 Shuji Mizumoto*
Shuji Mizumoto* Shuhei Yamada
Shuhei Yamada- Department of Pathobiochemistry, Faculty of Pharmacy, Meijo University, Nagoya, Japan
Glycosaminoglycans (GAGs) including chondroitin sulfate, dermatan sulfate, and heparan sulfate are covalently attached to specific core proteins to form proteoglycans, which are distributed at the cell surface as well as in the extracellular matrix. Proteoglycans and GAGs have been demonstrated to exhibit a variety of physiological functions such as construction of the extracellular matrix, tissue development, and cell signaling through interactions with extracellular matrix components, morphogens, cytokines, and growth factors. Not only connective tissue disorders including skeletal dysplasia, chondrodysplasia, multiple exostoses, and Ehlers-Danlos syndrome, but also heart and kidney defects, immune deficiencies, and neurological abnormalities have been shown to be caused by defects in GAGs as well as core proteins of proteoglycans. These findings indicate that GAGs and proteoglycans are essential for human development in major organs. The glycobiological aspects of congenital disorders caused by defects in GAG-biosynthetic enzymes including specific glysocyltransferases, epimerases, and sulfotransferases, in addition to core proteins of proteoglycans will be comprehensively discussed based on the literature to date.
Introduction
Glycosaminoglycans (GAGs), including chondroitin sulfate (CS), dermatan sulfate (DS), and heparan sulfate (HS), are linear polysaccharides that are covalently attached to core proteins, forming proteoglycans (PGs). PGs are ubiquitously distributed on the cell surface and in the extracellular matrix (Iozzo, 1998; Bishop et al., 2007; Thelin et al., 2013; Mizumoto et al., 2015a). GAGs are critically involved in a variety of biological functions including cell adhesion, cellular signaling, and the architecture of the extracellular matrix. A large number of studies have reported analyses of GAGs from various types of cells, nematodes, fruit flies, zebrafish, chicks, and mice (Häcker et al., 2005; Bülow and Hobert, 2006; Mizumoto et al., 2014). After the mapping of the of human genome, congenital disorders caused by defects in GAG biosynthesis have been revealed by many research groups (Mizumoto et al., 2013, 2014, 2015b; Mizumoto, 2018). This review provides a comprehensive overview of genetic disorders with symptoms affecting various areas of the body such as bone, skin, brain, heart, and immune system caused by defects in the biosynthesis of GAG side chains of PGs.
Biosynthetic Pathway of Glycosaminoglycans
Biosynthesis of Donor Substrates for GAGs and Transporters of Uridine 5′-Diphosphate-Sugars, Sulfate Ions, and 3′-Phosphoadenosine 5′-Phosphosulfate
Most glycosyltransferases and sulfotransferases utilize donor substrates such as uridine 5′-diphosphate (UDP)-sugars and 3′-phosphoadenosine 5′-phosphosulfate (PAPS), respectively. The nucleotide sugars, UDP-Glc, UDP-GlcA, UDP-GlcNAc, UDP-GalNAc, UDP-Gal, and UDP-Xyl, where Glc, GlcA, GlcNAc, GalNAc, Gal, and Xyl, represent D-glucose, D-glucuronic acid, N-acetyl-D-glucosamine, N-acetyl-D-galactosamine, D-galactose, and D-xylose, respectively, are produced predominantly from Glc, D-glucosamine (GlcN), and D-Gal (Figure 1). UDP-GlcA is formed by the action of the UDP-Glc dehydrogenase on UDP-Glc in the cytosol (Spicer et al., 1998). UDP-Xyl is formed by the action of UDP-GlcA decarboxylase/UDP-xylose synthase in the endoplasmic reticulum and Golgi apparatus (Moriarity et al., 2002). These UDP-sugars mainly synthesized in the cytosol, except for UDP-Xyl, are incorporated into the endoplasmic reticulum and Golgi lumen through nucleotide sugar transporters (Figure 1; Berninsone and Hirschberg, 2000).

Figure 1. Biosynthetic pathways of UDP-sugars and PAPS. 1, Hexokinase; 2, GlcNH2 acetyltransferase; 3, GlcNH2 6-phosphate N-acetyltransferase; 4, GlcNAc kinase; 5, GlcNAc phosphomutase; 6, UDP-GlcNAc pyrophosphorylase: 7, UDP-GalNAc 4-epimerase; 8, glutamine-fructose-6-phosphate aminotransferase; 9, Glc-6-phosphate isomerase; 10, hexokinase; 11, phosphoglucomutase; 12, UDP-Glc pyrophosphorylase; 13, UDP-Glc dehydrogenase; 14, UDP-Glc 4-epimerase; 15, galactokinase; 16, Gal-1-phosphate uridylyltransferase; 17, PAPS synthase; 18, UDP-GlcA decarboxylase; 19, sulfotransferases; 20, adenosine-3′, 5′-bisphosphate 3′-phosphatase; 21, GalNAc transferases; 22, nucleoside 5′-diphosphate phosphatase; 23, GlcNAc transferases; 24, GlcA transferases; 25, Xyl transferases; 26, Gal transferases.
Various GAG sulfotransferases catalyze the transfer of the sulfate group from the donor substrate, PAPS, to respective acceptor substrates (Kusche-Gullberg and Kjellén, 2003). PAPS is formed from adenosine 5′-triphosphate (ATP) and inorganic sulfate, which is incorporated into the cytosol through the sulfate transporter at the plasma membrane (Figure 1; Hästbacka et al., 1994). PAPS synthase (PAPSS) is a dual enzyme containing ATP sulfurylase and adenosine 5′-phosphosulfate kinase domains at C- and N-terminals, respectively (Venkatachalam, 2003).
Glycosaminoglycan Backbones
The biosynthesis of the linker region tetrasaccharide in CS, DS, and HS, but not keratan sulfate (KS) is initiated by the transfer of a xylose (Xyl) from UDP-Xyl to the specific serine residues on the core proteins of PGs by xylosyltransferase (XylT) encoded by XYLT1 and XYLT2 in the endoplasmic reticulum (Table 1; Götting et al., 2000; Pönighaus et al., 2007). Thereafter, galactosyltransferase-I (GalT-I), galactosyltransferase-II (GalT-II), and glucuronosyltransferase-I (GlcAT-I), which are encoded by B4GALT7, B3GALT6, and B3GAT3, respectively, transfer two galactoses (Gals) and a glucuronic acid (GlcA) from UDP-Gal and UDP-GlcA to the Xyl residues in the Golgi apparatus (Figure 2A; Kitagawa et al., 1998; Almeida et al., 1999; Okajima et al., 1999; Bai et al., 2001). Modifications in the linker region tetrasaccharide, including the 2-O-phosphorylation of the Xyl residue as well as sulfation at the C-6 position of the first Gal and at C-4 or C-6 of the second Gal residue, have been reported (Sugahara and Kitagawa, 2000). These modifications are catalyzed by GAG-Xyl 2-O-kinase, Xyl 2-O-phosphatase, and Gal-6-O-sulfotransferase encoded by FAM20B, ACPL2, and CHST3, respectively (Kitagawa et al., 2008; Koike et al., 2009, 2014), and affect the glycosyltransferase reactions of GalT-I and GlcAT-I in vitro, which may regulate the biosynthesis of GAG chains (Gulberti et al., 2005; Tone et al., 2008).
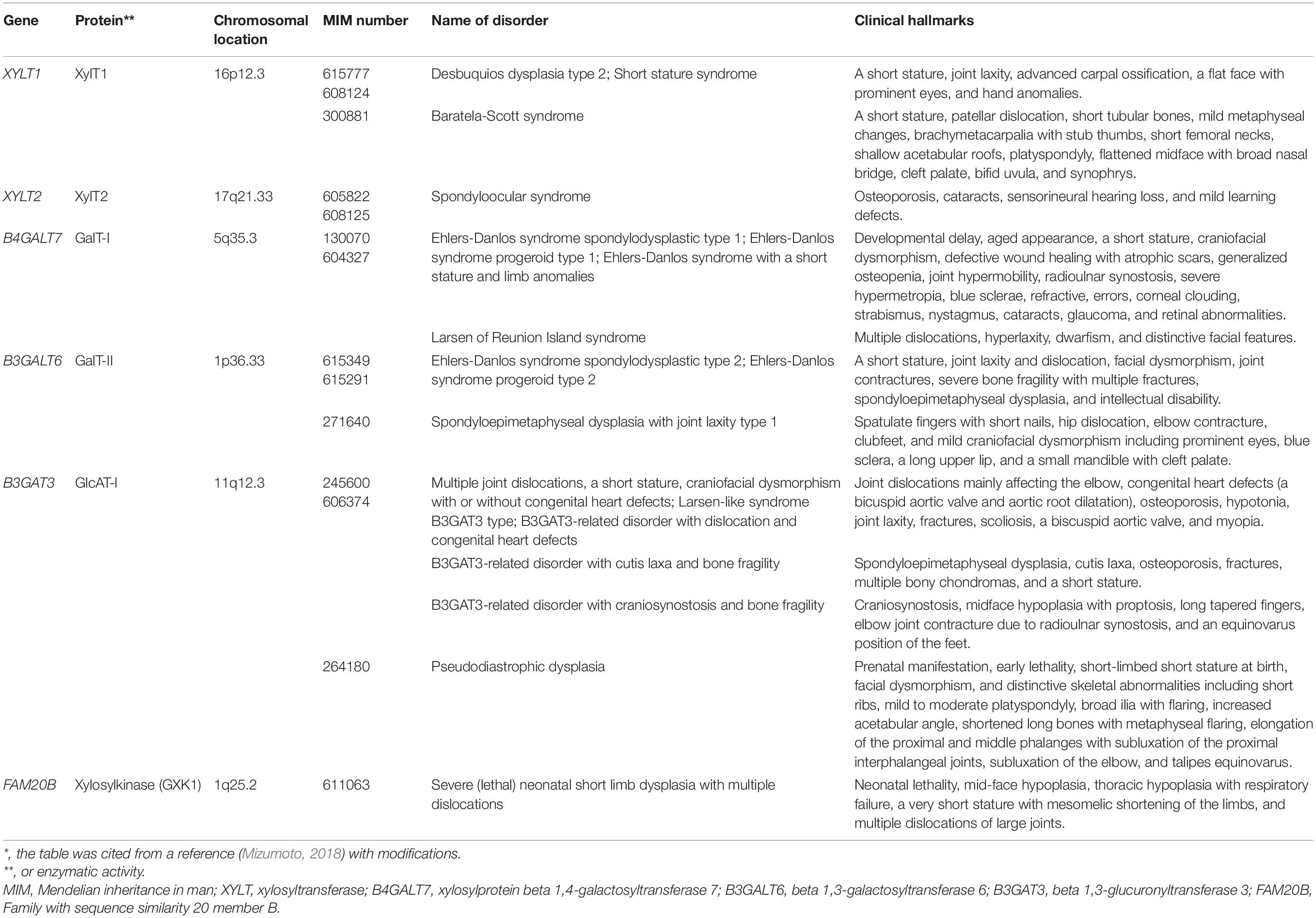
Table 1. Congenital disorders of GAG-linker region tetrasaccharide deficiency*.
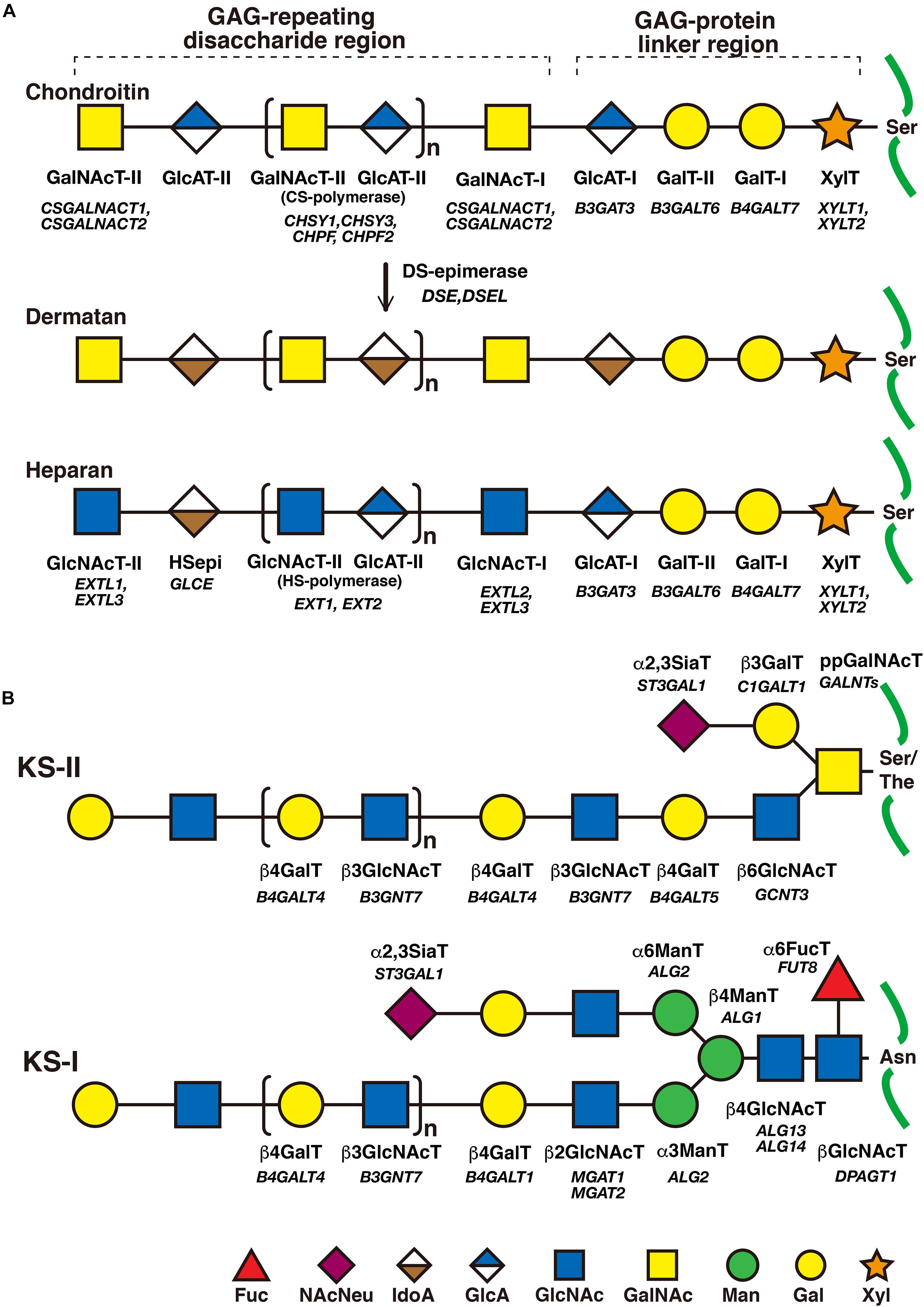
Figure 2. The biosynthetic assembly of GAGs. (A) Schematic presentation of the biosynthesis of CS, DS, and HS. Following the synthesis of specific core proteins of proteoglycans, the common GAG-linker region tetrasaccharide (CS, DS, and HS except for KS), GlcA-Gal-Gal-Xyl-O-, is built up by the consecutive addition of each sugar moiety from the respective UDP-sugar, including UDP-Xyl, UDP-Gal, or UDP-GlcA, by XylT, GalT-I, GalT-II, and GlcAT-I. The first GalNAc residue is then transferred to the GlcA residue at the non-reducing end in the linker region by GalNAcT-I, which initiates the construction of the chondroitin backbone. The repeating disaccharide region (chondroitin backbone of CS), [-3GalNAcβ1–4GlcAβ1-]n, is elongated by alternate additions of GlcA and GalNAc residues from UDP-GlcA and UDP-GalNAc catalyzed by CS-GlcAT-II and GalNAcT-II activities, respectively. After or during the formation of the chondroitin backbone, DSE converts GlcA into IdoA by epimerizing the C-5 carboxy group, resulting in the formation of the dermatan backbone of DS, [-3GalNAcβ1–4IdoUAα1-]n. Alternatively, the first GlcNAc residue is transferred to the GlcA residue at the non-reducing end in the linker region by GlcNAcT-I, which initiates the construction of the heparan backbone. The repeating disaccharide region (heparan backbone of HS), [-4GlcNAcα1–4GlcAβ1-]n, is elongated by alternate additions of the GlcA and GlcNAc residues from UDP-GlcA and UDP-GlcNAc catalyzed by HS-GlcAT-II and GlcNAcT-II activities, respectively. (B) Schematic presentation of the biosynthesis of KS. After the formation of the linkage region of KS, O- and N-linked oligosaccharides, the repeating disaccharide region, [-4GlcNAcβ1–3Galβ1-]n, is elongated by alternate additions of the Gal and GlcNAc residues from UDP-Gal and UDP-GlcNAc catalyzed by β4GalT and β3GlcNAcT activities, respectively. Each enzyme and its coding gene are described under the respective sugar symbols from the top of each line.
The CS-repeating disaccharide region, [GlcA–N-acetyl-galactosamine (GalNAc)]n, is constructed by chondroitin synthases (CHSYs) including CHSY1, CHSY3, chondroitin polymerizing factor (CHPF), CHPF2, CS N-acetylgalactosaminyltransferase 1 (CSGALNACT1), and CSGALNACT2 using UDP-GlcA and UDP-GalNAc as donor substrates in the Golgi apparatus (Figure 2A and Table 2; Kitagawa et al., 2001, 2003; Uyama et al., 2002, 2003; Izumikawa et al., 2007, 2008).
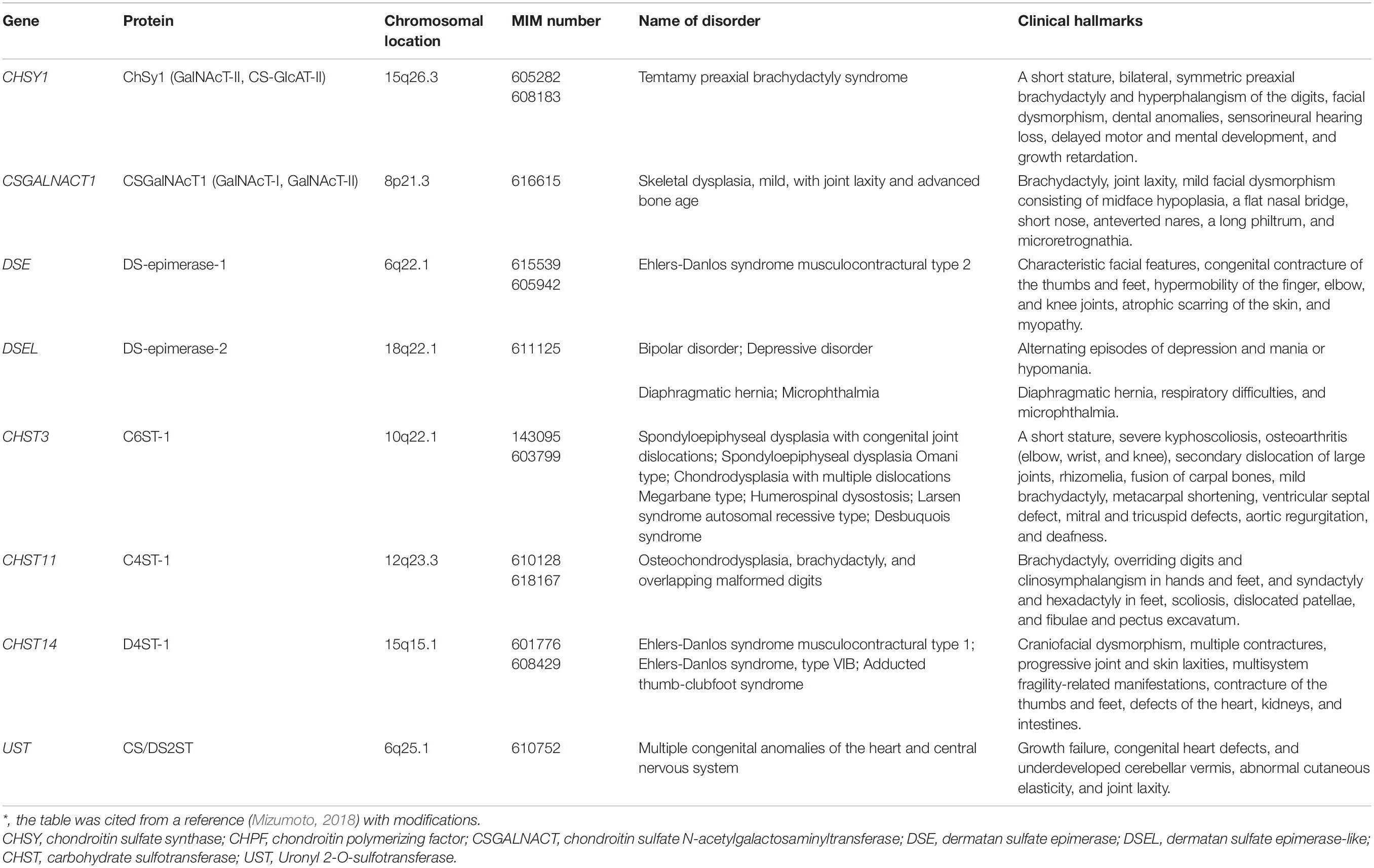
Table 2. Congenital disorders of CS and DS deficiencies*.
The DS-repeating disaccharide region, [iduronic acid (IdoA)–GalNAc]n, is formed by the epimerization of the C-5 position of GlcA residues in a chondroitin as a precursor molecule (Figure 2A; Maccarana et al., 2006; Pacheco et al., 2009).
The repeating disaccharide region of HS, [GlcA-GlcNAc]n, is assembled by exostosin (EXT) family proteins such as EXT1, EXT2, EXT-like 1 (EXTL1), EXTL2, and EXTL3 using UDP-GlcA and UDP-GlcNAc as donor substrates in the Golgi apparatus (Figure 2A and Table 3; Lind et al., 1998; McCormick et al., 1998; Kitagawa et al., 1999; Kim et al., 2001, 2003). After the formation of these polymers, various sulfotransferases transfer a sulfate group to the respective hydroxy and/or amino group in CS, DS, and HS, which gives rise to functional domains to interact with a wide range of proteins (Figure 3 and Tables 2, 3; Kusche-Gullberg and Kjellén, 2003).

