
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 02 December 2021
Sec. Livestock Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.723599
This article is part of the Research TopicPhenotypic Characterization, Genetics and Genomics of Livestock in Low Input SystemsView all 16 articles
 Abdessamad Ouhrouch1,2*
Abdessamad Ouhrouch1,2* Simon Boitard3
Simon Boitard3 Frédéric Boyer4
Frédéric Boyer4 Bertrand Servin5Anne Da Silva6François Pompanon4Abdelmajid Haddioui2
Bertrand Servin5Anne Da Silva6François Pompanon4Abdelmajid Haddioui2 Badr Benjelloun1*
Badr Benjelloun1*Sheep farming is a major source of meat in Morocco and plays a key role in the country’s agriculture. This study aims at characterizing the whole-genome diversity and demographic history of the main Moroccan sheep breeds, as well as to identify selection signatures within and between breeds. Whole genome data from 87 individuals representing the five predominant local breeds were used to estimate their level of neutral genetic diversity and to infer the variation of their effective population size over time. In addition, we used two methods to detect selection signatures: either for detecting selective sweeps within each breed separately or by detecting differentially selected regions by contrasting different breeds. We identified hundreds of genomic regions putatively under selection, which related to several biological terms involved in local adaptation or the expression of zootechnical performances such as Growth, UV protection, Cell maturation or Feeding behavior. The results of this study revealed selection signatures in genes that have an important role in traits of interest and increased our understanding of how genetic diversity is distributed in these local breeds. Thus, Moroccan local sheep breeds exhibit both a high genetic diversity and a large set of adaptive variations, and therefore, represent a valuable genetic resource for the conservation of sheep in the context of climate change.
Sheep were among the first domesticated animals about 11,000 years B.P (Vigne, 2011; Demirci et al., 2013). They are one of the main sources of meat and milk around the world (Ritchie and Roser, 2017). According to the FAO, the worldwide stocks of sheep reached 1,238 M heads in 2019 with proportions of 42.6% in Asia, 32.9% in Africa, 10.3% in Europe, 7.5% in Oceania and 6.7% in the Americas (FAOSTAT, 2020). The world production increase of sheep products was about 13.7% for meat and about 9.9% for milk between 2009 and 2019 (FAOSTAT, 2020). In the context of environmental changes, the improvement and conservation of this species are major challenges to sustainably meet the growing needs of human populations for meat and dairy products, both at the national and international levels (Bruford et al., 2015). Furthermore, in order to achieve the United Nations SDG#2 (Sustainable Development Goals), the target #2.5 is directly concerned by maintaining genetic diversity in domestic species, and two associated indicators are related to the proportion of endangered breeds (#2.5.1 & #2.5.2). Thus, local traditionally-managed sheep breeds represent an officially recognized valuable genetic resource for the conservation of this species on a global scale.
Technological advances during the last decade have made it possible to produce and process Whole Genome data for hundreds of individuals (Kulski, 2016). Similarly, bioinformatic improvement has incredibly advanced (Mangul et al., 2019). The DNA sequence of an individual is the most comprehensive collection of its genetic variation, and today’s sequencing technology that is much more increasingly efficient, faster and cheaper than ever (Kulski, 2016) allows access to this genome-wide variation for many individuals from the same population or breed. This allows a precise characterization of genetic resources, by characterizing properly their demographic dynamics and geographic structuration, as well as their adaptive diversity (Benjelloun et al., 2019).
In Morocco, sheep are marked by a high global genetic diversity indicating a high adaptive potential (Benjelloun, 2015; Benjelloun et al., 2021). The main local breeds currently farmed have significant ability to adapt to their breeding system and environment. However, the genetic diversity within each breed and the demographic and adaptive history that shaped this diversity are not yet very well known. Only a few hypotheses have been emitted to describe their origin (Boujenane, 1999).
Sheep farming in Morocco plays an important economic and sociological role. It is practiced all over the country where it is often one of the main sources of farmers’ income. Thus, sheep are bred under various environmental conditions and anthropogenic pressures. One of the main threats to these breeds is the unsupervised crossing practiced by farmers under increasing economic pressure as recently demonstrated by Belabdi et al. (2019). These practices, evaluated by the FAO to establish risk status of breeds, have led to genetic dilution (FAO, 2007), to which is added the risk of replacement of local breeds by cosmopolitan breeds (Taberlet et al., 2008).
The Moroccan sheep population is made up of about 1% of exogenous breeds and 99% of local breeds (Boujenane, 2006), among which the most important ones are Sardi, Dman, Timahdite, Beni Guil and Boujaad. Since 1980, five main local sheep breeds have been standardized and officially recognized by a large management program named the National Sheep Plan (MARA, 1980) which was based on assigning theses breeds to their exclusive specific habitat or area named “cradle of the breed”. Thus, Beni Guil is bred in the Eastern plateaus in large herds using pastoralism as main feeding sources. The Dman breed has been bred in the palm groves of the pre-Saharan regions of the South-Eastern Morocco for a long time and is mainly located in the oases. Dman is considered as the most isolated and phenotypically distinguished breed in Morocco. The Sardi breed belongs to the sheep population of the western plateaus in Morocco. It is considered as a large-body sheep and very appreciated in religious events. Timahdite is a rustic breed well adapted to mountainous areas with low-input systems. Finally, the Ouled Jellal breed, which is not considered indigenous in Morocco, was shown to be among the main breeds farmed in the Eastern region of the country together with Beni Guil since a long time (Belabdi et al., 2019), and is considered as a true breed of the steppe, well adapted to nomadism.
Many characteristics that condition the fitness of livestock are associated with the production performances. Thus, the identification of signatures of selection is a valuable approach to identify the genes and polymorphisms underlying the phenotypic variation of these traits that are subjected to both anthropogenic selection (Rochus et al., 2018) and natural selection related to e.g., climatic or ecological constraints. While previous studies using WGS have identified many genome-environment associations in Moroccan sheep (Benjelloun, 2015), the genomic bases of traits specific to local breeds are still undetermined.
In this context, this study aims at characterizing the main five Moroccan breeds by describing their genomic diversity, inferring their demographic history and identifying shared and breed-specific selective sweeps using whole genome data. The data obtained enabled us to distinguish genomic regions putatively involved in local adaptation from those more directly related to anthropogenic pressures and associated to breed-specific zootechnical performances.
We sampled 87 unrelated sheep representatives of the geographic distribution of five local Moroccan breeds (Sardi, Dman, Timahdite, Beni Guil and Ouled Jellal; Supplementary Table S1) across the Northern half of Morocco (North of latitude 28°) between January 2008 and March 2012 in accordance with ethical regulations of the European Union Directive 86/609/EEC, as described in Benjelloun et al. (2021) and Benjelloun (2015). Tissue samples were taken from the distal part of the ear and then placed in alcohol for 1 day, after which they were transferred to a silica gel tube pending the extraction of DNA.
Additionally, a worldwide breed panel consisting of 20 sheep representing 20 different worldwide breeds was provided by the International Sheep Genome Consortium. The panel represents sheep from Asia, Africa Australia, America and Europe (Supplementary Table S1). Similarly, 13 wild Asiatic mouflons (O. orientalis) were collected either from captive or recently hunted animals, and from frozen samples available at the Iranian Department of Environment. These worldwide sheep (n = 20) and Asiatic mouflons (n = 13) were previously used in Alberto et al. (2018) and their whole genome sequence data were included here for comparisons and for increasing power when identifying selection signatures in Moroccan sheep (see section 2.3 and section 2.4 in the Methods).
As described by Benjelloun et al. (2021), Alberto et al., 2018 and Benjelloun et al. (2019), Illumina reads were aligned to the sheep reference genome (OAR v3.1, GenBank assembly GCA_000317765.1 (Jiang et al., 2014), and variant discovery was performed using three different algorithms: Samtools Mpileup (Li et al., 2009), GATK UnifiedGenotyper (McKenna et al., 2010) and Freebayes (Garrison and Marth, 2012). Variants were called using a larger dataset than that used in this study (i.e., 160 sheep). Then two successive rounds of filtering variant sites were run. Filtering stage 1 merged together calls from the three algorithms, whilst filtering out the lowest-confidence calls. A variant site passed if it was called by at least two different calling algorithms with variant phred-scaled quality>30. An alternate allele at a site passed if it was called by any one of the calling algorithms, and the genotype count >0. Filtering stage 2 used Variant Quality Score Recalibration by GATK. First, a training set was generated of the highest-confidence variant sites where 1) the site was called by all three variant callers with variant phredscaled quality >100; 2) the site was biallelic, and 3) the minor allele count was at least three, counting only samples with genotype phredscaled quality >30. The training set was used to build a Gaussian model using the tool GATK VariantRecalibrator using the following variant annotations from UnifiedGenotyper: QD, HaplotypeScore, MQRankSum, ReadPosRankSum, FS, DP, Inbreeding Coefficient.
The Gaussian model was applied to the full data set, generating a VQSLOD (log odds ratio of being a true variant). Sites were filtered out if VQSLOD < cutoff value. The cutoff value was set for each population by the following: Minimum VQSLOD = {the median value of VQSLOD for training set variants}—3 * {the median absolute deviation VQSLOD of training set variants}. Measures of the transition/transversion ratio of SNPs suggest that this chosen cutoff criterion gives the best balance between selectivity and sensitivity.
Genotypes were improved and phased by Beagle 4 (Browning and Browning, 2013), and then filtered out where the genotype probability calculated by Beagle was <0.95. The resulting dataset was constituted of 47,622,950 variants.
