 Lijuan Zhu
Lijuan Zhu Xiaoji Su
Xiaoji Su- Children's Hospital of Fudan University Anhui Hospital, Hefei, China
Microduplications and reciprocal microdeletions of chromosome 1q21. 1 and/or 1q21.2 have been linked to variable clinical features, but the underlying pathogenic gene(s) remain unclear. Here we report that distinct microduplications were detected on chromosome 1q21.2 (GRCh37/hg19) in a mother (255 kb in size) and her newborn daughter (443 kb in size), while the same paternal locus was wild-type. Although the two microduplications largely overlap in genomic sequence (183 kb overlapping), the mother showed no clinical phenotype while the daughter presented with several features that are commonly observed on 1q21 microduplication or microdeletion patients, including developmental delay, craniofacial dysmorphism, congenital heart disease and sensorineural hearing loss. NBPF15 and NBPF16, two involved genes that are exclusively duplicated in the proband, may be the cause of the clinical manifestations. This study supports an association between NBPF genes and 1q21 copy number variation disorders.
Introduction
Extensive DNA repeats on chromosome 1q21.1 and 1q21.2 predispose the regions to recurrent rearrangement through non-allelic homologous recombination, which generates microduplication and microdeletion events. Despite variable penetrance, affected patients tend to present with developmental delays (Cooper et al., 2011), congenital heart disease (Christiansen et al., 2004; Mefford et al., 2008), intellectual disability (Sharp et al., 2006), macrocephaly/microcephaly (Brunetti-Pierri et al., 2008), autism spectrum disorder (Dolcetti et al., 2013), schizophrenia (International Schizophrenia Consortium, 2008; Stefansson et al., 2008), as well as neuroblastoma (Diskin et al., 2009). Hearing impairment is also commonly noticed on patients bearing either 1q21 microduplication (Rosenfeld et al., 2012; Dolcetti et al., 2013; Bernier et al., 2016; Pang et al., 2020) or the microdeletion (Brunetti-Pierri et al., 2008; Harvard et al., 2011; Rosenfeld et al., 2012), however this phenotype/genotype association has not been investigated specifically.
Since sizes of the 1q21 microduplications and reciprocal microdeletions can range from hundreds of kilobases to megabase, and dozens of genes may be involved, it is difficult to establish an association of a clinical feature with a specific gene. The GJA5 gene, encoding Cx40, has been linked to cardiac malformation in 1q21.1 microdeletion and microduplication carriers (Christiansen et al., 2004; Soemedi et al., 2012; Guida et al., 2013). Also, a role for Cx40 in heart development had also been validated in genetic knockout mice (Gu et al., 2003). Regarding the macrocephaly/microcephaly feature, the HYDIN2 gene, the paralog of HYDIN, was believed to be the cause of the abnormality (Brunetti-Pierri et al., 2008). However, genetic involvement in hearing impairment remains unclear in 1q21 copy number variation carriers. Here, we report a case observation where a newborn infant bearing 1q21.2 microduplication presents with mild developmental delays, facial dysmorphism, atrial septal defect and sensorineural hearing loss, with only two NBPF genes involved.
Results
A 3-month-old female infant, presenting with mild developmental delay and facial dysmorphism (Figure 1A), was admitted due to a failed newborn hearing screen. To confirm the patient's hearing capabilities, an auditory steady-state response (ASSR) and auditory brainstem response (ABR) tests were performed. The ASSR thresholds for the patient's left ear were measured as 80, 80, and 70 dB at carrier frequencies of 500, 1,000, and 2,000 Hz, respectively. The thresholds for the right ear were all 100 dB at the same measured frequencies. Consistently, in the ABR test, evoked potentials were observed for the left ear, but not the right ear, at 80 and 90 dB stimuli. A computed tomography (CT) scan showed that both ears, but primarily the right ear, presented with tympanosclerosis, in addition to a blockage of the right ear canal (Figure 1B). In addition to the hearing deficit, an echocardiogram revealed a 1.1 cm atrial septal defect (ASD) and hypertrophy of right atrium and right ventricle, indicating congenital heart disease (Figure 1C).
