 Duo Yang1†
Duo Yang1† Yong Chen
Yong Chen Wanqin Xie
Wanqin Xie- 1Department of Ophthalmology, The Jili Hospital of Liuyang and the Eye Hospital of Liuyang, Changsha, China
- 2National Health Committee Key Laboratory of Birth Defects for Research and Prevention, Hunan Provincial Maternal and Child Health Care Hospital, Changsha, China
- 3State Key Laboratory of Medicinal Chemical Biology, Tianjin Key Laboratory of Protein Science, College of Life Sciences, Nankai University, Tianjin, China
The signal-induced proliferation-associated 1-like 3 (SIPA1L3) gene that encodes a putative Rap GTPase-activating protein (RapGAP) has been associated with congenital cataract and eye development abnormalities. However, our current understanding of the mutation spectrum of SIPA1L3 associated with eye defects is limited. By using whole-exome sequencing plus Sanger sequencing validation, we identified a novel heterozygous c.1871A > G (p.Lys624Arg) variation within the predicted RapGAP domain of SIPA1L3 in the proband with isolated juvenile-onset cataracts from a three-generation Chinese family. In this family, the proband's father and grandmother were also heterozygous for the c.1871A > G variation and affected by cataracts varying in morphology, severity, and age of onset. Sequence alignment shows that the Lys 624 residue of SIPA1L3 is conserved across the species. Based on the resolved structure of Rap1–Rap1GAP complex, homology modeling implies that the Lys 624 residue is structurally homologous to the Lys 194 of Rap1GAP, a highly conserved lysine residue that is involved in the interface between Rap1 and Rap1GAP and critical for the affinity to Rap·GTP. We reasoned that arginine substitution of lysine 624 might have an impact on the SIPA1L3-Rap·GTP interaction, thereby affecting the regulatory function of SIPA1L3 on Rap signaling. Collectively, our finding expands the mutation spectrum of SIPA1L3 and provides new clues to the molecular mechanisms of SIPA1L3-related cataracts. Further investigations are warranted to validate the functional alteration of the p.Lys624Arg variant of SIPA1L3.
Introduction
Cataract, clinically characterized by opacity in the ocular lens, is a common eye disease that may cause vision impairment or blindness. Congenital cataract is usually diagnosed in the first year of life, with an average prevalence of 72 per 100,000 births worldwide (Shiels and Hejtmancik, 2019). About 30% of congenital cataract cases have a genetic etiology (Li et al., 2020). The most common inheritance pattern of congenital cataract is autosomal dominant, but it can be autosomal recessive or X-linked inheritance in some cases (Berry et al., 2020). Notably, while most inherited cataracts present at birth or early childhood, some can have an onset as late as adulthood (Shiels and Hejtmancik, 2019). More than 40 genes have been associated with congenital/inherited cataract. These genes mainly encode crystallins, membrane proteins, transcription or developmental factors, cytoskeletal proteins, and other proteins in the lens (Shiels and Hejtmancik, 2019; Berry et al., 2020).
The signal-induced proliferation-associated 1-like 3 (SIPA1L3) gene [Online Mendelian Inheritance in Man (OMIM) *616655 and #616851], located on chromosome 19, encodes a putative GTPase-activating protein (GAP) specific for the small GTPases of Rap family. The full-length human SIPA1L3 protein consists of 1,781 amino acid residues, with the predicted Rap GTPase-activating protein (RapGAP) domain (residues 611–828), PDZ domain (residues 966–1,042), and C-terminal coiled–coiled domain (residues 1,720–1,774). The RapGAP domain of the SIPA1L3 homolog SIPA1L1 has been shown to interact with Rap1, facilitating the switch of Rap1 from an active GTP-bound state to an inactive GDP-bound state (Evers et al., 2015).
