 Xianfu Zhang1*
Xianfu Zhang1* Chuzhao Lei
Chuzhao Lei- 1Key Laboratory of Applied Technology on Green-Eco-Healthy Animal Husbandry of Zhejiang Province, Zhejiang Provincial Engineering Laboratory for Animal Health Inspection and Internet Technology, College of Animal Science and Technology, College of Veterinary Medicine, Zhejiang A&F University, Hangzhou, China
- 2Yunnan Academy of Grassland and Animal Science, Kunming, China
- 3Key Laboratory of Animal Genetics, Breeding and Reproduction of Shaanxi Province, College of Animal Science and Technology, Northwest A&F University, Xianyang, China
Dianzhong cattle is a classic Chinese indigenous cattle breed with historical records dating back to 200 BC. But with its genomic differences having not been clearly elucidated, the quest for genomic characterization will be an essential step towards understanding the genomic basis of productivity and adaptation to survival under Chinese farming systems. Here we compared 10 Dianzhong cattle (four newly sequenced and six downloaded) with 29 published genomes of three underlying ancestral populations (Chinese zebu, Indian zebu, and Yanbian cattle) to characterize the genomic variations of Dianzhong cattle. Dianzhong cattle has a high nucleotide diversity (0.0034), second only to Chinese zebu. Together with analyses of linkage disequilibrium decay and runs of homozygosity, Dianzhong cattle displayed higher genomic diversity and weaker artificial selection compared with Yanbian cattle. From a selective sweep analysis by four methods (Fst, π-ratio, XP-CLR, and XP-EHH), the positive selective signals were mainly manifested in candidate genes and pathways related to heat resistance, growth and development, fat deposition, and male reproduction. Missense mutations were detected in candidate genes, SDS (c.944C > A and p.Ala315Glu), PDGFD (c.473A > G and p.Lys158Arg), and DDX4 (rs460251486, rs722912933, and rs517668236), which related to heat resistance, fat deposition, and spermatogenesis, respectively. Our findings unravel, at the genome-wide level, the unique diversity of Dianzhong cattle while emphasizing the opportunities for improvement of livestock productivity in further breeding programs.
Introduction
Domestic cattle generally refers to two subspecies, Bos taurus and Bos taurus indicus. They were domesticated in the Fertile Crescent (∼10,000 years ago) and the Indus Valley (∼8,000 years ago), respectively (Utsunomiya et al., 2019). These two subspecies can be interbred without barriers, unlike other bovine subspecies (buffalo, American bison, etc.) that have reproductive isolation (Wu et al., 2018). According to previous genomic study, the domestic cattle in the world could be divided into six major groups: European taurine, Eurasian taurine, East Asian taurine, African taurine, Indian indicine, and Chinese indicine (Chen et al., 2018). Besides that, as one of the important routes for the migration of Indian indicine into China, there are many hybrid cattle with different lineages in the Yunnan region of China.
Dianzhong cattle is one of the most widely distributed indigenous breeds in Yunnan. It has a long history of breeding, with historical records dating back to 200 BC (China National Commission of Animal Genetic Resources, 2011). In addition to agricultural use, it also has social importance, including during marriage, birth, death, and sacrificial ceremonies, as well as being regarded as representatives of wealth, prestige, and status (China National Commission of Animal Genetic Resources, 2011). Due to the hot and humid climate of the original area, Dianzhong cattle display superior heat tolerance and resistance to parasites. Although small in size, its labor performance is excellent, owing to its long-term use as the main farming and transportation livestock (Wen et al., 2014). Moreover, as a classic indigenous cattle breed, it also shows advantageous characteristics of high intramuscular fat, strong disease resistance, and crude feed tolerance (Hao et al., 2017). In recent years, on account of its slow growth and low reproductive performance, which cannot meet the growing needs of beef, local people have blindly and massively introduced commercial cattle for hybrid improvement (Hao et al., 2017). However, blind hybridization as well as the lack of breed conservation planning caused the threat of breed degradation in Dianzhong cattle.
Based on whole-genome sequencing, many studies initially focused on the genetic architecture and economic traits under positive selection in commercial breeds (Bovine et al., 2009; Stothard et al., 2011), and then the focus gradually shifted to the adaptation of indigenous breeds, such as climate tolerance and disease resistance (Kim et al., 2017; Taye et al., 2017; Kim et al., 2020). With the development of next-generation sequencing technology and the enrichment of re-sequencing databases, genome-wide genetic analysis studies play an increasingly powerful role in the investigation of germplasm resources of landraces, such as cold tolerance of Yanbian cattle (Shen et al., 2020), excellent meat quality of Mongolian cattle (Chen et al., 2021), heat tolerance and parasite resistance of African cattle (Kim et al., 2017), and growth rate and feed conversion of Jiaxian red cattle (Xia et al., 2021). An earlier study on Dianzhong cattle using Illumina BovineHD BeadChip (777K) demonstrated that Dianzhong cattle contained mainly indicine ancestry mixed with taurine ancestry. Based on the estimate of observed heterozygosity and expected heterozygosity, Dianzhong cattle had the maximum level of inbreeding coefficients in Yunnan region (Li et al., 2019). In fact, single-nucleotide polymorphism (SNP) array data with only limited and known SNPs might make many important genetic information hard to detect. In order to further explore the genetic potential of Dianzhong cattle, we compared the genome re-sequencing data of 10 Dianzhong cattle (including four that were newly sequenced) with 29 reference cattle from Yanbian, Jiangxi, and India to search for the candidate signatures of positive selection by four methods (Fst, π-ratio, XP-CLR, and XP-EHH).
