 Duo Chen
Duo Chen Xuehai Zheng
Xuehai Zheng Zhen Huang
Zhen Huang Youqiang Chen
Youqiang Chen Ting Xue
Ting Xue Gang Lin
Gang Lin- The Public Service Platform for Industrialization Development Technology of Marine Biological Medicine and Products of the State Oceanic Administration, Fujian Key Laboratory of Special Marine Bioresource Sustainable Utilization, Southern Institute of Oceanography, College of Life Sciences, Fujian Normal University, Fuzhou, China
Introduction
A pedunculate barnacle Capitulum mitella (Linnaeus, 1758) is a dominant intertidal cirripede (Lee et al., 2000). C. mitella distribute in the mid- and high-intertidal zone of tropical and subtropical coasts and is common in the East Sea and South Sea of China. C. mitella has a great commercial value in China, the petiole muscle of which has been a source of traditional delicious seafood (Yuan et al., 2016). The local market demand led to high-intensity catching activity, and this behavior caused significant decrease in quantity and habitat destruction in recent years. Artificial aquaculture of fishery resources will help to reduce overexploitation of natural stocks and meet market needs for fishery products.
In general, the barnacle life cycle consists of six nauplius instars and a nonfeeding cyprid instar. In the barnacle life history, the cyprid is a transitory phase between the pelagic and sessile lifestyle. The cyprid role is responsible for locating, exploring, and attaching to a suitable substratum; subsequently, a complex metamorphosis and permanent settlement happen (Lagersson and Høeg, 2002). Cypris attachment and metamorphosis are also known as “cypris settlement” (Clare and Matsumura, 2000; Franco et al., 2016). The cypris settlement is a crucial event for the survival and development of both adults and subsequent generations. Within the last three decades, the basic biology of C. mitella such as reproductive characteristics, sperm ultrastructure, and larval culture conditions was investigated in our lab. C. mitella cyprids have been cultured on a relatively large scale. However, they cannot attach and metamorphose into juveniles in an artificial environment, so they would die eventually and interrupt their life history.
In recent years, the cypris morphology of C. mitella has been depicted minutely using SEM (Rao and Lin, 2014). After we had successfully induced metamorphosis of the C. mitella cyprids into juveniles, we described a time line and specific morphological changes during the metamorphosis under microscopy and SEM (Lin and Rao, 2017). The impacts of external (i.e., water temperature and salinity) and internal (i.e., cyprid age) factors on the metamorphosis of C. mitella were researched (Rao and Lin, 2020). These efforts are possible for completing the life history in an artificial environment and aquaculture of C. mitella. Not only is the artificial aquaculture for C. mitella a production activity but also it helps to reduce the stress on overexploited populations.
Barnacles are frequently dominant marine foulings and widely distributed around the world that play crucial roles in marine ecology (Maréchal and Hellio, 2011). Recently, there are a few transcriptome researches on barnacles, such as a model animal Amphibalanus amphitrite (Chen et al., 2011; Yan et al., 2012; Chandramouli et al., 2015; Sarah et al., 2016), a giant barnacle Megabalanus volcano (Yan et al., 2017) and a stalked barnacle Neolepas marisindica (Ryu et al., 2019). These efforts showed that the transcriptome databases could greatly promote the research on molecular mechanisms of barnacle settlement. Genome sequencing has been performed for an acorn barnacle Amphibalanus amphitrite, but there is still no genome sequencing information for a stalked barnacle. Numerous reports on C. mitella were mainly related to mitochondrial DNA sequencing and quantitative proteomics analyses (Song and Yoon, 2013; Yoon et al., 2013; Yuan et al., 2016; Tian et al., 2020). So far, no genome literature on the C. mitella is available. Whereas a lack of genome information on the barnacles has obstructed further inquiry on the molecular mechanisms with development, attachment, metamorphosis, genetic evolution, and so on, so does on C. mitella.
Accordingly, in this study, we aimed to sequence and assemble the genome of C. mitella. The baseline data obtained from this study will be very useful in taxonomical identification, phylogenetic analysis, larval settlement mechanisms, artificial breeding and aquaculture, and species protection for C. mitella.
Materials and Methods
Sample Preparation and Genome Sequencing
C. mitella specimens were sampled from the intertidal zone of Dinghai (26.2487°N, 119.7987°E), Fujian Province, China, and immediately frozen in liquid nitrogen. Total DNA was isolated from fresh muscle samples with the DNeasy® Tissue Kit (Qiagen, Hilden, Germany) according to the DNeasy® Protocol for animal tissues. Qubit 3.0 (Thermo Fisher Scientific, Inc., Carlsbad, CA, USA) was used to assess the DNA concentration and quality before downstream sequencing. Approximately 5 μg of high-quality genomic DNA was sheared and size-selected (~40 kb) for PacBio library construction followed by sequencing on the PacBio Sequel II platform (PacBio Biosciences, Menlo Park, CA, USA). Sequence libraries with inserts of average 350 bp were constructed for Illumina paired-end (PE) sequencing according to the manufacturer's instructions, and sequencing was subsequently performed on Illumina HiSeq X Ten platform (Illumina, San Diego, CA, USA).
