 Catherine Mathé
Catherine Mathé Christophe Dunand
Christophe Dunand- Laboratoire de Recherche en Sciences Végétales, Université de Toulouse, CNRS, UPS, Toulouse INP, Auzeville-Tolosane, France
Introduction
In the last few decades, the explosion of genomic projects has produced huge sets of predicted genes and annotated sequences. The prediction of a gene structure can be defined as the capacity to determine the start and the stop of the gene as well as the positions of introns, if present. Despite the number of performant gene prediction programs combining ab initio and homology-based approaches (Mathe et al., 2002; Hoff and Stanke, 2015), the rate of mis-predicted genes is not negligible and can be due to several factors (Scalzitti et al., 2020). For example, unusually long introns, short exons or long genes can generate incomplete or partially predicted gene structure; short intergenic regions can lead to gene fusion; DNA sequencing errors (nucleotide deletions or insertions) introducing frameshifts can affect predictions; non-canonical splice sites, overlapping genes and genes located within introns are also a source of erroneous predictions. Due to high sequence identity and duplication rate, the risks of mis-prediction are exacerbated in the case of multigenic families (Figure 1, Fawal et al., 2014). In addition, protein annotation or function assignment, based on the presence of a hypothetical protein domain or on homology with known proteins, can also lead to an inappropriate annotation. The risk of mis-annotations is high for proteins containing multiple domains or small domain(s) common to several classes of proteins. For example, the PFAM domain PF07992 (Pyridine nucleotide-disulphide oxidoreductase) is detected in MonoDehydroAscorbate Reductases (MDARs), Glutathione Reductases (GRs), and in the Thioredoxin family (Trx) but does not discriminate between these three different families (Table 1). Mis-annotations are also observed for proteins belonging to superfamilies with conserved domain and large number of protein families and classes. As an example, 198 genes of the MYB superfamily have been detected in Arabidopsis thaliana (Yanhui et al., 2006), but the PFAM domain PF00249 (Myb_DNA-binding) does not discriminate between the R2R3-MYB, the R1R2R3-MYB, the MYB-related, and the atypical MYB families. In addition, the PF00249 entry also contains the SANT domain, which has a strong structural similarity to the Myb domain but is functionally divergent. Therefore, using this PFAM entry to extract MYB proteins returns many false positives (total of 326 sequences from A. thaliana).
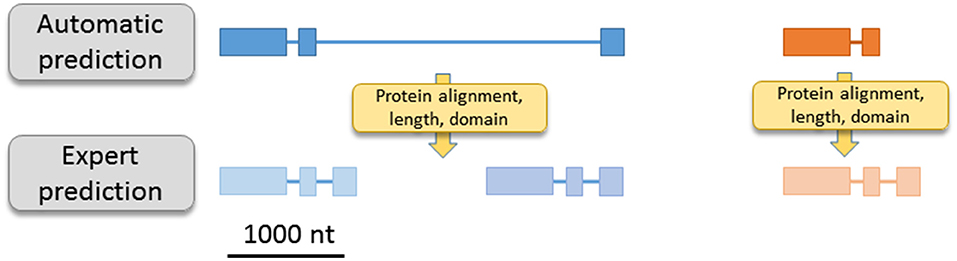
Figure 1. Illustration of two usual mis-prediction. For example, automatic prediction can generate one ORF result of two independent ORF fusion (left part) or partial ORF with a missing/partial exon (right part). Sequence homology, sequence length, domain detection… allow a large reduction of automatic bias.
