 Elena-Raluca Nicoli1
Elena-Raluca Nicoli1 Ida Annunziata2
Ida Annunziata2 Alessandra d’Azzo2,3
Alessandra d’Azzo2,3 Frances M. Platt4
Frances M. Platt4 Cynthia J. Tifft1,5
Cynthia J. Tifft1,5 Karolina M. Stepien6,7*
Karolina M. Stepien6,7*- 1Glycosphingolipid and Glycoprotein Disorders Unit, Medical Genetics Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, United States
- 2Department of Genetics, St. Jude Children’s Research Hospital, Memphis, TN, United States
- 3Department of Anatomy and Neurobiology, College of Graduate Health Sciences, University of Tennessee Health Science Center, Memphis, TN, United States
- 4Department of Pharmacology, University of Oxford, Oxford, United Kingdom
- 5Office of the Director, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, United States
- 6Adult Inherited Metabolic Disorders, Salford Royal NHS Foundation Trust, Salford, United Kingdom
- 7Division of Diabetes, Endocrinology and Gastroenterology, University of Manchester, Manchester, United Kingdom
GM1 gangliosidosis is a progressive, neurosomatic, lysosomal storage disorder caused by mutations in the GLB1 gene encoding the enzyme β-galactosidase. Absent or reduced β-galactosidase activity leads to the accumulation of β-linked galactose-containing glycoconjugates including the glycosphingolipid (GSL) GM1-ganglioside in neuronal tissue. GM1-gangliosidosis is classified into three forms [Type I (infantile), Type II (late-infantile and juvenile), and Type III (adult)], based on the age of onset of clinical symptoms, although the disorder is really a continuum that correlates only partially with the levels of residual enzyme activity. Severe neurocognitive decline is a feature of Type I and II disease and is associated with premature mortality. Most of the disease-causing β-galactosidase mutations reported in the literature are clustered in exons 2, 6, 15, and 16 of the GLB1 gene. So far 261 pathogenic variants have been described, missense/nonsense mutations being the most prevalent. There are five mouse models of GM1-gangliosidosis reported in the literature generated using different targeting strategies of the Glb1 murine locus. Individual models differ in terms of age of onset of the clinical, biochemical, and pathological signs and symptoms, and overall lifespan. However, they do share the major abnormalities and neurological symptoms that are characteristic of the most severe forms of GM1-gangliosidosis. These mouse models have been used to study pathogenic mechanisms, to identify biomarkers, and to evaluate therapeutic strategies. Three GLB1 gene therapy trials are currently recruiting Type I and Type II patients (NCT04273269, NCT03952637, and NCT04713475) and Type II and Type III patients are being recruited for a trial utilizing the glucosylceramide synthase inhibitor, venglustat (NCT04221451).
Introduction
Glycoconjugates play a key role in cellular function and are tightly regulated both in terms of biosynthesis and catabolism. β-Galactosidase (β-GAL) is a lysosomal hydrolase that cleaves β-linked galactose residues from the non-reducing end of glycan moieties found in various glycoconjugates. Reduction in β-GAL activity leads to the accumulation of GM1 ganglioside and its asialo derivative GA1, primarily in lysosomes of neuronal tissue (Caciotti et al., 2011; Jarnes Utz et al., 2017). The first description of GM1 and GA1 storage was made in a case of amaurotic idiocy (as it was then called), now called GM1 gangliosidosis (Jatzkewitz and Sandhoff, 1963). In addition to the storage of GM1 ganglioside, other glycoconjugates with β-galactose at the non-reducing end are detectable in high concentration in patients’ urine, including N-linked glycans, and various O-linked glycans (Lawrence et al., 2019). These disease-related β-linked galactose-terminal oligosaccharides arise from the lysosomal breakdown of glycoproteins that are stored in the brain (Tsay and Dawson, 1976), liver (Holmes and O’Brien, 1978), and other biological fluids including urine (Brunetti-Pierri and Scaglia, 2008; Bruggink et al., 2012) and amniotic fluid (Piraud et al., 2017). The significant proportion of these soluble glycans are metabolites of incompletely degraded N-linked glycans, such as A1G1, A2G2, A3G3, and A4G4. The N-glycan metabolite A2G2 was proposed to be a surrogate glycan biomarker of GM1 gangliosidosis (Lawrence et al., 2019).
GM1 gangliosidosis is an example of a family of inherited metabolic disorders termed lysosomal storage diseases. The estimated incidence of GM1 gangliosidosis is 1:100,000–200,000 live births (Brunetti-Pierri and Scaglia, 2008). However, some forms of the disease appear to be more prevalent in specific geographical areas, including Southern Brazil and Japan and among the Roma (Yoshida et al., 1991; Severini et al., 1999; Sinigerska et al., 2006).
GM1 presents with a continuum of disease severity but patients are loosely classified based on the age of onset of the symptoms into Type I (infantile), Type II (late-infantile and juvenile) and Type III (adult). In principle, disease severity should inversely correlate with residual enzyme activity levels. However, β-GAL activity is mostly measured with the synthetic fluorogenic substrate 4-methyl-umbelliferyl-β-D-galactopyranoside, which may not be accurate enough to estimate the clinical course of the disease on the basis of the measured residual enzyme activity (Inui et al., 1990; Caciotti et al., 2011; Ferreira and Gahl, 2017; Figure 1).
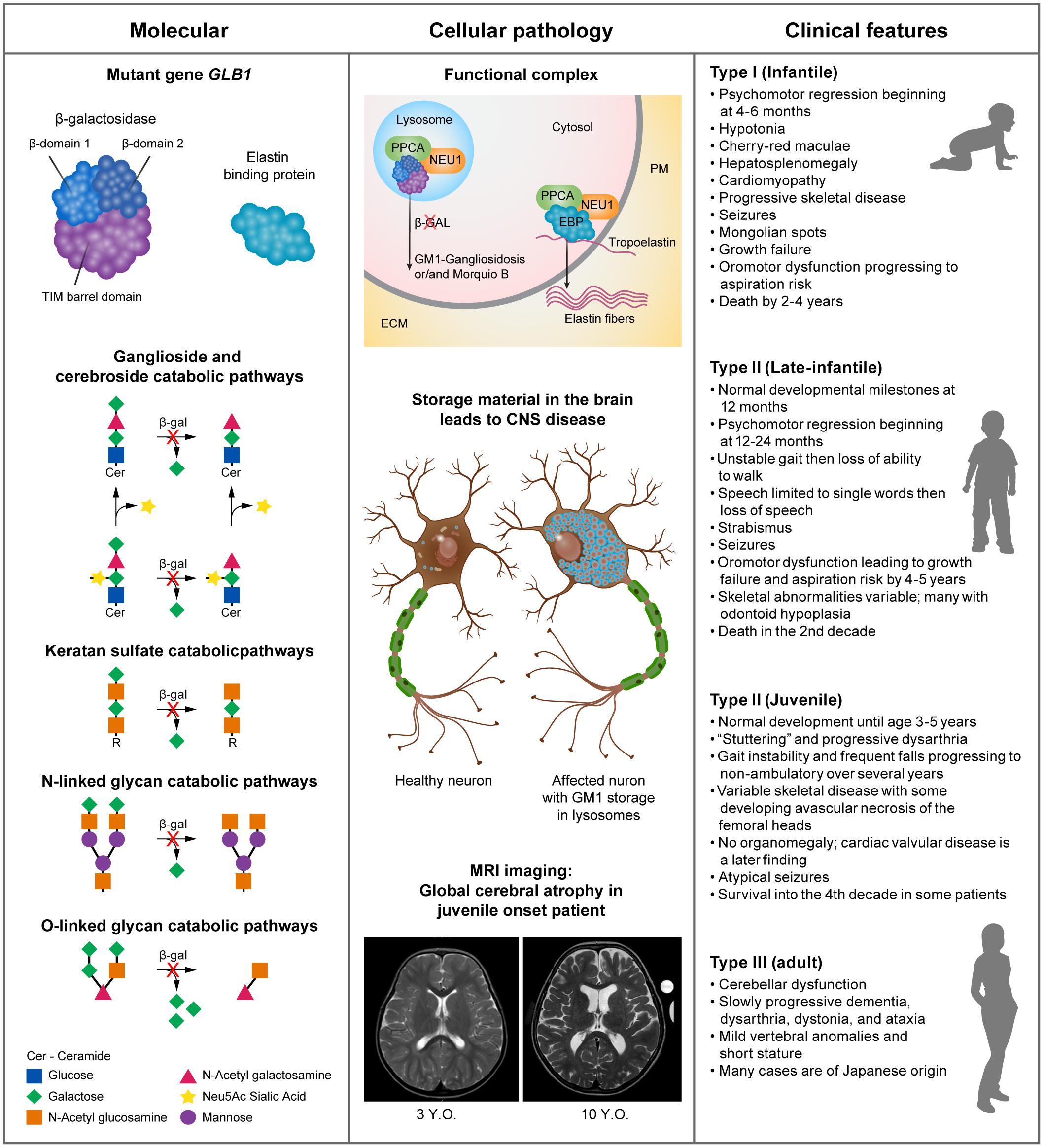
Figure 1. Pathogenesis and clinical manifestations. Panel 1: human β-GAL is composed of a catalytic TIM barrel domain followed by β-domain 1 and β-domain 2 (Ohto et al., 2012). Mutations in the GLB1 gene lead to impaired enzyme activity, which results in the progressive accumulation of complex gangliosides, specifically GM1. This, in turn, initiates a series of pathogenic events that ultimately lead to neurodegeneration (Kolter, 2012; Annunziata et al., 2018). Panel 2: through alternative splicing, the GLB1 gene gives rise to two transcripts, one encoding the hydrolytic enzyme β-galactosidase and the other the elastin binding protein (EBP). The primary role of EBP is to chaperone the deposition of elastin fibers in the extracellular matrix (ECM). β-galactosidase (GLB1) and EBP are found in complex with PPCA and NEU1 in lysosomes and the plasma membrane (PM), respectively (Caciotti et al., 2005; Bonten et al., 2014). Panel 3: although GM1 gangliosidosis is a disease continuum it can be loosely divided into 3 types, with Type II having 2 subtypes. The common use of a synthetic fluorogenic substrate to measure β-GAL activity makes it difficult to establish an accurate correlation between residual enzyme activity and clinical outcome. This may also be complicated by the regulatory and post posttranslational mechanisms that influence GM1-ganglioside catabolism and may vary among patients (Breiden and Sandhoff, 2019). The main symptoms of the disease commonly found in each type/subtype are summarized.