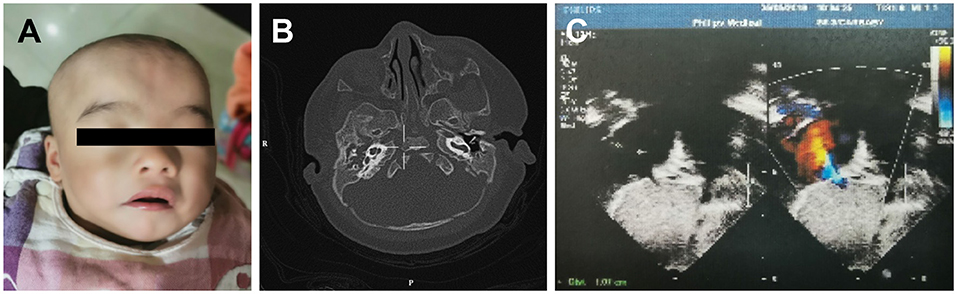
Figure 1. The developmental anomalies of the proband. (A) Patient shows facial dysmorphism. (B) Computerized tomography scan shows tympanosclerosis and blockage of the ear canals. (C) Echocardiogram showing a 1.1 cm atrial septal defect.
Based on the observations of these unrelated developmental defects, a gene mutation was suspected as the cause of the malformations. Mutations of mitochondrial DNA have been linked to developmental abnormalities such as deafness and congenital heart disease (Taylor and Turnbull, 2005). We thus sequenced mitochondrial DNA of peripheral blood cells (PBCs) from both affected proband and her non-affected mother, by means of long range PCR and next generation sequencing (Cui et al., 2013) with a depth coverage of 50,000x. However, the 3 identified mutations (m.6962G>A on COX1 gene, m.7447A>G on TRSN1 gene and m.16129G>A on Dloop_e region) were all maternally inherited, making them unlikely candidates for the proband's developmental abnormalities (Table 1).

Table 1. The three mitochondrial DNA mutations identified in proband and her mother.
To further investigate the involved pathogenesis, whole exome sequencing was performed using PBCs derived from the proband as well as her non-affected parents. Those point mutations in the proband with allele frequency below 0.2 or allele depth below 4 are regarded as unreliable and excluded. The remaining variants are then classified, according to ACMG practice guidelines (Richards et al., 2015) and OMIM database (https://www.omim.org/), as benign or potentially pathogenic. Finally, 11 missense point mutations of 8 proteins in the proband that could be potentially related to the developmental disorders were identified (Table 2). However, none of these mutations were likely to be the cause of the congenital anomalies, as they are all heterozygous and inherited from either of the non-affected parents who are also heterozygotes.
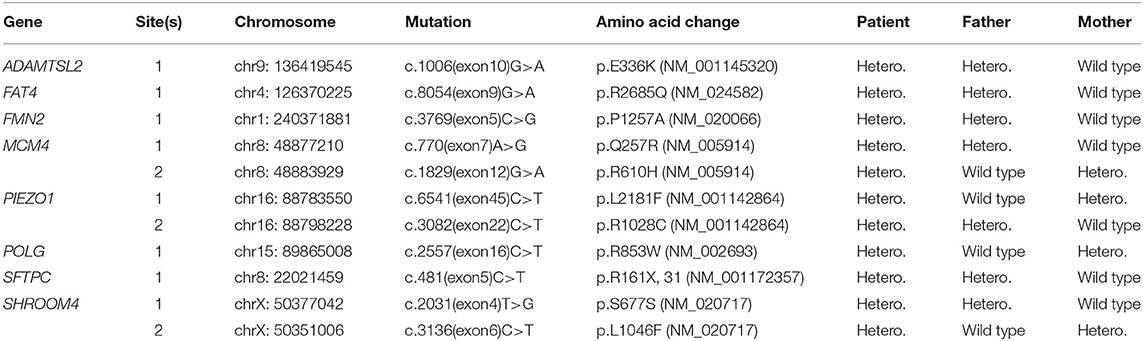
Table 2. Identified missense mutations in proband and their status in her parents.
