 Rachel Adihe Lokanga
Rachel Adihe Lokanga Daman Kumari
Daman Kumari Karen Usdin
Karen Usdin- 1Cancer Genetics Branch, National Cancer Institute, Bethesda, MD, United States
- 2Laboratory of Cell and Molecular Biology, National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD, United States
The human genome has many chromosomal regions that are fragile, demonstrating chromatin breaks, gaps, or constrictions on exposure to replication stress. Common fragile sites (CFSs) are found widely distributed in the population, with the largest subset of these sites being induced by aphidicolin (APH). Other fragile sites are only found in a subset of the population. One group of these so-called rare fragile sites (RFSs) is induced by folate stress. APH-inducible CFSs are generally located in large transcriptionally active genes that are A + T rich and often enriched for tracts of AT-dinucleotide repeats. In contrast, all the folate-sensitive sites mapped to date consist of transcriptionally silenced CGG microsatellites. Thus, all the folate-sensitive fragile sites may have a very similar molecular basis that differs in key ways from that of the APH CFSs. The folate-sensitive FSs include FRAXA that is associated with Fragile X syndrome (FXS), the most common heritable form of intellectual disability. Both CFSs and RFSs can cause chromosomal abnormalities. Recent work suggests that both APH-inducible fragile sites and FRAXA undergo Mitotic DNA synthesis (MiDAS) when exposed to APH or folate stress, respectively. Interestingly, blocking MiDAS in both cases prevents chromosome fragility but increases the risk of chromosome mis-segregation. MiDAS of both APH-inducible and FRAXA involves conservative DNA replication and POLD3, an accessory subunit of the replicative polymerase Pol δ that is essential for break-induced replication (BIR). Thus, MiDAS is thought to proceed via some form of BIR-like process. This review will discuss the recent work that highlights the similarities and differences between these two groups of fragile sites and the growing evidence for the presence of many more novel fragile sites in the human genome.
Introduction
Fragile sites are apparent as chromatin gaps, constrictions, or breaks in cells exposed to replication stress (Sutherland, 1991). These sites are typically classified based on the reagent that induces their expression most effectively. They are also classified as common or rare, depending on their frequency in the population (Feng and Chakraborty, 2017). The largest known group of fragile sites are most efficiently induced by aphidicolin (APH), an inhibitor of DNA polymerases α, δ, and ε. FRA3B and FRA16D are among the best known APH inducible CFSs. FRA3B is associated with the fragile histidine triad (FHIT) gene, a tumor suppressor gene located on chromosome 3p14.2 and FRA16D is associated with the WW domain-containing oxidoreductase (WWOX) gene, a tumor suppressor gene located on chromosome 16 (Bednarek et al., 2000). Another group of fragile sites are referred to as being folate-sensitive since they are induced by either too much or too little folate, with both situations resulting in nucleotide pool imbalances (Glover, 1981; James et al., 1993). Perhaps, the best known of the folate-sensitive fragile sites is the rare fragile site, FRAXA, a site on the X chromosome that is seen in individuals with fragile X syndrome (FXS), the most common heritable cause of intellectual disability and autism spectrum disorder (Lozano et al., 2014). Other fragile sites are induced by agents, such as 5-azacytidine, 5-bromo-2-deoxyuridine, or distamycin A that can be incorporated or intercalated into DNA (Schmid et al., 1980; Sutherland et al., 1985; Hori et al., 1988). Interestingly, FRA16B and FRA10B, two rare distamycin-inducible fragile sites, are AT-rich minisatellites (Yu et al., 1997; Hewett et al., 1998) that are expansions of the AT microsatellites normally present in the CFSs FRA16C and FRA10E, respectively (Zlotorynski et al., 2003). As such, they may share common features with the APH-inducible sites. While most fragile sites replicate late in the cell cycle, early replicating fragile sites (ERFSs) have also been identified that are readily induced by hydroxyurea, a reagent that causes depletion of deoxynucleotide pools (Barlow et al., 2013).
Fragile sites are all thought to be regions of the genome that for some reason are slow to complete replication, and their presence is associated with a variety of chromosome abnormalities. Genome instability at CFSs is thought to be a driving force for tumorigenesis with APH-CFSs being associated with copy number variations, including a variety of recurrent cancer deletions (Le Tallec et al., 2013; Wilson et al., 2015; Zheglo et al., 2019). Some CFS-associated CNVs are also associated with neurological disorders (Denison et al., 2003; Ambroziak et al., 2015; Zheglo et al., 2019). CFSs are also frequent sites of viral integration associated with cancer (Thorland et al., 2000; Yu et al., 2005). In contrast to the CNVs associated with CFSs, ERFSs are associated with recurrent chromosomal rearrangements during lymphomagenesis (Barlow et al., 2013). The RFS FRAXA is associated with a high frequency loss of the affected X chromosome in vitro in response to folate stress (Bjerregaard et al., 2018) and in vivo (Dobkin et al., 2009), and many cases of Jacobsen (11q-) syndrome, a chromosomal deletion disorder affecting chromosome 11, have been attributed to the presence of folate-sensitive fragile sites on that chromosome (Jones et al., 1994).
The Molecular Basis of the Replication Problems At Cfss and Folate-Sensitive Fss
Unlike ERFSs which are located in early replicating G + C-rich, gene-dense regions with high numbers of activated origins of replication (ORIs) (Barlow et al., 2013), many APH-inducible CFSs are located in active, A + T-rich genes that are >300kb in size, replicate late, and are frequently ORI-poor (Glover et al., 2017; Debatisse and Rosselli, 2019). CFSs have been reported to be located at topologically associated domains (TADs) in some studies (Sarni et al., 2020), but not others (Ji et al., 2020). Some CFSs are associated with the expression of different oncogenes that can modulate replication stress (Miron et al., 2015). Transcription is required for CFS expression (Helmrich et al., 2011; Park et al., 2021), although higher transcription rates are associated with reduced fragility, perhaps due to the associated shift of the locus to replication earlier in the cell cycle (Blin et al., 2019). The relationship to transcription likely explains the reported tissue specificity of CFS expression.
Many different models have been proposed to account for the replication difficulties of CFSs, including those invoking replication-transcription collisions that promote R-loop formation and ultimately the stalling of the replication fork (Helmrich et al., 2011) and/or structural blocks to replication fork progression resulting from hairpin or cruciform formation by the AT-dinucleotide-rich regions embedded within many CFSs (Zlotorynski et al., 2003; Ozeri-Galai et al., 2011; Irony-Tur Sinai et al., 2019; Van Wietmarschen et al., 2020). In addition, TAD boundaries located between different replication timing zones are known to be prone to replication fork stalling (Lombardi and Tarsounas, 2020). Since ORIs are only licensed in G1 and bound pre-replication complexes can be displaced by RNA Pol II, at least in yeast (Snyder et al., 1988), it has also been suggested that transcription of long genes results in a paucity of active ORIs within the gene body that delays the completion of replication (Brison et al., 2019). Parenthetically, while a paucity of ORIs is associated with replication stress at CFSs, it has been suggested that increased ORI initiation at ERFSs also causes replication stress, perhaps by prematurely depleting nucleotide pools or by increasing replication-transcription collisions (Barlow et al., 2013).
