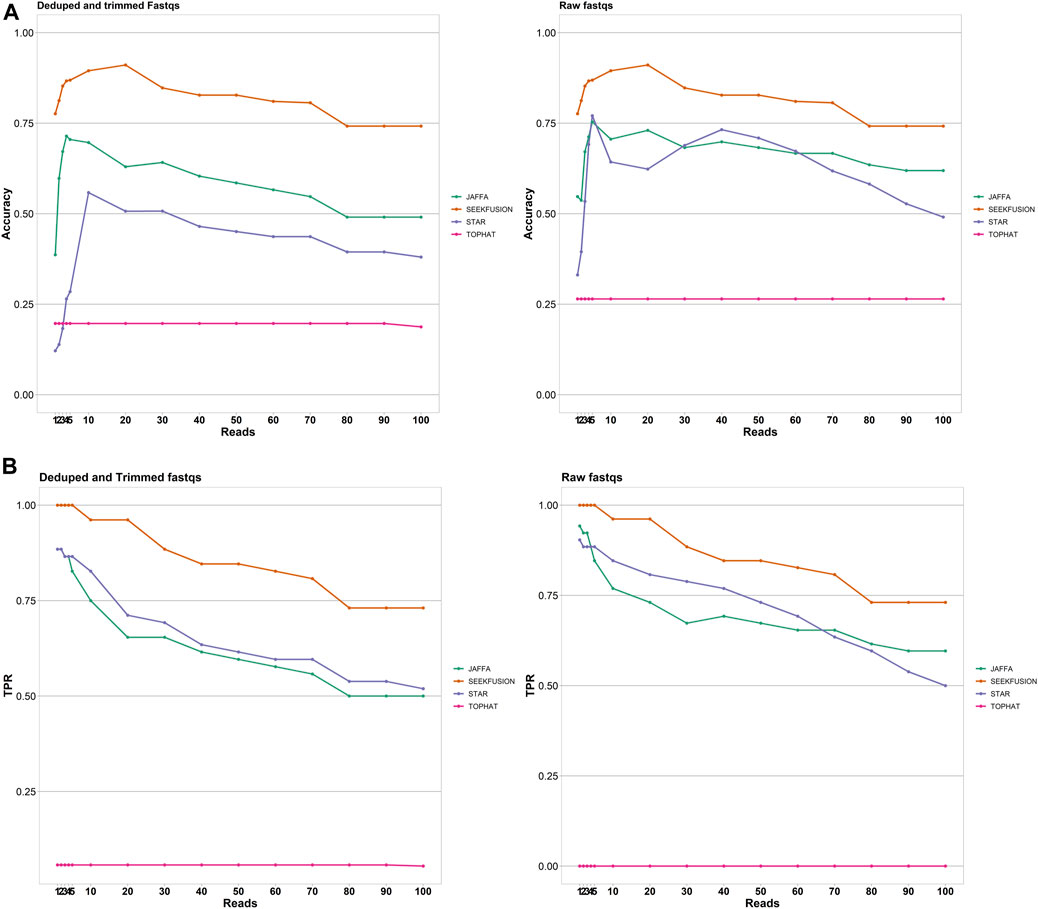
FIGURE 4. (A) Accuracy of the tools was assessed for the tools JAFFA, STAR-Fusion, Tophat Fusion and SeekFusion at varying read support thresholds. SeekFusion demonstrated highest accuracy in detecting the gene fusions at all levels of read support. The median accuracy of SeekFusion was 82% followed by JAFFA-Hybrid at 60%, STAR-Fusion at 39% and Tophat-Fusion at 20% for Mode1. SeekFusion was the only pipeline that demonstrated high accuracy in detecting fusions from raw FASTQs. (B) True Positive Rate of the tools was assessed for the tools JAFFA, STAR-Fusion, Tophat Fusion and SeekFusion at varying read support thresholds. SeekFusion demonstrated the highest sensitivity in detecting the gene fusions at all levels of read support. For the deduped and trimmed FASTQs, we observed that the median true positive rate of SeekFusion was 89% followed by STAR-Fusion at 69% and JAFFA-Hybrid at 65%. Tophat Fusion performed least favorably among the tools assessed with a median true positive rate of 1%. SeekFusion was the only tool that successfully detected true positives in the raw FASTQs.
The number of gene fusion-supporting reads detected by the four tools was assessed for the true positive gene fusions. Deduped and trimmed FASTQs were utilized in the analysis since only SeekFusion demonstrated the ability to successfully detect the true positive fusions from raw FASTQs. SeekFusion reported more gene fusion supporting reads than the other tools in 83% of cases (Figure 5). SeekFusion was followed by JAFFA, STAR-Fusion and Tophat Fusion for the number of fusion supporting reads.
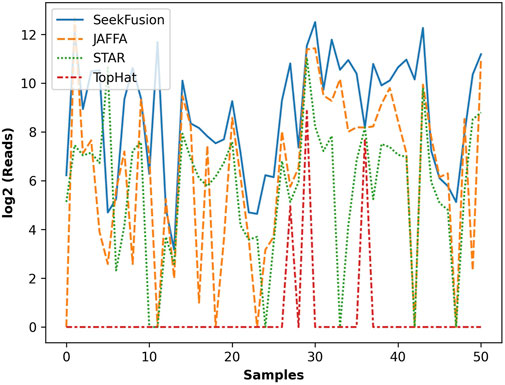
FIGURE 5. The gene fusion read support in log scale for each fusion detected by SeekFusion, STAR-Fusion, Tophat Fusion and JAFFA. In 83% of cases, SeekFusion demonstrated the highest read support amongst the algorithms tested.
