 Chaofan Zhu
Chaofan Zhu Meiying Zhang2
Meiying Zhang2 Jin Jen
Jin Jen Mingzhou Guo
Mingzhou Guo- 1Department of Head and Neck Surgery, Peking University Cancer Hospital and Institute, Beijing, China
- 2Department of Gastroenterology and Hepatology, Chinese PLA General Hospital, Beijing, China
- 3Genome Analysis Core, Medical Genome Facility, Center for Individualized Medicine, Mayo Clinic, Rochester, MN, United States
- 4State Key Laboratory of Kidney Diseases, Chinese PLA General Hospital, Beijing, China
Background: Thyroid cancer (TC) is the most common endocrine malignancy, and the incidence is increasing very fast. Surgical resection and radioactive iodine ablation are major therapeutic methods, however, around 10% of differentiated thyroid cancer and all anaplastic thyroid carcinoma (ATC) are failed. Comprehensive understanding the molecular mechanisms may provide new therapeutic strategies for thyroid cancer. Even though genetic heterogeneity is rigorously studied in various cancers, epigenetic heterogeneity in human cancer remains unclear.
Methods: A total of 405 surgical resected thyroid cancer samples were employed (three spatially isolated specimens were obtained from different regions of the same tumor). Twenty-four genes were selected for methylation screening, and frequently methylated genes in thyroid cancer were used for further validation. Methylation specific PCR (MSP) approach was employed to detect the gene promoter region methylation.
Results: Five genes (AP2, CDH1, DACT2, HIN1, and RASSF1A) are found frequently methylated (>30%) in thyroid cancer. The five genes panel is used for further epigenetic heterogeneity analysis. AP2 methylation is associated with gender (P < 0.05), DACT2 methylation is associated with age, gender and tumor size (all P < 0.05), HIN1 methylation is associated to tumor size (P < 0.05) and extra-thyroidal extension (P < 0.01). RASSF1A methylation is associated with lymph node metastasis (P < 0.01). For heterogeneity analysis, AP2 methylation heterogeneity is associated with tumor size (P < 0.01), CDH1 methylation heterogeneity is associated with lymph node metastasis (P < 0.05), DACT2 methylation heterogeneity is associated with tumor size (P < 0.01), HIN1 methylation heterogeneity is associated with tumor size and extra-thyroidal extension (all P < 0.01). The multivariable analysis suggested that the risk of lymph node metastasis is 2.5 times in CDH1 heterogeneous methylation group (OR = 2.512, 95% CI 1.135, 5.557, P = 0.023). The risk of extra-thyroidal extension is almost 3 times in HIN1 heterogeneous methylation group (OR = 2.607, 95% CI 1.138, 5.971, P = 0.023).
Conclusion: Five of twenty-four genes were found frequently methylated in human thyroid cancer. Based on 5 genes panel analysis, epigenetic heterogeneity is an universal event. Epigenetic heterogeneity is associated with cancer development and progression.
Introduction
Thyroid cancer (TC) is the most common endocrine malignancy and the incidence is 3.4% of all cancers (Seib and Sosa, 2019). Papillary thyroid cancer (PTC) and follicular thyroid cancer (FTC) are two most common thyroid cancer types, account for 80% and 15% of all thyroid cancer cases. Poorly differentiated thyroid carcinoma (PDTC) and anaplastic thyroid carcinoma (ATC) are account for 5% and 1%, respectively. Surgical resection and radioactive iodine ablation are major therapeutic strategies for thyroid cancer. Treatment failure was observed in about 10% of differentiated thyroid cancer and all ATC patients (Laha et al., 2020; Prete et al., 2020). In the last 30 years, genetic study suggests that the frequency of somatic mutations is relatively low in thyroid cancer (Mazzaferri, 1999). The MAPK and PI3K/Akt pathways were reported to be activated by somatic mutations in thyroid cancer (Mercer and Pritchard, 2003; Ball et al., 2007; Leboeuf et al., 2008; Salerno et al., 2009), and targeted therapies were applied to advanced cancers by tyrosine kinase inhibitors and anti-angiogenic drugs (Yamamoto et al., 2014; Valerio et al., 2017). With the understanding of cancer biology and molecular mechanism, precision medicine gets into the main stage of the era. The key to tailor the management of cancer, including thyroid cancer, is based on the better understanding of the molecular pathways. Landscape of cancer genome study has provided a lot of information of genetic alterations and therapeutic targets for cancer therapy. Mutations in different signaling pathways were found by genomic study in thyroid cancer (Vogelstein et al., 2013; Cha and Koo, 2016). DNA methylation was found frequently in human cancers, including esophageal, colorectal, lung, gastric, and hepatic cancers (Esteller et al., 2002; Brock et al., 2003; House et al., 2003; Guo et al., 2006a,b, 2007, 2008; Licchesi et al., 2008; Wang et al., 2009, 2017; Jia et al., 2012, 2013; Hu et al., 2015; Li et al., 2015; Zheng et al., 2017). However, epigenetics was not extensively studied in thyroid cancer. Pan-cancer landscape of aberrant DNA methylation across 24 cancers demonstrated that the methylation frequency was lowest in PTC (Saghafinia et al., 2018). Aberrant methylation of SOX17 and DACT2, the key components of Wnt signaling, were found frequently in thyroid cancer (Li et al., 2012; Zhao et al., 2014). Methylation of GPX3 was found associated with thyroid cancer metastasis (Zhao et al., 2015). Phenotypic and functional heterogeneity are hallmarks of human cancers (Visvader, 2011). Subpopulations of cancer cells with distinct phenotypic and molecular features within a tumor are called intratumor heterogeneity (ITH) (Bedard et al., 2013). For cancer genetic study, researchers mainly focused on mutational activation of oncogenes or inactivation of tumor-suppressor genes (TSGs). The theory of Darwinian-like clonal evolution of a single tumor was employed to explain the phenomenon of ITH (Nowell, 1976). However, the dominance of gene-centric views is challenged by cancer stem cell hypothesis, and phenotypic variability becomes the major topic of cancer research (Marusyk et al., 2012). The concept of epigenetic silencing being involved in Knudson’s two-hit theory was accepted and the causal relevance of epigenetic changes in cancer is being recognized (Nagasaka et al., 2010). The occurrence of abnormal epigenetic change is more frequently than driver mutations in human cancer. In the process of cancer initiation and development, disruption of the “epigenetic machinery” plays an important role. The disruption of “epigenetic machinery” may contribute to tumor phenotype heterogeneity (Guo et al., 2019).
ITH plays an important role in chemotherapeutic resistance (Guo et al., 2019). The best regimen for cancer therapy is to target all different subpopulations of cancer cells at the same time, to avoid chemo-resistance and reduce relapse (Pribluda et al., 2015). Therapeutic failures are often attributed to adaptive responses of cancer stem cells. Environmental and therapeutic pressures may drive transcriptional plasticity through the response of epigenetic regulators to cause durable disease remission in patients for many cancer therapeutic drugs (Dawson, 2017). Epigenetic heterogeneity is more dynamic compare to genetic heterogeneity. Cancer epigenetic heterogeneity is in its infancy and there are very limited studies involved in epigenetic heterogeneity (Dawson, 2017; Guo et al., 2019).
In this study, we evaluated epigenetic heterogeneity by examining promoter region methylation in a panel of genes in primary thyroid cancer. These genes are frequently methylated in thyroid and other cancers, and they are involved in different cancer-related signaling.