We used vcftools (Danecek et al., 2011) to estimate the heterozygosity (Ho), inbreeding coefficient (F) using polymorphic diploid bi-allelic SNPs and nucleotide diversity (π) for all diploid SNPs without missingness. We also calculated the FST index (Weir and Cockerham, 1984) using the same program (VCFtools) for each variant and their average over the whole genome between each two of the five Moroccan breeds, and between the 87 Moroccan individuals and the 22 sheep representing 12 cosmopolitan breeds.
In order to determine the demographic history of the studied breeds, we used the approximate Bayesian computation (ABC) approach implemented in PopSizeABC (Boitard et al., 2016a) to estimate the effective population size (Ne) through time from 130 K years to the present. PopSizeABC determines how Ne changes through time, by estimating empirical summary statistics from our VCF files and matching them to the simulated summary statistics obtained from simulated genomic data. The simulations explore a large set of Ne possible values for each of 21 pre-defined time windows, together with several values of the per generation per bp recombination rate (r), while assuming a fixed and pre-defined per generation per site mutation rate (μ). In order to compare the demographic history Moroccan sheep with the main worldwide breeds/populations, we included, in the analysis, genomic data of the worldwide sheep and Asiatic mouflons. The latters represent the closest wild descents of the species from which the current domestic sheep (O. aries) diverged since their domestication. The worldwide breeds were subjected to the improvement of their production performances by intensive selection (Alberto et al., 2018).
We estimated the recombination rate by using a uniform prior interval from 8e−9 to 13e−9 with an approximation of r = 10.65e−9, and then empirical data for each population. The minor allele count threshold (mac) for the Allele Frequency Spectrum (AFS) and Identity By State (IBS) statistics computation was set for each population to about 5% × N (where N is the number of samples) and the minor allele count threshold for LD statistics computation (mac_ld) was set to about 20% × N as recommended by Boitard et al., (2016a). The number of simulated datasets was nb_rep = 10,000, the number of independent segments in each dataset was nb_seg = 50 and the size of each segment in bp was L = 2,000,000, which overall gives a total of 1 Terabp of simulated data.
To identify selective sweeps, we have defined an integrated framework based on two complementary approaches as proposed in Boitard et al., (2016b). The first one (freqHMM) detects selection occurring within a single population while the second (FLK & hapFLK) is for detecting selection events differentiating populations.
A- A genome scan using the freqHMM program (Boitard et al., 2012) allowed identifying independently the putative regions under selection within each of the five studied breeds. This method is based on contrasting the local and genome-wide distributions of allele frequencies using data from a single population by assuming each SNP to have a hidden state which can take three different values: 1) “3 = Selection”; attributed to SNPs that are located in a selective sweep, 2) “intermediate”, for SNPs not selected but located close to a selective sweep and 3) “1 = neutral”, for SNPs that are far from any selective sweep. It aims at identifying ancient selection signatures that arose from new variants (hard sweeps) as described by Boitard et al. (2012). Ancestral alleles at each marker were defined using the homozygotes of the reference goat genome (CHIR_1.0; Dong et al., 2013) for the corresponding loci, by assuming that these are the ancestral alleles that mutated in Ovis to produce SNPs. Those ancestral alleles were added to the input VCF files using an in-house shell script. Afterwards, five different datasets representing whole genome variants with a total of polymorphic 31,442,046 SNPs (for which ancestral alleles were identified) of each of the five breeds were extracted using vcftools (Danecek et al., 2011) and data related to each chromosome was put in a separate file for each breed. Then, we calculated the number of derived and ancestral alleles for each SNP using vcftools. Parameter K of freqHMM was calibrated for each breed by simulating data using ms (Hudson, 2002) under neutrality, accounting for the demographic history previously estimated for the breed by PopSizeABC, and running freqHMM on these neutral data, as previously described in Boitard et al., (2016b). The value of K obtained by this approach was then used to run the freqHMM analysis on the empirical data for each one of the five breeds.
B- The second approach to detect selective sweeps was a combination of the FLK (Bonhomme et al., 2010) and hapFLK (Fariello et al., 2013) methods implemented in the hapFLK (1.4 version) program (https://forge-dga.jouy.inra.fr/projects/hapflk/files). It was used to detect selection signatures by differentiating haplotypes among hierarchically structured populations, as described by Fariello et al. (2013). This has been done using all the genomic autosomes with no missingness allowed. FLK is a test for inbreeding coefficient heterogeneity that uses phylogenetically estimated relationships between populations. The allele frequencies are rescaled using a population kinship matrix which is estimated from the genomic data, measuring the amount of genetic drift expected under neutral evolution. HapFLK uses the differences in the frequencies of allele haplotypes between populations and the hierarchical structure of subpopulations.
The shared variants between the five Moroccan breeds and the 19 Ovis Orientalis (included as outgroup) were combined using the option “-T CombineVariants” of GATK (McKenna et al., 2010). The resulting dataset consisting of 31,721,507 polymorphic SNPs was used for FLK analysis to calculate Reynolds distances and the resulting Kinship Matrix. Then, this dataset was subdivided into several files each of which has a window of 10 Mbp with an overlap of 1 Mbp between each two successive windows. Subsequently, we launched FLK/hapFLK on all files one by one with a specified number of clusters set to K = 25. The analysis was launched with the option of keeping the Outgroup when computing FLK/hapFLK scores. We applied the approach of Storey and Tibshirani (2003), implemented in the q-value R package (Storey et al., 2015) to control the false discovery rate (FDR) based on FLK/hapFLK p-values.
Lists of genes that include or less than 5 kb away from the identified candidate SNPs (Downstream 5′-end and upstream 3′-end) were established and used for the Gene Ontology (GO) enrichment analyses. Similarly, outlier genomic regions were constituted of 50 kb segments surrounding outlier SNPs. GO enrichment analyses were performed using GOwinda (Kofler and Schlötterer, 2012) in order to explore the biological processes in which the identified genes under selection are involved. Bos Taurus was used as the reference species for that analysis. GOwinda effectively corrects for the gene length bias while identifying clearly over-represented GO categories, considering only SNPs being located in an exon are associated with the corresponding gene (using option: -gene-definition exon). A 5% FDR threshold was applied on GOwinda outputs. The identified GO terms in each population were clustered into homogenous groups using REVIGO (Supek et al., 2011).
The individual observed heterozygosity (Ho) in the five breeds was 0.28 on average, varying from 0.22 in Dman to 0.35 in Beni Guil (Table 1). The inbreeding coefficient varied thus from 0.0007 in Beni Guil to 0.095 in Dman which was the most inbred of the five Moroccan breeds.
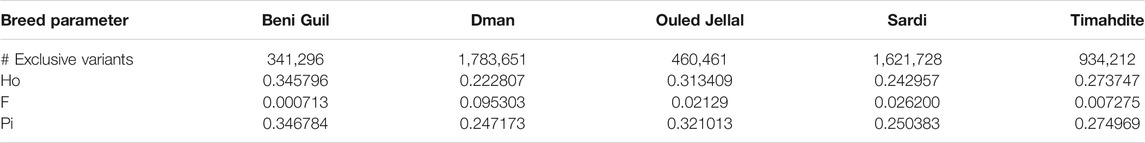
TABLE 1. Number of the specific variants and neutral genetic diversity parameters, for five Moroccan breeds.
The whole genome nucleotide diversity (π), calculated over the 87 Moroccan sheep was 0.29. The Dman and Sardi breeds showed close π values of 0.247 and 0.25, respectively. Timahdite, Ouled Jellal and Beni Guil showed slightly higher π values of 0.274, 0.32 and 0.346, respectively (Table 1).
The number of breed-specific SNPs strongly varied among breeds, from 341 k and 460 k in Beni Guil and Ouled Jellal, respectively, to 1.6 and 1.78 M in Sardi and Dman, respectively (Table 1).
Regarding genetic distances, the pairwise FST was low, from 0 between Beni Guil and both Dman and Ouled Jellal to 0.004 between Sardi and both Dman and Ouled Jellal (Table 2). Furthermore, the pairwise FST between these five breeds and the cosmopolitan sheep was 0.027.
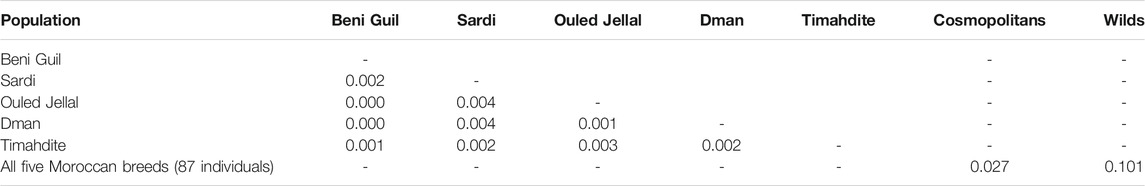
TABLE 2. Pairwise FST values between breeds.
According to FLK tree (Figure S1), Timahdite is closely grouped with Sardi and Dman, Beni Guil is the most divergent, with rather long branches, among the Moroccan breeds. As expected, wilds were distant from all domestic breeds.