MAPE and SMAPE are widely used measures of forecast accuracy; they aid in assessing accuracy based on percentage errors from the actual. MAPE (de Myttenaere et al., 2016) and SMAPE (Tofallis, 2015) values indicate that SeekFusion performed the best in terms of observed to expected gene fusion-supporting reads amongst the four tools (Table 3). A Wilcoxon test (Wilcoxon, 1945) on the SMAPE values between SeekFusion and other tools supported the hypothesis that SeekFusion retrieved the highest proportion of known fusion-supporting reads in the in-silico samples (Table 3). Each tool considered has a different method of fusion transcript identification and these account for differences in levels of read support.

TABLE 3. MAPE and SMAPE values for the 4 tools and the p-values based on the Wilcoxon test on SMAPE values between SeekFusion and other tools.
In addition to analytical performance, clinical bioinformatics pipelines are required to perform in a time-efficient manner to maintain necessary assay turnaround time. Benchmarking of runtimes was performed to assess the suitability of the algorithms for real-world clinical use. The single in-silico sample was excluded from the benchmarking since it does not represent a true patient sample. For both Analysis 1, JAFFA exhibited time-requirements that exceeded all other tools (Figure 6). The remaining three tools were comparable in their runtimes and were all considered amenable to routine clinical use, with STAR-Fusion performing most favorably in terms of time-requirements alone. In Analysis 2 due to scalability challenges posed by JAFFA-hybrid mode, analysis was performed in JAFFA-Direct mode and it was found that JAFFA-Direct mode was most favorable in terms of time-requirements alone.
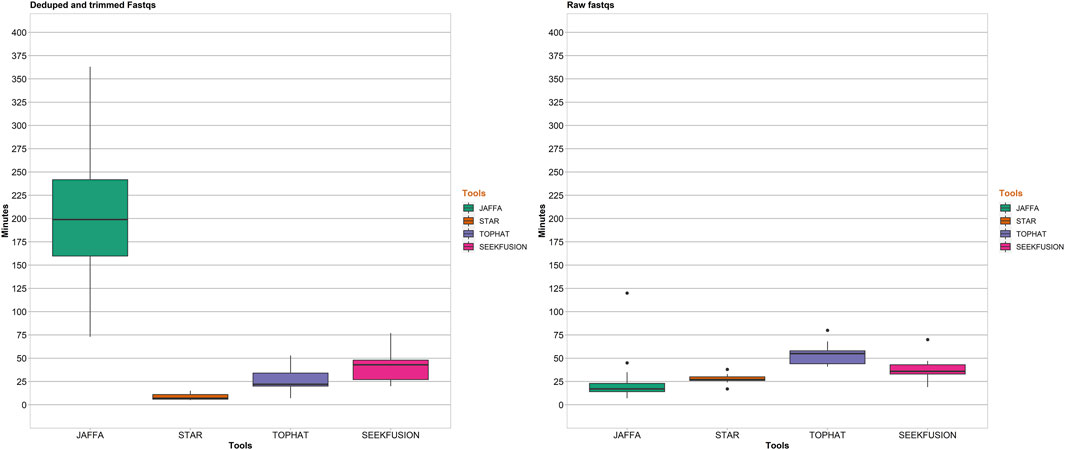
FIGURE 6. Pipeline completion time in minutes for the 4 tools. For Analysis 1, STAR-Fusion was the fastest among the 4 tools with median completion time of 6 min followed by Tophat Fusion at 20 min, SeekFusion at 45 min and JAFFA-Hybrid at 196 min. For Analysis 2, STAR-Fusion had a median completion time of 29 min, SeekFusion had a median completion time of 36 min, Tophat-Fusion had a median completion time of 53 min and JAFFA-Hybrid exhibited scalability issues withe pipeline failing due to memory issues after running for over 24 h (so the data was capped at 400 min for JAFFA Hybrid in the raw FASTQ mode). To circumvent the scalability issues, analysis in direct mode of JAFFA was performed and this mode demonstrated the fastest median completion time of 20 min.
Peak memory utilization of the individual tools benchmarked were assessed. The maximum input memory for each of the pipelines were capped at 180 GB. For each sample, the maximum memory used by the tool is used for calculating a per tool median memory utilization for analysis 1 and analysis 2. The median is rounded to the nearest GB. JAFFA utilized the most memory with a median memory utilization of 171 GB in analysis 1 and 172 GB in analysis 2. SeekFusion peaked at a median memory of 58 GB in analysis 1 and 64 GB in analysis 2, with CAP3 assembly being the most memory consuming module in the SeekFusion pipeline. STAR-Fusion peaked at a median memory of 30 GB in analysis 1 and 34 GB in analysis 2. Tophat-Fusion used the least memory; with peak of median memory in analysis 1 and analysis 2 of 2 GB.
Taken together, while considering high accuracy, high true positive rate, ease of installation, ease of usage in a clinical setting and turnaround time for the pipeline, SeekFusion was identified as the method for use of clinical assay development. Neurological oncology assay and sarcoma assay were developed, verified and approved as detailed in the Methods, Clinical Testing On Neurological Oncology and Sarcoma Samples section.
