 Hualin Liu
Hualin Liu Vimalkumar Prajapati
Vimalkumar Prajapati Shobha Prajapati3
Shobha Prajapati3 Harsh Bais
Harsh Bais Jianguo Lu
Jianguo Lu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 30 September 2021
Sec. Computational Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.724217
This article is part of the Research TopicComputational Genomics and structural Bioinformatics in Microbial ScienceView all 12 articles
Bacillus amyloliquefaciens is a gram-positive, nonpathogenic, endospore-forming, member of a group of free-living soil bacteria with a variety of traits including plant growth promotion, production of antifungal and antibacterial metabolites, and production of industrially important enzymes. We have attempted to reconstruct the biogeographical structure according to functional traits and the evolutionary lineage of B. amyloliquefaciens using comparative genomics analysis. All the available 96 genomes of B. amyloliquefaciens strains were curated from the NCBI genome database, having a variety of important functionalities in all sectors keeping a high focus on agricultural aspects. In-depth analysis was carried out to deduce the orthologous gene groups and whole-genome similarity. Pan genome analysis revealed that shell genes, soft core genes, core genes, and cloud genes comprise 17.09, 5.48, 8.96, and 68.47%, respectively, which demonstrates that genomes are very different in the gene content. It also indicates that the strains may have flexible environmental adaptability or versatile functions. Phylogenetic analysis showed that B. amyloliquefaciens is divided into two clades, and clade 2 is further dived into two different clusters. This reflects the difference in the sequence similarity and diversification that happened in the B. amyloliquefaciens genome. The majority of plant-associated strains of B. amyloliquefaciens were grouped in clade 2 (73 strains), while food-associated strains were in clade 1 (23 strains). Genome mining has been adopted to deduce antimicrobial resistance and virulence genes and their prevalence among all strains. The genes tmrB and yuaB codes for tunicamycin resistance protein and hydrophobic coat forming protein only exist in clade 2, while clpP, which codes for serine proteases, is only in clade 1. Genome plasticity of all strains of B. amyloliquefaciens reflects their adaption to different niches.
Since the 19th century, Bacilli is one of the most well-documented and preeminently characterized bacterial genera comprising classical microbiology, biochemistry, and advanced genomic and proteomic approaches (Alcaraz et al., 2010). Among the various species of bacilli, Bacillus amyloliquefaciens gains lots of research interest and has wide application in agriculture, pharmaceuticals, food industry, environmental industry, etc. (Sharma and Satyanarayana, 2013). Various strains of B. amyloliquefaciens are common habitants and frequently screened from various ecological niches, including soil, animal feces, human food, aquatic environments, and many more, reflecting its versatile metabolic capabilities (Earl et al., 2008). During evolution, the bacterial population acclimatized to their respective ecological niches, which lead to the differentiation as evidenced by various genomic and physiological characteristics (De Wit et al., 2012).
Versatility of nature and metabolic competencies of different strains of B. amyloliquefaciens provoke to expedite the comparative genomic analysis to address more in detail the life style of bacteria, their adaptation to various niches and how they overcome contenders, as well as to catch clear revelation on their biochemistry, physiology, and genetics (Sharma and Satyanarayana, 2013; Owusu-Darko et al., 2020). B. amyloliquefaciens have been known to promote plant growth via a variety of mechanisms (Baghaee Ravari and Heidarzadeh, 2014; Shao et al., 2015; Liu et al., 2016), act as biocontrol against numerous plant diseases caused by soil-borne microorganisms (Tan et al., 2016), be widely used as biofertilizers and biopesticides (Wu et al., 2015), antagonize plant pathogens by competing essential nutrient (Wu et al., 2016), produce antibiotic compounds (Srivastava et al., 2016), as well induce systemic acquired resistance (Ng et al., 2016). Moreover, it is well documented that B. amyloliquefaciens can be tailored for numerous environmental and industrial applications such as degradation of crude oil from oil-contaminated sites (Zhang J. et al., 2016) and feather degradation (Yang et al., 2016); can produce various enzymes like proteases (Wang et al., 2016), feruloyl esterase (Wang et al., 2017), phytase (Verma et al., 2016), and amylases (Prajapati et al., 2015); and can be employed as a biosorbent for the removal of pollutants (Sun et al., 2016) and their degradation (Zühlke et al., 2016), production of biosurfactant and AMPs, probiotics, etc. (Perez et al., 2017).
The number of bacterial genome sequences has almost doubled over the decades due to the decreasing cost of the sequencing with advancement in high-throughput sequencing technology. The generated sequences data are available freely in the public domain, which ultimately stimulate researchers to do more on genomic analysis. Comparative genome analysis always sharpens our understanding of the bacterial genome structure and its diversity at a particular niche. Moreover, the pan-genome of species includes analysis of all core genes, dispensable genes, and strain-specific genes, which need to be comprehensively investigated as they reveal the essential functions for the species or laterally transferred functions in specific strains (Vernikos et al., 2015). Bacillus is one of the most extensively studied species with prevalent sets of genome sequences to date; however, very few reports are available on core genes and strain-specific genes in the Bacillus species (Alcaraz et al., 2010). Kim et al. (2017) have reported the core gene data of multiple Bacillus species through pan-genome analysis to explore the Bacillus species in food microbiome.
In the present investigation, we have curated all the 96 genome sequences of B. amyloliquefaciens available in the NCBI database to carry out comparative genomic analysis. Based on contextual information, we were trying to understand the distribution of all strains of B. amyloliquefaciens with respect to their ecological niches and their source of isolation and location to get better insights into their phylogenetic position using the core genome. PAN genome analysis of all strains of B. amyloliquefaciens was conducted to acquire better impression on their functional difference, which affects their dynamic evolutionary processes. We were also interested in understanding the comparative account of various antimicrobial and virulence genes presented among all B. amyloliquefaciens strains. The consensus information and conclusion drawn from this presented comparative genomic study can be used as a benchmark for designing wet-lab experimentation and validation as well as to formulate new hypothesis.
In total, 96 genome sequences of B. amyloliquefaciens having an N50 size greater than 50 k were downloaded from the NCBI database (detailed in Supplementary Table 1). Pan-genome analysis was conducted by Roary (Page et al., 2015) embedded in the “Pan” module of PGCGAP v1.0.21 (Liu et al., 2020). Single-copy core proteins calling, alignment of sequences, sequences concatenating, best model chosen, and phylogenetic tree constructing were performed with the “CoreTree” module of PGCGAP v1.0.21. The pairwise similarity of genomes was calculated by Mash (Ondov et al., 2016) embedded in the module “MASH” of PGCGAP v1.0.21. COG annotation was performed with the module “pCOG” of PGCGAP v1.0.21 (Liu et al., 2020). The antimicrobial resistance and virulence genes were mined against the databases of argannot (Gupta et al., 2014), card (Jia et al., 2017), NCBI (Feldgarden et al., 2019), resfinder (Zankari et al., 2012), vfdb (Chen et al., 2016), and EcOH (Ingle et al., 2016) by the module “AntiRes” of PGCGAP v1.0.21 (Liu et al., 2020).
A total of 16,198 gene clusters were found by pan-genome analysis, of which 1,448 (8.95%) are single-copy and code for core proteins. Shell genes, soft-core genes, core genes, and cloud genes comprise 17.09, 5.48, 8.96, and 68.47%, respectively, which demonstrates that the genomes are very different in the gene content (Supplementary Table 2). The pan-genome curve shows that the number of total genes increased with the increase in the genome number; this indicates that B. amyloliquefaciens has an open pan-genome (Figure 1).