The PopsizeABC analysis highlighted that before 10,000 years ago, the estimated effective population size (Ne) was similar in all populations (Figure 1). Then, the variations of Ne differed in domestic and wild animals. For domestic populations, there was a strong bottleneck probably matching the time of domestication, which was not observed for the wilds. For Moroccan sheep, the low Ne persisted until 1 K years before present, and then increased. All Moroccan breeds had a very similar trajectory, with the exception of Timahdite which showed a slightly higher Ne in the recent past. Conversely, in cosmopolitan sheep, the bottleneck was first less pronounced but during the last 1 K years, the Ne regularly decreased until about 300 at present. However, we do not exclude that using a pool of individuals representing several breeds may impact the accuracy of the estimates.
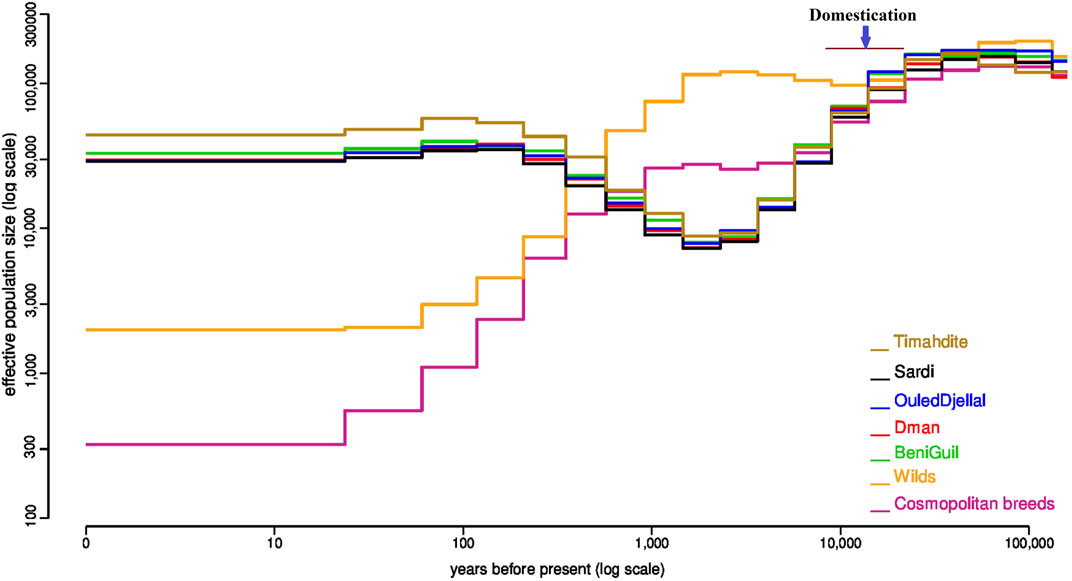
FIGURE 1. Variation of the effective population size in five Moroccan breeds in comparison with the wild Asiatic mouflon and a multi-breed group of cosmopolitan sheep. Estimates were obtained independently for each group by the popsizeABC algorithm.
The analysis of the whole genome variants of the 87 Moroccan sheep belonging to five breeds, detected selection signatures both within (FreqHMM, Boitard et al., 2012) and between (hapFLK, Fariello et al., 2013) breeds.
We identified 182,337 SNPs in 364 genomic regions (Supplementary Table S3) under selection. Comparing the studied breeds, we found that Dman displayed the higher number of regions under selection (203) followed by Ouled Jellal (109), Sardi (96), Beni Guil (95), and Timahdite in which we identified 59 genomic regions only (Table 3).
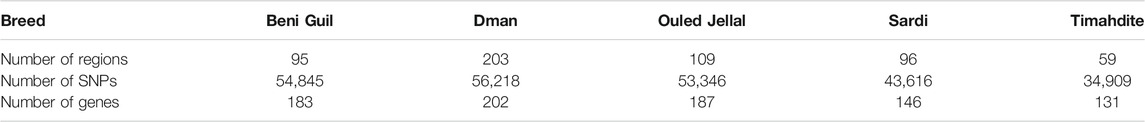
TABLE 3. Number of candidate regions and SNPs under selection, with the corresponding genes, within each Moroccan sheep breed (intra-breed selection, using freqHMM).
Regarding the shared selection signatures, we noticed that Dman share more SNPs/genes under selection with Sardi and Timahdite (respectively 14 K SNPs/70 genes and 9 K SNPs/60 genes; Table 4). A total of 219 SNPs were identified under selection in all five breeds, among which 203 were intergenic and 16 associated with three genes: HMGA2 (3 SNPs), RCOR1 (9 SNPs) and SBF2 (4 SNPs).
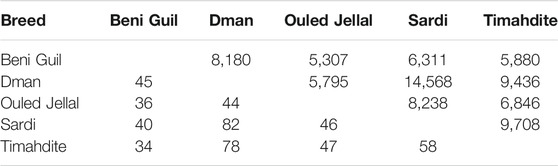
TABLE 4. Number of selected SNPs (above the diagonal) and genes (below the diagonal) in common between two Moroccan sheep breeds (intra-breed selection, using freqHMM).
An FDR and local FDR framework was applied to the hapFLK results in order to determine a reliable selection threshold, which was set to 0.1% FDR (q-values <0.001) in order to be sufficiently conservative. The whole process identified 8,887 SNPs in 27 different regions, of which 4,131 were associated with 20 genes and 4,756 were intergenic. Among the all outliers we identified 3,908 SNPs as top candidates with p-values lower than 10−12 (Figure 2). They were distributed across five regions associated with 9 genes (of which 7 genes have 31 missense variants). Among these regions, the greatest selective sweep was located on Chr10: 29,363,691-29,806,294. It was related to a high differentiation between Dman and the other breeds as shown in the cluster-plot (Figure 2B) where Dman had a different haplotype from that of the other breeds, and in the local tree (Figure 2C) where Dman displayed a stronger p-value in both SNP local tree (FLK scores) and haplotype local tree (hapFLK scores). This selective sweep was associated with the genes RXFP2 and ENSOARG00000011616. The other strongest selective sweeps were related to 1) the region located on Chr14: 13,329,709-14,250,423 differentiating Sardi and associated to 8 genes; 2) the region located on Chr19: 2,143,797-2,261,064 differentiating Sardi and Beni Guil from the other breeds and associated with intergenic SNPs; 3) the region located on Chr16: 34,551,942-34,646,856 differentiating Timahdite and associated with intergenic SNPs; 4) the region located on Chr2: 84,619,111-84,759,276 differentiating Timahdite and associated to the BNC2 gene and some intergenic SNPs (Figure 2A).
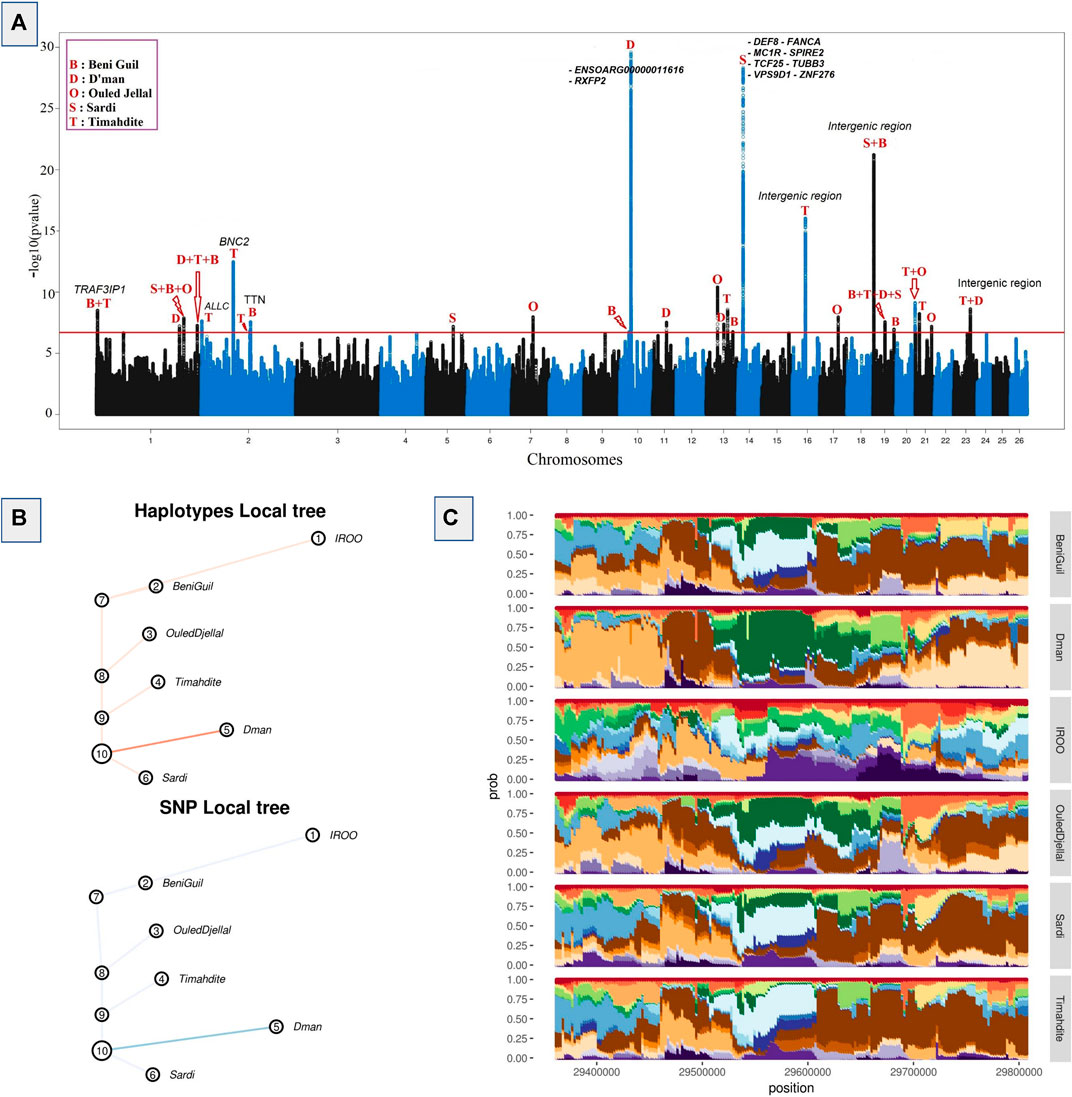
FIGURE 2. Selective sweeps detected by hapFLK and plots for an example candidate region. (A) Manhattan plot depicting hapFLK scores in five Moroccan sheep breeds along autosomal chromosomes. Each dot represents a SNP. The horizontal red line represents the 0.1% FDR threshold of significance. (B) Local tree for the example region on Chromosome 10 in Dman. (C) Haplotype cluster plot of the same example region on chromosome 10 in Dman (Cluster frequencies plot).