However, while some studies support a role of R-loops in replication stress at fragile sites, including FRA3B (Helmrich et al., 2011), others do not (Park et al., 2021). Furthermore, while molecular combing has demonstrated replication stalling at FRA16C (Ozeri-Galai et al., 2011) and at FRA16D and FRA6E in FANCD2−/− cells (Madireddy et al., 2016), combing studies of FRA3B and FRA6E showed no evidence of abnormal fork speed or replication fork stalling in normal APH-treated cells (Palumbo et al., 2010; Letessier et al., 2011). The lack of stalling at FRA3B together with the fact that transcription inhibition in S phase did not affect fragile site expression would be consistent with the idea that stalled replication forks and/or replication-transcription collisions are not a major source of replication stress at all CFSs (Brison et al., 2019). In addition, while delayed replication and their presence within large, transcriptionally active genes are consistent features of CFSs, these features are not sufficient for fragility, since a number of active, large genes that replicate late are not fragile (Wilson et al., 2015; Sarni et al., 2020; Park et al., 2021). Thus, the precise nature of the replication problem or problems at CFSs remains enigmatic and current thinking is that a combination of different factors may contribute to replication stress at different loci.
Unlike CFSs, many of the RFSs involve a much shorter region of DNA, usually 0.6–5kb. Of the 10 folate-sensitive RFSs characterized to date, all consist of a single tract of >200 CGG repeats (Table 1). In most cases, the repeat is in the 5' UTR of a gene that is epigenetically silenced (Lukusa and Fryns, 2008). Thus, fragility of these sites is likely to have a similar molecular basis. These sites are often associated with human disease, most commonly intellectual disability and autism spectrum disorder. However, it is not the fragile site itself that is responsible for this pathology, but rather the silencing-associated loss of the affected gene product. In the case of FRAXA and its associated disorder, FXS, the CGG repeat tract is located in the 5' UTR of the X-linked FMR1 gene. The CGG repeat tract is prone to two forms of instability, the tendency to gain repeats with time, a hallmark of the repeat expansion diseases (Paulson, 2018) and the propensity to show fragility and sex chromosome aneuploidy (Dobkin et al., 2009). Both CGG repeats and the complementary CCG repeat form secondary structures, including hairpins and either G4 quadruplexes or i-motif structures [reviewed in Mirkin (2006)]. In vitro the CGG repeats show a K+ specific block to DNA synthesis consistent with the underlying problem being the formation of a G4 structure (Usdin and Woodford, 1995). The repeats also stall DNA synthesis in mammalian model systems (Voineagu et al., 2009) and in the endogenous FMR1 locus (Gerhardt et al., 2014). In contrast to APH-inducible sites, the expression of FRAXA requires transcriptional silencing since those rare FXS alleles that escape silencing are not fragile (Yudkin et al., 2014). DNA methylation associated with gene silencing could increase the stability of fork blocking structures (Hardin et al., 1993; Lin et al., 2013). However, it is probable that silencing per se is not the trigger for fragility, but rather the delayed replication associated with silencing; transcribed FMR1 alleles replicate late in the cell cycle, in S4 or G2 (Hansen et al., 2010), with silenced FXS alleles replicating even later (Webb, 1992; Hansen et al., 1997). Folate stress would delay this even further. FXS alleles lack the association with a TAD boundary that is seen in normal alleles (Sun et al., 2018). Thus, while APH CFSs and FRAXA share some common features, the underlying problems responsible for these different classes of fragile sites are likely to also be different.

Table 1. Folate-sensitive rare fragile sites known to be associated with CGG microsatellites.
The Downstream Consequences of the Replication Problems At Fragile Sites
Both APH-inducible CFSs and the FRAXA locus undergo mitotic DNA synthesis (MiDAS; Minocherhomji et al., 2015; Bhowmick et al., 2016; Garribba et al., 2020), a salvage pathway that ensures that regions of the genome that have not completed replication by the start of mitosis are successfully duplicated before the cell divides (Minocherhomji et al., 2015). Given that folate-sensitive fragile sites are all comprised of long CGG microsatellites, it is reasonable to think that other folate-sensitive fragile sites undergo MiDAS as well. MiDAS at both APH-inducible CFSs and FRAXA shares some common features. Both proceed via conservative DNA replication, in which DNA synthesis is confined to just one of the sister chromatids. They both also require POLD3 (Bhowmick et al., 2016; Garribba et al., 2020). POLD3, an accessory subunit of the replicative polymerase Pol δ, is not required for normal chromosomal replication, but is required for break-induced replication (BIR) (Costantino et al., 2014), a form of homologous recombination involved in the repair of one-sided double-strand breaks (DSBs) arising at collapsed replication forks. Thus, MiDAS has many of the hallmarks of a BIR-related process. BIR usually proceeds via the cleavage of the leading strand template by one of the structure-selective endonucleases: XPF-ERCC1, MUS81-EME1, or SLX1-SLX4. Cleavage results in a free 3' DNA tail that can strand-invade the sister chromatid to create a D-loop thus allowing POLD3-dependent DNA synthesis to proceed using the sister chromatid as a template. Successful completion of BIR at fragile site loci results in completely replicated chromatids that can be properly segregated into daughter cells in anaphase. Inhibition of BIR, on the other hand, results in the reduced expression of both CFSs and FRAXA, consistent with the idea that fragility is an active process resulting from MiDAS that has not been completed by the time normal chromatin condensation begins (Minocherhomji et al., 2015; Garribba et al., 2020). BIR frequently involves multiple rounds of strand invasion, DNA synthesis, and dissociation (Smith et al., 2007). Dissociation at one interspersed repeat and reinvasion into a different one could produce deletions, if the second repeat was downstream of the first one, and duplications if upstream. This could contribute to the high incidence of CNVs associated with fragile sites. Repeated mispriming within the repeat tract during BIR could also account for the tendency of CGG repeat tracts to expand (Kononenko et al., 2018). However, in the case of CGG repeats at the FMR1 locus expansions occur in cells like ova that do not replicate (Yrigollen et al., 2014; Zhao and Usdin, 2018) and, in contrast to fragility, expansion at this locus requires transcription (Lokanga et al., 2014). Thus, the trigger for fragility and expansion of CGG repeats may differ.
