 Linya Ma1†
Linya Ma1† Jianjian Zhu
Jianjian Zhu Jing Wang
Jing Wang Dan Peng
Dan Peng- 1Department of Medical Genetics, Changde First People’s Hospital, Changde, China
- 2Changsha Kingmed Center for Clinical Laboratory, Changsha, China
- 3Affiliated Hospital of Changde City, University of South China, Hengyang, China
Background: Tyrosinase-positive oculocutaneous albinism (OCA, type II, OCA2) is an autosomal recessive genetic disease in which the biosynthesis of melanin decreases in the skin, hair, and eyes. OCA2 disease is caused by mutations in OCA2 gene. The gene product plays a role in regulating the pH of melanosomes. Up to now, hundreds of OCA2 mutations have been reported and novel variants are still being discovered.
Methods: In this study, we reviewed the records of OCA2 patients who had conducted albinism genetic testing, and then analyzed the clinical and genetic information of 28 OCA2 patients who had been genetically diagnosed by using Sanger sequencing and next-generation sequencing.
Results: In this study, we reported 31 variants screened from 28 Chinese OCA2 families, and characterized the detailed molecular and clinical presentations. There were 12 novel variants among all detected variants, including 3 missense variants (p.G393V, p.T482A, and p.R720P), 4 frameshift variants (p.R53Gfs∗49, p.N279Kfs∗17, p.I469Lfs∗4, p.I655Nfs∗12), 2 splicing variants (c.1637-2A > G, c.1951 + 1G > C), 2 stopgain variants (p.L278X, p.W652X) and 1 insertion variants (p.P315LinsT). One potential cluster of missense variants was implicated indicating the important roles of the underlying domains in OCA2 pathogenesis.
Conclusion: Our results were beneficial for diagnosis and precision clinical management for OCA2-related disorder, and this study expanded the mutation spectrum of oculocutaneous albinism.
Introduction
Albinism is a heterogeneous genetic disorder in which a group of genes related to pigment synthesis have mutated, leading to melanin deficiency. According to its clinical symptoms, it can be divided into oculocutaneous albinism (OCA) and ocular albinism (OA). Oculocutaneous albinism is a crowd of inherited disorders of melanin biosynthesis characterized by a generalized reduction in pigmentation of hair, skin and eyes (Gronskov et al., 2007), whereas in OA patients only the ocular pigment is deficient. The degree of hypopigmentation of skin and hair varies with the types of OCA. Oculocutaneous albinism is classified into non-syndromic oculocutaneous albinism and syndrome oculocutaneous albinism based on symptoms such as the presence of bleeding diathesis, immunodeficiency and neurological dysfunction (Simeonov et al., 2013). Mutations in TYR, OCA2, TYRP1, SLC45A2, SLC24A5, and LRMDA lead to six different non-syndromic OCA subtypes, they are OCA1, OCA2, OCA3, OCA4, OCA6, and OCA7, respectively (Arveiler et al., 2017). The prevalence of all forms of albinism varies considerably worldwide, estimated at approximately 1/17000, the mutation carrier rate is about 1 in 65 (Montoliu et al., 2014).
Tyrosinase-positive oculocutaneous albinism (OCA, type II, OMIM 203200) is an autosomal recessive genetic disease that reduces the biosynthesis of melanin in the skin, hair and eyes. Patients with OCA2 have characteristic visual abnormalities associated with albinism, including decreased vision and nystagmus. These symptoms are usually milder than those with OCA1 (Lee et al., 1994). OCA2 was first identified in 1993, in a patient with tyrosinase-positive oculocutaneous albinism. According to epidemiological survey, OCA2 is the most prevalent subtype in Africa and accounts for 30% of OCA cases worldwide. Studies have revealed that OCA2 is caused by mutations in OCA2, the human cDNA contains 3143 bases which encodes a protein that corresponds to the “pink-eyed dilution” (p) mouse mutant. In addition, the gene product plays a role in regulating the pH of melanosomes (Yuasa et al., 2007). Up to now, lots of OCA2 mutations have been reported and novel variants are still being discovered (Maurano et al., 2012; Martinez-Garcia and Montoliu, 2013; Wang et al., 2016; Qiu et al., 2018; Lin et al., 2019; Luo et al., 2019; Yang et al., 2019; Zhong et al., 2019; Chuan et al., 2021; Xu et al., 2021). In recent years, multiple groups of researchers have conducted epidemiological surveys on Chinese albinism patients. Although the proportions of each type are different, OCA1 is recognized as the main type of albinism, and exons 1 and 2 are mutation hotspots. The proportions of OCA2, OCA4, HPS1 and unknown mutations are behind (Lin et al., 2019; Luo et al., 2019; Chuan et al., 2021; Xu et al., 2021).
In this study, we performed mutational screening of OCA2 in 28 Chinese OCA2 patients. After literature search and database retrieval, 12 novel variants in OCA2 were identified. Our results provide new insight into the underlying mechanisms of OCA2 and at the same time, provide important information for genetic testing and counseling.
Materials and Methods
Patients
We reviewed the records of patients undergoing genetic testing for albinism in the Changde First People’s Hospital and Changsha Kingmed Center for Clinical Laboratory in recent years. We recruited 28 OCA2 families from Hunan Province, China. Among the 28 families, there are 28 patients, including 15 females and 13 males. All patients denied family history of consanguinity. These patients all have obvious albinism phenotype, but due to the incompletely medical records, the clinical data did not record their skin color, hair color, iris color and ophthalmic phenotype in detail.
Ethics Statement
A written informed consent was obtained from each subject or their guardians to participate in this study. The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Ethics Committee of Changde First People’s Hospital (protocol code 2020-014-01) and approved on January 23, 2020.