When combining both approaches, from a total of 31,721,507 analyzed SNPs, we identified as putatively under selection 46,799 variants associated with 155 genes in Sardi, 37,511 SNPs associated with 138 genes in Timahdite, 59,351 variants associated with 206 genes in Dman, 56,995 associated with 186 genes in the Beni Guil breed and 53,990 variants associated with 189 genes in Ouled Jellal. The Venn diagrams in Figure 3 illustrate the number of regions, SNPs and associated genes that are specific or common to the studied breeds. We identified 219 SNPs within 7 genomic regions under selection in all 5 breeds, which are associated with 3 genes: HMGA2, RCOR1 and SBF2.
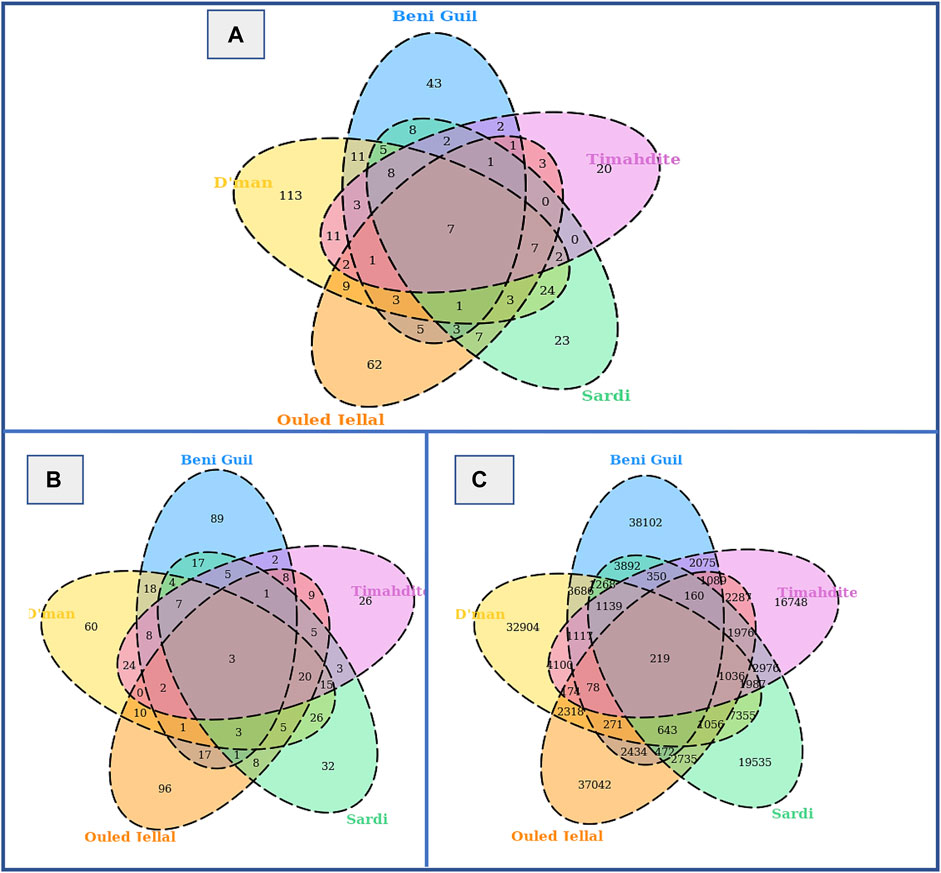
FIGURE 3. Selective sweeps, SNPs and genes identified by both freqHMM and hapFLK methods. (A) Venn diagram of candidate regions under selection. (B) Venn diagram of genes under selection. (C) Venn diagram of SNPs under selection.
The selection signatures detected by both methods (i.e., FreqHMM & HapFLK) allowed identifying a total of 7,735 GO categories enriched in genes under selection (Table 5), based on a threshold of FDR<5% (Supplementary Tables S4–S7). The lowest number of enriched GO categories per breed was for Timahdite, where 1,465 categories clustered in 223 homogenous groups of biological processes. The Sardi breed exhibited the highest number of enriched GO term, with 1,611 categories clustering in 229 groups of processes. In regard to these high numbers of enriched biological processes, we’ll limit our discussion to those which roles are the most straightforward given the related phenotypic traits and husbandry practices.
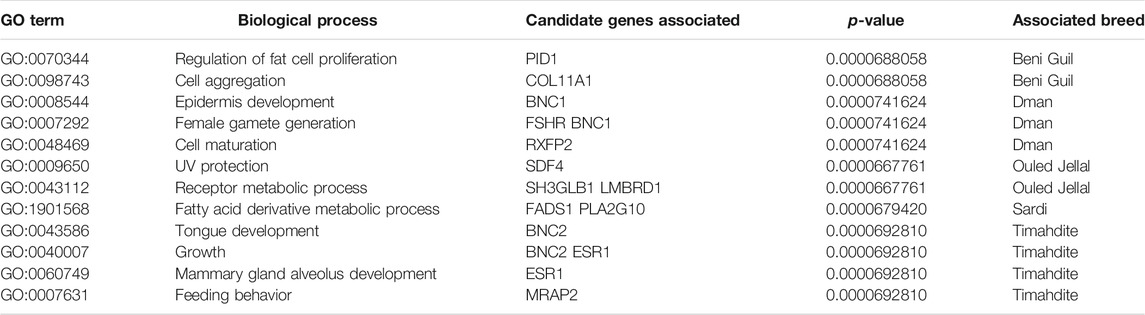
TABLE 5. Examples of biological processes enriched in candidate genes in the five sheep breeds.
The heterozygosity measured in Moroccan breeds was generally moderate (mean of 0.28) and globally lower than the values reported for Iranian sheep breeds (Eydivandi et al., 2021) and Welsh breeds (Beynon et al. (2015). Dman and Sardi breeds have even much lower heterozygosity values. Our results showed low inbreeding in Moroccan sheep, comparably to those reported by Eydivandi et al. (2021) except for Dman which was the most inbred (F = 0.1). FST values showed no clear differentiation between Moroccan breeds with an average of ∼0.002. These FST values are lower than those found in Welsh local sheep (Beynon et al., 2015) but still comparable to that of Russian local sheep (Deniskova et al., 2018), which were both estimated from WGS data.
Based on nucleotide diversity (π), we found that Dman was the most diversified of the Moroccan breeds. This could be explained by a lower intensity of selection, a higher founder population and wider breeding area for this breed. The slightly higher inbreeding in Dman would be explained by its sub-structuration in isolated Oases that leads to the mating of closely related individuals and by the longstanding use reproductive rams (Bouix and Kadiri, 1974; Darfaoui, 2000). However, these levels are still much lower than those measured by Beynon et al. (2015) on Welsh sheep breeds.
Furthermore, the past demographic dynamics (Figure 1), inferred with PopsizeABC and associating a pool of wild animals, was consistent with the occurrence of a domestication event 10,500 years ago in the Fertile Crescent (Vigne, 2011; Demirci et al., 2013). Indeed, earlier than 10.5 K years ago, effective sizes of all wild and domestic populations were similar, which is consistent with a common origin from the same ancestral species of Ovis Orientalis (Vigne, 2011). Around 10 K years B.P, the strong bottleneck linked to domestication is observed in all domestic breeds, Moroccan and cosmopolitan (Figure 1). The lasting decline in Moroccan breeds sizes between around 10 K and 2 K years would correspond to a long-term migration process consisting in colonizing gradually new constraining environments. Indeed, the colonization process lasted for millennia (Taberlet et al., 2008) and Morocco, which is located at the end of several migration routes, was marked by two main arrivals of domestic sheep; the first one occurred around 8,600 years B.P and would be associated with the first Berbers who settled in Morocco, while the second originated from Iberia around 7,100 years B.P (kandoussi et al., 2020).
Between 2,000- and 1,000-years B.P, all Moroccan breeds gradually increased in size which corroborates their common history and the absence of isolation obetween breeds at that time. The Timahdite breed shows a higher increase in effective population size for about five centuries in comparison with the other breeds. Inversely, the industrial breeds show a continuous decline during the last millennium which has been accentuated during the last century. Similarly, the effective size of the wild mouflon gradually declined for about a millennium, which is in line with the degradation of their habitat by humans and the intensification of hunting activity (Rezaei, et al., 2010). More recently, over the last 700 years, the Ne of Moroccan and cosmopolitan breeds evolves in different ways with a huge drop of the cosmopolitan populations, which is consistent with the evolution of breeding practices which can go as far as the intensive use of artificial insemination. Finally, if we except Timahdite which behaved differently for the last 500 years, the other Moroccan breeds remain very close, which suggests their recent foundation.