Materials and Methods
Sample Collection and Sequencing
We sampled four Dianzhong cattle from Chuxiong, Yunnan, China. Genomic DNA was extracted from the ear tissue samples as previously described (Jia et al., 2019). The pair-end libraries were constructed for each individual (500 bp insert size), and the DNA was subjected to Illumina NovaSeq sequencing using 2 × 150 bp model at Novogene Bioinformatics Institute (Beijing, China). Additionally, genomes of 35 publicly available representative groups, including Dianzhong cattle (n = 6), Chinese zebu (Jiangxi cattle, n = 10), Indian zebu (Brahman, Gir, and Nelore, n = 10), and Asian taurine (Yanbian cattle, n = 9), were used for the combined analysis (Supplementary Table S1). To obtain clean reads with high quality, the raw data was modified using Trimmomatic (LEADING:20 TRAILING:20 SLIDINGWINDOW:3:15 AVGQUAL:20 MINLEN:35 TOPHRED33) (Bolger et al., 2014).
Alignments and Variant Identification
Clean reads were aligned against the latest B. taurus reference genome (ARS-UCD1.2) using Burrows-Wheeler Aligner BWA-MEM (v0.7.13-r1126) with default parameters (Li et al., 2009). The Picard tool (http://broadinstitute.github.io/picard) was used to filter potential duplicate reads (REMOVE_DUPLICATES = true). SNP calling was performed by Genome Analysis Toolkit 3.8 (GATK) (Nekrutenko et al., 2012; Haplotype Caller, Genotype GVCFs and Select Variants module). After SNP calling, we used the module “Variant Filtration” of GATK to obtain high-quality SNPs with the parameters (“DP < 156 (1/3-fold total sequence depth for all individuals) || DP > 1404 (3-fold of total sequence depth for all individuals) || QD < 2.0 || FS > 60.0 || MQ < 40.0 || MQRankSum < −12.5 || ReadPosRankSum < −8.0 || SOR > 3.0”). Finally, we used ANNOVAR (Wang et al., 2010) to annotate the functions of the SNPs based on the B. taurus reference assembly ARS-UCD1.2.
Population Genetic Diversity
Nucleotide diversity for each group was investigated by VCFtools (Danecek et al., 2011) with a 50 kb non-overlapping window across all autosomes. The genetic relationship among four groups was performed by principal component analysis (PCA). Autosomal SNPs in high levels of pair-wise linkage disequilibrium (LD) were pruned using PLINK (Purcell et al., 2007) with the parameter (--indep-pair-wise 50 5 0.2) and were conducted using the smartPCA program in the EIGENSOFT v5.0 package (Patterson et al., 2006).
The LD decay for each group was measured using PopLDdecay (Zhang et al., 2019) with default parameters. Runs of homozygosity (ROHs) were identified using the Runs of Homozygosity program implemented in PLINK, which slides a window of 50 SNPs (-homozyg-window-snp 50) across the genome in estimating homozygosity (Xia et al., 2021). The following settings were performed for ROH identification: -homozyg-density 50 -homozyg-window-het 1 -homozyg-window-missing 2. The number and length of ROH for each group were estimated, and the length of ROH was divided into three categories: 0.5–1, 1–2, and 2–4 Mb reflecting ancient, historical, and recent inbreeding, respectively (Bhati et al., 2020). The strong linkage disequilibrium between parental genomic loci formed ROH, with long haplotype segments derived from recent ancestors and short haplotype segments derived from earlier ancestors (Jane et al., 2006).
Selective Sweep Identification
To identify selective sweep regions, genetic differentiation (Fst), genetic diversity (π-ratio), cross-population composite likelihood ratio test (XP-CLR), and cross-population extended haplotype homozygosity test (XP-EHH) were performed with 50 kb sliding window and 20 kb step between Dianzhong cattle and each other reference group separately. Selection scanning was performed using these four methods based on allele frequency, linkage imbalance, and population differentiation. The overlap of the top 1% windows in each method was considered as candidate signatures of selection. Genomic regions identified by at least two methods were considered to be candidate regions of positive selection (Sun et al., 2020). In addition, π and Fst were computed with 5 kb sliding window and 2 kb step by using VCFtools for each candidate gene. To gain a better understanding of the gene functions and signaling pathways of the identified candidate genes, online Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology (GO) analyses were conducted using DAVID 6.8 (Huang et al., 2009). The analyses for LD and construction of the haplotypes were performed with the online SHEsis software (http://analysis.bio-x.cn/myAnalysis.php) (Yong et al., 2005).
Results
Genome Resequencing and SNP Identification
Individual genomes of four Dianzhong cattle were generated to an average of ×∼9.8 coverage each and aligned to the B. taurus reference genome ARS-UCD1.2 with an average alignment rate of 99.72%. To reveal the diversity of Dianzhong cattle, 10 Dianzhong cattle (four new and six downloaded) were jointly genotyped with 29 publicly available genomes from three representative groups (10 Indian zebu, 10 Chinese zebu, and nine Yanbian cattle) (Supplementary Table S1). In total, 49,094,970 bi-allelic autosomal SNPs were detected, and the average alignment rate and sequencing depth of the final set reached 99.50% and ×∼12, respectively. After functional annotation of the polymorphic sites, the largest number of SNPs in the exon region was Dianzhong cattle (616,110). That presented the richer genetic diversity of Dianzhong cattle (Supplementary Table S2). Meanwhile, the analyses of unique SNPs and non-synonymous SNPs displayed that the genetic diversity of Dianzhong cattle was second only to Chinese zebu (Supplementary Figure S1).
Population Genetic Diversity and Relationships
On a genome-wide window scale of 50 kb, Yanbian cattle (East Asian taurine) showed a reduced level of nucleotide diversity compared to other groups (Figure 1A). The nucleotide diversity of Dianzhong cattle (0.0034) was second only to Chinese zebu (0.0039) and close to Indian zebu (0.0030). To explore relatedness among Dianzhong cattle and other cattle groups, PCA was conducted through using autosomal SNPs. The analysis ignored breed membership; nevertheless, it revealed clear breed structures as samples from the same group cluster together. PC1 and PC2 accordingly explained 9.12 and 4.78% of the total variations and separated taurine from indicine and Chinese zebu from Indian zebu, respectively (Figure 1B).