Of the 2,979 cases tested clinically using the neurological oncology assay, 569 cases were reported with clinically relevant gene fusions after careful variant review and orthogonal confirmation using RT-PCR (Figure 7A). Out of the 1,505 cases tested clinically using the sarcoma assay, 476 cases were reported (Figure 7B).
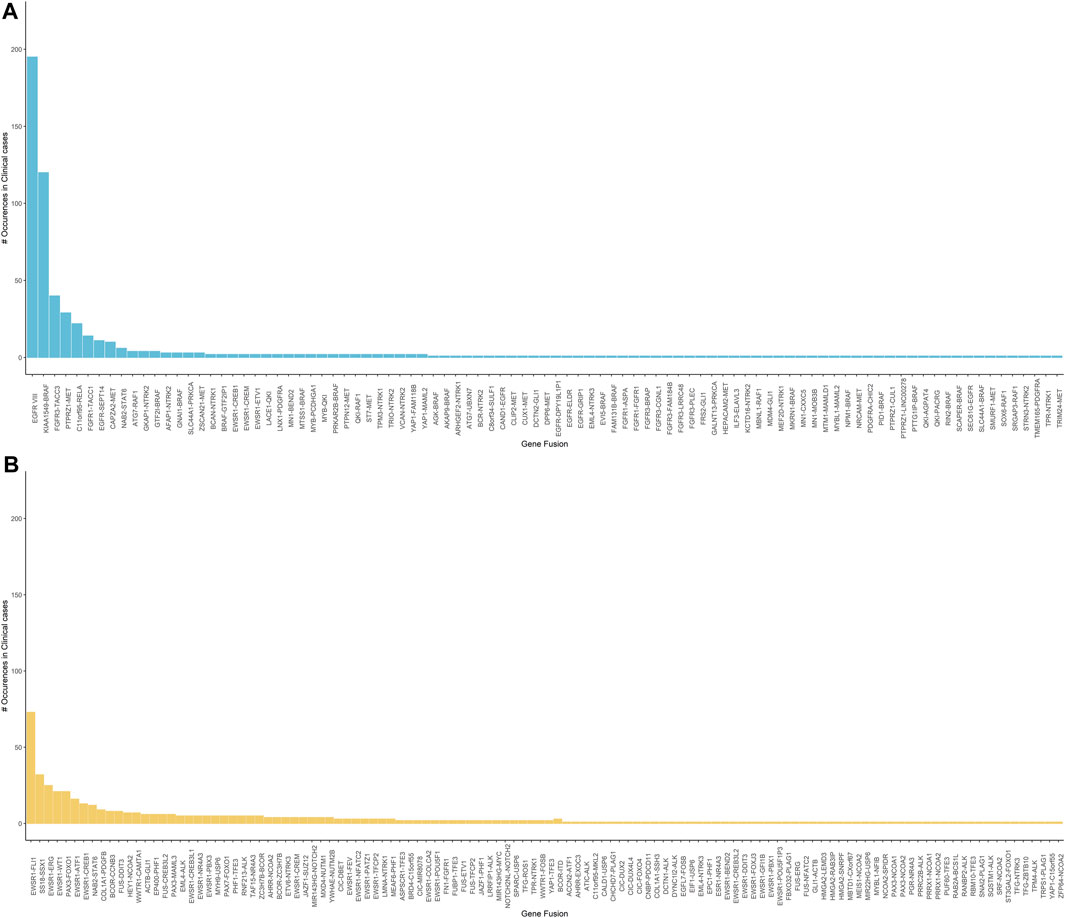
FIGURE 7. (A) Clinically reported gene fusions in neurological oncology cases indicating frequency of common occurring events in the tested cohort. It is observed that the EGFRvIII is the most observed event followed by the KIAA1549-BRAF and the FGFR3-TACC3 fusion. (B) Clinically reported gene fusions in sarcoma assay indicating frequency of common occurring events in the tested cohort. EWSR1 related fusions are the most commonly observed in our tested cohort.
In the neurological oncology assay, EGFRvIII was the most commonly found rearrangement, 195 out of 2,979 patients tested were identified with this rearrangement. EGFRvIII has been established to be present in up to 28–30% of the glioblastoma cells and constitutes to be a therapeutic target (Rutkowska et al., 2019). KIAA1549-BRAF was identified in 120 patients and this rearrangement has been established to be frequently found in pilocytic astrocytoma (Chen et al., 2019). In addition, our pipeline picked up several established fusions related to glioblastoma such as FGFR3-TACC3 and PTPRZ1-MET. Confirming the presence of NTRK gene fusions can guide treatment of the solid tumors (Filippi et al., 2021). Using our SeekFusion method on this assay, we reported NTRK gene fusions for 46 cases.
In the sarcoma assay, EWSR1 was the most commonly rearranged gene in our case series, with 3′ partners including FLI1, ERG, WT1, CREB1, ATF, PBX3, CREB3L1, TFCP, NR4A3, NFATC2, POU5F1, DDIT3, CREM, PATZ1, and COLCA2 in a wide variety of tumor types. SS18-SSX1 was reported in 32 synovial sarcoma cases. PAX3 was the 5′ fusion partner in 29 cases with 3′ partners being FOXO1 fusions identified in alveolar rhabdomyosarcoma cases, MAML3 and NCOA1 fusions in two different biphenotypic sinonasal sarcoma cases.