Materials and Methods
Patients and Specimens
A total of 405 samples from 135 cases of thyroid cancer (46 cases were served as discovery group for methylation screening and all cases were served as validation group) were obtained in Beijing Cancer Hospital from 2018 to 2020 (Supplementary Table 1). For heterogeneity analysis, a total of 405 surgical resected thyroid cancer samples were employed (three spatially isolated specimens were obtained from different regions of the same tumor). The median age was 42 years old (range 24–79 years old), including 104 cases of female and 31 cases of male patients. Cancer samples were classified according to the TNM staging system (AJCC2018), including tumor stage I (n = 130), stage II (n = 4), and stage III (n = 1). All of the tissue samples were immediately snap frozen in liquid nitrogen and preserved at −80°C before analysis. All samples were collected following the guidelines approved by the Institutional Review Board of the Beijing Cancer Hospital.
DNA Extraction, Bisulfite Modification, Methylation Specific PCR, and Screening for Representative Genes
Genomic DNA was extracted from frozen tissues, digested with protease K and then extracted using the standard phenol/chloroform procedure (Cao et al., 2013). Methylation-specific PCR (MSP) primers were designed according to genomic sequences around transcriptional start sites (TSS) and synthesized to detect unmethylated (U) and methylated (M) alleles. Bisulfite treatment was performed as previously described (Herman et al., 1996). MSP primers were listed in Supplementary Table 2. MSP amplification conditions were as follows: 95°C 5 min, 1 cycle; 95°C 30 s, 60°C 30 s, 72°C 40 s, 35 cycles; 72°C 5 min, 1 cycle.
To screen representative genes for heterogeneity analysis, twenty-four genes were selected as discovery group, including MGMT, TIMP3, DAPK, MLH1, TMEM176A, DIRAS1, SFRP1, SFRP2, HIN1, AP2, ER, DACT2, CDH1, RASSF1A, SOX17, GATA4, BCL6B, GPX3, CRBP-1, p16, RUNX3, RARβ, CDX2, and WIF1. All these genes were found frequently methylated in esophageal, colorectal, lung, gastric and hepatic cancers by our previous studies and others, except for AP2 (an important transcription factor). These genes were well characterized and reported to be involved in different signaling pathways. Among which, AP2, CDH1, DACT2, HIN1, and RASSF1A genes were found frequently methylated (>30%) and selected for heterogeneity analysis. The workflow is shown in Supplementary Figure 1.
Statistical Analysis
The Chi-square or Fisher exact-test was used to analyze the association of gene methylation status and clinical factors. Logistic regression analysis was used to analyze the association of methylation heterogeneity or clinical factors with lymph node Metastasis and Extrathyroidal extension. P < 0.05∗, P < 0.01∗∗, or P < 0.001∗∗∗ was regarded as statistically significant. Data were analyzed by SPSS 22.0 software.
Results
Selection of Frequently Methylated Genes in Human Thyroid Cancer
To explore epigenetic heterogeneity in thyroid cancer, 24 genes, which were found frequently methylated in other cancers, were selected as discovery group to detect 46 cases of thyroid cancer. As shown in Table 1 and Figure 1, the methylation rate is 0% (0/46)–56.52% (26/46). Five genes (AP2, CDH1, DACT2, HIN1, and RASSF1A) are methylated more than 30% in thyroid cancer. These five genes panel is selected as validation group for methylation heterogeneity analysis.
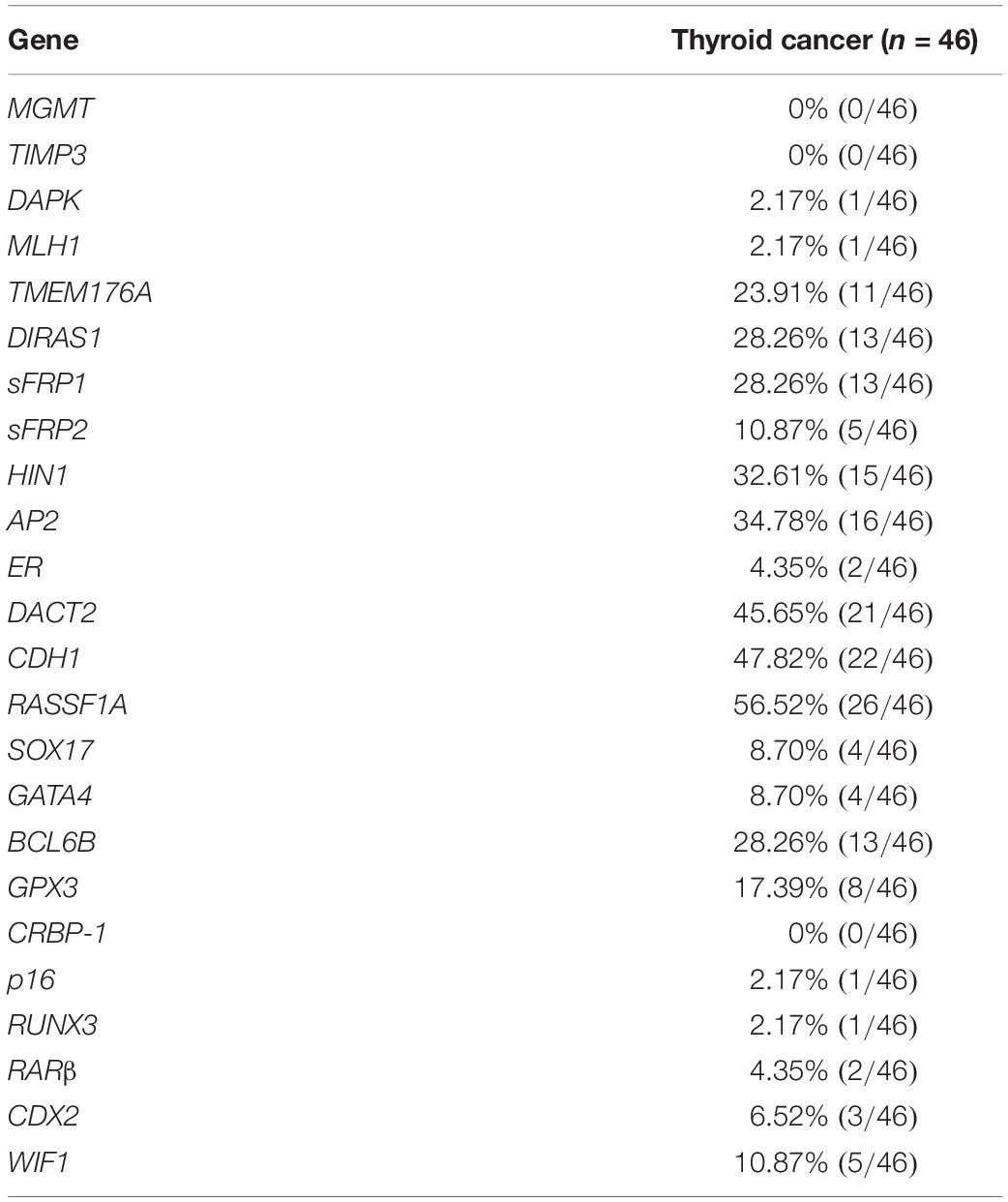
Table 1. Methylation status of 24 genes in discovery group.