Bi-allelic mutations in GLB1 result in a reduction in β-GAL activity and the build-up of GM1 ganglioside in multiple tissues including the brain (Brunetti-Pierri and Scaglia, 2008; Karimzadeh et al., 2017; Rha et al., 2021) leading to severe neurodegeneration resulting in morbidity and premature mortality (Ferreira and Gahl, 2017). More than 200 disease-causing mutations have been identified across the GLB1 gene, particularly in exons 2, 6, 15, and 16 (Figure 2).
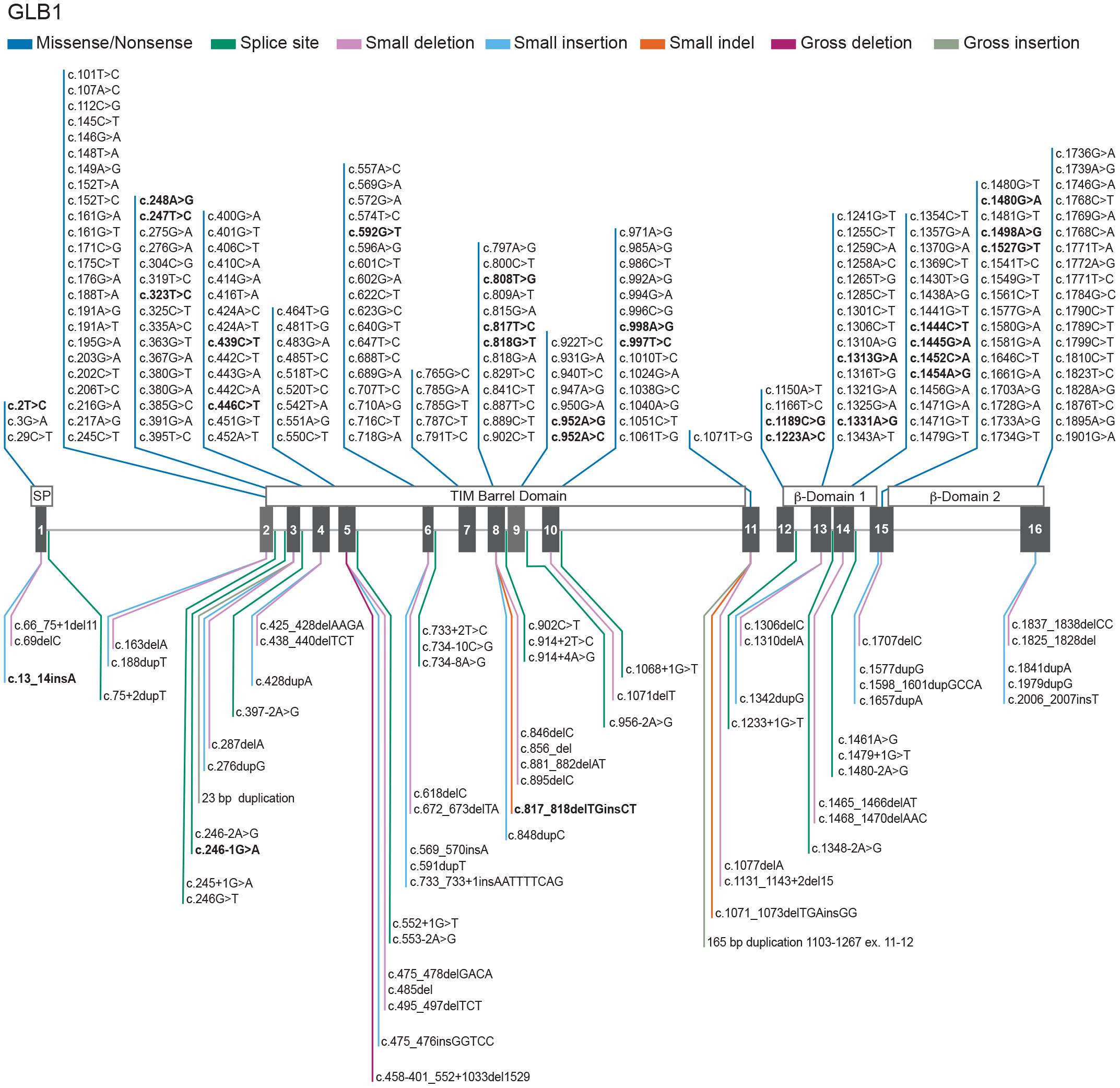
Figure 2. Genotypes in GM1 gangliosidosis. Schematic representation of 261 GLB1 variants with a reported phenotype of GM1-gangliosidosis and/or Morquio B registered in the database HGMD (2021) updated with the novel variants from Tebani et al. (2021). The GLB1 gene is located on the short arm of chromosome 3 (3p21.33). GM1 gangliosidosis and Morquio B disease result from biallelic mutations in GLB1 gene. Mutations affecting the catalytical site of the β-GAL enzyme may reduce or eliminate the degradation of GM1 substrate. GLB1 cDNA sequence NM_000404.4; Ref SeqGene NG_009005.1; 194 Missense/Nonsense (dark blue), 20 Splicing substitutions (dark green), 25 small deletions (pink), 17 small insertion/duplications (light blue), 2 small indels (orange), 1 gross deletion (purple), 2 gross insertion/duplications (light green). Bold text represents GM1 gangliosidosis and/or Morquio B reported phenotypes. A summary of the 261 reported variants with a phenotype of GM1 gangliosidosis and/or Morquio B disease can be found at GLB1 (HGMD, 2021; Tebani et al., 2021). The main domains of the protein are shown above the exons. For a full description on the clinical pathogenicity of each variant the database ClinVar (Landrum et al., 2018) provides information about genomic variation and its relationship to human health at GLB1 (ClinVar, 2021).
A smaller subset of mutations predominantly near the 3′ end of GLB1 can result in another lysosomal storage disease, Morquio B disease (mucopolysaccharidosis type IVB), which is characterized by primary lysosomal accumulation of the glycosaminoglycan keratan sulfate. Morquio B patients have predominantly skeletal abnormalities without the neurodegenerative aspects of GM1 gangliosidosis (Ferreira and Gahl, 2017). Cases with phenotypic features of both GM1 gangliosidosis and Morquio B disease have also been described (Mayer et al., 2009).
Distinct GLB1 gene mutations that affect the affinity of the enzyme for the two main substrates, GM1 ganglioside and keratan sulfate, explain at least in part the different clinical manifestations and the degree and type of storage material (Figure 2).
Five murine models have been developed that mimic the human phenotype and have formed that basis of our current understanding of disease pathogenesis. They have also been instrumental in the trialing of new therapies.
Comprehensive reviews of GM1 gangliosidosis have been recently published (Regier et al., 1993; Breiden and Sandhoff, 2019; Lawrence et al., 2019; Lang et al., 2020; Rha et al., 2021). In this mini review, we will focus on the clinical spectrum of disease and genetic variants, our current understanding of disease pathogenesis, the utility of available animal models and approaches to therapy for these devastating disorders.
Clinical Manifestations
Among the three subtypes of GM1 gangliosidosis, the infantile form is the most severe, with onset of symptoms before 6 months of age and death early in childhood (Jarnes Utz et al., 2017; Lang et al., 2020). One of the earliest signs that can be seen in infantile disease prenatally is hydrops fetalis, which is associated with many severe cases of LSDs (Stone and Sidransky, 1999), and should stimulate an investigation into the underlying causes of this phenotype (Iyer et al., 2021). The early diagnosis of infantile onset disease would have the greatest chance of successful therapeutic intervention.