As described previously the assay chemistry is gene-partner agnostic, enabling identification of novel gene fusions, and many novel fusions were detected in the clinical patient cohort. We have summarized all the neurological cancer and sarcoma gene fusions identified by our method in the Supplementary Table S7. We present two cases here in detail.
Case 1 is a 45-year-old male who presented with a lower left extremity mass suggestive of a low-grade mesenchymal neoplasm. By immunohistochemistry (IHC) the neoplastic cells were focally positive for desmin, epithelial membrane antigen and STAT6 and showed limited expression of TRK. ALK, OSCAR keratin and TLE1 were negative. Histologically the neoplasm was a very unusual appearing, multicystic proliferation of rather bland small round epithelioid cells, in areas showing the presence of an apparent second cell population, consisting of flattened cells with more abundant eosinophilic cytoplasm. This morphologic appearance was suggestive of at some level an adnexal lesion however a variety of epithelial markers are essentially negative. It also appeared to be related to angiomatoid fibrous histiocytoma although the morphologic features of this lesion are atypical and although it showed very limited coexpression of desmin and EMA it was negative for the EWSR1 and FUS gene fusions that characterize this tumor. Based on the very bland morphology of this lesion and the extremely low mitotic rate it is believed that the lesion is either entirely benign or at most has some limited capacity for local recurrence. NGS testing identified a novel TFG-ZBTB10 fusion (Figure 8A). The specific fusion TFG-ZBTB10 has not been described as a recurrent oncogenic event. However, both genes have been involved in fusion events with several other partners. TFG (Trafficking for Endoplasmic Reticulum to Golgi Regulator) has been reported as a 5′ partner in several fusions genes in acute leukemias, sarcomas and carcinomas. In contrast, ZBTB10 (Zinc Finger and BTB Domain-Containing Protein 10), which encodes for a zinc finger protein, has been found in isolated fusion events with other partner genes in breast and colonic adenocarcinomas.
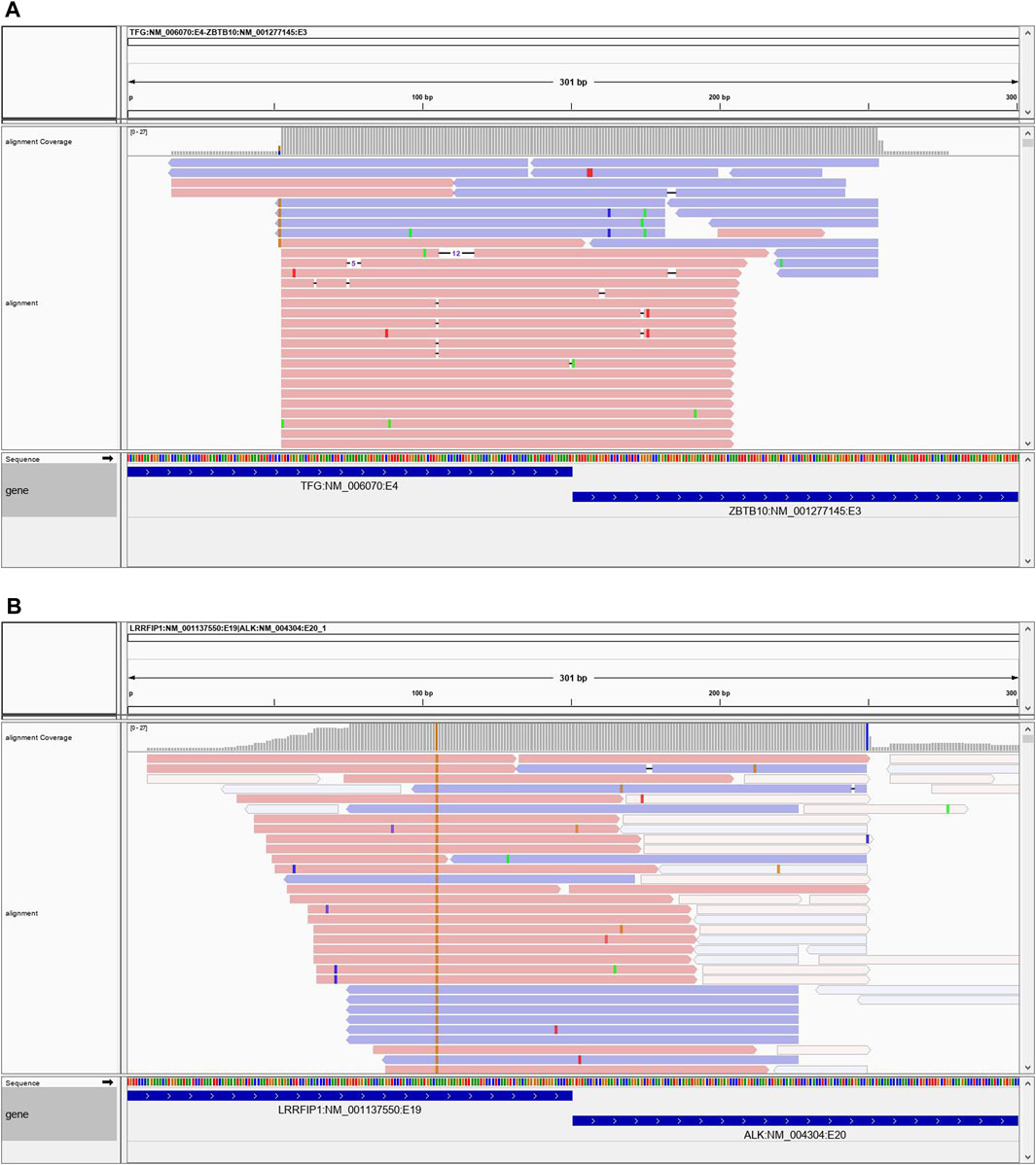
FIGURE 8. (A) Case 1 showing the reads spanning the TFG-ZBTB10 fusion. (B) Case 2 showing spanning reads for the LRRFIP1-ALK fusion.