Table 3. Congenital disorders of HS deficiency*.
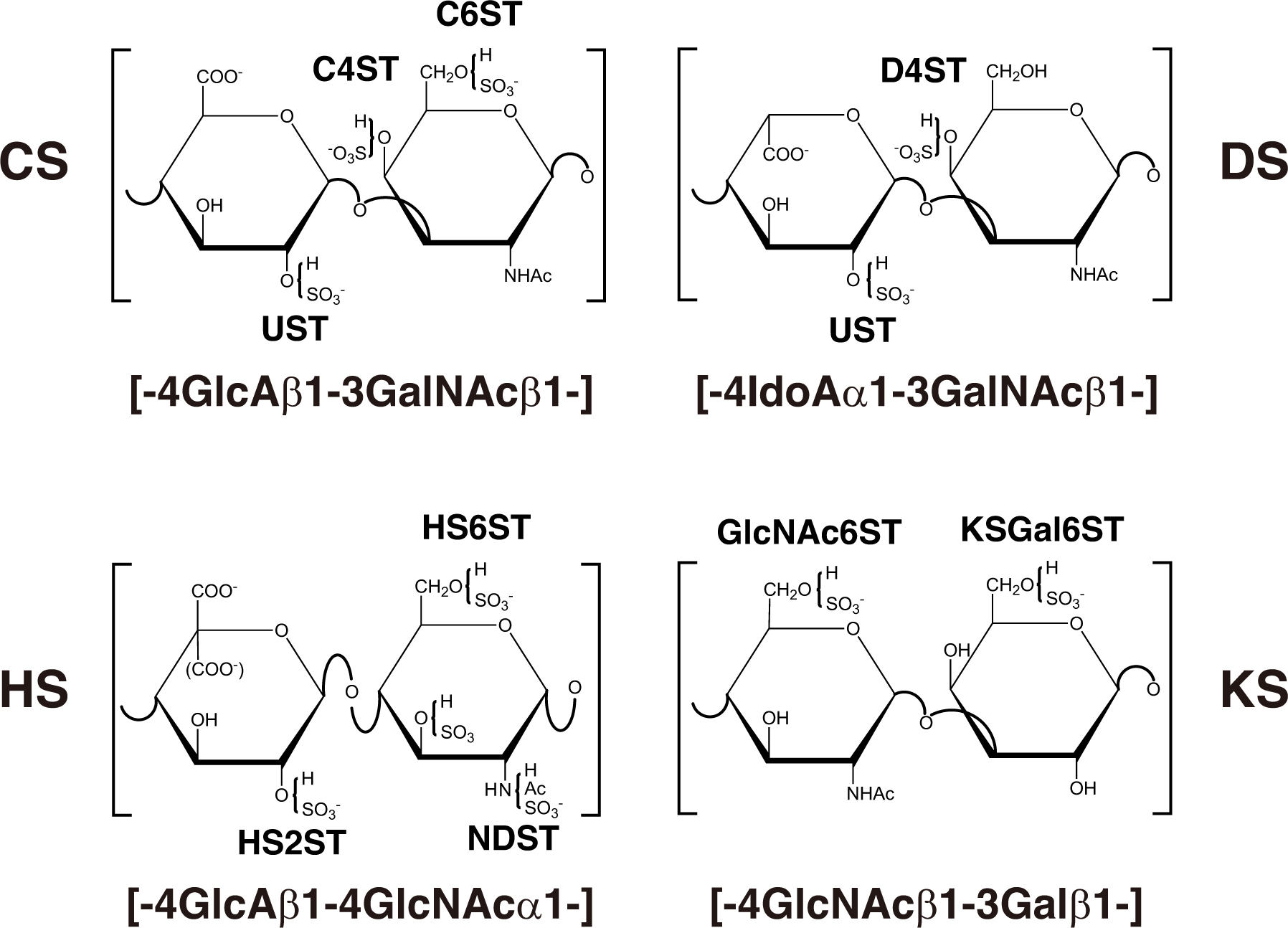
Figure 3. Typical repeating disaccharide units in GAGs and the respective sulfotransferase. The CS backbone consists of GlcA and GalNAc, whereas DS is a stereoisomer of CS that includes IdoA instead of GlcA. HS consist of uronic acids (IdoA and GlcA) and GlcNAc/GlcNH2 residues with varying proportions of IdoA and GlcA. These sugar residues can be esterified by sulfate at various positions as indicated in the figure by action of the respective sulfotransferase, which transfers the sulfate group from the sulfate donor 3′-phosphoadenosine 5′-phosphosulfate (PAPS) to the respective hydroxygroup of each residue.
The repeating disaccharide region of KS, [Gal-GlcNAc]n, is assembled by GalT and GlcNAcT encoded by B4GALT4 and B3GNT7, respectively (Figure 2B; Seko et al., 2003; Seko and Yamashita, 2004; Kitayama et al., 2007). Further modification by sulfation on Gal and GlcNAc residues occurs by the action of KS Gal 6-O-sulfotransferase and GlcNAc 6-O-sulfotransferase encoded by CHST1 and CHST5 as well as CHST6, respectively (Figure 3 and Table 4; Fukuta et al., 1997; Akama et al., 2001, 2002; Narentuya et al., 2019).

Table 4. Congenital disorders of KS deficiency.
Proteoglycan Linkeropathy
The disturbance of construction of the linker region tetrasaccharide, -O-Xyl-Gal-Gal-GlcA, in CS, DS, and HS by mutations in XYLT1, XYLT2, B4GALT7, B3GALT6, and B3GAT3 causes the hereditary disease “Proteoglycan linkeropathy,” which is characterized by abnormalities in connective tissue, bone, skin, and heart (Table 1). Proteoglycan linkeropathy, designated by Dr. Shiro IKEGAWA, is a collective term for diverse connective tissue disorders caused by mutations in these genes (Nakajima et al., 2013). Nosology and classification of skeletal disorders and Ehlers-Danlos syndrome (EDS) are referred to in the Nosology and classification of genetic skeletal disorders: 2019 revision and 2017 International Classification of the Ehlers–Danlos syndromes, respectively (Malfait et al., 2017, 2020; Mortier et al., 2019).
Disorders Caused by Mutations of XYLT1 and XYLT2
XYLT1 transfers a Xyl residue from UDP-Xyl to the serine residue on the specific core proteins of PGs (Figure 2A; Götting et al., 2000). Desbuquois dysplasia type 2 is an autosomal recessive disorder that is characterized by severe pre- and postnatal growth retardation, a short stature, joint laxity, the dislocation of large joints, and a flat face with prominent eyes (Table 1; Desbuquois et al., 1966), and is caused by mutations in XYLT1 (Bui et al., 2014; Silveira et al., 2016; Al-Jezawi et al., 2017; Guo et al., 2017a). Fibroblasts from patients (p.Arg147∗) show reduced CS rather than HS (Bui et al., 2014), which may indicate that the loss-of-function in XYLT1 affects the biosynthesis of CS rather than HS.
Autosomal recessive short stature syndrome was also caused by a homozygous mutation (p.Arg481Trp) in XYLT1 (Schreml et al., 2014). Although XYLT1 in control cells was located in the Golgi apparatus, p.Arg481Trp-XYLT1 was diffusely distributed throughout the cytoplasm with partial localization to the Golgi apparatus in patients’ fibroblasts (Schreml et al., 2014). Furthermore, decorin-PG from the fibroblasts of a patient was partially deglycanated (Schreml et al., 2014).
Baratela-Scott syndrome is an autosomal recessive disorder characterized by a short stature, patellar dislocation, short tubular bones, mild metaphyseal changes, brachymetacarpalia with stub thumbs, short femoral necks, shallow acetabular roofs, platyspondyly, a flattened midface with broad nasal bridge, cleft palate, bifid uvula, and synophrys (Baratela et al., 2012). This disease is caused by homozygous mutations in XYLT1, homozygous hypermethylation of exon 1 in XYLT1 that is caused by a GGC repeat expansion in the XYLT1 promoter region, or a heterozygous 3.1 Mbp deletion on 16p13 encompassing XYLT1 with hypermethylation (LaCroix et al., 2019).
Homozygous mutations in XYLT2 cause spondyloocular syndrome that is characterized by retinal detachment, amblyopia, nystagmus, hearing loss, heart septal defects, bone fragility, and mild learning difficulties (Table 1; Munns et al., 2015; Taylan et al., 2016, 2017; Umair et al., 2018). The levels of GAGs and XYLT activity in the dermal fibroblasts of a patient were lower than those of healthy subjects (Munns et al., 2015).
The different clinical manifestations between Desbuquois skeletal dysplasia type 2 and spondyloocular syndrome may indicate that XYLT1 and XYLT2 do not compensate for each other, and that the serine residue(s) of the respective core protein as a substrate for each XYLT and spatiotemporal expression may be distinct between XYLT1 and XYLT2.
Severe (Lethal) Neonatal Short Limb Dysplasia With Multiple Dislocations Caused by Mutations in FAM20B
FAM20B encodes a kinase, which phosphorylates a Xyl residue in the linker region tetrasaccharide of GAGs (Koike et al., 2009). Compound heterozygous mutations (p.Thr59Alafs∗19/p.Asn347Metfs∗4) in FAM20B cause neonatal short limb dysplasia resembling Desbuquois dysplasia (Table 1; Kuroda et al., 2019). The clinical hallmarks are mid-face hypoplasia, thoracic hypoplasia with respiratory failure, a very short stature with mesomelic shortening of the limbs, multiple dislocations of large joints, and preaxial digital hypoplasia as well as syndactyly, being similar to Desbuquois dysplasia (Kuroda et al., 2019).
Ehlers-Danlos Syndrome Spondylodysplastic Type 1 and Larsen Syndrome Reunion Island Type Caused by Mutations in B4GALT7
B4GALT7 encodes GalT-I, which transfers the second Gal residue in the linker region tetrasaccharide from UDP-Gal to the serine-O-Xyl (Figure 2A; Almeida et al., 1999; Okajima et al., 1999). Compound heterozygous and homozygous mutations in B4GALT7 cause Ehlers-Danlos syndrome (EDS) spondylodysplastic type 1 (Table 1; Almeida et al., 1999; Okajima et al., 1999; Malfait et al., 2017, 2020). Patients show various clinical symptoms including an aged appearance, craniofacial dysmorphism, hypermobile joints, generalized osteopenia, a short stature, hypotonic muscles, defective wound healing, global developmental delays, and scoliosis (Almeida et al., 1999; Guo et al., 2013; Arunrut et al., 2016; Salter et al., 2016; Ritelli et al., 2017; Mihalic Mosher et al., 2019). Skin fibroblasts of patients were shown to be defective for the DS side chain of DS-PGs, decorin and biglycan, and show a marked reduction in GalT-I activity in vitro (Quentin et al., 1990; Seidler et al., 2006). Furthermore, fibroblasts from patients exhibited reduced levels of sulfation on HS chains (Götte et al., 2008). Hence, various clinical manifestations in EDS spondylodysplastic type 1 might partially result from defects in not only DS, but also HS. Namely, the reasons why these patients with the same B4GALT7 gene mutations exhibited the wide range of symptoms might be due to the distinct mutational positions in B4GALT7, resulting in the different influence of enzymatic activity and/or intracellular localization of mutant enzymes and in the immature GAGs produced by the mutant enzymes.
Larsen syndrome Reunion Island type is caused by a homozygous mutation in B4GALT7 (Table 1; Cartault et al., 2015). The characteristic symptoms of Larsen syndrome are congenital large-joint dislocations including the hip, elbow, and knee, craniofacial abnormalities, and foot deformities (Larsen et al., 1950). Patients showed similar hallmarks to EDS spondylodysplastic type 1 caused by a mutation in B4GALT7, including dwarfism, distinctive facial features, and hyperlaxity, in addition to the classical symptoms of Larsen syndrome (Cartault et al., 2015). Thus, EDS spondylodysplastic type 1 and Larsen syndrome Reunion Island type may share clinical spectra.
Ehlers-Danlos Syndrome Spondylodysplastic Type 2 and Spondyloepimetaphysial Dysplasia With Joint Laxity Type 1 Caused by Mutations in B3GALT6
B3GALT6 encodes GalT-II, which transfers the third Gal residue in the linker region tetrasaccharide from UDP-Gal to serine-O-Xyl-Gal (Figure 2A; Bai et al., 2001). Compound heterozygous mutations in B3GALT6 cause EDS spondylodysplastic type 2, which is characterized by kyphoscoliosis, clubfeet, elbow malalignment, muscle hypotonia, wrinkled skin, and a characteristic facial appearance (Table 1; Malfait et al., 2013, 2020; Nakajima et al., 2013). Skin fibroblast cultures from patients produced less CS, DS, and HS than those from control cells (Malfait et al., 2013).
Spondyloepimetaphyseal dysplasia with joint laxity type 1 is also caused by compound heterozygous mutations in B3GALT6 (Table 1; Nakajima et al., 2013; Ritelli et al., 2014; Vorster et al., 2015; Alazami et al., 2016). Spondyloepimetaphyseal dysplasia with joint laxity type 1 is characterized by kyphoscoliosis, joint laxity, hip dislocation, elbow contracture, platyspondyly, clubfeet, and craniofacial dysmorphisms (Beighton et al., 1984). The recombinant mutant enzymes showed a significantly lower GalT-II activity than wild-type B3GALT6 (Nakajima et al., 2013). Furthermore, the production of HS and CS/DS in cultured lymphoblastoid cells from patients was shown to be lower and higher, respectively, than that in normal cells (Nakajima et al., 2013). In addition, the levels of HS and HS-PG, perlecan, were markedly lower in cultured fibroblasts from patients than those from normal subjects, whereas the levels of CS-PG and DS-PG, versican and decorin, respectively, in the fibroblasts of patients corresponded to the fibroblasts of normal subjects (Ritelli et al., 2014). These findings suggest that the causative mutations of spondyloepimetaphyseal dysplasia with joint laxity type 1 may affect the biosynthesis of only HS, and not CS or DS, which results in the skeletal and joint hallmarks of both disorders.
Disorders Caused From B3GAT3 Mutations
B3GAT3 encodes GlcAT-I, which transfers the 4th GlcA residue in the linker region tetrasaccharide, Xyl-Gal-Gal-GlcA, from UDP-GlcA to the serine-O-Xyl-Gal-Gal (Figure 2A; Kitagawa et al., 1998). Homozygous mutations in B3GAT3 cause multiple joint dislocations, a short stature, kyphoscoliosis, craniofacial dysmorphism, and heart defects including bicuspid aortic valve and aortic root dilatation (Table 1; Baasanjav et al., 2011; von Oettingen et al., 2014; Budde et al., 2015). GlcAT-I activity and the levels of CS, DS, and HS in fibroblasts from these patients were significantly lower than those in the fibroblasts of control cells (Baasanjav et al., 2011; Budde et al., 2015). Other homozygous mutations in B3GAT3 caused distinct hallmarks such as a short stature, spondyloepimetaphyseal dysplasia, osteoporosis, fractures, multiple bony chondromas, cutis laxa, blue sclerae, glaucoma, bilateral radio-ulnar synostosis, severe osteopenia, bilateral clubfeet, atrial and ventricular septal defects, hypertelorism, a small chest, diaphragmatic hernia, arachnodactyly, hypotonia, hearing loss, and perinatal cerebral infarction with bilateral supra- and infratentorial subdural hematomas (Jones et al., 2015; Alazami et al., 2016). Moreover, a patient with the compound heterozygous mutation in B3GAT3 exhibited severe osteoporosis, fractures, scoliosis, joint laxity, hypotonia, a bicuspid aortic valve, and myopia (Job et al., 2016).
Compound heterozygous mutation in B3GAT3 causes pseudodiastrophic dysplasia, which is associated with prenatal manifestation and early lethality (Table 1; Byrne et al., 2020). Pseudodiastrophic dysplasia is characterized by a short stature at birth, facial dysmorphism, and distinctive skeletal abnormalities including short ribs, platyspondyly, broad ilia with flaring, an increased acetabular angle, shortened long bones with metaphyseal flaring, elongation of the proximal and middle phalanges with subluxation of the proximal interphalangeal joints, subluxation of the elbow, and talipes equinovarus (Eteson et al., 1986; Fischetto et al., 1997; Yap et al., 2016). GlcAT-I activity of fibroblasts from a patient was markedly lower than that from healthy control cells (Byrne et al., 2020). Accordingly, the levels of CS, DS, and HS from the patient cells were significantly decreased compared with control cells (Byrne et al., 2020). Thus, this mutation, p.Arg169Trp/Arg225X, may be a complete loss of function of B3GAT3, which is responsible for the most severe and lethal manifestation of skeletal dysplasia.
Yauy et al. (2018) proposed the classification of B3GAT3-related disorders based on the diverse symptoms of patients with distinct mutations in B3GAT3 (Table 1): (1) “B3GAT3-related disorder with joint dislocation and congenital heart defect,” which is similar to Larsen syndrome; (2) “B3GAT3-related disorder with craniosynostosis and bone fragility,” which is similar to Antley-Bixler and Shprintzen-Goldberg syndromes; (3) “B3GAT3-related disorder with cutis laxa and bone fragility,” which is similar to geroderma osteodysplastica syndrome; and (4) “B3GAT3-related disorder with intermediate phenotype,” which is similar to Larsen syndrome and Antley-Bixler/Shprintzen-Goldberg syndromes (Yauy et al., 2018). Patients with a broad spectrum of hallmarks caused by mutations in the same B3GAT3 may show various degree of affected biosynthesis of CS, DS, and HS, which is consistent with other linkeropathies. Further analyses using cells from patients with the four types of mutations in B3GAT3 are required to understand the underlying pathogenic mechanisms.
Congenital Disorders of Chondroitin Sulfate Deficiency
Mild Skeletal Dysplasia With Joint Laxity and Advanced Bone Age by Mutations in CSGALNACT1
CSGALNACT1 encodes N-acetylgalactosaminyltransferase-I (GalNAcT-I) and GalNAcT-II, which transfer the 5th GalNAc and subsequent GalNAc residues (7, 9, 11th- - -) from UDP-GalNAc to serine-O-Xyl-Gal-Gal-GlcA and [GalNAc-GlcA]n, respectively (Figure 2A; Uyama et al., 2002, 2003). Homozygous or compound heterozygous mutations in CSGALNACT1 cause mild skeletal dysplasia, joint laxity, a short stature with an advanced bone age, facial dysmorphism, and mild language delay (Table 2; Vodopiutz et al., 2017; Mizumoto et al., 2020). GalNAcT activity was significantly reduced in fibroblasts from patients. In addition, the amount of CS was decreased in patients’ fibroblasts compared with control fibroblasts (Mizumoto et al., 2020).
Temtamy Preaxial Brachydactyly Syndrome Caused by Mutations in CHSY1
CHSY1 encodes a dual glycosyltransferase, which functions as both GalNAcT-II and CS-GlcAT-II, transferring GalNAc and GlcA residues from UDP-GalNAc and UDP-GlcA, respectively (Figure 2A; Kitagawa et al., 2001, 2003; Izumikawa et al., 2007, 2008). Homozygous or compound heterozygous mutations in CHSY1 cause Temtamy preaxial brachydactyly syndrome, which is characterized by preaxial brachydactyly, hyperphalangism of digits, facial dysmorphism, dental anomalies, sensorineural hearing loss, delayed motor and mental development, and growth retardation (Table 2; Temtamy et al., 1998, 2012; Li et al., 2010; Tian et al., 2010; Sher and Naeem, 2014). The level of CS in a skin section from a patient was lower than that in control skin (Tian et al., 2010). Furthermore, the up-regulated expressions of JAG1 and HES1, whose gene products are ligands of NOTCH and induced by NOTCH signaling, respectively, have been observed (Tian et al., 2010). NOTCH signaling is involved in appendicular skeletal development (Tian et al., 2010). Therefore, a decrease in CS by mutations in CHSY1 may influence changes in the expression of JAG1 and HES1, thereby leading to brachydactyly.
Spondyloepiphyseal Dysplasia With Congenital Joint Dislocations, Larsen Syndrome, and Humero-Spinal Dysostosis Caused by Mutations in CHST3
CHST3 encodes chondroitin 6-O-sulfotransferase 1 (C6ST1), which transfers a sulfate group from 3′-phosphoadenosine 5′-phosphosulfate (PAPS) to the C-6 hydroxy group of GalNAc residues in CS chains (Figure 3; Fukuta et al., 1998). Homozygous mutations in CHST3 cause spondyloepiphyseal dysplasia with congenital joint dislocations, which is characterized by a short stature, severe kyphoscoliosis, mild brachydactyly, arthritic joints, joint dislocation, rhizomelia, fusion of the carpal bones, metacarpal shortening, osteoarthritis of the elbow, deafness, and ventricular septal, mitral, and/or tricuspid defects (Table 2; Rajab et al., 2004; Thiele et al., 2004; van Roij et al., 2008; Tuysuz et al., 2009; Tanteles et al., 2013; Muys et al., 2016; Waryah et al., 2016; Srivastava et al., 2017). Loss of C6ST activity as well as the decrease of 6-O-sulfated CS were demonstrated in skin fibroblasts from patients (Thiele et al., 2004; van Roij et al., 2008). These findings suggest that chondroitin 4-O-sulfotransferase (C4ST) and/or 4-O-sulfation of CS cannot compensate for the loss-of-function in C6ST1.
Homozygous or heterozygous mutations in CHST3 cause Larsen syndrome and humero-spinal dysostosis, which are characterized by joint and knee dislocations, bilateral clubfeet, and elbow joint and spinal dysplasias (Table 2; Hermanns et al., 2008). Although there are a wide range of diagnoses for multiple dislocations in a newborn, Larsen syndrome is one of the most common entities (Hermanns et al., 2008). Hence, the different pathological diagnoses by mutations in CHST3 may be due to age-related descriptions of the same conditions (Hermanns et al., 2008). Twenty-four patients from 23 families with mutations in CHST3 were diagnosed with Larsen syndrome (15 families), humero-spinal dysostosis (four cases), chondrodysplasia with multiple dislocations (Megarbane type; two cases), Desbuquois syndrome (one case), and spondyloepiphyseal dysplasia (one case) (Unger et al., 2010).