Genomic DNA Preparation and Next-Generation Sequencing
Genomic DNA was extracted using whole blood DNA extraction kit (Tiangen Biotechnology, Beijing, China). The library was prepared by randomly fragmenting 2 μg of genomic DNA into fragments by ultrasonic shearing. According to the standard Illumina protocol, a HiSeq2500 sequencer was used for sample sequencing. The amplified DNA was captured with a Genodermatosis-related Gene Panel, which can capture all exons and splicing sites of more than 100 related genes. The raw reads were filtered to obtain high-quality clean reads, and mapped to the human reference genome (UCSC hg19), and the sention software suite was used to call Single Nucleotide Variants (SNVs) and small insertions or deletions (InDels).
Validation of Variants and Inheritance Analysis
All candidate pathogenic variants were verified by Sanger sequencing in the reported families to verify the heritability of the variants. We designed specific primers (Supplementary Table 1) to amplify the region containing the corresponding variation by polymerase chain reaction (PCR). The PCR products were then sequenced on ABI 3730XL Genetic Analyzer (Applied Biosystems Life Technologies) according to manufacturer s protocols.
Variant Classification and in silico Analysis
The data obtained by sequencing was screened according to the following criteria. First, the variants with minor allele frequency (MAF) >0.01 in the following three SNP databases were excluded. Including gnomAD,1 1000 Genome project,2 dbSNP3 and ESP6500.4 Second, we used the ClinVar database,5 the albinism database,6 the human gene mutation database (HGMD7) and OMIM8 to annotate variants. Third, in order to determine the pathogenicity of novel mutations, we in silico analyzed the pathogenicity of novel mutations with various tools, which included the programs of Sorting Intolerant From Tolerant (SIFT9), Polymorphism Phenotyping v2 (PolyPhen210), Mutation Assessor,11 Protein Variation Effect Analyzer (PROVEAN12) and CADD.13 Subsequently, we used I Mutant2.014 to evaluate the protein stability changes upon novel variants and clustalX2 for the protein conservation analysis. The structure changes of protein caused by amino acid substitutions were simulated by I-TASSER.15 Finally, according to the American Medical Genetics and Genomics (ACMG) guidelines, all detected novel variants were classified into pathogenic, likely pathogenic, uncertain significance, and likely benign or benign.
Results
Clinical Manifestation
By reviewing the records of patients who have performed albinism genetic testing in the First People’s Hospital of Changde and Changsha Kingmed Center for Clinical Laboratory in recent years, the clinical information of 28 patients who had been diagnosed as OCA2 through genetic testing was collected. All patients had typical OCA symptoms, including varying degrees of hypopigmentation on the skin, hair and iris. Moreover, patients did not show any other symptoms involving other systems. Among 28 patients, only 7 parents had tested for albinism-related genes (Supplementary Table 1). The clinical characteristics and mutant alleles of these 28 patients were shown in Table 1.
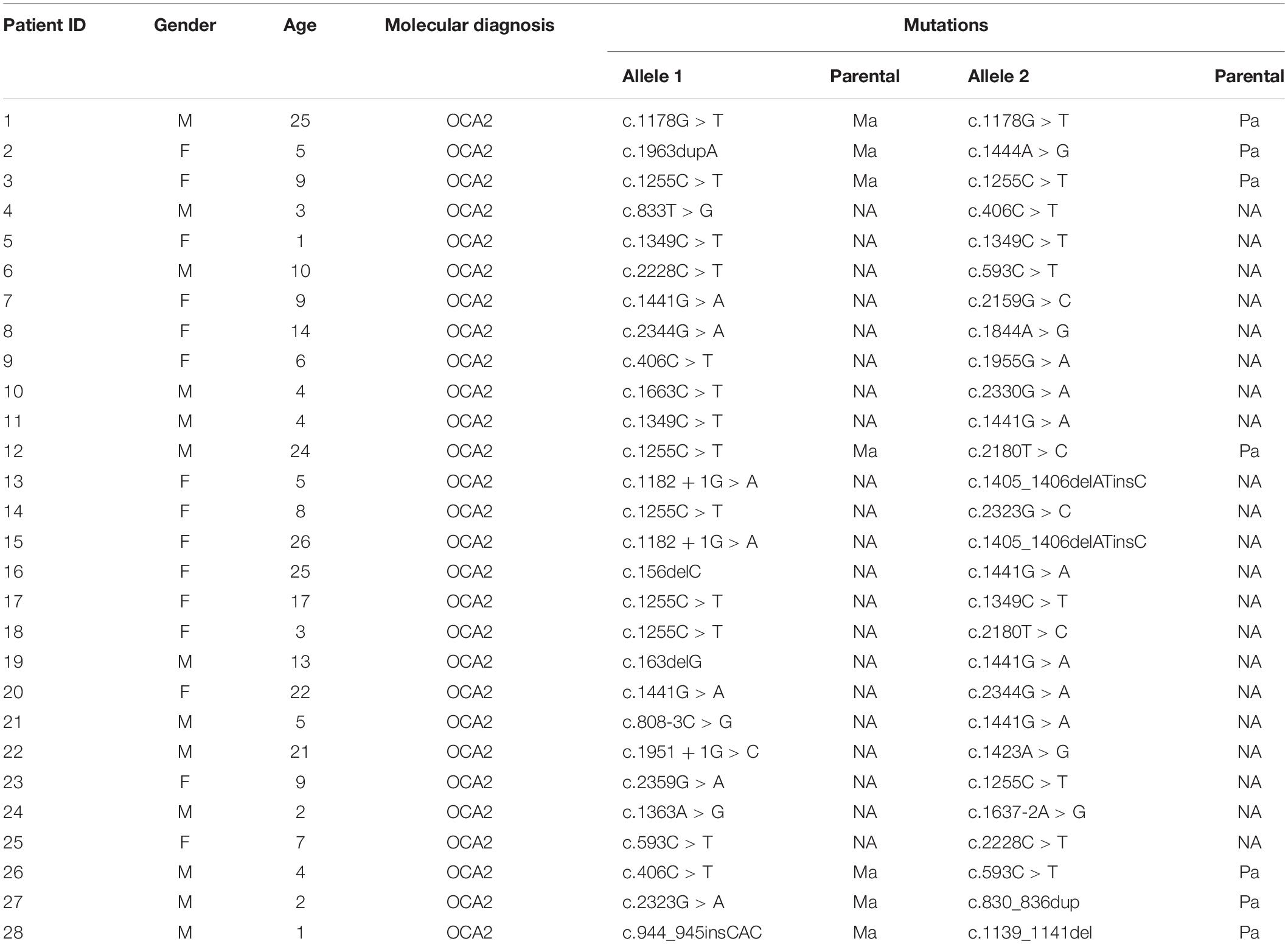
Table 1. Clinical characteristics and genotypes of the 28 patients.