Rap signaling plays crucial roles in cell growth, differentiation, and organization of the cytoskeleton in the human lens (Spilker and Kreutz, 2010). Hence, mutations in SIPA1L3 may disturb the regulation of Rap signaling during embryonic eye development and cause eye defects (Greenlees et al., 2015). SIPA1L3 was first identified as a causative gene for congenital cataract in a distantly consanguineous German family with two sisters homozygous for the c.4489C > T (p.R1497X) non-sense mutation (Evers et al., 2015). This mutation results in a truncated protein lacking the last 284 amino acids and is thought to be loss of function. In another independent study, two unrelated patients who carried distinct mutations in SIPA1L3 were documented. Patient 1 who manifested severe lens and ocular anterior segment abnormalities with cataracts carried a de novo balanced translocation, 46,XY,t(2;19)(q37.3;q13.1), which generated a variant of SIPA1L3 with a transection of 5′ un-translated region. Reduced SIPA1L3 expression was detected in the patient's lymphoblast. Patient 2 from the same study was found with bilateral congenital cataracts and heterozygous for a missense variation c.442G > T (p.Asp148Tyr). The p.Asp148Tyr mutation resides in the putative actin-binding domain of SIPA1L3, and the mutant has been shown to cause abnormal cytoskeleton organization and cell adhesion in epithelial cells (Greenlees et al., 2015).
Though SIPA1L3 has been known as a causative gene for cataracts and eye anterior dysgenesis, overall, our current understanding of the mutation spectrum of SIPA1L3 related to eye defects is limited. In this study, we report a novel missense variant, namely, the SIPA1L3 c.1871A > G (p.Lys624Arg) variant that co-segregated with cataracts in a three-generation Chinese family.
Materials and Methods
Ethical Compliance
This study was approved by the Ethics Committee of Hunan Provincial Maternal and Child Health Care Hospital. Written informed consent was obtained from the patient or the patient's legal representative for genetic tests.
Patients
A 17-year-old male presented complaining of loss of vision of the left eye. At the age of 14, he sensed a gradual blurring of vision of the left eye with fixed central visual occlusion, and he left it untreated at that time. The symptoms of the left eye became aggravated when he aged 15. At the age of 17, he incidentally found that the left eye had lost vision. He visited the ophthalmic outpatient at the Eye Hospital of Liuyang with his grandmother. Physical examination showed that the patient had normal cardiopulmonary function with no remarkable congenital anomalies or developmental disorders. Opacity of the lens in the left eye was readily visible. Hand movement test, count finger test, and light perception of the left eye were passed by the patient, and color vision was normal. Distant vision of the right eye was 20/80, and near vision of the right eye was 0.5/33 cm. The corneas of both eyes were transparent and iris texture was clear. No abnormality was seen in routine blood tests. During the outpatient visit, the patient's grandmother (aged 71) reported that she used to suffer from severe cataracts and had received surgery in the hospital and that the patient's father (aged 40) also had cataracts, but his vision in both eyes was still acceptable and had not received treatment yet. The index patient was treated by cataract surgery. Vision acuity examination after surgery showed distant vision 20/100 and near vision 0.7/33 cm for the left eye and no vision change for the right eye. To find out the potential causative mutation for cataract in the family, the index patient and his parents and grandmother volunteered for genetic tests.
Genetic Analysis
Genomic DNA extraction from blood was performed using QIAamp DNA Blood Mini Kit (QIAGEN, Hilden, Germany). Chromosomal microarray analysis (CMA) was performed using Affymetrix CytoScan®750 K Array (Affymetrix Inc., CA, USA) according to the manufacturer's instructions.
Whole-exome sequencing (WES) was performed following the pipeline protocols at BGI Clinical Testing Center (Shenzhen, Guangdong, China). In this clinic-oriented service, the annotation of sequence variants including single-nucleotide variation (SNV) and insertion–deletion (InDel) was limited to those derived from the definitive disease-associated genes in the OMIM database. Among the annotated variants, further filtering for identification of candidate variants was based on phenotypic overlap with genes in OMIM; minor allele frequency [<1% in public databases including NHLBI GO Exome Sequencing Project (ESP6500), 1000 Genomes, Exome Aggregation Consortium (ExAC), Genome Aggregation Database (GnomAD), and/or GnomAD-EAS]; and predictive results from bioinformatics tools including SIFT, Mutation Taster, and PolyPhen-2. Sanger sequencing was conducted at RuiBiotech (Beijing, China), and the PCR primers were designed according to the reference genomic sequence of SIPA1L3 (GenBank accession number: NG_046730.1).