Osteochondrodysplasia, Brachydactyly, and Overlapping Malformed Digits Caused by Mutations in CHST11
CHST11 encodes chondroitin 4-O-sulfotransferase 1 (C4ST1), which transfers a sulfate group from PAPS to the C-4 hydroxy group of GalNAc residues in CS chains (Figure 3; Hiraoka et al., 2000; Yamauchi et al., 2000). Homozygous in-frame deletion of 15 nucleotides in CHST11, which results in the deletion of amino acids (Lys156-Asn160), causes limb malformations including brachydactyly, overriding digits, clino-symphalangism, syndactyly, and hexadactyly, and skeletal defects including scoliosis, dislocation of patellae and fibulae, and pectus excavatum (Table 2; Shabbir et al., 2018). Biochemical analyses using a recombinant enzyme as well as patients’ cells have not been conducted to our knowledge.
The deletion of a 55-kb region within chromosome 12q23 that encompasses a part of CHST11 and an embedded microRNA, MIR3922, causes defects of the digits of the upper and lower limbs and malignant lymphoproliferative disease (Chopra et al., 2015). Although the digital malformations are consistent with the hallmarks of the proband who was homozygous for an in-frame deletion of 15 nucleotides in CHST11, as described above, the lymphoproliferative disorder is not. Hence, it remains unclear whether the deletion in CHST11 contributes to T-cell lymphoproliferative disorder. It should be noted that compound heterozygous variants in MBD3L5 and PRND and homozygous missense variants in EPHA10 and OLA1 were also identified in the proband (Chopra et al., 2015). Further analysis of CHST11 and/or 4-O-sulfation of CS in the differentiation as well as proliferation of lymphocytes is required.
Congenital Disorders of Dermatan Sulfate Deficiency
Ehlers-Danlos Syndrome Musculocontractural Type 2 Caused by Mutations in DSE
DSE encodes dermatan sulfate epimerase (DSE), which converts GlcA into IdoA by epimerizing the C-5 carboxy group of GlcA residues in the repeating disaccharide region of the chondroitin precursor chain, [GlcA-GalNAc]n, to form the disaccharide region of DS, [IdoA-GalNAc]n (Figure 2A; Maccarana et al., 2006). The homozygous mutations in DSE cause EDS musculocontractural type 2 (Table 2; Müller et al., 2013; Syx et al., 2015; Lautrup et al., 2020; Malfait et al., 2020). The clinical hallmarks of the patients exhibited characteristic facial features including hypertelorism, blue sclera, midfacial hypoplasia, contracture of the thumbs and feet, atrophic scars on the skin, and hypermobility of the finger, elbow, and knee joints (Müller et al., 2013; Syx et al., 2015). DSE activity of the fibroblasts from the patient was markedly decreased compared with healthy control cells, which resulted in a lower level of DS, being accompanied by an increase in the level of CS (Müller et al., 2013). These findings indicate that mutations in DSE may cause a decrease in DS, leading to EDS musculocontractural type 2. In order to elucidate the underlying pathogenic mechanism of EDS, it will be necessary to analyze the signaling pathway(s) regulated by DS as well as functional protein(s) interacting with DS chains of DS-PGs.
Bipolar Disorder and Diaphragmatic Hernia Caused by Mutations in DSEL
DSEL encodes dermatan sulfate epimerase-like (DSEL)/DSE2, which is a homolog of DSE and can catalyze the reaction converting GlcA into IdoA (Pacheco et al., 2009). The 113 probands with bipolar disorder characterized by alternating episodes of depression were identified as caused by various homozygous as well as heterozygous mutations in DSEL, such as the substitution of adenine to guanine in the 5′-non-coding region 546 bp upstream of the coding region, p.Val287Ile, p.Pro673Ser, p.Tyr730Cys, p.Pro942Ser, and p.Ile1113Met, in DSEL (Table 2; Goossens et al., 2003). Moreover, the recurrent early-onset major depressive disorder was caused by single nucleotide polymorphism (SNP), rs17077540, which was detected 75 kbp upstream of DSEL (Shi et al., 2011). DSEL but not DSE was demonstrated to be expressed predominantly in the brain (Table 2; Nakao et al., 2000; Maccarana et al., 2006; Akatsu et al., 2011; Bartolini et al., 2012). Functional analyses of DSEL using neuronal cells and model organisms such as the mouse and zebrafish, may lead to elucidation of the pathogenic mechanism of this disorder.
The deletion of 2.7 Mbp at chromosome 18q22.1, which contains three genes, CDH19, DSEL, and TXNDC10/TMX3 encoding cadherin 19, DSEL/DSE2, and thioredoxin domain-containing protein10/thioredoxin related transmembrane protein 3, respectively, causes diaphragmatic hernia (Zayed et al., 2010). Furthermore, the heterozygous substitution, c.1515G > A (p.Met14Ile), in DSEL also results in the disease (Table 2; Zayed et al., 2010). Thus, DSEL and/or DS might contribute to the development of the diaphragm, although the possibility of the involvement of CDH19 and TXNDC10/TMX3 in this disorder cannot be excluded.
Ehlers-Danlos Syndrome Musculocontractural Type 1 Caused by Mutations in CHST14
CHST14 encodes dermatan 4-O-sulfotransferase 1 (D4ST1), which transfers a sulfate group from PAPS to the C-4 hydroxy group of GalNAc residues in DS chains (Figure 3; Evers et al., 2001; Mikami et al., 2003). Homozygous, complex homozygous, or compound heterozygous mutations in CHST14 cause EDS musculocontractural type 1 (Table 2; Dündar et al., 2009; Malfait et al., 2010, 2020; Miyake et al., 2010), which is characterized by kyphoscoliosis, muscular hypotonia, hyperextensible, thin, and bruisable skin, atrophic scarring, joint hypermobility, multiple joint contracture, characteristic craniofacial features, joint laxities, and recurrent dislocations (Kosho et al., 2005, 2010; Dündar et al., 2009; Malfait et al., 2010, 2020; Miyake et al., 2010). Fibroblasts from probands with the mutations in CHST14 showed markedly lower D4ST activity than that from the respective control (Miyake et al., 2010). Surprisingly, CS, instead of non-sulfated DS (dermatan), was produced on the decorin core protein from skin fibroblasts of probands (Miyake et al., 2010). Collagen fibrils were dispersed in the patient dermis; in contrast, they were regularly and tightly assembled in the healthy controls (Hirose et al., 2019). The affected GAG side chain on decorin was altered from DS to CS. Since CS has a linear conformation, the CS side chain on decorin stretched from the outer surface of collagen fibrils to adjacent fibrils (Hirose et al., 2019). In contrast, DS has a curved conformation, and the DS side chain on decorin maintained close contact with attached collagen fibrils. Therefore, alteration of the side chain on decorin from DS to CS caused by loss-of-function in CHST11 results in the spatial disorganization of collagen networks, which may disrupt the appropriate structure of GAG side chains surrounding collagen fibrils (Hirose et al., 2019). Functional analyses of CHST14 using knockout mice are ongoing (Yoshizawa et al., 2018; Hirose et al., 2021; Nitahara-Kasahara et al., submitted).
Homozygous mutations or compound heterozygous mutation in CHST14 caused adducted thumb-clubfoot syndrome in 11 patients from 4 families (Table 2; Dündar et al., 2009). This syndrome is characterized by congenital contracture of the thumbs and feet, joint instability, a typical facial appearance, facial clefting, coagulopathy, thin and translucent skin, connective tissue fragility with aging, and defects in the heart, kidneys, or intestines (Dundar et al., 1997; Sonoda and Kouno, 2000; Janecke et al., 2001). Five out of eleven probands with adducted thumb-clubfoot syndrome died in early infancy or childhood (Dundar et al., 1997). Malfait et al. (2010) proposed to unify EDS and the adducted thumb-clubfoot syndrome, caused by mutations in CHST14, as “musculocontractural EDS,” which was adopted in the 2017 International Classification of the Ehlers–Danlos syndromes as “EDS musculocontractural type 1” (Table 2; Malfait et al., 2017).
Congenital Disorders of Heparan Sulfate Deficiency
Multiple Osteochondromas Caused by EXT1 or EXT2 Mutations
EXT1 encodes exostosin 1 (EXT1), a dual glycosyltransferase (GlcNAcT-II and HS-GlcAT-II), which transfers the GlcNAc and GlcA residues from UDP-GlcNAc and UDP-GlcA, respectively, to [GlcA-GlcNAc]n (Figure 2A; Lind et al., 1998; McCormick et al., 1998). EXT2 also encodes a dual glycosyltransferase, which has activities of both GlcNAcT-II and HS-GlcAT-II (Lind et al., 1998; McCormick et al., 1998). The hetero-dimeric complex of EXT1 and EXT2 shows polymerization activity to construct repeating disaccharides in HS, [GlcA-GlcNAc]n, by alternating addition of GlcA and GlcNAc from UDP-GlcA and UDP-GlcNAc, respectively, in vitro (Figure 2A; Kim et al., 2003). Heterozygous mutations in EXT1 or EXT2 cause autosomal dominant disorder, osteochondroma, which is characterized by cartilaginous capped bony outgrowths located at the growth plates of long bones, a short stature, camptomelic dysplasia including the forearm and crus, early-onset osteoarthritis, and malignant transformation in approximately 1% of patients (Table 3; Solomon, 1963; Hennekam, 1991; Ahn et al., 1995; Stickens et al., 1996; Philippe et al., 1997; Wuyts et al., 1998; Bovée et al., 1999; Wuyts and Van Hul, 2000; Cheung et al., 2001; Faiyaz-Ul-Haque et al., 2004). To our knowledge, 436 and 223 mutations in EXT1 and EXT2, respectively, have been identified to date.
Langer-Giedion syndrome (trichorhinophalangeal syndrome type II) is an autosomal dominant disorder caused by a deletion in chromosome 8q23.3-q24.11, which includes EXT1 and TRPS1 encoding exostosin 1 and transcriptional repressor GATA binding 1, respectively (Table 3; Lüdecke et al., 1995). Langer-Giedion syndrome is characterized by multiple osteochondromas, cone-shaped epiphyses, a short stature, bulbous nose, sparse scalp hair, a long philtrum, and bushy eyebrows (Lüdecke et al., 1995). A deletion in chromosome 8q24.11 or 8q23.3-q24.13, which contains EXT1, but not TRPS1, also causes Langer-Giedion syndrome (Wuyts et al., 2002; McBrien et al., 2008; Pereza et al., 2012).
A heterozygous deletion of 2.3 Mbp in chromosome 11p11.21 containing EXT2 causes Potocki-Shaffer-syndrome that is characterized by multiple osteochondromas, developmental delays, biparietal foramina, craniofacial abnormalities, and intellectual disability (Table 3; Swarr et al., 2010; Palka et al., 2012). These hallmarks, except for multiple osteochondromas, may be caused by the lack of eight other genes in the chromosome 11p11.21.
Seizures-Scoliosis-Macrocephaly Syndrome Caused by Mutations in EXT2
The complex homozygous mutations in EXT2 cause seizures-scoliosis-macrocephaly syndrome, but not multiple osteochondromas (Table 3; Farhan et al., 2015). The seizures-scoliosis-macrocephaly syndrome is characterized by seizures, intellectual disability, hypotonia, scoliosis, macrocephaly, hypertelorism, and renal dysfunction (Farhan et al., 2015). The protein level of the mutant EXT2, p.Met87Arg/p.Arg95Cys, in fibroblasts from patients was lower than that from control fibroblasts (Farhan et al., 2015). These findings indicate that EXT2 and/or HS side chains of HS-PGs may contribute to normal brain development, possibly by regulating the signaling pathway of Wnt as well as fibroblast growth factor (FGF) and/or by controlling the assembly of extracellular matrix. Wnt and FGF signalings were shown to be affected during nervous system development in zebrafish by mutants in ext2/dackel (Lee et al., 2004; Fischer et al., 2011).
Immunoskeletal Dysplasia With Neurodevelopmental Abnormalities Caused by Mutations in EXTL3
EXTL3 encodes exostosin-like-3 (EXTL3), a dual glycosyltransferase (GlcNAcT-I and GlcNAcT-II), which transfers a GlcNAc residue from UDP-GlcNAc to the GlcA residue at the non-reducing end of the common GAG-linker region tetrasaccharide and to the GlcA residue in the repeating disaccharide region of HS chains, respectively (Figure 2A; Kim et al., 2001). The homozygous mutations in EXTL3 cause immunoskeletal dysplasia with neurodevelopmental abnormalities that is characterized by severe platyspondyly, kyphoscoliosis, pelvic distortion, constriction of the proximal femora and brachydactyly, neurological abnormalities including generalized seizures, opisthotonus, hyperreflexia, intellectual disability, nystagmus, muscular hypotonia, and a decrease in T-lymphocyte subsets as well as immunoglobulins (Table 3; Guo et al., 2017b; Oud et al., 2017; Volpi et al., 2017). Although wild-type EXTL3 from a healthy subject localized to the Golgi apparatus, p.Arg513Cys-EXTL3 from a patient did not (Oud et al., 2017). Therefore, the mislocalization of the mutant EXTL3 (p.Arg513Cys) may cause loss-of-function of EXTL3. Moreover, HS levels in the fibroblasts of patients were markedly lower than those in healthy controls (Oud et al., 2017). Up-regulation of FGF signaling as well as increase in the 2-O- and 6-O-sulfation of HS were observed in the fibroblasts of a patient with a homozygous mutation in EXTL3 (Volpi et al., 2017), being consistent with the reports that FGF signaling requires both 2-O- and 6-O-sulfation on HS. The reduced expansion of lymphohematopoietic progenitor cells and defects in the differentiation of thymic epithelial progenitor cells were also detected in the induced pluripotent stem (iPS) cells of the patient (Volpi et al., 2017). These findings suggest that EXTL3 and/or HS-PGs play roles in not only skeletal development, but also cell differentiation of the nervous and immune systems. Their functions in specific organs such as the brain and thymus are under investigation using conditional knockout mice.
Intellectual Disability Due to Autosomal Recessive State Caused by Mutations in NDST1
NDST1 encodes GlcNAc N-deacetylase/N-sulfotransferase 1 (NDST1), which has bifunctional domains including an N-deacetylase and N-sulfotransferase, acting on GlcNAc residues in HS chains (Figure 3; Hashimoto et al., 1992; Dixon et al., 1995). The homozygous mutations in NDST1 cause autosomal recessive intellectual disability, muscular hypotonia, epilepsy, and postnatal growth deficiency (Table 3; Najmabadi et al., 2011; Reuter et al., 2014). Furthermore, the compound heterozygous mutation in NDST1 causes developmental delay, a short stature, seizures, cranial nerve palsies, ataxia, severe respiratory difficulties in infancy, and distinctive facial features including a long nose and bifid uvula (Table 3; Armstrong et al., 2017). These findings suggest that NDST1 and/or N-sulfation in HS chains may contribute to development of the brain and lungs and maintenance of their functions.
Neurofacioskeletal Syndrome With or Without Renal Agenesis Caused by Mutations in HS2ST1
HS2ST1 encodes HS 2-O-sulfotransferase 1 (HS2ST1), which transfers a sulfate group from PAPS to the C-2 hydroxy group of GlcA as well as IdoA residues in HS chains (Figure 3; Kobayashi et al., 1997). Complex heterozygous mutations in HS2ST1 cause the brachydactyly of hands and feet, flexion contractures, corpus callosum agenesis or hypoplasia, intellectual disability, uni- or bilateral renal agenesis, developmental delay, and facial dysmorphism such as a coarse face, upslanted palpebral fissures, a broad nasal tip, and wide mouth (Table 3; Schneeberger et al., 2020). HS chains of skin fibroblasts from patients lacked 2-O-sulfation of GlcA/IdoA residues, resulting in an increase in N- and 6-O-sulfation of glucosamine residue (Schneeberger et al., 2020). Furthermore, decreased activation of mitogen-activated protein kinase, ERK1/2, by FGF2, was detected in the cell cultures of affected individuals (Schneeberger et al., 2020). These findings indicate that HS2ST1 and 2-O-sulfated HS are required for skeletal and renal development in humans.
Hypogonadotropic Hypogonadism With or Without Anosmia and Kallmann Syndrome Caused by Mutations in HS6ST1
HS6ST1 encodes HS 6-O-sulfotransferase 1 (HS6ST1), which transfers a sulfate group from PAPS to the C-6 hydroxy group of GlcNAc and glucosamine residues in HS chains (Figure 3; Habuchi et al., 1998, 2000). The homozygous or heterozygous mutations cause Kallmann syndrome and idiopathic hypogonadotrophic hypogonadism (Table 3; Tornberg et al., 2011). Congenital hypogonadotrophic hypogonadism is characterized by the impaired production or action of gonadotropin-releasing hormone (GnRH), which results in dysfunction of the hypothalamic-pituitary-gonadal hormone axis, leading to low testosterone levels and impaired fertility (Boehm et al., 2015; Millar et al., 2021). Furthermore, congenital hypogonadotrophic hypogonadism is a GnRH deficiency that is associated with other developmental anomalies including a cleft lip or palate, dental agenesis, ear anomalies, hearing impairment, renal agenesis, bimanual synkinesis, and skeletal anomalies (Boehm et al., 2015). Kallmann syndrome is characterized by anosmia or hyposmia in addition to the manifestations of hypogonadotrophic hypogonadism, which results from abnormal embryonic migration of GnRH neurons from their origin in the olfactory placode to the forebrain (Boehm et al., 2015). Congenital hypogonadotrophic hypogonadism and Kallmann syndrome are caused by mutations in KAL1/ANOS1, FGF8, FGF17, and FGF receptor 1 (FGFR1) (Bick et al., 1992; Dodé et al., 2003; Falardeau et al., 2008; Miraoui et al., 2013). Anosmin-1, an extracellular matrix protein, encoded by KAL1/ANOS1 enhances FGF signaling through interaction with the FGF-FGFR-HS-PG complex on the cell surface (González-Martínez et al., 2004). These findings suggest that 6-O-sulfation of GlcNAc and/or glucosamine residues in HS chains is required for their interaction with anosmin-1 and/or FGF, thereby allowing the regulation of FGF signaling.
Congenital Disorders of Keratan Sulfate Deficiency
Macular Corneal Dystrophy Caused by Mutations in CHST6
CHST6 encodes a corneal GlcNAc 6-O-sulfotransferase (C-GlcNAc6ST), which transfers a sulfate group from PAPS to the C-6 hydroxy group of GlcNAc in KS chains (Figure 3; Akama et al., 2001, 2002). Homozygous, heterozygous, or compound heterozygous mutations in CHST6 cause macular corneal dystrophy characterized by an autosomal recessive hereditary disease, progressive punctate opacities in the cornea, bilateral loss of vision (Table 4; Akama et al., 2000; El-Ashry et al., 2002; Aldave et al., 2004; Sultana et al., 2005). However, the influence of a defect in CHST6 on KS biosynthesis remains unclear. Hence, further analyses of the sulfated modification as well as level of KS are required to elucidate the underlying pathogenic mechanisms.
Congenital Disorders Caused by Deficiency of Uridine Diphosphate-Glucuronic Acid and 3′-Phosphoadenosine 5′-Phosphosulfate Synthase
Developmental and Epileptic Encephalopathy Caused by Mutations in UGDH
UGDH encodes UDP-Glc dehydrogenase (UGDH), which is an oxidoreductase that converts UDP-Glc to UDP-GlcA in the cytosol (Figure 1; Spicer et al., 1998). Compound heterozygous and homozygous mutations in UGDH cause developmental and epileptic encephalopathy, characterized by intractable epileptic seizures and developmental delay (Table 5; Freeze et al., 2015; McTague et al., 2016; Hengel et al., 2020). The recombinant mutant enzyme of UGDH, p.Ala44Val and p.Ala82Thr, mainly forms a dimer or remains a monomer species, respectively, whereas the wild-type UGDH predominantly forms a hexamer (Hengel et al., 2020). Furthermore, the values of Vmax of p.Ala44Val and p.Ala82Thr enzymes were less than 50% and at an undetermined level, respectively, compared with that of the wild-type (Hengel et al., 2020). The probands of 36 cases from 25 families showed epileptic encephalopathy with developmental delay and hypotonia (Hengel et al., 2020). These hallmarks are similar to those probands with mutations in CHSY1, EXTL3, or NDST1 (Li et al., 2010; Tian et al., 2010; Najmabadi et al., 2011; Reuter et al., 2014; Guo et al., 2017b; Oud et al., 2017; Volpi et al., 2017). Thus, UGDH and/or UDP-GlcA is responsible for not only neuronal development but also the biosynthesis of GAGs. Although the level of GAGs in patients with mutations in UGDH has not been investigated, the embryos from mutant mice, lazy mesoderm, with mutations in UGDH showed less staining with anti-CS or anti-HS antibodies, compared with the wild-type embryos for both antibodies (García-García and Anderson, 2003). Further analyses including quantification of GAGs from patients and glucuronokinase as well as UDP-GlcA pyrophosphrylase that form GlcA-1-phosphate and UDP-GlcA, respectively, which have been identified in zebrafish but not mammals, are required.