Mutation Pattern and Potential Missense Variant Clusters in OCA2
Based on the analysis and statistics of the sequencing results, 2 of the 28 patients were single homozygous (Patient 1 and 3 in Table 1), and the rest 26 probands were compound heterozygous. Moreover, missense variants (54.8%, 17/31) were the most prevalent mutation. To investigate the mutation pattern of OCA2, we aggregated the mutations information of all patients. There were 31 variants in 28 patients, including twelve likely gene-disrupting (LGD) variants (6 frameshift, 2 stopgain, 4 splice-site), 1 deletion,1 insertion and seventeen missense variants (Table 2 and Figure 1). We observed one potential missense cluster, 16 out of 17 missense variants were clustered in P-permease domain (Figure 1).

Table 2. Summary of OCA2 variants identified in this study.
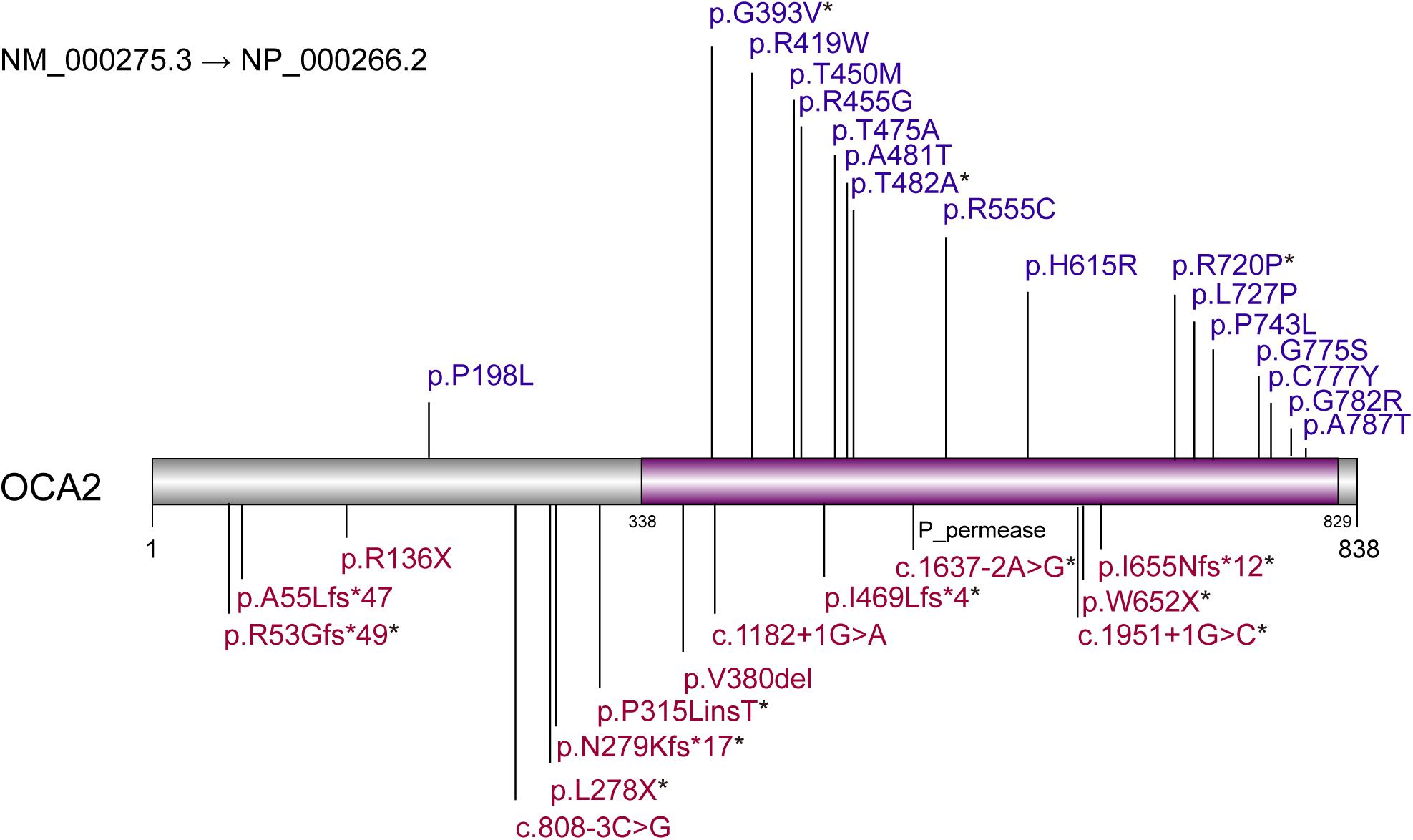
Figure 1. Variants pattern of OCA2. Loss-of-function variants are labeled above the diagram. Missense variants are labeled below the diagram. Purple region represents P-permease domain. * represents the novel variants.