While both the expression of CFSs and FRAXA likely involve some form of BIR, the process at these sites differs with respect to some of the proteins involved as illustrated in Figure 1. Specifically, initiation of BIR at APH-inducible sites involves cleavage of the replication intermediates by Mus81-EME1 (Ying et al., 2013) acting in conjunction with the scaffolding protein, SLX4 (Minocherhomji et al., 2015). Processing of the cleavage product requires Rad52 (Ying et al., 2013). In contrast, BIR at FRAXA requires the RAD51 recombinase and the SLX1-SLX4 endonuclease (Garribba et al., 2020). It has been suggested that the DNA secondary structures formed by the CGG repeat tract result in an atypical stalled fork that is a poor substrate for MUS81-EME1 (Garribba et al., 2020), a complex that specifically nicks duplex DNA on the 5'-side of a single-stranded/double-stranded DNA branch point (Wyatt et al., 2013). In contrast, the SLX1 endonuclease, which is activated by binding to SLX4, has a wider range of possible substrates and can incise duplex or single-stranded DNA on either the 5'- or 3'-sides of the branch point, thus allowing SLX1-SLX4 to nick either the leading or lagging strand template (Wyatt et al., 2013). This difference in substrates may account for the involvement of different enzymes for processing stalled replication forks in the case of CFSs and FRAXA (Garribba et al., 2020). Interestingly, in a tissue culture reporter system, siRNA knockdown of either RAD51 or RAD52 significantly reduced BIR-associated mutagenesis of the region flanking a CGG repeat tract (Kononenko et al., 2018). Whether this reflects two different BIR subpathways operating in these cells or some sort of hybrid process is unclear.
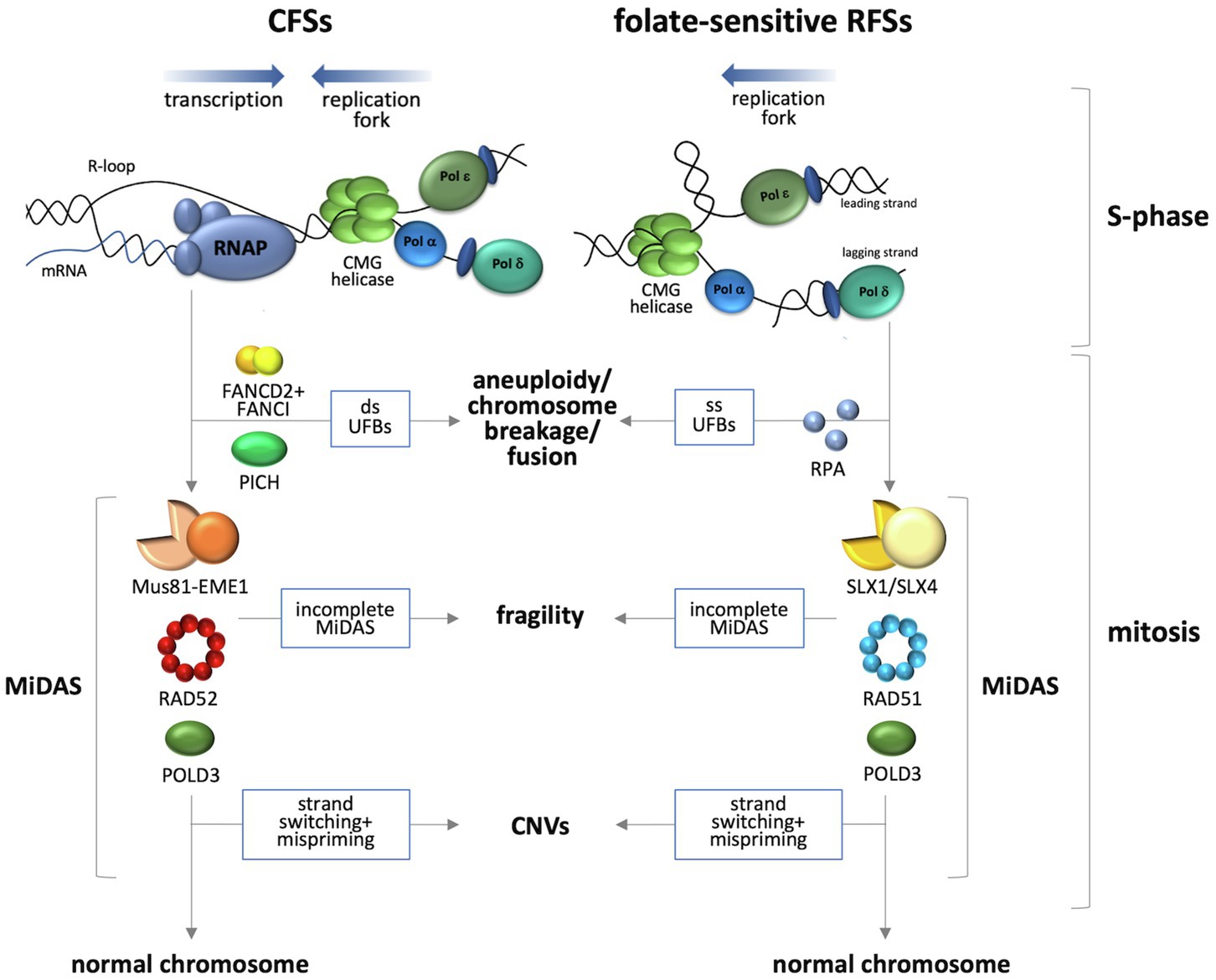
Figure 1. Current models for events occurring at loci containing APH-inducible and folate-sensitive FSs. APH-inducible sites are difficult to replicate, a situation that may be exacerbated by collisions between the replication fork and the transcription complex. A head-on collision is depicted here since it is associated with elevated levels of DSBs as well as the formation of R-loops (Hamperl et al., 2017) suggested to be important for fragility of these sites (Helmrich et al., 2011). Folate-sensitive RFSs are associated with replication problems resulting from the formation of a fork blocking lesion at a locus that is transcriptionally silent. Rescue of the stalled replication forks occurs by MiDAS that involves two different BIR subpathways (Wyatt et al., 2013; Ying et al., 2013; Minocherhomji et al., 2015; Bhowmick et al., 2016; Garribba et al., 2020). Completion of MiDAS produces a normal chromosome, while failure to do so results in chromosome fragility. Strand switching during BIR that results in mispriming can result in CNVs. Failure to initiate MiDAS results in either double-stranded UFBs in the case of the APH sites (Chan et al., 2007) or single-stranded UFBs in the case of FRAXA (Bjerregaard et al., 2018).