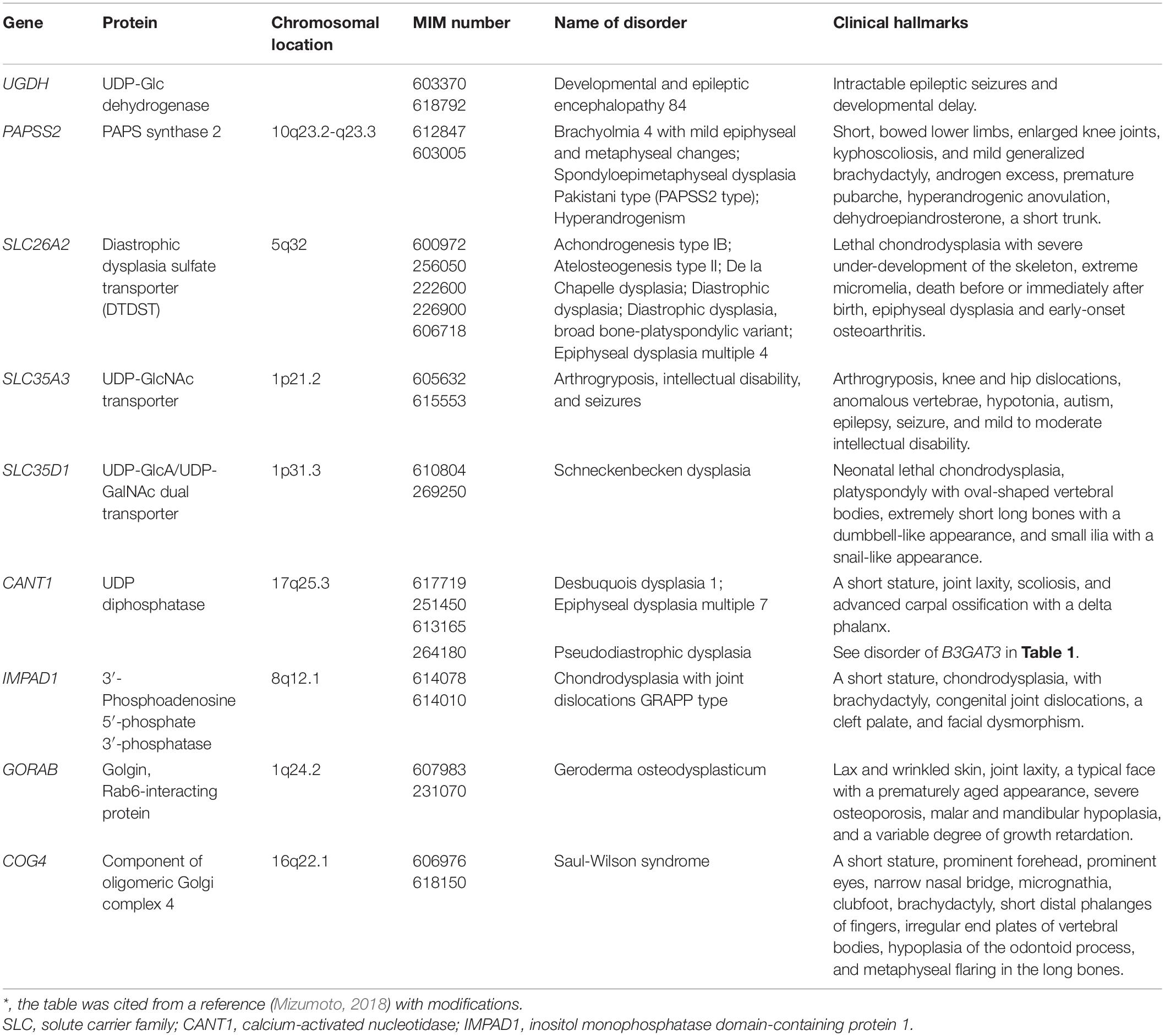
Table 5. Congenital disorders of UDP-sugars, PAPS, and related protein deficiencies*.
Spondyloepimetaphyseal Dysplasia of the Pakistani Type and Brachyolmia 4 With Mild Epiphyseal and Metaphyseal Changes Caused by Mutations in PAPSS2
PAPSS2 encodes PAPS synthase 2 (PAPSS2), which is a dual enzyme containing ATP sulfurylase and adenosine 5′-phosphosulfate kinase domains at C- and N-terminals, respectively (Figure 1; Fuda et al., 2002; Venkatachalam, 2003). Spondyloepimetaphyseal dysplasia of the Pakistani type that is characterized by a short stature at birth, short-bowed lower limbs, mild brachydactyly, kyphoscoliosis, enlarged knee joints, and early-onset degenerative joint disease of the hands and knees, is caused by homozygous mutations in PAPSS2 (Table 5; Ahmad et al., 1998; Tüysüz et al., 2013).
Compound heterozygous mutations in PAPSS2 cause hyperandrogenism, premature pubarche, and hyperandrogenic anovulation, in addition to a short stature and skeletal dysplasia, which may result from a low level of serum dehydroepiandrosterone sulfate with increasing androgen (Table 5; Noordam et al., 2009).
Furthermore, brachyolmia, which is characterized by a short stature, short trunk, irregular endplates, narrow intervertebral disks, precocious calcification of rib cartilages, a short femoral neck, mildly shortened metacarpals, and a normal intelligence and facies, are also caused by compound heterozygous and homozygous mutations in PAPSS2 (Table 5; Miyake et al., 2012; Iida et al., 2013; Bownass et al., 2019). PAPSS2 mutation might present a gradation of the phenotypic spectrum from brachyolmia to spondylo-epiphyseal and spondylo-epimetaphyseal dysplasia of the Pakistani type (Miyake et al., 2012). Brachymorphic mice, which have a missense mutation, p.Gly79Arg, in PAPSS2, are characterized by short stature as well as low level of CS (Sugahara and Schwartz, 1979; Pennypacker et al., 1981; Kurima et al., 1998). These findings indicate that sulfated modification of CS is required for cartilage development.
Congenital Disorders by Deficiency of Uridine Diphosphate-Sugar and Sulfate Transporters
Diastrophic Dysplasia and Achondrogenesis Type IB Caused by Mutations in SLC26A2
SLC26A2 encodes a sulfate transporter, which incorporates sulfate anions into the cytosol at the plasma membrane (Figure 1; Hästbacka et al., 1994; Satoh et al., 1998; Seidler and Nikolovska, 2019). The incorporated sulfate is activated to adenosine-phosphosulfate and then to PAPS by PAPS synthase (Venkatachalam, 2003). Mutations in SLC26A2 cause diastrophic dysplasia, achondrogenesis 1B, and atelosteogenesis type 2 (Table 5; Hästbacka et al., 1994, 1996; Superti-Furga et al., 1996; Rossi and Superti-Furga, 2001). Diastrophic dysplasia is an osteochondrodysplasia characterized by a short-limbed short stature, kyphoscoliosis, generalized dysplasia of the joints, hitchhiker thumbs, metatarsus adductus deformity of the feet, deformation of the ear lobes, and cleft plate (Superti-Furga et al., 1996). Achondrogenesis 1B is also a recessively inherited chondrodysplasia, which is characterized by extremely poor skeletal development and perinatal death (Table 5; Superti-Furga et al., 1996). Furthermore, atelosteogenesis type 2 is characterized by severely shortened limbs, a small chest, scoliosis, a club foot, abducted thumbs and great toes, and cleft palate (Table 5; Hästbacka et al., 1996). The incorporation of sulfate and formation of adenosine-phosphosulfate as well as PAPS were impaired in cultured cells from the proband of achondrogenesis 1B, thereby resulting in the sulfation of PGs (Superti-Furga et al., 1996). These findings suggest that mutations in SLC26A2 cause low sulfation of GAGs, thereby disturbing the assembly of the extracellular matrix as well as cellular signaling.
Arthrogryposis, Intellectual Disability, and Seizures Caused by Mutations in SLC35A3
SLC35A3 encodes UDP-GlcNAc transporter, which incorporates UDP-GlcNAc into the Golgi apparatus from the cytosol (Figure 1; Ishida et al., 1999; Szulc et al., 2020). Compound heterozygous and homozygous mutations of SLC35A31 cause neuroskeletal disorder, which is characterized by arthrogryposis, knee and hip dislocations, anomalous vertebrae, hypotonia, autism, epilepsy, seizure, and mild to moderate intellectual disability (Table 5; Edvardson et al., 2013; Edmondson et al., 2017; Miyake et al., 2021). Moreover, the probands showed deficiency of several N-glycans, no difference in the biosynthesis of keratan sulfate, and hallmarks such as cleft palate, micrognathia, a patent foramen ovale, patent ductus arteriosus, posterior embryotoxon, short limbs, camptodactyly, talipes valgus, rocker-bottom feet, and facial dysmorphism (Edvardson et al., 2013; Edmondson et al., 2017). Notably, a CS side chain of serum bikunin, CS-PG, from a patient with mutation in SLC35A3 was shorter than that from a control subject (Haouari et al., 2020). Because CS did not contain the GlcNAc residue, the finding indicates several possibilities that SLC35A3 transports UDP-GalNAc in addition to UDP-GlcNAc, and that UDP-GlcNAc-4′-epimerase/UDP-Gal-4′-epimerase encoded by GALE is located in the Golgi apparatus in addition to cytosol (Thoden et al., 2001; Broussard et al., 2020; Fushinobu, 2021).
Schneckenbecken Dysplasia Caused by Mutations in SLC35D1
SLC35D1 encodes UDP-GlcA/UDP-GalNAc dual transporter, which incorporates UDP-GlcA and UDP-GalNAc into the Golgi apparatus from the cytosol (Figure 1; Muraoka et al., 2001). Mutations of SLC35D1 cause Schneckenbecken dysplasia, which is characterized by perinatally lethal skeletal dysplasia, thoracic hypoplasia, severe flattening of the vertebral bodies and short, thick long bones (Table 5; Hiraoka et al., 2007; Furuichi et al., 2009). The German term “Schneckenbecken” refers to the snail-like appearance of the ilia that results from a medial bone projection from the inner iliac margin (Nikkels et al., 2001). Mice deficient in Slc35d1–/– showed decreased CS in the cartilage and no change in HS (Hiraoka et al., 2007). These findings suggest that SLC35D1 predominantly transports UDP-GalNAc rather than UDP-GlcA in vitro to form CS polysaccharides in the Golgi apparatus.
Congenital Disorders Caused by Glycosaminoglycan-Related Genes
Desbuquois Dysplasia 1 Caused by Mutations in CANT1
CANT1 encodes a calcium-dependent nucleoside diphosphatase that hydrolyzes UDP, which is a reaction product after transfer of a sugar residue by a glycosyltransferase, into uridine 5′-monophosphate (UMP) and an inorganic phosphate in the Golgi apparatus (Figure 1; Failer et al., 2002; Smith et al., 2002). The compound heterozygous and homozygous mutations cause Desbuquois dysplasia type 1, which is characterized by severe growth retardation, joint laxity, short extremities, and progressing scoliosis (Table 5; Huber et al., 2009; Faden et al., 2010; Dai et al., 2011; Furuichi et al., 2011; Laccone et al., 2011; Nizon et al., 2012b; Singh et al., 2015; Balasubramanian et al., 2017; Menzies et al., 2019). The phenotypes of this disorder are common to those of Desbuquois dysplasia type 2 caused by mutations in XYLT1 (Bui et al., 2014; Silveira et al., 2016; Al-Jezawi et al., 2017; Guo et al., 2017a).
Homozygous mutation in CANT1 causes pseudodiastrophic dysplasia, which is associated with prenatal manifestation and early lethality (Table 5; Byrne et al., 2020). The probands showed similar hallmarks with the probands with a compound heterozygous mutation in B3GAT3 (Byrne et al., 2020).
These findings suggest that the accumulation of UDP in the Golgi apparatus might inhibit the enzyme activity of XYLT1, B3GAT3 and UDP-GlcA decarboxylase/UDP-Xyl synthase, and the decrease in UMP might lead to impairment of UDP-Xyl or UDP-GlcA transporters, which are antiporters for UMP and UDP-XYl or UDP-GlcA, respectively, thereby resulting in defects in the biosynthesis of GAGs.
Chondrodysplasia With Joint Dislocations of GPAPP Type Caused by Mutations in IMPAD1
The sulfate donor substrate, PAPS, is converted into adenosine-3′, 5′-bisphosphate after a reaction with a sulfotransferase in the Golgi apparatus. IMPAD1 encodes inositol monophosphatase domain-containing protein 1/Golgi 3′-phosphoadenosine 5′-phosphate 3′-phosphatase (IMPAD1/GPAPP), which hydrolyzes the phosphate from 3′-phosphoadenosine 5′-phosphate to adenosine 5′-phosphate (Figure 1; Vissers et al., 2011; Nizon et al., 2012a). Homozygous mutations in IMPAD1 cause chondrodysplasia with joint dislocations (Table 5; Vissers et al., 2011; Nizon et al., 2012a), which is characterized by a short stature, brachydactyly, joint dislocations, micrognathia, cleft palate, and facial dysmorphism (Vissers et al., 2011; Nizon et al., 2012a). The knockout mice, Impad1–/–, exhibited a reduction of 4-O-sulfated CS with increasing non-sulfated CS, and a small decrease in total sulfated HS with a corresponding increase of non-sulfated HS in the lung (Frederick et al., 2008). Therefore, the catabolic abnormality in the reaction product of donor substrate including 3′-phosphoadenosine 5′-phosphate may result in the inhibition of GAG-sulfotransferases, and such skeletal disorders develop.
Geroderma Osteodysplasticum Caused by Mutations in GORAB
GORAB encodes Rab6-interacting Golgi protein (Hennies et al., 2008; Takeda et al., 2017). The homozygous mutations in GORAB cause gerodermia osteodysplastica that is characterized by skin laxity and early-onset osteoporosis (Table 5; Rajab et al., 2008). The knockout mice, GORAB–/–, showed a reduction in the level of DS, but not CS and HS, in skin and cartilage (Chan et al., 2018). These findings suggest that the loss of Gorab results in a defect of the DS side chain in PGs due to disturbed transport in the Golgi compartment, leading to the development of gerodermia osteodysplastica.
Saul-Wilson Syndrome Caused by Mutations in COG4
COG4 encodes the component of oligomeric Golgi complex 4, which is involved in intracellular vesicular transport (Climer et al., 2018). The heterozygous mutation in COG4 causes a rare skeletal dysplasia, Saul-Wilson syndrome, which is characterized by a short stature, prominent forehead, prominent eyes, a narrow nasal bridge, micrognathia, clubfoot, brachydactyly, short distal phalanges of fingers, irregular end plates of vertebral bodies, hypoplasia of the odontoid process, and metaphyseal flaring in the long bones (Table 5; Saul and Wilson, 1990; Ferreira et al., 2018). Although protein N-glycosylation in sera and fibroblasts from probands was not notably altered, DS-PG, decorin, was affected. A smaller population of glycanated decorin and longer CS/DS side chain on decorin in the affected fibroblasts were observed, compared with those in healthy subjects (Ferreira et al., 2018). These findings suggest that specific transport by oligomeric Golgi complex might affect the cellular localization and/or polymerization as well as sulfation of specific PGs.
Congenital Disorders Caused by Mutations in Core Proteins of Proteoglycans
Chondroitin Sulfate-Proteoglycans
Aggrecan encoded by ACAN consists of a protein with 2,431 amino acids (∼220 kDa), which is modified by CS (>100 chains per monomer), KS, and other oligosaccharides (Doege et al., 1991). The autosomal dominant spondyloepiphyseal dysplasia Kimberley type, which is characterized by short stature, stocky build, early-onset osteoarthritis of joints, flattened vertebral bodies with sclerosis and end-plate irregularity, and flattened femoral epiphyses, is caused by a single–base-pair insertion in ACAN, which introduces a frameshift of 212 amino acids, including 22 cysteine residues, followed by a premature stop codon (Table 6; Gleghorn et al., 2005). Furthermore, homozygous or heterozygous mutations in ACAN cause spondyloepimetaphyseal dysplasia Aggrecan type or dominant familial osteochondritis dissecans, respectively (Tompson et al., 2009; Stattin et al., 2010).
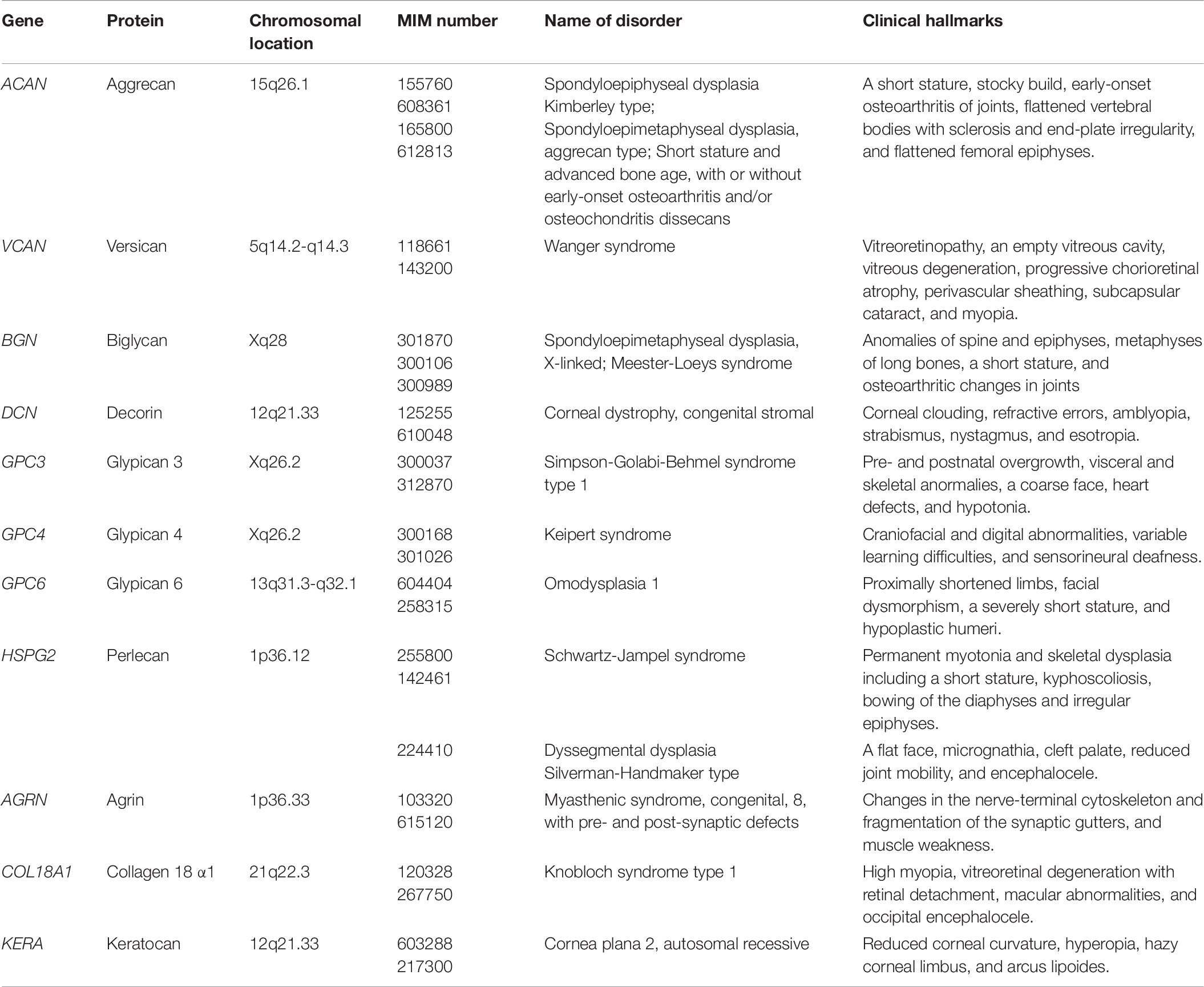
Table 6. Congenital disorders of core proteins of PGs.
Versican encoded by VCAN consists of a protein with 3,396 amino acids, which has ∼20 putative CS attachment sites as well as potential N- and O-glycosylation sites (Dours-Zimmermann and Zimmermann, 1994). The autosomal dominant Wanger syndrome, which is characterized by vitreoretinopathy, an empty vitreous cavity, vitreous degeneration, progressive chorioretinal atrophy, perivascular sheathing, subcapsular cataract, and myopia, is caused by heterozygous mutations in VCAN (Table 6; Miyamoto et al., 2005; Kloeckener-Gruissem et al., 2006, 2013).
Dermatan Sulfate-Proteoglycans
Biglycan encoded by BGN consists of a protein with 368 amino acids, which has 2 putative DS attachment sites (Fisher et al., 1989). Two missense mutations in BGN cause the X-linked form of spondyloepimetaphyseal dysplasia, which is characterized by anomalies of the spine as well as the epiphyses and metaphyses of the long bones, thereby resulting in a short stature and osteoarthritic changes in joints (Table 6; Cho et al., 2016). Thoracic aortic aneurysms and dissections are also caused by missense and non-sense mutations and 21 or 28-kb deletions in BGN (Meester et al., 2017). The clinical hallmarks are characterized by the early-onset of aortic aneurysm and dissection, joint hypermobility and contractures, deformities of the skin striae and pectus, craniofacial dysmorphisms including dolichocephaly, hypertelorism, down-slanting eyes, a high-arched plate, proptosis, malar hypoplasia, and frontal bossing as well as manifestations of Loeys-Dietz syndrome such as a bifid uvula and cervical spine instability (Meester et al., 2017). Moreover, TGFβ signaling is up-regulated in patients, thereby being similar to probands with Loeys-Dietz syndrome caused by mutations in TGFβ signaling-related proteins (Van Laer et al., 2014).
Decorin encoded by DCN consists of a protein with 359 amino acids, which has a putative DS attachment site (Fisher et al., 1989). Autosomal dominant congenital stromal corneal dystrophy is caused by mutations in DCN, and is characterized by corneal clouding with fine opacities observed as small flakes and spots, refractive errors, amblyopia, strabismus, nystagmus, and esotropia (Table 6; Van Ginderdeuren et al., 2002; Bredrup et al., 2005; Rødahl et al., 2006; Kim et al., 2011; Jing et al., 2014). All mutations in DCN causing dominant corneal dystrophy result in the truncated forms of decorin core protein, which may affect matrix assembly in the corneal stroma in a dominant-negative manner, supported by analyses using model mice with a frameshift mutation in Dcn (Chen et al., 2011).
Heparan Sulfate-Proteoglycans
Glypicans encoded by GPC1 to GPC6 consist of proteins with 550–580 amino acids, which have two putative HS attachment sites at the C-terminus. Glypicans are bound to the plasma membrane by a glycosylphosphatidylinositol (GPI) anchor (Filmus et al., 2008; Filmus and Capurro, 2014). Simpson-Golabi-Behmel syndrome is an X-linked syndrome, caused by mutations in GPC3, and characterized by pre- and postnatal overgrowth, visceral and skeletal anormalies, a coarse face, heart defects, and hypotonia (Table 6; Veugelers et al., 2000; Sakazume et al., 2007). Keipert syndrome, known as nasodigitoacoustic syndrome, is also an X-linked syndrome, caused by mutations in GPC4, and characterized by craniofacial and digital abnormalities, variable learning difficulties, and sensorineural deafness (Table 6; Amor et al., 2019). Autosomal-recessive omodysplasia is caused by homozygous mutations in GPC6, and is characterized by proximally shortened limbs, facial dysmorphism, a severely short stature, and hypoplastic humeri (Table 6; Campos-Xavier et al., 2009).