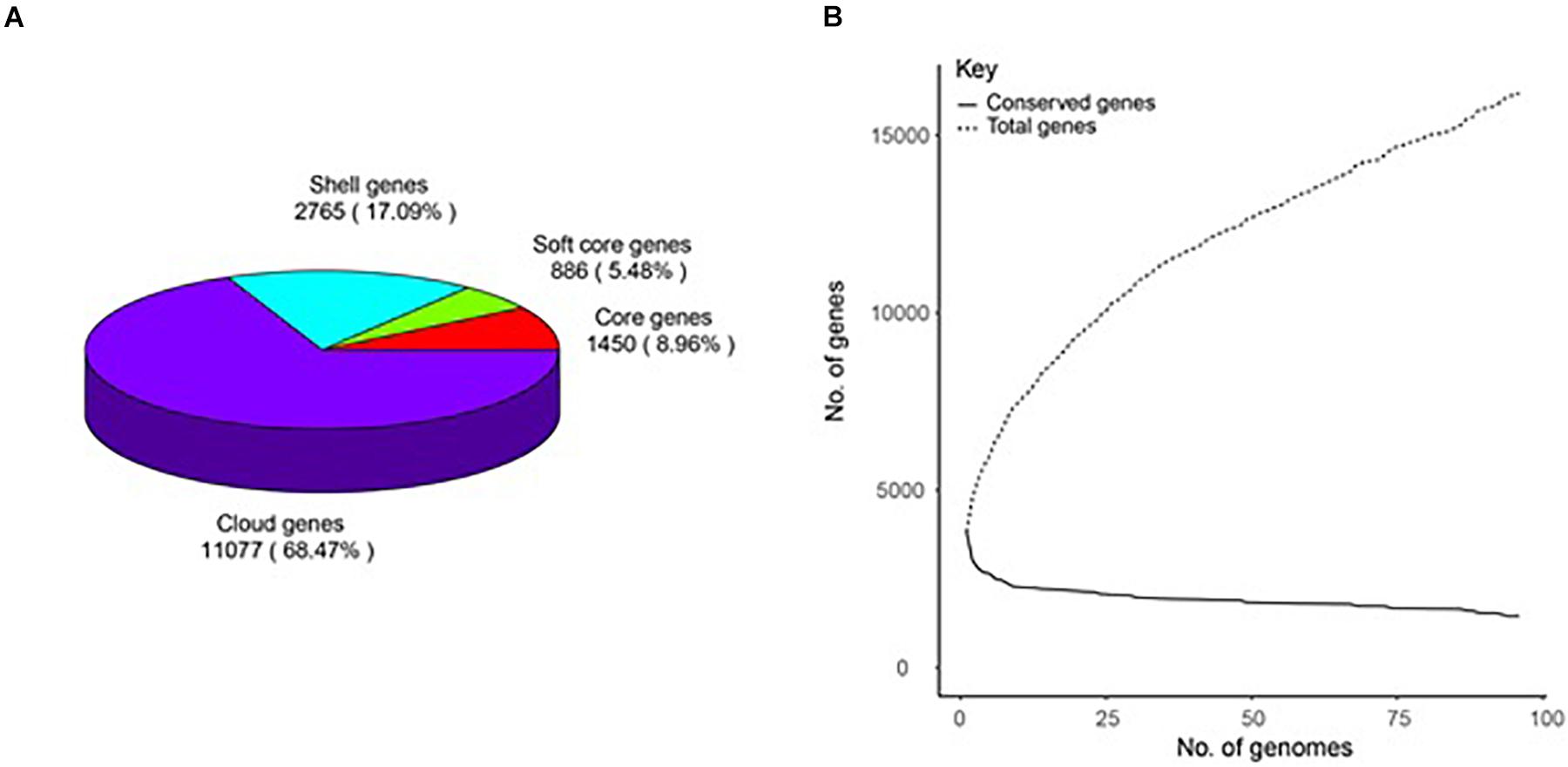
Figure 1. Pan-genome of 96 B. amyloliquefaciens. (A) A pie chart of the breakdown of genes (core: 99% ≤ strains ≤ 100%, soft-core: 95% ≤ strains < 99%, shell: 15% ≤ strains < 95%, and cloud: 0% ≤ strains < 15%). (B) The pan-genome curve of the 96 genomes.
The evolutionary relationship between the 96 B. amyloliquefaciens strains was investigated by the construction of a phylogenetic tree based on the alignment sequences of 1,154 concatenation core proteins (Figure 2). Bacillus pumilus SAFR-032 (Gioia et al., 2007) was used as the outgroup. The strains are divided into two clades, and clade 2 consists of two clusters. The location where the strain was isolated was mapped outside of the tree as the color strip. Strains from America are mainly located in cluster 2 of clade 2, while strains from Asia and Europe are scattered in all clades. The isolation source of the strain was also marked on the tree. According to known information, almost all the plant-associated strains are located in clade 2, and strains isolated from food are mainly located in clade 1. The above result implies that B. amyloliquefaciens has differentiated mainly into plant-associated and food-associated, as it clearly showed in the clades. However, some species of B. amyloliquefaciens isolated from water, soil, etc. are scattered in clade 2.
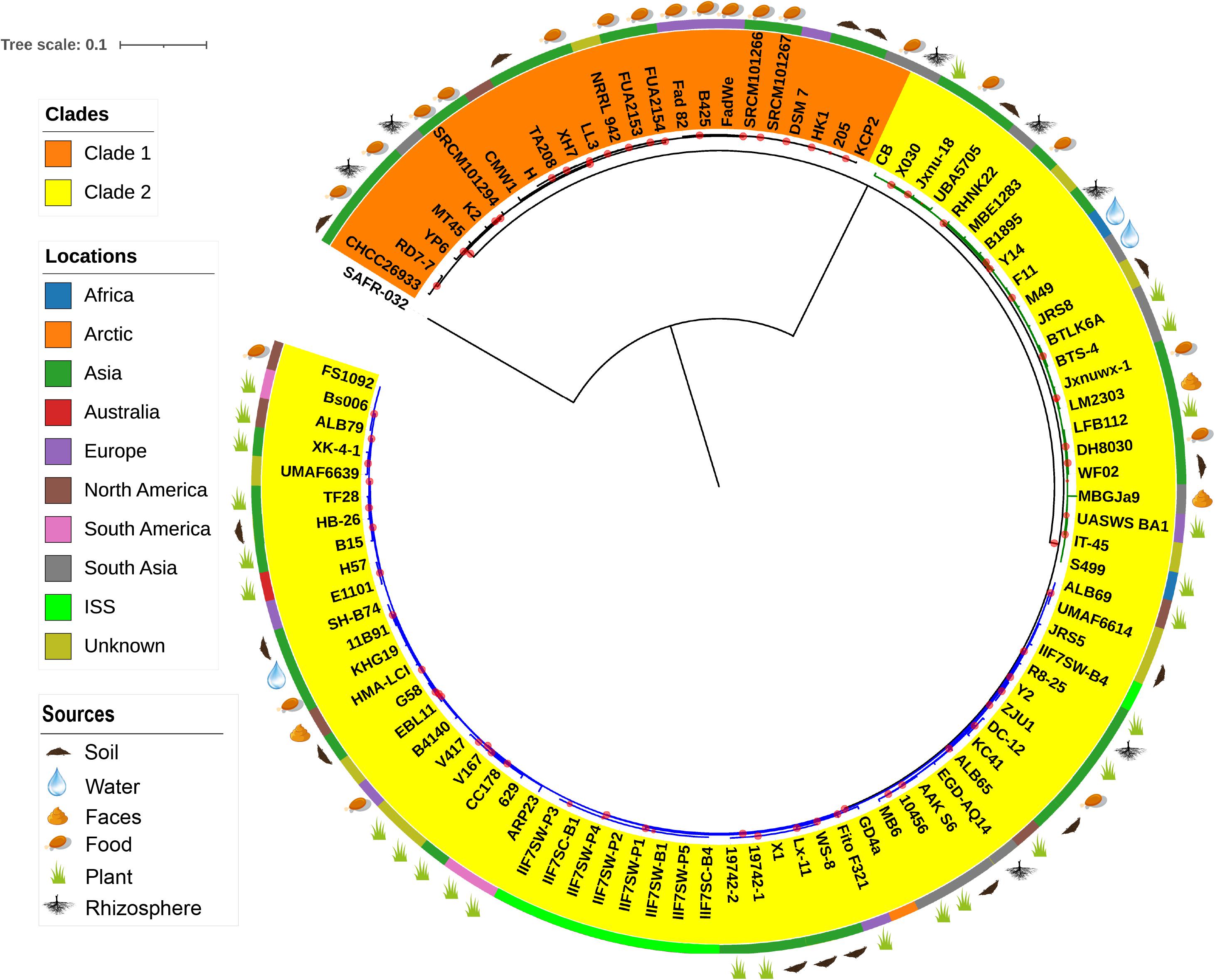
Figure 2. The phylogenetic tree of single-copy core proteins. The maximum-likelihood tree was constructed by the module “CoreTree” of PGCGAP v1.0.21 (Liu et al., 2020) with the best fit model JTT+F+R4. The tree was rooted against Bacillus pumilus SAFR-032 as an appropriate outgroup. Red circles on the branch represent bootstrap values larger than 80%. The background color of the labels (colored ranges) represents the clades, and the green and blue branches represent cluster 1 and cluster 2 of clade 2, respectively. The color strip outside the tree describes where the strain was isolated; ISS means International Space Station. The cartoons outside the tree indicate that the strain was isolated from soil, food, rhizosphere, water, or faces of herbivores, or is associated with the plant.