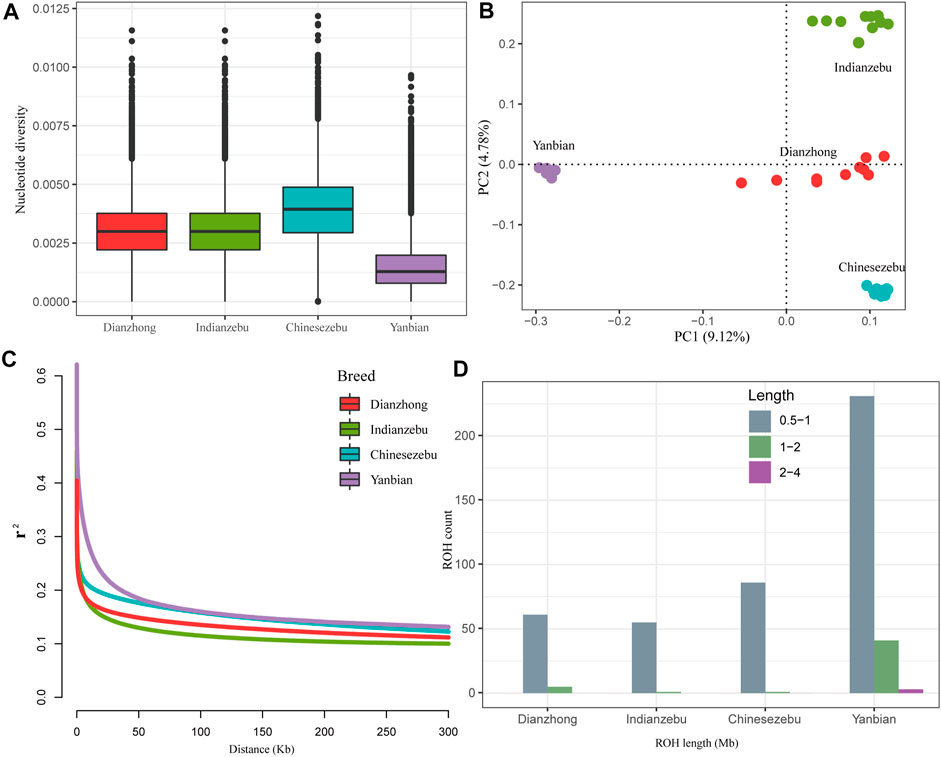
FIGURE 1. Summary statistics for genomic diversity. (A) Genome-wide distribution of nucleotide diversity of each group. The horizontal line inside the box indicated the median of this distribution; box limits indicated the first and the third quartiles, points showed outliers. Data points outside the whiskers can be considered as outliers. (B) Principal component analysis of four cattle groups. (C) Genome-wide average LD decay estimated from each group. (D) The estimation of number and length of ROH for each group.
Demographic inferences in cattle population were made based on the analyses of ROH and LD decay. For LD patterns, at short distances, Indian zebu showed lower LD level, and the highest LD level was found in Yanbian cattle, followed by Chinese zebu and Dianzhong cattle. Yanbian cattle continued to have a higher LD than all other breeds when the distances were larger, while a contrary trend was observed in indicine (Indian zebu and Chinese zebu) and hybrid (Dianzhong) (Figure 1C). To evaluate the ROH pattern of Dianzhong cattle and other cattle groups, we divided the length of ROH into three size classes: 0.5–1, 1–2, and 2–4 Mb (Figure 1D). The vast majority of ROH identified in all groups were between 0.5 and 1 Mb in length, but apparently Yanbian cattle had more medium (1–2 Mb) and long (2–4 Mb) ROHs, showing stronger inbreeding. As expected, the pattern of ROH profile was nearly consistent with the result of LD decay.
Candidate Regions and Genes Under Positive Selection
Due to the genetic separation between Dianzhong cattle and three reference groups, we compared Dianzhong cattle with the other three reference populations using four selective sweep methods respectively in three groups.
In the Dianzhong cattle vs. Yanbian cattle comparison, the top 1% of candidate genes in four selection methods were extracted at the intersection (Supplementary Figure S2): MARS2 (Webb et al., 2015) and ARL6IP6 (Abumansour et al., 2015) (growth and development), RFTN2 (Wei et al., 2018) (DNA damage response), and N6AMT1 (Wang et al., 2020) (feed efficiency). GO and KEGG analyses were performed on candidate genes scanned twice or more by four methods. From DAVID gene ontology, 12 significant (p < 0.05) GO biological process (BP) terms were enriched (Supplementary Table S16). Among them, “oxidation–reduction process, GO:0055114” (n = 18) contained more genes than other GO terms. The other candidate genes were mainly enriched in hot resistance (“cardiac muscle cell differentiation, GO:0055007,” “lipid catabolic process, GO:0016042,” “positive regulation of ATPase activity, GO:0032781,” “mitochondrial DNA repair, GO:0043504,” and “regulation of oxidative stress-induced intrinsic apoptotic signaling pathway, GO:1902175”). Moreover, we noticed a region of about 0.06 Mb scanned by XP-CLR on chromosome 17 (including TRNAG-CCC, SDSL, SDS, and PLBD2), showing a strong positive selection signal. Thereinto, a missense mutation (c.944C > A, p.Ala315Glu) was found at the SDS gene. This mutation presented a huge divergence between Dianzhong cattle (allele C frequency = 1) and Yanbian cattle (allele A frequency = 1).