Perlecan encoded by HSPG2 consists of a protein with 4,392 amino acids (∼467 kDa), which is a major HS-PG with two or three HS as well as one or two CS, distributed to the basement membrane as well as the extracellular matrix of muscle, cartilage, and bone marrow (Cohen et al., 1993; Nicole et al., 2000; Melrose, 2020). Schwartz-Jampel syndrome is an autosomal recessive disorder, caused by homozygous or compound heterozygous mutations in HSPG2, and characterized by permanent myotonia and skeletal dysplasia including a short stature, kyphoscoliosis, bowing of the diaphysis, and irregular epiphyses (Table 6; Nicole et al., 2000; Arikawa-Hirasawa et al., 2002; Stum et al., 2006). Dyssegmental dysplasia Silverman-Handmaker type is an autosomal recessive skeletal dysplasia with anisospondyly and micromelia, and is caused by homozygous or heterozygous mutations in HSPG2 (Table 6; Arikawa-Hirasawa et al., 2001). The probands showed the hallmarks including a flat face, micrognathia, cleft palate, reduced joint mobility, and encephalocele.
Agrin encoded by AGRN consists of a protein with 2,068 amino acids, which has three potential HS attachments sites, and an extracellular matrix molecule released by the nerve and critical for formation of the neuromuscular junction (Rupp et al., 1991, 1992; Denzer et al., 1995). Myasthenic syndrome is a neuromuscular transmission disorder characterized by a major disorganization of the neuromuscular junction such as changes in the nerve-terminal cytoskeleton and fragmentation of the synaptic gutters, thereby leading to muscle weakness, caused by homozygous or heterozygous mutations in AGRN (Table 6; Huzé et al., 2009; Maselli et al., 2012).
Collagen18α1 encoded by COL18A1 consists of a protein with 1,339 amino acids, which is a subfamily of collagen predominantly expressed in the extracellular matrix of the liver, lung, and kidney, and is a subfamily of collagen distributed in the extracellular matrix (Oh et al., 1994). Knobloch syndrome type 1 is an autosomal recessive disorder defined by the occurrence of high myopia, vitreoretinal degeneration with retinal detachment, macular abnormalities, and occipital encephalocele, which is caused by homozygous or compound heterozygous mutations in COL18A1 (Table 5; Sertié et al., 2000; Suzuki et al., 2002; Caglayan et al., 2014).
Kepatan Sulfate-Proteoglycans
Keratocan encoded by KERA consists of a protein core of 352 amino acids with three covalent KS attachment sites at asparagine residues (Corpuz et al., 1996). Cornea plana type 2 is autosomal recessive and a more severe disease than type 1, characterized by a reduced corneal curvature, hyperopia, hazy corneal limbus, and arcus lipoides, and is caused by homozygous mutations in KERA (Table 6; Tahvanainen et al., 1996; Pellegata et al., 2000).
Conclusion and Perspectives
Genomic sequences have led to the identification of a variety of congenital disorders caused by defects in anabolism and catabolism of GAGs as well as PGs. Some of them have common clinical features such as skeletal, skin, heart, neuronal, and immune abnormalities. However, the molecular mechanisms of the onsets underlying these congenital diseases remain to be elucidated. To clarify the molecular pathogeneses including analyses of the functional domains of GAGs, identifications of the affected core proteins of PGs, the functional proteins interacting with GAGs, and the signaling pathways transduced by GAGs, are required. Achievement of these analyses lead to new therapeutics including the applications of adeno-associated virus, enzyme-replacement therapy, oligosaccharides derived from GAGs, and mimetics of GAGs for these disorders.
Author Contributions
SM wrote the manuscript. SY provided fruitful comments. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by a Grant-in Aid for Scientific Research (C) 19K07054 (to SM) from the Japan Society for the Promotion of Science, Japan. Grant-in Aid for Research Center for Pathogenesis of Intractable Diseases from the Research Institute of Meijo University (SM and SY).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with the authors SM and SY.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Medical English Service for editing the English text of the draft of this manuscript.
References
Ahmad, M., Faiyaz Ul Haque, M., Ahmad, W., Abbas, H., Haque, S., Krakow, D., et al. (1998). Distinct, autosomal recessive form of spondyloepimetaphyseal dysplasia segregating in an inbred Pakistani kindred. Am. J. Med. Genet. 78, 468–473. doi: 10.1002/(sici)1096-8628(19980806)78:5<468::aid-ajmg13>3.0.co;2-d
Ahn, J., Lüdecke, H. J., Lindow, S., Horton, W. A., Lee, B., Wagner, M. J., et al. (1995). Cloning of the putative tumour suppressor gene for hereditary multiple exostoses (EXT1). Nat. Genet. 11, 137–143. doi: 10.1038/ng1095-137
Akama, T. O., Misra, A. K., Hindsgaul, O., and Fukuda, M. N. (2002). Enzymatic synthesis in vitro of the disulfated disaccharide unit of corneal keratan sulfate. J. Biol. Chem. 277, 42505–42513. doi: 10.1074/jbc.m207412200
Akama, T. O., Nakayama, J., Nishida, K., Hiraoka, N., Suzuki, M., McAuliffe, J., et al. (2001). Human corneal GlcNac 6-O-sulfotransferase and mouse intestinal GlcNac 6-O-sulfotransferase both produce keratan sulfate. J. Biol. Chem. 276, 16271–16278. doi: 10.1074/jbc.m009995200
Akama, T. O., Nishida, K., Nakayama, J., Watanabe, H., Ozaki, K., Nakamura, T., et al. (2000). Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nat. Genet. 26, 237–241. doi: 10.1038/79987
Akatsu, C., Mizumoto, S., Kaneiwa, T., Maccarana, M., Malmström, A., Yamada, S., et al. (2011). Dermatan sulfate epimerase 2 is the predominant isozyme in the formation of the chondroitin sulfate/dermatan sulfate hybrid structure in postnatal developing mouse brain. Glycobiology 21, 565–574. doi: 10.1093/glycob/cwq208
Alazami, A. M., Al-Qattan, S. M., Faqeih, E., Alhashem, A., Alshammari, M., Alzahrani, F., et al. (2016). Expanding the clinical and genetic heterogeneity of hereditary disorders of connective tissue. Hum. Genet. 135, 525–540. doi: 10.1007/s00439-016-1660-z
Aldave, A. J., Yellore, V. S., Thonar, E. J., Udar, N., Warren, J. F., Yoon, M. K., et al. (2004). Novel mutations in the carbohydrate sulfotransferase gene (CHST6) in American patients with macular corneal dystrophy. Am. J. Ophthal. 137, 465–473. doi: 10.1016/j.ajo.2003.09.036
Al-Jezawi, N. K., Ali, B. R., and Al-Gazali, L. (2017). Endoplasmic reticulum retention of xylosyltransferase 1 (XYLT1) mutants underlying Desbuquois dysplasia type II. Am. J. Med. Genet. A 173, 1773–1781. doi: 10.1002/ajmg.a.38244
Almeida, R., Levery, S. B., Mandel, U., Kresse, H., Schwientek, T., Bennett, E. P., et al. (1999). Cloning and expression of a proteoglycan UDP-galactose:beta-xylose β1,4-galactosyltransferase I. A seventh member of the human β4-galactosyltransferase gene family. J. Biol. Chem. 274, 26165–26171.
Amor, D. J., Stephenson, S. E. M., Mustapha, M., Mensah, M. A., Ockeloen, C. W., Lee, W. S., et al. (2019). Pathogenic variants in GPC4 cause Keipert syndrome. Am. J. Hum. Genet. 104, 914–924. doi: 10.1016/j.ajhg.2019.02.026
Arikawa-Hirasawa, E., Le, A. H., Nishino, I., Nonaka, I., Ho, N. C., Francomano, C. A., et al. (2002). Structural and functional mutations of the perlecan gene cause Schwartz-Jampel syndrome, with myotonic myopathy and chondrodysplasia. Am. J. Hum. Genet. 70, 1368–1375. doi: 10.1086/340390
Arikawa-Hirasawa, E., Wilcox, W. R., Le, A. H., Silverman, N., Govindraj, P., Hassell, J. R., et al. (2001). Dyssegmental dysplasia, Silverman-Handmaker type, is caused by functional null mutations of the perlecan gene. Nat. Genet. 27, 431–434. doi: 10.1038/86941
Armstrong, L., Tarailo-Graovac, M., Sinclair, G., Seath, K. I., Wasserman, W. W., Ross, C. J., et al. (2017). A girl with developmental delay, ataxia, cranial nerve palsies, severe respiratory problems in infancy-Expanding NDST1 syndrome. Am. J. Med. Genet. A 173, 712–715. doi: 10.1002/ajmg.a.37621
Arunrut, T., Sabbadini, M., Jain, M., Machol, K., Scaglia, F., and Slavotinek, A. (2016). Corneal clouding, cataract, and colobomas with a novel missense mutation in B4GALT7-a review of eye anomalies in the linkeropathy syndromes. Am. J. Med. Genet. A 170, 2711–2718. doi: 10.1002/ajmg.a.37809
Baasanjav, S., Al-Gazali, L., Hashiguchi, T., Mizumoto, S., Fischer, B., Horn, D., et al. (2011). Faulty initiation of proteoglycan synthesis causes cardiac and joint defects. Am. J. Hum. Genet. 89, 15–27. doi: 10.1016/j.ajhg.2011.05.021
Bai, X., Zhou, D., Brown, J. R., Crawford, B. E., Hennet, T., and Esko, J. D. (2001). Biosynthesis of the linkage region of glycosaminoglycans: cloning and activity of galactosyltransferase II, the sixth member of the β1,3-galactosyltransferase family (β3GalT6). J. Biol. Chem. 276, 48189–48195.
Balasubramanian, K., Li, B., Krakow, D., Nevarez, L., Ho, P. J., Ainsworth, J. A., et al. (2017). MED resulting from recessively inherited mutations in the gene encoding calcium-activated nucleotidase CANT1. Am. J. Med. Genet. A 173, 2415–2421. doi: 10.1002/ajmg.a.38349
Baratela, W. A., Bober, M. B., Tiller, G. E., Okenfuss, E., Ditro, C., Duker, A., et al. (2012). A newly recognized syndrome with characteristic facial features, skeletal dysplasia, and developmental delay. Am. J. Med. Genet. A 158A, 1815–1822. doi: 10.1002/ajmg.a.35445
Bartolini, B., Thelin, M. A., Rauch, U., Feinstein, R., Oldberg, A., Malmström, A., et al. (2012). Mouse development is not obviously affected by the absence of dermatan sulfate epimerase 2 in spite of a modified brain dermatan sulfate composition. Glycobiology 22, 1007–1016. doi: 10.1093/glycob/cws065
Beighton, P., Gericke, G., Kozlowski, K., and Grobler, L. (1984). The manifestations and natural history of spondylo-epi-metaphyseal dysplasia with joint laxity. Clin. Genet. 26, 308–317. doi: 10.1111/j.1399-0004.1984.tb01065.x
Berninsone, P. M., and Hirschberg, C. B. (2000). Nucleotide sugar transporters of the Golgi apparatus. Curr. Opin. Struct. Biol. 10, 542–547. doi: 10.1016/s0959-440x(00)00128-7
Bick, D., Franco, B., Sherins, R. J., Heye, B., Pike, L., Crawford, J., et al. (1992). Brief report: intragenic deletion of the KALIG-1 gene in Kallmann’s syndrome. New Eng. J. Med. 326, 1752–1755.
Bishop, J. R., Schuksz, M., and Esko, J. D. (2007). Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446, 1030–1037. doi: 10.1038/nature05817
Boehm, U., Bouloux, P. M., Dattani, M. T., de Roux, N., Dodé, C., Dunkel, L., et al. (2015). Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism–pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 11, 547–564. doi: 10.1038/nrendo.2015.112
Bovée, J. V., Cleton-Jansen, A. M., Wuyts, W., Caethoven, G., Taminiau, A. H., Bakker, E., et al. (1999). EXT-mutation analysis and loss of heterozygosity in sporadic and hereditary osteochondromas and secondary chondrosarcomas. Am. J. Hum. Genet. 65, 689–698. doi: 10.1086/302532
Bownass, L., Abbs, S., Armstrong, R., Baujat, G., Behzadi, G., Berentsen, R. D., et al. (2019). PAPSS2-related brachyolmia: Clinical and radiological phenotype in 18 new cases. Am. J. Med. Genet. A 179, 1884–1894.
Bredrup, C., Knappskog, P. M., Majewski, J., Rødahl, E., and Boman, H. (2005). Congenital stromal dystrophy of the cornea caused by a mutation in the decorin gene. Invest. Ophthalmol. Vis. Sci. 46, 420–426. doi: 10.1167/iovs.04-0804
Broussard, A., Florwick, A., Desbiens, C., Nischan, N., Robertson, C., Guan, Z., et al. (2020). Human UDP-galactose 4′-epimerase (GALE) is required for cell-surface glycome structure and function. J. Biol. Chem. 295, 1225–1239. doi: 10.1016/s0021-9258(17)49882-6
Budde, B. S., Mizumoto, S., Kogawa, R., Becker, C., Altmüller, J., Thiele, H., et al. (2015). Skeletal dysplasia in a consanguineous clan from the island of Nias/Indonesia is caused by a novel mutation in B3GAT3. Hum. Genet. 134, 691–704. doi: 10.1007/s00439-015-1549-2
Bui, C., Huber, C., Tuysuz, B., Alanay, Y., Bole-Feysot, C., Leroy, J. G., et al. (2014). XYLT1 mutations in Desbuquois dysplasia type 2. Am. J. Hum. Genet. 94, 405–414. doi: 10.1016/j.ajhg.2014.01.020
Bülow, H. E., and Hobert, O. (2006). The molecular diversity of glycosaminoglycans shapes animal development. Annu. Rev. Cell Dev. Biol. 22, 375–407. doi: 10.1146/annurev.cellbio.22.010605.093433
Byrne, A. B., Mizumoto, S., Arts, P., Yap, P., Feng, J., Schreiber, A. W., et al. (2020). Pseudodiastrophic dysplasia expands the known phenotypic spectrum of defects in proteoglycan biosynthesis. J. Med. Genet. 57, 454–460. doi: 10.1136/jmedgenet-2019-106700
Caglayan, A. O., Baranoski, J. F., Aktar, F., Han, W., Tuysuz, B., Guzel, A., et al. (2014). Brain malformations associated with Knobloch syndrome–review of literature, expanding clinical spectrum, and identification of novel mutations. Pediatr. Neurol. 51, 806–813. doi: 10.1016/j.pediatrneurol.2014.08.025
Campos-Xavier, A. B., Martinet, D., Bateman, J., Belluoccio, D., Rowley, L., Tan, T. Y., et al. (2009). Mutations in the heparan-sulfate proteoglycan glypican 6 (GPC6) impair endochondral ossification and cause recessive omodysplasia. Am. J. Hum. Genet. 84, 760–770. doi: 10.1016/j.ajhg.2009.05.002
Cartault, F., Munier, P., Jacquemont, M. L., Vellayoudom, J., Doray, B., Payet, C., et al. (2015). Expanding the clinical spectrum of B4GALT7 deficiency: homozygous p.R270C mutation with founder effect causes Larsen of Reunion Island syndrome. Eur. J. Hum. Genet. 23, 49–53. doi: 10.1038/ejhg.2014.60
Chan, W. L., Steiner, M., Witkos, T., Egerer, J., Busse, B., Mizumoto, S., et al. (2018). Impaired proteoglycan glycosylation, elevated TGF-β signaling, and abnormal osteoblast differentiation as the basis for bone fragility in a mouse model for gerodermia osteodysplastica. PLoS Genet. 14:e1007242. doi: 10.1371/journal.pgen.1007242
Chen, S., Sun, M., Meng, X., Iozzo, R. V., Kao, W. W., and Birk, D. E. (2011). Pathophysiological mechanisms of autosomal dominant congenital stromal corneal dystrophy: C-terminal-truncated decorin results in abnormal matrix assembly and altered expression of small leucine-rich proteoglycans. Am. J. Pathol. 179, 2409–2419.
Cheung, P. K., McCormick, C., Crawford, B. E., Esko, J. D., Tufaro, F., and Duncan, G. (2001). Etiological point mutations in the hereditary multiple exostoses gene EXT1: a functional analysis of heparan sulfate polymerase activity. Am. J. Hum. Genet. 69, 55–66. doi: 10.1086/321278
Cho, S. Y., Bae, J. S., Kim, N. K. D., Forzano, F., Girisha, K. M., Baldo, C., et al. (2016). BGN mutations in X-linked spondyloepimetaphyseal dysplasia. Am. J. Hum. Genet. 98, 1243–1248.
Chopra, S. S., Leshchiner, I., Duzkale, H., McLaughlin, H., Giovanni, M., Zhang, C., et al. (2015). Inherited CHST11/MIR3922 deletion is associated with a novel recessive syndrome presenting with skeletal malformation and malignant lymphoproliferative disease. Mol. Genet. Genomic Med. 3, 413–423. doi: 10.1002/mgg3.152
Climer, L. K., Pokrovskaya, I. D., Blackburn, J. B., and Lupashin, V. V. (2018). Membrane detachment is not essential for COG complex function. Mol. Biol. Cell 29, 964–974. doi: 10.1091/mbc.e17-11-0694
Cohen, I. R., Grässel, S., Murdoch, A. D., and Iozzo, R. V. (1993). Structural characterization of the complete human perlecan gene and its promoter. Proc. Natl. Acad. Sci. U. S. A. 90, 10404–10408. doi: 10.1073/pnas.90.21.10404
Corpuz, L. M., Funderburgh, J. L., Funderburgh, M. L., Bottomley, G. S., Prakash, S., and Conrad, G. W. (1996). Molecular cloning and tissue distribution of keratocan. Bovine corneal keratan sulfate proteoglycan 37A. J. Biol. Chem. 271, 9759–9763.
Dai, J., Kim, O. H., Cho, T. J., Miyake, N., Song, H. R., Karasugi, T., et al. (2011). A founder mutation of CANT1 common in Korean and Japanese Desbuquois dysplasia. J. Hum. Genet. 56, 398–400. doi: 10.1038/jhg.2011.28
Denzer, A. J., Gesemann, M., Schumacher, B., and Ruegg, M. A. (1995). An amino-terminal extension is required for the secretion of chick agrin and its binding to extracellular matrix. J. Cell Biol. 131, 1547–1560. doi: 10.1083/jcb.131.6.1547
Desbuquois, G., Grenier, B., Michel, J., and Rossignol, C. (1966). Nanisme chondrodystrophique avec ossification anarchique et polymalformations chez deux sœurs. Arch. Franc. Pediat. 23, 573–587.
Dixon, J., Loftus, S. K., Gladwin, A. J., Scambler, P. J., Wasmuth, J. J., and Dixon, M. J. (1995). Cloning of the human heparan sulfate-N-deacetylase/N-sulfotransferase gene from the treacher Collins syndrome candidate region at 5q32-q33.1. Genomics 26, 239–244. doi: 10.1016/0888-7543(95)80206-2
Dodé, C., Levilliers, J., Dupont, J. M., De Paepe, A., Le Dû, N., Soussi-Yanicostas, N., et al. (2003). Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat. Genet. 33, 463–465.
Doege, K. J., Sasaki, M., Kimura, T., and Yamada, Y. (1991). Complete coding sequence and deduced primary structure of the human cartilage large aggregating proteoglycan, aggrecan. Human-specific repeats, and additional alternatively spliced forms. J. Biol. Chem. 266, 894–902. doi: 10.1016/s0021-9258(17)35257-2
Dours-Zimmermann, M. T., and Zimmermann, D. R. (1994). A novel glycosaminoglycan attachment domain identified in two alternative splice variants of human versican. J. Biol. Chem. 269, 32992–32998. doi: 10.1016/s0021-9258(20)30089-2
Dundar, M., Demiryilmaz, F., Demiryilmaz, I., Kumandas, S., Erkilic, K., Kendirci, M., et al. (1997). An autosomal recessive adducted thumb-club foot syndrome observed in Turkish cousins. Clin. Genet. 51, 61–64. doi: 10.1111/j.1399-0004.1997.tb02417.x
Dündar, M., Müller, T., Zhang, Q., Pan, J., Steinmann, B., Vodopiutz, J., et al. (2009). Loss of dermatan-4-sulfotransferase 1 function results in adducted thumb-clubfoot syndrome. Am. J. Hum. Genet. 85, 873–882. doi: 10.1016/j.ajhg.2009.11.010
Edmondson, A. C., Bedoukian, E. C., Deardorff, M. A., McDonald-McGinn, D. M., Li, X., He, M., et al. (2017). A human case of SLC35A3-related skeletal dysplasia. Am. J. Med. Genet. A 173, 2758–2762. doi: 10.1002/ajmg.a.38374
Edvardson, S., Ashikov, A., Jalas, C., Sturiale, L., Shaag, A., Fedick, A., et al. (2013). Mutations in SLC35A3 cause autism spectrum disorder, epilepsy and arthrogryposis. J. Med. Genet. 50, 733–739. doi: 10.1136/jmedgenet-2013-101753
El-Ashry, M. F., Abd El-Aziz, M. M., Wilkins, S., Cheetham, M. E., Wilkie, S. E., Hardcastle, A. J., et al. (2002). Identification of novel mutations in the carbohydrate sulfotransferase gene (CHST6) causing macular corneal dystrophy. Invest. Ophthal. Vis. Sci. 43, 377–382.
Eteson, D. J., Beluffi, G., Burgio, G. R., Belloni, C., Lachman, R. S., and Rimoin, D. L. (1986). Pseudodiastrophic dysplasia: a distinct newborn skeletal dysplasia. J. Pediatr. 109, 635–641. doi: 10.1016/s0022-3476(86)80227-x
Evers, M. R., Xia, G., Kang, H. G., Schachner, M., and Baenziger, J. U. (2001). Molecular cloning and characterization of a dermatan-specific N-acetylgalactosamine 4-O-sulfotransferase. J. Biol. Chem. 276, 36344–36353.
Faden, M., Al-Zahrani, F., Arafah, D., and Alkuraya, F. S. (2010). Mutation of CANT1 causes Desbuquois dysplasia. Am. J. Med. Genet. A 152A, 1157–1160.