The similarity of genome pairs has been compared within and between clades and clusters (Figure 3). Genomes in clade 1 are found to be more similar than those that are observed in clade 2 (p < 0.001), while the similarity between genomes of the two clades is found to be very low, which indicates that strains in clade 2 undergone more differentiation, which may be related to their adaption to specific plants and other associated niches. When focusing on clade 2, genomes in cluster 1 are more similar than genomes in cluster 2 (p < 0.001), and the genome similarity between the two clusters is seen to be relatively low. Comparison of the genome size between both clades and its associated cluster has been carried out and depicted in Figure 3B. It has been observed that the genome size of clade 2 is slightly greater than that of clade 1, while the GC% content of clade 2 is significantly greater than that of clade 1 (p < 0.001; Figure 3C).
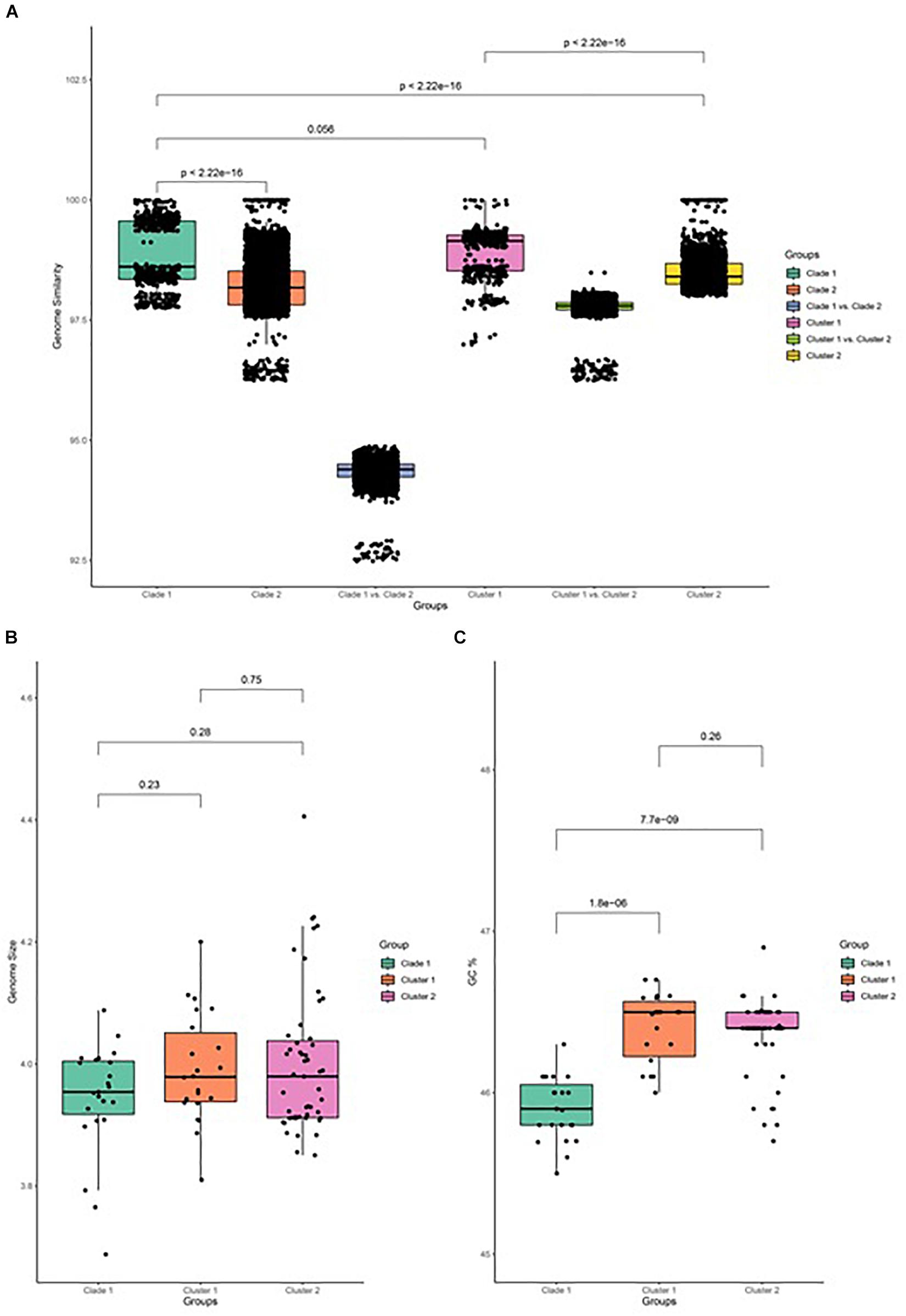
Figure 3. Genome feature of B. amyloliquefaciens. (A) Genome similarity between all pairs of strains in clade 1, between all pairs of strains in clade 2, between strains in clade 1 and those in clade 2, between strains in cluster 1, between strains in cluster 2, and between strains in cluster 1 and those in cluster 2. (B) Genome size of clade 1 and cluster 1 and cluster 2 of clade 2. Wilcox test was performed and marked on top of the box plot. (C) GC percent of clade 1, cluster 1, and cluster 2 of clade 2. Wilcox test was performed and the p-value was marked on top of the box plot.
Compared with the genomes of clade 2, the genomes of clade 1 have a unique gene composition (Figure 4A). It was observed that all the species in clade I have lost 335 genes (Supplementary Table 2 lines 2,592–2,926), which exists in all the genomes of clade 2 and have 490 unique core genes (Supplementary Table 2 lines 3,969–4,458). To reveal the difference of gene contents between the two clades, the gene family analysis has been performed with module “OrthoF” of PGCGAP v1.0.21. A total of 9,245 orthogroups are found, out of which 4,872 orthogroups are observed to be common between the two clades, while 1,055 are unique to clade 1, and the remaining 3,363 are unique to clade 2. The functions of these unique orthogroups are revealed through COG annotation as shown in Figure 4B. The relative abundance of functional classes I (lipid transport and metabolism), G (carbohydrate transport and metabolism), and Q (secondary metabolites biosynthesis, transport, and catabolism) is found to be higher in clade 2 compared to that in clade 1, while the relative abundance of classes D (cell cycle control, cell division, chromosome partitioning), E (amino acid transport and metabolism), H (coenzyme transport and metabolism), L (replication, recombination, and repair), M (cell wall/membrane/envelope biogenesis), and X (Mobilome: prophages, transposons) is higher in clade 1 than that in clade 2 (Figure 4B).