Genome Assembly
Canu (V2.0, https://github.com/marbl/canu/) (Koren et al., 2017) was used to perform the genome de novo assembly based on the PacBio long reads. After the initial assembly, primary contigs were corrected for three cycles with the Illumina reads using Pilon (V1.24, https://github.com/broadinstitute/pilon) (Belton et al., 2012). We used fresh muscle tissue from the same individual as in genome sequencing for the Hi-C library construction and sequencing. The Hi-C library was generated using Mbo I restriction enzyme following in situ ligation protocols (Peng et al., 2021). In brief, fresh muscle tissue was sampled and frozen in liquid nitrogen. Tissues were cross-linked with fresh formaldehyde and quenched with glycine. The total DNA was extracted from the nuclei in the lysis buffer [10 mM Tris-HCl (pH 8.0), 10 mM NaCl, 0.2% NP40, and complete protease inhibitors (Roche)]. The purified DNA was digested with Mbo I and was labeled by Biotin-14-dATP (Thermo Fisher Scientific) and then ligated by T4 DNA Ligase. After ligation, the cross-linking was incubated with proteinase K overnight; the ligated DNA was sheared into 200–600-bp fragments and then blunt-end repaired and A-tailed, followed by purification through biotin-streptavidin-mediated pull-down. Finally, the Hi-C libraries were quantified and sequenced on the Illumina HiSeq X Ten platform using a PE-150 module. The Hi-C sequencing data were then aligned to the scaffolds using BWA-MEM alignment algorithm (V1.2.2, https://github.com/lh3/bwa). Two ends of reads were independently aligned to the genome and only selected the read pairs for which both ends were uniquely aligned to the genome. The hiclib software (V0.5.7, http://github.com/hiclib) and a previously reported method (Gong et al., 2018) were applied to filter the Hi-C reads, and the interaction frequency was quantified and normalized among contigs. Lachesis (Burton et al., 2013) with default parameters was then applied to cluster contigs with the agglomerative hierarchical clustering method using the interaction matrix between sequences. To construct a high-quality assembly, we sequenced the genome by utilizing a combination of Illumina (~78.52 Gb of raw data), PacBio single-molecule real-time (SMRT) sequencing (~60.23 Gb of raw data), and Hi-C chromosome-scale scaffolding (~44.40 Gb of raw data) (Belton et al., 2012). The raw reads were filtered out by certain low-quality scrap reads and/or by minimum CLR length. After read quality filtering by PacBio Quality checking tool (http://github.com/PacificBiosciences/), a total of 60.17 Gb of long-read sequence data were produced, comprising 4,342,961 subreads with a read N50 of 19.979 Kb. This initial genome assembly was 463.09 Mb in length with a contig N50 of 3.14 Mb (Table 1; Figures 1, 2). A total of 259,360,062 reads (87.63 Gb) with a Q30 of 93.23% were generated for Hi-C analysis. Finally, 16 chromosomes anchored by the contigs were generated with the guidance of HiC reads; this result consists of the karyotype analysis (Supplementary Figure 1). This chromosome-level assembly has an average length of 28.11 Mb, with the shortest chromosome of 19.66 Mb (Chr16) and the longest one of 45.43 Mb (Chr1) (Figure 2). The scaffold N50 reached 3.14–31.03 Mb (Table 1), providing a chromosome-level genome assembly for C. mitella. The completeness of this assembled genome was assessed based on BUSCO analysis, which revealed that nearly 94.4% of the Arthropoda orthologs were included in the assembled (Table 2). These results indicated a high-quality assembly of C. mitella genome.
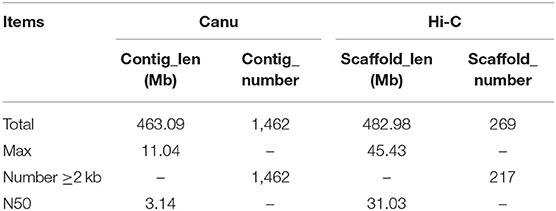
Table 1. Assembly statistics for Capitulum mitella.