A total of 42 candidate genes were overlapped in four selection methods by comparing Dianzhong cattle with Indian zebu (Supplementary Figure S2), and three of them were significantly enriched in “kinase binding, GO:0019900” (p-value = 0.02). In the functional prediction of all candidate genes, eight GO BP terms were significantly enriched (p < 0.05) (Supplementary Table S17). The candidate genes were mainly enriched in growth (“post-embryonic development, GO:0009791,” “skeletal muscle tissue development, GO:0007519”; and “embryonic skeletal system morphogenesis, GO:0048704”) and heat stress resistance (“DNA recombination, GO:0006310,” “inflammatory response, GO:0006954,” “response to hydrogen peroxide, GO:0042542,” and “respiratory gaseous exchange, GO:0007585”). Most genes (n = 11) were included in “inflammatory response, GO:0006954.” Furthermore, we detected a missense mutation (c.473A > G, p.Lys158Arg) at the fat deposition-related gene PDGFD. Allele A displayed a rare distribution (frequency 0.2) in Dianzhong cattle, whereas it showed an opposite pattern (frequency 0.9) in Indian zebu.
In the selection signal analysis between Dianzhong cattle and Chinese zebu, 36 common candidate genes were scanned by four selection methods. There were two genes enriched in the “response to retinoic acid, GO:0032526” (p-value = 0.04). The strongest signal was found in KIT gene, which was associated with sperm differentiation [33]. In KEGG analysis, four digestion-related pathways were enriched (Supplementary Table S18): “gastric acid secretion, bta04971,” “pancreatic secretion, bta04972,” “salivary secretion, bta04970,” and “carbohydrate digestion and absorption, bta04973.” In addition, three missense mutations (rs460251486, rs722912933, and rs517668236) with strong linkage effect were detected at candidate gene DDX4. According to the analyses for LD and haplotype construction of the three mutations, they exhibited a low recombination rate with r2 values ranging from 0.31 to 0.61 (Supplementary Figure S3) (Ardlie et al., 2002). DDX4 gene was the candidate gene selected by Fst (Figure 2A), and through the calculation of Fst and π with a smaller window (5 kb), a significant differentiation was observed between Dianzhong cattle and Chinese zebu (Figure 2B). Simultaneously, according to the calculation of the combined haplotype frequencies at the three loci, the haplotype with the most distribution in Dianzhong cattle was the homozygous mutational haplotype (frequency 0.601), while the homozygous wild haplotype (frequency 0.700) was the commonest one in Chinese zebu (Figure 2C).
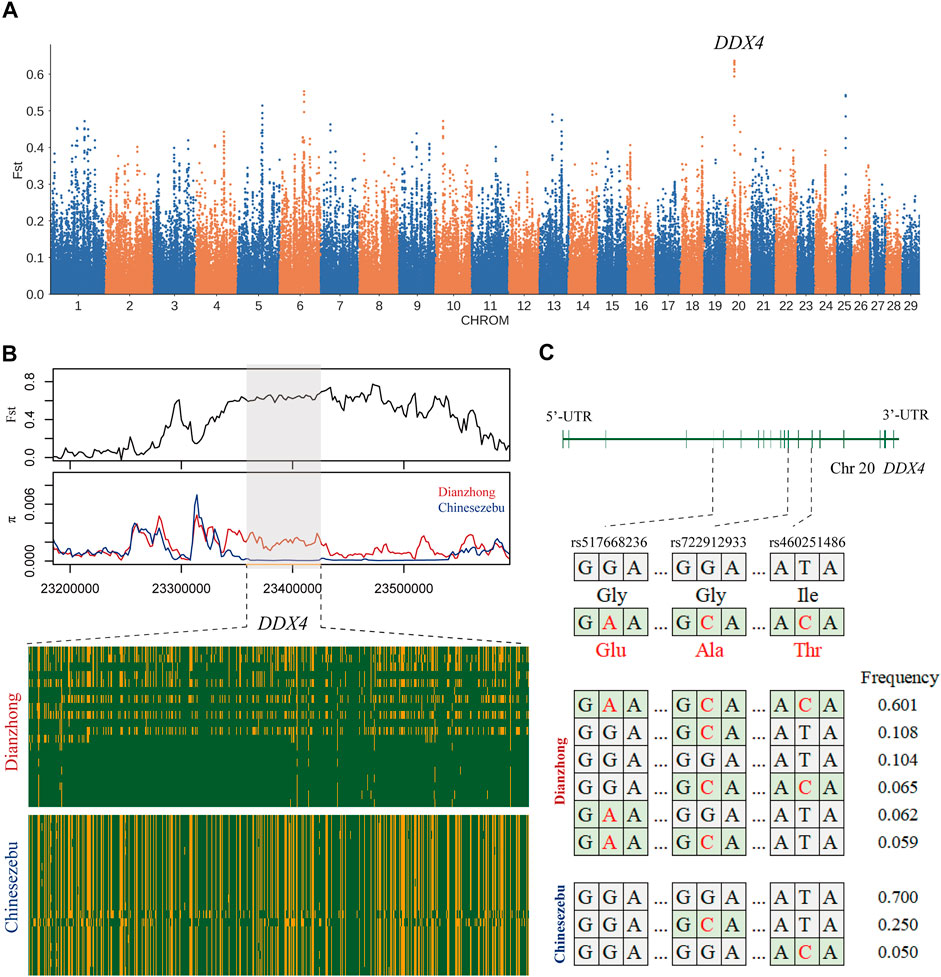
FIGURE 2. Analysis of the signatures of positive selection between Dianzhong cattle and Chinese zebu. (A) Manhattan plot of selective sweeps. (B) Fst and π plots and haplotype patterns heatmap of the DDX4 gene region. (C) Structure of DDX4 with exons indicated by vertical bars. Three missense mutations and their haplotypes were highlighted in red.