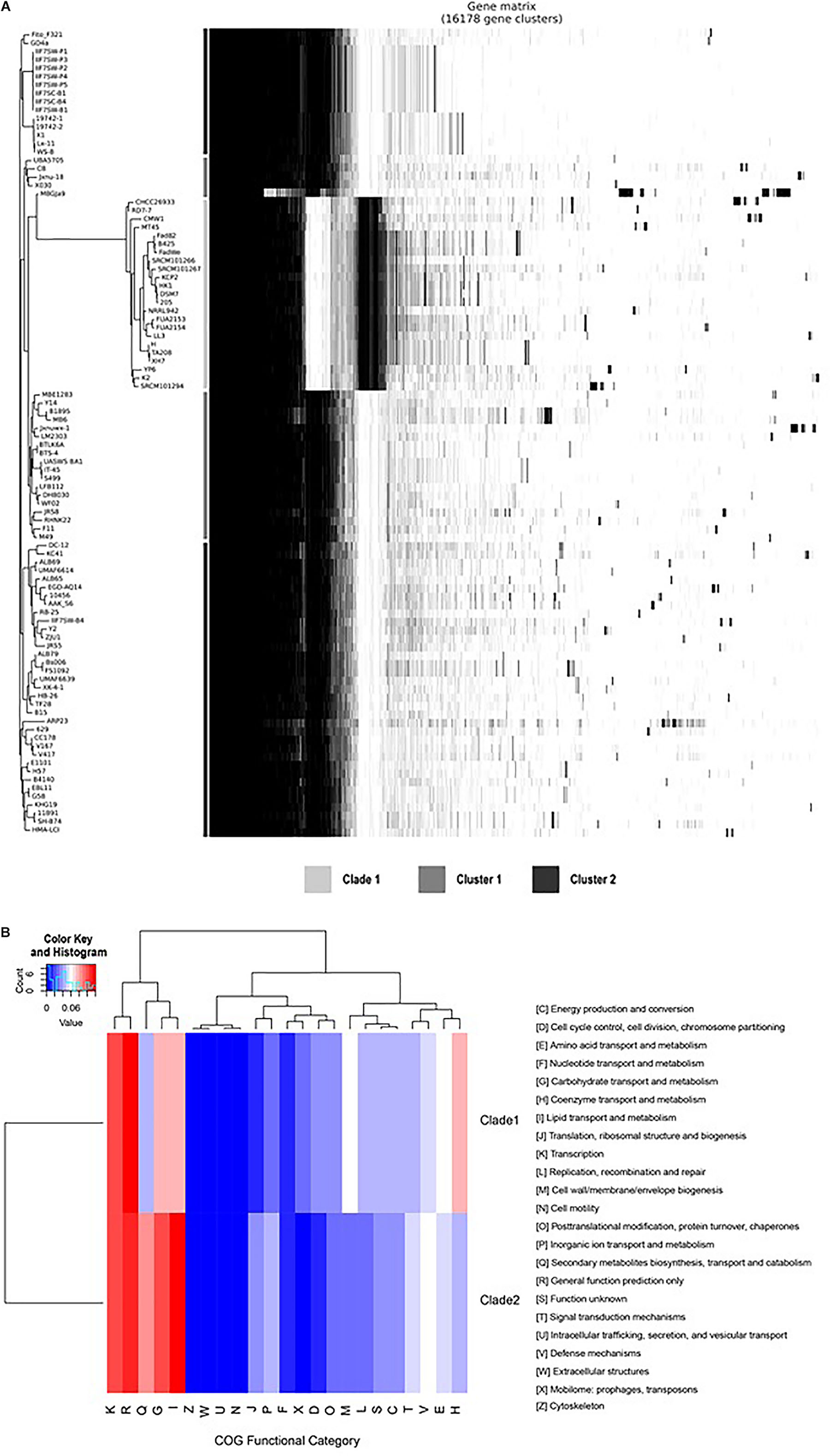
Figure 4B. Comparison of gene families between clade 1 and clade 2. The plot shows the tree compared to a matrix with the presence and absence of core and accessory genes (A); the bars between the tree and the matrix show to which clade/cluster the strain belongs. The heat map shows the relative abundance of the function classes corresponding to the unique orthogroups of clade 1 and clade 2 (B).
It is well documented that antimicrobial resistance and virulence genes are disseminated in the environment according to the function of the respective ecological niche; therefore, we have investigated the distribution of these genes in B. amyloliquefaciens. The antimicrobial resistance and virulence genes from different databases have been mined and mapped on the phylogenetic tree (Figure 5). To demonstrate the topological structure of the tree more clearly, the outgroup strain has been removed and the tree presented on midpoint rooted. All strains of B. amyloliquefaciens including those from foods contain more than one virulence factor. It is observed that tmrB and yuaB are only existing in clade 2, while clpP is prevailing only in clade 1. The gene tmrB is intending an ATP-binding tunicamycin resistance protein found in B. subtilis (Noda et al., 1995), while yuaB can form a highly hydrophobic coat around B. subtilis biofilms (Kobayashi and Iwano, 2012). The gene clpP codes for a serine protease involved in proteolysis and is required for growth under stress conditions (Gaillot et al., 2000, 2001). Interestingly, the B. amyloliquefaciens strain MBGJa9 has more virulence factors than other strains, and it is seen that isdA, isdB, isdC, isdD, isdE, isdF, isdG, and srtB form a gene cluster, whose productions participated in the uptake of iron and heme (Skaar and Schneewind, 2004; Skaar et al., 2004).
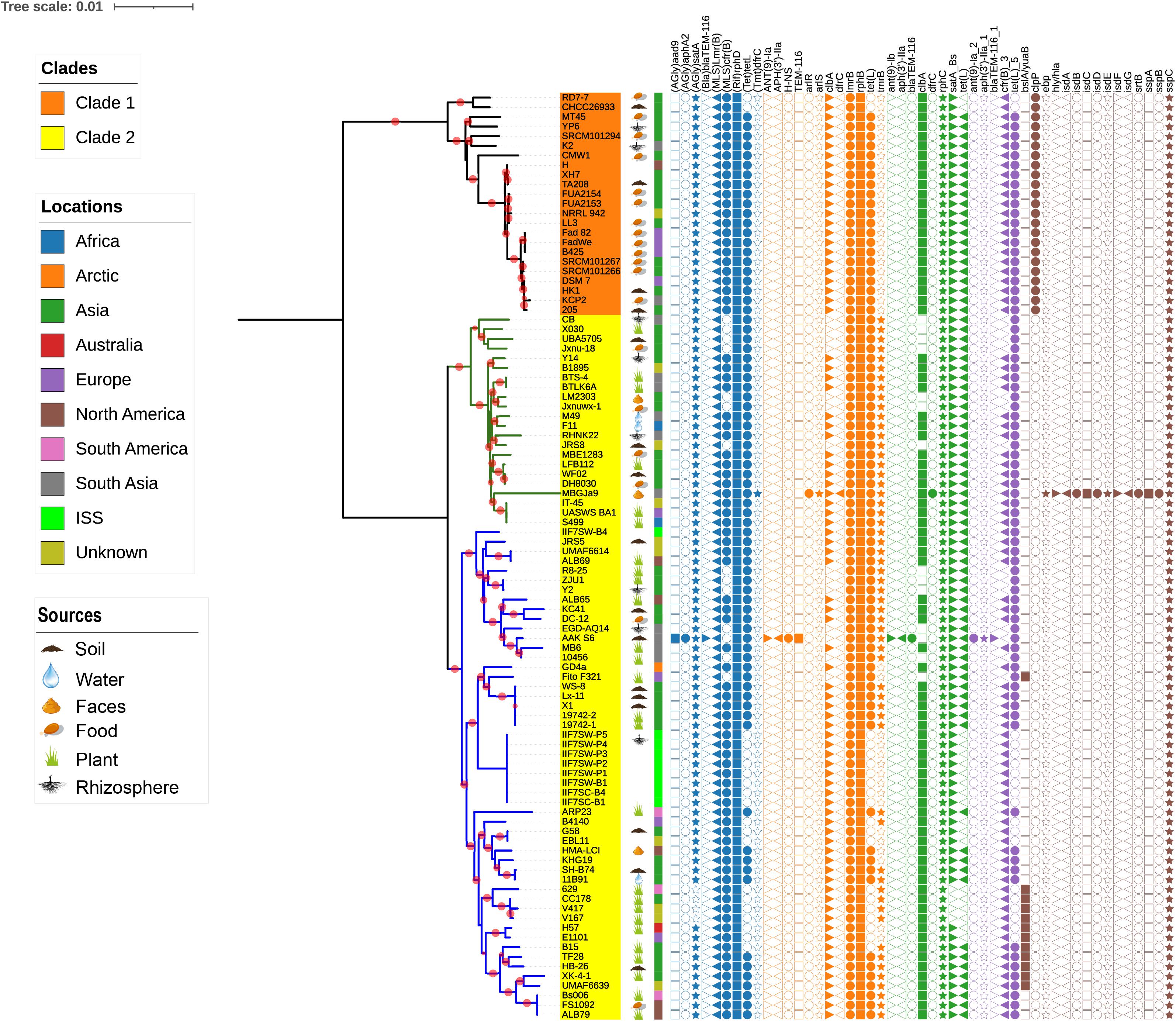
Figure 5. The midpoint rooted phylogenetic tree of single-copy core proteins. The tree was constructed by the module “CoreTree” of PGCGAP v1.0.21 (Liu et al., 2020) with the best fit model JTT+F+R4. Red circles on the branch represent bootstrap values larger than 80%. The background color of the labels represents the clades, and the green and blue branches represent cluster 1 and cluster 2 of clade 2, respectively. The cartoons after the strain name indicate that the strain was isolated from soil, food, rhizosphere, water, or faces of herbivores, or is associated with the plant. The color strip outside the tree describes where the strain was isolated; ISS means International Space Station. The symbols on the right side of the tree represent antibiotic resistance genes or virulence genes from each database [blue: argannot (Gupta et al., 2014), orange: card (Jia et al., 2017), green: NCBI (Michael Feldgarden et al., 2019), purple: resfinder (Zankari et al., 2012), and gray: vfdb (Chen et al., 2016)].