Pathogenic Novel Variant Pattern of OCA2-Related OCA2 Genes
There were 12 novel variants among all variants, including 3 missense variants. Of the remaining 19 reported mutations, six were reported for the first time in the Chinese population, including p.A55Lfs∗47, p.L727P, p.P743L, p.G775S, p.C777Y, and p.G782R. By comparing the alignments of OCA2 orthologous peptide sequences of the three novel missense variants (p.G393V, p.T482A, p.R720P), most variants show evolutionary conservation (Figure 2A) and predicted to be damaging or probably damaging by PolyPhen2, PROVEAN, SIFT and had a high CADD and Mutation Assessor score (Table 3). The p.T482A and p.R720P were assessed to decrease the OCA2 protein stability by I Mutant2.0, which was consistent with the simulated result by I-TASSER that amino acid substitutions resulted in the structure changes of protein (Figure 2B).
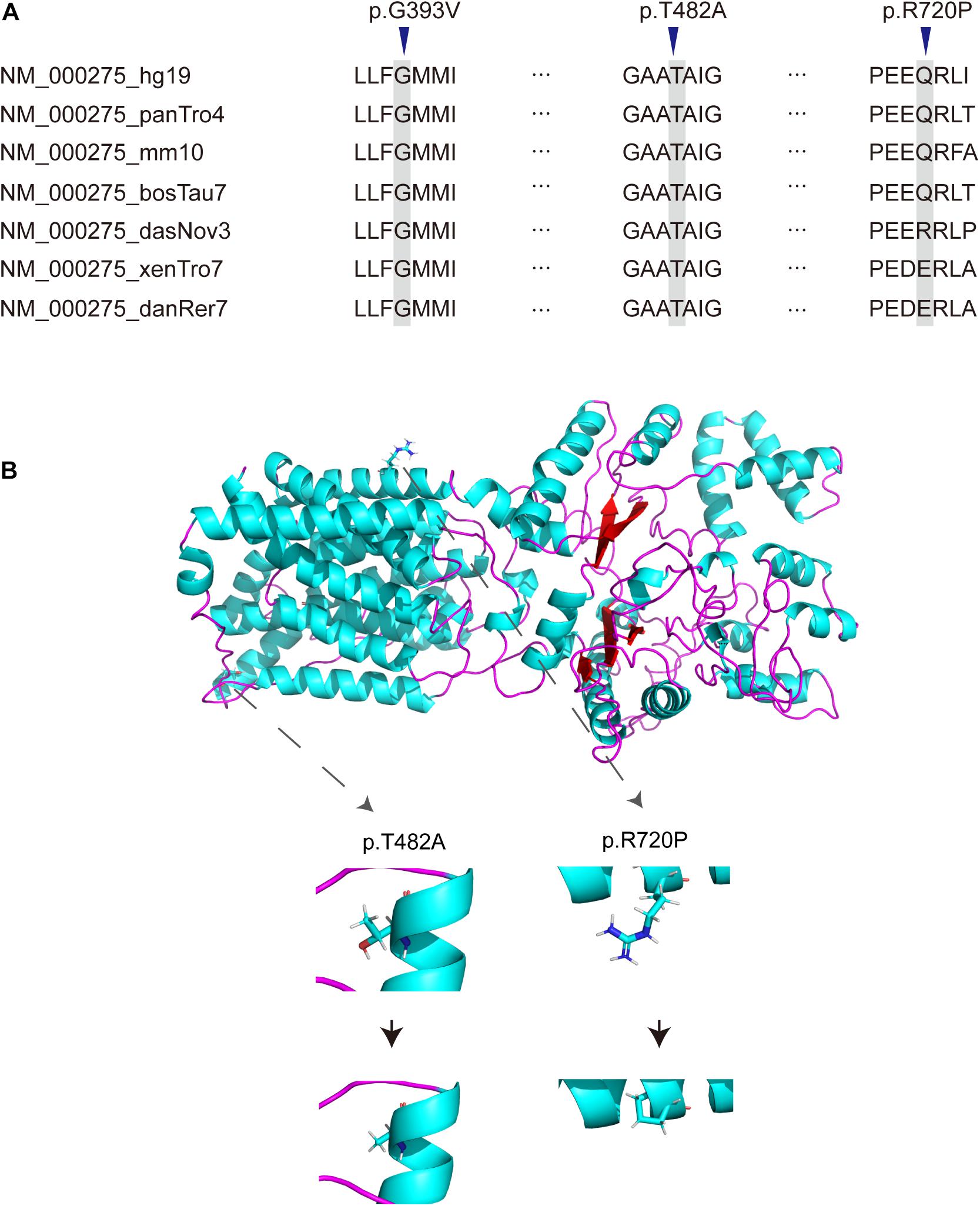
Figure 2. Novel missense variants of OCA2. (A) Conservation analysis of OCA2 missense variants. (B) Simulation of the amino acids conformation changes by I-TASSER.