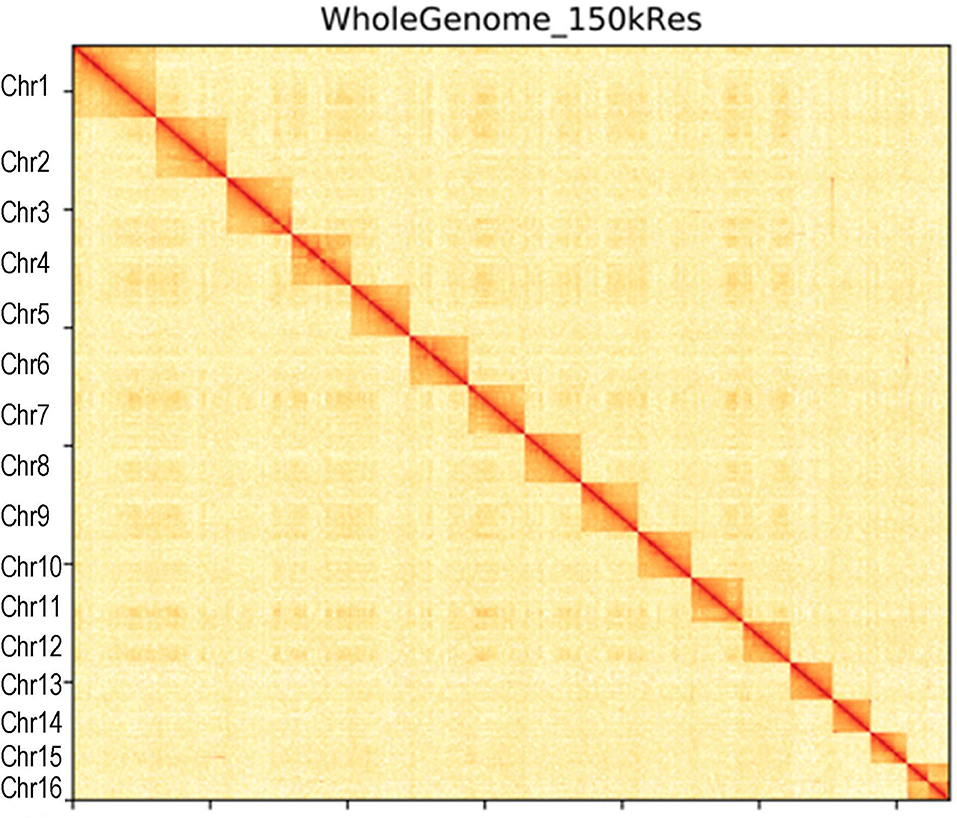
Figure 1. Hi-C interaction heatmap of Capitulum mitella genome. The heatmap shows that the genome was split into 100-kb bins, and the interaction links of individual chromosomes were validated.
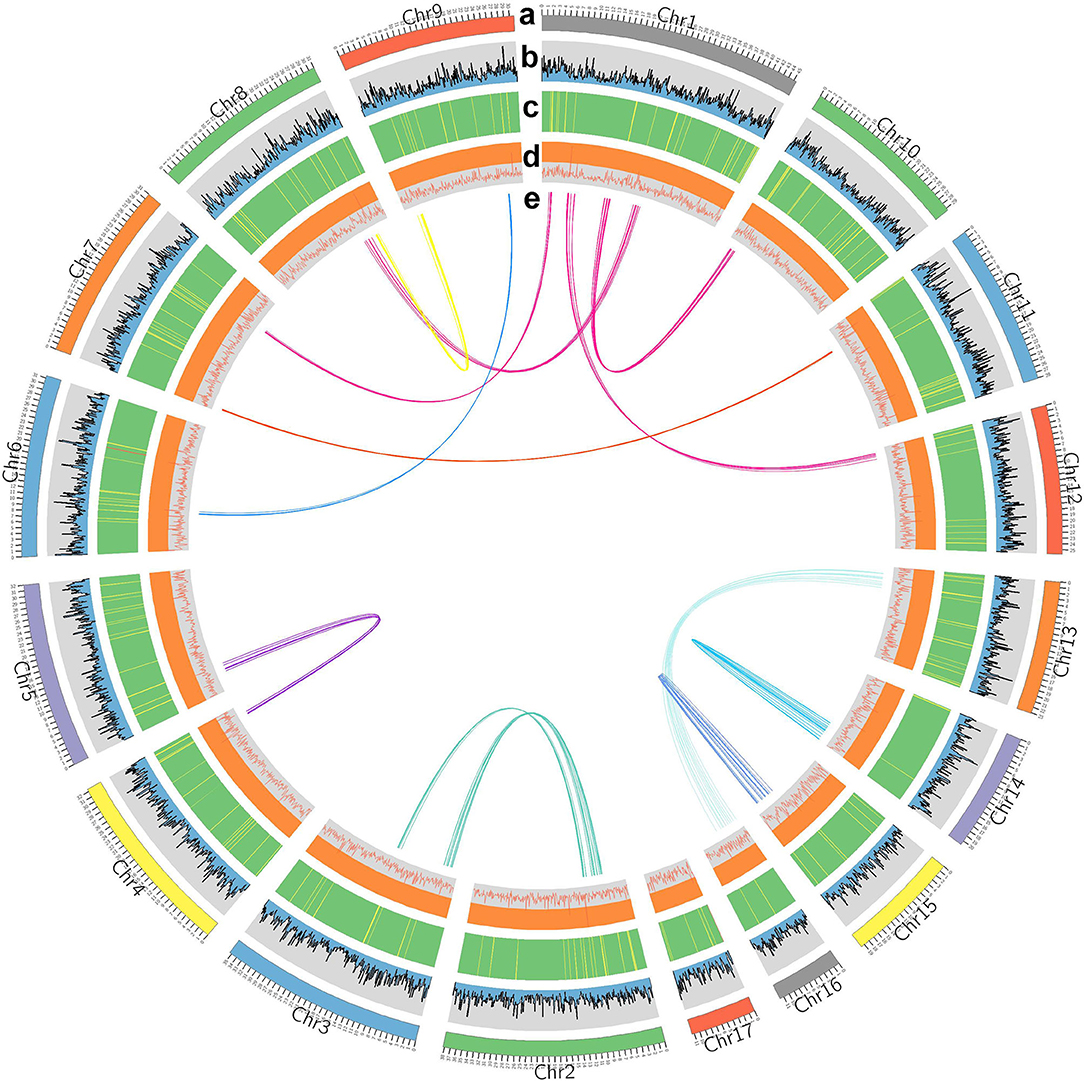
Figure 2. Characterization of Capitulum mitella. Genome elements are arranged in the Circos plot. (a) Eight assembled chromosomes of the genome. (b) The distribution of GC content. (c) Distribution of gene density (green). (d) Statistics for coverage of TEs. (e) Circos diagram of major interchromosomal relationships in the C. mitella genome. Each line represents a syntenic block; block size = 3 kb.
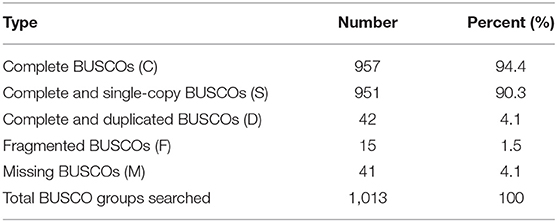
Table 2. Assessment of the completeness of the Capitulum mitella genome assembly by BUSCO.
Genome Annotation
The assembled C. mitella genome was homology-based annotated using a collection of protein-coding genes and RNA sequencing data from nine animal species: Apis mellifera, Caenorhabditis elegans, Crassostrea gigas, Daphnia pulex, Drosophila melanogaster, Mizuhopecten yessoensis, Limulus polyphemus, Penaeus vannamei, and Plutella xylostella. D. pulex and P. vannamei belong to subphylum Crustacea. D. melanogaster, P. xylostella, and A. mellifera belong to the subphylum Hexapoda, which is closely related to C. mitella. M. yessoensis and C. gigas belong to phylum Mollusca, and their ontogeny is similar to that of C. mitella through larval metamorphosis and attachment. De novo gene predictions were carried out by the AUGUSTUS package (V2.0) provided by BRAKER (V2.1.4) (Hoff et al., 2015). The MAKER pipeline was employed to identify the 21,899 protein-coding genes (Holt and Yandell, 2011), integrating protein sequences and transcript genes from the de novo assembly of C. mitella transcriptome data. All protein-coding genes were functionally annotated BLASTP (V2.2.3) (Anoop and Agostinho, 2015) against the public protein sequence databases EggNOG, Gene Ontology (GO), Cluster of Orthologous Groups (COG), and Kyoto Encyclopedia of Genes and Genomes (KEGG) with an E-value ≤ 1e−5. We functionally annotated 14,204, 9,312, 14,204, and 5,368 genes to EggNOG, GO, COG, and KEGG, respectively (Table 3). Small RNAs (sRNAs) and noncoding RNAs (ncRNAs) were predicted by Rfam and miRNA databases using tRNAsan-SE (V2.0) (Bernard et al., 2014) and BLASTN (V2.2.3), respectively. Some types of ncRNAs consisting of microRNAs (miRNAs) and small nuclear RNAs (snRNAs) were identified using INFERNAL (V.1.1.4) by searching the Rfam database (http://infernal.janelia.org/). In total, 136 miRNAs, 748 tRNAs, 56 rRNAs, and 87 snRNAs were identified in C. mitella genome (Supplementary Table 1). Tandem Repeats Finder (V4.09.1), LTR_FINDER (V1.07), and RepeatMasker (V4.0) (González and Deyholos, 2012) were combined to predict repeat sequences in the C. mitella genome. In the C. mitella genome, repetitive sequences accounted for 3.53%, and long terminal repeat (LTR) retrotransposons accounted for 0.28% of the genome, including 0.18% Ty3/gypsy, 0.03% Ty1/copia, and 0.06% other (Supplementary Table 2).