Case 2 is a 23-year-old male who presented with a 14 × 4.8 × 7.8 cm right chest mass which was eroding the underlying clavicle. Microscopically, this is a spindle cell neoplasm showing a whorling, fascicular growth pattern with scattered chronic inflammatory cells and foci of hyalinized fibrosis. The somewhat enlarged nuclei raise concern for the possibility of a low-grade sarcoma, but they maintain a rather uniform appearance and lack significant atypia. IHC studies show the tumor cells to be positive for FLI-1 and negative for S100 protein, SOX10, desmin, myogenin, WT1, SMA, CD34 and pan-cytokeratin. Given these findings, the differential diagnosis included follicular dendritic sarcoma and angiomatoid fibrous histiocytoma. Additional stains performed locally showed the cells to be negative for CD31, CD35 and CD21. NGS testing revealed a novel RRFIP1-ALK gene fusion (Figure 8B). ALK fusions have been identified in a variety of neoplasms, including inflammatory myofibroblastic tumor, many subtypes of lymphomas and leukemias, adenocarcinomas, benign fibrous histiocytoma, and several other tumors. LRRFIP1 encodes the leucine-rich repeat flightless-interacting protein 1 (FLI-1 interacting protein), a DNA binding transcriptional repressor and may regulate expression of TNF, EGFR and PDGFA. Additionally, it may regulate smooth muscle cell proliferation following arterial damage through PDGFA repression. Previously, an LRRFIP1-MET fusion was identified in an atypical Spitz tumor (PMID: 26013381). There is also another report of an LRRFIP1-FGFR1 fusion in a myeloid neoplasm (PMID: 19369959). While the LRRFIP1-ALK gene fusion observed in this case has not been previously reported in the literature, tumors with ALK fusions have been shown to respond to ALK-inhibitor therapies. Given the histologic findings, it was felt that the tumor was within the spectrum of an inflammatory myofibroblastic tumor.
We have developed a novel bioinformatics pipeline to enable accurate and precise gene fusion detection for commonly used RNA UMI-based amplicon NGS assays. A combination of traditional alignment and de-novo assembly-based approaches enables SeekFusion to increase accuracy of fusion calling over other published and widely used gene fusion-calling algorithms, while maintaining reasonable computational expense. We have demonstrated that SeekFusion exhibits analytical sensitivity with minimal false positive calls and produces higher read support per gene fusion call when compared to other benchmarked bioinformatics tools. Furthermore, SeekFusion is computationally efficient and capable of facilitating realistic turnaround times in a clinical setting. The bioinformatics pipeline and quality check turnaround times are less than a day for the neurological oncology assay and the sarcoma assays mentioned in our study, which enables an analytic time of 14 days for the assay, from sample collection, until clinical report.
Although STAR-Fusion, JAFFA-Hybrid and TOPHAT-Fusion have previously been benchmarked to perform adequately for gene fusion detection using standard RNA-Seq assays (Haas et al., 2019), our study demonstrates these tools lack in performance for RNA UMI-based amplicon NGS assays. SeekFusion provides a tailored solution capable of sensitive and specific fusion gene detection from commonly used assays such as QIAseq RNAscan or Archer FusionPlex, thus making it widely applicable for clinical use. SeekFusion pipeline’s applicability to routine clinical use is further aided by its DOCKER-based deployment. Despite the pipeline’s complex multi-step operation, this packaging is compatible with a wide range of deployment systems including high performance clusters and cloud computing platforms. The implementation conceals the minutiae of the pipeline architecture from end-users and ensures ease of configuration with limited requirements for computational expertise.
In terms of computational run-time, all tested algorithms performed at a level that is amenable to clinical turnaround times, with the possible exception of JAFFA, which demonstrated obvious scalability issues and required as much as 6 hours per sample to complete a single analysis. STAR-Fusion, Tophat-Fusion and SeekFusion performed similarly, although STAR-Fusion showed overall lower run-times than the other tools. Considered in totality, however, the overall performance characteristics of SeekFusion combined with clinically favorable run-times demonstrate it to be the optimal solution for fusion transcript identification.
SeekFusion pipeline’s use of common output file formats represent another advantage for its implementation. Most gene fusion calling algorithms utilize arbitrary formats which present compatibility issues with downstream tools. The use of a standard VCF output for candidate events enables more seamless integration with existing tools and workflows. The generation of an IGV session file further facilitates the utilization of SeekFusion’s outputs with common downstream components. The ability to readily visualize the level of support and alignment quality for fusion-supporting reads is a key functionality. The visualization functionality is currently unavailable in the other tested gene fusion calling software and allows end-users to visually judge the quality of the supporting reads and infer confidence of a gene fusion call.