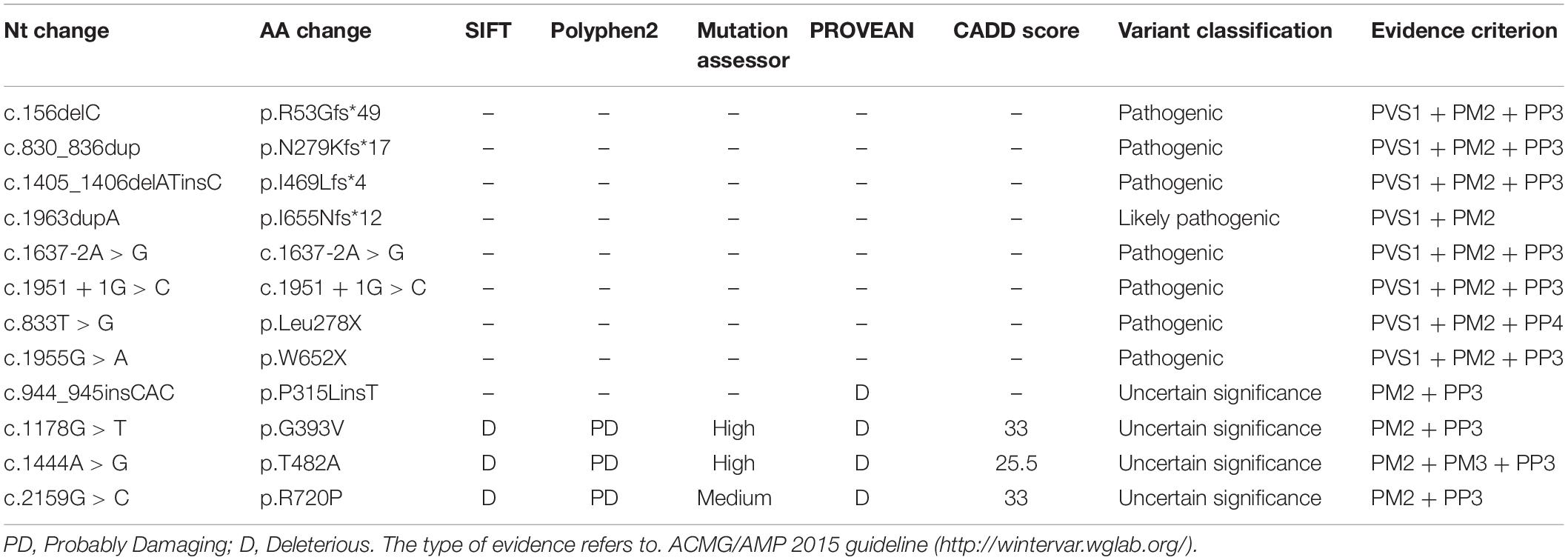
Table 3. Pathogenicity analysis of novel variants.
Discussion
Oculocutaneous albinism is a group of autosomal recessive genetic diseases caused by the reduction or lack of melanin synthesis in melanocytes that affects the pigmentation of hair, skin and eyes (Yang et al., 2019). Different types of OCA cannot be completely distinguished by clinical phenotype, therefore, molecular diagnosis have become a useful tool and a necessary condition for genetic consultation (Gronskov et al., 2007). OCA2 is one of the main subtypes of OCA, which is caused by mutations in OCA2 (Yuasa et al., 2007). The protein encoded by this gene has 12 transmembrane domains and is an intact membrane protein involved in the transport of small molecules, especially tyrosine, which is the precursor of melanin synthesis (Rinchik et al., 1993; Gronskov et al., 2007).
In this study, we collected 28 oculocutaneous albinism patients with compound heterozygous or homozygous OCA2 variants identified by sequencing. All patients have typical clinical phenotypes of oculocutaneous albinism (Table 1). Thirty one variants have been identified in all patients, including 19 reported variants and 12 novel variants. Among all the variants, missense variants (54.8%, 17/31) were the most common mutation (Chumakov and Kronrod, 1990; Spritz et al., 1997; Sviderskaya et al., 1997; Passmore et al., 1999; Suzuki et al., 2003; Duan et al., 2006; Hongyi et al., 2007; Yuasa et al., 2007; Chiang et al., 2008; Dai et al., 2008; Rooryck et al., 2008; Gronskov et al., 2009; Wei et al., 2010, 2011, 2013; Gargiulo et al., 2011). The missense variants, p.R419W and p.A481T were the most general OCA2 variants, six patients have inherited these variants, respectively. R A Spritz (Spritz et al., 1997) have found p.R419W in OCA2 patients, but the variant classification of the HGMD database and ClinVar database are inconsistent. According to the comprehensive analysis of ACMG, the variant was consistent with uncertain significance. The high frequency of p.R419W in our study could provide more evidence for its pathogenicity. In addition, the pathogenicity of p.A481T has not been determined, it was recorded as benign in the ClinVar database, but reported to be an Asian-specific hypopigmentation allele in 2007 (Yuasa et al., 2007). The p.P743L variant in the OCA2 gene has been reported in the homozygous state or in the compound heterozygous state in multiple unrelated individuals with OCA2 (Lee et al., 1994; King et al., 2003; Hutton and Spritz, 2008; Sengupta et al., 2010; Jaworek et al., 2012; Richards et al., 2015; Shahzad et al., 2017). Variant p.P743L is a semi-conservative amino acid substitution. Due to the differences in physiochemical properties between the two residues, the switch of proline to leucine residue will have significant impact on protein structure, indicating the pathogenic effect of the mutation. In our study, 2 patients inherited this missense substitution, most of the previous reports for this substitution were in European, American and African, this was the first report in Chinese. The splice variant c.1182 + 1G > A was also found in two patients, in 2019, Dan Luo et al., found four patients carrying this mutation, and one of them was homozygous (Luo et al., 2019). In addition, a research showed that the variant was expected to affect splicing following exon 11, thereby leading to abnormal splicing of the transcript (Montoliu et al., 2014).