Type II consists of late infantile and juvenile subtypes (Figure 1). Late infantile disease patients meet developmental milestones at 12 months but have onset of symptoms generally between 12 and 24 months. Patients quickly lose the ability to ambulate and have difficulty swallowing and handling secretions necessitating gastrostomy placement. Late infantile patients succumb to their illness by the mid second decade.
Juvenile patients have onset at 3–5 years of age, having learned to walk, run, and speak in sentences. The first symptoms may be unsteady gait, frequent falls, and “slurring” of speech progressing to ataxia and dysarthria. Children become unable to walk without assistance then lose the ability to walk. Dysarthria progresses to inability to swallow effectively and subsequent weight loss, necessitating gastrostomy tube placement. Children with juvenile onset disease can live into their 4th decade of life (Regier et al., 1993, 2016; Jarnes Utz et al., 2017).
Adult onset GM1 represents with an attenuated form with slower disease progression and very mild dysmorphic features (Suzuki et al., 2001) and greater clinical variability (Tonin et al., 2019). The onset of symptoms occurs in early childhood to late teens, as described in individuals of Japanese descent (Inui et al., 1990; Arash-Kaps et al., 2019).
Patients with all types of GM1 gangliosidosis can have variable skeletal disease including pectus carinatum with spine and rib changes that lead to restrictive lung disease (Ferreira et al., 2020). Radiographic changes can allow differentiation between the late-infantile and juvenile forms of Type II disease. Odontoid hypoplasia, as observed in all late infantile patients but not juvenile patients, requires cervical spine evaluation in late infantile patients prior to anesthesia, and anesthetic care optimally provided by pediatric anesthesiologists with experience in patients with cervical spine pathology to prevent perioperative morbidity or mortality (Ferreira et al., 2020).
The progressive nature of the neurological disease in GM1 gangliosidosis requires close monitoring. Biomarkers in blood, urine, and CSF biomarkers may be potentially useful in this regard (Gray-Edwards et al., 2017a,b; Rha et al., 2021). Disease progression can also be monitored using minimally invasive neuroimaging. In Type II disease both late infantile and juvenile patients demonstrate progressive atrophy in the cerebrum and cerebellum with greater variability and slower progression in the juvenile subtype. Quantitative magnetic resonance spectroscopy (MRS) shows increasing deficits of N-acetyl aspartate (NAA) across many brain regions with greater and swifter deficits seen in the more rapidly progressive late infantile subtype (Regier et al., 2016). These biomarkers of disease progression also correlate with developmental progression of disease and may serve as useful outcome measures for clinical trials.
In contrast to GM1 gangliosidosis, Morquio B disease (Figure 2) patients do not have CNS disease but can have neurologic compromise due to underlying skeletal disease, such as spinal nerve compression (Abumansour et al., 2020).
Genotypes
There is a poor genotype-phenotype correlation in GM1 gangliosidosis demonstrated by the clinical variability in age of onset and progression of disease even between siblings with the same genotype. One can speculate that polymorphisms or mutations in the other genes of the β-GAL complex, protective protein/cathepsin A (PPCA) and neuraminidase 1 (NEU1) (see Figure 1) may account for this variability. An additional limitation to reach an accurate genotype-phenotype correlation is posed by the way the enzyme assay is commonly performed using a soluble fluorogenic substrate that does not reflect the topology and membrane microenvironment of the natural substrate, GM1 ganglioside. Moreover, regulatory, and post-translational mechanisms that modulate GM1 catabolism further hamper an accurate prediction of the clinical course of the disease in patients, if based only on residual enzyme activity against the synthetic substrate (Breiden and Sandhoff, 2019).
Most patients with GM1 gangliosidosis are compound heterozygotes and aside from biallelic null mutations that produce type I disease, it is difficult to attribute specific phenotypes to any single mutation. Generalizations based on crystallographic structure of the β-GAL enzyme have been attempted (Ohto et al., 2012). Mutations associated with type I/infantile onset GM1 gangliosidosis, for the most part, are located in the core protein region causing β-gal instability, whereas mutations associated with milder phenotypes, such as types II and III GM1 gangliosidosis, tend to be on the protein surface (Morita et al., 2009; Ohto et al., 2012; Rha et al., 2021). Recently, the determination of the 3D structure of murine β-gal in complex with PPCA has revealed that some mutations at conserved amino acid residues found in GM1 gangliosidosis patients affect formation of the complex (Gorelik et al., 2021). These findings further complicate genotype-phenotype correlation, in relation to the penetrance of specific disease phenotypes.
Out of the total 261 reported pathogenic variants associated with a phenotype of GM1 gangliosidosis and/or Morquio B disease, most of them are missense/nonsense (194), and the rest are splicing substitutions (20), small deletions (25), small insertion/duplications (17), small indels (2), gross insertion/duplications (2), and a single large deletion (Figure 2). The largest number of mutations are found in exons 2 (26 variants), 6 (23 variants), 15 (21 variants) and 16 (24 variants). Previous reports implicate exons 2, 6, and 15 (Brunetti-Pierri and Scaglia, 2008) as hot spots for mutations, however exon 16 also harbors multiple pathogenic variants.
Mouse Models
The first two genetically engineered mouse models of GM1 gangliosidosis were reported in 1997 by two groups (Hahn et al., 1997) and (Matsuda et al., 1997a,b). These knockout mice were generated by homologous recombination at the murine Glb1 locus that disrupted the gene by introducing a selectable marker cassette in either exon 6 or exon 15 (Hahn et al., 1997; Matsuda et al., 1997a,b). Both mouse models closely recapitulated the infantile/juvenile onset form of GM1 gangliosidosis and have been used extensively for studying disease pathogenesis and for testing therapeutic modalities (Table 1).
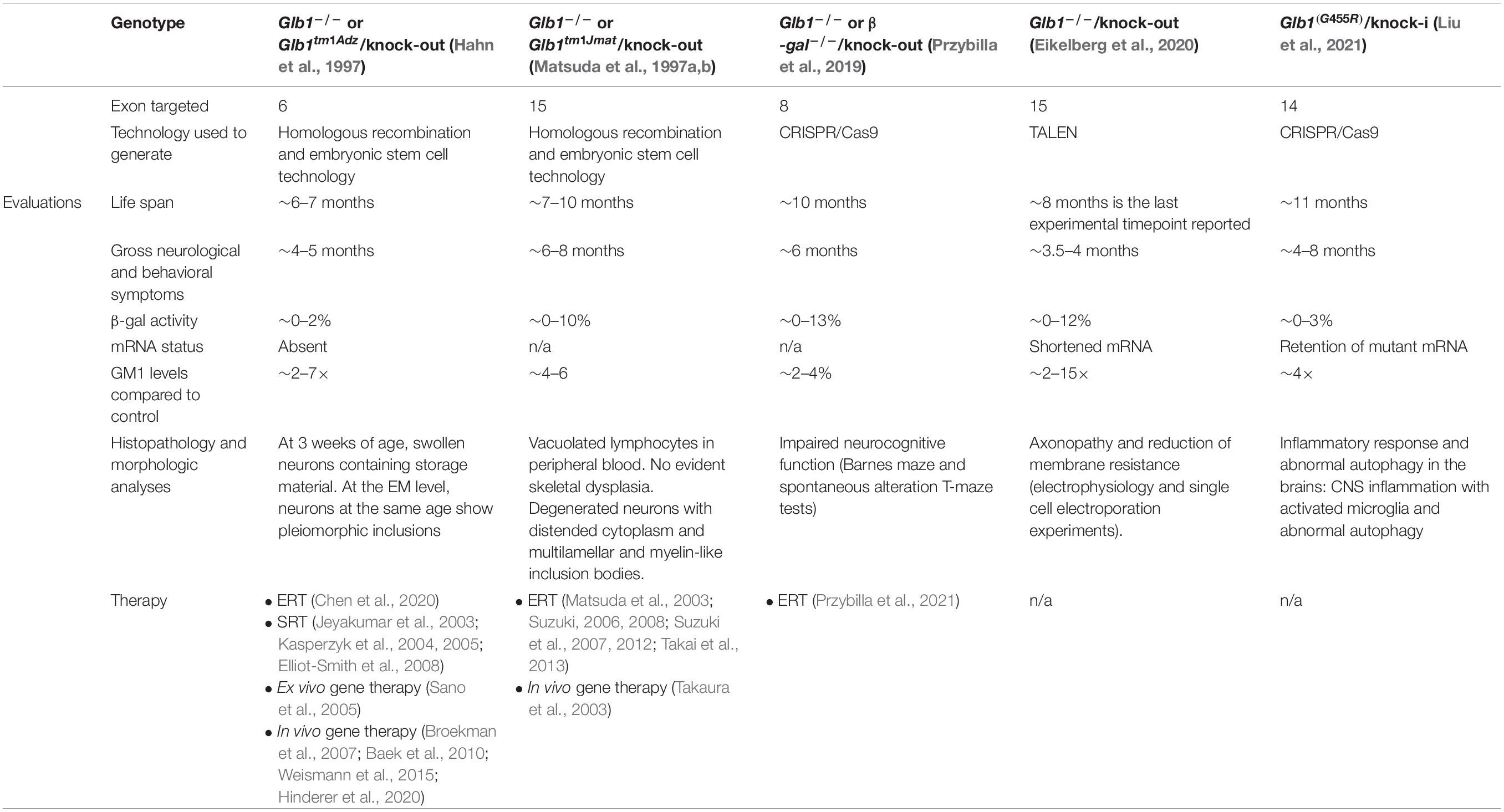
Table 1. Overview of developed GM1 gangliosidosis mouse models: characteristics and therapies tested.