Discussion
Genomic features may reflect a variety of multifarious historical events, including climate change, species introduction, and artificial selection (Chen et al., 2021). The characteristics of population genetic diversity are essential for assessing the genetic potential of breeds as well as for the utilization and protection of cattle breed resources (Xia et al., 2021). In this study, three underlying ancestors of Dianzhong cattle were selected as reference groups to explore the genetic information of Dianzhong cattle. The nucleotide diversity ranking calculated in this study (Chinese zebu > Dianzhong cattle > Indian zebu > Yanbian cattle) was basically consistent with the earlier results (Chen et al., 2018). Dianzhong cattle, belonging to hybrid cattle, obtained more abundant genetic information from multiple genetic sources, while Chinese zebu had high nucleotide diversity which may be due to previous reception of introgression from Banteng (Bos javanicus) (Chen et al., 2018). The monotonous environment and high degree of breeding may be the reasons why the nucleotide diversity of Yanbian cattle is lower than that of indicine and hybrid cattle (Singh et al., 2021; Xia et al., 2021). In addition, more 2–4 MB and 1–2 MB ROHs and more rapid LD decayed in Yanbian cattle confirmed this once again. The nucleotide diversity in our Indian zebu was higher than those in other groups, and the LD level was lower than others, suggesting that Indian zebu had a higher effective population size (Chen et al., 2021). In summary, the LD decay pattern and ROH distribution of each group were roughly consistent with the results of nucleotide diversity.
Owing to the humid and hot living environment, Dianzhong cattle evolved to be prominently heat resistant (China National Commission of Animal Genetic Resources, 2011). In the comparative analysis with Yanbian cattle living in a cold environment, functional enrichment analysis revealed that heat resistance-related pathways were significantly overrepresented in candidate genes under positive selection. Moreover, a putatively selected gene, SDS, encodes serine dehydratase, an enzyme that catalyzes the degradation of serine to pyruvate, which was functionally involved in heat resistance (Ogawa et al., 1989). SDS activated inflammasomes by mediating mitochondrial membrane potential, thereby affecting individual immune function (Çağdaş et al., 2020). Inflammasomes are important in the defense against microbial infections and have roles in shaping the adaptive immune responses (Joly et al., 2012; Yu et al., 2015).
The typical characteristic of Dianzhong cattle is small in body size, approximately 101 cm tall and weighing 170 kg in adulthood (Wen et al., 2014). Compared with the tall Indian zebu by selection analysis, there were many growth- and development-related items in GO enrichment. What is more, because of the preference of East Asians for good meat quality (high marbling score), Dianzhong cattle have high potential for accumulating intramuscular fat and producing highly marbled beef (Hao et al., 2017). The PDGFD gene selected by XP-CLR was related to perivascular adipose tissue generation (Dong et al., 2020). Members of the PDGF family have an anti-fat effect and inhibit the differentiation of preadipocytes (Ma et al., 2018; Pan et al., 2019). PDGFD played a pivotal role in inhibiting the differentiation of white adipocytes by regulating the expression of PPARγ2 and C/EBPα (Li et al., 2020). Early studies have reported that the PDGFD gene was hypothesized to be one of the candidate genes leading to fat tail formation in indigenous sheep (Zhao et al., 2020; Zhu et al., 2021). In addition to the possible involvement of intermuscular fat deposition, we ventured to speculate that the PDGFD gene may also contribute to the formation of indicine hump. However, this is just a conjecture, and more theoretical and experimental supports were required.
Poor fertility is one of the most important factors restricting the development of Dianzhong cattle (China National Commission of Animal Genetic Resources, 2011). The process of admixture among the Bos subspecies brought adaptability but caused a cost of reduced reproductive fitness due to genomic incompatibility at the same time (Kim et al., 2020). Jiangxi cattle (Chinese zebu) is a precocious puberty cattle breed that can breed for the first time at the age of 10 months (Shuanping et al., 2019), while Dianzhong bulls do so later than 30 months old (China National Commission of Animal Genetic Resources, 2011). In comparison between Dianzhong cattle and Chinese zebu, some candidate genes were significantly enriched in “response to retinoic acid, GO:0032526” (p = 0.04). Retinoic acid promoted the differentiation of male germ cells and induced the differentiation of mouse pluripotent cell bodies into outer male germ cells (Mahabadi et al., 2020). In addition, candidate gene KIT, as the highest-ranked in overlap, is a transmembrane protein receptor related to germ cell maturation (Rossi et al., 2000). It is also a sign of the loss of efficacy of spermatogonial stem cells (Schrans-Stassen et al., 1999). In general, the presence of multiple linkage loci associated with a specific phenotype in a gene indicated a highly probable connection between the gene and this phenotype (Jia et al., 2019; Cao et al., 2020). Hence, the haplotypes formed by the three strongly linked missense mutations in the DDX4 gene were obviously different between Dianzhong cattle and Chinese zebu, implying that the spermatogenesis-related DDX4 gene may be related to the weak reproductive performance of Dianzhong cattle. DDX4 gene encodes an ATP-dependent RNA helicase (Hay et al., 1988). In DDX4-knockout mice, germ cell development was normal in female homozygous null mice, but male mice were sterile due to the failure of their germ cells to progress from leptotene to zygotene of meiotic prophase I, and the cells underwent apoptosis (Tanaka et al., 2000). The expression of DDX4 in germ cells of male mice has been described in humans, dogs, cattle, pigs, and stallions (Lee et al., 2018a). Interestingly, KIT gene, together with DDX4 gene, was considered to be a marker for undifferentiated spermatogonia before puberty and differentiated spermatogonia after puberty in porcine testis (Lee et al., 2018b).
In conclusion, our population genomic analyses of Dianzhong cattle and other three reference groups provide novel insights into their genetic diversity and selective sweep. This will point out the direction for genetic assessment and development of reasonable improvement of Dianzhong cattle. Moreover, we identified a series of candidate genes that may be important for the heat resistance, intermuscular fat deposition, and reproductive barriers of this breed. These results provide a basis for further research on the genome characteristics of other important indigenous beef cattle in the future (Gao et al., 2017).