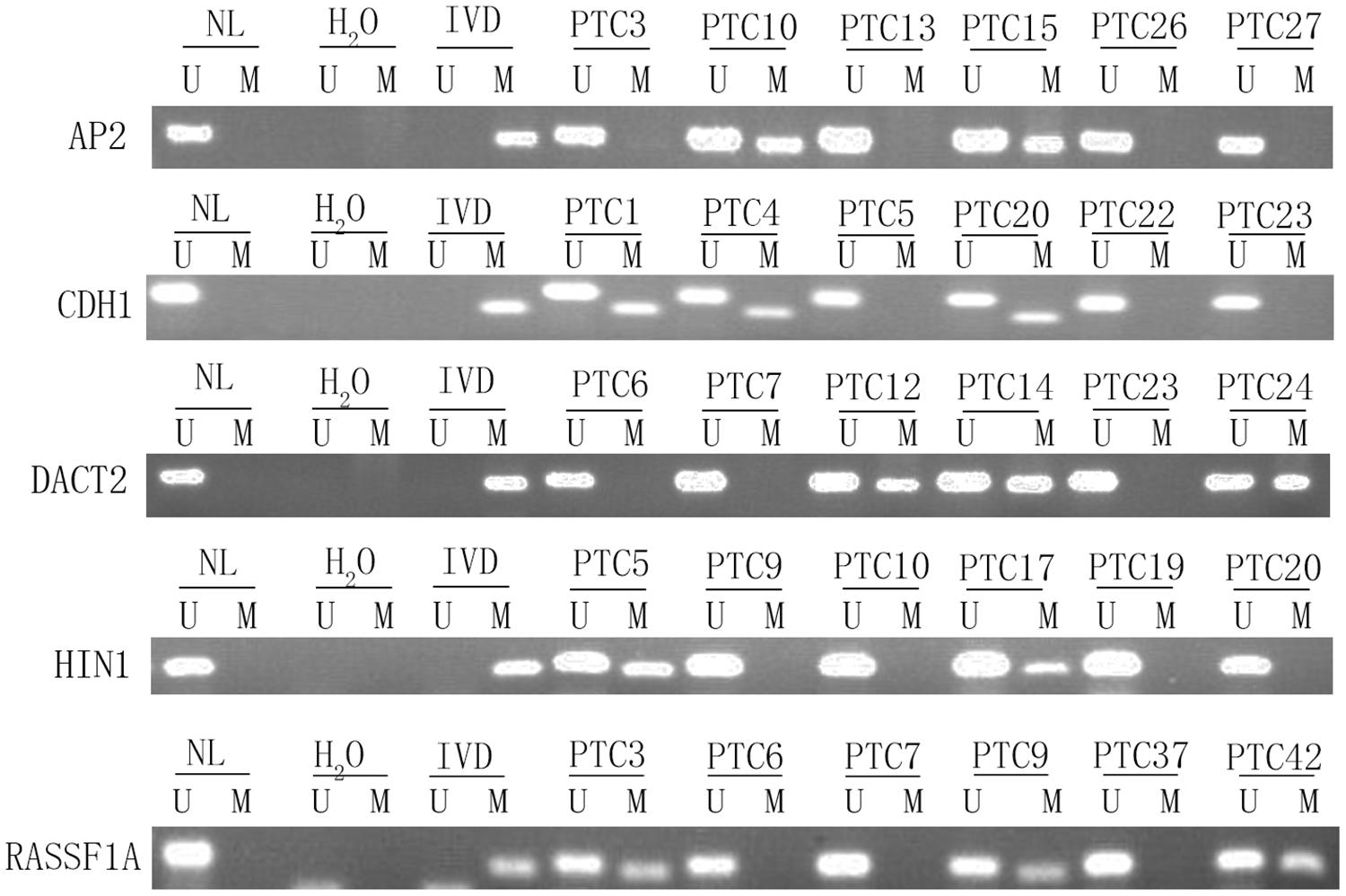
Figure 1. Representative methylation results of AP2, HIN1, DACT2, RASSF1A, and CDH1 in thyroid cancer (discovery group). IVD, in vitro-methylated DNA (methylation control); NL, normal lymphocyte DNA (unmethylation control); H2O, double distilled water; U, unmethylation; M, methylation; PTC, papillary thyroid cancer.
Intratumor Epigenetic Heterogeneity in Thyroid Cancer
To evaluate the intratumor epigenetic heterogeneity in thyroid cancer, three different tumor samples are obtain from isolated locations in the same patient. Totally 405 cancer samples are gained from 135 patients. The methylation status of AP2, CDH1, DACT2, HIN1, and RASSF1A genes is examined by MSP.
The association of promoter region methylation and clinical factors is analyzed by Chi-square tests, including gender, age, tumor size, tumor location, TNM stage, lymph node metastasis, and extra-thyroidal extension. As shown in Table 2, AP2 methylation is associated with gender (P < 0.05), while no association were found between AP2 methylation and age, tumor size, tumor location, TNM stage, lymph node metastasis, and extra-thyroidal extension (all P > 0.05). No association are found between CDH1 methylation and gender, age, tumor size, tumor location, TNM stage, lymph node metastasis, and extra-thyroidal extension (all P > 0.05). DACT2 methylation is associated with age, gender and tumor size (all P < 0.05), while no association is found between DACT2 methylation and tumor location, TNM stage, lymph node metastasis, and extra-thyroidal extension (all P > 0.05). HIN1 methylation is associated with tumor size (P < 0.05) and extra-thyroidal extension (P < 0.01), while no association is found between HIN1 methylation and age, tumor location, TNM stage, and lymph node metastasis (all P > 0.05). RASSF1A methylation is associated with lymph node metastasis (P < 0.01), while no association is found between RASSF1A methylation and age, tumor size, tumor location, TNM stage, and extra-thyroidal extension (all P > 0.05).
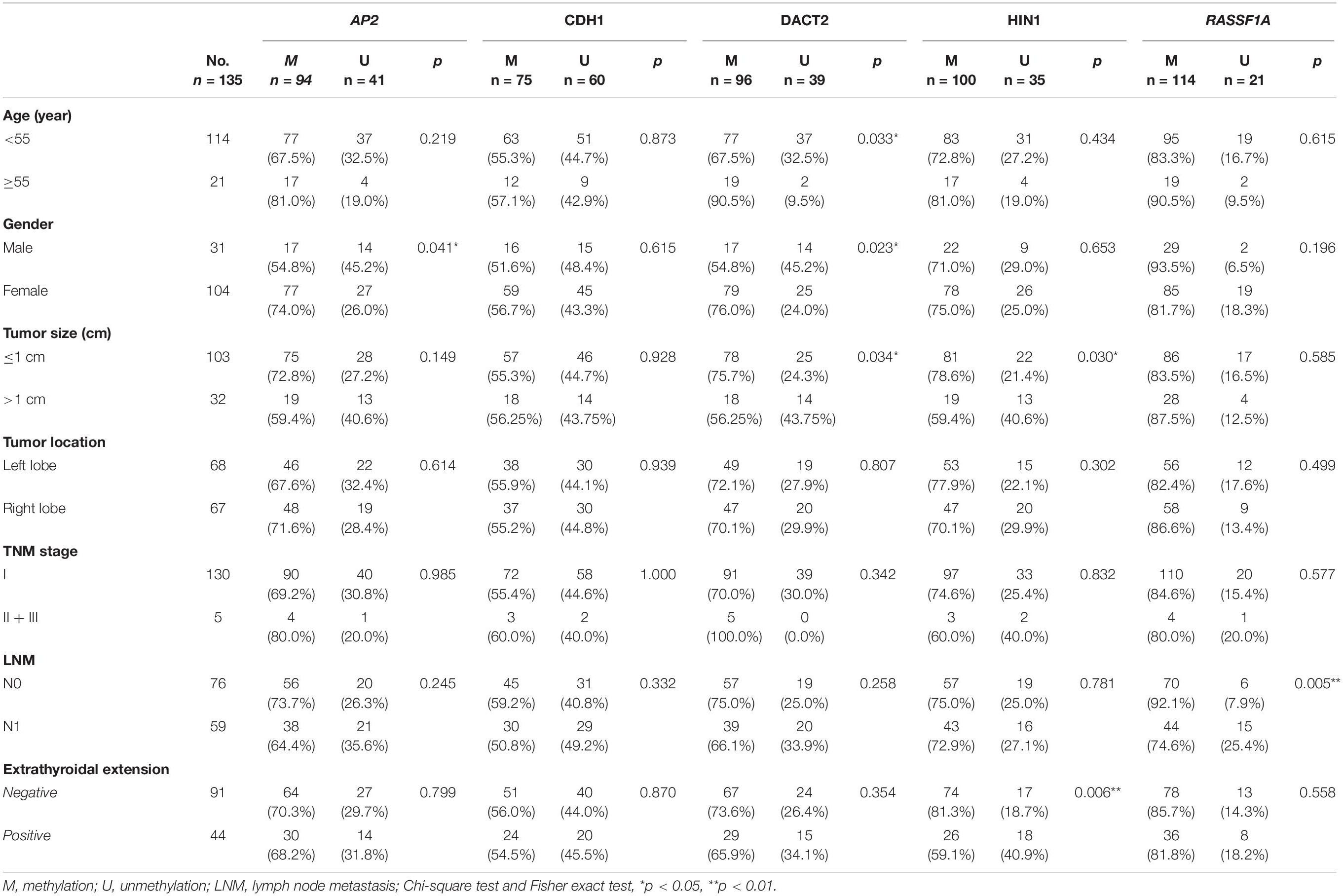
Table 2. The association of gene methylation and clinical factors in validation group.