Present investigation using the 96 strains of B. amyloliquefaciens revealed that it has an extensive pan-genome, and it represents an ample number of genes that were observed to be uniquely associated with each of the divergent species. Population size and respective ecological niche versatility of B. amyloliquefaciens are considered to be the most influential factors in determining the pan-genome size, and it can be seen that total genes against the total number of genome sequences are edified up so it is impossible to envisage the size of the full pan-genome. The resulted open pan-genome of B. amyloliquefaciens is unsurprising because of the source of isolation and its geographical location, which always adds up new genes to the entire gene pool of species. This species divergence could be an attribute of different mechanisms such as horizontal transfer, transposition, and transformation (Konstantinidis and Tiedje, 2005; Tettelin et al., 2005). On the contrary, observed few core genes in the investigation might be due to the higher number of genomes, the incorporation of genomes from other genera, as well as the inclusion of draft genomes in the data set (Lefebure et al., 2010; Inglin et al., 2018). It is well documented that incomplete, unfinished, or partially assembled genomes have a large impact on the occurrence of core genomes in the analysis as core genomes seem to be very sensitive to the heterogenous data set and poor quality sequences (Mendes-Soares et al., 2014).
16s rRNA has been widely used for the taxonomy assessment of prokaryotes and has served as the broad context, though the better taxonomic resolution of the microbial species is achieved through “polyphasic approach” and is highly effective (Rosselló-Mora and Amann, 2001; Na et al., 2018). 16s rRNA has limitations as it hampers the phylogenic resolution at the species or subspecies level. The application of genome sequences is highly recommended for the taxonomic understanding of microbial species instead of routinely used DNA–DNA hybridization and 16s rRNA phylogeny (Chun et al., 2018). Therefore, instead of a single gene, genome-based phylogeny called phylogenomics has set up better taxonomic positioning as it uses sets of core genes (Eisen and Fraser, 2003). The genome sequences of all B. amyloliquefaciens strains are accessible in the Gene Bank NCBI database, which allows us to determine the degree of genome variability among all species as well as distinct out the taxonomic validity of all the isolates and reconstruct their phylogenetic relationship. Two distinct clades were observed when phylogeny was inferred using single copy core protein. Clade 1 comprises 23 strains of B. amyloliquefaciens, out of which 56% were food-associated, 17.39% were from soil, and 8.69% were rhizospheric. Clade 2 comprises 73 strains, and it is distinguished into two different clusters, where clusters 1 and 2 comprise 22 and 51 strains of B. amyloliquefaciens, respectively. Clade 2 was more enriched with the species of plant origin/host and comprised ∼35.61%, while the strains of soil, food, indoor biome, and rhizosphere origin were 16.43, 10.95, 12.32, and 6.84%, respectively. Two distinct clades were demarcated, one of which was food-associated (clade 1) and the other one plant-associated (clade 2). The selection of core gene sets for accurate phylogeny analysis may vary with the availability of the genome sequences at the time of analysis.
Comparison of the genome similarity between the strains of both clades indicates that the strains grouped together in clade 1 are more similar than those of clade 2. The majority of the plant-associated strains of B. amyloliquefaciens are grouped under clade 2, while nonplant-associated strains are mainly found in clade 1, though some scattering is seen with respect to some other ecological niches. Plant-associated strains of B. amyloliquefaciens have adopted more modification in their genome, which is directly related to their adaption to the specific plant. Hence, it is believed that the genome size of the plant-associated strains of B. amyloliquefaciens is always greater than that of the nonplant-associated B. amyloliquefaciens and so the GC % content. Zhang N. et al. (2016) reported that the core genomes of the plant-associated strains of B. amyloliquefaciens have more gene contents related to the intermediary metabolism and secondary metabolite biosynthesis as compared to those of nonplant-associated strains. Plant-associated strains also possess specific genes for the synthesis of antibiotics as well as for the utilization of plant-derived substrates.
During the assessment of the core and accessory genes, it was observed that the strains of B. amyloliquefaciens grouped in clade 1 have lost many genes that are present in the strains of clade 2 (Figure 4A). Exopolysaccharides (EPSs) play very important role in bacteria, specifically those that are plant-associated and have a variety of functions. It helps microorganisms in adherence, pathogenesis, and symbiosis as well as protects from desiccation in some adverse condition (Stingele et al., 1999). The glycosyltransferase gene region comprises the EPS gene cluster, i.e., epsF-2, epsD, epsI, epsM, epsL, and epsJ, which are involved in the biosynthesis of EPS, and has a profound role in plant-associated strains of B. amyloliquefaciens, while it was missing in the strains belonging to clade 1. Plant-associated B. amyloliquefaciens strains (clade 2) harbor a certain gene cluster absent in clade 1, which is involved in the biosynthesis of lipopeptides through nonribosomal peptide synthetases (NRPS) including fengycin (fen). Gene clusters involved in the synthesis of bacillaene (bae) are responsible for the profound antimicrobial activity and are lost in all strains of B. amyloliquefaciens in clade 1. The PKS gene cluster, which includes pksI_2, pksG_2, pksN_2, and pksS, was also found to be present in clade 2 but lost in clade 1 (Figure 4A). Some of the genes such as cystathionine beta-lyase (patB), putative multidrug resistance ABC transporter ATP-binding/permease protein (yheI), cold shock protein (cspC), spermidine/spermine N(1)-acetyltransferase (paiA), putative sugar phosphate isomerase (ywlF), 3-dehydroshikimate dehydratase (asbF), putative ABC transporter substrate-binding lipoprotein (yhfQ), sirohydrochlorin ferrochelatase (sirB), putative metallo-hydrolase (yflN), dipeptidyl-peptidase 5 (ddp5), L-aspartate oxidase (nadB), putative sporulation hydrolase (cotR), stress response kinase A (srkA), sortase D (srtD), ATP-dependent dethiobiotin synthetase (bioD 1), glycerophosphodiester phosphodiesterase (glypQ), folylpolyglutamate synthase (fpgS), and putative ABC transporter permease (ytrC) were found to be uniquely associated to the strain of B. amyloliquefaciens that belongs to clade 1. Hence, the presence of certain gene clusters in clade 2 and their absence in clade 1 conclude that plant-associated strains of B. amyloliquefaciens have more abundant gene clusters for intermediary metabolism as well as for antibiotic production compared to the nonplant-associated strains. Niazi et al. (2014) have reported that B. amyloliquefaciens subsp. plantarum, a rhizobacterium that mends plant growth and stress management, also possesses the more abundant gene cluster that is actively involved in the production of certain hydrolytic enzymes as well as secondary metabolites. It is well documented that the rhizosphere environment has a very dynamic microbial community because of the effect of root exudates and the constant interaction and competition among microbes, as they need to contend with each other for various resources such as nutrient supply, which ultimately leads them to produce various metabolites such as antibiotic and extracellular hydrolases (Bais et al., 2006).