While initiation of MiDAS is required for cytogenetic expression of the fragile site, failure to initiate MiDAS at both sets of loci leads to increased formation of ultrafine bridges (UFBs) in anaphase (Minocherhomji et al., 2015; Bhowmick et al., 2016; Garribba et al., 2020). These UFBs are anaphase bridges that do not stain with conventional DNA stains like Hoechst or DAPI and are not associated with histones (Chan et al., 2009). Failure to resolve these UFBs results the formation of micronuclei and chromosome mis-segregation (Fernandez-Casanas and Chan, 2018). Perhaps not surprisingly given the differences in the underlying cause of replication fork stalling, CFSs and the FRAXA locus also differ in the nature of the UFBs that are formed when MiDAS does not occur. The UFBs formed at APH-inducible sites are coated with PICH, a DNA translocase, are double-stranded (Liu et al., 2014), and are bounded by FANCD2/FANCI foci (Chan et al., 2009). The absence of an effect of topoisomerase II inhibition on the frequency of these UFBs suggests that they reflect the presence of under-replicated DNA or unresolved replication intermediates rather than dsDNA catenanes (Chan et al., 2009). The UFBs associated with FRAXA on the other hand are not associated with FANCD2, FANCI, or PICH. Instead, they are coated with RPA (Bjerregaard et al., 2018) and are thus likely to be single-stranded, consistent with unresolved HR intermediates (Fernandez-Casanas and Chan, 2018).
Concluding Remarks
Lessons learnt from these two groups of fragile sites have allowed many more potential fragile sites to be identified. For example, genome-wide mapping of loci that undergo MiDAS in the presence of APH has identified hundreds of potential new CFSs (Ji et al., 2020; Macheret et al., 2020). Genome-wide studies of loci showing a fragility signature consisting of a TAD boundary that overlaps a highly transcribed, large gene with APH-induced replication delay, also suggest the presence of additional sites (Sarni et al., 2020). In addition, folate stress induces MiDAS or γ-H2AX foci, a marker of DSBs, at many genomic loci in normal human cells (Kumari et al., 2009; Garribba et al., 2020), suggesting that there are also several common folate-sensitive fragile sites that are as yet uncharacterized. Furthermore, a recent study of epigenetic variation in the human genome suggests the existence of at least 19 rare, long, and silenced CGG repeat tracts that could well also be fragile (Garg et al., 2020).
In addition to the CGG repeat diseases associated with folate-sensitive RFSs, many other repeat expansion diseases are known (Paulson, 2018). The CTG repeats responsible for a subset of these disorders, block replication (Samadashwily et al., 1997; Pelletier et al., 2003), induce BIR (Kim et al., 2017), and cause chromosome fragility in yeast (Freudenreich et al., 1998; Freudenreich and Lahiri, 2004). They also block replication in human cells (Liu et al., 2012). Furthermore, when cells containing a reporter construct with (CTG)100 repeats were treated with hydroxyurea replication-dependent DSBs were seen close to the replication fork (Gadgil et al., 2020). Increased fragility as evidenced by the loss of an adjacent fluorescent reporter was also seen along with evidence of BIR. Interestingly, unlike BIR at the FRAXA locus, this BIR was dependent on MUS81 (Gadgil et al., 2020). CTG and CAG repeats form hairpins (reviewed in Mirkin, 2006), like the CGG and CCG repeats responsible for FRAXA. However, they do not form G4 or i-motif structures. The MUS81 requirement for fragility may reflect this difference. Since some of the CTG/CAG expansion disorders can involve thousands of repeats (Fu et al., 1992; Mahadevan et al., 1992; Van Kuilenburg et al., 2019), they may also be fragile. However, no mitotic fragility has been reported for individuals with one such disorder, myotonic dystrophy type 1 (DM1; Jalal et al., 1993; Wenger et al., 1996). This may reflect the fact that, according to the ENCODE dataset, DMPK, the affected gene, replicates in the G1 or G1b phase of the cell cycle (Thurman et al., 2007; Hansen et al., 2010). Similarly, the GAA repeat tract responsible for Friedreich ataxia has key hallmarks of a mammalian fragile site: It blocks DNA synthesis (Krasilnikova and Mirkin, 2004; Gerhardt et al., 2016; Murat et al., 2020), is fragile in yeast (Kim et al., 2008), and is prone to chromosomal duplications in culture (Kumari et al., 2015). It is also associated with a high frequency of de novo mutations in the flanking regions (Bidichandani et al., 1999), a hallmark of BIR. However, as with the DM1 repeats, these loci are not apparent as gaps or constrictions in the chromatin in metaphase spreads, perhaps because they too replicate early in S phase (Kumari et al., 2015).
Thus, the repeats responsible for the repeat expansion diseases, may represent an unappreciated double threat to the human genome: the first threat being mediated via the deleterious effects of having large numbers of repeats in the DNA, RNA, and/or protein encoded by the affected loci (Paulson, 2018) and the second posed by the difficulty of replicating the repeats, with downstream effects on genome integrity, including aneuploidy, translocations, and CNVs. In addition to the repeats currently known to be associated with pathology, many thousands of other microsatellites with potential to stall DNA replication are known to be present in the human genome (Murat et al., 2020). Thus, the number of potentially fragile sites in the human genome could well be much higher than currently appreciated.
Author Contributions
All authors contributed equally to this manuscript.
Funding
Funding for this work is provided by a grant from the Intramural Program of NIDDK, NIH to KU (1ZIADK057808).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ambroziak, W., Koziorowski, D., Duszyc, K., Gorka-Skoczylas, P., Potulska-Chromik, A., Slawek, J., et al. (2015). Genomic instability in the PARK2 locus is associated with Parkinson's disease. J. Appl. Genet. 56, 451–461. doi: 10.1007/s13353-015-0282-9
Barlow, J. H., Faryabi, R. B., Callen, E., Wong, N., Malhowski, A., Chen, H. T., et al. (2013). Identification of early replicating fragile sites that contribute to genome instability. Cell 152, 620–632. doi: 10.1016/j.cell.2013.01.006
Bednarek, A. K., Laflin, K. J., Daniel, R. L., Liao, Q., Hawkins, K. A., and Aldaz, C. M. (2000). WWOX, a novel WW domain-containing protein mapping to human chromosome 16q23.3-24.1, a region frequently affected in breast cancer. Cancer Res. 60, 2140–2145.