Molecular Modeling
In terms of the structural data of Rap1–Rap1GAP complex deposited in the protein data bank (PDB entry: 1SRQ), homology modeling of the RapGAP domain (residues 611 to 828) of SIPA1L3 (UniProKB: O60292) was performed using the SWISS-MODEL (https://swissmodel.expasy.org/) (Waterhouse et al., 2018). All structure figures were prepared using PyMOL (http://pymol.sourceforge.net/). The schematic diagram of Rap1–Rap1GAP interactions was generated by using LigPlus (http://www.ebi.ac.uk/thornton-srv/software/LigPlus/) (Laskowski and Swindells, 2011).
Results
Characteristics of the Cataracts in the Index Patient
The left eye of the index patient presented with cortical and nuclear white opacities of the lens (Figure 1A). The lens posterior capsule and the fundus of the left eye could not be seen clearly (Figure 1B). His right eye showed gray–white opacities at the Y suture region of the lens nucleus, slight deposition of granular cloudy substances at the lens posterior capsule, and clear optic disc in fundus (Figures 1C,D). During the cataract surgery, slight liquefaction of the lens cortex and softness in the lens nucleus of the right eye were noticed.
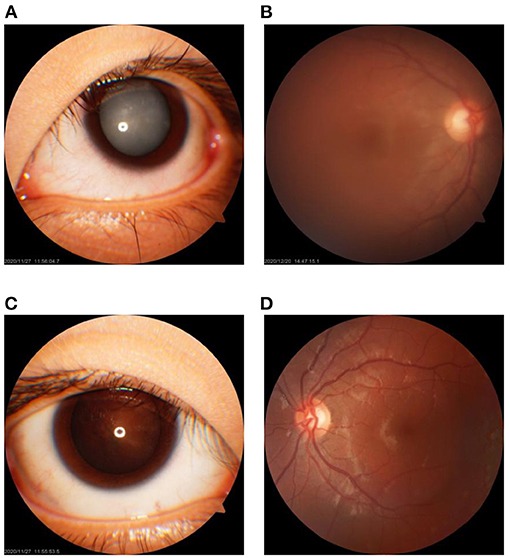
Figure 1. Clinical feature of the cataracts in the 17-year-old male proband. (A,B) The cortical cataract of the lens and the fundus of the left eye, respectively. (C,D) The nuclear cataract of the lens of the right eye with cloudiness at the posterior lens capsule and the fundus of the right eye, respectively.
Genetic Variation
CMA detected no common pathogenic copy number variations in the index patient and his grandmother (III-1 and II-2, Figure 2A) (data not shown). However, the index patient carried a heterozygous c.1871A > G/p.Lys624Arg variation in SIPA1L3 that is thought to be associated with cataract as revealed by WES (Supplementary Data Sheet 1). This variation was also detected in his grandmother (II-2) with two additional heterozygous variations related to cataracts, namely, the DNMBP c.3410G > A/p.Arg1137Gln on chromosome 10 and the NHS c.350C>G/p.Ala117Gly on chromosome X (Supplementary Data Sheet 2). Considering the previous findings that the DNMBP and NHS are associated with autosomal recessive infantile cataract (Ansar et al., 2018) and X-linked congenital cataract (Burdon et al., 2003; Ling et al., 2019), respectively, and the fact that the DNMBP c.3420G > A and NHS c.350C > G variations were absent from the index patient, we thus chose to focus on the SIPA1L3 c.1871A > G variation for further analysis thereafter.