Unmethylation or methylation in all three samples from the same individual is regarded as homogeneity, while unmethylation or methylation in one or two samples from the same individual is regarded as methylation heterogeneity. As shown in Figures 2, 3, methylation heterogeneity is found in 25.93% (35/135), 37.78% (51/135), 44.44% (60/135), 44.44% (60/135), and 41.48% (56/135) of cases for RASSF1A, CDH1, AP2, HIN1, and DACT2 genes. The results suggest that methylation heterogeneity is a common event in human thyroid cancer.

Figure 2. Methylation pattern of 3 different samples for each patient. M, methylation; U, unmethylation; Colum (A–C) are 3 different samples for each case; PTC, papillary thyroid cancer.
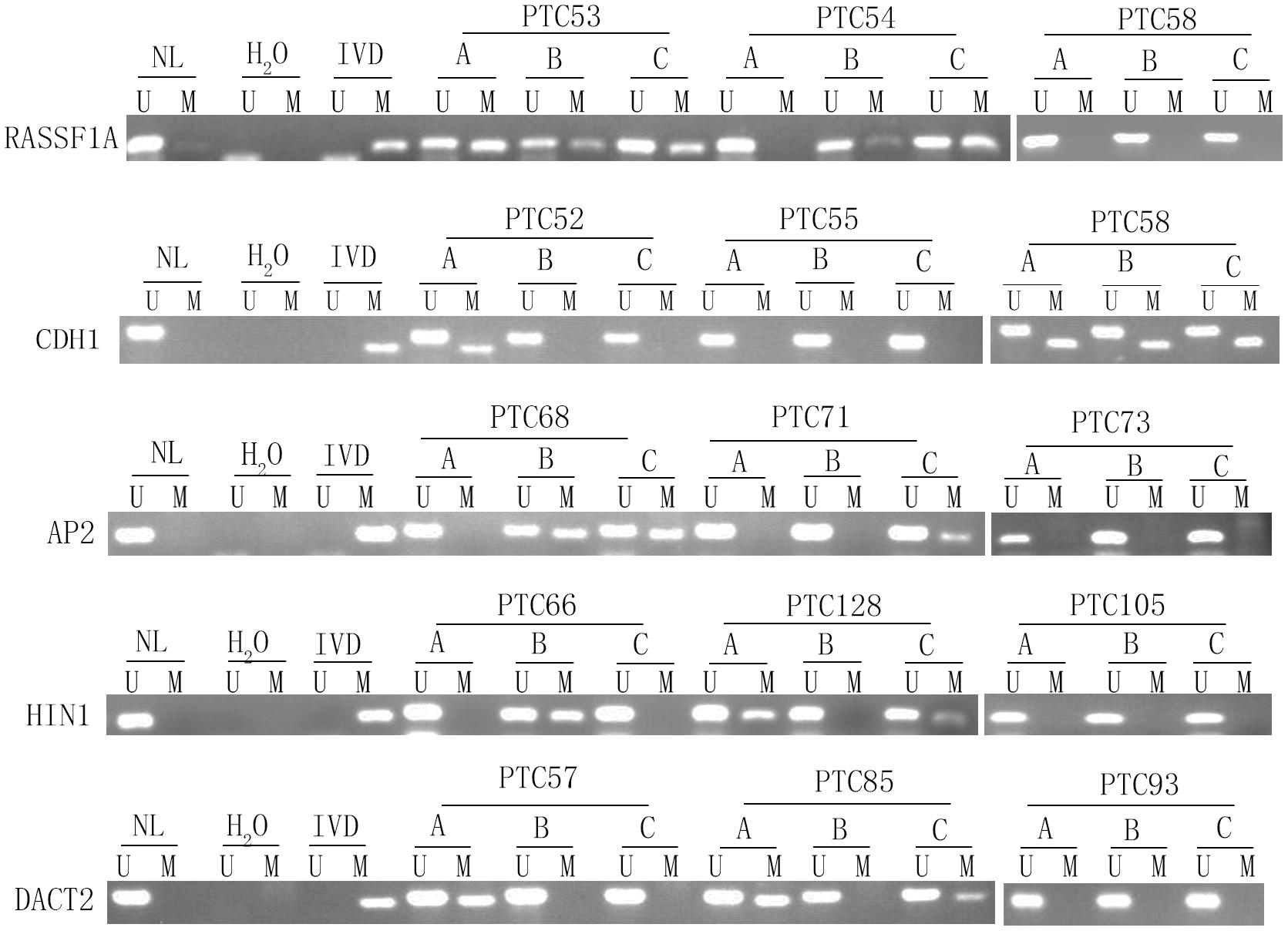
Figure 3. Representative methylation heterogeneity results for each of five genes. IVD, in vitro-methylated DNA (methylation control); NL, normal lymphocyte DNA (unmethylation control); H2O, double distilled water; U, unmethylation; M, methylation; PTC, papillary thyroid cancer.
The association of methylation heterogeneity and clinical factors is further analyzed. As shown in Table 3, AP2 methylation heterogeneity is associated with tumor size (P < 0.01), no association are found between AP2 methylation heterogeneity and gender, age, tumor location, TNM stage, lymph node metastasis and extra-thyroidal extension (all P > 0.05). CDH1 methylation heterogeneity is associated with lymph node metastasis (P < 0.05), no association are found between CDH1 methylation heterogeneity and gender, age, tumor size, tumor location, TNM stage and extra-thyroidal extension (all P > 0.05). DACT2 methylation heterogeneity is associated with tumor size (P < 0.01), no association are found between DACT2 methylation heterogeneity and gender, age, tumor location, TNM stage, lymph node metastasis and extra-thyroidal extension (all P > 0.05). HIN1 methylation heterogeneity is associated with tumor size and extra-thyroidal extension (all P < 0.01), no association are found between HIN1 methylation heterogeneity and gender, age, tumor location, TNM stage and lymph node metastasis (all P > 0.05). No association are found between RASSF1A methylation heterogeneity and gender, age, tumor size, tumor location, TNM stage, lymph node metastasis and extra-thyroidal extension (all P > 0.05). Above results demonstrate that methylation heterogeneity of three genes (AP2, DACT2, and HIN1) is associated with tumor size.