Among the 31 different mutations in the OCA2 gene, there are 12 novel mutations (4 frameshift, 2 stopgain, 2 splice-site,1 insertion and 3 missense). Frameshift mutations p.R53Gfs∗49, p.N279Kfs∗17, p.I469Lfs∗4, p.I655Nfs∗12 and stopgain mutations p.Leu278X, p.W652X were expected to affect the original protein OCA2 function by changing the protein sequence or leading to early termination of protein translation. The truncated protein lacks intact transmembrane domain, which could cause the dysfunction of tyrosine transport and precursor melanin synthesis, and lead to the location of the protein in the nucleus. Splicing mutations c.1637-2A > G and c.1951 + 1G > C were expected to affect splicing of the transcript, thereby leading to abnormal protein function. The literature research showed that most missense mutations occur in the loop between the transmembrane domains (Spritz, 1994). In addition, most of the missense variants found in this study were located in the P_permease domain, which was linked to human melanosomal P gene. Variants in P gene were responsible for classic phenotype of OCA2 (Lee et al., 1995; Puri et al., 2000; Brilliant, 2001; Staleva et al., 2002). Missense variants p.G393V occurred in the fourth transmembrane domain of P protein, which would lead to inactivity of the P protein. Three novel missense variants p.G393V, p.T482A and p.R720P were evolutionary conservation and in silico predicted to be damaging or probably damaging. The T482A and R720P are assessed to decrease the OCA2 protein stability, thereby affect to protein function possibly. According to the ACMG, these missense variants are recorded as uncertain significance, therefore, the pathogenicity of these missense mutations needs in-depth exploration.
At present, due to the lack of effective treatment for albinism, prenatal diagnosis is particularly important to prevent the birth of patients. On the basis of prenatal genetic testing and diagnosis, genetic counseling and inspection guidance can effectively prevent the birth of severely patients (Bao et al., 2019; Geng et al., 2019; Li et al., 2019).
In summary, we identified 31 distinct variants of OCA2 by next-generation sequencing, in addition, 12 variants were novel ones. We characterized the molecular and phenotypic data for patients with OCA2 variants and revealed one potential missense variant cluster by curating published data. Our findings will benefit not only for the genetic diagnosis and counseling but also provide motivation for further functional characterizations of OCA2 variants.
Data Availability Statement
The datasets generated for this study can be found in the China National GeneBank with the accession code of CNP0002229.
Ethics Statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
LM and JZhu analyzed the data and prepared the manuscript. JW and YH collected the data. JZha and CW rechecked the data. DP and YZ provided theoretical guidance. DP revised the manuscript and read and approved the final manuscript. All authors approved the final manuscript.
Funding
This work was supported by the 2020 Hunan Provincial Clinical Medical Technology Innovation Guidance Project (2020SSK50201).
Conflict of Interest
JW was employed by Changsha Kingmed Center for Clinical Laboratory.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all the health care technicians in the Department of Medical Genetics, Affiliated Hospital of Changde city of South China University.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.715437/full#supplementary-material
Supplementary Figure 1 | Pedigree drawings of the seven families.
Footnotes
- ^ https://gnomad.broadinstitute.org/
- ^ http://www.internationalgenome.org
- ^ http://www.ncbi.nlm.nih.gov/dbvar
- ^ http://evs.gs.washington.edu/EVS
- ^ http://www.ncbi.nlm.nih.gov/clinvar
- ^ http://www.ifpcs.org/albinism
- ^ http://www.biobase-international.com/product/hgmd
- ^ https://omim.org/
- ^ http://sift.jcvi.org
- ^ http://genetics.bwh.harvard.edu/pph2
- ^ http://mutationassessor.org/
- ^ http://provean.jcvi.org/index.php
- ^ http://cadd.gs.washington.edu/download
- ^ http://folding.biofold.org/cgi-bin/i-mutant2.0.cgi
- ^ https://zhanglab.ccmb.med.umich.edu/I-TASSER/
References
Arveiler, B., Lasseaux, E., and Morice-Picard, F. (2017). [Clinical and genetic aspects of albinism]. Presse Med. 46, 648–654. doi: 10.1016/j.lpm.2017.05.020
Bao, Y., Suo, L., Qian, P., Huang, H., Yang, Y., Tang, J., et al. (2019). Clinical and genetic analysis of Dent disease with nephrotic range albuminuria in Shaanxi. China. Sci. China Life Sci. 62, 1590–1593.
Brilliant, M. H. (2001). The mouse p (pink-eyed dilution) and human P genes, oculocutaneous albinism type 2 (OCA2), and melanosomal pH. Pigment Cell Res. 14, 86–93. doi: 10.1034/j.1600-0749.2001.140203.x
Chiang, P. W., Fulton, A. B., Spector, E., and Hisama, F. M. (2008). Synergistic interaction of the OCA2 and OCA3 genes in a family. Am. J. Med. Genet. A 146A, 2427–2430. doi: 10.1002/ajmg.a.32453
Chuan, Z., Yan, Y., Hao, S., Zhang, Q., Zhou, B., Feng, X., et al. (2021). Mutation Analysis of 63 Northwest Chinese Probands with Oculocutaneous Albinism. Curr. Eye Res. 46, 140–143. doi: 10.1080/02713683.2020.1781192
Chumakov, A. A., and Kronrod, B. A. (1990). [The status and outlook of the training of pathologists in the RSFSR]. Arkh. Patol. 52, 50–52.
Dai, C., Li, W., Gao, B., Li, L. Y., and Lu, G. X. (2008). [Mutation screening of the TYR and P gene in three patients with oculocutaneous albinism]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 25, 373–377.
Duan, H. L., Li, H. Y., Wu, W. Q., Zheng, H., and Chen, Z. (2006). [A novel P gene mutation in a Chinese family with oculocutaneous albinism]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 23, 614–617.