Our estimates of the current Ne (Supplementary Table S1) and diversity parameters (Table 1) would illustrate the uniqueness of Moroccan local sheep breeds. The current Ne values for domestics (highest for Timahdite with Ne ∼ 44 K) are very high when compared to what is reported in the literature for sheep (e.g., Maiwashe and Blackburn, 2004; Tapio et al., 2005; Beynon et al., 2015), while the estimates for mouflon populations (Ne ∼ 2 K) are comparable to those reported by Beynon et al. (2015) in Welsh domestic sheep. We should however consider that estimates for recent time using popsize ABC can sometimes reach 5 to 10 times those based on pedigree or molecular data. This has been observed in a cattle breed (Boitard et al., 2016b). These results confirm actual measures of diversity using metrics such as nucleotide diversity. In any case, these results would also show the strong adaptive potential of Moroccan sheep breeds and the opportunity of implementing efficient programs for their breeding while maintaining this richness. Indeed, a high effective population size would be associated to a high intra-breed genomic diversity that include both adaptive and standing genetic variation. Furthermore, the latter is known to be a main driver of new adaptive traits that can be needed/useful in the context of new environmental pressures (Savolainen et al., 2013). Inversely, the very low effective size (Ne = 317) estimated from a mix of cosmopolitan breeds, despite their presence in very large numbers worldwide, show the huge threat they represent if their use to replace local breeds continues (Taberlet et al., 2008).
The selection signatures identified would shed light on the biological processes underlying both adaptive and zootechnical traits selected in each breed. Most of the regions identified under selection were intergenic (64%). This would illustrate the important role of regulation in the realization of biological processes and the expression of traits as reported by ENCODE Project Consortium et al. (2007). However, this could sometimes be due to hitch-hiking mechanisms and also to some limitations in the functional annotation of genomes (Benjelloun et al., 2015).
From the literature, the three genes identified as under selection in all five studied breeds (i.e., HMGA2, RCOR1 and SBF2) are generally involved in cellular functioning. The expression of HMGA2 is strongly associated with body size and growth in mice, humans and dogs (Webster et al., 2015; Vignali and Marracci, 2020; Yang et al., 2010). Furthermore, the inactivation of HMGA2 in pigs resulted in a huge body-size reduction (Chung et al., 2018). The RCOR1 gene has a role in transcriptional regulation, and is involved in repressing neuronal gene expression in non-neuronal cells (Coulson, 2005). Mutations in the SBF2 gene were associated with autosomal recessive Charcot-Marie-Tooth Disease type 4B2 in humans (Senderek et al., 2003), and has been associated with growing traits in cattle (Jahuey-Martínez et al., 2016) and horse (Al Abri et al., 2018).
Besides, we identified many selection signatures (589 candidate regions) specific to one or a few breeds. Several GO Terms were enriched in genes putatively selected (Supplementary Tables S4–S8); they were associated with large categories such as: organ development, pigmentation pathways, proliferation and lipid metabolism. We discuss here the genes which role appears to be quite straightforward, which includes participation to functional processes related to morphology, pigmentation or skin coloration, zootechnical performance and prolificacy.
The MC1R gene, which is candidate in the Sardi breed, has been associated with a large panel of skin or coat colors (Sturm and Duffy, 2012). It may be involved in the particular coloration pattern specific to this breed and preferred by consumers (Supplementary Figure S1C) in relation to the religious ceremony of Aid Al Adha. This breed is characterized by a white head devoid of wool with black spots around the eyes, muzzle, paws (feet) and at the tips of the ears (Boujenane 1999, http://www.anoc.ma/les-races/races-ovines/sardi/, January 2021). Another candidate specific of Sardi is SLC9A3. The deficiency of this gene causes severe obstructive azoospermia and infertility in male mice (Wang et al., 2017).
The BNC2 and EDN3 genes, identified in Timahdite, have been reported as potentially associated with skin pigmentation (Hider et al., 2013; Fariello et al., 2014), and may be involved in the specific skin coloration of this breed (Supplementary Figure S1D): a brown head, without spots neither black nor yellow, always very clear, sometimes, and extending behind the ears and into the trough area (Boujenane, 1999).
We also identified ESR1 as a candidate, which is a major mediator of estrogen action and is strongly linked to bone mass and osteoporosis in mice (Nakamura et al., 2007; Börjesson et al., 2010). Some alleles were significantly associated with adult human height (Wood et al., 2014). This gene would play a role in growth, as well as another candidate, MRAP2, which modulates melanocortin receptor signaling and have been associated with severe obesity in human (Asai et al., 2013; Schonnop et al., 2016).
RXFP2 has been associated with the horned/polled phenotype in many sheep breeds (Johnston et al., 2011; Dominik et al., 2012; Kijas et al., 2012; Wang et al., 2014). This gene was identified under selection only in Dman which is the only Moroccan breed (Supplementary Figure S1E) with both polled males and females (Boujenane 2006; Boujenane et al., 2013). Also, the seasonal expression of this gene was correlated with the differentiation of Leydig cells in the testis (Hombach-Klonisch et al., 2004; Serranito et al., 2021), which would suggest a possible involvement in fertility. Similarly, BNC1 and FSHR were identified, while Dman is known for its exceptional prolificacy in comparison with the other Moroccan breeds. It has an ovulation rate of 2.8 versus a maximum of 1.3 in the other breeds, with the ability to breed all year long with 190–350 days of interval between two lambings (Boujenane, 2006) depending on husbandry practices. It is also characterized by its early puberty (210 days in average, Boujenane 2006). The FSHR gene would be related to fertility, as the protein it encodes for is located in the testis and granulosa cells of the ovaries (Kwok et al., 2005), and is involved in follicle maturation and proliferation of granulosa cells (Sudo et al., 2002). Also, mutations in this gene were associated with the Polycystic Ovary Syndrome in humans which is characterized by obesity and anovulatory infertility (Gu et al., 2010; Laven, 2019). Besides, BNC1 is a candidate which encodes for a zinc finger protein present, among other places, in the basal cell layer of the epidermis and in hair follicles and which is also expressed in the germ cells of testis and ovary (Luchi and Green, 1999). This protein is thought to play a regulatory role in keratinocyte proliferation and has been shown to be involved in premature ovarian failure and testicular premature aging (Zhang et al., 2018; Li et al., 2020). It is a crucial transcription factor for spermatogenesis and male fertility (Li et al., 2020). Thus, we could hypothesize thus that these last two genes play a role in the reproductive performance of the Dman breed.
The TTN gene was identified under selection (with 5 missense variants) exclusively in the Beni Guil breed. It has been associated with meat and carcass traits in pigs (Braglia et al., 2013), meat colour, pH and conductivity in loin 24 h postmortem (Wimmers et al., 2007). Similarly, PID1 is identified as a candidate. This gene modulates insulin signaling and mitochondrial function in adipocytes and muscle cells, and was reported as a candidate gene for fat deposition, in humans, based on its high expression in adipose tissue of obese subjects in comparison with normal subjects (Wang et al., 2006). These two genes may be responsible for meat quality and the important fat percentage known of Beni Guil. Indeed, this breed is one of the best meat breeds in Morocco. Its carcass scores a high quality and a high fatness state with a white and firm cover fat, what makes it well appreciated by professionals and consumers (Belhaj et al., 2020). Since 2011, it has been certified by the PGI (Protected Geographical Indication) label (Belhaj et al., 2018), which represents the excellence of European agricultural food production.
The SDF4 (also called CAB45) found in Ouled Jellal was involved in protecting against UV-induced damages (Zhu et al., 2008) and was revealed as a modulator of cell proliferation and tumor growth (Chen et al., 2016). Mutations in this gene may have occurred, as an adaptation, in response to the effect of sunlight (i.e., exposure to Ultraviolets) on the entirely white skin color of the Ouled Jellal breed (Chellig, 1992). in fact, fair/clear types of skin color are found, in humans, to be significantly more sensitive to UV rays than darker skin types (Halder and Bridgeman-Shah 1995; Hemminki et al., 2002).
Besides specific-breed candidates, the Microphthalmia-associated transcription factor (MITF) is a selected gene in both Beni Guil, Dman, Sardi and Timahdite. It is a basic transcription factor which regulates the differentiation and development of melanocytes and pigment cell-specific transcription of the melanogenesis enzyme genes (Saravanaperumal et al., 2014). This gene was also associated with eye and coat spotting color in some dog breeds (Rothschild et al., 2006; Stritzel et al., 2009). We could hypothesize that this gene may interact with other genes to produce different coat color patterns.
We characterized the neutral diversity, demographic history and selection signatures using whole genome variants in the five main sheep breeds from Morocco. Globally, these five breeds are not very genetically differentiated, but they show a high number of specific variants and very high effective population sizes unlike cosmopolitan breeds. This illustrates that Moroccan indigenous breeds are highly diversified and have thus the potential to develop key adaptive characteristics to face upcoming climate changes. This makes them valuable resources for conservation and future preservation of sheep even at the worldwide scale.