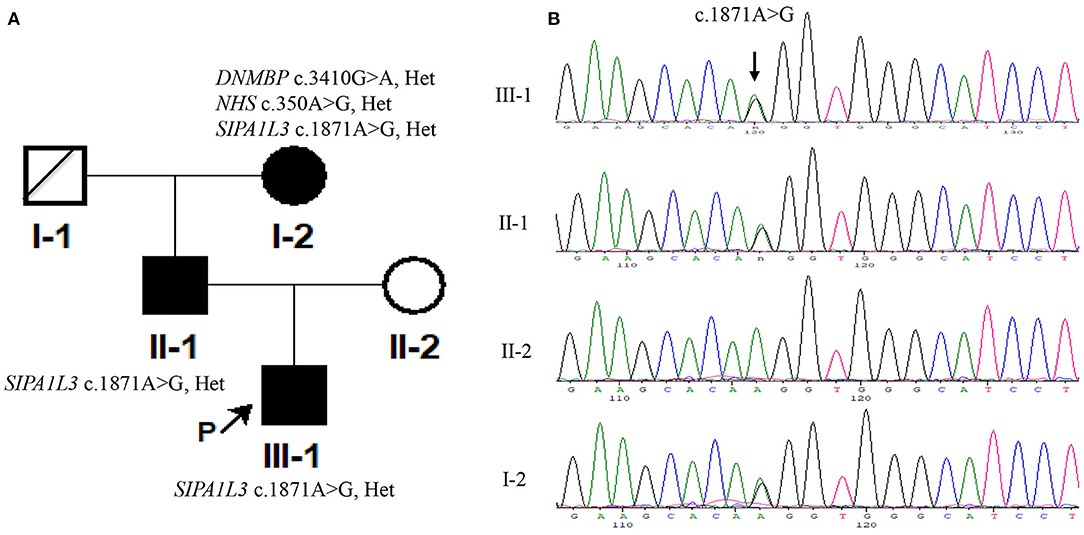
Figure 2. The signal-induced proliferation-associated 1-like 3 (SIPA1L3) c.1871A > G variation in the patients was detected by using Sanger sequencing. (A) The pedigree of the Chinese family in the current study. (B) Sanger sequencing results of c.1871A > G variation in the Chinese family. The genomic DNA used for PCR amplification was extracted using peripheral blood (III-1 and I-2) or oral swabs (II-1 and II-2). The sequencing was performed in both forward and reverse direction on a genetic analyzer 3730XL (Applied Biosystems). The results of forward sequencing are representatively shown.
The SIPA1L3 c.1871A > G missense variation possessed an allele frequency of 0.000009 in GnomAD (http://gnomad.broadinstitute.org) and was predicted to be deleterious by using SIFT (http://sift.jcvi.org) and Mutation Taster (http://www.mutationtaster.org). Sanger sequencing confirmed that the index patient and his father and grandmother (III-1, II-1, and I-2; Figure 2B) were heterozygous for the c.1871A > G variation and that this variation was absent from his mother (II-2, Figure 2B) and 200 unrelated controls comprising of 100 couples for assisted reproduction technology in Hunan Provincial Maternal and Child Health Care Hospital (data not shown). Moreover, the DNMBP c.3420G > A and NHS c.350C > G variations identified in the grandmother (I-2) were not detected in the index patient (III-1) and his father (II-1) (Figure 2A). According to ACMG guidelines (Richards et al., 2015), rules PM2, PP1, and PP3 were applied, and the c.1871A > G variant was considered to be of uncertain significance.
Molecular Modeling
Sequence alignment reveals that the RapGAP domain (residues 611–828) of SIPA1L3 is highly conserved across the species and highly homologous to the RapGAP domain (residues 181–397) of human Rap1GAP (UniProKB: P47736) (Figure 3A). Molecular modeling indicates that the Lys 624 of SIPA1L3 is structurally homologous to the Lys 194 of Rap1GAP (Figures 3B,C). In terms of the resolved structure, the Lys 194 of Rap1GAP is involved in the interface between Rap1 and Rap1GAP, where the Lys 194 of Rap1GAP forms a non-covalent bond with the threonine residue 61 (Thr61) in the switch II region of Rap1 (Figure 3D). Based on homology, it is speculated that arginine substitution of Lys 624 of SIPA1L3 may have a potential impact on the SIPA1L3–Rap interaction.