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by the Administration of Affairs Concerning Experimental Animals of China (Protocol number, WAFAC1008).
Author Contributions
XZ and PJ contributed equally towards the construction and execution of this manuscript. KQ helped in sample collection. JZ and JL revised the manuscript and provided valuable suggestion. CL and BH contributed in the funding for the research.
Funding
This work was supported by the China Agriculture Research System of MOF and MARA (CARS-37), the Program of Yunling Scholar, the Young and Middle-Aged Academic Technology Leader Backup Talent Cultivation Program in Yunnan Province, China (no. 2018HB045), and the Yunnan Provincial Major S&T Project (no. 2019ZG007 and 2019ZG011).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Thanks to the High-Performance Computing of Northwest A&F University for providing computing resources.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.719215/full#supplementary-material
References
Abumansour, I. S., Hijazi, H., Alazmi, A., Alzahrani, F., Bashiri, F. A., Hassan, H., et al. (2015). ARL6IP6, a Susceptibility Locus for Ischemic Stroke, Is Mutated in a Patient with Syndromic Cutis Marmorata Telangiectatica Congenita. Hum. Genet. 134 (8), 815–822. doi:10.1007/s00439-015-1561-6
Ardlie, K. G., Kruglyak, L., and Seielstad, M. (2002). Patterns of Linkage Disequilibrium in the Human Genome. Nat. Rev. Genet. 3 (4), 299–309. doi:10.1038/nrg777
Bhati, M., Kadri, N. K., Crysnanto, D., and Pausch, H. (2020). Assessing Genomic Diversity and Signatures of Selection in Original Braunvieh Cattle Using Whole-Genome Sequencing Data. BMC Genomics 21 (1), 27. doi:10.1186/s12864-020-6446-y
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics (Oxford, England) 30 (15), 2114–2120. doi:10.1093/bioinformatics/btu170
Bovine, H. C., Gibbs, R. A., Taylor, J. F., Van Tassell, C. P., Barendse, W., Eversole, K. A., et al. (2009). Genome-wide Survey of SNP Variation Uncovers the Genetic Structure of Cattle Breeds. Science 324 (5926), 528–532. doi:10.1126/science.1167936
Cao, Y., Jia, P., Wu, Z., Huang, M., Chen, S., Zhang, J., et al. (2020). A Novel SNP of MYO1A Gene Associated with Heat-Tolerance in Chinese Cattle. Anim. Biotechnol. 4, 1–6. doi:10.1080/10495398.2020.1837147
Çağdaş, D., Sürücü, N., Tan, Ç., Kayaoğlu, B., Özgül, R. K., Akkaya-Ulum, Y. Z., et al. (2020). Autoinflammation in Addition to Combined Immunodeficiency: SLC29A3 Gene Defect. Mol. Immunol. 121, 28–37. doi:10.1016/j.molimm.2020.02.014
Chen, N., Cai, Y., Chen, Q., Li, R., Wang, K., Huang, Y., et al. (2018). Whole-genome Resequencing Reveals World-wide Ancestry and Adaptive Introgression Events of Domesticated Cattle in East Asia. Nat. Commun. 9 (1), 2337. doi:10.1038/s41467-018-04737-0
Chen, Q., Shen, J., Hanif, Q., Chen, N., Huang, Y., Dang, R., et al. (2021). Whole Genome Analyses Revealed Genomic Difference between European Taurine and East Asian Taurine. J. Anim. Breed. Genet. 138 (1), 56–68. doi:10.1111/jbg.12501
China National Commission of Animal Genetic Resources (2011). Animal Genetic Resources in China Bovines. Beijing: Chinese Agricultural Press.
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The Variant Call Format and VCFtools. Bioinformatics 27 (15), 2156–2158. doi:10.1093/bioinformatics/btr330
Dong, K., Yang, M., Han, J., Ma, Q., Han, J., Song, Z., et al. (2020). Genomic Analysis of Worldwide Sheep Breeds Reveals PDGFD as a Major Target of Fat-Tail Selection in Sheep. BMC Genomics 21 (1), 800. doi:10.1186/s12864-020-07210-9
Gao, Y., Gautier, M., Ding, X., Zhang, H., Wang, Y., Wang, X., et al. (2017). Species Composition and Environmental Adaptation of Indigenous Chinese Cattle. Sci. Rep. 7 (1), 16196. doi:10.1038/s41598-017-16438-7
Gibson, J., Morton, N. E., and Collins, A. (2006). Extended Tracts of Homozygosity in Outbred Human Populations. Hum. Mol. Genet. 15 (5), 789–795. doi:10.1093/hmg/ddi493
Hao, W., Zhixian, X., Peichang, Y., Chunfeng, L., Xiangsheng, S., Rongfa, L., et al. (2017). Protection, Exploitation and Utilization of Genetic Resources in Dianzhong Cattle. Curr. Anim. husbandry 403 (35), 6–7.