The β-gal–/– mice described by Hahn et al. (1997) showed substantial early neuronal loss in the brain and spinal cord (Tessitore et al., 2004). GM1 is abundant in the neuronal plasma membrane (PM) and is the only ganglioside that can influence Ca2++ transfer across membranes by interacting with Ca2+-binding proteins (Ledeen and Wu, 2015; Annunziata et al., 2018). In β-gal–/– neurons impaired lysosomal degradation of GM1 results in the abnormal accumulation of the ganglioside in internal membranes, specifically those of the ER resulting in two pathogenic effects: (1) it enhances the flux of Ca2+ out of the ER, thereby altering ER Ca2+ levels, which activates an unfolded protein response (UPR) (Tessitore et al., 2004); and (2) it increases the number of membrane contact sites between the ER and mitochondria, known as mitochondria associated ER membranes (MAMs). These GM1-enriched microdomains mediate the abnormal flux of Ca2+ from the ER to the mitochondria, which ultimately results in mitochondria Ca2+ overload (Sano et al., 2009). The combination of these Ca2+-dependent pathogenic events steers the simultaneous activation of UPR and mitochondria-mediated neuronal apoptosis (Tessitore et al., 2004; Sano et al., 2009). In addition, the progressive neurodegeneration in these mice elicits a widespread neuroinflammatory response, accompanied by the release of cytokines and chemokines in the brain interstitial fluid and the CSF (Jeyakumar et al., 2003; Sano et al., 2005), which likely accelerates CNS disease. This neuroinflammatory response was shown to favor the recruitment of genetically modified BM monocytes expressing a therapeutic β-gal enzyme, following ex vivo gene therapy in β-gal–/– mice (Sano et al., 2005) (see below).
With the development of new gene editing approaches, additional β-gal deficient mouse models have been more recently generated using either TALEN or CRISPR/Cas9 technologies (Przybilla et al., 2019; Eikelberg et al., 2020; Liu et al., 2021). Using CRISPR/Cas9 Przybilla et al. (2019) engineered a knock-out mouse model by introducing a deletion in exon 8 of the Glb1 gene. Phenotypic alterations in these mice were evaluated using behavioral tests that showed profound neurocognitive impairment (Przybilla et al., 2019). In Eikelberg et al. (2020) described a knock-out model using TALENs to target exon 15 of Glb1. These mice display brain and spinal cord pathology, characterized by swelling of axons and loss of myelin, leading to abnormal electrophysiological activity of neurons (Eikelberg et al., 2020). This is the first electrophysiological study performed in a mouse model of the disease, showing abnormalities that the authors attribute to increased neuronal cell size and reduced membrane resistance. The most recent β-gal mutant mouse generated using CRISPR/Cas9 is a knock-in model that introduces a human missense mutation in exon 14 of Glb1, described in a patient with late-infantile GM1 gangliosidosis (Liu et al., 2021). The CNS phenotype in this model included impaired motor function, as well as extensive microgliosis, accompanied by activation of autophagy (Liu et al., 2021). The characteristics of available mouse models are summarized in Table 1.
Therapeutic Strategies
Until very recently, therapy for GM1 gangliosidosis was limited to symptomatic management. However, several experimental therapies have been trialed in murine (Table 1) and feline models (Gray-Edwards et al., 2017a,b, 2020). Since GM1 primarily affects the brain, targeted delivery must traverse the blood-brain barrier (BBB) or be delivered directly to the brain. Experimental therapies are discussed below.
Substrate Reduction Therapy
The rationale of Substrate Reduction Therapy (SRT) is to use small molecule inhibitors of enzymes responsible for the biosynthesis of stored substrates (Jeyakumar et al., 2002; Platt et al., 2018). For example miglustat, is a N-alkylated iminosugar that is a reversible competitive inhibitor of glucosylceramide synthase, the enzyme catalyzing the first committed step in the biosynthesis of most glycosphingolipids, including gangliosides (Platt et al., 1994). This approach aims to balance the rate of glycosphingolipid biosynthesis with the impaired rate of glycosphingolipid catabolism (Platt et al., 2001). Miglustat crosses the BBB and so can in principle be applied to treat glycosphingolipid storage diseases affecting the periphery and the brain (Cox et al., 2000; Kasperzyk et al., 2004, 2005; Patterson et al., 2007; Treiber et al., 2007; Ficicioglu, 2008). Miglustat was approved for treating type 1 Gaucher disease in 2002 and for Niemann-Pick disease type C in 2009 (Platt et al., 2018). Miglustat has also been proposed for the treatment of GM1 gangliosidosis. Indeed, miglustat reduced GM1 ganglioside in the central nervous system of a mouse model of GM1 gangliosidosis (Kasperzyk et al., 2004), and led to functional improvements and a decrease in brain inflammation (Kasperzyk et al., 2004; Treiber et al., 2007; Elliot-Smith et al., 2008).
Despite the demonstrated effectiveness of miglustat in other storage disorders, its use in GM1 gangliosidosis type II has been tested only in a few patients. In 2007, Tifft et al. (2007) reported that miglustat administration improved neurological functions in two patients with juvenile GM1 gangliosidosis (Tifft et al., 2007). Deodato et al. (2017) described similar neurological improvement in the juvenile form of the disease (Deodato et al., 2017). Stabilization and/or slowing of neurological progression in three of four patients was observed by Fischetto et al. (2020).
Miglustat combined with a ketogenic diet has been used to treat children with GM1 and GM2 gangliosidosis (ClinicalTrials.gov Identifier NCT02030015). The aim of this study was to learn if synergistic enteral regimen for treatment of the gangliosidoses will show improvement in overall survival and clinical benefits in neurodevelopmental abilities in children with gangliosidosis diseases. The Syner-G regimen may have prolonged lifespan, however, the small sample size and variability in other palliative care measures used by families prevented definitive conclusions to be drawn (Jarnes Utz et al., 2017).
Venglustat, another SRT drug chemically distinct from miglustat and designed specifically to cross the BBB is an orally available inhibitor of the enzyme glucosylceramide synthase (Peterschmitt et al., 2021). It is currently under study for GM1 gangliosidosis and several other LSDs in the same degradation pathway including late-onset GM2-gangliosidosis (Tay-Sachs and Sandhoff diseases), Fabry disease, and neuronopathic Gaucher disease (type III) (NCT04221451).
Enzyme Enhancement Therapy
Enzyme enhancement therapy (EET), also termed pharmacological chaperone therapy, has been proposed for GM1 (Matsuda et al., 2003; Suzuki, 2006). The aim is to use small molecules to stabilize potentially unstable or misfolded mutant proteins in the endoplasmic reticulum to enhance lysosomal delivery and increase half-life (Parenti, 2009). Small molecule chaperones that cross the BBB, would be a prerequisite for disorders with CNS involvement (Begley et al., 2008).
Several pharmacological chaperones including galactose, N-octyl-4-epi-β-valienamine (NOEV)alkylated or fluorinated derivates of N-butyldeoxynojirimycin (NB-DNJ), and (5aR)-5a-C-Pentyl-4-epiisofagomine have been tested against numerous GLB1 mutant enzymes (Matsuda et al., 2003; Suzuki et al., 2007, 2012; Suzuki, 2008, 2014; Fantur et al., 2010; Takai et al., 2013; Parenti et al., 2015; Thonhofer et al., 2016; Front et al., 2017).
Treatment with NOEV, a galactose analog, at the early stage of the disease reduced disease progression and prolonged survival in a murine model of GM1 gangliosidosis (see Table 1; Suzuki, 2014; Parenti et al., 2015). The compound was determined to cross the BBB for CNS delivery (Suzuki, 2014).
Collectively, dozens of patient cell lines with missense mutations have been shown to be responsive to the chaperones listed above, in some cases resulting in greater than the 10–15% residual β-gal activity sufficient to avoid substrate accumulation (Leinekugel et al., 1992; Iwasaki et al., 2006).
Pharmacological chaperones have broad tissue distribution and can be given orally; major advantages for treatment (Front et al., 2017). In addition, they have been shown to work synergistically with other therapies, such as ERT (Kishnani et al., 2017).