Failer, B. U., Braun, N., and Zimmermann, H. (2002). Cloning, expression, and functional characterization of a Ca2+-dependent endoplasmic reticulum nucleoside diphosphatase. J. Biol. Chem. 277, 36978–36986. doi: 10.1074/jbc.m201656200
Faiyaz-Ul-Haque, M., Ahmad, W., Zaidi, S. H., Hussain, S., Haque, S., Ahmad, M., et al. (2004). Novel mutations in the EXT1 gene in two consanguineous families affected with multiple hereditary exostoses (familial osteochondromatosis). Clin. Genet. 66, 144–151. doi: 10.1111/j.1399-0004.2004.00275.x
Falardeau, J., Chung, W. C., Beenken, A., Raivio, T., Plummer, L., Sidis, Y., et al. (2008). Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J. Clin. Invest. 118, 2822–2831. doi: 10.1172/jci34538
Farhan, S. M., Wang, J., Robinson, J. F., Prasad, A. N., Rupar, C. A., Siu, V. M., et al. (2015). Old gene, new phenotype: mutations in heparan sulfate synthesis enzyme, EXT2 leads to seizure and developmental disorder, no exostoses. J. Med. Genet. 52, 666–675. doi: 10.1136/jmedgenet-2015-103279
Ferreira, C. R., Xia, Z. J., Clément, A., Parry, D. A., Davids, M., Taylan, F., et al. (2018). A Recurrent De Novo Heterozygous COG4 substitution leads to Saul-Wilson syndrome, disrupted vesicular trafficking, and altered proteoglycan glycosylation. Am. J. Hum. Genet. 103, 553–567. doi: 10.1016/j.ajhg.2018.09.003
Filmus, J., and Capurro, M. (2014). The role of glypicans in Hedgehog signaling. Matrix Biol. 35, 248–252. doi: 10.1016/j.matbio.2013.12.007
Filmus, J., Capurro, M., and Rast, J. (2008). Glypicans. Genome Biol. 9:224. doi: 10.1186/gb-2008-9-5-224
Fischer, S., Filipek-Gorniok, B., and Ledin, J. (2011). Zebrafish Ext2 is necessary for Fgf and Wnt signaling, but not for Hh signaling. BMC Dev. Biol. 11:53. doi: 10.1186/1471-213X-11-53
Fischetto, R., Causio, F., Corso, G., Lillo, V., Natale, B., and Papadia, F. (1997). Pseudodiastrophic dysplasia type Burgio in a newborn. Am. J. Med. Genet. 71, 222–225. doi: 10.1002/(sici)1096-8628(19970808)71:2<222::aid-ajmg20>3.0.co;2-f
Fisher, L. W., Termine, J. D., and Young, M. F. (1989). Deduced protein sequence of bone small proteoglycan I (biglycan) shows homology with proteoglycan II (decorin) and several nonconnective tissue proteins in a variety of species. J. Biol. Chem. 264, 4571–4576. doi: 10.1016/s0021-9258(18)83781-4
Frederick, J. P., Tafari, A. T., Wu, S. M., Megosh, L. C., Chiou, S. T., Irving, R. P., et al. (2008). A role for a lithium-inhibited Golgi nucleotidase in skeletal development and sulfation. Proc. Natl. Acad. Sci. U. S. A. 105, 11605–11612.
Freeze, H. H., Eklund, E. A., Ng, B. G., and Patterson, M. C. (2015). Neurological aspects of human glycosylation disorders. Annu. Rev. Neurosci. 38, 105–125. doi: 10.1146/annurev-neuro-071714-034019
Fuda, H., Shimizu, C., Lee, Y. C., Akita, H., and Strott, C. A. (2002). Characterization and expression of human bifunctional 3′-phosphoadenosine 5′-phosphosulphate synthase isoforms. Biochem. J. 365, 497–504. doi: 10.1042/bj20020044
Fukuta, M., Inazawa, J., Torii, T., Tsuzuki, K., Shimada, E., and Habuchi, O. (1997). Molecular cloning and characterization of human keratan sulfate Gal-6-sulfotransferase. J. Biol. Chem. 272, 32321–32328. doi: 10.1074/jbc.272.51.32321
Fukuta, M., Kobayashi, Y., Uchimura, K., Kimata, K., and Habuchi, O. (1998). Molecular cloning and expression of human chondroitin 6-sulfotransferase. Biochim. Biophys. Acta 1399, 57–61. doi: 10.1016/s0167-4781(98)00089-x
Furuichi, T., Dai, J., Cho, T. J., Sakazume, S., Ikema, M., Matsui, Y., et al. (2011). CANT1 mutation is also responsible for Desbuquois dysplasia, type 2 and Kim variant. J. Med. Genet. 48, 32–37. doi: 10.1136/jmg.2010.080226
Furuichi, T., Kayserili, H., Hiraoka, S., Nishimura, G., Ohashi, H., Alanay, Y., et al. (2009). Identification of loss-of-function mutations of SLC35D1 in patients with Schneckenbecken dysplasia, but not with other severe spondylodysplastic dysplasias group diseases. J. Med. Genet. 46, 562–568. doi: 10.1136/jmg.2008.065201
Fushinobu, S. (2021). Molecular evolution and functional divergence of UDP-hexose 4-epimerases. Curr. Opin. Chem. Biol. 61, 53–62. doi: 10.1016/j.cbpa.2020.09.007
García-García, M. J., and Anderson, K. V. (2003). Essential role of glycosaminoglycans in Fgf signaling during mouse gastrulation. Cell 114, 727–737. doi: 10.1016/s0092-8674(03)00715-3
Gleghorn, L., Ramesar, R., Beighton, P., and Wallis, G. (2005). A mutation in the variable repeat region of the aggrecan gene (AGC1) causes a form of spondyloepiphyseal dysplasia associated with severe, premature osteoarthritis. Am. J. Hum. Genet. 77, 484–490. doi: 10.1086/444401
González-Martínez, D., Kim, S. H., Hu, Y., Guimond, S., Schofield, J., Winyard, P., et al. (2004). Anosmin-1 modulates fibroblast growth factor receptor 1 signaling in human gonadotropin-releasing hormone olfactory neuroblasts through a heparan sulfate-dependent mechanism. J. Neurosci. 24, 10384–10392. doi: 10.1523/jneurosci.3400-04.2004
Goossens, D., Van Gestel, S., Claes, S., De Rijk, P., Souery, D., Massat, I., et al. (2003). A novel CpG-associated brain-expressed candidate gene for chromosome 18q-linked bipolar disorder. Mol. Psychiatry 8, 83–89. doi: 10.1038/sj.mp.4001190
Götte, M., Spillmann, D., Yip, G. W., Versteeg, E., Echtermeyer, F. G., van Kuppevelt, T. H., et al. (2008). Changes in heparan sulfate are associated with delayed wound repair, altered cell migration, adhesion and contractility in the galactosyltransferase I (β4GalT-7) deficient form of Ehlers-Danlos syndrome. Hum. Mol. Genet. 17, 996–1009. doi: 10.1093/hmg/ddm372
Götting, C., Kuhn, J., Zahn, R., Brinkmann, T., and Kleesiek, K. (2000). Molecular cloning and expression of human UDP-D-Xylose:proteoglycan core protein β-D-xylosyltransferase and its first isoform XT-II. J. Mol. Biol. 304, 517–528. doi: 10.1006/jmbi.2000.4261
Gulberti, S., Lattard, V., Fondeur, M., Jacquinet, J. C., Mulliert, G., Netter, P., et al. (2005). Phosphorylation and sulfation of oligosaccharide substrates critically influence the activity of human β1,4-galactosyltransferase 7 (GalT-I) and β1,3-glucuronosyltransferase I (GlcAT-I) involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. J. Biol. Chem. 280, 1417–1425. doi: 10.1074/jbc.m411552200
Guo, L., Elcioglu, N. H., Iida, A., Demirkol, Y. K., Aras, S., Matsumoto, N., et al. (2017a). Novel and recurrent XYLT1 mutations in two Turkish families with Desbuquois dysplasia, type 2. J. Hum. Genet. 62, 447–451. doi: 10.1038/jhg.2016.143
Guo, L., Elcioglu, N. H., Mizumoto, S., Wang, Z., Noyan, B., Albayrak, H. M., et al. (2017b). Identification of biallelic EXTL3 mutations in a novel type of spondylo-epi-metaphyseal dysplasia. J. Hum. Genet. 62, 797–801. doi: 10.1038/jhg.2017.38
Guo, M. H., Stoler, J., Lui, J., Nilsson, O., Bianchi, D. W., Hirschhorn, J. N., et al. (2013). Redefining the progeroid form of Ehlers-Danlos syndrome: report of the fourth patient with B4GALT7 deficiency and review of the literature. Am. J. Med. Genet. A. 161A, 2519–2527.
Habuchi, H., Kobayashi, M., and Kimata, K. (1998). Molecular characterization and expression of heparan-sulfate 6-sulfotransferase. Complete cDNA cloning in human and partial cloning in Chinese hamster ovary cells. J. Biol. Chem. 273, 9208–9213.
Habuchi, H., Tanaka, M., Habuchi, O., Yoshida, K., Suzuki, H., Ban, K., et al. (2000). The occurrence of three isoforms of heparan sulfate 6-O-sulfotransferase having different specificities for hexuronic acid adjacent to the targeted N-sulfoglucosamine. J. Biol. Chem. 275, 2859–2868.
Häcker, U., Nybakken, K., and Perrimon, N. (2005). Heparan sulphate proteoglycans: the sweet side of development. Nat. Rev. Mol. Cell Biol. 6, 530–541. doi: 10.1038/nrm1681
Haouari, W., Dubail, J., Lounis-Ouaras, S., Prada, P., Bennani, R., Roseau, C., et al. (2020). Serum bikunin isoforms in congenital disorders of glycosylation and linkeropathies. J. Inherit. Metab. Dis. 43, 1349–1359. doi: 10.1002/jimd.12291
Hashimoto, Y., Orellana, A., Gil, G., and Hirschberg, C. B. (1992). Molecular cloning and expression of rat liver N-heparan sulfate sulfotransferase. J. Biol. Chem. 267, 15744–15750. doi: 10.1016/s0021-9258(19)49598-7
Hästbacka, J., de la Chapelle, A., Mahtani, M. M., Clines, G., Reeve-Daly, M. P., Daly, M., et al. (1994). The diastrophic dysplasia gene encodes a novel sulfate transporter: positional cloning by fine-structure linkage disequilibrium mapping. Cell 78, 1073–1087. doi: 10.1016/0092-8674(94)90281-x
Hästbacka, J., Superti-Furga, A., Wilcox, W. R., Rimoin, D. L., Cohn, D. H., and Lander, E. S. (1996). Atelosteogenesis type II is caused by mutations in the diastrophic dysplasia sulfate-transporter gene (DTDST): evidence for a phenotypic series involving three chondrodysplasias. Am. J. Hum. Genet. 58, 255–262.
Hengel, H., Bosso-Lefèvre, C., Grady, G., Szenker-Ravi, E., Li, H., Pierce, S., et al. (2020). Loss-of-function mutations in UDP-glucose 6-dehydrogenase cause recessive developmental epileptic encephalopathy. Nat. Commun. 11:595. doi: 10.1038/s41467-020-14360-7
Hennies, H. C., Kornak, U., Zhang, H., Egerer, J., Zhang, X., Seifert, W., et al. (2008). Gerodermia osteodysplastica is caused by mutations in SCYL1BP1, a Rab-6 interacting golgin. Nat. Genet. 40, 1410–1412. doi: 10.1038/ng.252
Hermanns, P., Unger, S., Rossi, A., Perez-Aytes, A., Cortina, H., Bonafe, L., et al. (2008). Congenital joint dislocations caused by carbohydrate sulfotransferase 3 deficiency in recessive Larsen syndrome and humero-spinal dysostosis. Am. J. Hum. Genet. 82, 1368–1374. doi: 10.1016/j.ajhg.2008.05.006
Hiraoka, N., Nakagawa, H., Ong, E., Akama, T. O., Fukuda, M. N., and Fukuda, M. (2000). Molecular cloning and expression of two distinct human chondroitin 4-O-sulfotransferases that belong to the HNK-1 sulfotransferase gene family. J. Biol. Chem. 275, 20188–20196. doi: 10.1074/jbc.m002443200
Hiraoka, S., Furuichi, T., Nishimura, G., Shibata, S., Yanagishita, M., Rimoin, D. L., et al. (2007). Nucleotide-sugar transporter SLC35D1 is critical to chondroitin sulfate synthesis in cartilage and skeletal development in mouse and human. Nat. Med. 13, 1363–1367. doi: 10.1038/nm1655
Hirose, T., Mizumoto, S., Hashimoto, A., Takahashi, Y., Yoshizawa, T., Nitahara-Kasahara, Y., et al. (2021). Systematic investigation of the skin in Chst14–/– mice: A model for skin fragility in musculocontractural Ehlers-Danlos syndrome caused by CHST14 variants (mcEDS-CHST14). Glycobiology 31, 137–150. doi: 10.1093/glycob/cwaa058
Hirose, T., Takahashi, N., Tangkawattana, P., Minaguchi, J., Mizumoto, S., Yamada, S., et al. (2019). Structural alteration of glycosaminoglycan side chains and spatial disorganization of collagen networks in the skin of patients with mcEDS-CHST14. Biochim. Biophys. Acta Gen. Subj. 1863, 623–631. doi: 10.1016/j.bbagen.2018.12.006
Huber, C., Oulès, B., Bertoli, M., Chami, M., Fradin, M., Alanay, Y., et al. (2009). Identification of CANT1 mutations in Desbuquois dysplasia. Am. J. Hum. Genet. 85, 706–710. doi: 10.1016/j.ajhg.2009.10.001
Huzé, C., Bauché, S., Richard, P., Chevessier, F., Goillot, E., Gaudon, K., et al. (2009). Identification of an agrin mutation that causes congenital myasthenia and affects synapse function. Am. J. Hum. Genet. 85, 155–167.
Iida, A., Simsek-Kiper, P. Ö, Mizumoto, S., Hoshino, T., Elcioglu, N., Horemuzova, E., et al. (2013). Clinical and radiographic features of the autosomal recessive form of brachyolmia caused by PAPSS2 mutations. Hum. Mutat. 34, 1381–1386. doi: 10.1002/humu.22377
Iozzo, R. V. (1998). Matrix proteoglycans: from molecular design to cellular function. Annu. Rev. Biochem. 67, 609–652. doi: 10.1146/annurev.biochem.67.1.609
Ishida, N., Yoshioka, S., Chiba, Y., Takeuchi, M., and Kawakita, M. (1999). Molecular cloning and functional expression of the human Golgi UDP-N-acetylglucosamine transporter. J. Biochem. 126, 68–77. doi: 10.1093/oxfordjournals.jbchem.a022437
Izumikawa, T., Koike, T., Shiozawa, S., Sugahara, K., Tamura, J., and Kitagawa, H. (2008). Identification of chondroitin sulfate glucuronyltransferase as chondroitin synthase-3 involved in chondroitin polymerization: chondroitin polymerization is achieved by multiple enzyme complexes consisting of chondroitin synthase family members. J. Biol. Chem. 283, 11396–11406. doi: 10.1074/jbc.m707549200
Izumikawa, T., Uyama, T., Okuura, Y., Sugahara, K., and Kitagawa, H. (2007). Involvement of chondroitin sulfate synthase-3 (chondroitin synthase-2) in chondroitin polymerization through its interaction with chondroitin synthase-1 or chondroitin-polymerizing factor. Biochem. J. 403, 545–552. doi: 10.1042/bj20061876
Janecke, A. R., Unsinn, K., Kreczy, A., Baldissera, I., Gassner, I., Neu, N., et al. (2001). Adducted thumb-club foot syndrome in sibs of a consanguineous Austrian family. J. Med. Genet. 38, 265–269. doi: 10.1136/jmg.38.4.265
Jing, Y., Kumar, P. R., Zhu, L., Edward, D. P., Tao, S., Wang, L., et al. (2014). Novel decorin mutation in a Chinese family with congenital stromal corneal dystrophy. Cornea 33, 288–293. doi: 10.1097/ico.0000000000000055
Job, F., Mizumoto, S., Smith, L., Couser, N., Brazil, A., Saal, H., et al. (2016). Functional validation of novel compound heterozygous variants in B3GAT3 resulting in severe osteopenia and fractures: expanding the disease phenotype. BMC Med. Genet. 17:86. doi: 10.1186/s12881-016-0344-9
Jones, K. L., Schwarze, U., Adam, M. P., Byers, P. H., and Mefford, H. C. (2015). A homozygous B3GAT3 mutation causes a severe syndrome with multiple fractures, expanding the phenotype of linkeropathy syndromes. Am. J. Med. Genet. A 167A, 2691–2696. doi: 10.1002/ajmg.a.37209
Kim, B. T., Kitagawa, H., Tamura, J., Saito, T., Kusche-Gullberg, M., Lindahl, U., et al. (2001). Human tumor suppressor EXT gene family members EXTL1 and EXTL3 encode α1,4-N-acetylglucosaminyltransferases that likely are involved in heparan sulfate/heparin biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 98, 7176–7181. doi: 10.1073/pnas.131188498
Kim, B. T., Kitagawa, H., Tanaka, J., Tamura, J., and Sugahara, K. (2003). In vitro heparan sulfate polymerization: crucial roles of core protein moieties of primer substrates in addition to the EXT1-EXT2 interaction. J. Biol. Chem. 278, 41618–41623.
Kim, J. H., Ko, J. M., Lee, I., Kim, J. Y., Kim, M. J., and Tchah, H. (2011). A novel mutation of the decorin gene identified in a Korean family with congenital hereditary stromal dystrophy. Cornea 30, 1473–1477. doi: 10.1097/ico.0b013e3182137788
Kitagawa, H., Izumikawa, T., Uyama, T., and Sugahara, K. (2003). Molecular cloning of a chondroitin polymerizing factor that cooperates with chondroitin synthase for chondroitin polymerization. J. Biol. Chem. 278, 23666–23671. doi: 10.1074/jbc.m302493200
Kitagawa, H., Shimakawa, H., and Sugahara, K. (1999). The tumor suppressor EXT-like gene EXTL2 encodes an α1, 4-N-acetylhexosaminyltransferase that transfers N-acetylgalactosamine and N-acetylglucosamine to the common glycosaminoglycan-protein linkage region. The key enzyme for the chain initiation of heparan sulfate. J. Biol. Chem. 274, 13933–13937. doi: 10.1074/jbc.274.20.13933
Kitagawa, H., Tone, Y., Tamura, J., Neumann, K. W., Ogawa, T., Oka, S., et al. (1998). Molecular cloning and expression of glucuronyltransferase I involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. J. Biol. Chem. 273, 6615–6618. doi: 10.1074/jbc.273.12.6615
Kitagawa, H., Tsutsumi, K., Ikegami-Kuzuhara, A., Nadanaka, S., Goto, F., Ogawa, T., et al. (2008). Sulfation of the galactose residues in the glycosaminoglycan-protein linkage region by recombinant human chondroitin 6-O-sulfotransferase-1. J. Biol. Chem. 283, 27438–27443. doi: 10.1074/jbc.m803279200
Kitagawa, H., Uyama, T., and Sugahara, K. (2001). Molecular cloning and expression of a human chondroitin synthase. J. Biol. Chem. 276, 38721–38726. doi: 10.1074/jbc.m106871200
Kitayama, K., Hayashida, Y., Nishida, K., and Akama, T. O. (2007). Enzymes responsible for synthesis of corneal keratan sulfate glycosaminoglycans. J. Biol. Chem. 282, 30085–30096. doi: 10.1074/jbc.m703695200
Kloeckener-Gruissem, B., Bartholdi, D., Abdou, M. T., Zimmermann, D. R., and Berger, W. (2006). Identification of the genetic defect in the original wagner syndrome family. Mol. Vis. 12, 350–355.
Kloeckener-Gruissem, B., Neidhardt, J., Magyar, I., Plauchu, H., Zech, J. C., Morlé, L., et al. (2013). Novel VCAN mutations and evidence for unbalanced alternative splicing in the pathogenesis of wagner syndrome. Eur. J. Hum. Genet. 21, 352–356. doi: 10.1038/ejhg.2012.137
Kobayashi, M., Habuchi, H., Yoneda, M., Habuchi, O., and Kimata, K. (1997). Molecular cloning and expression of Chinese hamster ovary cell heparan-sulfate 2-sulfotransferase. J. Biol. Chem. 272, 13980–13985. doi: 10.1074/jbc.272.21.13980
Koike, T., Izumikawa, T., Sato, B., and Kitagawa, H. (2014). Identification of phosphatase that dephosphorylates xylose in the glycosaminoglycan-protein linkage region of proteoglycans. J. Biol. Chem. 289, 6695–6708. doi: 10.1074/jbc.m113.520536
Koike, T., Izumikawa, T., Tamura, J., and Kitagawa, H. (2009). FAM20B is a kinase that phosphorylates xylose in the glycosaminoglycan-protein linkage region. Biochem. J. 421, 157–162. doi: 10.1042/bj20090474
Kosho, T., Miyake, N., Hatamochi, A., Takahashi, J., Kato, H., Miyahara, T., et al. (2010). A new Ehlers-Danlos syndrome with craniofacial characteristics, multiple congenital contractures, progressive joint and skin laxity, and multisystem fragility-related manifestations. Am. J. Med. Genet. 152A, 1333–1346.