Having ruled out the possibilities that mutations of mitochondrial DNA or genomic coding sequence were the cause of the clinical manifestations, we next evaluated if copy number changes apply to the patient's genomic DNA. To this end, whole genome sequencing of PBCs derived from the proband and her parents was performed and the copy number variation was analyzed. A microduplication (copy number = 3) on chromosome 1 was observed in the proband and maternal genomic DNA, but was lacking from the paternal genome (Figure 2A). The microduplication in the proband (chr1:148,511,359–148,954,460) was distinct from that of her mother (chr1:148,771,265–149,026,439), although both duplications arose on chromosome 1q21.2 and the duplicated fragments largely overlapped (Figure 2B). Chromosome microarray of the proband's genomic DNA confirmed the 1q21.2 microduplication (Figure 2C). NBPF15 and NBPF16 are the only involved genes specific to the proband, suggesting that these two genes might be the causative agents of the congenital abnormalities. Consistent with this observation, and expanding upon it, NBPF genes are repeatedly found to be involved in the 1q21 microduplication or microdeletion in patients with congenital hearing deficits (Figure 2B).
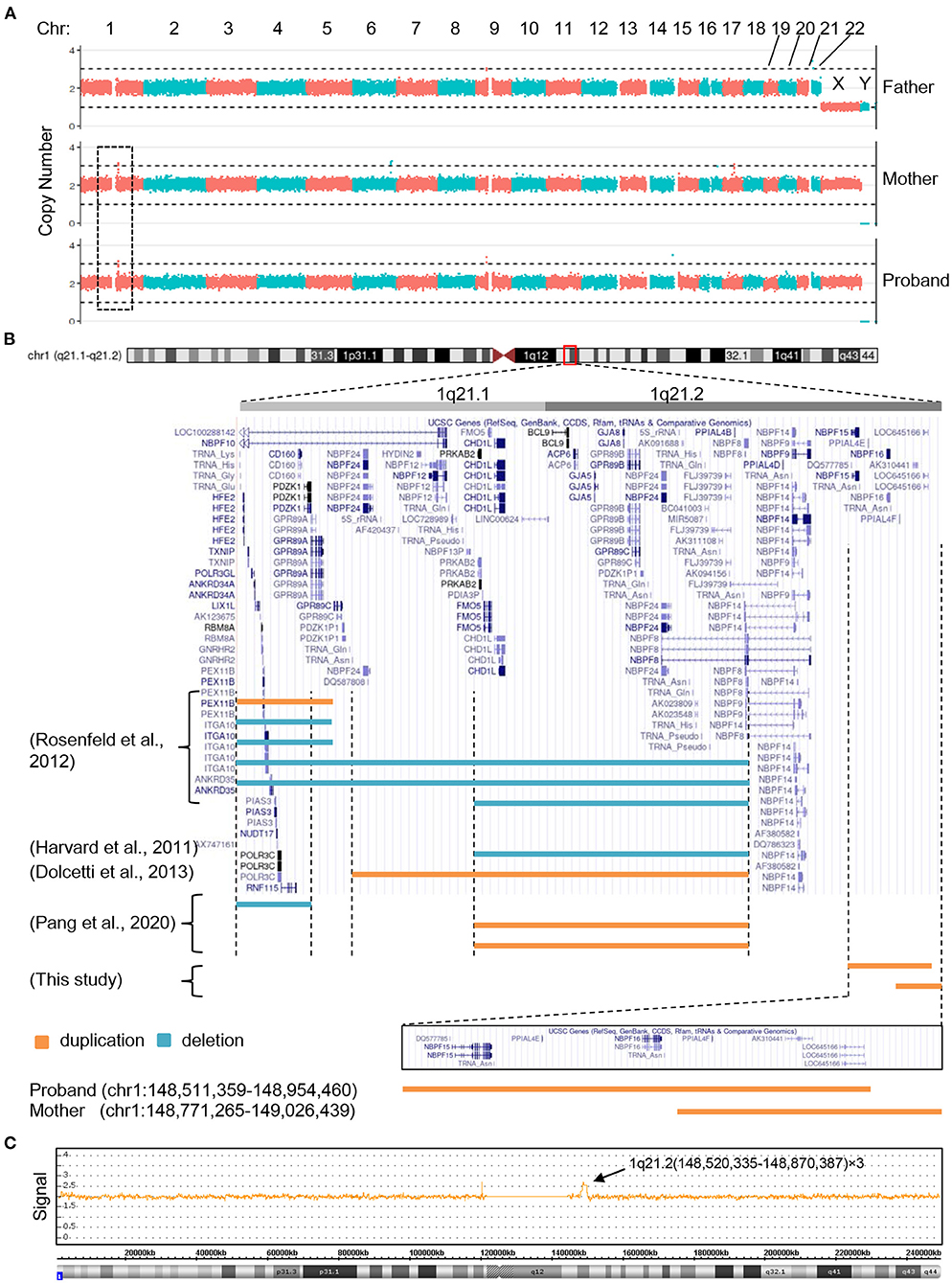
Figure 2. The 1q21 microduplications or microdeletions of the proband, her mother and other reported patients with hearing problem. (A) Copy number analysis of the proband and her parents by next generation sequencing. (B) Positional plots of the microduplications and microdeletions on 1q21.1 and 1q21.2 regions (http://genome.ucsc.edu). (C) Confirmation of the proband's microduplication by CytoScan HD array.
Discussion
Although this study only involves one patient, it is unique in several ways; first, the microduplicated segment of the proband is 1q21.2 specific, contrary to the majority of reported 1q21 copy number changes, which primarily occur in the 1q21.1 region (Brunetti-Pierri et al., 2008; Mefford et al., 2008; Diskin et al., 2009; Harvard et al., 2011; Rosenfeld et al., 2012; Soemedi et al., 2012; Dolcetti et al., 2013; Bernier et al., 2016; Pang et al., 2020). Despite this difference, the proband shares similar clinical features with 1q21.1 microduplication or microdeletion carriers, such as developmental delays, craniofacial dysmorphism, congenital heart disease and hearing loss. The