Bhowmick, R., Minocherhomji, S., and Hickson, I. D. (2016). RAD52 facilitates mitotic DNA synthesis following replication stress. Mol. Cell 64, 1117–1126. doi: 10.1016/j.molcel.2016.10.037
Bidichandani, S. I., Purandare, S. M., Taylor, E. E., Gumin, G., Machkhas, H., and Harati, Y. (1999). Somatic sequence variation at the Friedreich ataxia locus includes complete contraction of the expanded GAA triplet repeat, significant length variation in serially passaged lymphoblasts and enhanced mutagenesis in the flanking sequence. Hum. Mol. Genet. 8, 2425–2436. doi: 10.1093/hmg/8.13.2425
Bjerregaard, V. A., Garribba, L., Mcmurray, C. T., Hickson, I. D., and Liu, Y. (2018). Folate deficiency drives mitotic missegregation of the human FRAXA locus. Proc. Natl. Acad. Sci 115, 13003–13008. doi: 10.1073/pnas.1808377115
Blin, M., Le Tallec, B., Nahse, V., Schmidt, M., Brossas, C., Millot, G. A., et al. (2019). Transcription-dependent regulation of replication dynamics modulates genome stability. Nat. Struct. Mol. Biol. 26, 58–66. doi: 10.1038/s41594-018-0170-1
Brison, O., El-Hilali, S., Azar, D., Koundrioukoff, S., Schmidt, M., Nahse, V., et al. (2019). Transcription-mediated organization of the replication initiation program across large genes sets common fragile sites genome-wide. Nat. Commun. 10:5693. doi: 10.1038/s41467-019-13674-5
Chan, K. L., North, P. S., and Hickson, I. D. (2007). BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 26, 3397–3409. doi: 10.1038/sj.emboj.7601777
Chan, K. L., Palmai-Pallag, T., Ying, S., and Hickson, I. D. (2009). Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 11, 753–760. doi: 10.1038/ncb1882
Costantino, L., Sotiriou, S. K., Rantala, J. K., Magin, S., Mladenov, E., Helleday, T., et al. (2014). Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 343, 88–91. doi: 10.1126/science.1243211
Debacker, K., Winnepenninckx, B., Longman, C., Colgan, J., Tolmie, J., Murray, R., et al. (2007). The molecular basis of the folate-sensitive fragile site FRA11A at 11q13. Cytogenet. Genome Res. 119, 9–14. doi: 10.1159/000109612
Debatisse, M., and Rosselli, F. (2019). A journey with common fragile sites: From S phase to telophase. Genes Chromosomes Cancer 58, 305–316. doi: 10.1002/gcc.22704
Denison, S. R., Callahan, G., Becker, N. A., Phillips, L. A., and Smith, D. I. (2003). Characterization of FRA6E and its potential role in autosomal recessive juvenile parkinsonism and ovarian cancer. Genes Chromosomes Cancer 38, 40–52. doi: 10.1002/gcc.10236
Dobkin, C., Radu, G., Ding, X. H., Brown, W. T., and Nolin, S. L. (2009). Fragile X prenatal analyses show full mutation females at high risk for mosaic turner syndrome: fragile X leads to chromosome loss. Am. J. Med. Genet. A 149A, 2152–2157. doi: 10.1002/ajmg.a.33011
Feng, W., and Chakraborty, A. (2017). Fragility extraordinaire: unsolved mysteries of chromosome fragile sites. Adv. Exp. Med. Biol. 1042, 489–526. doi: 10.1007/978-981-10-6955-0_21
Fernandez-Casanas, M., and Chan, K. L. (2018). The unresolved problem of DNA bridging. Genes 9:623. doi: 10.3390/genes9120623
Freudenreich, C. H., Kantrow, S. M., and Zakian, V. A. (1998). Expansion and length-dependent fragility of CTG repeats in yeast. Science 279, 853–856. doi: 10.1126/science.279.5352.853
Freudenreich, C. H., and Lahiri, M. (2004). Structure-forming CAG/CTG repeat sequences are sensitive to breakage in the absence of Mrc1 checkpoint function and S-phase checkpoint signaling: implications for trinucleotide repeat expansion diseases. Cell Cycle 3, 1370–1374. doi: 10.4161/cc.3.11.1246
Fu, Y. H., Pizzuti, A., Fenwick, R. G., King, J., Rajnarayan, S., Dunne, P. W., et al. (1992). An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 255, 1256–1258. doi: 10.1126/science.1546326
Gadgil, R. Y., Romer, E. J., Goodman, C. C., Rider, S. D., Damewood, F. J., Barthelemy, J. R., et al. (2020). Replication stress at microsatellites causes DNA double-strand breaks and break-induced replication. J. Biol. Chem. 295, 15378–15397. doi: 10.1074/jbc.RA120.013495
Garg, P., Jadhav, B., Rodriguez, O. L., Patel, N., Martin-Trujillo, A., Jain, M., et al. (2020). A survey of rare epigenetic variation in 23,116 human genomes identifies disease-relevant Epivariations and CGG expansions. Am. J. Hum. Genet. 107, 654–669. doi: 10.1016/j.ajhg.2020.08.019
Garribba, L., Bjerregaard, V. A., Goncalves Dinis, M. M., Ozer, O., Wu, W., Sakellariou, D., et al. (2020). Folate stress induces SLX1- and RAD51-dependent mitotic DNA synthesis at the fragile X locus in human cells. Proc. Natl. Acad. Sci 117, 16527–16536. doi: 10.1073/pnas.1921219117
Gerhardt, J., Bhalla, A. D., Butler, J. S., Puckett, J. W., Dervan, P. B., Rosenwaks, Z., et al. (2016). Stalled DNA replication forks at the endogenous GAA repeats drive repeat expansion in Friedreich's ataxia cells. Cell Rep. 16, 1218–1227. doi: 10.1016/j.celrep.2016.06.075
Gerhardt, J., Tomishima, M. J., Zaninovic, N., Colak, D., Yan, Z., Zhan, Q., et al. (2014). The DNA replication program is altered at the FMR1 locus in fragile X embryonic stem cells. Mol. Cell 53, 19–31. doi: 10.1016/j.molcel.2013.10.029
Glover, T. W. (1981). FUdR induction of the X chromosome fragile site: evidence for the mechanism of folic acid and thymidine inhibition. Am. J. Hum. Genet. 33, 234–242.
Glover, T. W., Wilson, T. E., and Arlt, M. F. (2017). Fragile sites in cancer: more than meets the eye. Nat. Rev. Cancer 17, 489–501. doi: 10.1038/nrc.2017.52
Hamperl, S., Bocek, M. J., Saldivar, J. C., Swigut, T., and Cimprich, K. A. (2017). Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell 170:e719. doi: 10.1016/j.cell.2017.07.043
Hansen, R. S., Canfield, T. K., Fjeld, A. D., Mumm, S., Laird, C. D., and Gartler, S. M. (1997). A variable domain of delayed replication in FRAXA fragile X chromosomes: X inactivation-like spread of late replication. Proc. Natl. Acad. Sci 94, 4587–4592.