Enzyme Replacement Therapy
Enzyme replacement therapy (ERT) as potential treatment for GM1 gangliosidosis was first tested using either a purified (Reynolds et al., 1978) or a recombinant (Samoylova et al., 2008) feline β-gal enzyme in vitro. Since β-gal cannot cross the BBB, several therapeutic “Trojan horse” strategies have been utilized, including creating fusion proteins of the enzyme with lectin subunit ribosome-inactivating toxin B (RTB) of ricin (Condori et al., 2016), and to the carboxyl terminus of the heavy chain of a mouse chimeric monoclonal antibody against the mouse transferrin receptor (mTfR-GLB1) (Przybilla et al., 2021).
Encapsulation of β-gal enzyme into artificial nanoparticles to traverse the BBB has also been experimented in vitro (Gupta et al., 2017; Kelly et al., 2017). Mechanically breaching the BBB has been described by Chen et al. (2020) who used direct intracerebroventricular (ICV) injection of rhβ-gal to β-gal–/– mice, which led to normalization of neuropathology.
Stem Cell Transplantation
Stem cell transplantation (SCT) early in the course of disease may ameliorate symptoms in GM1 gangliosidosis (Sawada et al., 2009), although for optimal benefit, like Krabbe disease, the transplant would need to be undertaken in the first few weeks of life; a strong argument for universal newborn screening for GM1 disease. Although it could be successfully utilized in GM1 gangliosidosis and may reduce visceral features, the long-term correction of neurological symptoms is less likely. Improvement was observed in a 7-month GM1 gangliosidosis baby who after SCT developed normally until regression was noted at the age of 20–25 months (Shield et al., 2005). The risk of procedure-related mortality with transplantation has decreased with improvements in chemotherapy regimens and should be considered in cases of very early diagnosis with limited therapeutic options.
Gene Therapy
Pre-clinical studies in mouse models resulted in extended life expectancy, β-gal activity restoration and decreased storage levels in the CNS and peripheral organs (see Table 1). After the successful treatment in the mouse (Takaura et al., 2003; Broekman et al., 2007, 2009; Baek et al., 2010; Weismann et al., 2015) studies were extended to the feline model with dramatic response in widespread distribution of β-gal enzyme, improved function, and greatly extended lifespan (McCurdy et al., 2014; Gray-Edwards et al., 2017a,b). The dramatic improvement observed in the murine and feline models paved the way for in vitro studies in human cerebral organoids (Latour et al., 2019) and subsequent phase I clinical trials in patients with Type I and Type II disease. Three trials are currently recruiting: intravenous delivery of AAV9-GLB1 (ClinicalTrials.gov Identifier NCT03952637), LYS-GM101 administered via cisterna magna (ClinicalTrials.gov Identifier NCT04273269), and PBGM01 delivered via cisterna magna (ClinicalTrials.gov Identifier NCT04713475).
Conclusion
GM1 gangliosidosis is a severe LSD underpinned by complex pathophysiological mechanisms. Numerous disease causing GLB1 mutations have been reported. However, genotype-phenotype correlation has been difficult to establish due in part to the way the enzyme is commonly assayed in patients’ samples. In addition, the clinical outcome of the disease in patients may be strongly influenced by post-translational and regulatory mechanisms controlling GM1 catabolism that may vary from patient to patient. These caveats ask for further elucidation of the cellular pathophysiology underlying this disease that may improve our understanding of the fundamental cell biology of GM1 ganglioside and the enzyme complex that regulates its catabolism in the lysosome. Multiple mouse models of this disorder have been instrumental for the pre-clinical testing of multiple therapies, several of which are currently in clinical trials.
Author Contributions
E-RN, KS, FP, CT, Ad’A, and IA were involved in designing the concept of the review and oversight. KS and E-RN drafted the manuscript. All authors reviewed the manuscript, read, and approved the final manuscript.
Funding
E-RN and CT work was supported by the Intramural Research Program of the National Human Genome Research Institute, NIH. FP was a Royal Society Wolfson Merit Award recipient and a Wellcome Trust Investigator in Science. Ad’A and IA work was funded in part by NIH grants CA021764, GM104981; the Assisi Foundation of Memphis and the American Lebanese Syrian Associated Charities (ALSAC).
Conflict of Interest
Ad’A holds the Jewelers for Children Endowed Chair in Genetics and Gene Therapy.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Julia Fekecs and Darryl Leja—NHGRI medical illustrators.
Abbreviations
BBB, Blood-brain barrier; β -GAL, β-galactosidase; EBP, Elastin binding protein; PPCA, Protective protein/cathepsin A; NEU1, α -neuraminidase 1; CNS, Central nervous system; CRISPR/Cas9, Clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9; TALEN, Transcription activator-like effector nucleases; ERT, Enzyme replacement therapy; EET, Enzyme enhancement therapy; SRT, Substrate reduction therapy; AAV, Adeno-associated virus; ICV, Intracerebroventricular; mTfR, Mouse transferrin receptor; NB-DNJ, N-butyldeoxynojirimycin; miglustat; NOEV, N-octyl -4-epi-β-valienamine; HGMD, Human Gene Mutation Database; Cer, Ceramide; GlcCer, Glucosylceramide; LacCer, Lactosylceramide; GA2, Asialo-GM2 (GalNAc1,4Gal1,4Glc1Cer); GA1, Asialo-GM1 (Gal1,3GalNAc1,4Gal1,4Glc1Cer); GM3, Neu5Ac2,3Gal1,4Glc1Cer; GM2, GalNAc1,4(Neu5Ac2,3)Gal1,4Glc1Cer; GM1a, GM1 [Gal1,3GalNAc1,4(Neu5Ac2,3)Gal1,4Glc1Cer]; Glc, Glucose; Gal, Galactose.
References
Abumansour, I. S., Yuskiv, N., Paschke, E., and Stockler-Ipsiroglu, S. (2020). Morquio-B disease: clinical and genetic characteristics of a distinct GLB1-related dysostosis multiplex. JIMD Rep. 51, 30–44. doi: 10.1002/jmd2.12065
Annunziata, I., Sano, R., and d’Azzo, A. (2018). Mitochondria-associated ER membranes (MAMs) and lysosomal storage diseases. Cell Death Dis. 9:328. doi: 10.1038/s41419-017-0025-4
Arash-Kaps, L., Komlosi, K., Seegraber, M., Diederich, S., Paschke, E., Amraoui, Y., et al. (2019). The Clinical and Molecular Spectrum of GM1 Gangliosidosis. J. Pediatr. 215, 152–157e153. doi: 10.1016/j.jpeds.2019.08.016
Baek, R. C., Broekman, M. L., Leroy, S. G., Tierney, L. A., Sandberg, M. A., d’Azzo, A., et al. (2010). AAV-mediated gene delivery in adult GM1-gangliosidosis mice corrects lysosomal storage in CNS and improves survival. PLoS One 5:e13468. doi: 10.1371/journal.pone.0013468
Begley, D. J., Pontikis, C. C., and Scarpa, M. (2008). Lysosomal storage diseases and the blood-brain barrier. Curr. Pharm. Des. 14, 1566–1580. doi: 10.2174/138161208784705504
Bonten, E. J., Annunziata, I., and d’Azzo, A. (2014). Lysosomal multienzyme complex: pros and cons of working together. Cell. Mol. Life Sci. 71, 2017–2032. doi: 10.1007/s00018-013-1538-3
Breiden, B., and Sandhoff, K. (2019). Lysosomal glycosphingolipid storage diseases. Annu. Rev. Biochem. 88, 461–485. doi: 10.1146/annurev-biochem-013118-111518
Broekman, M. L., Baek, R. C., Comer, L. A., Fernandez, J. L., Seyfried, T. N., and Sena-Esteves, M. (2007). Complete correction of enzymatic deficiency and neurochemistry in the GM1-gangliosidosis mouse brain by neonatal adeno-associated virus-mediated gene delivery. Mol. Ther. 15, 30–37. doi: 10.1038/sj.mt.6300004
Broekman, M. L., Tierney, L. A., Benn, C., Chawla, P., Cha, J. H., and Sena-Esteves, M. (2009). Mechanisms of distribution of mouse beta-galactosidase in the adult GM1-gangliosidosis brain. Gene Ther. 16, 303–308. doi: 10.1038/gt.2008.149
Bruggink, C., Poorthuis, B. J., Deelder, A. M., and Wuhrer, M. (2012). Analysis of urinary oligosaccharides in lysosomal storage disorders by capillary high-performance anion-exchange chromatography-mass spectrometry. Anal. Bioanal. Chem. 403, 1671–1683. doi: 10.1007/s00216-012-5968-9
Brunetti-Pierri, N., and Scaglia, F. (2008). GM1 gangliosidosis: review of clinical, molecular, and therapeutic aspects. Mol. Genet. Metab. 94, 391–396. doi: 10.1016/j.ymgme.2008.04.012
Caciotti, A., Donati, M. A., Boneh, A., d’Azzo, A., Federico, A., Parini, R., et al. (2005). Role of beta-galactosidase and elastin binding protein in lysosomal and nonlysosomal complexes of patients with GM1-gangliosidosis. Hum. Mutat. 25, 285–292. doi: 10.1002/humu.20147
Caciotti, A., Garman, S. C., Rivera-Colon, Y., Procopio, E., Catarzi, S., Ferri, L., et al. (2011). GM1 gangliosidosis and Morquio B disease: an update on genetic alterations and clinical findings. Biochim. Biophys. Acta 1812, 782–790. doi: 10.1016/j.bbadis.2011.03.018
Chen, J. C., Luu, A. R., Wise, N., Angelis, R., Agrawal, V., Mangini, L., et al. (2020). Intracerebroventricular enzyme replacement therapy with beta-galactosidase reverses brain pathologies due to GM1 gangliosidosis in mice. J. Biol. Chem. 295, 13532–13555. doi: 10.1074/jbc.RA119.009811
ClinVar (2021). Xxxxxx. Available online at: https://www.ncbi.nlm.nih.gov/clinvar/?term=GLB1[gene] (accessed June 25, 2021).