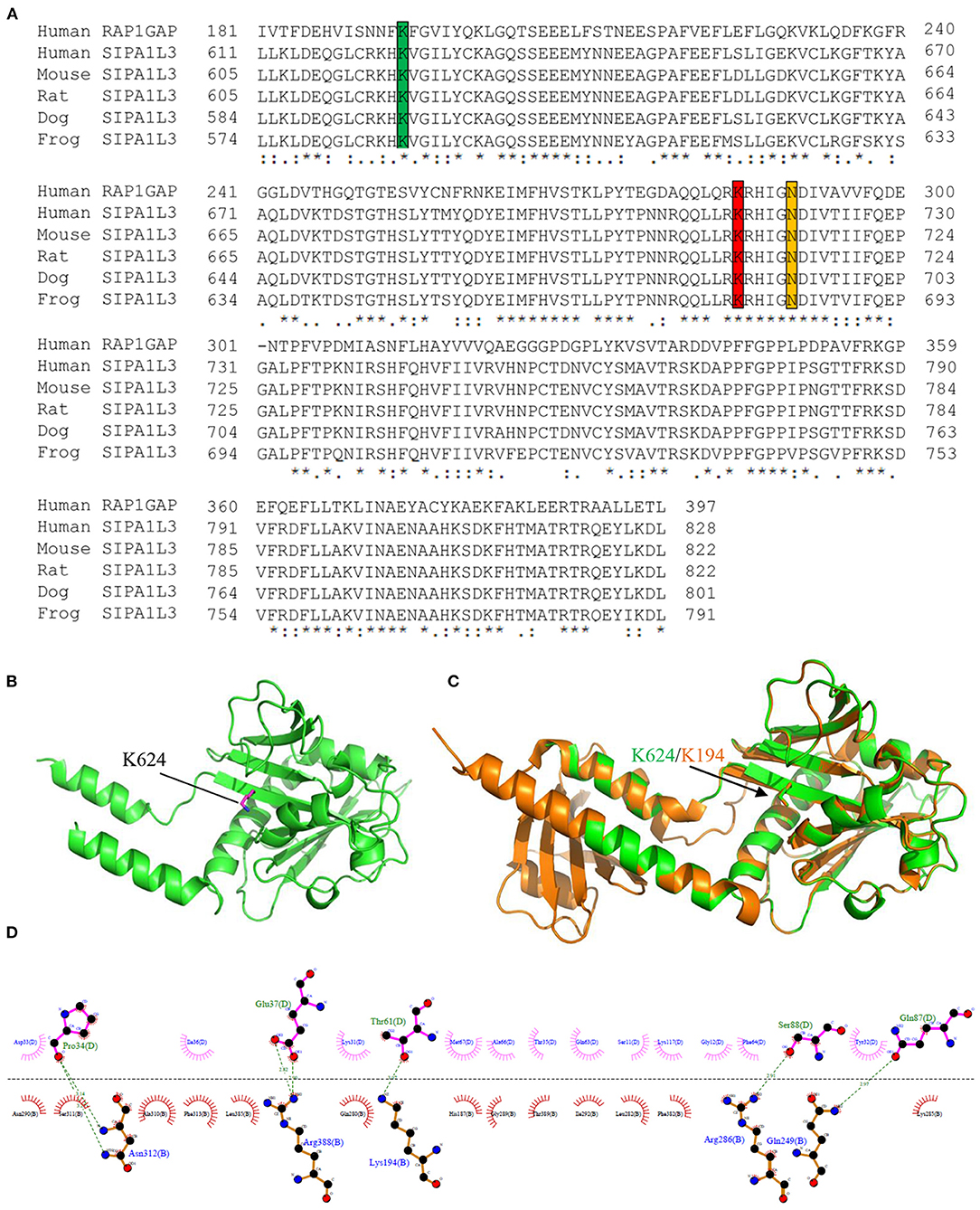
Figure 3. The lysine residue 624 within the Rap GTPase-activating protein (RapGAP) domain of SIPA1L3 is highly conserved and shares homology with the lysine residue 194 of human Rap1GAP. (A) Alignment of RapGAP domain of SIPA1L3 of different species with the human Rap1GAP. The highly conserved lysine residues 194 and 285 and the key catalytic asparagine residue (Asn) 290 of Rap1GAP are marked by green, red, and yellow, respectively. (B) Homology modeling shows the conformation of Lys 624 (purple) within the RapGAP domain of SIPA1L3. (C) The Lys 624 (green) is superimposed over the Lys 194 of Rap1GAP (brown). The crystal structure data of Rap1GAP was retrieved from Protein Data Bank (PDB Entry: 1SRQ). (D) In terms of the structural data of Rap1–Rap1GAP complex (1SRQ), the interface between Rap1 (molecule B) and Rap1GAP (molecule D) that involves the Lys 194 was generated using LigPlus software.