Hansen, R. S., Thomas, S., Sandstrom, R., Canfield, T. K., Thurman, R. E., Weaver, M., et al. (2010). Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl. Acad. Sci 107, 139–144. doi: 10.1073/pnas.0912402107
Hardin, C. C., Corregan, M., Brown, B. A. 2nd, and Frederick, L. N. (1993). Cytosine-cytosine+ base pairing stabilizes DNA quadruplexes and cytosine methylation greatly enhances the effect. Biochemistry 32, 5870–5880. doi: 10.1021/bi00073a021
Helmrich, A., Ballarino, M., and Tora, L. (2011). Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 44, 966–977. doi: 10.1016/j.molcel.2011.10.013
Hewett, D. R., Handt, O., Hobson, L., Mangelsdorf, M., Eyre, H. J., Baker, E., et al. (1998). FRA10B structure reveals common elements in repeat expansion and chromosomal fragile site genesis. Mol. Cell 1, 773–781. doi: 10.1016/S1097-2765(00)80077-5
Hori, T., Takahashi, E., and Murata, M. (1988). Nature of distamycin A-inducible fragile sites. Cancer Genet. Cytogenet. 34, 189–194. doi: 10.1016/0165-4608(88)90258-0
Irony-Tur Sinai, M., Salamon, A., Stanleigh, N., Goldberg, T., Weiss, A., Wang, Y. H., et al. (2019). AT-dinucleotide rich sequences drive fragile site formation. Nucleic Acids Res. 47, 9685–9695. doi: 10.1093/nar/gkz689
Jalal, S. M., Lindor, N. M., Michels, V. V., Buckley, D. D., Hoppe, D. A., Sarkar, G., et al. (1993). Absence of chromosome fragility at 19q13.3 in patients with myotonic dystrophy. Am. J. Med. Genet. 46, 441–443. doi: 10.1002/ajmg.1320460419
James, S. J., Miller, B. J., Cross, D. R., Mcgarrity, L. J., and Morris, S. M. (1993). The essentiality of folate for the maintenance of deoxynucleotide precursor pools, DNA synthesis, and cell cycle progression in PHA-stimulated lymphocytes. Environ. Health Perspect. 101 (Suppl 5), 173–178.
Ji, F., Liao, H., Pan, S., Ouyang, L., Jia, F., Fu, Z., et al. (2020). Genome-wide high-resolution mapping of mitotic DNA synthesis sites and common fragile sites by direct sequencing. Cell Res. 30, 1009–1023. doi: 10.1038/s41422-020-0357-y
Jones, C., Slijepcevic, P., Marsh, S., Baker, E., Langdon, W. Y., Richards, R. I., et al. (1994). Physical linkage of the fragile site FRA11B and a Jacobsen syndrome chromosome deletion breakpoint in 11q23.3. Hum. Mol. Genet. 3, 2123–2130. doi: 10.1093/hmg/3.12.2123
Kim, H. M., Narayanan, V., Mieczkowski, P. A., Petes, T. D., Krasilnikova, M. M., Mirkin, S. M., et al. (2008). Chromosome fragility at GAA tracts in yeast depends on repeat orientation and requires mismatch repair. EMBO J. 27, 2896–2906. doi: 10.1038/emboj.2008.205
Kim, J. C., Harris, S. T., Dinter, T., Shah, K. A., and Mirkin, S. M. (2017). The role of break-induced replication in large-scale expansions of (CAG)n /(CTG)n repeats. Nat. Struct. Mol. Biol. 24, 55–60. doi: 10.1038/nsmb.3334
Knight, S. J., Flannery, A. V., Hirst, M. C., Campbell, L., Christodoulou, Z., Phelps, S. R., et al. (1993). Trinucleotide repeat amplification and hypermethylation of a CpG island in FRAXE mental retardation. Cell 74, 127–134. doi: 10.1016/0092-8674(93)90300-F
Kononenko, A. V., Ebersole, T., Vasquez, K. M., and Mirkin, S. M. (2018). Mechanisms of genetic instability caused by (CGG)n repeats in an experimental mammalian system. Nat. Struct. Mol. Biol. 25, 669–676. doi: 10.1038/s41594-018-0094-9
Krasilnikova, M. M., and Mirkin, S. M. (2004). Replication stalling at Friedreich's ataxia (GAA)n repeats in vivo. Mol. Cell. Biol. 24, 2286–2295. doi: 10.1128/MCB.24.6.2286-2295.2004
Kumari, D., Hayward, B., Nakamura, A. J., Bonner, W. M., and Usdin, K. (2015). Evidence for chromosome fragility at the frataxin locus in Friedreich ataxia. Mutat. Res. 781, 14–21. doi: 10.1016/j.mrfmmm.2015.08.007
Kumari, D., Somma, V., Nakamura, A. J., Bonner, W. M., D'ambrosio, E., and Usdin, K. (2009). The role of DNA damage response pathways in chromosome fragility in fragile X syndrome. Nucleic Acids Res. 37, 4385–4392. doi: 10.1093/nar/gkp391
Le Tallec, B., Millot, G. A., Blin, M. E., Brison, O., Dutrillaux, B., and Debatisse, M. (2013). Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep. 4, 420–428. doi: 10.1016/j.celrep.2013.07.003
Letessier, A., Millot, G. A., Koundrioukoff, S., Lachages, A. M., Vogt, N., Hansen, R. S., et al. (2011). Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 470, 120–123. doi: 10.1038/nature09745
Lin, J., Hou, J. Q., Xiang, H. D., Yan, Y. Y., Gu, Y. C., Tan, J. H., et al. (2013). Stabilization of G-quadruplex DNA by C-5-methyl-cytosine in bcl-2 promoter: implications for epigenetic regulation. Biochem. Biophys. Res. Commun. 433, 368–373. doi: 10.1016/j.bbrc.2012.12.040
Liu, G., Chen, X., Gao, Y., Lewis, T., Barthelemy, J., and Leffak, M. (2012). Altered replication in human cells promotes DMPK (CTG)(n). (CAG)(n) repeat instability. Mol. Cell. Biol. 32, 1618–1632. doi: 10.1128/MCB.06727-11
Liu, Y., Nielsen, C. F., Yao, Q., and Hickson, I. D. (2014). The origins and processing of ultra fine anaphase DNA bridges. Curr. Opin. Genet. Dev. 26, 1–5. doi: 10.1016/j.gde.2014.03.003
Lokanga, A. R., Zhao, X. N., Entezam, A., and Usdin, K. (2014). X inactivation plays a major role in the gender bias in somatic expansion in a mouse model of the fragile X-related disorders: implications for the mechanism of repeat expansion. Hum. Mol. Genet. 23, 4985–4994. doi: 10.1093/hmg/ddu213
Lombardi, E. P., and Tarsounas, M. (2020). Topologically associating domain boundaries are enriched in early firing origins and restrict replication fork progression. bioRxiv [Preprint]. doi: 10.1101/2020.10.21.348946
Lozano, R., Rosero, C. A., and Hagerman, R. J. (2014). Fragile X spectrum disorders. Intractable Rare Dis Res 3, 134–146. doi: 10.5582/irdr.2014.01022