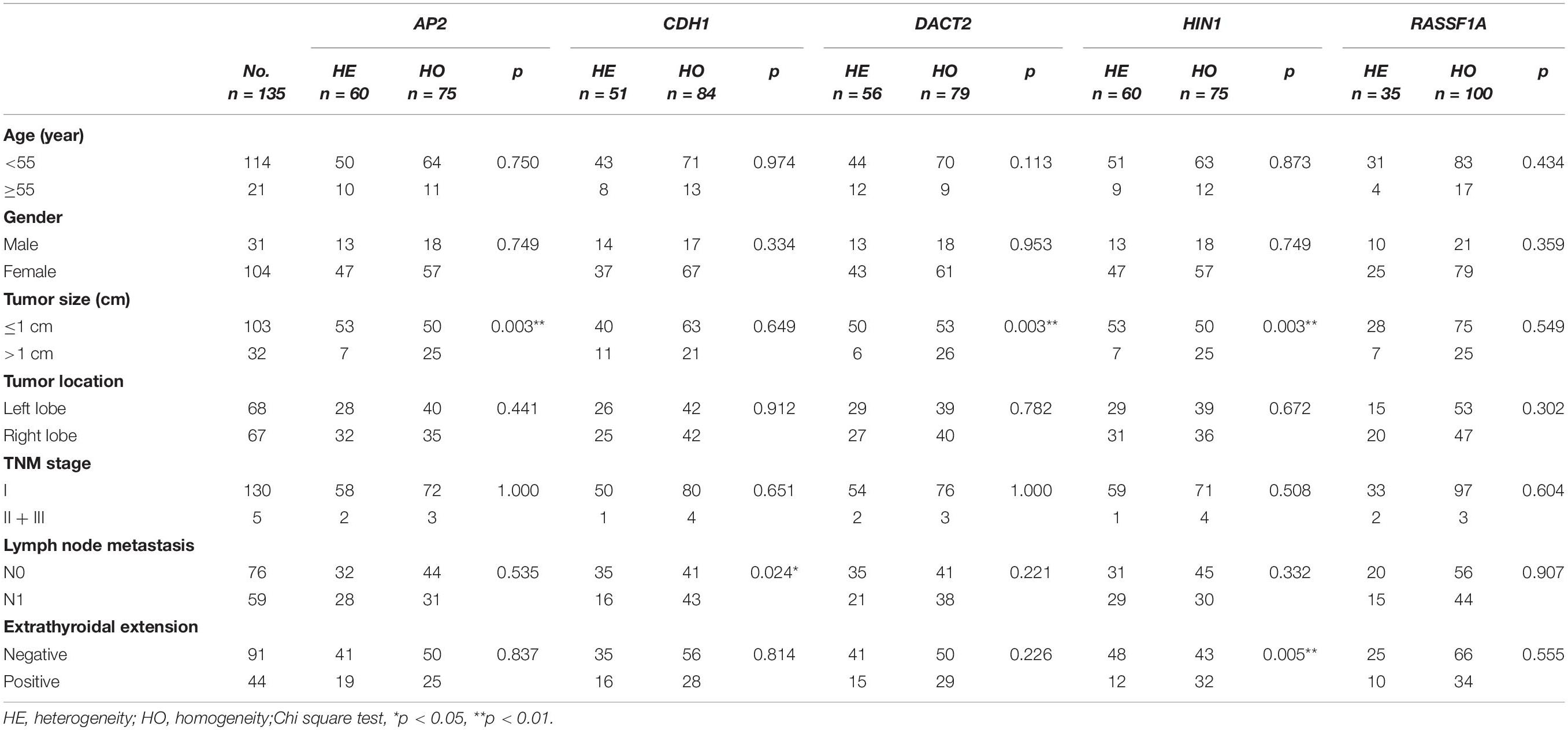
Table 3. The association of methylation heterogeneity and clinical factors.
The association of DNA methylation heterogeneity and lymph node metastasis or extra-thyroidal extension is further analyzed by logistic regression model. Univariate logistic analysis indicated that CDH1 methylation heterogeneity, age and tumor size are associated with lymph node metastasis, independently (P = 0.026, P = 0.000, P = 0.040, Table 4). HIN1 heterogeneous methylation, tumor size and tumor location are associated with extra-thyroidal extension, independently (P = 0.006, P = 0.004, P = 0.025, Table 5). Interesting, the multivariable analysis suggested that the risk of lymph node metastasis is 2.5 times in CDH1 heterogeneous methylation group compare to CDH1 homogeneous methylation group (OR = 2.512, 95% CI 1.135, 5.557, P = 0.023, Table 4). The risk of extra-thyroidal extension is almost 3 times in HIN1 heterogeneous methylation group than in HIN1 homogeneous methylation group (OR = 2.607, 95% CI 1.138, 5.971, P = 0.023, Table 5). The results indicate that with the growing of tumor size, the phenotype of tumor cell becomes diversity. Epigenetic herterogeneity may reflect different phenotypes of population of tumor cells and increased the risk of tumor metastasis significantly. Epigenetic herterogeneity may provide more information for tumor therapeutic strategies.
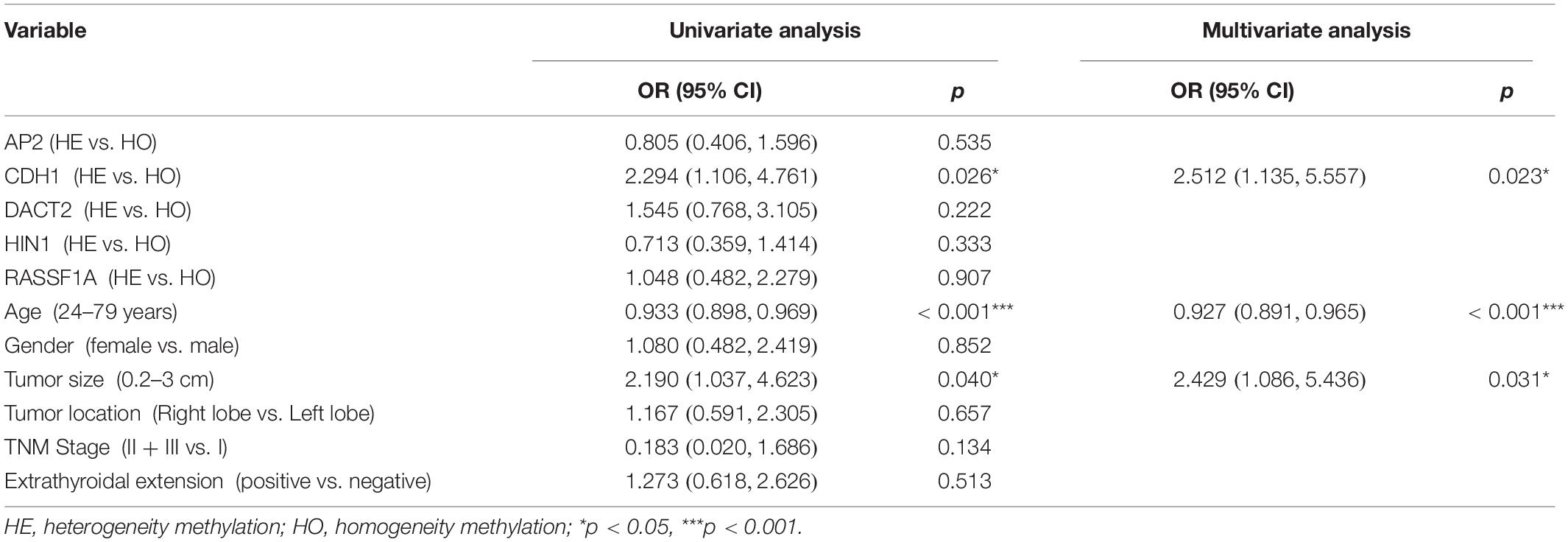
Table 4. Logistic regression model for lymph node metastasis.