second unique feature of this study is that the proband's microduplication is distinct from that of her mother. While both microduplications are largely overlap, the difference seen in the proband and her mother may be indicative of de novo 1q21.2 rearrangements. Last but not the least, there are only two genes from the same gene family (NBPF15 and NBPF16) specifically involved in the proband's 1q21.2 microduplication, which strongly suggests the genetic pathogenesis.
NBPF genes contain multiple copies of DUF1220/Olduvai domain which has been linked to human brain evolution (Popesco et al., 2006; O'Bleness et al., 2012). Of the ~300 haploid copies of human Olduvai domain, about 80% localize in 1q21.1 and 1q21.2 chromosomal regions; the driver of Olduvai domain expansion is thought to remain active in the general population (Heft et al., 2020), which is consistent with patients' recurrent rearrangement of the 1q21 region. Clinically, an increase in Olduvai copy number shows a linear association with severity of autism symptoms (Davis et al., 2014, 2015, 2019), while a decrease in the copy number is associated with schizophrenia (Searles Quick et al., 2015). These reported observations are in line with the associations of 1q21 microduplications and microdeletions with autism and schizophrenia, respectively (International Schizophrenia Consortium, 2008; Stefansson et al., 2008; Dolcetti et al., 2013). Olduvai copy number has also been proven to be linearly associated with brain size (Dumas et al., 2012), which justifies the macrocephaly/microcephaly feature in 1q21 copy number variation carriers.
In conclusion, our case observation supports an association between NBPF genes and 1q21 microduplication and microdeletion syndromes. However, it does not rule out the potential role of other genes involved in the observed pathogenesis.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of Children's Hospital of Fudan University Anhui Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
LZ and XS conducted the research. LZ analyzed data and drafted the manuscript. Both authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Hospital's Internal Research fund to LZ.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the patient and her parents for their participation in this research. We also thank Zhikai Wang, Philip Moresco, and Haiwei Mou for proof-reading the manuscript.