Investigations on selection signatures provided valuable insights about genes and biological processes targeted by selection, which are essentially involved in pigmentation, zootechnical performance, adaptation and reproduction traits. Many genomic regions highlighted here need further investigations to decipher their specific roles in the expression of phenotypes specific to the studied breeds. However, the outlier genomic variants identified here represent valuable candidates and should be conserved in priority when conceiving the upcoming genomic breeding programs. Those programs have to consider improving production as well as adaptation traits while maintaining the diversity of these local sheep breeds.
Publicly available datasets were analyzed in this study. This data can be found here: ftp://ftp.ebi.ac.uk/pub/databases/nextgen/ovis/variants/genus_snps/.
The animal study was reviewed and approved by Data analyzed here have been produced previously within the EU FP7 NextGen project in respect of all ethical requirements.
BB, FP, AH, BS, and ADS designed and supervised the study. AO analyzed the data in the light of discussions with SB, BB and FB. AO together with BB wrote the manuscript. FP, SB, and ADS revised the manuscript. All authors contributed to discussions about the study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.723599/full#supplementary-material
Al Abri, M. A., Posbergh, C., Palermo, K., Sutter, N. B., Eberth, J., Hoffman, G. E., et al. (2018). Genome-Wide Scans Reveal a Quantitative Trait Locus for Withers Height in Horses Near the ANKRD1 Gene. J. Equine Vet. Sci. 60, 67–73.e1. doi:10.1016/j.jevs.2017.05.008
Alberto, F. J., Boyer, F., Orozco-terWengel, P., Streeter, I., Servin, B., de Villemereuil, P., et al. (2018). Convergent Genomic Signatures of Domestication in Sheep and Goats. Nat. Commun. 9, 813. doi:10.1038/s41467-018-03206-y
Asai, M., Ramachandrappa, S., Joachim, M., Shen, Y., Zhang, R., Nuthalapati, N., et al. (2013). Loss of Function of the Melanocortin 2 Receptor Accessory Protein 2 Is Associated with Mammalian Obesity. Science 341 (6143), 275–278. doi:10.1126/science.1233000
Association Nationale des Ovins et Caprins (2021). Sardi. Availale at: http://www.anoc.ma/les-races/races-ovines/sardi/ (Accessed January 2021).
Baek, B., Park, J., and Baek, K. (2010). Genetic Variations of Follicle Stimulating Hormone Receptor Are Associated with Polycystic Ovary Syndrome. Int. J. Mol. Med. 26, 107–112. doi:10.3892/ijmm_00000441
Belabdi, I., Ouhrouch, A., Lafri, M., Gaouar, S. B. S., Ciani, E., Benali, A. R., et al. (2019). Genetic Homogenization of Indigenous Sheep Breeds in Northwest Africa. Sci. Rep. 9, 7920. doi:10.1038/s41598-019-44137-y
Belhaj, K., Mansouri, F., Ben Moumen, A., Fauconnier, M.-L., Boukharta, M., Caid, H. S., et al. (2018). Physicochemical and Nutritional Characteristics of Béni Guil Lamb Meat Raised in Eastern Morocco. Mnm 11, 175–185. doi:10.3233/MNM-17195
Belhaj, K., Mansouri, F., Tikent, A., Ouchatbi, A., Boukharta, M., Hana Serghini, C., et al. (2020). Quality Characteristics of the Carcass of Beni-Guil Sheep, a Protected Geographical Indication Certified Product of Eastern Morocco: Preliminary Study. Rev. Elev. Med. Vet. Pays Trop. 73 (1), 21–26. doi:10.19182/remvt.31843
Benjelloun, B., Alberto, F. J., Streeter, I., Boyer, F. d. r., Coissac, E., Stucki, S., et al. (2015). Characterizing Neutral Genomic Diversity and Selection Signatures in Indigenous Populations of Moroccan Goats (Capra hircus) Using WGS Data. Front. Genet. 6, 107. doi:10.3389/fgene.2015.00107
Benjelloun, B., Boyer, F., Streeter, I., Zamani, W., Engelen, S., Alberti, A., et al. (2019). An Evaluation of Sequencing Coverage and Genotyping Strategies to Assess Neutral and Adaptive Diversity. Mol. Ecol. Resour. 19, 1497–1515.13070. doi:10.1111/1755-0998.13070
Benjelloun, B., Leempoel, K., Boyer, F., Stucki, S., Streeter, I., Orozco ter Wengel, P., et al. (2021). Multiple Adaptive Solutions to Face Climatic Constraints: Novel Insights in the Debate Over the Role of Convergence in Local Adaptation. bioRxiv [Preprint]. doi:10.1101/2021.11.18.469099
Benjelloun, B. (2015). Diversité des génomes et adaptation locale des petits ruminants d’un pays méditerranéen: le Maroc. Thèse de doctorat. Biodiversité, Ecologie, Environnement. Grenoble, France: Université Grenoble Alpes.
Beynon, S. E., Slavov, G. T., Farré, M., Sunduimijid, B., Waddams, K., Davies, B., et al. (2015). Population Structure and History of the Welsh Sheep Breeds Determined by Whole Genome Genotyping. BMC Genet. 16, 65. doi:10.1186/s12863-015-0216-x
ENCODE Project Consortium Birney, E., Birney, E., Stamatoyannopoulos, J. A., Dutta, A., Guigó, R., Gingeras, T. R., et al. (2007). Identification and Analysis of Functional Elements in 1% of the Human Genome by the ENCODE Pilot Project. Nature 447 (7146), 799–816. doi:10.1038/nature05874
Boitard, S., Boussaha, M., Capitan, A., Rocha, D., and Servin, B. (2016b). Uncovering Adaptation from Sequence Data: Lessons from Genome Resequencing of Four Cattle Breeds. Genetics 203 (1), 433–450. doi:10.1534/genetics.115.181594
Boitard, S., Rodríguez, W., Jay, F., Mona, S., and Austerlitz, F. (2016a). Inferring Population Size History from Large Samples of Genome-wide Molecular Data - an Approximate Bayesian Computation Approach. Plos Genet. 12 (3), e1005877. doi:10.1371/journal.pgen.1005877
Boitard, S., Schlotterer, C., Nolte, V., Pandey, R. V., and Futschik, A. (2012). Detecting Selective Sweeps from Pooled Next-Generation Sequencing Samples. Mol. Biol. Evol. 29, 2177–2186. doi:10.1093/molbev/mss090
Bonhomme, M., Chevalet, C., Servin, B., Boitard, S., Abdallah, J., Blott, S., et al. (2010). Detecting Selection in Population Trees: The Lewontin and Krakauer Test Extended. Genetics 186 (1), 241–262. doi:10.1534/genetics.110.117275
Börjesson, A. E., Lagerquist, M. K., Liu, C., Shao, R., Windahl, S. H., Karlsson, C., et al. (2010). The Role of Estrogen Receptor α in Growth Plate Cartilage for Longitudinal Bone Growth. J. Bone Miner Res. 25 (12), 2690–2700. doi:10.1002/jbmr.156
Bouix, J., and Kadiri, M. (1975). Un des éléments majeurs de la mise en valeur des palmeraies: la race ovine DMan (One of the important factors in development of oases : the DMan breed of sheep). Options Mdditerr 26, 87–93.
Boujenane, I., Chikhi, A., Lakcher, O., and Ibnelbachyr, M. (2013). Genetic and Environmental Factors Affecting Perinatal and Preweaning Survival of D'man Lambs. Trop. Anim. Health Prod. 45, 1391–1397. doi:10.1007/s11250-013-0376-5
Boujenane, I. (1999). Les ressources génétiques ovines au Maroc. First Edition. Publisher: Actes Editions, Rabat, Morocco. 9981-801-41-0.
Boujenane, I. (2006). Reproduction and Production Performance of Moroccan Sheep Breeds. CAB Rev. 1, 014. doi:10.1079/PAVSNNR20061014
Braglia, S., Davoli, R., Zappavigna, A., Zambonelli, P., Buttazzoni, L., Gallo, M., et al. (2013). SNPs of MYPN and TTN Genes Are Associated to Meat and Carcass Traits in Italian Large White and Italian Duroc Pigs. Mol. Biol. Rep. 40, 6927–6933. doi:10.1007/s11033-013-2812-z
Bruford, M. W., Ginja, C., Hoffmann, I., Joost, S., Orozco-terWengel, P., Alberto, F. J., et al. (2015). Prospects and Challenges for the Conservation of Farm Animal Genomic Resources, 2015-2025. Front. Genet. 6, 314. doi:10.3389/fgene.2015.00314
Chellig, R. (1992). Les Races Ovines Algériennes. Ben-AknounAlger: Office des Publications Universitaires.
Chen, L., Xu, S., Xu, Y., Lu, W., Liu, L., Yue, D., et al. (2016). Cab45S Promotes Cell Proliferation through SERCA2b Inhibition and Ca2+ Signaling. Oncogene 35, 35–46. doi:10.1038/onc.2015.56
Chung, J., Zhang, X., Collins, B., Sper, R. B., Gleason, K., Simpson, S., et al. (2018). High Mobility Group A2 (HMGA2) Deficiency in Pigs Leads to Dwarfism, Abnormal Fetal Resource Allocation, and Cryptorchidism. PNAS 115 (21), 5420–5425. doi:10.1073/pnas.1721630115
Coulson, J. M. (2005). Transcriptional Regulation: Cancer, Neurons and the REST. Curr. Biol. 15, R665–R668. doi:10.1016/j.cub.2005.08.032
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The Variant Call Format and VCFtools. Bioinformatics 27, 2156–2158. doi:10.1093/bioinformatics/btr330
Darfaoui, E. M. (2000). D’man Sheep Breeding Programme in Morocco. ICAR Technical Series 3. Rome, Italy: International Committee for Animal Recording, 319–330.