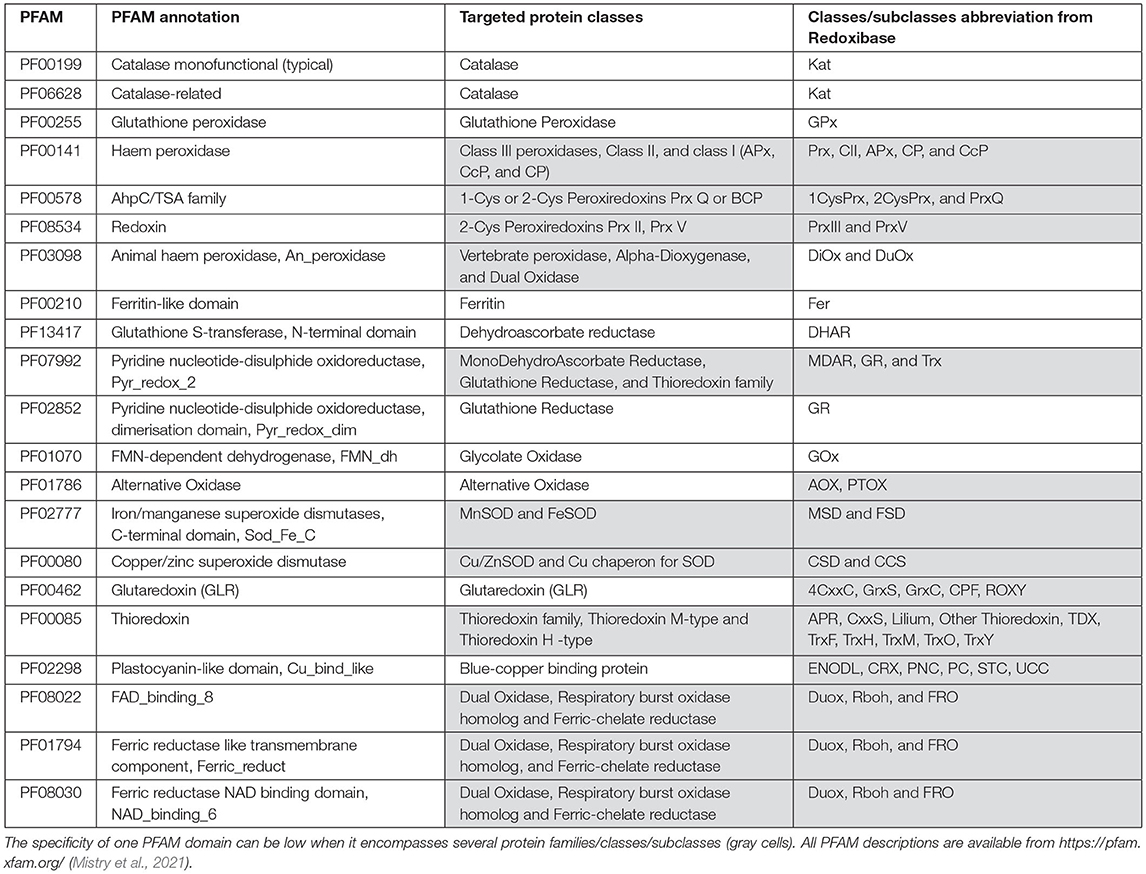
Table 1. Illustration of PFAM domains diversities and specificities.
ROS Gene Network, Contrasted Situations
Reactive Oxygen Species (ROS) are constitutively produced in plants during photosynthesis, respiration, and photorespiration but also produced in a control manner as signal or active molecules. In all cases, ROS homeostasis can be controlled by a large set of proteins described as ROS gene network (Inupakutika et al., 2016). Most of the proteins of this network are members of large superfamilies characterized by PFAM domains that are more or less specific. Indeed, one PFAM entry may encompass several classes or subclasses of proteins (Table 1, gray cells) and lead to mis-annotations.