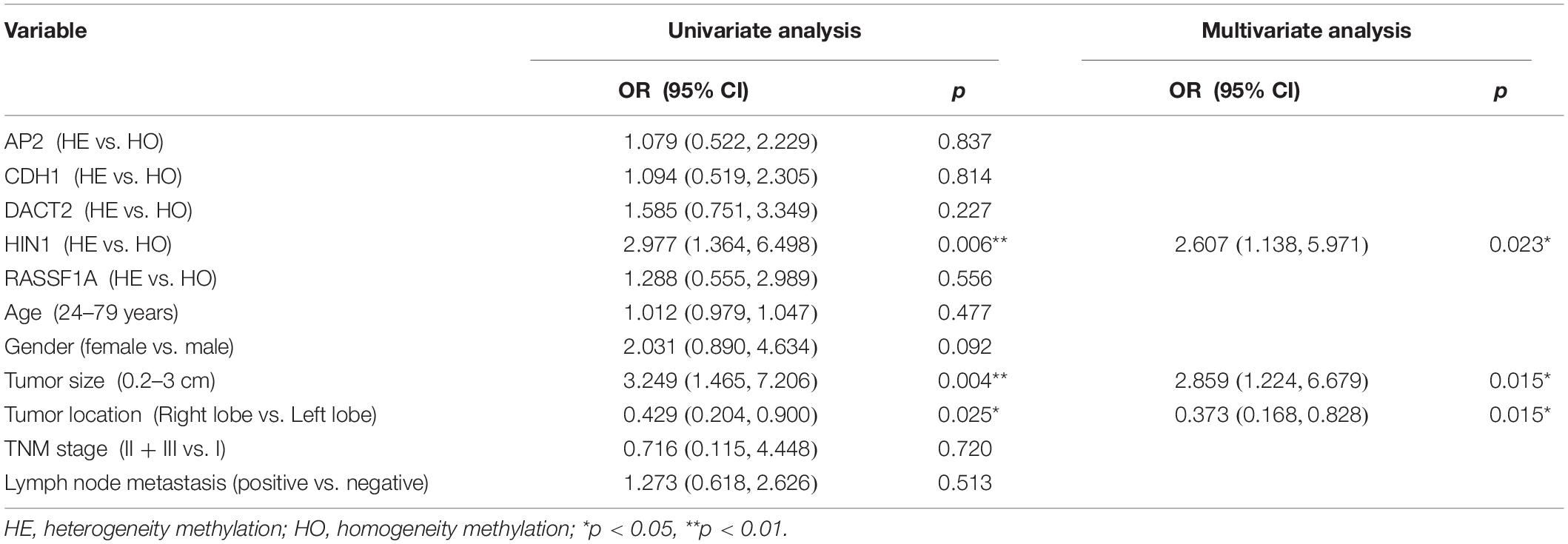
Table 5. Logistic regression analysis for extrathyroidal extension.
Discussion
Genomic heterogeneity has been well studied in various cancers (Burrell et al., 2013; Hiley et al., 2016; Maura et al., 2021; Nicholson and Fine, 2021; Wand and Lambert, 2021). However, there are a very few reports about epigenetic heterogeneity in human cancers. Our previous study found that “a field defect of epigenetic changes” was presented in bronchial margins of surgical resected lung cancer samples (Guo et al., 2004). By detecting the methylation status of five genes (RASSF1A, p16, DAPK, MGMT, and Rb) in 34 tumors (including 15 melanoma primaries, 19 metastases), heterogeneous methylation was found in 70% of the cases (Rastetter et al., 2007). Among 9 MSI-positive primary endometrial cancers lack of MLH1 expression, which was evaluated by immunohistochemistry, Varley et al. (2009) found that 8 cases were methylated in the promoter region. In the 8 cases of methylated patients, four cases were heterogeneously methylated and four tumors were homogeneously methylated. In the discovery group, we select 24 genes, which were well characterized and frequently methylated in various cancers. Five genes are found frequently methylated (> 30%) in 46 cases of human thyroid cancer. In the validation group, total of 405 samples are included (135 cases of patients and 3 samples from different location of each tumor). Among these five genes, DACT2 methylation is associated with gender, age, and tumor size (all P < 0.05), HIN1 methylation is associated with tumor size (P < 0.05) and extra-thyroidal extension (P < 0.01), RASSF1A methylation is associated with lymph node metastasis (P < 0.01). The results suggest that methylation of DACT2, HIN1, and RASSF1A increases the malignance of thyroid cancer. Further analysis find that methylation heterogeneity of AP2, DACT2, and HIN1 are associated with tumor size. The results suggest that epigenetic heterogeneity is increasing with tumor growth. It indicates that the phenotype of cancer cells is varied in different tumor stages. Thus, the therapeutic strategies need according to the phenotypes of cancer cells, which are determined by epigenetic changes. The multivariable analysis suggested that CDH1 methylation heterogeneity is associated with lymph node metastasis and HIN1 methylation heterogeneity is associated with extra-thyroidal extension. The results suggest that CDH1 and HIN1 methylation heterogeneity may increase tumor metastasis. Methylation heterogeneity of these two genes may serve as prognostic marker for thyroid cancer.
The conventional clinical therapeutics is the “one-size-fits-all-approach.” However, the ultimate aim of precision medicine is to enable clinicians to accurately and efficiently identify the most effective preventative or therapeutic intervention for a specific patient. Epigenetic switches play important roles in carcinogenesis and tumor progression, and epigenetic switches are reversible (Guo et al., 2019). In this study, we focus mainly on the epigenetic heterogeneity of transcriptional regulators, which were found involved in different cancer-related signaling pathways. Epigenetic heterogeneity is found in four (CDH1, AP2, HIN1, and DACT2) of the five detected genes in thyroid cancer. The results suggest that epigenetic heterogeneity is a universal mechanism of cancer development. It is notable that epigenetic heterogeneity is associated with tumor size or tumor metastasis. For the first time, we find that epigenetic heterogeneity is related to cancer development. Based on “BRCAness” principle, our recent study found that methylation of NRN1 was a novel synthetic lethal marker for PI3K-Akt-mTOR and ATR inhibitors in human esophageal cancer (McLornan et al., 2014; Lord and Ashworth, 2016, 2017; Du et al., 2021). It is reasonable to tailor the regimen for each individual of cancer patients by epigenetic changes or epigenetic heterogeneity.
In conclusion, 5 of 24 genes were found frequently methylated in human thyroid cancer. Based on the 5 genes panel analysis, epigenetic heterogeneity is an universal event. Epigenetic heterogeneity is associated with cancer development and progression.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
All samples were collected following the guidelines approved by the Institutional Review Board of the Beijing Cancer Hospital. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
CZ, MZ, and QW designed the project and performed the experiments. MZ and MG wrote the manuscript. MG has made significant contributions to the concept of this study. BL and JJ take part in data interpretation. All authors read and approved the final manuscript.