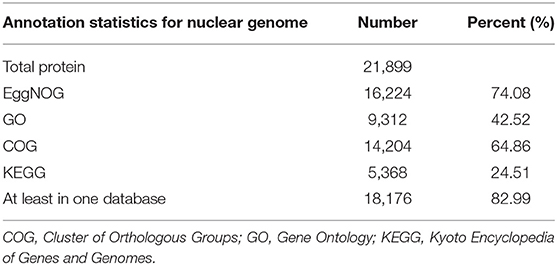
Table 3. General function annotation results statistics.
Genome Evolution and Gene Family Analysis
In the current study, in order to investigate the evolution relationships among C. mitella and nine other species, we also perform phylogenomic analysis based on the amino acid sequences. These 10 species were A. mellifera, C. mitella, C. elegans, C. gigas, D. melanogaster, D. pulex, M. yessoensis, L. polyphemus, P. vannamei, and P. xylostella. Their protein sequences were acquired from the NCBI database, and single-copy genes were obtained by OrthoFinder (V2.27) (Emms and Kelly, 2015). Protein sequences of single-copy gene orthologs were aligned using MUSCLE software (V3.8.425). The alignments of Coding DNA Sequences (CDSs) that were guided by protein alignments were concatenated into the superalignment of nucleotide sequences. The phylogenetic relationships were then constructed using the maximum likelihood (ML) methods. On the basis of the protein alignments, the CDSs were aligned and then concatenated into a superalignment matrix for each family. The cluster size differences between the ancestor and each species were compared to analyze the expansion and contraction of the gene families using CAFE (V2.1) (Koichiro et al., 2007). The phylogenetic tree showed that C. mitella and P. vannamei phylogenetically were clustered into an independent ~530 million years ago (Mya), which was earlier than those of Insecta (325 Mya) (Figure 3). Comparative genomic analyses were performed among abovementioned 10 animal species, and we detected 15,620 families of homologous genes, among which 7,232 gene families were identified in C. mitella, including 694 and 6,538 gene families showing contraction and expansion, respectively (Figure 3). GO analysis revealed that the 694 expanded orthogroups were involved in the metabolic process, developmental process, stimulus response, transporter activity, and catalytic activity (Supplementary Table 3). The analysis of KEGG pathways manifested that most of the 694 expanded genes were annotated to the signaling molecules and interaction, cell growth and death, glycine, environmental adaptation, and serine and threonine metabolism (Supplementary Table 4). The C. mitella is a common species in middle tidal areas of intertidal zones, distributed in rock surfaces or crevices. Therefore, C. mitella have evolved a complex mechanism to adapt to dramatically changing environments such as large temperature differences and desiccation. So, the expanded orthogroups in C. mitella are related to the metabolic process, stimulus process, environmental adaptation, and so on. By contrast, the group of contracted gene families was related to lipid metabolism, phenylalanine metabolism, signal transduction, immune system, environmental adaptation, and cell growth and death (Supplementary Table 5). The GO terms annotation of the contracted genes showed that they were most related to the stimulus response, biological regulation, catalytic activity, growth, reproductive process, signaling, and developmental process (Supplementary Table 6). The contracted gene families in this process related to the development in the insect, suggesting that C. mitella and the other compared species share the core signal regulation network in the developmental process. The comparison of A. mellifera, C. mitella, C. elegans, C. gigas, D. melanogaster, D. pulex, M. yessoensis, L. polyphemus, P. vannamei, and P. xylostella revealed that 3,374 (28.26%) of the 11,935 C. mitella gene families were shared by the other nine species, whereas 8,561 gene families were unique to C. mitella (Figure 4). GO functional analysis revealed that these 8,561 unique families were enriched in the immune system process, developmental process, signaling, aging, stimulus response, and calcium signaling pathway (Supplementary Table 7), while KEGG annotation presented that most of the 8,561 unique families were gathered in signal transduction and cysteine and methionine metabolism (Supplementary Table 8).