Hay, B., Jan, L. Y., and Jan, Y. N. (1988). A Protein Component of Drosophila Polar Granules Is Encoded by Vasa and Has Extensive Sequence Similarity to ATP-dependent Helicases. Cell 55 (4), 577–587. doi:10.1016/0092-8674(88)90216-4
Huang, D. W., Sherman, B. T., and Lempicki, R. A. (2009). Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 4 (1), 44–57. doi:10.1038/nprot.2008.211
Jia, P., Cai, C., Qu, K., Chen, N., Jia, Y., Hanif, Q., et al. (2019). Four Novel SNPs of MYO1A Gene Associated with Heat-Tolerance in Chinese Cattle. Animals 9 (11), 964. doi:10.3390/ani9110964
Joly, S., Eisenbarth, S. C., Olivier, A. K., Williams, A., Kaplan, D. H., Cassel, S. L., et al. (2012). Cutting Edge: Nlrp10 Is Essential for Protective Antifungal Adaptive Immunity against Candida Albicans, J Immunol, 10, 189. Baltimore, Md. 1950, 4713–7. doi:10.4049/jimmunol.1201715
Kim, J., Hanotte, O., Mwai, O. A., Dessie, T., Bashir, S., Diallo, B., et al. (2017). The Genome Landscape of Indigenous African Cattle. Genome Biol. 18 (1), 34. doi:10.1186/s13059-017-1153-y
Kim, K., Kwon, T., Dessie, T., Yoo, D., Mwai, O. A., Jang, J., et al. (2020). The Mosaic Genome of Indigenous African Cattle as a Unique Genetic Resource for African Pastoralism. Nat. Genet. 52 (10), 1099–1110. doi:10.1038/s41588-020-0694-2
Lee, R., Lee, W.-Y., Park, H.-J., Ha, W.-T., Woo, J.-S., Chung, H.-J., et al. (2018a). Stage-specific Expression of DDX4 and C-Kit at Different Developmental Stages of the Porcine Testis. Anim. Reprod. Sci. 190, 18–26. doi:10.1016/j.anireprosci.2017.12.020
Lee, R., Lee, W.-Y., Park, H.-J., Ha, W.-T., Woo, J.-S., Chung, H.-J., Lee, J.-H., Hong, K., and Song, H. (2018b). Stage-specific expression of DDX4 and c-kit at different developmental stages of the porcine testis. Animal Reproduction Science 190, 18–26. doi:10.1016/j.anireprosci.2017.12.020
Li, H., and Durbin, R. (2009). Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, R., Li, C., Chen, H., Liu, X., Xiao, H., and Chen, S. (2019). Genomic Diversity and Admixture Patterns Among Six Chinese Indigenous Cattle Breeds in Yunnan. Asian-australas J. Anim. Sci. 32 (8), 1069–1076. doi:10.5713/ajas.18.0605
Li, X., Yang, J., Shen, M., Xie, X.-L., Liu, G.-J., Xu, Y.-X., et al. (2020). Whole-genome Resequencing of Wild and Domestic Sheep Identifies Genes Associated with Morphological and Agronomic Traits. Nat. Commun. 11 (1), 2815. doi:10.1038/s41467-020-16485-1
Ma, L., Li, Z., Cai, Y., Xu, H., Yang, R., and Lan, X. (2018). Genetic Variants in Fat- and Short-Tailed Sheep from High-Throughput RNA-Sequencing Data. Anim. Genet. 49 (5), 483–487. doi:10.1111/age.12699
Mahabadi, J. A., Tameh, A. A., Talaei, S. A., Karimian, M., Rahiminia, T., Enderami, S. E., et al. (2020). Retinoic Acid And/or Progesterone Differentiate Mouse Induced Pluripotent Stem Cells into Male Germ Cells In Vitro. J. Cel. Biochem. 121 (3), 2159–2169. doi:10.1002/jcb.29439
Nekrutenko, A., and Taylor, J. (2012). Next-generation Sequencing Data Interpretation: Enhancing Reproducibility and Accessibility. Nat. Rev. Genet. 13 (9), 667–672. doi:10.1038/nrg3305
Ogawa, H., Gomi, T., Konishi, K., Date, T., Nakashima, H., Nose, K., et al. (1989). Human Liver Serine Dehydratase. J. Biol. Chem. 264 (27), 15818–15823. doi:10.1016/S0021-9258(18)71550-0
Pan, Z., Li, S., Liu, Q., Wang, Z., Zhou, Z., Di, R., et al. (2019). Rapid Evolution of a Retro-Transposable Hotspot of Ovine Genome Underlies the Alteration of BMP2 Expression and Development of Fat Tails. BMC Genomics 20 (1), 261. doi:10.1186/s12864-019-5620-6
Patterson, N., Price, A. L., and Reich, D. (2006). Population Structure and Eigenanalysis. Plos Genet. 2, e190–e2093. doi:10.1371/journal.pgen.0020190
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 81 (3), 559–575. doi:10.1086/519795
Rossi, P., Sette, C., Dolci, S., and Geremia, R. (2000). Role of C-Kit in Mammalian Spermatogenesis. J. Endocrinol. Invest. 23 (9), 609–615. doi:10.1007/BF03343784
Schrans-Stassen, B. H. G. J., van de Kant, H. J. G., de Rooij, D. G., and van Pelt, A. M. M. (1999). Differential Expression of C-Kit in Mouse Undifferentiated and Differentiating Type a Spermatogonia. Endocrinology 140 (12), 5894–5900. doi:10.1210/endo.140.12.7172
Shen, J., Hanif, Q., Cao, Y., Yu, Y., Lei, C., Zhang, G., et al. (2020). Whole Genome Scan and Selection Signatures for Climate Adaption in Yanbian Cattle. Front. Genet. 11, 94. doi:10.3389/fgene.2020.00094
Shuanping, Z., Hai, J., Lei, X., Juan, W., Mo, L., and Yutang, J. (2019). Analysis on the Characteristics of Wannan Cattle Germplasm Resources and Hybridization Improvement Effect. Chin. Cattle Sci. 45 (05), 21–23.