Demirci, S., Koban Baştanlar, E., Dağtaş, N. D., Pişkin, E., Engin, A., Özer, F., et al. (2013). Mitochondrial DNA Diversity of Modern, Ancient and Wild Sheep (Ovis Gmelinii Anatolica) from Turkey: New Insights on the Evolutionary History of Sheep. PLoS ONE 8 (12), e81952. doi:10.1371/journal.pone.0081952
Deniskova, T. E., Dotsev, A. V., Selionova, M. I., Kunz, E., Medugorac, I., Reyer, H., et al. (2018). Population Structure and Genetic Diversity of 25 Russian Sheep Breeds Based on Whole-Genome Genotyping. Genet. Sel. Evol. 50, 29. doi:10.1186/s12711-018-0399-5
Dominik, S., Henshall, J. M., and Hayes, B. J. (2012). A Single Nucleotide Polymorphism on Chromosome 10 Is Highly Predictive for the Polled Phenotype in Australian Merino Sheep. Anim. Genet. 43 (4), 468–470. doi:10.1111/j.1365-2052.2011.02271.x
Dong, Y., Xie, M., Jiang, Y., Xiao, N., Du, X., Zhang, W., et al. (2013). Sequencing and Automated Whole-Genome Optical Mapping of the Genome of a Domestic Goat (Capra hircus). Nat. Biotechnol. 31 (2), 135–141. doi:10.1038/nbt.2478
Eydivandi, S., Roudbar, M. A., Karimi, M. O., and Sahana, G. (2021). Genomic Scans for Selective Sweeps through Haplotype Homozygosity and Allelic Fixation in 14 Indigenous Sheep Breeds from Middle East and South Asia. Sci. Rep. 11, 2834. doi:10.1038/s41598-021-82625-2
Fariello, M.-I., Servin, B., Tosser-Klopp, G., Rupp, R., Moreno, C., Cristobal, M. S., et al. (2014). Selection Signatures in Worldwide Sheep Populations. PLOS ONE 9 (8), e103813. doi:10.1371/journal.pone.0103813
Fariello, M. I., Boitard, S., Naya, H., SanCristobal, M., and Servin, B. (2013). Detecting Signatures of Selection through Haplotype Differentiation Among Hierarchically Structured Populations. Genetics 193 (3), 929–941. doi:10.1534/genetics.112.147231
Food and Agriculture Organization of the United Nations (2020). Compare Data. Available at: http://www.fao.org/faostat/en/#compare/(Accessed in December, 2020).
Food and agriculture organization of the United Nations (2007). Commission on Genetic Resources for Food and Agriculture. Rome.The State of the World’s Animal Genetic Resources for Food and Agriculture – in Brief
Garrison, E., and Marth, G. (2012). Haplotype-based Variant Detection from Short-Read Sequencing. arXiv:1207.3907.
Halder, R. M., and Bridgeman-Shah, S. (1995). Skin Cancer in African Americans. Cancer 75, 667–673. doi:10.1002/1097-0142(19950115)75:2+<667:aid-cncr2820751409>3.0.co;2-i
Ritchie, Hannah., and Roser, Max. (2017). "Meat and Dairy Production". Published online at OurWorldInData.org
Hemminki, K., Xu, G., Kause, L., Koulu, L. M., Zhao, C., and Jansen, C. T. (2002). Demonstration of UV-Dimers in Human Skin DNA In Situ 3 Weeks after Exposure. Carcinogenesis 23 (4), 605–609. doi:10.1093/carcin/23.4.605
Hider, J. L., Gittelman, R. M., Shah, T., Edwards, M., Rosenbloom, A., Akey, J. M., et al. (2013). Exploring Signatures of Positive Selection in Pigmentation Candidate Genes in Populations of East Asian Ancestry. BMC Evol. Biol. 13, 150. doi:10.1186/1471-2148-13-150
Hombach-Klonisch, S., Schön, J., Kehlen, A., Blottner, S., and Klonisch, T. (2004). Seasonal Expression of INSL3 and Lgr8/Insl3 Receptor Transcripts Indicates Variable Differentiation of Leydig Cells in the Roe Deer Testis. Biol. Reprod. 71 (4), 1079–1087. doi:10.1095/biolreprod.103.024752
Hudson, R. R. (2002). Generating Samples under a Wright-Fisher Neutral Model of Genetic Variation, Bioinformatics, vol. 18 (pg. 337–338). doi:10.1093/bioinformatics/18.2.337
Iuchi, S., and Green, H. (1999). Basonuclin, a Zinc finger Protein of Keratinocytes and Reproductive Germ Cells, Binds to the rRNA Gene Promoter. Proc. Natl. Acad. Sci. 96 (17), 9628–9632. doi:10.1073/pnas.96.17.9628
Jahuey-Martínez, F. J., Parra-Bracamonte, G. M., Sifuentes-Rincón, A. M., Martínez-González, J. C., Gondro, C., García-Pérez, C. A., et al. (2016). Genomewide Association Analysis of Growth Traits in Charolais Beef Cattle1. J. Anim. Sci. 94 (11), 4570–4582. doi:10.2527/jas.2016-0359
Jiang, Y., Xie, M., Chen, W., Talbot, R., Maddox, J. F., Faraut, T., et al. (2014). The Sheep Genome Illuminates Biology of the Rumen and Lipid Metabolism. Science 344, 1168–1173. doi:10.1126/science.1252806
Johnston, S. E., McEwan, J. C., Pickering, N. K., Kijas, J. W., Beraldi, D., Pilkington, J. G., et al. (20112011). Genome-wide Association Mapping Identifies the Genetic Basis of Discrete and Quantitative Variation in Sexual Weaponry in a Wild Sheep Population. Mol. Ecol. 20, 2555–2566. doi:10.1111/j.1365-294x.2011.05076.x
Kandoussi, A., Boujenane, I., Auger, C., Serranito, B., Germot, A., Piro, M., et al. (2020). The Origin of Sheep Settlement in Western Mediterranean. Sci. Rep. 10, 10225. doi:10.1038/s41598-020-67246-5
Kijas, J. W., Lenstra, J. A., Hayes, B., Boitard, S., Porto Neto, L. R., San Cristobal, M., et al. (2012). Genome-Wide Analysis of the World's Sheep Breeds Reveals High Levels of Historic Mixture and Strong Recent Selection. Plos Biol. 10 (2), e1001258. doi:10.1371/journal.pbio.1001258
Kofler, R., and Schlotterer, C. (2012). Gowinda: Unbiased Analysis of Gene Set Enrichment for Genome-wide Association Studies. Bioinformatics 28, 2084–2085. doi:10.1093/bioinformatics/bts315
Kulski, J. K. (2016). Next-Generation Sequencing - an Overview of the History, Tools, and "Omic" Applications. Next Generation Sequencing - Advances, Applications and Challenges. doi:10.5772/61964
Kwok, H.-F., So, W.-K., Wang, Y., and Ge, W. (2005). Zebrafish Gonadotropins and Their Receptors: I. Cloning and Characterization of Zebrafish Follicle-Stimulating Hormone and Luteinizing Hormone Receptors- Evidence for Their Distinct Functions in Follicle Development1. Biol. Reprod. 72 (6), 1370–1381. doi:10.1095/biolreprod.104.038190
Laven, J. S. E. (2019). Follicle Stimulating Hormone Receptor (FSHR) Polymorphisms and Polycystic Ovary Syndrome (PCOS). Front. Endocrinol. 10, 23. doi:10.3389/fendo.2019.00023
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map Format and SAMtools. Bioinformatics 25, 2078–2079. doi:10.1093/bioinformatics/btp352
Li, J.-Y., Liu, Y.-F., Xu, H.-Y., Zhang, J.-Y., Lv, P.-P., Liu, M.-E., et al. (2020). Basonuclin 1 Deficiency Causes Testicular Premature Aging: BNC1 Cooperates with TAF7L to Regulate Spermatogenesis. J. Mol. Cel Biol 12 (1), 71–83. doi:10.1093/jmcb/mjz035
Li, J.-Y., Ying, Y.-Y., Qian, Y.-L., Chen, J.-P., Huang, Y., Liu, J., et al. (2021). BNC1 Promotes Spermatogenesis by Regulating Transcription of Ybx2 and Papolb via Direct Binding to Their Promotor Elements. Reprod. Sci. 28 (3), 785–793. doi:10.1007/s43032-020-00342-z
Maiwashe, A. N., and Blackburn, H. D. (2004). Genetic Diversity in and Conservation Strategy Considerations for Navajo Churro Sheep1. J. Anim. Sci. 82, 2900–2905. doi:10.2527/2004.82102900x
Mangul, S., Martin, L. S., Eskin, E., and Blekhman, R. (2019). Improving the Usability and Archival Stability of Bioinformatics Software. Genome Biol. 20, 47. doi:10.1186/s13059-019-1649-8
MARA (Ministère de l’Agriculture et de la Réforme Agraire) (1980). Le Plan Moutonnier. Rabat: Direction de l’Élevage.