A limitation of the previously published tools profiled in this study, beyond core performance metrics, is the lack of ability to detect clinically relevant single gene events (Supplementary Table S4), such as the EGFR vIII transcript variant that is a relatively frequent event in glioblastomas (An et al., 2018). The ability of SeekFusion to detect aberrant single-gene events expands its clinical utility and offers additional advantages over pre-existing algorithms. Customization of filter settings in the SeekFusion pipeline enables the inclusion of specific single gene events as required by clinical end-users, while maintaining a low rate of false positives by excluding normal transcript variants. Furthermore, we have demonstrated SeekFusion’s ability to detect novel fusions leveraging the gene-partner agnostic Qiaseq RNA, or similar chemistry, further evidencing its clinical utility. It should be noted that SeekFusion has been tailored to utilize the outputs from common PCR and UMI-based RNA assays such as the Qiaseq assay. It is therefore not likely to natively generalize to alternative, clinically utilized, capture-based methods. While these alternative approaches may be fundamentally similar, further customization and benchmarking will be required to assess the potential applicability of SeekFusion to other chemistries and represents a foundation for future development.
A potential limitation of our study was the relatively small number of clinical samples that were available for initial benchmarking. This reflected the difficulties in obtaining broader research consent, which we overcame by supplementing real clinical samples with oligonucleotide spike-ins and in-silico simulated data. Despite the diversity of the test data utilized, SeekFusion demonstrated high analytical sensitivity and specificity across all datatypes when compared to other available tools, further supporting SeekFusion pipeline’s accuracy and versatility. SeekFusion pipeline’s packaging allows it to be installed and deployed in a clinical setting with minimal efforts. The pipeline enables customization of thresholds, tools, and settings through a configuration file. The setup of SeekFusion for another clinical assay is easily enabled through simple user configuration, as mentioned in the readme instructions in https://hub.docker.com/repository/docker/jagadhesh89/seekfusion. To validate SeekFusion and establish analytical sensitivity and specificity for a similar clinical assay, it is recommended to run the pipeline on a robust set of known positives and negatives, including positive in-silico controls and a positive control with spiked in oligonucleotides.
In summary, SeekFusion represents an accurate, time-efficient and versatile solution for the clinical detection of fusion transcripts from common RNA UMI-based amplicon NGS assays. It is our hope that the tool’s public availability and cross-platform capabilities will enable ready deployment and facilitate accurate gene fusion identification in a range of clinical laboratory and disease settings.
Datasets used for benchmarking analyses in this study is available in the NCBI’s SRA website. The docker shared in article has test samples with real fusions and the entire code base. Any fusion sequence or breakpoint information for identified fusion transcripts in clinical cases can be shared on request. Requests to access the datasets should be directed to YmFsYW4uamFnYWRoZXNod2FyQG1heW8uZWR1.
The studies involving human participants were reviewed and approved by Minimal risk IRB at the Mayo Clinic. The study involves no patient interaction or counseling. The study involves using clinical sample data and processing it through pipeline developed during the study and reporting results. There is no PHI or personal identifiers revealed in the study and the results are completely anonymized. The IRB approved study is titled “Investigation of residual samples using next-generation sequencing, conventional karyotyping, FISH, Array, PCR and Sanger sequencing.” The IRB number is 15-007359. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was funded by the Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, Minnesota.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Dr. Jaime Davila from QHS (Health Sciences Research) - Biomedical Statistics and Informatics, Mayo Clinic, Rochester, Minnesota for valuable advice for the pipeline development. We also thank Jason Sinnwell and Poulami Barman from QHS (Health Sciences Research) - Biomedical Statistics and Informatics, Mayo Clinic, Rochester, Minnesota for their valuable advice for statistics.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.739054/full#supplementary-material