References
Bernier, R., Steinman, K. J., Reilly, B., Wallace, A. S., Sherr, E. H., Pojman, N., et al. (2016). Clinical phenotype of the recurrent 1q21.1 copy-number variant. Genet. Med. 18, 341–349. doi: 10.1038/gim.2015.78
Brunetti-Pierri, N., Berg, J. S., Scaglia, F., Belmont, J., Bacino, C. A., Sahoo, T., et al. (2008). Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 40, 1466–1471. doi: 10.1038/ng.279
Christiansen, J., Dyck, J. D., Elyas, B. G., Lilley, M., Bamforth, J. S., Hicks, M., et al. (2004). Chromosome 1q21.1 contiguous gene deletion is associated with congenital heart disease. Circ. Res. 94, 1429–1435. doi: 10.1161/01.RES.0000130528.72330.5c
Cooper, G. M., Coe, B. P., Girirajan, S., Rosenfeld, J. A., Vu, T. H., Baker, C., et al. (2011). A copy number variation morbidity map of developmental delay. Nat. Genet. 43, 838–846. doi: 10.1038/ng.909
Cui, H., Li, F., Chen, D., Wang, G., Truong, C. K., Enns, G. M., et al. (2013). Comprehensive next-generation sequence analyses of the entire mitochondrial genome reveal new insights into the molecular diagnosis of mitochondrial DNA disorders. Genet. Med. 15, 388–394. doi: 10.1038/gim.2012.144
Davis, J. M., Heft, I., Scherer, S. W., and Sikela, J. M. (2019). A third linear association between Olduvai (DUF1220) copy number and severity of the classic symptoms of inherited autism. Am. J. Psychiatry 176, 643–650. doi: 10.1176/appi.ajp.2018.18080993
Davis, J. M., Searles Quick, V. B., and Sikela, J. M. (2015). Replicated linear association between DUF1220 copy number and severity of social impairment in autism. Hum. Genet. 134, 569–575. doi: 10.1007/s00439-015-1537-6
Davis, J. M., Searles, V. B., Anderson, N., Keeney, J., Dumas, L., and Sikela, J. M. (2014). DUF1220 dosage is linearly associated with increasing severity of the three primary symptoms of autism. PLoS Genet. 10:e1004241. doi: 10.1371/journal.pgen.1004241
Diskin, S. J., Hou, C., Glessner, J. T., Attiyeh, E. F., Laudenslager, M., Bosse, K., et al. (2009). Copy number variation at 1q21.1 associated with neuroblastoma. Nature 459, 987–991. doi: 10.1038/nature08035
Dolcetti, A., Silversides, C. K., Marshall, C. R., Lionel, A. C., Stavropoulos, D. J., Scherer, S. W., et al. (2013). 1q21.1 Microduplication expression in adults. Genet. Med. 15, 282–289. doi: 10.1038/gim.2012.129
Dumas, L. J., O'Bleness, M. S., Davis, J. M., Dickens, C. M., Anderson, N., Keeney, J. G., et al. (2012). DUF1220-domain copy number implicated in human brain-size pathology and evolution. Am. J. Hum. Genet. 91, 444–454. doi: 10.1016/j.ajhg.2012.07.016
Gu, H., Smith, F. C., Taffet, S. M., and Delmar, M. (2003). High incidence of cardiac malformations in connexin40-deficient mice. Circ. Res. 93, 201–206. doi: 10.1161/01.RES.0000084852.65396.70
Guida, V., Ferese, R., Rocchetti, M., Bonetti, M., Sarkozy, A., Cecchetti, S., et al. (2013). A variant in the carboxyl-terminus of connexin 40 alters GAP junctions and increases risk for tetralogy of Fallot. Eur. J. Hum. Genet. 21, 69–75. doi: 10.1038/ejhg.2012.109
Harvard, C., Strong, E., Mercier, E., Colnaghi, R., Alcantara, D., Chow, E., et al. (2011). Understanding the impact of 1q21.1 copy number variant. Orphanet J. Rare Dis. 6:54. doi: 10.1186/1750-1172-6-54