Gargiulo, A., Testa, F., Rossi, S., Di Iorio, V., Fecarotta, S., de Berardinis, T., et al. (2011). Molecular and clinical characterization of albinism in a large cohort of Italian patients. Invest. Ophthalmol. Vis. Sci. 52, 1281–1289. doi: 10.1167/iovs.10-6091
Geng, J., Liu, Y., Guo, Y., Wang, H., Tai, J., Jin, Y., et al. (2019). Correlation between TERT C228T and clinicpathological features in pediatric papillary thyroid carcinoma. Sci. China Life Sci. 62, 1563–1571.
Gronskov, K., Ek, J., and Brondum-Nielsen, K. (2007). Oculocutaneous albinism. Orphanet. J. Rare Dis. 2:43. doi: 10.1186/1750-1172-2-43
Gronskov, K., Ek, J., Sand, A., Scheller, R., Bygum, A., Brixen, K., et al. (2009). Birth prevalence and mutation spectrum in danish patients with autosomal recessive albinism. Invest. Ophthalmol. Vis. Sci. 50, 1058–1064. doi: 10.1167/iovs.08-2639
Hongyi, L., Haiyun, W., Hui, Z., Qing, W., Honglei, D., Shu, M., et al. (2007). Prenatal diagnosis of oculocutaneous albinism type II and novel mutations in two Chinese families. Prenat. Diagn. 27, 502–506. doi: 10.1002/pd.1713
Hutton, S. M., and Spritz, R. A. (2008). Comprehensive analysis of oculocutaneous albinism among non-Hispanic caucasians shows that OCA1 is the most prevalent OCA type. J. Invest. Dermatol. 128, 2442–2450. doi: 10.1038/jid.2008.109
Jaworek, T. J., Kausar, T., Bell, S. M., Tariq, N., Maqsood, M. I., Sohail, A., et al. (2012). Molecular genetic studies and delineation of the oculocutaneous albinism phenotype in the Pakistani population. Orphanet. J. Rare Dis. 7:44. doi: 10.1186/1750-1172-7-44
King, R. A., Willaert, R. K., Schmidt, R. M., Pietsch, J., Savage, S., Brott, M. J., et al. (2003). MC1R mutations modify the classic phenotype of oculocutaneous albinism type 2 (OCA2). Am. J. Hum. Genet. 73, 638–645. doi: 10.1086/377569
Lee, S. T., Nicholls, R. D., Bundey, S., Laxova, R., Musarella, M., and Spritz, R. A. (1994). Mutations of the P gene in oculocutaneous albinism, ocular albinism, and Prader-Willi syndrome plus albinism. N. Engl. J. Med. 330, 529–534. doi: 10.1056/NEJM199402243300803
Lee, S. T., Nicholls, R. D., Jong, M. T., Fukai, K., and Spritz, R. A. (1995). Organization and sequence of the human P gene and identification of a new family of transport proteins. Genomics 26, 354–363. doi: 10.1016/0888-7543(95)80220-g
Li, Z., Zhu, P., Huang, H., Pan, Y., Han, P., Cui, H., et al. (2019). Identification of a novel COL4A5 mutation in the proband initially diagnosed as IgAN from a Chinese family with X-linked Alport syndrome. Sci. China Life Sci. 62, 1572–1579.
Lin, Y., Chen, X., Yang, Y., Che, F., Zhang, S., Yuan, L., et al. (2019). Mutational analysis of TYR, OCA2, and SLC45A2 genes in chinese families with oculocutaneous albinism. Mol. Genet. Genomic Med. 7:e00687. doi: 10.1002/mgg3.687
Luo, D., Linpeng, S., Zeng, L., Tan, H., Li, Z., and Wu, L. (2019). Molecular genetic study of 59 Chinese Oculocutaneous albinism families. Eur. J. Med. Genet. 62:103709. doi: 10.1016/j.ejmg.2019.103709
Martinez-Garcia, M., and Montoliu, L. (2013). Albinism in Europe. J. Dermatol. 40, 319–324. doi: 10.1111/1346-8138.12170
Maurano, M. T., Wang, H., Kutyavin, T., and Stamatoyannopoulos, J. A. (2012). Widespread site-dependent buffering of human regulatory polymorphism. PLoS Genet. 8:e1002599. doi: 10.1371/journal.pgen.1002599
Montoliu, L., Gronskov, K., Wei, A. H., Martinez-Garcia, M., Fernandez, A., Arveiler, B., et al. (2014). Increasing the complexity: new genes and new types of albinism. Pigment Cell Melanoma Res. 27, 11–18. doi: 10.1111/pcmr.12167
Passmore, L. A., Kaesmann-Kellner, B., and Weber, B. H. (1999). Novel and recurrent mutations in the tyrosinase gene and the P gene in the German albino population. Hum. Genet. 105, 200–210. doi: 10.1007/s004390051090
Puri, N., Gardner, J. M., and Brilliant, M. H. (2000). Aberrant pH of melanosomes in pink-eyed dilution (p) mutant melanocytes. J. Invest. Dermatol. 115, 607–613. doi: 10.1046/j.1523-1747.2000.00108.x
Qiu, B., Ma, T., Peng, C., Zheng, X., and Yang, J. (2018). Identification of five novel variants in chinese oculocutaneous albinism by targeted next-generation sequencing. Genet. Test. Mol. Biomarkers 22, 252–258. doi: 10.1089/gtmb.2017.0211
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Rinchik, E. M., Bultman, S. J., Horsthemke, B., Lee, S. T., Strunk, K. M., Spritz, R. A., et al. (1993). A gene for the mouse pink-eyed dilution locus and for human type II oculocutaneous albinism. Nature 361, 72–76. doi: 10.1038/361072a0