Macheret, M., Bhowmick, R., Sobkowiak, K., Padayachy, L., Mailler, J., Hickson, I. D., et al. (2020). High-resolution mapping of mitotic DNA synthesis regions and common fragile sites in the human genome through direct sequencing. Cell Res. 30, 997–1008. doi: 10.1038/s41422-020-0358-x
Madireddy, A., Kosiyatrakul, S. T., Boisvert, R. A., Herrera-Moyano, E., Garcia-Rubio, M. L., Gerhardt, J., et al. (2016). FANCD2 facilitates replication through common fragile sites. Mol. Cell 64, 388–404. doi: 10.1016/j.molcel.2016.09.017
Mahadevan, M., Tsilfidis, C., Sabourin, L., Shutler, G., Amemiya, C., Jansen, G., et al. (1992). Myotonic dystrophy mutation: an unstable CTG repeat in the 3' untranslated region of the gene. Science 255, 1253–1255. doi: 10.1126/science.1546325
Metsu, S., Rainger, J. K., Debacker, K., Bernhard, B., Rooms, L., Grafodatskaya, D., et al. (2014a). A CGG-repeat expansion mutation in ZNF713 causes FRA7A: association with autistic spectrum disorder in two families. Hum. Mutat. 35, 1295–1300. doi: 10.1002/humu.22683
Metsu, S., Rooms, L., Rainger, J., Taylor, M. S., Bengani, H., Wilson, D. I., et al. (2014b). FRA2A is a CGG repeat expansion associated with silencing of AFF3. PLoS Genet. 10:e1004242. doi: 10.1371/journal.pgen.1004242
Minocherhomji, S., Ying, S., Bjerregaard, V. A., Bursomanno, S., Aleliunaite, A., Wu, W., et al. (2015). Replication stress activates DNA repair synthesis in mitosis. Nature 528, 286–290. doi: 10.1038/nature16139
Mirkin, S. M. (2006). DNA structures, repeat expansions and human hereditary disorders. Curr. Opin. Struct. Biol. 16, 351–358. doi: 10.1016/j.sbi.2006.05.004
Miron, K., Golan-Lev, T., Dvir, R., Ben-David, E., and Kerem, B. (2015). Oncogenes create a unique landscape of fragile sites. Nat. Commun. 6:7094. doi: 10.1038/ncomms8094
Murat, P., Guilbaud, G., and Sale, J. E. (2020). DNA polymerase stalling at structured DNA constrains the expansion of short tandem repeats. Genome Biol. 21:209. doi: 10.1186/s13059-020-02124-x
Naim, V., Wilhelm, T., Debatisse, M., and Rosselli, F. (2013). ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 15, 1008–1015. doi: 10.1038/ncb2793
Nancarrow, J. K., Kremer, E., Holman, K., Eyre, H., Doggett, N. A., Le Paslier, D., et al. (1994). Implications of FRA16A structure for the mechanism of chromosomal fragile site genesis. Science 264, 1938–1941. doi: 10.1126/science.8009225
Ozeri-Galai, E., Lebofsky, R., Rahat, A., Bester, A. C., Bensimon, A., and Kerem, B. (2011). Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol. Cell 43, 122–131. doi: 10.1016/j.molcel.2011.05.019
Palumbo, E., Matricardi, L., Tosoni, E., Bensimon, A., and Russo, A. (2010). Replication dynamics at common fragile site FRA6E. Chromosoma 119, 575–587. doi: 10.1007/s00412-010-0279-4
Park, S. H., Bennett-Baker, P., Ahmed, S., Arlt, M. F., Ljungman, M., Glover, T. W., et al. (2021). Locus-specific transcription silencing at the FHIT gene suppresses replication stress-induced copy number variant formation and associated replication delay. Nucleic Acids Res. 49, 7507–7524. doi: 10.1093/nar/gkab559
Parrish, J. E., Oostra, B. A., Verkerk, A. J., Richards, C. S., Reynolds, J., Spikes, A. S., et al. (1994). Isolation of a GCC repeat showing expansion in FRAXF, a fragile site distal to FRAXA and FRAXE. Nat. Genet. 8, 229–235. doi: 10.1038/ng1194-229
Paulson, H. (2018). Repeat expansion diseases. Handb. Clin. Neurol. 147, 105–123. doi: 10.1016/B978-0-444-63233-3.00009-9
Pelletier, R., Krasilnikova, M. M., Samadashwily, G. M., Lahue, R., and Mirkin, S. M. (2003). Replication and expansion of trinucleotide repeats in yeast. Mol. Cell. Biol. 23, 1349–1357. doi: 10.1128/MCB.23.4.1349-1357.2003
Samadashwily, G. M., Raca, G., and Mirkin, S. M. (1997). Trinucleotide repeats affect DNA replication in vivo. Nat. Genet. 17, 298–304. doi: 10.1038/ng1197-298
Sarafidou, T., Kahl, C., Martinez-Garay, I., Mangelsdorf, M., Gesk, S., Baker, E., et al. (2004). Folate-sensitive fragile site FRA10A is due to an expansion of a CGG repeat in a novel gene, FRA10AC1, encoding a nuclear protein. Genomics 84, 69–81. doi: 10.1016/j.ygeno.2003.12.017
Sarni, D., Sasaki, T., Irony Tur-Sinai, M., Miron, K., Rivera-Mulia, J. C., Magnuson, B., et al. (2020). 3D genome organization contributes to genome instability at fragile sites. Nat. Commun. 11:3613. doi: 10.1038/s41467-020-17448-2
Schmid, M., Klett, C., and Niederhofer, A. (1980). Demonstration of a heritable fragile site in human chromosome 16 with distamycin A. Cytogenet. Cell Genet. 28, 87–94. doi: 10.1159/000131516
Shaw, M. A., Chiurazzi, P., Romain, D. R., Neri, G., and Gecz, J. (2002). A novel gene, FAM11A, associated with the FRAXF CpG island is transcriptionally silent in FRAXF full mutation. Eur. J. Hum. Genet. 10, 767–772. doi: 10.1038/sj.ejhg.5200881
Smith, C. E., Llorente, B., and Symington, L. S. (2007). Template switching during break-induced replication. Nature 447, 102–105. doi: 10.1038/nature05723
Snyder, M., Sapolsky, R. J., and Davis, R. W. (1988). Transcription interferes with elements important for chromosome maintenance in Saccharomyces cerevisiae. Mol. Cell. Biol. 8, 2184–2194. doi: 10.1128/mcb.8.5.2184-2194.1988
Sun, J. H., Zhou, L., Emerson, D. J., Phyo, S. A., Titus, K. R., Gong, W., et al. (2018). Disease-associated short tandem repeats co-localize with chromatin domain boundaries. Cell 175:e215. doi: 10.1016/j.cell.2018.08.005
Sutherland, G. R., Parslow, M. I., and Baker, E. (1985). New classes of common fragile sites induced by 5-azacytidine and bromodeoxyuridine. Hum. Genet. 69, 233–237. doi: 10.1007/BF00293031
Thorland, E. C., Myers, S. L., Persing, D. H., Sarkar, G., Mcgovern, R. M., Gostout, B. S., et al. (2000). Human papillomavirus type 16 integrations in cervical tumors frequently occur in common fragile sites. Cancer Res. 60, 5916–5921.