Discussion
SIPA1L3 is regarded as a negative regulator of Rap signaling that plays important roles in neuronal function and lens development (Spilker and Kreutz, 2010; Dolnik et al., 2016; Rothe et al., 2017). Previous studies have demonstrated that homozygous or heterozygous mutations in the SIPA1L3 gene may cause isolated cataracts or eye development abnormalities including microphthalmia and anterior segment dysgenesis with cataracts (Evers et al., 2015; Greenlees et al., 2015). Of note, the reported SIPA1L3-related cataracts were characterized by an infantile or early onset with high severity (Table 1). The present study reports the co-segregation of SIPA1L3 Lys624Arg mutation with isolated cataracts in a three-generation Chinese family. The 17-year-old male proband and his father and grandmother were affected by cataracts varying in morphology, severity, and age of onset. Even within the proband, the development of cataracts in his two eyes was asymmetric (Table 1). Though the detailed ages of onset could not be identified for the father and grandmother in this Chinese family (II-1 and I-2 in the pedigree, Figure 2A), it is unlikely that their cataracts had a very early onset in terms of their description. Though the mutation status in the grandmother (I-2, Figure 2A) was complicated by the concomitant presence of missense variants in DNMBP and NHS, Sanger sequencing confirmed that the proband and his father (III-1 and II-1, Figure 2A) only carried the candidate variation SIPA1L3 c.1871A > G (p.Lys624Arg). Based on the clinical findings in our study, the cataracts associated with SIPA1L3 p.Lys624Arg mutation can be heterogeneous within and between patients. As this kind of cataracts was found to affect the Y suture and the posterior lens capsule, central vision and development of visual system might be severely affected, leading to amblyopia. Early diagnosis and treatment may offer better vision and visual function to the affected individuals.
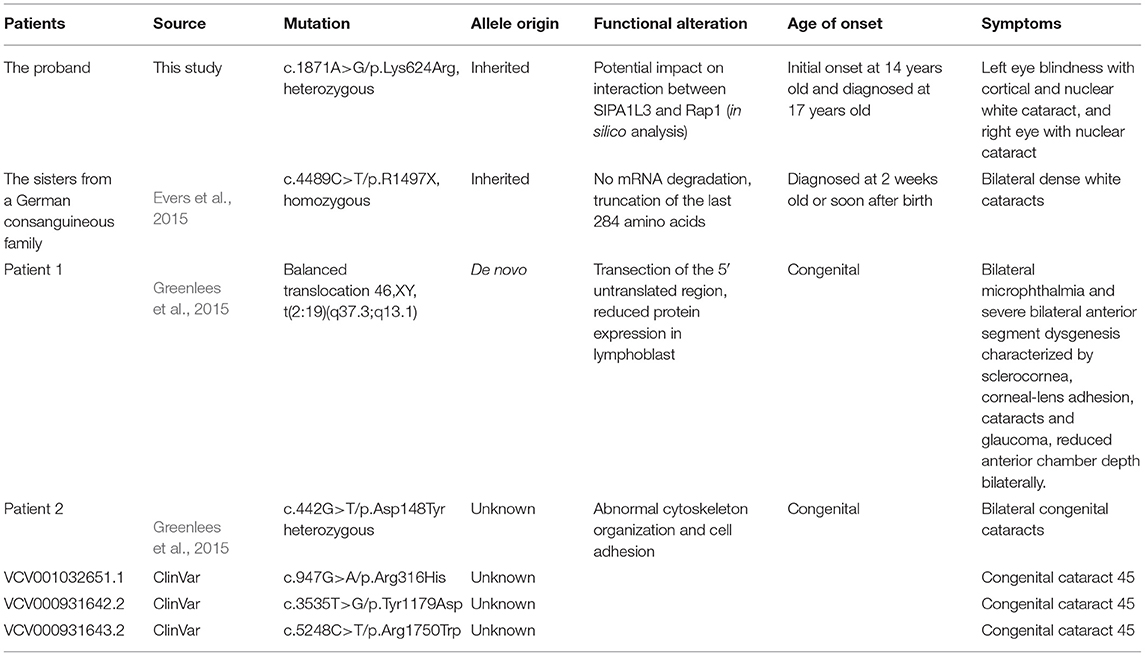
Table 1. Comparison of genotypes and phenotypes of patients with SIPA1L3-associated cataracts.