Rooryck, C., Morice-Picard, F., Elcioglu, N. H., Lacombe, D., Taieb, A., and Arveiler, B. (2008). Molecular diagnosis of oculocutaneous albinism: new mutations in the OCA1-4 genes and practical aspects. Pigment Cell Melanoma Res. 21, 583–587. doi: 10.1111/j.1755-148X.2008.00496.x
Sengupta, M., Mondal, M., Jaiswal, P., Sinha, S., Chaki, M., Samanta, S., et al. (2010). Comprehensive analysis of the molecular basis of oculocutaneous albinism in Indian patients lacking a mutation in the tyrosinase gene. Br. J. Dermatol. 163, 487–494. doi: 10.1111/j.1365-2133.2010.09830.x
Shahzad, M., Yousaf, S., Waryah, Y. M., Gul, H., Kausar, T., Tariq, N., et al. (2017). Molecular outcomes, clinical consequences, and genetic diagnosis of Oculocutaneous Albinism in Pakistani population. Sci. Rep. 7:44185. doi: 10.1038/srep44185
Simeonov, D. R., Wang, X., Wang, C., Sergeev, Y., Dolinska, M., Bower, M., et al. (2013). DNA variations in oculocutaneous albinism: an updated mutation list and current outstanding issues in molecular diagnostics. Hum. Mutat. 34, 827–835. doi: 10.1002/humu.22315
Spritz, R. A. (1994). Molecular genetics of oculocutaneous albinism. Hum. Mol. Genet. 3, 1469–1475. doi: 10.1093/hmg/3.suppl_1.1469
Spritz, R. A., Lee, S. T., Fukai, K., Brondum-Nielsen, K., Chitayat, D., Lipson, M. H., et al. (1997). Novel mutations of the P gene in type II oculocutaneous albinism (OCA2). Hum. Mutat. 10, 175–177.
Staleva, L., Manga, P., and Orlow, S. J. (2002). Pink-eyed dilution protein modulates arsenic sensitivity and intracellular glutathione metabolism. Mol. Biol. Cell 13, 4206–4220. doi: 10.1091/mbc.e02-05-0282
Suzuki, T., Miyamura, Y., Matsunaga, J., Shimizu, H., Kawachi, Y., Ohyama, N., et al. (2003). Six novel P gene mutations and oculocutaneous albinism type 2 frequency in Japanese albino patients. J. Invest. Dermatol. 120, 781–783. doi: 10.1046/j.1523-1747.2003.12127.x
Sviderskaya, E. V., Bennett, D. C., Ho, L., Bailin, T., Lee, S. T., and Spritz, R. A. (1997). Complementation of hypopigmentation in p-mutant (pink-eyed dilution) mouse melanocytes by normal human P cDNA, and defective complementation by OCA2 mutant sequences. J. Invest. Dermatol. 108, 30–34. doi: 10.1111/1523-1747.ep12285621
Wang, X., Zhu, Y., Shen, N., Peng, J., Wang, C., Liu, H., et al. (2016). Mutation analysis of a Chinese family with oculocutaneous albinism. Oncotarget 7, 84981–84988. doi: 10.18632/oncotarget.13109
Wei, A., Wang, Y., Long, Y., Wang, Y., Guo, X., Zhou, Z., et al. (2010). A comprehensive analysis reveals mutational spectra and common alleles in Chinese patients with oculocutaneous albinism. J. Invest. Dermatol. 130, 716–724. doi: 10.1038/jid.2009.339
Wei, A., Yang, X., Lian, S., and Li, W. (2011). Implementation of an optimized strategy for genetic testing of the Chinese patients with oculocutaneous albinism. J. Dermatol. Sci. 62, 124–127. doi: 10.1016/j.jdermsci.2011.02.009
Wei, A. H., Yang, X. M., Lian, S., and Li, W. (2013). Genetic analyses of Chinese patients with digenic oculocutaneous albinism. Chin. Med. J. 126, 226–230.
Xu, C., Xiang, Y., Li, H., Xu, Y., Mao, Y., Zhou, L., et al. (2021). Genetic analysis and prenatal diagnosis of 20 Chinese families with oculocutaneous albinism. J. Clin. Lab. Anal. 35:e23647. doi: 10.1002/jcla.23647
Yang, Q., Yi, S., Li, M., Xie, B., Luo, J., Wang, J., et al. (2019). Genetic analyses of oculocutaneous albinism types 1 and 2 with four novel mutations. BMC Med. Genet. 20:106. doi: 10.1186/s12881-019-0842-7
Yuasa, I., Umetsu, K., Harihara, S., Miyoshi, A., Saitou, N., Park, K. S., et al. (2007). OCA2 481Thr, a hypofunctional allele in pigmentation, is characteristic of northeastern Asian populations. J. Hum. Genet. 52, 690–693. doi: 10.1007/s10038-007-0167-9
Keywords: OCA2 gene, missense variants, oculocutaneous albinism, next-generation sequencing, novel variants
Citation: Ma L, Zhu J, Wang J, Huang Y, Zhang J, Wang C, Zhou Y and Peng D (2021) Genetic Analysis of 28 Chinese Families With Tyrosinase-Positive Oculocutaneous Albinism. Front. Genet. 12:715437. doi: 10.3389/fgene.2021.715437
Received: 28 May 2021; Accepted: 30 July 2021;
Published: 11 October 2021.
Edited by:
Tieliu Shi, East China Normal University, ChinaReviewed by:
Lu Xie, Shanghai Center For Bioinformation Technology, ChinaYaqiong Jin, Capital Medical University, China
Copyright © 2021 Ma, Zhu, Wang, Huang, Zhang, Wang, Zhou and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dan Peng, ZGFzYW4wOTg4QDEyNi5jb20=
†These authors share first authorship