Singh, A. K., Liu, W., Zakari, S., Wu, J., Yang, B., Jiang, X. J., et al. (2021). A Global Review of Rubber Plantations: Impacts on Ecosystem Functions, Mitigations, Future Directions, and Policies for Sustainable Cultivation. Sci. Total Environ. 796, 148948. doi:10.1016/j.scitotenv.2021.148948
Stothard, P., Choi, J.-W., Basu, U., Sumner-Thomson, J. M., Meng, Y., Liao, X., et al. (2011). Whole Genome Resequencing of Black Angus and Holstein Cattle for SNP and CNV Discovery. BMC Genomics 12, 559. doi:10.1186/1471-2164-12-559
Sun, T., Huang, G.-y., Wang, Z.-h., Teng, S.-h., Cao, Y.-h., Sun, J.-l., et al. (2020). Selection Signatures of Fuzhong Buffalo Based on Whole-Genome Sequences. BMC Genomics 21 (1), 674. doi:10.1186/s12864-020-07095-8
Tanaka, S. S., Toyooka, Y., Akasu, R., Katoh-Fukui, Y., Nakahara, Y., Suzuki, R., et al. (2000). The Mouse Homolog of Drosophila Vasa Is Required for the Development of Male Germ Cells. Genes Dev. 14 (7), 841–853. doi:10.1101/gad.14.7.841
Taye, M., Kim, J., Yoon, S. H., Lee, W., Hanotte, O., Dessie, T., et al. (2017). Whole Genome Scan Reveals the Genetic Signature of African Ankole Cattle Breed and Potential for Higher Quality Beef. BMC Genet. 18 (1), 11. doi:10.1186/s12863-016-0467-1
Utsunomiya, Y. T., Milanesi, M., Fortes, M. R. S., Porto‐Neto, L. R., Utsunomiya, A. T. H., Silva, M. V. G. B., et al. (2019). Genomic Clues of the Evolutionary History of Bos indicus Cattle. Anim. Genet. 50 (6), 557–568. doi:10.1111/age.12836
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 38 (16), e164. doi:10.1093/nar/gkq603
Wang, X., and Kadarmideen, H. N. (2020). Metabolite Genome-wide Association Study (mGWAS) and Gene-Metabolite Interaction Network Analysis Reveal Potential Biomarkers for Feed Efficiency in Pigs. Metabolites 10 (5), 201. doi:10.3390/metabo10050201
Webb, B. D., Wheeler, P. G., Hagen, J. J., Cohen, N., Linderman, M. D., Diaz, G. A., et al. (2015). Novel, Compound Heterozygous, Single-Nucleotide Variants inMARS2Associated with Developmental Delay, Poor Growth, and Sensorineural Hearing Loss. Hum. Mutat. 36 (6), 587–592. doi:10.1002/humu.22781
Wei, F., Hao, P., Zhang, X., Hu, H., Jiang, D., Yin, A., et al. (2018). Etoposide-induced DNA Damage Affects Multiple Cellular Pathways in Addition to DNA Damage Response. Oncotarget 9 (35), 24122–24139. doi:10.18632/oncotarget.24517
Wen, L., Deliang, C., Xiaofang, O., Jincheng, Z., Xiangguang, M., and Jiacai, Z. (2014). Determination and Analysis of Growth Performance of Hybrid Progeny of Dianzhong Cattle. Shanghai Anim. husbandry Vet. Newsl. 01, 32–35.
Wu, D.-D., Ding, X.-D., Wang, S., Wójcik, J. M., Zhang, Y., Tokarska, M., et al. (2018). Pervasive Introgression Facilitated Domestication and Adaptation in the Bos Species Complex. Nat. Ecol. Evol. 2 (7), 1139–1145. doi:10.1038/s41559-018-0562-y
Xia, X., Zhang, S., Zhang, H., Zhang, Z., Chen, N., Li, Z., et al. (2021). Assessing Genomic Diversity and Signatures of Selection in Jiaxian Red Cattle Using Whole-Genome Sequencing Data. BMC Genomics 22 (1), 43. doi:10.1186/s12864-020-07340-0
Yong, Y., and He, L. (2005). SHEsis, a Powerful Software Platform for Analyses of Linkage Disequilibrium, Haplotype Construction, and Genetic Association at Polymorphism Loci. Cell Res 15 (2), 97–98. doi:10.1038/sj.cr.7290272
Yu, S.-X., Du, C.-T., Chen, W., Lei, Q.-Q., Li, N., Qi, S., et al. (2015). Genipin Inhibits NLRP3 and NLRC4 Inflammasome Activation via Autophagy Suppression. Sci. Rep. 5, 17935. doi:10.1038/srep17935
Zhang, C., Dong, S.-S., Xu, J.-Y., He, W.-M., and Yang, T.-L. (2019). PopLDdecay: A Fast and Effective Tool for Linkage Disequilibrium Decay Analysis Based on Variant Call Format Files. Bioinformatics 35 (10), 1786–1788. doi:10.1093/bioinformatics/bty875
Zhao, F., Deng, T., Shi, L., Wang, W., Zhang, Q., Du, L., et al. (2020). Genomic Scan for Selection Signature Reveals Fat Deposition in Chinese Indigenous Sheep with Extreme Tail Types. Animals 10 (5), 773. doi:10.3390/ani10050773
Keywords: Dianzhong cattle, hybrid, genetic diversity, selection signatures, DDX4
Citation: Zhang X, Qu K, Jia P, Zhang J, Liu J, Lei C and Huang B (2021) Assessing Genomic Diversity and Productivity Signatures in Dianzhong Cattle by Whole-Genome Scanning. Front. Genet. 12:719215. doi: 10.3389/fgene.2021.719215
Received: 02 June 2021; Accepted: 10 September 2021;
Published: 05 October 2021.
Edited by:
Johann Sölkner, University of Natural Resources and Life Sciences Vienna, AustriaReviewed by:
Gábor Mészáros, University of Natural Resources and Life Sciences Vienna, AustriaNina Moravčíková, Slovak University of Agriculture, Slovakia
Copyright © 2021 Zhang, Qu, Jia, Zhang, Liu, Lei and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xianfu Zhang, MjAwNjAxNTlAemFmdS5lZHUuY24=; Bizhi Huang, aGJ6QHluYnAuY24=