Funding
This work was supported by grants from the National Key Research and Development Program of China (Grant Nos. 2018YFA0208902 and 2020YFC2002705), the National Science Foundation of China (NSFC Nos. U1604281 and 81672138), the Beijing Science Foundation of China (BJSFC No. 7171008), and the National Key Scientific Instrument Special Program of China (Grant No. 2011YQ03013405).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.714071/full#supplementary-material
Supplementary Figure 1 | The workflow of epigenetic heterogeneity analysis.
Supplementary Table 1 | Clinical factors of patients.
Supplementary Table 2 | Primers used for methylation analysis.
References
Ball, D. W., Jin, N., Rosen, D. M., Dackiw, A., Sidransky, D., Xing, M., et al. (2007). Selective growth inhibition in BRAF mutant thyroid cancer by the mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244. J. Clin. Endocrinol. Metab. 92, 4712–4718. doi: 10.1210/jc.2007-1184
Bedard, P. L., Hansen, A. R., Ratain, M. J., and Siu, L. L. (2013). Tumour heterogeneity in the clinic. Nature 501, 355–364. doi: 10.1038/nature12627
Brock, M. V., Gou, M., Akiyama, Y., Muller, A., Wu, T. T., Montgomery, E., et al. (2003). Prognostic importance of promoter hypermethylation of multiple genes in esophageal adenocarcinoma. Clin. Cancer Res. 9:2912.
Burrell, R. A., McGranahan, N., Bartek, J., and Swanton, C. (2013). The causes and consequences of genetic heterogeneity in cancer evolution. Nature 501, 338–345. doi: 10.1038/nature12625
Cao, B., Yang, Y., Pan, Y., Jia, Y., Brock, M. V., Herman, J. G., et al. (2013). Epigenetic silencing of CXCL14 induced colorectal cancer migration and invasion. Discov. Med. 16, 137–147.
Cha, Y. J., and Koo, J. S. (2016). Next-generation sequencing in thyroid cancer. J. Transl. Med. 14:322.
Dawson, M. A. (2017). The cancer epigenome: concepts, challenges, and therapeutic opportunities. Science (New York, NY) 355, 1147–1152. doi: 10.1126/science.aam7304
Du, W., Gao, A., Herman, J. G., Wang, L., Zhang, L., Jiao, S., et al. (2021). Methylation of NRN1 is a novel synthetic lethal marker of PI3K-Akt-mTOR and ATR inhibitors in esophageal cancer. Cancer Sci. 112, 2870–2883. doi: 10.1111/cas.14917
Esteller, M., Guo, M., Moreno, V., Peinado, M. A., Capella, G., Galm, O., et al. (2002). Hypermethylation-associated inactivation of the cellular retinol-binding-protein 1 gene in human cancer. Cancer Res. 62, 5902–5905.
Guo, M., House, M. G., Akiyama, Y., Qi, Y., Capagna, D., Harmon, J., et al. (2006a). Hypermethylation of the GATA gene family in esophageal cancer. Int. J. Cancer 119, 2078–2083. doi: 10.1002/ijc.22092
Guo, M., House, M. G., Hooker, C., Han, Y., Heath, E., Gabrielson, E., et al. (2004). Promoter hypermethylation of resected bronchial margins: a field defect of changes? Clin. Cancer Res. 10, 5131–5136. doi: 10.1158/1078-0432.ccr-03-0763
Guo, M., House, M. G., Suzuki, H., Ye, Y., Brock, M. V., Lu, F., et al. (2007). Epigenetic silencing of CDX2 is a feature of squamous esophageal cancer. Int. J. Cancer 121, 1219–1226. doi: 10.1002/ijc.22828
Guo, M., Peng, Y., Gao, A., Du, C., and Herman, J. G. (2019). Epigenetic heterogeneity in cancer. Biomark. Res. 7:23.
Guo, M., Ren, J., Brock, M. V., Herman, J. G., and Carraway, H. E. (2008). Promoter methylation of HIN-1 in the progression to esophageal squamous cancer. Epigenetics 3, 336–341. doi: 10.4161/epi.3.6.7158
Guo, M., Ren, J., House, M. G., Qi, Y., Brock, M. V., and Herman, J. G. (2006b). Accumulation of promoter methylation suggests epigenetic progression in squamous cell carcinoma of the esophagus. Clin. Cancer Res. 12, 4515–4522. doi: 10.1158/1078-0432.ccr-05-2858
Herman, J. G., Graff, J. R., Myöhänen, S., Nelkin, B. D., and Baylin, S. B. (1996). Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. U.S.A. 93, 9821–9826. doi: 10.1073/pnas.93.18.9821
Hiley, C. T., Le Quesne, J., Santis, G., Sharpe, R., de Castro, D. G., Middleton, G., et al. (2016). Challenges in molecular testing in non-small-cell lung cancer patients with advanced disease. Lancet (Lond. Engl.) 388, 1002–1011.
House, M. G., Guo, M., Efron, D. T., Lillemoe, K. D., Cameron, J. L., Syphard, J. E., et al. (2003). Tumor suppressor gene hypermethylation as a predictor of gastric stromal tumor behavior. J. Gastrointes. Surg. 7, 1004–1014; discussion 1014.
Hu, S., Cao, B., Zhang, M., Linghu, E., Zhan, Q., Brock, M. V., et al. (2015). Epigenetic silencing BCL6B induced colorectal cancer proliferation and metastasis by inhibiting P53 signaling. Am. J. Cancer Res. 5, 651–662.
Jia, Y., Yang, Y., Brock, M. V., Zhan, Q., Herman, J. G., and Guo, M. (2013). Epigenetic regulation of DACT2, a key component of the Wnt signalling pathway in human lung cancer. J. Pathol. 230, 194–204. doi: 10.1002/path.4073
Jia, Y., Yang, Y., Zhan, Q., Brock, M. V., Zheng, X., Yu, Y., et al. (2012). Inhibition of SOX17 by microRNA 141 and methylation activates the WNT signaling pathway in esophageal cancer. J. Mol. Diagn. 14, 577–585. doi: 10.1016/j.jmoldx.2012.06.004
Laha, D., Nilubol, N., and Boufraqech, M. (2020). New therapies for advanced thyroid cancer. Front. Endocrinol. 11:82. doi: 10.3389/fendo.2020.00082
Leboeuf, R., Baumgartner, J. E., Benezra, M., Malaguarnera, R., Solit, D., Pratilas, C. A., et al. (2008). BRAFV600E mutation is associated with preferential sensitivity to mitogen-activated protein kinase kinase inhibition in thyroid cancer cell lines. J. Clin. Endocrinol. Metab. 93, 2194–2201. doi: 10.1210/jc.2007-2825
Li, J. Y., Han, C., Zheng, L. L., and Guo, M. Z. (2012). Epigenetic regulation of Wnt signaling pathway gene SRY-related HMG-box 17 in papillary thyroid carcinoma. Chin. Med. J. 125, 3526–3531.