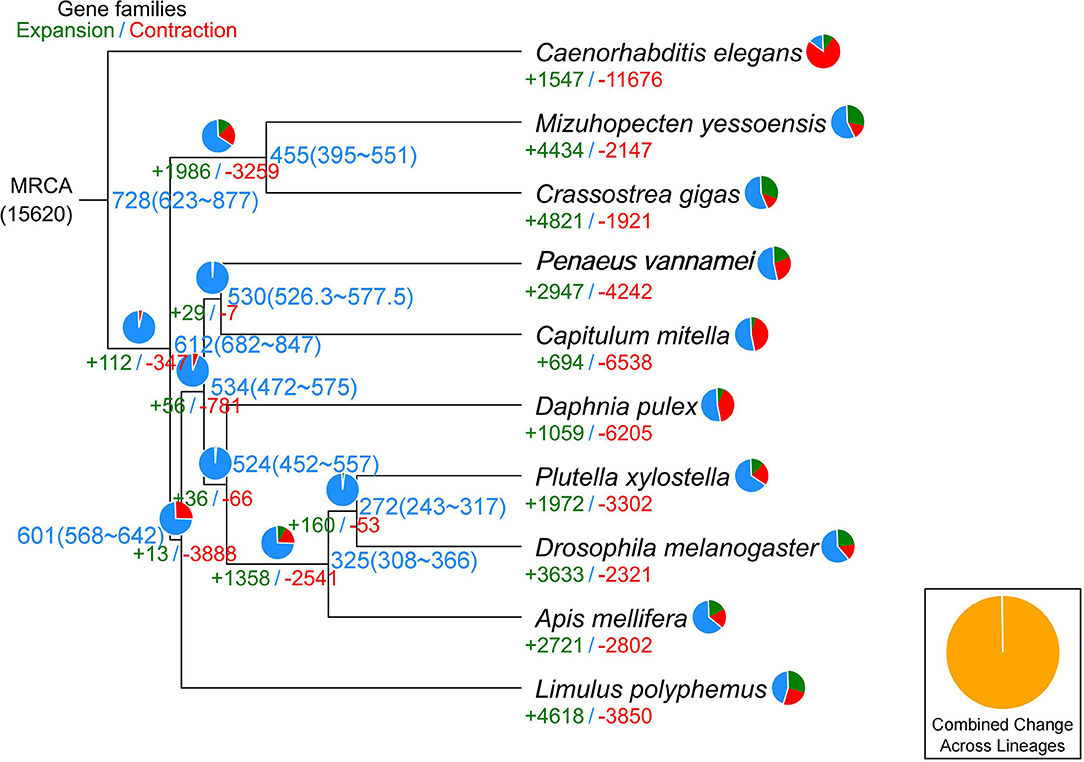
Figure 3. Phylogenetic tree showing the number of gene families displaying expansion (green) and contraction (red) among 10 animal species. The pie charts show the proportions of expanded (green), contracted (red), and conserved (blue) gene families among all gene families. The estimated divergence time (million years ago) is displayed in the phylogenetic tree in black. MRCA, most recent common ancestor.
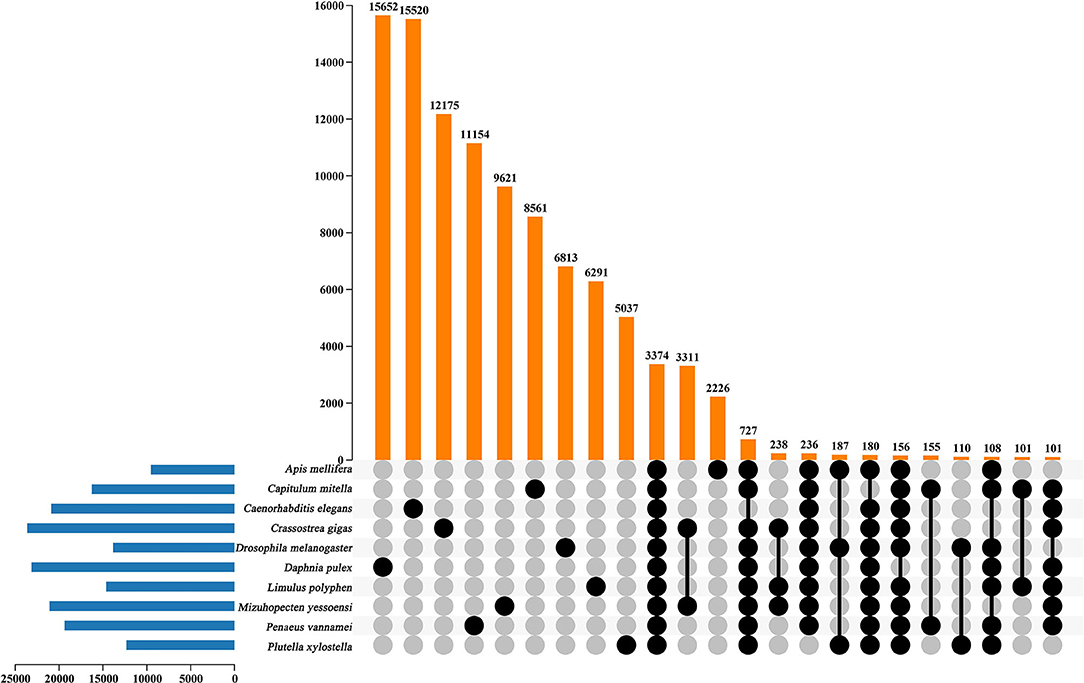
Figure 4. UpSet plot of the intersection of gene families in A. mellifera, C.mitella, C. elegans, C. gigas, D.melanogaster, D. pulex, M. yessoenisis, L. polyphemus, P. vannamei and P. xylostella. The numbers of gene families (clusters) are indicated for each species and species intersection.