Peroxidases, which belong to this network, participate in oxidation-reduction reactions using hydrogen peroxide (H2O2) as an electron acceptor and various substrates as electron donors. They may or may not contain a prosthetic group also called haem, justifying further subdivision into two major protein families, namely “haem peroxidases” and “non-haem peroxidases.” The haem peroxidases, such as the non-animal peroxidase family, are found in all kingdoms (Passardi et al., 2007). This family was first described thanks to structural homology (Welinder et al., 1992). It includes three classes of peroxidases: Class I (CI Prxs), Class II (CII Prxs), and Class III (CIII Prxs). This family is grouped under a unique PFAM entry (PF00141) (Table 1), which describes the conserved peroxidase domain (mainly the heam binding sites). This PFAM domain can extract most of the non-animal encoded sequences from any annotated genome, but unfortunately, it does not discriminate between the three classes (Figure 2B) and may produce erroneous annotations that require correction by experts. Over the past 5 years, 12 global phylogenetic and expression analysis of CIII Prxs from different plant species have been published, including four in 2020 (Ren et al., 2014; Wang et al., 2015; Cao et al., 2016; Moural et al., 2017; Duan et al., 2019; Wu et al., 2019; Yan et al., 2019; Zhu et al., 2019; Li et al., 2020; Xiao et al., 2020; Yang et al., 2020; Cai et al., 2021). These studies, based on available plant genomes, mostly contain incorrect predictions and annotations that may lead to erroneous or incomplete conclusions. Partial and longer sequences or pseudogenes were considered as complete sequences and APx sequences, which are CI Prxs, were annotated as CIII Prxs.

Figure 2. Two CIII Prxs (AtPrx41 and AtPrx42) and 4 APxs (AtAPx03, AtAPx06, AtAPx07, and AtAPx-R) sequences from A. thaliana have been compared using various available tools. (A) Structure intron/exon. Exons (blue boxes) and introns (gray lines) are drawn in scale. (B) Location of the unique PFAM domain PF00141 (green boxes), di-sulfide bridges (gray links) and conserved amino acid residues (pink diamonds, purple, and other disks) (Blázquez et al., 2003). (C) Conserved motifs in CIII Prxs and APxs. The MEME program was used to identify conserved motifs. The maximum number of motifs was set to 10, the optimum width of motifs to 15–50 amino acid residues (Brown et al., 2013). Each colored box represents a different motif found in CIII Prxs and/or APxs. CIII: motifs only found in CIII Prx protein sequences; CI: motifs only found in CI Prx protein sequences; *: motifs in common between CIII and CI Prx protein sequences. All the sequences are available from the Redoxibase (Savelli et al., 2019).
Plant NADPH oxidases, also known as Respiratory Burst Oxidase Homologs (RBOHs) catalyze the production of superoxide, . They belong to a large gene family containing NADPH Oxidases (NOXs), found in animals and fungi, and the bifunctional proteins Dual Oxidases (DUOXs), present in animals. Due to the multi-domain organization, the family encompasses three PFAM accessions (PF08022, PF01794, PF08030) (Table 1). In addition, as the RBOH family is composed of a reduced number of copies (about 10), the risk of mis-annotation is reduced compared to CIII Prxs. Otherwise, the high number of introns, together with the short length of some introns and exons, are a source of mis-prediction. Since 2019, more than 10 articles dealing with the global phylogenetic and expression analysis of RBOHs from different plant species have been published (Cheng et al., 2013, 2019; Kaur et al., 2018; Chang et al., 2020; Wang et al., 2020; Yu et al., 2020). Despite their multi-domain composition and long length, few mis-predictions were detected. This may be due to the low duplication rate and to the low sequence conservation.
Solutions to Improve Prediction and Annotation Errors
If this situation is extrapolated to all multigenic families (2,024 gene families in A. thaliana involving 17,481 genes) and to all available and annotated plant genomes (up to date, 134 publicly available from Phytozome, https://phytozome-next.jgi.doe.gov/), we are afraid that a hundred published studies already led to partial or incorrect conclusions.
The guarantee of an exhaustive and qualitative set of sequences is necessary to perform reliable studies, especially phylogeny, comparative genomic, and integrative analysis. Thus, efforts to provide high quality gene prediction and protein annotation are required, especially as mis-prediction and mis-annotation are rapidly amplified with subsequent articles that refer to incorrect results.