It is documented that bacteria have produced antibiotics for millions of years, which results in the evolution and induction of resistance genes. More precisely, the intensive nonmedical use of antibiotics such as in agricultural and in some industrial applications is not certain and has led to significant dissemination of resistance genes in the environment (Pawlowski et al., 2018). The genomes of all the strains of B. amyloliquefaciens were mapped to different databases to evaluate the distribution of antibiotic resistance genes and virulence genes. Many different genes were perceived and were scattered among all the strains of B. amyloliquefaciens; also, the observed genes belonged to a variety of resistance classes. The gene (AGlu) satA codes for the enzyme aminoglycoside acetyltransferase, and it belongs to the class aminoglycosidese, which is present in almost all the strains independent of its host environment. Aminoglycoside is considered to be part of the broad spectrum of antibiotics, and it acts by inhibiting the protein synthesis, though it works best in synergy with other antimicrobials (Krause et al., 2016). Two genes, lmrB and cfrB, belonging to the class Macrolide-Lincosamide-Streptogramin B (MLS) were present in most of the strains considered in the investigation. The gene product of lmrB and cfrB confers specific resistance to lincosamides, such as lincomycin and clindamycin, and synthetic antibiotic linezolid, respectively, (Kim et al., 2001; Toh et al., 2007). The advent of new and more stable macrolide and its vague use could be the key reasons for the induction of such resistance imparting genes, and it provides an opportunity for microbial populations to acquire MLS resistance (Roberts et al., 1999). (Rif) rphD, rphB, and rphC genes code for trifamycin kinase (phosphotransferase), which confers resistance against rifampin, the most commonly used rifamycin. The enzyme rifampin phosphotransferase present in many environmental bacteria, which used to be induced by selective pressure and nonclinical use of antibiotics, has led to the inactivation of rifampin and ATP to phosphor-rifampin and AMP+Pi (Stogios et al., 2016). More than 40 different tetracycline resistance genes have been reported in numerous bacterial genera of agricultural and industrial use. The dispersion of the tetL gene among the bacterial genera was much higher than any other tet resistant genes (Roberts, 2005). In the present investigation, the genome sequences of all the strains were mapped against six different databases, i.e., argannot, NCBI, plasmidfinder, card, resfinder, and yfdb, and they reveal the presence of tetL genes among all the strains. Colibactin is a genotoxic molecule coded by the clb gene cluster in many enteric bacteria, and it is widely distributed in nature (Kawanishi et al., 2020). clbA is a plasmid-encoded cfr gene under the control of an inducible promoter reported in B. velezensis (B. amyloliquefaciens subsp. plantarum), while clbB and clbC are found in Brevibacillus brevis and B. clausii, respectively, (Hansen et al., 2012). The gene sspC codes for cytoplasmic protein known as staphostatin and is present in all the strains of B. amyloliquefaciens. It is a very specific and tightly binding inhibitor of staphopain B (SspB). The main function of sspC is to protect the cytosolic protein from the degradation executed by misfolded or activated SspB. Shaw et al. (2005) reported that in the absence of sspC protein, major alteration in cellular physiology occurred, and the growth and viability of the microbial cells were impaired. The gene clpP is prevailing only in the strains that belong to clade 1, and it codes for the caseinolytic protease proteolytic subunit (ClpP) serine proteases. The ClpP protein confers certain advantages to the microorganisms to sustain in varying environmental conditions as well as stress conditions. ClpC and ClpP are heat shock proteins and are subunits of ATP-dependent proteases reported in B. subtilis. The transcription of genes clpC and clpP is always negatively regulated under nonstressed condition (Krüger et al., 2001). The virulence and infectivity of a number of microorganisms/pathogens are affected due to the alteration of the ClpP protein function. Clp proteins are highly conserved and have played a very important role in the proteolysis of prokaryotic cell and eukaryotic organelles, though only few reports are available describing the importance of Clp-mediated proteolysis in organisms (Krüger et al., 2001; Moreno-Cinos et al., 2019). Tunicamycin, a nucleoside antibiotic, kills most of the gram-positive bacteria, and it acts by inhibiting the important cell wall component called teichoic acid, which drives the physiology and pathogenesis of microorganisms. The exposure of bacteria toward the sub-inhibitory concentration of tunicamycin leads to the reduction in biofilm production, virulence protein, as well bacterial adhesion and invasion (Zhu et al., 2018). The presence of the tmrB gene leads to the production of the TmrB protein, which imparts tunicamycin resistance to B. subtilis. The TmrB protein is present in both cytoplasmic and membrane fractions, though it is completely hydrophilic, and it attaches to the membrane by its C-terminal amphiphilic alpha-helix (Noda et al., 1995). Many plant growth promoting bacteria reported to produce biofilm, which is their key strategy to survive successfully in some harsh conditions as well as in plant rhizosphere. Biofilm formation capability of microorganisms makes them a good biocontrol agent as it leads to the reduction in infection caused by fungal and bacterial pathogens (Hobley et al., 2013). The bslA/yuaB gene present in many of the plant-associated strains of B. amyloliquefaciens codes for unique surface active protein BslA, which forms a hydrophobic surface layer called hydrophobins. The surface layer regulates the diffusion of various molecules, perception of signaling molecules from other microbial community, as well as nutrient uptake, in addition to imparting the protection to the bacterial cell. The contextual information of ecological and evolutionary facts as well as the application of comparative genomics and the dropping cost of genome sequencing collectively aid to understanding more precisely the structure of microbial diversity and its ecological distribution. Phylogenomics reveals the segregation of all 96 strains of B. amyloliquefaciens into two clades. Majority of the plant-associated B. amyloliquefaciens strains are grouped in clade 2, while clade 1 accomplishes mostly food-associated strains. The distribution of resistance and virulence genes among all the strains of B. amyloliquefaciens has been reported, and it will serve as a benchmark and resourceful information to deduce the hypothesis or conclusion as well as to exploit the potential of any strains through wet-lab experimentation. In future prospectus, we will try to dig out some temporal genes and their occurrence pattern in order to comprehend the significant role of microorganisms as well as the structure of the entire microbial community with its respective environmental niches.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author/s.
VP conceived and modeled the study. VP and HL analyzed the data and prepared the methods and results. VP and SP prepared the manuscript. HB and JL corrected the manuscript and inputs. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.724217/full#supplementary-material
Supplementary Figure 1 | A flow chart representation of analyses conducted by different modules of PGCGAP v1.0.21, which integrates some popular software and in-house scripts (Liu et al., 2020).
Supplementary Table 1 | Details of 96 B. amyloliquefaciens strains, including their genome size, GC%, scaffolds, CDS, its host and geographical location of the isolates, etc., retrieved from the NCBI database for comparative genome analysis.
Supplementary Table 2 | Data analysis of pan-genome segregation to identify single-copy core proteins, shell genes, soft-core genes, core genes, and cloud genes, including its annotation, genome fragment and order within fragments, accessory order, and accessory order with fragment.
Alcaraz, L. D., Moreno-Hagelsieb, G., Eguiarte, L. E., Souza, V., Herrera-Estrella, L., and Olmedo, G. (2010). Understanding the evolutionary relationships and major traits of Bacillus through comparative genomics. BMC Genomics 11:332. doi: 10.1186/1471-2164-11-332
Baghaee Ravari, S., and Heidarzadeh, N. (2014). Isolation and characterization of rhizosphere auxin producing Bacilli and evaluation of their potency on wheat growth improvement. Arch. Agron. Soil Sci. 60, 895–905. doi: 10.1080/03650340.2013.856003
Bais, H. P., Weir, T. L., Perry, L. G., Gilroy, S., and Vivanco, J. M. (2006). The role of root exudates in rhizosphere interactions with plants and other organisms. Annu. Rev. Plant Biol. 57, 233–266. doi: 10.1146/annurev.arplant.57.032905.105159
Chen, L., Zheng, D., Liu, B., Yang, J., and Jin, Q. (2016). VFDB 2016: hierarchical and refined dataset for big data analysis–10 years on. Nucleic Acids Res. 44, D694–D697. doi: 10.1093/nar/gkv1239
Chun, J., Oren, A., Ventosa, A., Christensen, H., Arahal, D. R., da Costa, M. S., et al. (2018). Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68, 461–466. doi: 10.1099/ijsem.0.002516
De Wit, P. J. G. M., Van Der Burgt, A., Ökmen, B., Stergiopoulos, I., Abd-Elsalam, K. A., Aerts, A. L., et al. (2012). The genomes of the fungal plant pathogens Cladosporium fulvum and Dothistroma septosporum reveal adaptation to different hosts and lifestyles but also signatures of common ancestry. PLoS Genet 8:e1003088.
Earl, A. M., Losick, R., and Kolter, R. (2008). Ecology and genomics of Bacillus subtilis. Trends Microbiol. 16, 269–275.