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: A MapReduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res. 20, 1297–1303. doi:10.1101/gr.107524.110
Nakamura, T., Imai, Y., Matsumoto, T., Sato, S., Takeuchi, K., Igarashi, K., et al. (2007). Estrogen Prevents Bone Loss via Estrogen Receptor α and Induction of Fas Ligand in Osteoclasts. Cell 130, 811–823. doi:10.1016/j.cell.2007.07.025
Rezaei, H. R., Naderi, S., Chintauan-Marquier, I. C., Taberlet, P., Virk, A. T., Naghash, H. R., et al. (2010). Evolution and Taxonomy of the Wild Species of the Genus Ovis (Mammalia, Artiodactyla, Bovidae). Mol. Phylogenet. Evol. 54 (2), 315–326. doi:10.1016/j.ympev.2009.10.037
Rochus, C. M., Tortereau, F., Plisson-Petit, F., Restoux, G., Moreno-Romieux, C., Tosser-Klopp, G., et al. (2018). Revealing the Selection History of Adaptive Loci Using Genome-wide Scans for Selection: An Example from Domestic Sheep. BMC Genomics 19, 71. doi:10.1186/s12864-018-4447-x
Rothschild, M. F., Van Cleave, P. S., Glenn, K. L., Carlstrom, L. P., and Ellinwood, N. M. (2006). Association of MITF with white Spotting in Beagle Crosses and Newfoundland Dogs. Anim. Genet. 37, 606–607. doi:10.1111/j.1365-2052.2006.01534.x
Saravanaperumal, S. A., Pediconi, D., Renieri, C., and La Terza, A. (2014). Alternative Splicing of the Sheep MITF Gene: Novel Transcripts Detectable in Skin. Gene 552 (1), 165–175. 15. doi:10.1016/j.gene.2014.09.031
Savolainen, O., Lascoux, M., and Merilä, J. (2013). Ecological Genomics of Local Adaptation. Nat. Rev. Genet. 14, 807–820. doi:10.1038/nrg3522
Schonnop, L., Kleinau, G., Herrfurth, N., Volckmar, A.-L., Cetindag, C., Müller, A., et al. (2016). Decreased Melanocortin-4 Receptor Function Conferred by an Infrequent Variant at the Human Melanocortin Receptor Accessory Protein 2 Gene. Obesity 24 (9), 1976–1982. doi:10.1002/oby.21576
Senderek, J., Bergmann, C., Weber, S., Ketelsen, U. P., Schorle, H., Rudnik-Schöneborn, S., et al. (2003). Mutation of the SBF2 Gene, Encoding a Novel Member of the Myotubularin Family, in Charcot-Marie-Tooth Neuropathy Type 4B2/11p15. Hum. Mol. Genet. 12 (Issue 3,1), 349–356. doi:10.1093/hmg/ddg030
Serranito, B., Cavalazzi, M., Vidal, P., Taurisson-Mouret, D., Ciani, E., Bal, M., et al. (2021). Local Adaptations of Mediterranean Sheep and Goats through an Integrative Approach. BioRxiv - the Preprint Server for Biology [Preprint]. Available at: https://www.biorxiv.org/content/10.1101/2021.01.22.427461v1.full.pdf. doi:10.1038/s41598-021-00682-z
Storey, J. D., Bass, A. J., Dabney, A., and Robinson, D. (2015). Qvalue: Q-Value Estimation for False Discovery Rate Control. R package version 2.2.2. Available at: http://github.com/jdstorey/qvalue.
Storey, J. D., and Tibshirani, R. (2003). Statistical Significance for Genomewide Studies. Proc. Natl. Acad. Sci. 100, 9440–9445. doi:10.1073/pnas.1530509100
Stritzel, S., Wöhlke, A., and Distl, O. (2009). A Role of Themicrophthalmia-Associated Transcription Factorin Congenital Sensorineural Deafness and Eye Pigmentation in Dalmatian Dogs. J. Anim. Breed. Genet. 126, 59–62. doi:10.1111/j.1439-0388.2008.00761.x
Sturm, R. A., and Duffy, D. L. (2012). Human Pigmentation Genes under Environmental Selection. Genome Biol. 13 (139), 248. doi:10.1186/gb-2012-13-9-248
Sudo, S., Kudo, M., Wada, S., Sato, O., Hsueh, A. J., and Fujimoto, S. (2002). Genetic and Functional Analyses of Polymorphisms in the Human FSH Receptor Gene. Mol. Hum. Reprod. 8, 893–899. doi:10.1093/molehr/8.10.893
Supek, F., Bošnjak, M., Škunca, N., and Šmuc, T. (2011). REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 6, e21800. doi:10.1371/journal.pone.0021800
Taberlet, P., Valentini, A., Rezaei, H. R., Naderi, S., Pompanon, F., Negrini, R., et al. (2008). Are Cattle, Sheep, and Goats Endangered Species? Mol. Ecol. 17 (1), 275–284. doi:10.1111/j.1365-294x.2007.03475.x
Tapio, M., Tapio, I., Grislis, Z., Holm, L.-E., Jeppsson, S., Kantanen, J., et al. (2005). Native Breeds Demonstrate High Contributions to the Molecular Variation in Northern European Sheep. Mol. Ecol. 14, 3951–3963. doi:10.1111/j.1365-294X.2005.02727.x
Vignali, R., and Marracci, S. (2020). HMGA Genes and Proteins in Development and Evolution. Ijms 1921 (2), 654. doi:10.3390/ijms21020654
Vigne, J.-D. (2011). The Origins of Animal Domestication and Husbandry: a Major Change in the History of Humanity and the Biosphere. Comptes Rendus Biologies 334, 171–181. doi:10.1016/j.crvi.2010.12.009
Wang, B., Zhang, M., Ni, Y.-h., Liu, F., Fan, H.-q., Fei, L., et al. (2006). Identification and Characterization of NYGGF4, a Novel Gene Containing a Phosphotyrosine-Binding (PTB) Domain that Stimulates 3T3-L1 Preadipocytes Proliferation. Gene 379, 132–140. doi:10.1016/j.gene.2006.05.008
Wang, X., Zhou, G., Li, Q., Zhao, D., and Chen, Y. (2014). Discovery of SNPs in RXFP2 Related to Horn Types in Sheep. Small Ruminant Res. 116, 133–136. doi:10.1016/j.smallrumres.2013.10.022
Wang, Y.-Y., Lin, Y.-H., Wu, Y.-N., Chen, Y.-L., Lin, Y.-C., Cheng, C.-Y., et al. (2017). Loss of SLC9A3 Decreases CFTR Protein and Causes Obstructed Azoospermia in Mice. Plos Genet. 13 (4), e1006715. doi:10.1371/journal.pgen.1006715
Webster, M. T., Kamgari, N., Perloski, M., Hoeppner, M. P., Axelsson, E., Hedhammar, Å., et al. (2015). Linked Genetic Variants on Chromosome 10 Control Ear Morphology and Body Mass Among Dog Breeds. BMC Genomics 16 (1), 474. doi:10.1186/s12864-015-1702-2
Weir, B. S., and Cockerham, C. C. (1984). Estimating F -Statistics for the Analysis of Population Structure. Evolution 38, 1358–1370. doi:10.2307/240864110.1111/j.1558-5646.1984.tb05657.x
Wimmers, K., Murani, E., Te Pas, M. F. W., Chang, K. C., Davoli, R., Merks, J. W. M., et al. (2007). Associations of Functional Candidate Genes Derived from Gene-Expression Profiles of Prenatal Porcine Muscle Tissue with Meat Quality and Muscle Deposition. Int. Soc. Anim. Genet. Anim. Genet. 38, 474–484. doi:10.1111/j.1365-2052.2007.01639.x
Wood, A. R., Esko, T., Yang, J., Vedantam, S., Pers, T. H., Gustafsson, S., et al. (2014). Defining the Role of Common Variation in the Genomic and Biological Architecture of Adult Human Height. Nat. Genet. 46, 1173–1186. doi:10.1038/ng.3097
Yang, T.-L., Guo, Y., Zhang, L.-S., Tian, Q., Yan, H., Guo, Y.-F., et al. (2010). HMGA2 Is Confirmed to Be Associated with Human Adult Height. Ann. Hum. Genet. 74 (1), 11–16. doi:10.1111/j.1469-1809.2009.00555.x
Zhang, D., Liu, Y., Zhang, Z., Lv, P., Liu, Y., Li, J., et al. (2018). Basonuclin 1 Deficiency Is a Cause of Primary Ovarian Insufficiency. Hum. Mol. Genet. 27 (21), 3787–3800. doi:10.1093/hmg/ddy261
Keywords: sheep, whole genome sequences, local breeds, demography, selection signatures
Citation: Ouhrouch A, Boitard S, Boyer F, Servin B, Da Silva A, Pompanon F, Haddioui A and Benjelloun B (2021) Genomic Uniqueness of Local Sheep Breeds From Morocco. Front. Genet. 12:723599. doi: 10.3389/fgene.2021.723599
Received: 11 June 2021; Accepted: 09 November 2021;
Published: 02 December 2021.
Edited by:
Olivier Hubert Hanotte, University of Nottingham, United KingdomReviewed by:
Lubos Vostry, Czech University of Life Sciences Prague, CzechiaCopyright © 2021 Ouhrouch, Boitard, Boyer, Servin, Da Silva, Pompanon, Haddioui and Benjelloun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abdessamad Ouhrouch, b3Vocm91Y2gxOTkxQGdtYWlsLmNvbQ==; Badr Benjelloun, YmFkci5iZW5qZWxsb3VuQGlucmEubWE=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.