Condori, J., Acosta, W., Ayala, J., Katta, V., Flory, A., Martin, R., et al. (2016). Enzyme replacement for GM1-gangliosidosis: uptake, lysosomal activation, and cellular disease correction using a novel beta-galactosidase:RTB lectin fusion. Mol. Genet. Metab. 117, 199–209. doi: 10.1016/j.ymgme.2015.12.002
Cox, T., Lachmann, R., Hollak, C., Aerts, J., van Weely, S., Hrebicek, M., et al. (2000). Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet 355, 1481–1485. doi: 10.1016/S0140-6736(00)02161-9
Deodato, F., Procopio, E., Rampazzo, A., Taurisano, R., Donati, M. A., Dionisi-Vici, C., et al. (2017). The treatment of juvenile/adult GM1-gangliosidosis with Miglustat may reverse disease progression. Metab. Brain Dis. 32, 1529–1536. doi: 10.1007/s11011-017-0044-y
Eikelberg, D., Lehmbecker, A., Brogden, G., Tongtako, W., Hahn, K., Habierski, A., et al. (2020). Axonopathy and reduction of membrane resistance: key features in a new murine model of human GM1-gangliosidosis. J. Clin. Med. 9:1004. doi: 10.3390/jcm9041004
Elliot-Smith, E., Speak, A. O., Lloyd-Evans, E., Smith, D. A., van der Spoel, A. C., Jeyakumar, M., et al. (2008). Beneficial effects of substrate reduction therapy in a mouse model of GM1 gangliosidosis. Mol. Genet. Metab. 94, 204–211. doi: 10.1016/j.ymgme.2008.02.005
Fantur, K., Hofer, D., Schitter, G., Steiner, A. J., Pabst, B. M., Wrodnigg, T. M., et al. (2010). DLHex-DGJ, a novel derivative of 1-deoxygalactonojirimycin with pharmacological chaperone activity in human G(M1)-gangliosidosis fibroblasts. Mol. Genet. Metab. 100, 262–268. doi: 10.1016/j.ymgme.2010.03.019
Ferreira, C. R., and Gahl, W. A. (2017). Lysosomal storage diseases. Transl. Sci. Rare Dis. 2, 1–71. doi: 10.3233/TRD-160005
Ferreira, C. R., Regier, D. S., Yoon, R., Pan, K. S., Johnston, J. M., Yang, S., et al. (2020). The skeletal phenotype of intermediate GM1 gangliosidosis: clinical, radiographic and densitometric features, and implications for clinical monitoring and intervention. Bone 131:115142. doi: 10.1016/j.bone.2019.115142
Ficicioglu, C. (2008). Review of miglustat for clinical management in Gaucher disease type 1. Ther. Clin. Risk Manag. 4, 425–431. doi: 10.2147/tcrm.s6865
Fischetto, R., Palladino, V., Mancardi, M. M., Giacomini, T., Palladino, S., Gaeta, A., et al. (2020). Substrate reduction therapy with Miglustat in pediatric patients with GM1 type 2 gangliosidosis delays neurological involvement: a multicenter experience. Mol. Genet. Genomic Med. 8:e1371. doi: 10.1002/mgg3.1371
Front, S., Biela-Banas, A., Burda, P., Ballhausen, D., Higaki, K., Caciotti, A., et al. (2017). (5aR)-5a-C-Pentyl-4-epi-isofagomine: a powerful inhibitor of lysosomal beta-galactosidase and a remarkable chaperone for mutations associated with GM1-gangliosidosis and Morquio disease type B. Eur. J. Med. Chem. 126, 160–170. doi: 10.1016/j.ejmech.2016.09.095
Gorelik, A., Illes, K., Hasan, S. M. N., Nagar, B., and Mazhab-Jafari, M. T. (2021). Structure of the murine lysosomal multienzyme complex core. Sci. Adv. 7:eabf4155. doi: 10.1126/sciadv.abf4155
Gray-Edwards, H. L., Jiang, X., Randle, A. N., Taylor, A. R., Voss, T. L., Johnson, A. K., et al. (2017a). Lipidomic evaluation of feline neurologic disease after AAV gene therapy. Mol. Ther. Methods Clin. Dev. 6, 135–142. doi: 10.1016/j.omtm.2017.07.005
Gray-Edwards, H. L., Maguire, A. S., Salibi, N., Ellis, L. E., Voss, T. L., Diffie, E. B., et al. (2020). 7T MRI predicts amelioration of neurodegeneration in the brain after AAV gene therapy. Mol. Ther. Methods Clin. Dev. 17, 258–270. doi: 10.1016/j.omtm.2019.11.023
Gray-Edwards, H. L., Regier, D. S., Shirley, J. L., Randle, A. N., Salibi, N., Thomas, S. E., et al. (2017b). Novel biomarkers of human GM1 gangliosidosis reflect the clinical efficacy of gene therapy in a feline model. Mol. Ther. 25, 892–903. doi: 10.1016/j.ymthe.2017.01.009
Gupta, M., Pandey, H., and Sivakumar, S. (2017). Intracellular delivery of beta-galactosidase enzyme using arginase-responsive dextran sulfate/Poly-l-arginine capsule for lysosomal storage disorder. ACS Omega 2, 9002–9012. doi: 10.1021/acsomega.7b01230
Hahn, C. N., del Pilar Martin, M., Schroder, M., Vanier, M. T., Hara, Y., Suzuki, K., et al. (1997). Generalized CNS disease and massive GM1-ganglioside accumulation in mice defective in lysosomal acid beta-galactosidase. Hum. Mol. Genet. 6, 205–211. doi: 10.1093/hmg/6.2.205
HGMD (2021). The Human Gene Mutation Database at the Institute of Medical Genetics in Cardiff. Available online at: http://www.hgmd.cf.ac.uk/ac/gene.php?gene=GLB1. (Accessed June 25, 2021)
Hinderer, C., Nosratbakhsh, B., Katz, N., and Wilson, J. M. (2020). A single injection of an optimized adeno-associated viral vector into cerebrospinal fluid corrects neurological disease in a murine model of GM1 gangliosidosis. Hum. Gene. Ther. 31, 1169–1177. doi: 10.1089/hum.2018.206
Holmes, E. W., and O’Brien, J. S. (1978). Hepatic storage of oligosaccharides and glycolipids in a cat affected with GM1 gangliosidosis. Biochem. J. 175, 945–953. doi: 10.1042/bj1750945
Inui, K., Namba, R., Ihara, Y., Nobukuni, K., Taniike, M., Midorikawa, M., et al. (1990). A case of chronic GM1 gangliosidosis presenting as dystonia: clinical and biochemical studies. J. Neurol. 237, 491–493. doi: 10.1007/BF00314770
Iwasaki, H., Watanabe, H., Iida, M., Ogawa, S., Tabe, M., Higaki, K., et al. (2006). Fibroblast screening for chaperone therapy in beta-galactosidosis. Brain Dev. 28, 482–486. doi: 10.1016/j.braindev.2006.02.002
Iyer, N. S., Gimovsky, A. C., Ferreira, C. R., Critchlow, E. J., and Al-Kouatly, H. B. (2021). Lysosomal storage disorders as an etiology of nonimmune hydrops fetalis: a systematic review. Clin. Genet. 212, 281–290. doi: 10.1111/cge.14005
Jarnes Utz, J. R., Kim, S., King, K., Ziegler, R., Schema, L., Redtree, E. S., et al. (2017). Infantile gangliosidoses: mapping a timeline of clinical changes. Mol. Genet. Metab. 121, 170–179. doi: 10.1016/j.ymgme.2017.04.011
Jatzkewitz, H., and Sandhoff, K. (1963). On a biochemically special form of infantile amaturotic idiocy. Biochim. Biophys. Acta 70, 354–356. doi: 10.1016/0006-3002(63)90764-9
Jeyakumar, M., Butters, T. D., Dwek, R. A., and Platt, F. M. (2002). Glycosphingolipid lysosomal storage diseases: therapy and pathogenesis. Neuropathol. Appl. Neurobiol. 28, 343–357. doi: 10.1046/j.1365-2990.2002.00422.x
Jeyakumar, M., Thomas, R., Elliot-Smith, E., Smith, D. A., van der Spoel, A. C., d’Azzo, A., et al. (2003). Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain 126(Pt 4), 974–987. doi: 10.1093/brain/awg089
Karimzadeh, P., Naderi, S., Modarresi, F., Dastsooz, H., Nemati, H., Farokhashtiani, T., et al. (2017). Case reports of juvenile GM1 gangliosidosisis type II caused by mutation in GLB1 gene. BMC Med. Genet. 18:73. doi: 10.1186/s12881-017-0417-4
Kasperzyk, J. L., d’Azzo, A., Platt, F. M., Alroy, J., and Seyfried, T. N. (2005). Substrate reduction reduces gangliosides in postnatal cerebrum-brainstem and cerebellum in GM1 gangliosidosis mice. J. Lipid Res. 46, 744–751. doi: 10.1194/jlr.M400411-JLR200