Abel, H. J., Al-Kateb, H., Cottrell, C. E., Bredemeyer, A. J., Pritchard, C. C., Grossmann, A. H., et al. (2014). Detection of Gene Rearrangements in Targeted Clinical Next-Generation Sequencing. J. Mol. Diagn. 16 (4), 405–417. doi:10.1016/j.jmoldx.2014.03.006
An, Z., Aksoy, O., Zheng, T., Fan, Q.-W., and Weiss, W. A. (2018). Epidermal Growth Factor Receptor and EGFRvIII in Glioblastoma: Signaling Pathways and Targeted Therapies. Oncogene 37 (12), 1561–1575. doi:10.1038/s41388-017-0045-7
Bender, S., Gronych, J., Warnatz, H-J., Hutter, B., Gröbner, S., Ryzhova, M., et al. (2016). Recurrent MET Fusion Genes Represent a Drug Target in Pediatric Glioblastoma. Nat. Med. 22 (11), 1314–1320. doi:10.1038/nm.4204
Blessing, M. M., Blackburn, P. R., Krishnan, C., Harrod, V. L., Barr Fritcher, E. G., Zysk, C. D., et al. (2019). Desmoplastic Infantile Ganglioglioma: A MAPK Pathway-Driven and Microglia/Macrophage-Rich Neuroepithelial Tumor. J. Neuropathol. Exp. Neurol. 78 (11), 1011–1021. doi:10.1093/jnen/nlz086
Blomquist, T. M., Crawford, E. L., Lovett, J. L., Yeo, J., Stanoszek, L. M., Levin, A., et al. (2013). Targeted RNA-Sequencing with Competitive Multiplex-PCR Amplicon Libraries. PLoS One 8 (11). e79120. doi:10.1371/journal.pone.0079120
Carrara, M., Beccuti, M., Cavallo, F., Donatelli, S., Lazzarato, F., Cordero, F., et al. (2013). State of Art Fusion-Finder Algorithms Are Suitable to Detect Transcription-Induced Chimeras in normal Tissues. BMC Bioinformatics 14 Suppl 7 (Suppl. 7Suppl 7), S2–S. doi:10.1186/1471-2105-14-S7-S2
Carrara, M., Beccuti, M., Lazzarato, F., Cavallo, F., Cordero, F., Donatelli, S., et al. (2013). State-of-the-art Fusion-Finder Algorithms Sensitivity and Specificity. Biomed. Res. Int. 2013, 340620. doi:10.1155/2013/340620
Chen, R., Keoni, C., Waker, C. A., Lober, R. M., Chen, Y.-H., and Gutmann, D. H. (2019). KIAA1549-BRAF Expression Establishes a Permissive Tumor Microenvironment through NFκB-Mediated CCL2 Production. Neoplasia 21 (1), 52–60. doi:10.1016/j.neo.2018.11.007
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: an Ultra-fast All-In-One FASTQ Preprocessor. Bioinformatics 34 (17), i884–i890. doi:10.1093/bioinformatics/bty560
Davidson, N. M., Majewski, I. J., and Oshlack, A. (2015). JAFFA: High Sensitivity Transcriptome-Focused Fusion Gene Detection. Genome Med. 7 (1), 43. doi:10.1186/s13073-015-0167-x
de Myttenaere, A., Golden, B., Le Grand, B., and Rossi, F. (2016). Mean Absolute Percentage Error for Regression Models. Neurocomputing 192, 38–48. doi:10.1016/j.neucom.2015.12.114
Drilon, A., Wang, L., Arcila, M. E., Balasubramanian, S., Greenbowe, J. R., Ross, J. S., et al. (2015). Broad, Hybrid Capture-Based Next-Generation Sequencing Identifies Actionable Genomic Alterations in Lung Adenocarcinomas Otherwise Negative for Such Alterations by Other Genomic Testing Approaches. Clin. Cancer Res. 21 (16), 3631–3639. doi:10.1158/1078-0432.ccr-14-2683
Docker, Merkel D. (2014). Lightweight Linux Containers for Consistent Development and Deployment. Linux Journal 2014 (239), 2.
Filippi, R., Depetris, I., and Satolli, M. A. (2021). Evaluating Larotrectinib for the Treatment of Advanced Solid Tumors Harboring an NTRK Gene Fusion. Expert Opin. Pharmacother. 22 (6), 677–684. doi:10.1080/14656566.2021.1876664
Gliem, T. J., and Aypar, U. (2017). Development of a Chromosomal Microarray Test for the Detection of Abnormalities in Formalin-Fixed, Paraffin-Embedded Products of Conception Specimens. J. Mol. Diagn. 19 (6), 843–847. doi:10.1016/j.jmoldx.2017.07.001
Haas, B. J., Dobin, A., Li, B., Stransky, N., Pochet, N., and Regev, A. (2019). Accuracy Assessment of Fusion Transcript Detection via Read-Mapping and De Novo Fusion Transcript Assembly-Based Methods. Genome Biol. 20, 213. doi:10.1186/s13059-019-1842-9
Haynes, B. C., Blidner, R. A., Cardwell, R. D., Zeigler, R., Gokul, S., Thibert, J. R., et al. (2019). An Integrated Next-Generation Sequencing System for Analyzing DNA Mutations, Gene Fusions, and RNA Expression in Lung Cancer. Translational Oncol. 12 (6), 836–845. doi:10.1016/j.tranon.2019.02.012
Huang, X., and Madan, A. (1999). CAP3: A DNA Sequence Assembly Program. Genome Res. 9 (9), 868–877. doi:10.1101/gr.9.9.868
Jain, P., Silva, A., Han, H. J., Lang, S. S., Zhu, Y., Boucher, K., et al. (2017). Overcoming Resistance to Single-Agent Therapy for Oncogenic BRAF Gene Fusions via Combinatorial Targeting of MAPK and PI3K/mTOR Signaling Pathways. Oncotarget 8 (No 49), 84697–84713. doi:10.18632/oncotarget.20949
Kent, W. J. (2002). BLAT---The BLAST-like Alignment Tool. Genome Res. 12 (4), 656–664. doi:10.1101/gr.229202
Kim, D., and Salzberg, S. L. (2011). TopHat-Fusion: an Algorithm for Discovery of Novel Fusion Transcripts. Genome Biol. 12 (8), R72. doi:10.1186/gb-2011-12-8-r72