Kosho, T., Takahashi, J., Ohashi, H., Nishimura, G., Kato, H., and Fukushima, Y. (2005). Ehlers-Danlos syndrome type VIB with characteristic facies, decreased curvatures of the spinal column, and joint contractures in two unrelated girls. Am. J. Med. Genet. 138A, 282–287. doi: 10.1002/ajmg.a.30965
Kurima, K., Warman, M. L., Krishnan, S., Domowicz, M., Krueger, R. C. Jr., Deyrup, A., et al. (1998). A member of a family of sulfate-activating enzymes causes murine brachymorphism. Proc. Natl. Acad. Sci. U. S. A. 95, 8681–8685. doi: 10.1073/pnas.95.15.8681
Kuroda, Y., Murakami, H., Enomoto, Y., Tsurusaki, Y., Takahashi, K., Mitsuzuka, K., et al. (2019). A novel gene (FAM20B encoding glycosaminoglycan xylosylkinase) for neonatal short limb dysplasia resembling Desbuquois dysplasia. Clin. Genet. 95, 713–717. doi: 10.1111/cge.13530
Kusche-Gullberg, M., and Kjellén, L. (2003). Sulfotransferases in glycosaminoglycan biosynthesis. Curr. Opin. Struct. Biol. 13, 605–611. doi: 10.1016/j.sbi.2003.08.002
Laccone, F., Schoner, K., Krabichler, B., Kluge, B., Schwerdtfeger, R., Schulze, B., et al. (2011). Desbuquois dysplasia type I and fetal hydrops due to novel mutations in the CANT1 gene. Eur. J. Hum. Genet. 19, 1133–1137.
LaCroix, A. J., Stabley, D., Sahraoui, R., Adam, M. P., Mehaffey, M., Kernan, K., et al. (2019). GGC repeat expansion and exon 1 methylation of XYLT1 is a common pathogenic variant in Baratela-Scott syndrome. Am. J. Hum. Genet. 104, 35–44. doi: 10.1016/j.ajhg.2018.11.005
Larsen, L. J., Schottstaedt, E. R., and Bost, F. C. (1950). Multiple congenital dislocations associated with characteristic facial abnormality. J. Pediat. 37, 574–581. doi: 10.1016/s0022-3476(50)80268-8
Lautrup, C. K., Teik, K. W., Unzaki, A., Mizumoto, S., Syx, D., Sin, H. H., et al. (2020). Delineation of musculocontractural Ehlers-Danlos syndrome caused by dermatan sulfate epimerase deficiency. Mol. Genet. Genomic Med. 8:e1197. doi: 10.1002/mgg3.1197
Lee, J. S., von der Hardt, S., Rusch, M. A., Stringer, S. E., Stickney, H. L., Talbot, W. S., et al. (2004). Axon sorting in the optic tract requires HSPG synthesis by ext2 (dackel) and extl3 (boxer). Neuron 44, 947–960.
Li, Y., Laue, K., Temtamy, S., Aglan, M., Kotan, L. D., Yigit, G., et al. (2010). Temtamy preaxial brachydactyly syndrome is caused by loss-of-function mutations in chondroitin synthase 1, a potential target of BMP signaling. Am. J. Hum. Genet. 87, 757–767. doi: 10.1016/j.ajhg.2010.10.003
Lind, T., Tufaro, F., McCormick, C., Lindahl, U., and Lidholt, K. (1998). The putative tumor suppressors EXT1 and EXT2 are glycosyltransferases required for the biosynthesis of heparan sulfate. J. Biol. Chem. 273, 26265–26268. doi: 10.1074/jbc.273.41.26265
Lüdecke, H. J., Wagner, M. J., Nardmann, J., La Pillo, B., Parrish, J. E., Willems, P. J., et al. (1995). Molecular dissection of a contiguous gene syndrome: localization of the genes involved in the Langer-Giedion syndrome. Hum. Mol. Genet. 4, 31–36. doi: 10.1093/hmg/4.1.31
Maccarana, M., Olander, B., Malmström, J., Tiedemann, K., Aebersold, R., Lindahl, U., et al. (2006). Biosynthesis of dermatan sulfate: chondroitin-glucuronate C5-epimerase is identical to SART2. J. Biol. Chem. 281, 11560–11568.
Malfait, F., Castori, M., Francomano, C. A., Giunta, C., Kosho, T., and Byers, P. H. (2020). The Ehlers-Danlos syndromes. Nat. Rev. Dis. Primers 6:64. doi: 10.1038/s41572-020-0194-9
Malfait, F., Francomano, C., Byers, P., Belmont, J., Berglund, B., Black, J., et al. (2017). The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 175, 8–26. doi: 10.1002/ajmg.c.31552
Malfait, F., Kariminejad, A., Van Damme, T., Gauche, C., Syx, D., Merhi-Soussi, F., et al. (2013). Defective initiation of glycosaminoglycan synthesis due to B3GALT6 mutations causes a pleiotropic Ehlers-Danlos-syndrome-like connective tissue disorder. Am. J. Hum. Genet. 92, 935–945. doi: 10.1016/j.ajhg.2013.04.016
Malfait, F., Syx, D., Vlummens, P., Symoens, S., Nampoothiri, S., Hermanns-Lê, T., et al. (2010). Musculocontractural Ehlers-Danlos Syndrome (former EDS type VIB) and adducted thumb clubfoot syndrome (ATCS) represent a single clinical entity caused by mutations in the dermatan-4-sulfotransferase 1 encoding CHST14 gene. Hum. Mutat. 31, 1233–1239. doi: 10.1002/humu.21355
Maselli, R. A., Fernandez, J. M., Arredondo, J., Navarro, C., Ngo, M., Beeson, D., et al. (2012). LG2 agrin mutation causing severe congenital myasthenic syndrome mimics functional characteristics of non-neural (z-) agrin. Hum Genet. 131, 1123–1135. doi: 10.1007/s00439-011-1132-4
McBrien, J., Crolla, J. A., Huang, S., Kelleher, J., Gleeson, J., and Lynch, S. A. (2008). Further case of microdeletion of 8q24 with phenotype overlapping Langer-Giedion without TRPS1 deletion. Am. J. Med. Genet. A 146A, 1587–1592. doi: 10.1002/ajmg.a.32347
McCormick, C., Leduc, Y., Martindale, D., Mattison, K., Esford, L. E., Dyer, A. P., et al. (1998). The putative tumour suppressor EXT1 alters the expression of cell-surface heparan sulfate. Nat. Genet. 19, 158–161. doi: 10.1038/514
McTague, A., Howell, K. B., Cross, J. H., Kurian, M. A., and Scheffer, I. E. (2016). The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 15, 304–316. doi: 10.1016/s1474-4422(15)00250-1
Meester, J. A., Vandeweyer, G., Pintelon, I., Lammens, M., Van Hoorick, L., De Belder, S., et al. (2017). Loss-of-function mutations in the X-linked biglycan gene cause a severe syndromic form of thoracic aortic aneurysms and dissections. Genet. Med. 19, 386–395. doi: 10.1038/gim.2016.126
Melrose, J. (2020). Perlecan, a modular instructive proteoglycan with diverse functional properties. Int. J. Biochem. Cell Biol. 128:105849. doi: 10.1016/j.biocel.2020.105849
Menzies, L., Cullup, T., Calder, A., Wilson, L., and Faravelli, F. (2019). A novel homozygous variant in CANT1 in a patient with Kim-type Desbuquois dysplasia. Clin. Dysmorphol. 28, 219–223. doi: 10.1097/mcd.0000000000000291
Mihalic Mosher, T., Zygmunt, D. A., Koboldt, D. C., Kelly, B. J., Johnson, L. R., McKenna, D. S., et al. (2019). Expansion of B4GALT7 linkeropathy phenotype to include perinatal lethal skeletal dysplasia. Eur. J. Hum. Genet. 27, 1569–1577. doi: 10.1038/s41431-019-0464-8
Mikami, T., Mizumoto, S., Kago, N., Kitagawa, H., and Sugahara, K. (2003). Specificities of three distinct human chondroitin/dermatan N-acetylgalactosamine 4-O-sulfotransferases demonstrated using partially desulfated dermatan sulfate as an acceptor: implication of differential roles in dermatan sulfate biosynthesis. J. Biol. Chem. 278, 36115–36127. doi: 10.1074/jbc.m306044200
Millar, A. C., Faghfoury, H., and Bieniek, J. M. (2021). Genetics of hypogonadotropic hypogonadism. Transl. Androl. Urol. 10, 1401–1409.
Miraoui, H., Dwyer, A. A., Sykiotis, G. P., Plummer, L., Chung, W., Feng, B., et al. (2013). Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism. Am. J. Hum. Genet. 92, 725–743.
Miyake, N., de Oliveira Stephan, B., Kim, C. A., and Matsumoto, N. (2021). A Brazilian case arising from a homozygous canonical splice site SLC35A3 variant leading to an in-frame deletion. Clin. Genet. 99, 607–608. doi: 10.1111/cge.13909
Miyake, N., Elcioglu, N. H., Iida, A., Isguven, P., Dai, J., Murakami, N., et al. (2012). PAPSS2 mutations cause autosomal recessive brachyolmia. J. Med. Genet. 49, 533–538.
Miyake, N., Kosho, T., Mizumoto, S., Furuichi, T., Hatamochi, A., Nagashima, Y., et al. (2010). Loss-of-function mutations of CHST14 in a new type of Ehlers-Danlos syndrome. Hum. Mutat. 31, 966–974.
Miyamoto, T., Inoue, H., Sakamoto, Y., Kudo, E., Naito, T., Mikawa, T., et al. (2005). Identification of a novel splice site mutation of the CSPG2 gene in a Japanese family with Wagner syndrome. Invest. Ophthalmol. Vis. Sci. 46, 2726–2735. doi: 10.1167/iovs.05-0057
Mizumoto, S. (2018). Defects in biosynthesis of glycosaminoglycans cause hereditary bone, skin, heart, immune, and neurological disorders. Trends Glycosci. Glycotechnol. 30, E67–E89.
Mizumoto, S., Ikegawa, S., and Sugahara, K. (2013). Human genetic disorders caused by mutations in genes encoding biosynthetic enzymes for sulfated glycosaminoglycans. J. Biol. Chem. 288, 10953–10961.
Mizumoto, S., Janecke, A. R., Sadeghpour, A., Povysil, G., McDonald, M. T., Unger, S., et al. (2020). CSGALNACT1-congenital disorder of glycosylation: A mild skeletal dysplasia with advanced bone age. Hum. Mutat. 41, 655–667. doi: 10.1002/humu.23952
Mizumoto, S., Yamada, S., and Sugahara, K. (2014). Human genetic disorders and knockout mice deficient in glycosaminoglycan. Biomed Res. Int. 2014:495764. doi: 10.1155/2014/495764
Mizumoto, S., Yamada, S., and Sugahara, K. (2015a). Molecular interactions between chondroitin-dermatan sulfate and growth factors/receptors/matrix proteins. Curr. Opin. Struct. Biol. 34, 35–42.
Mizumoto, S., Yamada, S., and Sugahara, K. (2015b). Mutations in biosynthetic enzymes for the protein linker region of chondroitin/dermatan/heparan sulfate cause skeletal and skin dysplasias. Biomed. Res. Int. 2015:861752. doi: 10.1155/2015/861752
Moriarity, J. L., Hurt, K. J., Resnick, A. C., Storm, P. B., Laroy, W., Schnaar, R. L., et al. (2002). UDP-glucuronate decarboxylase, a key enzyme in proteoglycan synthesis. J. Biol. Chem. 277, 16968–16975. doi: 10.1074/jbc.m109316200
Mortier, G. R., Cohn, D. H., Cormier-Daire, V., Hall, C., Krakow, D., Mundlos, S., et al. (2019). Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. A 179, 2393–2419. doi: 10.1002/ajmg.a.61366
Müller, T., Mizumoto, S., Suresh, I., Komatsu, Y., Vodopiutz, J., Dundar, M., et al. (2013). Loss of dermatan sulfate epimerase (DSE) function results in musculocontractural Ehlers-Danlos syndrome. Hum. Mol. Genet. 22, 3761–3772. doi: 10.1093/hmg/ddt227
Munns, C. F., Fahiminiya, S., Poudel, N., Munteanu, M. C., Majewski, J., Sillence, D. O., et al. (2015). Homozygosity for frameshift mutations in XYLT2 result in a spondylo-ocular syndrome with bone fragility, cataracts, and hearing defects. Am. J. Hum. Genet. 96, 971–978. doi: 10.1016/j.ajhg.2015.04.017
Muraoka, M., Kawakita, M., and Ishida, N. (2001). Molecular characterization of human UDP-glucuronic acid/UDP-N-acetylgalactosamine transporter, a novel nucleotide sugar transporter with dual substrate specificity. FEBS Lett. 495, 87–93. doi: 10.1016/s0014-5793(01)02358-4
Muys, J., Blaumeiser, B., Jacquemyn, Y., and Janssens, K. (2016). Prenatal homozygosity mapping detects a novel mutation in CHST3 in a fetus with skeletal dysplasia and joint dislocations. Clin. Case Rep. 5, 440–445. doi: 10.1002/ccr3.800
Najmabadi, H., Hu, H., Garshasbi, M., Zemojtel, T., Abedini, S. S., Chen, W., et al. (2011). Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 478, 57–63.
Nakajima, M., Mizumoto, S., Miyake, N., Kogawa, R., Iida, A., Ito, H., et al. (2013). Mutations in B3GALT6, which encodes a glycosaminoglycan linker region enzyme, cause a spectrum of skeletal and connective tissue disorders. Am. J. Hum. Genet. 92, 927–934. doi: 10.1016/j.ajhg.2013.04.003
Nakao, M., Shichijo, S., Imaizumi, T., Inoue, Y., Matsunaga, K., Yamada, A., et al. (2000). Identification of a gene coding for a new squamous cell carcinoma antigen recognized by the CTL. J. Immunol. 164, 2565–2574. doi: 10.4049/jimmunol.164.5.2565
Narentuya, Takeda-Uchimura, Y., Foyez, T., Zhang, Z., Akama, T. O., Yagi, H., et al. (2019). GlcNAc6ST3 is a keratan sulfate sulfotransferase for the protein-tyrosine phosphatase PTPRZ in the adult brain. Sci. Rep. 9:4387. doi: 10.1038/s41598-019-40901-2
Nicole, S., Davoine, C. S., Topaloglu, H., Cattolico, L., Barral, D., Beighton, P., et al. (2000). Perlecan, the major proteoglycan of basement membranes, is altered in patients with Schwartz-Jampel syndrome (chondrodystrophic myotonia). Nat. Genet. 26, 480–483. doi: 10.1038/82638
Nikkels, P. G., Stigter, R. H., Knol, I. E., and van der Harten, H. J. (2001). Schneckenbecken dysplasia, radiology, and histology. Pediatr. Radio. 31, 27–30. doi: 10.1007/s002470000357
Nizon, M., Alanay, Y., Tuysuz, B., Kiper, P. O., Geneviève, D., Sillence, D., et al. (2012a). IMPAD1 mutations in two Catel-Manzke like patients. Am. J. Med. Genet. A 158A, 2183–2187. doi: 10.1002/ajmg.a.35504
Nizon, M., Huber, C., De Leonardis, F., Merrina, R., Forlino, A., Fradin, M., et al. (2012b). Further delineation of CANT1 phenotypic spectrum and demonstration of its role in proteoglycan synthesis. Hum. Mutat. 33, 1261–1266. doi: 10.1002/humu.22104
Noordam, C., Dhir, V., McNelis, J. C., Schlereth, F., Hanley, N. A., Krone, N., et al. (2009). Inactivating PAPSS2 mutations in a patient with premature pubarche. N. Engl. J. Med. 360, 2310–2318.
Oh, S. P., Kamagata, Y., Muragaki, Y., Timmons, S., Ooshima, A., and Olsen, B. R. (1994). Isolation and sequencing of cDNAs for proteins with multiple domains of Gly-Xaa-Yaa repeats identify a distinct family of collagenous proteins. Proc. Natl. Acad. Sci. U. S. A 91, 4229–4233. doi: 10.1073/pnas.91.10.4229
Okajima, T., Yoshida, K., Kondo, T., and Furukawa, K. (1999). Human homolog of Caenorhabditis elegans sqv-3 gene is galactosyltransferase I involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. J. Biol. Chem. 274, 22915–22918. doi: 10.1074/jbc.274.33.22915
Oud, M. M., Tuijnenburg, P., Hempel, M., van Vlies, N., Ren, Z., Ferdinandusse, S., et al. (2017). Mutations in EXTL3 cause neuro-immuno-skeletal dysplasia syndrome. Am. J. Hum. Genet. 100, 281–296.
Pacheco, B., Malmström, A., and Maccarana, M. (2009). Two dermatan sulfate epimerases form iduronic acid domains in dermatan sulfate. J. Biol. Chem. 284, 9788–9795. doi: 10.1074/jbc.m809339200
Palka, C., Alfonsi, M., Mohn, A., Guanciali Franchi, P., Chiarelli, F., and Calabrese, G. (2012). Delayed diagnosis of Potocki-Shaffer syndrome in a woman with multiple exostoses and mental retardation. Mol. Syndromol. 2, 259–261. doi: 10.1159/000337925
Pellegata, N. S., Dieguez-Lucena, J. L., Joensuu, T., Lau, S., Montgomery, K. T., Krahe, R., et al. (2000). Mutations in KERA, encoding keratocan, cause cornea plana. Nat. Genet. 25, 91–95. doi: 10.1038/75664
Pennypacker, J. P., Kimata, K., and Brown, K. S. (1981). Brachymorphic mice (bm/bm): a generalized biochemical defect expressed primarily cartilage. Dev. Biol. 81, 280–287. doi: 10.1016/0012-1606(81)90291-8
Pereza, N., Severinski, S., Ostojić, S., Volk, M., Maver, A., Dekanić, K. B., et al. (2012). Third case of 8q23.3-q24.13 deletion in a patient with Langer-Giedion syndrome phenotype without TRPS1 gene deletion. Am. J. Med. Genet. A 158A, 659–663. doi: 10.1002/ajmg.a.35201
Philippe, C., Porter, D. E., Emerton, M. E., Wells, D. E., Simpson, A. H., and Monaco, A. P. (1997). Mutation screening of the EXT1 and EXT2 genes in patients with hereditary multiple exostoses. Am. J. Hum. Genet. 61, 520–528. doi: 10.1086/515505
Pönighaus, C., Ambrosius, M., Casanova, J. C., Prante, C., Kuhn, J., Esko, J. D., et al. (2007). Human xylosyltransferase II is involved in the biosynthesis of the uniform tetrasaccharide linkage region in chondroitin sulfate and heparan sulfate proteoglycans. J. Biol. Chem. 282, 5201–5206. doi: 10.1074/jbc.m611665200
Quentin, E., Gladen, A., Rodén, L., and Kresse, H. (1990). A genetic defect in the biosynthesis of dermatan sulfate proteoglycan: galactosyltransferase I deficiency in fibroblasts from a patient with a progeroid syndrome. Proc. Natl. Acad. Sci. U. S. A. 87, 1342–1346. doi: 10.1073/pnas.87.4.1342
Rajab, A., Kornak, U., Budde, B. S., Hoffmann, K., Jaeken, J., Nürnberg, P., et al. (2008). Geroderma osteodysplasticum hereditaria and wrinkly skin syndrome in 22 patients from Oman. Am. J. Med. Genet. A. 146A, 965–976.
Rajab, A., Kunze, J., and Mundlos, S. (2004). Spondyloepiphyseal dysplasia Omani type: a new recessive type of SED with progressive spinal involvement. Am. J. Med. Genet. 126A, 413–419. doi: 10.1002/ajmg.a.20606
Reuter, M. S., Musante, L., Hu, H., Diederich, S., Sticht, H., Ekici, A. B., et al. (2014). NDST1 missense mutations in autosomal recessive intellectual disability. Am. J. Med. Genet. A 164A, 2753–2763.
Ritelli, M., Chiarelli, N., Zoppi, N., Dordoni, C., Quinzani, S., Traversa, M., et al. (2014). Insights in the etiopathology of galactosyltransferase II (GalT-II) deficiency from transcriptome-wide expression profiling of skin fibroblasts of two sisters with compound heterozygosity for two novel B3GALT6 mutations. Mol. Genet. Metab. Rep. 2, 1–15. doi: 10.1016/j.ymgmr.2014.11.005
Ritelli, M., Dordoni, C., Cinquina, V., Venturini, M., Calzavara-Pinton, P., and Colombi, M. (2017). Expanding the clinical and mutational spectrum of B4GALT7-spondylodysplastic Ehlers-Danlos syndrome. Orphanet. J. Rare Dis. 12:153. doi: 10.1186/s13023-017-0704-3
Rødahl, E., Van Ginderdeuren, R., Knappskog, P. M., Bredrup, C., and Boman, H. (2006). A second decorin frame shift mutation in a family with congenital stromal corneal dystrophy. Am. J. Ophthalmol. 142, 520–521. doi: 10.1016/j.ajo.2006.03.064
Rossi, A., and Superti-Furga, A. (2001). Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene (SLC26A2): 22 novel mutations, mutation review, associated skeletal phenotypes, and diagnostic relevance. Hum. Mutat. 17, 159–171. doi: 10.1002/humu.1
Rupp, F., Ozçelik, T., Linial, M., Peterson, K., Francke, U., and Scheller, R. (1992). Structure and chromosomal localization of the mammalian agrin gene. J. Neurosci. 12, 3535–3544. doi: 10.1523/jneurosci.12-09-03535.1992
Rupp, F., Payan, D. G., Magill-Solc, C., Cowan, D. M., and Scheller, R. H. (1991). Structure and expression of a rat agrin. Neuron 6, 811–823. doi: 10.1016/0896-6273(91)90177-2
Sakazume, S., Okamoto, N., Yamamoto, T., Kurosawa, K., Numabe, H., Ohashi, Y., et al. (2007). GPC3 mutations in seven patients with Simpson-Golabi-Behmel syndrome. Am. J. Med. Genet. A 143A, 1703–1707. doi: 10.1002/ajmg.a.31822
Salter, C. G., Davies, J. H., Moon, R. J., Fairhurst, J., Bunyan, D., Ddd Study, et al. (2016). Further defining the phenotypic spectrum of B4GALT7 mutations. Am. J. Med. Genet. A 170, 1556–1563.