Kasperzyk, J. L., El-Abbadi, M. M., Hauser, E. C., D’Azzo, A., Platt, F. M., and Seyfried, T. N. (2004). N-butyldeoxygalactonojirimycin reduces neonatal brain ganglioside content in a mouse model of GM1 gangliosidosis. J. Neurochem. 89, 645–653. doi: 10.1046/j.1471-4159.2004.02381.x
Kelly, J. M., Gross, A. L., Martin, D. R., and Byrne, M. E. (2017). Polyethylene glycol-b-poly(lactic acid) polymersomes as vehicles for enzyme replacement therapy. Nanomedicine (Lond.) 12, 2591–2606. doi: 10.2217/nnm-2017-0221
Kishnani, P., Tarnopolsky, M., Roberts, M., Sivakumar, K., Dasouki, M., Dimachkie, M. M., et al. (2017). Duvoglustat HCl increases systemic and tissue exposure of active acid alpha-glucosidase in pompe patients co-administered with alglucosidase alpha. Mol. Ther. 25, 1199–1208. doi: 10.1016/j.ymthe.2017.02.017
Landrum, M. J., Lee, J. M., Benson, M., Brown, G. R., Chao, C., Chitipiralla, S., et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067. doi: 10.1093/nar/gkx1153
Lang, F. M., Korner, P., Harnett, M., Karunakara, A., and Tifft, C. J. (2020). The natural history of Type 1 infantile GM1 gangliosidosis: a literature-based meta-analysis. Mol. Genet. Metab. 129, 228–235. doi: 10.1016/j.ymgme.2019.12.012
Latour, Y. L., Yoon, R., Thomas, S. E., Grant, C., Li, C., Sena-Esteves, M., et al. (2019). Human GLB1 knockout cerebral organoids: a model system for testing AAV9-mediated GLB1 gene therapy for reducing GM1 ganglioside storage in GM1 gangliosidosis. Mol. Genet. Metab. Rep. 21:100513. doi: 10.1016/j.ymgmr.2019.100513
Lawrence, R., Van Vleet, J. L., Mangini, L., Harris, A., Martin, N., Clark, W., et al. (2019). Characterization of glycan substrates accumulating in GM1 Gangliosidosis. Mol. Genet. Metab. Rep. 21:100524. doi: 10.1016/j.ymgmr.2019.100524
Ledeen, R. W., and Wu, G. (2015). The multi-tasked life of GM1 ganglioside, a true factotum of nature. Trends Biochem. Sci. 40, 407–418. doi: 10.1016/j.tibs.2015.04.005
Leinekugel, P., Michel, S., Conzelmann, E., and Sandhoff, K. (1992). Quantitative correlation between the residual activity of beta-hexosaminidase A and arylsulfatase A and the severity of the resulting lysosomal storage disease. Hum. Genet. 88, 513–523. doi: 10.1007/BF00219337
Liu, S., Feng, Y., Huang, Y., Jiang, X., Tang, C., Tang, F., et al. (2021). A GM1 gangliosidosis mutant mouse model exhibits activated microglia and disturbed autophagy. Exp. Biol. Med. (Maywood) 246, 1330–1341. doi: 10.1177/1535370221993052
Matsuda, J., Suzuki, O., Oshima, A., Ogura, A., Naiki, M., and Suzuki, Y. (1997a). Neurological manifestations of knockout mice with beta-galactosidase deficiency. Brain Dev. 19, 19–20.
Matsuda, J., Suzuki, O., Oshima, A., Ogura, A., Noguchi, Y., Yamamoto, Y., et al. (1997b). Beta-galactosidase-deficient mouse as an animal model for GM1-gangliosidosis. Glycoconj. J. 14, 729–736.
Matsuda, J., Suzuki, O., Oshima, A., Yamamoto, Y., Noguchi, A., Takimoto, K., et al. (2003). Chemical chaperone therapy for brain pathology in G(M1)-gangliosidosis. Proc. Natl. Acad. Sci. U.S.A. 100, 15912–15917. doi: 10.1073/pnas.2536657100
Mayer, F. Q., Pereira Fdos, S., Fensom, A. H., Slade, C., Matte, U., and Giugliani, R. (2009). New GLB1 mutation in siblings with Morquio type B disease presenting with mental regression. Mol. Genet. Metab. 96:148. doi: 10.1016/j.ymgme.2008.11.159
McCurdy, V. J., Johnson, A. K., Gray-Edwards, H. L., Randle, A. N., Brunson, B. L., Morrison, N. E., et al. (2014). Sustained normalization of neurological disease after intracranial gene therapy in a feline model. Sci. Transl. Med. 6:231ra248. doi: 10.1126/scitranslmed.3007733
Morita, M., Saito, S., Ikeda, K., Ohno, K., Sugawara, K., Suzuki, T., et al. (2009). Structural bases of GM1 gangliosidosis and Morquio B disease. J. Hum. Genet. 54, 510–515. doi: 10.1038/jhg.2009.70
Ohto, U., Usui, K., Ochi, T., Yuki, K., Satow, Y., and Shimizu, T. (2012). Crystal structure of human beta-galactosidase: structural basis of Gm1 gangliosidosis and morquio B diseases. J. Biol. Chem. 287, 1801–1812. doi: 10.1074/jbc.M111.293795
Parenti, G. (2009). Treating lysosomal storage diseases with pharmacological chaperones: from concept to clinics. EMBO Mol. Med. 1, 268–279. doi: 10.1002/emmm.200900036
Parenti, G., Andria, G., and Valenzano, K. J. (2015). Pharmacological chaperone therapy: preclinical development, clinical translation, and prospects for the treatment of lysosomal storage disorders. Mol. Ther. 23, 1138–1148. doi: 10.1038/mt.2015.62
Patterson, M. C., Vecchio, D., Prady, H., Abel, L., and Wraith, J. E. (2007). Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. Lancet Neurol. 6, 765–772. doi: 10.1016/S1474-4422(07)70194-1
Peterschmitt, M. J., Crawford, N. P. S., Gaemers, S. J. M., Ji, A. J., Sharma, J., and Pham, T. T. (2021). Pharmacokinetics, pharmacodynamics, safety, and tolerability of oral venglustat in healthy volunteers. Clin. Pharmacol. Drug Dev. 10, 86–98. doi: 10.1002/cpdd.865
Piraud, M., Pettazzoni, M., Menegaut, L., Caillaud, C., Nadjar, Y., Vianey-Saban, C., et al. (2017). Development of a new tandem mass spectrometry method for urine and amniotic fluid screening of oligosaccharidoses. Rapid Commun. Mass Spectrom. 31, 951–963. doi: 10.1002/rcm.7860
Platt, F. M., d’Azzo, A., Davidson, B. L., Neufeld, E. F., and Tifft, C. J. (2018). Lysosomal storage diseases. Nat. Rev. Dis. Primers 4:27. doi: 10.1038/s41572-018-0025-4
Platt, F. M., Jeyakumar, M., Andersson, U., Priestman, D. A., Dwek, R. A., Butters, T. D., et al. (2001). Inhibition of substrate synthesis as a strategy for glycolipid lysosomal storage disease therapy. J. Inh Metab. Dis. 24, 275–290. doi: 10.1023/a:1010335505357
Platt, F. M., Neises, G. R., Dwek, R. A., and Butters, T. D. (1994). N-butyldeoxynojirimycin is a novel inhibitor of glycolipid biosynthesis. J. Biol. Chem. 269, 8362–8365.
Przybilla, M. J., Ou, L., Tabaran, A. F., Jiang, X., Sidhu, R., Kell, P. J., et al. (2019). Comprehensive behavioral and biochemical outcomes of novel murine models of GM1-gangliosidosis and Morquio syndrome type B. Mol. Genet. Metab. 126, 139–150. doi: 10.1016/j.ymgme.2018.11.002
Przybilla, M. J., Stewart, C., Carlson, T. W., Ou, L., Koniar, B. L., Sidhu, R., et al. (2021). Examination of a blood-brain barrier targeting beta-galactosidase-monoclonal antibody fusion protein in a murine model of GM1-gangliosidosis. Mol. Genet. Metab. Rep. 27:100748. doi: 10.1016/j.ymgmr.2021.100748
Regier, D. S., Kwon, H. J., Johnston, J., Golas, G., Yang, S., Wiggs, E., et al. (2016). MRI/MRS as a surrogate marker for clinical progression in GM1 gangliosidosis. Am. J. Med. Genet. A 170, 634–644. doi: 10.1002/ajmg.a.37468
Regier, D. S., Tifft, C. J., and Rothermel, C. E. (1993). “GLB1-related disorders,” in GeneReviews, eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, G. Mirzaa, et al. (Seattle, WA: University of Washington).