Data Availability Statement
The whole genome sequence data reported in this paper available in the Genome Warehouse in National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences, under accession number GWHBAVK00000000.1 that is publicly accessible at https://bigd.big.ac.cn/gwh. The complete sequences generated in this study was deposited to the NCBI, under the accession PRJNA725059.
Author Contributions
DC, XZ, ZH, and TX contributed to genome assembly and annotations. TX, ZH, and GL contributed to manuscript preparation. KL, ZH, GL, XR, and YC contributed to sampling and sequencing. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by the cooperation project on production and education of the University of Fujian province, China (2018N5007).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.707546/full#supplementary-material
Supplementary Figure 1. The karyotype of C. mitella.
Supplementary Table 1. Repetitive element annotations in C. mitella.
Supplementary Table 2. Repetitive element annotations.
Supplementary Table 3. GO enrichment of genes in the expansion families in C. mitella genome.
Supplementary Table 4. KEGG enrichment of genes in the expansion families in C. mitella genome.
Supplementary Table 5. KEGG enrichment of genes in the contraction families in C. mitella genome.
Supplementary Table 6. GO enrichment of genes in the contraction families in C. mitella genome.
Supplementary Table 7. GO enrichment of genes that were specifically identified in C. mitella genome.
Supplementary Table 8. KEGG enrichment of genes that were specifically identified in C. mitella genome.
References
Anoop, G. A., and Agostinho, A. (2015). Whole genome sequencing of the symbiont pseudovibrio sp. from the intertidal marine sponge Polymastia penicillus revealed a gene repertoire for host-switching permissive lifestyle. Genome Biol. Evol. 7, 3022–3032. doi: 10.1093/gbe/evv199
Belton, J. M., Mccord, R. P., Gibcus, J. H., Naumova, N., Zhan, Y., and Dekker, J. (2012). Hi-c: a comprehensive technique to capture the conformation of genomes. Methods 58, 268–276. doi: 10.1016/j.ymeth.2012.05.001
Bernard, B., Zofia, N., Le, B. A., and Bénédicte, C. (2014). Computational prediction and experimental validation of micrornas in the brown alga Ectocarpus siliculosus. Nucleic Acids Res. 42, 417–429. doi: 10.1093/nar/gkt856
Burton, J. N., Adey, A., Patwardhan, R. P., Qiu, R., Kitzman, J. O., and Shendure, J. (2013). Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125. doi: 10.1038/nbt.2727
Chandramouli, K. H., Al-Aqeel, S., Ryu, T., Zhang, H., Serinda, L., Ghosheh, Y., et al. (2015). Transcriptome and proteome dynamics in larvae of the barnacle Balanus amphitrite from the red sea. BMC Genom. 16:1063. doi: 10.1186/s12864-015-2262-1
Chen, Z. F., Kiyotaka, M., Hao, W., Arellano, S. M., Yan, X., Intikhab, A., et al. (2011). Toward an understanding of the molecular mechanisms of barnacle larval settlement: a comparative transcriptomic approach. PLoS ONE. 6:e22913. doi: 10.1371/journal.pone.0022913
Clare, A. S., and Matsumura, K. (2000). Nature and perception of barnacle settlement pheromones. Biofouling 15, 57–71. doi: 10.1080/08927010009386298
Emms, D. M., and Kelly, S. (2015). Orthofinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 1–14. doi: 10.1186/s13059-015-0721-2
Franco, S. C., Aldred, N., Cruz, T., and Clare, A. S. (2016). Modulation of gregarious settlement of the stalked barnacle, Pollicipes pollicipes: a laboratory study. Sci. Mar. 80, 217–228. doi: 10.3989/scimar.04342.01A
Gong, G., Dan, C., Xiao, S., Guo, W., Huang, P., Xiong, Y., et al. (2018). Chromosomal-level assembly of yellow catfish genome using third-generation dna sequencing and hi-c analysis. GigaScience 7:giy120. doi: 10.1093/gigascience/giy120
González, L. G., and Deyholos, M. K. (2012). Identification, characterization and distribution of transposable elements in the flax (Linum usitatissimum L.) genome. BMC Genom. 13:644. doi: 10.1186/1471-2164-13-644
Hoff, K. J., Simone, L., Alexandre, L., Mark, B., and Mario, S. (2015). BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 5, 767–769. doi: 10.1093/bioinformatics/btv661