As implied by previous findings, the molecular mechanisms by which SIPA1L3 mutations cause cataracts could be diverse. The loss-of-function mechanism is implicated by the homozygous p.R1497X mutant identified in the two sisters with infantile bilateral dense white cataracts from a distantly consanguineous German family (Evers et al., 2015). The dominant-negative effect of the p.Asp148Tyr mutation in the putative actin-binding domain of SIPA1L3 is suggested by its heterozygosity in a patient with congenital bilateral cataracts, as well as the impact on cytoskeleton organization and cell adhesion upon transient expression of the mutant in epithelial cells (Greenlees et al., 2015). In addition, the possibility that haploinsufficiency may be causing cataracts could not be excluded. Reduced SIPA1L3 expression was detected in lymphoblast from a patient carrying a transaction of the 5′ untranslated region of SIPA1L3 resulting from a balanced translocation. The patient manifested lens and ocular anterior segment abnormalities including cataracts (Greenlees et al., 2015). Regarding the p.Lys624Arg missense mutation in the present study, the co-segregation of this mutation with cataracts in the three-generation Chinese family implies an autosomal dominant inheritance. Interestingly, this mutation is localized in the predicted RapGAP domain of SIPA1L3, a region that is thought to confer the GAP activity of the protein. However, as the lysine residue at codon 624 is replaced by arginine (an amino acid with similar properties), prediction of functional consequence of this mutation is more challenging.
The RapGAP domain of SIPA1L3 shares homology with Rap1GAP, a GTPase-activating protein specific for the well-studied Rap1 (Spilker and Kreutz, 2010). The crystal structure of Rap1–Rap1GAP complex has been resolved (Scrima et al., 2008). We have found that the Lys 624 within the RapGAP domain of SIPA1L3 shares high similarity with Lys 194 of Rap1GAP, a residue that is positioned at the interface between Rap1 and Rap1GAP and interacts with threonine residue 61 (Thr 61) of Rap1 to stabilize the complex. Based on homology, we infer that Lys 624 is likely involved in the interaction between SIPA1L3 and Rap. Although both lysine and arginine are positively charged amino acids, arginine substitution of Lys 624 may have an impact on the SIPA1L3–Rap interaction in view of the steric structure. However, to test this hypothesis, whether there is a functional conservation between SIPA1L3 and Rap1GAP should be addressed prior to determining how the Lys624Arg mutation would affect the affinity between SIPA1L3 and Rap·GTP. Alternatively, arginine substitution may block the post-translational modification (e.g., acetylation or ubiquitination) that is supposed to add to the Lys 624. Again, this hypothesis remains to be experimentally validated.
In summary, we report a novel SIPA1L3 missense variant, namely, the p.Lys624Arg mutant that co-segregated with isolated cataracts in a familial case. To the best of our knowledge, this is the first report regarding mutation in the putative RapGAP domain of SIPA1L3 that may be contributing to cataracts. Our finding expands the mutation spectrum of SIPA1L3 and provides new insights into the pathogenesis of SIPA1L3-related cataracts.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of Hunan Provincial Maternal and Child Health Care Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
DY and SZ recruited the patients and noted the clinical phenotypes of the patients. HaiZ and JL performed the experiments. HaoZ performed molecular modeling. ZY and BN interpreted the data of genetic tests. WX and YC designed the study and reviewed all the data. WX wrote the manuscript daft. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the Health Committee of Hunan Province (C20180704 and 225 Talents Project), the Natural Science Foundation of Hunan Province (2019JJ80078), and the Major Scientific and Technological Projects for Collaborative Prevention and Control of Birth Defects in Hunan Province (2019SK1010).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors sincerely thank the patients for participation in the present study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.715599/full#supplementary-material