Kim, S.-I., Lee, Y., Kim, S. K., Kang, H. J., and Park, S.-H. (2017). Aggressive Supratentorial Ependymoma, RELA Fusion-Positive with Extracranial Metastasis: A Case Report. J. Pathol. Transl Med. 51 (6), 588–593. doi:10.4132/jptm.2017.08.10
Kivioja, T., Vähärautio, A., Karlsson, K., Bonke, M., Enge, M., Linnarsson, S., et al. (2012). Counting Absolute Numbers of Molecules Using Unique Molecular Identifiers. Nat. Methods 9 (1), 72–74. doi:10.1038/nmeth.1778
Li, H., and Durbin, R. (2009). Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map Format and SAMtools. Bioinformatics 25 (16), 2078–2079. doi:10.1093/bioinformatics/btp352
Liu, S., Tsai, W.-H., Ding, Y., Chen, R., Fang, Z., Huo, Z., et al. (2016). Comprehensive Evaluation of Fusion Transcript Detection Algorithms and a Meta-Caller to Combine Top Performing Methods in Paired-End RNA-Seq Data. Nucleic Acids Res. 44 (5), e47. doi:10.1093/nar/gkv1234
Mitelman, F., Johansson, B., and Mertens, F. (2007). The Impact of Translocations and Gene Fusions on Cancer Causation. Nat. Rev. Cancer 7 (4), 233–245. doi:10.1038/nrc2091
Moskalev, E. A., Frohnauer, J., Merkelbach-Bruse, S., Schildhaus, H.-U., Dimmler, A., Schubert, T., et al. (2014). Sensitive and Specific Detection of EML4-ALK Rearrangements in Non-small Cell Lung Cancer (NSCLC) Specimens by Multiplex Amplicon RNA Massive Parallel Sequencing. Lung Cancer 84 (3), 215–221. doi:10.1016/j.lungcan.2014.03.002
Pekar-Zlotin, M., Hirsch, F. R., Soussan-Gutman, L., Ilouze, M., Dvir, A., Boyle, T., et al. (2015). Fluorescence In Situ Hybridization, Immunohistochemistry, and Next-Generation Sequencing for Detection of EML4-ALK Rearrangement in Lung Cancer. Oncologist 20 (3), 316–322. doi:10.1634/theoncologist.2014-0389
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative Genomics Viewer. Nat. Biotechnol. 29 (1), 24–26. doi:10.1038/nbt.1754
Rutkowska, A., Stoczyńska-Fidelus, E., Janik, K., Włodarczyk, A., and Rieske, P (2019). EGFRvIII: An Oncogene with Ambiguous Role. J. Oncol. 2019, 1092587. doi:10.1155/2019/1092587
Shaw, A. T., Solomon, B. J., Chiari, R., Riely, G. J., Besse, B., Soo, R. A., et al. (2019). Lorlatinib in Advanced ROS1-Positive Non-small-cell Lung Cancer: a Multicentre, Open-Label, Single-Arm, Phase 1–2 Trial. Lancet Oncol. 20 (12)–701.
Sze, S. H., Pimsler, M. L., Tomberlin, J. K., Jones, C. D., and Tarone, A. M. (2017). A Scalable and Memory-Efficient Algorithm for De Novo Transcriptome Assembly of Non-model Organisms. BMC Genomics 18 (4), 387. doi:10.1186/s12864-017-3735-1
Thilly, R. S. Ca. W. G. (1993). Specificity, Efficiency, and Fidelity of PCR. Genome Res. 3, S18–S29.
Tofallis, C. (2015). A Better Measure of Relative Prediction Accuracy for Model Selection and Model Estimation. J. Oper. Res. Soc. 66 (8), 1352–1362. doi:10.1057/jors.2014.103
Voss, K. (2017). Full-stack Genomics Pipelining with GATK4 + WDL + Cromwell. F1000Research. 6 (1381).
Vu, T. N., Deng, W., Trac, Q. T., Calza, S., Hwang, W., and Pawitan, Y. (2018). A Fast Detection of Fusion Genes from Paired-End RNA-Seq Data. BMC Genomics 19 (1), 786. doi:10.1186/s12864-018-5156-1
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-seq: a Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 10 (1), 57–63. doi:10.1038/nrg2484
Wilcoxon, F. (1945). Individual Comparisons by Ranking Methods. Biometrics Bull. 1 (6), 80–83. doi:10.2307/3001968
Wong, R. K. Y., MacMahon, M., Woodside, J. V., and Simpson, D. A. (2019). A Comparison of RNA Extraction and Sequencing Protocols for Detection of Small RNAs in Plasma. BMC Genomics 20 (1), 446. doi:10.1186/s12864-019-5826-7
Keywords: gene fusion, RNA, bioinformatics, UMI consensus, QiaSeq, sarcoma, neuro-oncolgy, neuro-oncological disease
Citation: Balan J, Jenkinson G, Nair A, Saha N, Koganti T, Voss J, Zysk C, Barr Fritcher E, Ross C, Giannini C, Raghunathan A, Kipp B, Jenkins R, Ida C, Halling K, Blackburn P, Dasari S, Oliver G and Klee E (2021) SeekFusion - A Clinically Validated Fusion Transcript Detection Pipeline for PCR-Based Next-Generation Sequencing of RNA. Front. Genet. 12:739054. doi: 10.3389/fgene.2021.739054
Received: 21 July 2021; Accepted: 24 September 2021;
Published: 22 October 2021.
Edited by:
Andrew J. Mungall, Canada’s Michael Smith Genome Sciences Centre, CanadaReviewed by:
Prashanth N. Suravajhala, Amrita Vishwa Vidyapeetham University, IndiaCopyright © 2021 Balan, Jenkinson, Nair, Saha, Koganti, Voss, Zysk, Barr Fritcher, Ross, Giannini, Raghunathan, Kipp, Jenkins, Ida, Halling, Blackburn, Dasari, Oliver and Klee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric W. Klee, S2xlZS5FcmljQG1heW8uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.