Holt, C., and Yandell, M. (2011). Maker2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 12:491. doi: 10.1186/1471-2105-12-491
Koichiro, T., Joel, D., Masatoshi, N., and Sudhir, K. (2007). MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 8, 1596–1599. doi: 10.1093/molbev/msm092
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Lagersson, N. C., and Høeg, J. T. (2002). Settlement behavior and antennulary biomechanics in cypris larvae of Balanus amphitrite (crustacea: thecostraca: cirripedia). Mar Biol.141, 513–526. doi: 10.1007/s00227-002-0854-1
Lee, C., Shim, J. M., and Kim, C. H. (2000). Larval development of Capitulum mitella (Cirripedia: Pedunculata) reared in the laboratory. J. Mar. Biol. Assoc. UK 80, 457–464. doi: 10.1017/S0025315400002150
Lin, G., and Rao, X. (2017). Metamorphosis of the pedunculate barnacle Capitulum mitella Linnaeus, 1758 (Cirripedia: Scalpelliformes). J. Mar. Biol. Assoc. UK. 97, 1643–1650. doi: 10.1017/S0025315416001089
Maréchal, J. P., and Hellio, C. (2011). Antifouling activity against barnacle cypris larvae: do target species matter (Amphibalanus amphitrite versus Semibalanus balanoides). Int. Biodeter. Biodegr. 65, 92–101. doi: 10.1016/j.ibiod.2010.10.002
Peng, Y., Li, H., Liu, Zh., Zhang, C., Li, K., Gong, Y., et al. (2021). Chromosome-level genome assembly of the Arctic fox (Vulpes lagopus) using PacBio sequencing and Hi-C technology. Mol. Ecol. Resour. 1, 1–16. doi: 10.1111/1755-0998.13397
Rao, X., and Lin, G. (2014). Scanning electron microscopy of the cypris larvae of Capitulum mitella (Cirripedia: Thoracica: Scalpellomorpha). J. Mar. Biol. Assoc. UK 94, 361–368. doi: 10.1017/S0025315413001173
Rao, X., and Lin, G. (2020). Effects of age, salinity and temperature on the metamorphosis and survival of Capitulum mitella cyprids (Cirripedia: Thoracica: Scalpellomorpha). J. Mar. Biol. Assoc. UK. 100, 55–62. doi: 10.1017/S0025315419001152
Ryu, T., Woo, S., and Lee, N. (2019). The first reference transcriptome assembly of the stalked barnacle, Neolepas marisindica, from the onnuri vent field on the central indian ridge. Mar. Genom. 48, 100679–100682. doi: 10.1016/j.margen.2019.04.004
Sarah, A. A., Taewoo, R., Zhang, H., Chandramouli, K. H., and Timothy, R. (2016). Transcriptome and proteome studies reveal candidate attachment genes during the development of the barnacle Amphibalanus amphitrite. Front. Mar. Sci. 3:171. doi: 10.3389/fmars.2016.00171
Song, Y. J., and Yoon, J. M. (2013). Genetic differences of three Pollicipesmitellapopulations identified by pcr analysis. J. Reprod. Dev. 17, 199–205. doi: 10.12717/DR.2013.17.3.199
Tian, M., Chen, P., Song, J., He, F., and Shen, X. (2020). The first mitochondrial genome of Capitulum mitella (Crustacea: Cirripedia) from China: revealed the phylogenetic relationship within Thoracica. Mitochondrial DNA B 5, 2573–2575. doi: 10.1080/23802359.2020.1781564
Yan, G., Zhang, G., Huang, J., Yi, L., Sun, J., Zeng, C., et al. (2017). Comparative transcriptomic analysis reveals candidate genes and pathways involved in larval settlement of the barnacle Megabalanus volcano. Int. J. Mol. Sci. 18, 2253–2267. doi: 10.3390/ijms18112253
Yan, X. C., Chen, Z. F., Jin, S., Matsumura, K., and Wu, R. (2012). Transcriptomic analysis of neuropeptides and peptide hormones in the barnacle Balanus amphitrite: Evidence of roles in larval settlement. PLoS ONE. 7:e46513. doi: 10.1371/journal.pone.0046513
Yoon, M., Jung, J. Y., and Dong, S. K. (2013). Genetic diversity and gene flow patterns in Pollicipesmitella in korea inferred from mitochondrial dna sequence analysis. Fish Aquat. Sci. 16, 243–251. doi: 10.5657/FAS.2013.0243
Keywords: Capitulum mitella, chromosome genome assembly, PacBio, Hi-C analysis, genome evolution analysis
Citation: Chen D, Zheng X, Huang Z, Chen Y, Xue T, Li K, Rao X and Lin G (2021) Chromosome Genome Assembly and Annotation of the Capitulum mitella With PacBio and Hi-C Sequencing Data. Front. Genet. 12:707546. doi: 10.3389/fgene.2021.707546
Received: 10 May 2021; Accepted: 12 July 2021;
Published: 18 August 2021.
Edited by:
Qiang Lin, Chinese Academy of Sciences, ChinaCopyright © 2021 Chen, Zheng, Huang, Chen, Xue, Li, Rao and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gang Lin, bGdmZnpAZmpudS5lZHUuY24=