Li, Y., Yang, Y., Lu, Y., Herman, J. G., Brock, M. V., Zhao, P., et al. (2015). Predictive value of CHFR and MLH1 methylation in human gastric cancer. Gastric Cancer 18, 280–287. doi: 10.1007/s10120-014-0370-2
Licchesi, J. D., Westra, W. H., Hooker, C. M., Machida, E. O., Baylin, S. B., and Herman, J. G. (2008). Epigenetic alteration of Wnt pathway antagonists in progressive glandular neoplasia of the lung. Carcinogenesis 29, 895–904. doi: 10.1093/carcin/bgn017
Lord, C. J., and Ashworth, A. (2016). BRCAness revisited. Nat. Rev. Cancer. 16, 110–120. doi: 10.1038/nrc.2015.21
Lord, C. J., and Ashworth, A. (2017). PARP inhibitors: synthetic lethality in the clinic. Science 355, 1152–1158. doi: 10.1126/science.aam7344
Marusyk, A., Almendro, V., and Polyak, K. (2012). Intra-tumour heterogeneity: a looking glass for cancer? Nat. Rev. Cancer 12, 323–334. doi: 10.1038/nrc3261
Maura, F., Landgren, O., and Morgan, G. J. (2021). Designing evolutionary-based interception strategies to block the transition from precursor phases to multiple myeloma. Clin. Cancer Res. 27, 15–23. doi: 10.1158/1078-0432.ccr-20-1395
Mazzaferri, E. L. (1999). An overview of the management of papillary and follicular thyroid carcinoma. Thyroid 9, 421–427. doi: 10.1089/thy.1999.9.421
McLornan, D. P., List, A., and Mufti, G. J. (2014). Applying synthetic lethality for the selective targeting of cancer. N. Engl. J. Med. 371, 1725–1735. doi: 10.1056/NEJMra1407390
Mercer, K. E., and Pritchard, C. A. (2003). Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim. Biophys. Acta 1653, 25–40. doi: 10.1016/S0304-419X(03)00016-7
Nagasaka, T., Rhees, J., Kloor, M., Gebert, J., Naomoto, Y., Boland, C. R., et al. (2010). Somatic hypermethylation of MSH2 is a frequent event in lynch syndrome colorectal cancers. Cancer Res. 70, 3098–3108. doi: 10.1158/0008-5472.CAN-09-3290
Nicholson, J. G., and Fine, H. A. (2021). Diffuse glioma heterogeneity and its therapeutic implications. Cancer Discov. 11, 575–590. doi: 10.1158/2159-8290.CD-20-1474
Nowell, P. C. (1976). The clonal evolution of tumor cell populations. Science (New York, NY) 194, 23–28. doi: 10.1126/science.959840
Prete, A., Borges de Souza, P., Censi, S., Muzza, M., Nucci, N., and Sponziello, M. (2020). Update on fundamental mechanisms of thyroid cancer. Front. Endocrinol. 11:102. doi: 10.3389/fendo.2020.00102
Pribluda, A., de la Cruz, C. C., and Jackson, E. L. (2015). Intratumoral heterogeneity: from diversity comes resistance. Clin. Cancer Res. 21, 2916–2923. doi: 10.1158/1078-0432.CCR-14-1213
Rastetter, M., Schagdarsurengin, U., Lahtz, C., Fiedler, E., Marsch, W., Dammann, R., et al. (2007). Frequent intra-tumoural heterogeneity of promoter hypermethylation in malignant melanoma. Histol. Histopathol. 22, 1005–1015.
Saghafinia, S., Mina, M., Riggi, N., Hanahan, D., and Ciriello, G. (2018). Pan-cancer landscape of aberrant DNA methylation across human tumors. Cell Rep. 25, 1066–1080.e8. doi: 10.1016/j.celrep.2018.09.082
Salerno, P., De Falco, V., Tamburrino, A., Nappi, T. C., Vecchio, G., Schweppe, R. E., et al. (2009). Cytostatic activity of adenosine triphosphate-competitive kinase inhibitors in BRAF mutant thyroid carcinoma cells. Endocr. Rev. 30:932. doi: 10.1210/edrv.30.7.9986
Seib, C. D., and Sosa, J. A. (2019). Evolving understanding of the epidemiology of thyroid cancer. Endocrinol. Metab. Clin. North Am. 48, 23–35. doi: 10.1016/j.ecl.2018.10.002
Valerio, L., Pieruzzi, L., Giani, C., Agate, L., Bottici, V., Lorusso, L., et al. (2017). Targeted therapy in thyroid cancer: state of the art. Clin. Oncol. (R. Coll. Radiol.) 29, 316–324. doi: 10.1016/j.clon.2017.02.009
Varley, K. E., Mutch, D. G., Edmonston, T. B., Goodfellow, P. J., and Mitra, R. D. (2009). Intra-tumor heterogeneity of MLH1 promoter methylation revealed by deep single molecule bisulfite sequencing. Nucleic Acids Res. 37, 4603–4612. doi: 10.1093/nar/gkp457
Vogelstein, B., Papadopoulos, N., Velculescu, V. E., Zhou, S., Diaz, L. A. Jr., and Kinzler, K. W. (2013). Cancer genome landscapes. Science (New York, NY) 339, 1546–1558. doi: 10.1126/science.1235122
Wand, H., and Lambert, S. A. (2021). Improving reporting standards for polygenic scores in risk prediction studies. Nature 591, 211–219. doi: 10.1038/s41586-021-03243-6
Wang, J. S., Guo, M., Montgomery, E. A., Thompson, R. E., Cosby, H., Hicks, L., et al. (2009). DNA promoter hypermethylation of p16 and APC predicts neoplastic progression in Barrett’s esophagus. Am. J. Gastroenterol. 104, 2153–2160. doi: 10.1038/ajg.2009.300
Wang, Y., Zhang, Y., Herman, J. G., Linghu, E., and Guo, M. (2017). Epigenetic silencing of TMEM176A promotes esophageal squamous cell cancer development. Oncotarget 8, 70035–70048. doi: 10.18632/oncotarget.19550
Yamamoto, Y., Matsui, J., Matsushima, T., Obaishi, H., Miyazaki, K., Nakamura, K., et al. (2014). Lenvatinib, an angiogenesis inhibitor targeting VEGFR/FGFR, shows broad antitumor activity in human tumor xenograft models associated with microvessel density and pericyte coverage. Vasc. Cell 6:18. doi: 10.1186/2045-824X-6-18
Zhao, H., Li, J., Li, X., Han, C., Zhang, Y., Zheng, L., et al. (2015). Silencing GPX3 expression promotes tumor metastasis in human thyroid cancer. Curr. Protein Pept. Sci. 16, 316–321. doi: 10.2174/138920371604150429154840
Zhao, Z., Herman, J. G., Brock, M. V., Sheng, J., Zhang, M., Liu, B., et al. (2014). Methylation of DACT2 promotes papillary thyroid cancer metastasis by activating Wnt signaling. PLoS One 9:e112336. doi: 10.1371/journal.pone.0112336
Keywords: epigenetic heterogeneity, DNA methylation, AP2, CDH1, DACT2, HIN1, RASSF1A
Citation: Zhu C, Zhang M, Wang Q, Jen J, Liu B and Guo M (2021) Intratumor Epigenetic Heterogeneity—A Panel Gene Methylation Study in Thyroid Cancer. Front. Genet. 12:714071. doi: 10.3389/fgene.2021.714071
Received: 24 May 2021; Accepted: 16 August 2021;
Published: 03 September 2021.
Edited by:
Obul Reddy Bandapalli, Hopp Children’s Cancer Center Heidelberg (KiTZ), GermanyReviewed by:
Jitian Li, Henan Luoyang Orthopedic Hospital, ChinaEnrique Ambrocio-Ortiz, Instituto Nacional de Enfermedades Respiratorias-México (INER), Mexico
Copyright © 2021 Zhu, Zhang, Wang, Jen, Liu and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Baoguo Liu, bGJnMjlAMTYzLmNvbQ==; Mingzhou Guo, bXpndW9AaG90bWFpbC5jb20=