Eisen, J. A., and Fraser, C. M. (2003). Phylogenomics: intersection of evolution and genomics. Science 300, 1706–1707. doi: 10.1126/science.1086292
Feldgarden, M., Brover, V., Haft, D. H., Prasad, A. B., Slotta, D. J., Tolstoy, I., et al. (2019). Validating the AMRFinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob. Agents Chemother. 63, e483–e419. doi: 10.1128/AAC.00483-19
Gaillot, O., Bregenholt, S., Jaubert, F., Di Santo, J. P., and Berche, P. (2001). Stress-induced ClpP serine protease of Listeria monocytogenes is essential for induction of listeriolysin O-dependent protective immunity. Infect. Immun. 69, 4938–4943. doi: 10.1128/iai.69.8.4938-4943.2001
Gaillot, O., Pellegrini, E., Bregenholt, S., Nair, S., and Berche, P. (2000). The ClpP serine protease is essential for the intracellular parasitism and virulence of Listeria monocytogenes. Mol. Microbiol. 35, 1286–1294. doi: 10.1046/j.1365-2958.2000.01773.x
Gioia, J., Yerrapragada, S., Qin, X., Jiang, H., Igboeli, O. C., Muzny, D., et al. (2007). Paradoxical DNA repair and peroxide resistance gene conservation in Bacillus pumilus SAFR-032. PLoS One 2:e928. doi: 10.1371/journal.pone.0000928
Gupta, S. K., Padmanabhan, B. R., Diene, S. M., Lopez-Rojas, R., Kempf, M., Landraud, L., et al. (2014). ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 58, 212–220. doi: 10.1128/aac.01310-13
Hansen, L. H., Planellas, M. H., Long, K. S., and Vester, B. (2012). The order Bacillales hosts functional homologs of the worrisome cfr antibiotic resistance gene. Antimicrob. Agents Chemother. 56, 3563–3567. doi: 10.1128/aac.00673-12
Hobley, L., Ostrowski, A., Rao, F. V., Bromley, K. M., Porter, M., Prescott, A. R., et al. (2013). BslA is a self-assembling bacterial hydrophobin that coats the Bacillus subtilis biofilm. Proc. Natl. Acad. Sci.U.S.A. 110:13600. doi: 10.1073/pnas.1306390110
Ingle, D. J., Valcanis, M., Kuzevski, A., Tauschek, M., Inouye, M., Stinear, T., et al. (2016). In silico serotyping of E. coli from short read data identifies limited novel O-loci but extensive diversity of O:H serotype combinations within and between pathogenic lineages. Microbial. Genomics 2:e000064. doi: 10.1099/mgen.0.000064
Inglin, R. C., Meile, L., and Stevens, M. J. A. (2018). Clustering of pan-and core-genome of Lactobacillus provides novel evolutionary insights for differentiation. BMC Genomics 19:284. doi: 10.1186/s12864-018-4601-5
Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., et al. (2017). CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–D573. doi: 10.1093/nar/gkw1004
Kawanishi, M., Shimohara, C., Oda, Y., Hisatomi, Y., Tsunematsu, Y., Sato, M., et al. (2020). Genotyping of a gene cluster for production of colibactin and in vitro genotoxicity analysis of Escherichia coli strains obtained from the Japan Collection of Microorganisms. Genes Environ. 42:12. doi: 10.1186/s41021-020-00149-z
Kim, H. J., Kim, Y., Lee, M. S., and Lee, H. S. (2001). Gene lmrB of Corynebacterium glutamicum confers efflux-mediated resistance to lincomycin. Mol. Cells 12, 112–116.
Kim, Y., Koh, I., Young, L. M., Chung, W. H., and Rho, M. (2017). Pan-genome analysis of Bacillus for microbiome profiling. Sci. Rep. 7:10984. doi: 10.1038/s41598-017-11385-9
Kobayashi, K., and Iwano, M. (2012). BslA(YuaB) forms a hydrophobic layer on the surface of Bacillus subtilis biofilms. Mol. Microbiol. 85, 51–66. doi: 10.1111/j.1365-2958.2012.08094.x
Konstantinidis, K. T., and Tiedje, J. M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 102, 2567–2572. doi: 10.1073/pnas.0409727102
Krause, K. M., Serio, A. W., Kane, T. R., and Connolly, L. E. (2016). Aminoglycosides: an overview. Cold Spring Harb. Perspect. Med. 6:a027029. doi: 10.1101/cshperspect.a027029
Krüger, E., Zühlke, D., Witt, E., Ludwig, H., and Hecker, M. (2001). Clp-mediated proteolysis in Gram-positive bacteria is autoregulated by the stability of a repressor. EMBO J. 20, 852–863. doi: 10.1093/emboj/20.4.852
Lefebure, T., Pavinski Bitar, P. D., Suzuki, H., and Stanhope, M. J. (2010). Evolutionary dynamics of complete Campylobacter pan-genomes and the bacterial species concept. Genome Biol. Evol. 2, 646–655.
Liu H., Xin, B., Zheng, J., Zhong, H., Yu, Y., Peng, D., et al. (2020). Build a bioinformatics analysis platform and apply it to routine analysis of microbial genomics and comparative genomics. Protocol. Exchange. doi: 10.21203/rs.2.21224/v3
Liu, Y., Chen, L., Zhang, N., Li, Z., Zhang, G., Xu, Y., et al. (2016). Plant-microbe communication enhances auxin biosynthesis by a root-associated bacterium, Bacillus amyloliquefaciens SQR9. Mol. Plant Microbe Interact. 29, 324–330. doi: 10.1094/mpmi-10-15-0239-r
Mendes-Soares, H., Suzuki, H., Hickey, R. J., and Forney, L. J. (2014). Comparative functional genomics of Lactobacillus spp. reveals possible mechanisms for specialization of vaginal lactobacilli to their environment. J. Bacteriol. 196, 1458–1470. doi: 10.1128/jb.01439-13
Moreno-Cinos, C., Goossens, K., Salado, I. G., Van Der Veken, P., De Winter, H., and Augustyns, K. (2019). ClpP Protease, a promising antimicrobial target. Int. J. Mol. Sci. 20:2232. doi: 10.3390/ijms20092232
Na, S. I., Kim, Y. O., Yoon, S. H., Ha, S. M., Baek, I., and Chun, J. (2018). UBCG: Up-to-date bacterial core gene set and pipeline for phylogenomic tree reconstruction. J. Microbiol. 56, 280–285. doi: 10.1007/s12275-018-8014-6
Ng, L. C., Sariah, M., Sariam, O., Radziah, O., and Zainal Abidin, M. A. (2016). PGPM-induced defense-related enzymes in aerobic rice against rice leaf blast caused by Pyricularia oryzae. Eur. J. Plant Pathol. 145, 167–175. doi: 10.1007/s10658-015-0826-1
Niazi, A., Manzoor, S., Asari, S., Bejai, S., Meijer, J., and Bongcam-Rudloff, E. (2014). Genome analysis of Bacillus amyloliquefaciens Subsp. plantarum UCMB5113: a rhizobacterium that improves plant growth and stress management. PLoS One 9:e104651. doi: 10.1371/journal.pone.0104651
Noda, Y., Takatsuki, A., Yoda, K., and Yamasaki, M. (1995). TmrB protein, which confers resistance to tunicamycin on Bacillus subtilis, binds tunicamycin. Biosci. Biotechnol. Biochem. 59, 321–322. doi: 10.1271/bbb.59.321
Ondov, B. D., Treangen, T. J., Melsted, P., Mallonee, A. B., Bergman, N. H., Koren, S., et al. (2016). Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 17:132. doi: 10.1186/s13059-016-0997-x
Owusu-Darko, R., Allam, M., Ismail, A., Ferreira, C. A. S., Oliveira, S. D. D., and Buys, E. M. (2020). Comparative genome analysis of Bacillus sporothermodurans with Its closest phylogenetic neighbor, Bacillus oleronius, and Bacillus cereus and Bacillus subtilis Groups. Microorganisms 8:1185.
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Pawlowski, A. C., Westman, E. L., Koteva, K., Waglechner, N., and Wright, G. D. (2018). The complex resistomes of Paenibacillaceae reflect diverse antibiotic chemical ecologies. ISME J. 12, 885–897. doi: 10.1038/s41396-017-0017-5
Perez, K. J., Viana, J. D. S., Lopes, F. C., Pereira, J. Q., dos Santos, D. M., Oliveira, J. S., et al. (2017). Bacillus spp. isolated from puba as a source of biosurfactants and antimicrobial lipopeptides. Front. Microbiol. 8:61.