Is there a solution to reduce the rate of mis-prediction and mis-annotation in global analysis studies of large multigenic families? In the case of haem peroxidases, there are several cues to discriminate between CI APxs and CIII Prxs and to determine whether the gene predictions and protein annotations are accurate. (i) The number of gene copies is high and variable between species in CIII Prxs due to recent duplications, while it is low and conserved within the green lineage in APxs. (ii) The intron/exon structure (positions, number, and lengths of introns) is conserved in CIII Prxs (between none to three introns as illustrated Figure 2A with the two first lines) and distinct from that of APxs (between 8 to 10 introns as illustrated with the four last lines). Identification of conserved intron position and sequence alignment are powerful in discriminating between the two classes. (iii) The CIII Prxs contain conserved cysteines involved in 4 disulfide bonds whereas CI Prxs do not (Figure 2B). (iv) The protein size is characteristic as well as the highly conserved amino acids (pink diamonds, purple, and oher disks, Figure 2B) and the motifs of 15–50 amino acids defined with the MEME program (Bailey et al., 2015) (Figure 2C). (v) The CIII Prxs mostly contain a signal peptide, which targets them to the secretion pathway, whereas APxs are found in the various chloroplastic compartments or in the cytoplasm. Therefore, the combination of automatic prediction/annotation with a minimal expert control of sequence alignment should allow to verify the points (iii), (iv), and (v) and reduce the amount of erroneous predictions and annotations. Recently, new programs were developed to specifically address annotation of gene family taking into account intron conservation (Keilwagen et al., 2019) or preliminary search for a target domain (Kim et al., 2020). The generalization of these uses should be very helpful and significantly improve the sensibility and specificity of predictions.
Conclusion
Expert annotations for large protein families and dedicated databases with manually verified proteins used as reference for prediction and annotation of additional genes are the solution. Currently, experts are already available for 166 families from The Arabidopsis Information Resource (TAIR) (https://www.arabidopsis.org/browse/genefamily/) and a few databases are dedicated to protein families. On the one hand, publications based on automatic annotations of genomes can still be done but, may lead to partial and error-prone conclusions. On the otherhand, expert annotation is a background work, time-consuming and not considered as an attractive task. This method has been experimented for some vertebrate genomes with the HAVANA group (https://www.sanger.ac.uk/group/vertebrate-annotation/) but it is hardly imaginable to extend it to the thousands of available genomes. However, expert annotation would reveal many incorrect predictions and annotations with a gain in terms of biological data, avoiding mis-interpretation in downstream analysis. An intermediary solution can be adopted, as in GENCODE (Frankish et al., 2021) which combines HAVANA manual expertise with automated annotation. In all cases, it remains the responsibility of the researchers to check the quality of annotation before drawing conclusions and formulating hypothesis. Despite the real progress made in annotating genomes as a whole, precautions are still crucial before interpretation, especially when gene families are involved.
Author Contributions
CM and CD contributed to the writing. CD prepared the first draft and made the figure. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors are thankful to the Paul Sabatier-Toulouse 3 University and to the Center National de la Recherche Scientifique (CNRS) for granting their work. The authors also thank Dr. Elisabeth Jamet for her critical reading and constructive comments.