Heft, I. E., Mostovoy, Y., Levy-Sakin, M., Ma, W., Stevens, A. J., Pastor, S., et al. (2020). The Driver of extreme human-specific olduvai repeat expansion remains highly active in the human genome. Genetics 214, 179–191. doi: 10.1534/genetics.119.302782
International Schizophrenia Consortium (2008). Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455, 237–241. doi: 10.1038/nature07239
Mefford, H. C., Sharp, A. J., Baker, C., Itsara, A., Jiang, Z., Buysse, K., et al. (2008). Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 359, 1685–1699. doi: 10.1056/NEJMoa0805384
O'Bleness, M. S., Dickens, C. M., Dumas, L. J., Kehrer-Sawatzki, H., Wyckoff, G. J., and Sikela, J. M. (2012). Evolutionary history and genome organization of DUF1220 protein domains. G3 2, 977–986. doi: 10.1534/g3.112.003061
Pang, H., Yu, X., Kim, Y. M., Wang, X., Jinkins, J. K., Yin, J., et al. (2020). Disorders associated with diverse, recurrent deletions and duplications at 1q21.1. Front. Genet. 11:577. doi: 10.3389/fgene.2020.00577
Popesco, M. C., Maclaren, E. J., Hopkins, J., Dumas, L., Cox, M., Meltesen, L., et al. (2006). Human lineage-specific amplification, selection, and neuronal expression of DUF1220 domains. Science 313, 1304–1307. doi: 10.1126/science.1127980
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Rosenfeld, J. A., Traylor, R. N., Schaefer, G. B., McPherson, E. W., Ballif, B. C., Klopocki, E., et al. (2012). Proximal microdeletions and microduplications of 1q21.1 contribute to variable abnormal phenotypes. Eur. J. Hum. Genet. 20, 754–761. doi: 10.1038/ejhg.2012.6
Searles Quick, V. B., Davis, J. M., Olincy, A., and Sikela, J. M. (2015). DUF1220 copy number is associated with schizophrenia risk and severity: implications for understanding autism and schizophrenia as related diseases. Transl. Psychiatry 5:e697. doi: 10.1038/tp.2015.192
Sharp, A. J., Hansen, S., Selzer, R. R., Cheng, Z., Regan, R., Hurst, J. A., et al. (2006). Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat. Genet. 38, 1038–1042. doi: 10.1038/ng1862
Soemedi, R., Topf, A., Wilson, I. J., Darlay, R., Rahman, T., Glen, E., et al. (2012). Phenotype-specific effect of chromosome 1q21.1 rearrangements and GJA5 duplications in 2436 congenital heart disease patients and 6760 controls. Hum. Mol. Genet. 21, 1513–1520. doi: 10.1093/hmg/ddr589
Stefansson, H., Rujescu, D., Cichon, S., Pietiläinen, O. P., Ingason, A., Steinberg, S., et al. (2008). Large recurrent microdeletions associated with schizophrenia. Nature 455, 232–236. doi: 10.1038/nature07229
Keywords: 1q21.2, microduplication, hearing loss, congenital heart disease, Olduvai domain
Citation: Zhu L and Su X (2021) Case Report: Neuroblastoma Breakpoint Family Genes Associate With 1q21 Copy Number Variation Disorders. Front. Genet. 12:728816. doi: 10.3389/fgene.2021.728816
Received: 22 June 2021; Accepted: 26 August 2021;
Published: 27 September 2021.
Edited by:
Mario Capasso, University of Naples Federico II, ItalyReviewed by:
Marzia Ognibene, Unità Genetica Medica - IRCCS G. Gaslini, ItalyAna Pilar Berbegall, Hospital Quirónsalud Valencia, Spain
Copyright © 2021 Zhu and Su. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lijuan Zhu, ljzhucell@126.com