References
Ansar, M., Chung, H. L., Taylor, R. L., Nazir, A., Imtiaz, S., Sarwar, M. T., et al. (2018). Bi-allelic loss-of-function variants in DNMBP cause infantile cataracts. Am. J. Hum. Genet. 103, 568–578. doi: 10.1016/j.ajhg.2018.09.004
Berry, V., Georgiou, M., Fujinami, K., Quinlan, R., Moore, A., and Michaelides, M. (2020). Inherited cataracts: molecular genetics, clinical features, disease mechanisms and novel therapeutic approaches. Br. J. Ophthalmol. 104, 1331–1337. doi: 10.1136/bjophthalmol-2019-315282
Burdon, K. P., McKay, J. D., Sale, M. M., Russell-Eggitt, I. M., Mackey, D. A., Wirth, M. G., et al. (2003). Mutations in a novel gene, NHS, cause the pleiotropic effects of Nance-Horan syndrome, including severe congenital cataract, dental anomalies, and mental retardation. Am. J. Hum. Genet. 73, 1120–1130. doi: 10.1086/379381
Dolnik, A., Kanwal, N., Mackert, S., Halbedl, S., Proepper, C., Bockmann, J., et al. (2016). Sipa1l3/SPAR3 is targeted to postsynaptic specializations and interacts with the Fezzin ProSAPiP1/Lzts3. J. Neurochem. 136, 28–35. doi: 10.1111/jnc.13353
Evers, C., Paramasivam, N., Hinderhofer, K., Fischer, C., Granzow, M., Schmidt-Bacher, A., et al. (2015). SIPA1L3 identified by linkage analysis and whole-exome sequencing as a novel gene for autosomal recessive congenital cataract. Eur. J. Hum. Genet. 23, 1627–1633. doi: 10.1038/ejhg.2015.46
Greenlees, R., Mihelec, M., Yousoof, S., Speidel, D., Wu, S. K., Rinkwitz, S., et al. (2015). Mutations in SIPA1L3 cause eye defects through disruption of cell polarity and cytoskeleton organization. Hum. Mol. Genet. 24, 5789–5804. doi: 10.1093/hmg/ddv298
Laskowski, R. A., and Swindells, M. B. (2011). LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786. doi: 10.1021/ci200227u
Li, J., Chen, X., Yan, Y., and Yao, K. (2020). Molecular genetics of congenital cataracts. Exp. Eye Res. 191:107872. doi: 10.1016/j.exer.2019.107872
Ling, C., Sui, R., Yao, F., Wu, Z., Zhang, X., and Zhang, S. (2019). Whole exome sequencing identified a novel truncation mutation in the NHS gene associated with Nance-Horan syndrome. BMC Med. Genet. 20:14. doi: 10.1186/s12881-018-0725-3
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Rothe, M., Kanwal, N., Dietmann, P., Seigfried, F. A., Hempel, A., Schütz, D., et al. (2017). An Epha4/Sipa1l3/Wnt pathway regulates eye development and lens maturation. Development 144, 321–333. doi: 10.1242/dev.147462
Scrima, A., Thomas, C., Deaconescu, D., and Wittinghofer, A. (2008). The Rap-RapGAP complex: GTP hydrolysis without catalytic glutamine and arginine residues. EMBO J. 27, 1145–1153. doi: 10.1038/emboj.2008.30
Shiels, A., and Hejtmancik, J. F. (2019). Biology of inherited cataracts and opportunities for treatment. Annu. Rev. Vis. Sci. 5, 123–149. doi: 10.1146/annurev-vision-091517-034346
Spilker, C., and Kreutz, M. R. (2010). RapGAPs in brain: multipurpose players in neuronal Rap signalling. Eur. J. Neurosci. 32, 1–9. doi: 10.1111/j.1460-9568.2010.07273.x
Keywords: Rap GTPase, GTPase-activating protein, SIPA1L3, RapGAP, cataract
Citation: Yang D, Zhou H, Lin J, Zhao S, Zhou H, Yin Z, Ni B, Chen Y and Xie W (2021) Case Report: A Novel Missense Variant in the SIPA1L3 Gene Associated With Cataracts in a Chinese Family. Front. Genet. 12:715599. doi: 10.3389/fgene.2021.715599
Received: 27 May 2021; Accepted: 19 August 2021;
Published: 16 September 2021.
Edited by:
Jian-Huan Chen, Jiangnan University, ChinaReviewed by:
Yanshan Liu, Wuxi Children's Hospital, ChinaHuang Yuqiang, Shantou University and the Chinese University of Hong Kong, China
Copyright © 2021 Yang, Zhou, Lin, Zhao, Zhou, Yin, Ni, Chen and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong Chen, Y2hlbnlvbmcwMDA4QHNpbmEuY29t; Wanqin Xie, d2FucWlueGllQDEyNi5jb20=
†These authors have contributed equally to this work and share first authorship
‡These authors share senior authorship