References
Bailey, T. L., Johnson, J., Grant, C. E., and Noble, W. S. (2015). The MEME suite. Nucleic Acids Res. 43, W39–49. doi: 10.1093/nar/gkv416
Blázquez, M. A., Ahn, J. H., and Weigel, D. (2003). A thermosensory pathway controlling flowering time in Arabidopsis thaliana. Nat. Genet. 33, 168–171. doi: 10.1038/ng1085
Brown, P., Baxter, L., Hickman, R., Beynon, J., Moore, J. D., and Ott, S. (2013). MEME-LaB: motif analysis in clusters. Bioinformatics 29, 1696–1697. doi: 10.1093/bioinformatics/btt248
Cai, K., Chen, S., Liu, Y., Zhao, X., and Chen, S. (2021). Genome-wide identification and analysis of class III peroxidases in Betula pendula. BMC Genomics 22:314. doi: 10.1186/s12864-021-07622
Cao, Y., Han, Y., Meng, D., Li, D., Jin, Q., Lin, Y., et al. (2016). Structural, evolutionary, and functional analysis of the class III peroxidase gene family in chinese pear. Front. Plant Sci.7:1874. doi: 10.3389/fpls.2016.01874
Chang, Y., Li, B., Shi, Q., Geng, R., Geng, S., Liu, J., et al. (2020). Comprehensive analysis of Respiratory Burst Oxidase Homologs (Rboh) gene family and function of. Front. Genet. 11:788. doi: 10.3389/fgene.2020.00788
Cheng, C., Xu, X., Gao, M., Li, J., Guo, C., Song, J., et al. (2013). Genome-wide analysis of respiratory burst oxidase homologs in grape (Vitis vinifera L.). Int. J. Mol. Sci. 14, 24169–24186. doi: 10.3390/ijms141224169
Cheng, X., Li, G., Manzoor, M. A., Wang, H., Abdullah, M., Su, X., et al. (2019). In silico genome-wide analysis of Respiratory Burst Oxidase Homolog (RBOH) family genes in five fruit-producing trees, and potential functional analysis on lignification of stone cells in chinese white pear. Cells 8:520. doi: 10.3390/cells8060520
Duan, P., Wang, G., Chao, M., Zhang, Z., and Zhang, B. (2019). Genome-wide identification and analysis of class III peroxidases in allotetraploid cotton. Genes 10:473. doi: 10.3390/genes10060473
Fawal, N., Li, Q., Mathé, C, and Dunand, C. (2014). Automatic multigenic family annotation: risks and solutions. Trends Genet. 30, 323–325. doi: 10.1016/j.tig.2014.06.004
Frankish, A., Diekhans, M., Jungreis, I., Lagarde, J., Loveland, J. E., Mudge, J. M., et al. (2021). GENCODE 2021. Nucleic Acids Res. 49, D916–D923. doi: 10.1093/nar/gkaa1087
Hoff, K. J., and Stanke, M. (2015). Current methods for automated annotation of protein-coding genes. Curr. Opin. Insect. Sci. 7, 8–14. doi: 10.1016/j.cois.2015.02.008
Inupakutika, M. A., Sengupta, S., Devireddy, A. R., Azad, R. K., and Mittler, R. (2016). The evolution of reactive oxygen species metabolism. J. Exp. Bot. 67, 5933–5943. doi: 10.1093/jxb/erw382
Kaur, G., Guruprasad, K., Temple, B. R. S., Shirvanyants, D. G., Dokholyan, N. V., and Pati, P. K. (2018). Structural complexity and functional diversity of plant NADPH oxidases. Amino Acids. 50, 79–94. doi: 10.1007/s00726-017-2491-5
Keilwagen, J., Hartung, F., and Grau, J. (2019). GeMoMa: homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol. Biol. 1962, 161–177. doi: 10.1007/978-1-4939-9173-0_9
Kim, S., Cheong, K., Park, J., Kim, M. S., Kim, J., Seo, M. K., et al. (2020). TGFam-Finder: a novel solution for target-gene family annotation in plants. New Phytol. 227, 1568–1581. doi: 10.1111/nph.16645
Li, Q., Dou, W., Qi, J., Qin, X., Chen, S., and He, Y. (2020). Genome wide analysis of the CIII peroxidase family in sweet orange. J. Genet. 99:10. doi: 10.1007/s12041-019-1163-5