Reynolds, G. C., Baker, H. J., and Reynolds, R. H. (1978). Enzyme replacement using liposome carriers in feline Gm1 gangliosidosis fibroblasts. Nature 275, 754–755. doi: 10.1038/275754a0
Rha, A. K., Maguire, A. S., and Martin, D. R. (2021). GM1 gangliosidosis: mechanisms and management. Appl. Clin. Genet. 14, 209–233. doi: 10.2147/TACG.S206076
Samoylova, T. I., Martin, D. R., Morrison, N. E., Hwang, M., Cochran, A. M., Samoylov, A. M., et al. (2008). Generation and characterization of recombinant feline beta-galactosidase for preclinical enzyme replacement therapy studies in GM1 gangliosidosis. Metab. Brain Dis. 23, 161–173. doi: 10.1007/s11011-008-9086-5
Sano, R., Annunziata, I., Patterson, A., Moshiach, S., Gomero, E., Opferman, J., et al. (2009). GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca(2+)-dependent mitochondrial apoptosis. Mol. Cell 36, 500–511. doi: 10.1016/j.molcel.2009.10.021
Sano, R., Tessitore, A., Ingrassia, A., and d’Azzo, A. (2005). Chemokine-induced recruitment of genetically modified bone marrow cellgliosidosis mice corrects neuronal pathology. Blood 106, 2259–2268. doi: 10.1182/blood-2005-03-1189
Sawada, T., Tanaka, A., Higaki, K., Takamura, A., Nanba, E., Seto, T., et al. (2009). Intracerebral cell transplantation therapy for murine GM1 gangliosidosis. Brain Dev. 31, 717–724. doi: 10.1016/j.braindev.2008.11.004
Severini, M. H., Silva, C. D., Sopelsa, A., Coelho, J. C., and Giugliani, R. (1999). High frequency of type 1 GM1 gangliosidosis in southern Brazil. Clin. Genet. 56, 168–169. doi: 10.1034/j.1399-0004.1999.560215.x
Shield, J. P., Stone, J., and Steward, C. G. (2005). Bone marrow transplantation correcting beta-galactosidase activity does not influence neurological outcome in juvenile GM1-gangliosidosis. J. Inherit. Metab. Dis. 28, 797–798. doi: 10.1007/s10545-005-0089-7
Sinigerska, I., Chandler, D., Vaghjiani, V., Hassanova, I., Gooding, R., Morrone, A., et al. (2006). Founder mutation causing infantile GM1-gangliosidosis in the Gypsy population. Mol. Genet. Metab. 88, 93–95. doi: 10.1016/j.ymgme.2005.12.009
Stone, D. L., and Sidransky, E. (1999). Hydrops fetalis: lysosomal storage disorders in extremis. Adv. Pediatr. 46, 409–440.
Suzuki, Y. (2006). Beta-galactosidase deficiency: an approach to chaperone therapy. J. Inherit. Metab. Dis. 29, 471–476. doi: 10.1007/s10545-006-0287-y
Suzuki, Y. (2008). Chemical chaperone therapy for GM1-gangliosidosis. Cell. Mol. Life Sci. 65, 351–353. doi: 10.1007/s00018-008-7470-2
Suzuki, Y. (2014). Emerging novel concept of chaperone therapies for protein misfolding diseases. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 90, 145–162. doi: 10.2183/pjab.90.145
Suzuki, Y., Ichinomiya, S., Kurosawa, M., Matsuda, J., Ogawa, S., Iida, M., et al. (2012). Therapeutic chaperone effect of N-octyl 4-epi-beta-valienamine on murine G(M1)-gangliosidosis. Mol. Genet. Metab. 106, 92–98. doi: 10.1016/j.ymgme.2012.02.012
Suzuki, Y., Ichinomiya, S., Kurosawa, M., Ohkubo, M., Watanabe, H., Iwasaki, H., et al. (2007). Chemical chaperone therapy: clinical effect in murine G(M1)-gangliosidosis. Ann. Neurol. 62, 671–675. doi: 10.1002/ana.21284
Suzuki, Y., Oshima, A., and Nanba, E. (2001). “β-Galactosidase deficiency (β-galactosidosis): GM1 gangliosidosis and Morquio B disease,” in The Metabolic and Molecular Bases of Inherited Disease, eds C. R. Shriver, A. L. Beaudet, W. S. Sly, and D. Valle (New York, NY: McGraw Hill), 3775–3809.
Takai, T., Higaki, K., Aguilar-Moncayo, M., Mena-Barragan, T., Hirano, Y., Yura, K., et al. (2013). A bicyclic 1-deoxygalactonojirimycin derivative as a novel pharmacological chaperone for GM1 gangliosidosis. Mol. Ther. 21, 526–532. doi: 10.1038/mt.2012.263
Takaura, N., Yagi, T., Maeda, M., Nanba, E., Oshima, A., Suzuki, Y., et al. (2003). Attenuation of ganglioside GM1 accumulation in the brain of GM1 gangliosidosis mice by neonatal intravenous gene transfer. Gene Ther. 10, 1487–1493. doi: 10.1038/sj.gt.3302033
Tebani, A., Sudrie-Arnaud, B., Dabaj, I., Torre, S., Domitille, L., Snanoudj, S., et al. (2021). Disentangling molecular and clinical stratification patterns in beta-galactosidase deficiency. J. Med. Genet. doi: 10.1136/jmedgenet-2020-107510 [Epub ahead of print].
Tessitore, A., del, P. M. M., Sano, R., Ma, Y., Mann, L., Ingrassia, A., et al. (2004). GM1-ganglioside-mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Mol. Cell 15, 753–766. doi: 10.1016/j.molcel.2004.08.029
Thonhofer, M., Weber, P., Santana, A. G., Fischer, R., Pabst, B. M., Paschke, E., et al. (2016). Synthesis of C-5a-chain extended derivatives of 4-epi-isofagomine: powerful beta-galactosidase inhibitors and low concentration activators of GM1-gangliosidosis-related human lysosomal beta-galactosidase. Bioorg. Med. Chem. Lett. 26, 1438–1442. doi: 10.1016/j.bmcl.2016.01.059
Tifft, C., Adams, D., and Morgan, C. (2007). 55 Miglustat improves function in patients with juvenile GM1 gangliosidosis. Mol. Genet. Metab. 4:24.
Tonin, R., Caciotti, A., Procopio, E., Fischetto, R., Deodato, F., Mancardi, M. M., et al. (2019). Pre-diagnosing and managing patients with GM1 gangliosidosis and related disorders by the evaluation of GM1 ganglioside content. Sci. Rep. 9:17684. doi: 10.1038/s41598-019-53995-5
Treiber, A., Morand, O., and Clozel, M. (2007). The pharmacokinetics and tissue distribution of the glucosylceramide synthase inhibitor miglustat in the rat. Xenobiotica 37, 298–314. doi: 10.1080/00498250601094543
Tsay, G. C., and Dawson, G. (1976). Oligosaccharide storage in brains from patients with fucosidosis, GM1-gangliosidosis and GM2-gangliosidosis (Sandhoff’s disease). J. Neurochem. 27, 733–740. doi: 10.1111/j.1471-4159.1976.tb10401.x
Weismann, C. M., Ferreira, J., Keeler, A. M., Su, Q., Qui, L., Shaffer, S. A., et al. (2015). Systemic AAV9 gene transfer in adult GM1 gangliosidosis mice reduces lysosomal storage in CNS and extends lifespan. Hum. Mol. Genet. 24, 4353–4364. doi: 10.1093/hmg/ddv168
Keywords: GM1 gangliosidosis, glycoconjugates metabolism, beta galactosidase, gene therapy, mouse model
Citation: Nicoli E-R, Annunziata I, d’Azzo A, Platt FM, Tifft CJ and Stepien KM (2021) GM1 Gangliosidosis—A Mini-Review. Front. Genet. 12:734878. doi: 10.3389/fgene.2021.734878
Received: 01 July 2021; Accepted: 16 August 2021;
Published: 03 September 2021.
Edited by:
Desheng Liang, Central South University, ChinaReviewed by:
Baoheng Gui, The Second Affiliated Hospital of Guangxi Medical University, ChinaYao Zhang, Peking University First Hospital, China
Konrad Sandhoff, University of Bonn, Germany
Copyright © 2021 Nicoli, Annunziata, d’Azzo, Platt, Tifft and Stepien. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karolina M. Stepien, a3N0ZXBpZW5AZG9jdG9ycy5vcmcudWs=