Thurman, R. E., Day, N., Noble, W. S., and Stamatoyannopoulos, J. A. (2007). Identification of higher-order functional domains in the human ENCODE regions. Genome Res. 17, 917–927. doi: 10.1101/gr.6081407
Usdin, K., and Woodford, K. J. (1995). CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res. 23, 4202–4209. doi: 10.1093/nar/23.20.4202
Van Kuilenburg, A. B. P., Tarailo-Graovac, M., Richmond, P. A., Drogemoller, B. I., Pouladi, M. A., Leen, R., et al. (2019). Glutaminase deficiency caused by short tandem repeat expansion in GLS. N. Engl. J. Med. 380, 1433–1441. doi: 10.1056/NEJMoa1806627
Van Wietmarschen, N., Sridharan, S., Nathan, W. J., Tubbs, A., Chan, E. M., Callen, E., et al. (2020). Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature 586, 292–298. doi: 10.1038/s41586-020-2769-8
Verkerk, A. J., Pieretti, M., Sutcliffe, J. S., Fu, Y. H., Kuhl, D. P., Pizzuti, A., et al. (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914. doi: 10.1016/0092-8674(91)90397-H
Voineagu, I., Surka, C. F., Shishkin, A. A., Krasilnikova, M. M., and Mirkin, S. M. (2009). Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat. Struct. Mol. Biol. 16, 226–228. doi: 10.1038/nsmb.1527
Webb, T. (1992). Delayed replication of Xq27 in individuals with the fragile X syndrome. Am. J. Med. Genet. 43, 1057–1062. doi: 10.1002/ajmg.1320430633
Wenger, S. L., Giangreco, C. A., Tarleton, J., and Wessel, H. B. (1996). Inability to induce fragile sites at CTG repeats in congenital myotonic dystrophy. Am. J. Med. Genet. 66, 60–63. doi: 10.1002/(SICI)1096-8628(19961202)66:1<60::AID-AJMG13>3.0.CO;2-O
Wilson, T. E., Arlt, M. F., Park, S. H., Rajendran, S., Paulsen, M., Ljungman, M., et al. (2015). Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res. 25, 189–200. doi: 10.1101/gr.177121.114
Winnepenninckx, B., Debacker, K., Ramsay, J., Smeets, D., Smits, A., Fitzpatrick, D. R., et al. (2007). CGG-repeat expansion in the DIP2B gene is associated with the fragile site FRA12A on chromosome 12q13.1. Am. J. Hum. Genet. 80, 221–231. doi: 10.1086/510800
Wyatt, H. D., Sarbajna, S., Matos, J., and West, S. C. (2013). Coordinated actions of SLX1-SLX4 and MUS81-EME1 for Holliday junction resolution in human cells. Mol. Cell 52, 234–247. doi: 10.1016/j.molcel.2013.08.035
Ying, S., Minocherhomji, S., Chan, K. L., Palmai-Pallag, T., Chu, W. K., Wass, T., et al. (2013). MUS81 promotes common fragile site expression. Nat. Cell Biol. 15, 1001–1007. doi: 10.1038/ncb2773
Yrigollen, C. M., Martorell, L., Durbin-Johnson, B., Naudo, M., Genoves, J., Murgia, A., et al. (2014). AGG interruptions and maternal age affect FMR1 CGG repeat allele stability during transmission. J. Neurodev. Disord. 6:24. doi: 10.1186/1866-1955-6-24
Yu, S., Mangelsdorf, M., Hewett, D., Hobson, L., Baker, E., Eyre, H. J., et al. (1997). Human chromosomal fragile site FRA16B is an amplified AT-rich minisatellite repeat. Cell 88, 367–374. doi: 10.1016/S0092-8674(00)81875-9
Yu, T., Ferber, M. J., Cheung, T. H., Chung, T. K., Wong, Y. F., and Smith, D. I. (2005). The role of viral integration in the development of cervical cancer. Cancer Genet. Cytogenet. 158, 27–34. doi: 10.1016/j.cancergencyto.2004.08.021
Yudkin, D., Hayward, B. E., Aladjem, M. I., Kumari, D., and Usdin, K. (2014). Chromosome fragility and the abnormal replication of the FMR1 locus in fragile X syndrome. Hum. Mol. Genet. 23, 2940–2952. doi: 10.1093/hmg/ddu006
Zhao, X. N., and Usdin, K. (2018). Timing of expansion of fragile X Premutation alleles During intergenerational transmission in a mouse model of the fragile X-related disorders. Front. Genet. 9:314. doi: 10.3389/fgene.2018.00314
Zheglo, D., Brueckner, L. M., Sepman, O., Wecht, E. M., Kuligina, E., Suspitsin, E., et al. (2019). The FRA14B common fragile site maps to a region prone to somatic and germline rearrangements within the large GPHN gene. Genes Chromosomes Cancer 58, 284–294. doi: 10.1002/gcc.22706
Keywords: break-induced DNA replication, mitotic DNA synthesis, SLX1-SLX4, MUS81/EME1, replication fork blockage, R-loops, origins of replication, secondary DNA structures
Citation: Lokanga RA, Kumari D and Usdin K (2021) Common Threads: Aphidicolin-Inducible and Folate-Sensitive Fragile Sites in the Human Genome. Front. Genet. 12:708860. doi: 10.3389/fgene.2021.708860
Edited by:
Advaitha Madireddy, Rutgers, The State University of New Jersey, United StatesReviewed by:
Jeannine Gerhardt, Cornell University, United StatesEmanuela Volpi, University of Westminster, United Kingdom
Batsheva Kerem, Hebrew University of Jerusalem, Israel
Copyright © 2021 Lokanga, Kumari and Usdin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karen Usdin, a2FyZW51QG5paC5nb3Y=