Satoh, H., Susaki, M., Shukunami, C., Iyama, K., Negoro, T., and Hiraki, Y. (1998). Functional analysis of diastrophic dysplasia sulfate transporter. Its involvement in growth regulation of chondrocytes mediated by sulfated proteoglycans. J. Biol. Chem. 273, 12307–12315. doi: 10.1074/jbc.273.20.12307
Saul, R. A., and Wilson, W. G. (1990). A“new”skeletaldysplasia in two unrelated boys. Am. J. Med. Genet. 35, 388–393. doi: 10.1002/ajmg.1320350315
Schneeberger, P. E., von Elsner, L., Barker, E. L., Meinecke, P., Marquardt, I., Alawi, M., et al. (2020). Bi-allelic pathogenic variants in HS2ST1 cause a syndrome characterized by developmental delay and corpus callosum, skeletal, and renal abnormalities. Am. J. Hum. Genet. 107, 1044–1061. doi: 10.1016/j.ajhg.2020.10.007
Schreml, J., Durmaz, B., Cogulu, O., Keupp, K., Beleggia, F., Pohl, E., et al. (2014). The missing “link”: an autosomal recessive short stature syndrome caused by a hypofunctional XYLT1 mutation. Hum. Genet. 133, 29–39. doi: 10.1007/s00439-013-1351-y
Seidler, D. G., Faiyaz-Ul-Haque, M., Hansen, U., Yip, G. W., Zaidi, S. H., Teebi, A. S., et al. (2006). Defective glycosylation of decorin and biglycan, altered collagen structure, and abnormal phenotype of the skin fibroblasts of an Ehlers-Danlos syndrome patient carrying the novel Arg270Cys substitution in galactosyltransferase I (β4GalT-7). J. Mol. Med. (Berl.) 84, 583–594. doi: 10.1007/s00109-006-0046-4
Seidler, U., and Nikolovska, K. (2019). Slc26 family of anion transporters in the gastrointestinal tract: expression, function, regulation, and role in disease. Compr. Physiol. 9, 839–872. doi: 10.1002/cphy.c180027
Seko, A., Dohmae, N., Takio, K., and Yamashita, K. (2003). β1,4-Galactosyltransferase (β4GalT)-IV is specific for GlcNAc 6-O-sulfate. β4GalT-IV acts on keratan sulfate-related glycans and a precursor glycan of 6-sulfosialyl-Lewis X. J. Biol. Chem. 278, 9150–9158. doi: 10.1074/jbc.m211480200
Seko, A., and Yamashita, K. (2004). β1,3-N-Acetylglucosaminyltransferase-7 (β3Gn-T7) acts efficiently on keratan sulfate-related glycans. FEBS Lett. 556, 216–220. doi: 10.1016/s0014-5793(03)01440-6
Sertié, A. L., Sossi, V., Camargo, A. A., Zatz, M., Brahe, C., and Passos-Bueno, M. R. (2000). Collagen XVIII, containing an endogenous inhibitor of angiogenesis and tumor growth, plays a critical role in the maintenance of retinal structure and in neural tube closure (Knobloch syndrome). Hum. Mol. Genet. 9, 2051–2058. doi: 10.1093/hmg/9.13.2051
Shabbir, R. M. K., Nalbant, G., Ahmad, N., Malik, S., and Tolun, A. (2018). Homozygous CHST11 mutation in chondrodysplasia, brachydactyly, overriding digits, clino-symphalangism and synpolydactyly. J. Med. Genet. 55, 489–496. doi: 10.1136/jmedgenet-2017-105003
Sher, G., and Naeem, M. (2014). A novel CHSY1 gene mutation underlies Temtamy preaxial brachydactyly syndrome in a Pakistani family. Eur. J. Med. Genet. 57, 21–24. doi: 10.1016/j.ejmg.2013.11.001
Shi, J., Potash, J. B., Knowles, J. A., Weissman, M. M., Coryell, W., Scheftner, W. A., et al. (2011). Genome-wide association study of recurrent early-onset major depressive disorder. Mol. Psychiatry 16, 193–201.
Silveira, C., Leal, G. F., and Cavalcanti, D. P. (2016). Desbuquois dysplasia type II in a patient with a homozygous mutation in XYLT1 and new unusual findings. Am. J. Med. Genet. A 170, 3043–3047. doi: 10.1002/ajmg.a.37858
Singh, A., Kim, O. H., Iida, A., Park, W. Y., Ikegawa, S., and Kapoor, S. (2015). A novel CANT1 mutation in three Indian patients with Desbuquois dysplasia kim type. Eur. J. Med. Genet. 58, 105–110. doi: 10.1016/j.ejmg.2014.11.006
Smith, T. M., Hicks-Berger, C. A., Kim, S., and Kirley, T. L. (2002). Cloning, expression, and characterization of a soluble calcium-activated nucleotidase, a human enzyme belonging to a new family of extracellular nucleotidases. Arch. Biochem. Biophys. 406, 105–115. doi: 10.1016/s0003-9861(02)00420-4
Sonoda, T., and Kouno, K. (2000). Two brothers with distal arthrogryposis, peculiar facial appearance, cleft palate, short stature, hydronephrosis, retentio testis, and normal intelligence: a new type of distal arthrogryposis? Am. J. Med. Genet. 91, 280–285. doi: 10.1002/(sici)1096-8628(20000410)91:4<280::aid-ajmg8>3.0.co;2-b
Spicer, A. P., Kaback, L. A., Smith, T. J., and Seldin, M. F. (1998). Molecular cloning and characterization of the human and mouse UDP-glucose dehydrogenase genes. J. Biol. Chem. 273, 25117–25124. doi: 10.1074/jbc.273.39.25117
Srivastava, P., Pandey, H., Agarwal, D., Mandal, K., and Phadke, S. R. (2017). Spondyloepiphyseal dysplasia Omani type: CHST3 mutation spectrum and phenotypes in three Indian families. Am. J. Med. Genet. A 173, 163–168. doi: 10.1002/ajmg.a.37996
Stattin, E. L., Wiklund, F., Lindblom, K., Onnerfjord, P., Jonsson, B. A., Tegner, Y., et al. (2010). A missense mutation in the aggrecan C-type lectin domain disrupts extracellular matrix interactions and causes dominant familial osteochondritis dissecans. Am. J. Hum. Genet. 86, 126–137. doi: 10.1016/j.ajhg.2009.12.018
Stickens, D., Clines, G., Burbee, D., Ramos, P., Thomas, S., Hogue, D., et al. (1996). The EXT2 multiple exostoses gene defines a family of putative tumour suppressor genes. Nat. Genet. 14, 25–32. doi: 10.1038/ng0996-25
Stum, M., Davoine, C. S., Vicart, S., Guillot-Noël, L., Topaloglu, H., Carod-Artal, F. J., et al. (2006). Spectrum of HSPG2 (Perlecan) mutations in patients with Schwartz-Jampel syndrome. Hum. Mutat. 27, 1082–1091. doi: 10.1002/humu.20388
Sugahara, K., and Kitagawa, H. (2000). Recent advances in the study of the biosynthesis and functions of sulfated glycosaminoglycans. Curr. Opin. Struct. Biol. 10, 518–527. doi: 10.1016/s0959-440x(00)00125-1
Sugahara, K., and Schwartz, N. B. (1979). Defect in 3’-phosphoadenosine 5’-phosphosulfate formation in brachymorphic mice. Proc. Natl. Acad. Sci. U. S. A. 76, 6615–6618. doi: 10.1073/pnas.76.12.6615
Sultana, A., Sridhar, M. S., Klintworth, G. K., Balasubramanian, D., and Kannabiran, C. (2005). Allelic heterogeneity of the carbohydrate sulfotransferase-6 gene in patients with macular corneal dystrophy. Clin. Genet. 68, 454–460. doi: 10.1111/j.1399-0004.2005.00517.x
Superti-Furga, A., Hästbacka, J., Wilcox, W. R., Cohn, D. H., van der Harten, H. J., Rossi, A., et al. (1996). Achondrogenesis type IB is caused by mutations in the diastrophic dysplasia sulphate transporter gene. Nat. Genet. 12, 100–102. doi: 10.1038/ng0196-100
Suzuki, O. T., Sertié, A. L., Der Kaloustian, V. M., Kok, F., Carpenter, M., Murray, J., et al. (2002). Molecular analysis of collagen XVIII reveals novel mutations, presence of a third isoform, and possible genetic heterogeneity in Knobloch syndrome. Am. J. Hum. Genet. 71, 1320–1329. doi: 10.1086/344695
Swarr, D. T., Bloom, D., Lewis, R. A., Elenberg, E., Friedman, E. M., Glotzbach, C., et al. (2010). Potocki-Shaffer syndrome: comprehensive clinical assessment, review of the literature, and proposals for medical management. Am. J. Med. Genet. A 152A, 565–572. doi: 10.1002/ajmg.a.33245
Syx, D., Van Damme, T., Symoens, S., Maiburg, M. C., van de Laar, I., Morton, J., et al. (2015). Genetic heterogeneity and clinical variability in musculocontractural Ehlers-Danlos syndrome caused by impaired dermatan sulfate biosynthesis. Hum. Mutat. 36, 535–547. doi: 10.1002/humu.22774
Szulc, B., Sosicka, P., Maszczak-Seneczko, D., Skurska, E., Shauchuk, A., Olczak, T., et al. (2020). Biosynthesis of GlcNAc-rich N- and O-glycans in the Golgi apparatus does not require the nucleotide sugar transporter SLC35A3. J. Biol. Chem. 295, 16445–16463. doi: 10.1074/jbc.ra119.012362
Tahvanainen, E., Forsius, H., Kolehmainen, J., Damsten, M., Fellman, J., and de la Chapelle, A. (1996). The genetics of cornea plana congenita. J. Med. Genet. 33, 116–119. doi: 10.1136/jmg.33.2.116
Takeda, R., Takagi, M., Shinohara, H., Futagawa, H., Narumi, S., Hasegawa, T., et al. (2017). Novel compound heterozygous mutations identified by whole exome sequencing in a Japanese patient with geroderma osteodysplastica. Eur. J. Med. Genet. 60, 635–638. doi: 10.1016/j.ejmg.2017.08.002
Tanteles, G. A., Dixit, A., Dhar, S., and Surim, M. (2013). Two Somali half-siblings with CHST3-related chondrodysplasia illustrating the phenotypic spectrum and intrafamilial variability. Am. J. Med. Genet. A 161A, 2588–2593.
Taylan, F., Costantini, A., Coles, N., Pekkinen, M., Héon, E., Şıklar, Z., et al. (2016). Spondyloocular syndrome: novel mutations in XYLT2 gene and expansion of the phenotypic spectrum. J. Bone Miner. Res. 31, 1577–1585. doi: 10.1002/jbmr.2834
Taylan, F., Yavaş Abalı, Z., Jäntti, N., Güneş, N., Darendeliler, F., Baş, F., et al. (2017). Two novel mutations in XYLT2 cause spondyloocular syndrome. Am. J. Med. Genet. A 173, 3195–3200. doi: 10.1002/ajmg.a.38470
Temtamy, S., Aglan, M., Topaloglu, A. K., Wollnik, B., Amr, K., El-Badry, T. H., et al. (2012). Definition of the phenotypic spectrum of Temtamy preaxial brachydactyly syndrome associated with autosomal recessive CHYS1 mutations. Middle East J. Med. Genet. 1, 64–70. doi: 10.1097/01.mxe.0000414918.78299.94
Temtamy, S. A., Meguid, N. A., Ismail, S. I., and Ramzy, M. I. (1998). A new multiple congenital anomaly, mental retardation syndrome with preaxial brachydactyly, hyperphalangism, deafness and orodental anomalies. Clin. Dysmorph. 7, 249–255. doi: 10.1097/00019605-199810000-00003
Thelin, M. A., Bartolini, B., Axelsson, J., Gustafsson, R., Tykesson, E., Pera, E., et al. (2013). Biological functions of iduronic acid in chondroitin/dermatan sulfate. FEBS J. 280, 2431–2446. doi: 10.1111/febs.12214
Thiele, H., Sakano, M., Kitagawa, H., Sugahara, K., Rajab, A., Höhne, W., et al. (2004). Loss of chondroitin 6-O-sulfotransferase-1 function results in severe human chondrodysplasia with progressive spinal involvement. Proc. Natl. Acad. Sci. U. S. A. 101, 10155–10160. doi: 10.1073/pnas.0400334101
Thoden, J. B., Wohlers, T. M., Fridovich-Keil, J. L., and Holden, H. M. (2001). Human UDP-galactose 4-epimerase. Accommodation of UDP-N-acetylglucosamine within the active site. J. Biol. Chem. 276, 15131–15136.
Tian, J., Ling, L., Shboul, M., Lee, H., O’Connor, B., Merriman, B., et al. (2010). Loss of CHSY1, a secreted FRINGE enzyme, causes syndromic brachydactyly in humans via increased NOTCH signaling. Am. J. Hum. Genet. 87, 768–778. doi: 10.1016/j.ajhg.2010.11.005
Tompson, S. W., Merriman, B., Funari, V. A., Fresquet, M., Lachman, R. S., Rimoin, D. L., et al. (2009). A recessive skeletal dysplasia, SEMD aggrecan type, results from a missense mutation affecting the C-type lectin domain of aggrecan. Am. J. Hum. Genet. 84, 72–79. doi: 10.1016/j.ajhg.2008.12.001
Tone, Y., Pedersen, L. C., Yamamoto, T., Izumikawa, T., Kitagawa, H., Nishihara, J., et al. (2008). 2-O-phosphorylation of xylose and 6-O-sulfation of galactose in the protein linkage region of glycosaminoglycans influence the glucuronyltransferase-I activity involved in the linkage region synthesis. J. Biol. Chem. 283, 16801–16807. doi: 10.1074/jbc.m709556200
Tornberg, J., Sykiotis, G. P., Keefe, K., Plummer, L., Hoang, X., Hall, J. E., et al. (2011). Heparan sulfate 6-O-sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotrophic hypogonadism. Proc. Natl. Acad. Sci. U. S. A. 108, 11524–11529. doi: 10.1073/pnas.1102284108
Tuysuz, B., Mizumoto, S., Sugahara, K., Celebi, A., Mundlos, S., and Turkmen, S. (2009). Omani-type spondyloepiphyseal dysplasia with cardiac involvement caused by a missense mutation in CHST3. Clin. Genet. 75, 375–383. doi: 10.1111/j.1399-0004.2009.01167.x
Tüysüz, B., Yılmaz, S., Gül, E., Kolb, L., Bilguvar, K., Evliyaoğlu, O., et al. (2013). Spondyloepimetaphyseal dysplasia Pakistani type: expansion of the phenotype. Am. J. Med. Genet. A 161A, 1300–1308.
Umair, M., Eckstein, G., Rudolph, G., Strom, T., Graf, E., Hendig, D., et al. (2018). Homozygous XYLT2 variants as a cause of spondyloocular syndrome. Clin. Genet. 93, 913–918. doi: 10.1111/cge.13179
Unger, S., Lausch, E., Rossi, A., Mégarbané, A., Sillence, D., Alcausin, M., et al. (2010). Phenotypic features of carbohydrate sulfotransferase 3 (CHST3) deficiency in 24 patients: congenital dislocations and vertebral changes as principal diagnostic features. Am. J. Med. Genet. 152A, 2543–2549. doi: 10.1002/ajmg.a.33641
Uyama, T., Kitagawa, H., Tamura, J., and Sugahara, K. (2002). Molecular cloning and expression of human chondroitin N-acetylgalactosaminyltransferase: the key enzyme for chain initiation and elongation of chondroitin/dermatan sulfate on the protein linkage region tetrasaccharide shared by heparin/heparan sulfate. J. Biol. Chem. 277, 8841–8846.
Uyama, T., Kitagawa, H., Tanaka, J., Tamura, J., Ogawa, T., and Sugahara, K. (2003). Molecular cloning and expression of a second chondroitin N-acetylgalactosaminyltransferase involved in the initiation and elongation of chondroitin/dermatan sulfate. J. Biol. Chem. 278, 3072–3078. doi: 10.1074/jbc.m209446200
Van Ginderdeuren, R., De Vos, R., Casteels, I., and Foets, B. (2002). Report of a new family with dominant congenital heredity stromal dystrophy of the cornea. Cornea 21, 118–120. doi: 10.1097/00003226-200201000-00025
Van Laer, L., Dietz, H., and Loeys, B. (2014). Loeys-Dietz syndrome. Adv. Exp. Med. Biol. 802, 95–105.
van Roij, M. H., Mizumoto, S., Yamada, S., Morgan, T., Tan-Sindhunata, M. B., Meijers-Heijboer, H., et al. (2008). Spondyloepiphyseal dysplasia, Omani type: further definition of the phenotype. Am. J. Med. Genet. A 146A, 2376–2384. doi: 10.1002/ajmg.a.32482
Venkatachalam, K. V. (2003). Human 3′-phosphoadenosine 5′-phosphosulfate (PAPS) synthase: biochemistry, molecular biology and genetic deficiency. IUBMB Life 55, 1–11.
Veugelers, M., Cat, B. D., Muyldermans, S. Y., Reekmans, G., Delande, N., Frints, S., et al. (2000). Mutational analysis of the GPC3/GPC4 glypican gene cluster on Xq26 in patients with Simpson-Golabi-Behmel syndrome: identification of loss-of-function mutations in the GPC3 gene. Hum. Mol. Genet. 9, 1321–1328. doi: 10.1093/hmg/9.9.1321
Vissers, L. E., Lausch, E., Unger, S., Campos-Xavier, A. B., Gilissen, C., Rossi, A., et al. (2011). Chondrodysplasia and abnormal joint development associated with mutations in IMPAD1, encoding the Golgi-resident nucleotide phosphatase, gPAPP. Am. J. Hum. Genet. 88, 608–615. doi: 10.1016/j.ajhg.2011.04.002
Vodopiutz, J., Mizumoto, S., Lausch, E., Rossi, A., Unger, S., Janocha, N., et al. (2017). Chondroitin sulfate N-acetylgalactosaminyltransferase-1 (CSGalNAcT-1) deficiency results in a mild skeletal dysplasia and joint laxity. Hum. Mutat. 38, 34–38. doi: 10.1002/humu.23070
Volpi, S., Yamazaki, Y., Brauer, P. M., van Rooijen, E., Hayashida, A., Slavotinek, A., et al. (2017). EXTL3 mutations cause skeletal dysplasia, immune deficiency, and developmental delay. J. Exp. Med. 214, 623–637. doi: 10.1084/jem.20161525
von Oettingen, J. E., Tan, W. H., and Dauber, A. (2014). Skeletal dysplasia, global developmental delay, and multiple congenital anomalies in a 5-year-old boy-report of the second family with B3GAT3 mutation and expansion of the phenotype. Am. J. Med. Genet. A 164A, 1580–1586. doi: 10.1002/ajmg.a.36487
Vorster, A. A., Beighton, P., and Ramesar, R. S. (2015). Spondyloepimetaphyseal dysplasia with joint laxity (Beighton type); mutation analysis in eight affected South African families. Clin. Genet. 87, 492–495. doi: 10.1111/cge.12413
Waryah, A. M., Shahzad, M., Shaikh, H., Sheikh, S. A., Channa, N. A., Hufnagel, R. B., et al. (2016). A novel CHST3 allele associated with spondyloepiphyseal dysplasia and hearing loss in Pakistani kindred. Clin. Genet. 90, 90–95. doi: 10.1111/cge.12694
Wuyts, W., Roland, D., Lüdecke, H. J., Wauters, J., Foulon, M., Van Hul, W., et al. (2002). Multiple exostoses, mental retardation, hypertrichosis, and brain abnormalities in a boy with a de novo 8q24 submicroscopic interstitial deletion. Am. J. Med. Genet. 113, 326–332. doi: 10.1002/ajmg.10845
Wuyts, W., and Van Hul, W. (2000). Molecular basis of multiple exostoses: mutations in the EXT1 and EXT2 genes. Hum. Mutat. 15, 220–227. doi: 10.1002/(sici)1098-1004(200003)15:3<220::aid-humu2>3.0.co;2-k
Wuyts, W., Van Hul, W., De Boulle, K., Hendrickx, J., Bakker, E., Vanhoenacker, F., et al. (1998). Mutations in the EXT1 and EXT2 genes in hereditary multiple exostoses. Am. J. Hum. Genet. 62, 346–354.
Yamauchi, S., Mita, S., Matsubara, T., Fukuta, M., Habuchi, H., Kimata, K., et al. (2000). Molecular cloning and expression of chondroitin 4-sulfotransferase. J. Biol. Chem. 275, 8975–8981. doi: 10.1074/jbc.275.12.8975
Yap, P., Liebelt, J. E., Amor, D. J., Moore, L., and Savarirayan, R. (2016). Pseudodiastrophic dysplasia: Two cases delineating and expanding the pre and postnatal phenotype. Am. J. Med. Genet. A 170A, 1363–1366. doi: 10.1002/ajmg.a.37548
Yauy, K., Tran Mau-Them, F., Willems, M., Coubes, C., Blanchet, P., Herlin, C., et al. (2018). B3GAT3-related disorder with craniosynostosis and bone fragility due to a unique mutation. Genet. Med. 20, 269–274. doi: 10.1038/gim.2017.109
Yoshizawa, T., Mizumoto, S., Takahashi, Y., Shimada, S., Sugahara, K., Nakayama, J., et al. (2018). Vascular abnormalities in the placenta of Chst14–/– fetuses: implications in the pathophysiology of perinatal lethality of the murine model and vascular lesions in human CHST14/D4ST1 deficiency. Glycobiology 28, 80–89. doi: 10.1093/glycob/cwx099
Zayed, H., Chao, R., Moshrefi, A., Lopezjimenez, N., Delaney, A., Chen, J., et al. (2010). A maternally inherited chromosome 18q22.1 deletion in a male with late-presenting diaphragmatic hernia and microphthalmia-evaluation of DSEL as a candidate gene for the diaphragmatic defect. Am. J. Med. Genet. 152A, 916–923. doi: 10.1002/ajmg.a.33341
Keywords: chondroitin sulfate, dermatan sulfate, heparan sulfate, proteoglycan, glycosaminoglycan, connective tissue disorder, skeletal disorder, skin disorder
Citation: Mizumoto S and Yamada S (2021) Congenital Disorders of Deficiency in Glycosaminoglycan Biosynthesis. Front. Genet. 12:717535. doi: 10.3389/fgene.2021.717535
Received: 31 May 2021; Accepted: 12 August 2021;
Published: 03 September 2021.
Edited by:
Fransiska Malfait, Ghent University, BelgiumReviewed by:
Robert Smigiel, Wrocław Medical University, PolandNicolina Cristina Sorrentino, Telethon Institute of Genetics and Medicine (TIGEM), Italy
Copyright © 2021 Mizumoto and Yamada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuji Mizumoto, bWl6dW1vdG9AbWVpam8tdS5hYy5qcA==