Prajapati, V. S., Trivedi, U. B., and Patel, K. C. (2015). A statistical approach for the production of thermostable and alklophilic alpha-amylase from Bacillus amyloliquefaciens KCP2 under solid-state fermentation. 3 Biotech 5, 211–220. doi: 10.1007/s13205-014-0213-1
Roberts, M. C. (2005). Update on acquired tetracycline resistance genes. FEMS Microbiol. Lett. 245, 195–203. doi: 10.1016/j.femsle.2005.02.034
Roberts, M. C., Sutcliffe, J., Courvalin, P., Jensen, L. B., Rood, J., and Seppala, H. (1999). Nomenclature for macrolide and macrolide-lincosamide-streptogramin B resistance determinants. Antimicrob. Agents Chemother. 43, 2823–2830. doi: 10.1128/AAC.43.12.2823
Rosselló-Mora, R., and Amann, R. (2001). The species concept for prokaryotes. FEMS Microbiol. Rev. 25, 39–67. doi: 10.1111/j.1574-6976.2001.tb00571.x
Shao, J., Li, S., Zhang, N., Cui, X., Zhou, X., Zhang, G., et al. (2015). Analysis and cloning of the synthetic pathway of the phytohormone indole-3-acetic acid in the plant-beneficial Bacillus amyloliquefaciens SQR9. Microbial. Cell Factories 14:130. doi: 10.1186/s12934-015-0323-4
Sharma, A., and Satyanarayana, T. (2013). Comparative genomics of Bacillus species and its relevance in industrial microbiology. Genomics Insights 6, 25–36.
Shaw, L. N., Golonka, E., Szmyd, G., Foster, S. J., Travis, J., and Potempa, J. (2005). Cytoplasmic control of premature activation of a secreted protease zymogen: deletion of staphostatin B (SspC) in Staphylococcus aureus 8325-4 yields a profound pleiotropic phenotype. J. Bacteriol. 187, 1751–1762. doi: 10.1128/JB.187.5.1751-1762.2005
Skaar, E. P., Gaspar, A. H., and Schneewind, O. (2004). IsdG and IsdI, heme-degrading enzymes in the cytoplasm of Staphylococcus aureus. J. Biol. Chem. 279, 436–443. doi: 10.1074/jbc.M307952200
Skaar, E. P., and Schneewind, O. (2004). Iron-regulated surface determinants (Isd) of Staphylococcus aureus: stealing iron from heme. Microbes Infect. 6, 390–397. doi: 10.1016/j.micinf.2003.12.008
Srivastava, S., Bist, V., Srivastava, S., Singh, P. C., Trivedi, P. K., Asif, M. H., et al. (2016). Unraveling aspects of Bacillus amyloliquefaciens mediated enhanced production of rice under biotic stress of Rhizoctonia solani. Front. Plant Sci. 7:587.
Stingele, F., Newell, J. W., and Neeser, J. R. (1999). Unraveling the function of glycosyltransferases in Streptococcus thermophilus Sfi6. J. Bacteriol. 181, 6354–6360. doi: 10.1128/JB.181.20.6354-6360.1999
Stogios, P. J., Cox, G., Spanogiannopoulos, P., Pillon, M. C., Waglechner, N., Skarina, T., et al. (2016). Rifampin phosphotransferase is an unusual antibiotic resistance kinase. Nat. Commun. 7:11343. doi: 10.1038/ncomms11343
Sun, P., Hui, C., Wang, S., Wan, L., Zhang, X., and Zhao, Y. (2016). Bacillus amyloliquefaciens biofilm as a novel biosorbent for the removal of crystal violet from solution. Colloids Surf. B Biointerfaces 139, 164–170. doi: 10.1016/j.colsurfb.2015.12.014
Tan, S., Gu, Y., Yang, C., Dong, Y., Mei, X., Shen, Q., et al. (2016). Bacillus amyloliquefaciens T-5 may prevent Ralstonia solanacearum infection through competitive exclusion. Biol. Fertility Soils 52, 341–351. doi: 10.1007/s00374-015-1079-z
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955. doi: 10.1073/pnas.0506758102
Toh, S.-M., Xiong, L., Arias, C. A., Villegas, M. V., Lolans, K., Quinn, J., et al. (2007). Acquisition of a natural resistance gene renders a clinical strain of methicillin-resistant Staphylococcus aureus resistant to the synthetic antibiotic linezolid. Mol. Microbiol. 64, 1506–1514. doi: 10.1111/j.1365-2958.2007.05744.x
Verma, A., Singh, V. K., and Gaur, S. (2016). Computational based functional analysis of Bacillus phytases. Comput. Biol. Chem. 60, 53–58. doi: 10.1016/j.compbiolchem.2015.11.001
Vernikos, G., Medini, D., Riley, D. R., and Tettelin, H. (2015). Ten years of pan-genome analyses. Curr. Opin. Microbiol. 23, 148–154.
Wang, H., Yang, L., Ping, Y., Bai, Y., Luo, H., Huang, H., et al. (2016). Engineering of a Bacillus amyloliquefaciens strain with high neutral protease producing capacity and optimization of its fermentation conditions. PLoS One 11:e0146373. doi: 10.1371/journal.pone.0146373
Wang, X., Bai, Y., Cai, Y., and Zheng, X. (2017). Biochemical characteristics of three feruloyl esterases with a broad substrate spectrum from Bacillus amyloliquefaciens H47. Process Biochem. 53, 109–115. doi: 10.1016/j.procbio.2016.12.012
Wu, B., Changjun, W., Dihong, X., Heng, Z., Huan, Y., Jinping, L., et al. (2016). Effects of Bacillus amyloliquefaciens ZM9 on bacterial wilt and rhizosphere microbial communities of tobacco. Appl. Soil Ecol. 103, 1–12. doi: 10.1016/j.apsoil.2016.03.002
Wu, L., Wu, H.-J., Qiao, J., Gao, X., and Borriss, R. (2015). Novel routes for improving biocontrol activity of Bacillus based bioinoculants. Front. Microbiol. 6:1395.
Yang, L., Wang, H., Lv, Y., Bai, Y., Luo, H., Shi, P., et al. (2016). Construction of a rapid feather-degrading bacterium by overexpression of a highly efficient alkaline keratinase in its parent strain Bacillus amyloliquefaciens K11. J. Agric. Food Chem. 64, 78–84. doi: 10.1021/acs.jafc.5b04747
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Zhang, J., Xue, Q., Gao, H., Lai, H., and Wang, P. (2016). Bacterial degradation of crude oil using solid formulations of bacillus strains isolated from oil-contaminated soil towards microbial enhanced oil recovery application. RSC Adv. 6, 5566–5574. doi: 10.1039/C5RA23772F
Zhang, N., Yang, D., Kendall, J. R. A., Borriss, R., Druzhinina, I. S., Kubicek, C. P., et al. (2016). Comparative genomic analysis of Bacillus amyloliquefaciens and Bacillus subtilis reveals evolutional traits for adaptation to plant-associated habitats. Front. Microbiol. 7:2039. doi: 10.3389/fmicb.2016.02039
Zhu, X., Liu, D., Singh, A. K., Drolia, R., Bai, X., Tenguria, S., et al. (2018). tunicamycin mediated inhibition of wall teichoic acid affects Staphylococcus aureus and listeria monocytogenes cell morphology, biofilm formation and virulence. Front. Microbiol. 9:1352.
Zühlke, M.-K., Schlüter, R., Henning, A.-K., Lipka, M., Mikolasch, A., Schumann, P., et al. (2016). A novel mechanism of conjugate formation of bisphenol A and its analogues by Bacillus amyloliquefaciens: Detoxification and reduction of estrogenicity of bisphenols. Int. Biodeterior. Biodegradation 109, 165–173. doi: 10.1016/j.ibiod.2016.01.019
Keywords: B. amyloliquefaciens, phylogenomics, genome evaluation, comparative genomics, functional traits, antimicrobial resistance and virulence genes
Citation: Liu H, Prajapati V, Prajapati S, Bais H and Lu J (2021) Comparative Genome Analysis of Bacillus amyloliquefaciens Focusing on Phylogenomics, Functional Traits, and Prevalence of Antimicrobial and Virulence Genes. Front. Genet. 12:724217. doi: 10.3389/fgene.2021.724217
Received: 12 June 2021; Accepted: 26 August 2021;
Published: 30 September 2021.
Edited by:
Saumya Patel, Gujarat University, IndiaReviewed by:
Archna Suman, Indian Agricultural Research Institute (ICAR), IndiaCopyright © 2021 Liu, Prajapati, Prajapati, Bais and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vimalkumar Prajapati, dmltYWxwcmFqYXBhdGlAbmF1Lmlu; orcid.org/0000-0003-4257-1728
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.