Mathe, C., Sagot, M. F., Schiex, T., and Rouze, P. (2002). Current methods of gene prediction, their strengths and weaknesses. Nucleic Acids Res. 30, 4103–4117. doi: 10.1093/nar/gkf543
Mistry, J., Chuguransky, S., Williams, L., Qureshi, M., Salazar, G. A., Sonnhammer, E. L. L., et al. (2021). Pfam: the protein families database in 2021. Nucleic Acids Res. 49, D412–D419. doi: 10.1093/nar/gkaa913
Moural, T. W., Lewis, K. M., Barnaba, C., Zhu, F., Palmer, N. A., Sarath, G., et al. (2017). Characterization of class III peroxidases from switchgrass. Plant Physiol. 173, 417–433. doi: 10.1104/pp.16.01426
Passardi, F., Bakalovic, N., Teixeira, F. K., Margis-Pinheiro, M., Penel, C., and Dunand, C. (2007). Prokaryotic origins of the non-animal peroxidase superfamily and organelle-mediated transmission to eukaryotes. Genomics 89, 567–579. doi: 10.1016/j.ygeno.2007.01.006
Ren, L. L., Liu, Y. J., Liu, H. J., Qian, T. T., Qi, L. W., Wang, X. R., et al. (2014). Subcellular relocalization and positive selection play key roles in the retention of duplicate genes of populus class III peroxidase family. Plant Cell. 26, 2404–2419. doi: 10.1105/tpc.114.124750
Savelli, B., Li, Q., Webber, M., Jemmat, A. M., Robitaille, A., Zamocky, M., et al. (2019). RedoxiBase: a database for ROS homeostasis regulated proteins. Redox Biol. 26:101247. doi: 10.1016/j.redox.2019.101247
Scalzitti, N., Jeannin-Girardon, A., Collet, P., Poch, O., and Thompson, J. D. (2020). A benchmark study of ab initio gene prediction methods in diverse eukaryotic organisms. BMC Genomics 21:293. doi: 10.1186/s12864-020-6707-9
Wang, W., Chen, D., Liu, D., Cheng, Y., Zhang, X., Song, L., et al. (2020). Comprehensive analysis of the Gossypium hirsutum L. respiratory burst oxidase homolog (Ghrboh) gene family. BMC Genomics 21:91. doi: 10.1186/s12864-020-6503-6
Wang, Y., Wang, Q., Zhao, Y., Han, G., and Zhu, S. (2015). Systematic analysis of maize class III peroxidase gene family reveals a conserved subfamily involved in abiotic stress response. Gene 566, 95–108. doi: 10.1016/j.gene.2015.04.041
Welinder, K. G., Mauro, J. M., and Nørskov-Lauritsen, L. (1992). Structure of plant and fungal peroxidases. Biochem. Soc. Trans. 20, 337–340. doi: 10.1042/bst0200337
Wu, C., Ding, X., Ding, Z., Tie, W., Yan, Y., Wang, Y., et al. (2019). The class III peroxidase (POD) gene family in Cassava: identification, phylogeny, duplication, and expression. Int. J. Mol. Sci. 20:2730. doi: 10.3390/ijms20112730
Xiao, H., Wang, C., Khan, N., Chen, M., Fu, W., and Guan, L. (2020). Genome-wide identification of the class III POD gene family and their expression profiling in grapevine (Vitis vinifera L). BMC Genomics 21:444. doi: 10.1186/s12864-020-06828-z
Yan, J., Su, P., Li, W., Xiao, G., Zhao, Y., Ma, X., et al. (2019). Genome-wide and evolutionary analysis of the class III peroxidase gene family in wheat and Aegilops tauschii reveals that some members are involved in stress responses. BMC Genomics 20:666. doi: 10.1186/s12864-019-6006-5
Yang, X., Yuan, J., Luo, W., Qin, M., Yang, J., Wu, W., et al. (2020). Genome-wide identification and expression analysis of the class III peroxidase gene family in potato. Front. Genet. 11:593577. doi: 10.3389/fgene.2020.593577
Yanhui, C., Xiaoyuan, Y., Kun, H., Meihua, L., Jigang, L., Zhaofeng, G., et al. (2006). The MYB transcription factor superfamily of Arabidopsis: expression analysis and phylogenetic comparison with the rice MYB family. Plant Mol. Biol. 60, 107–124. doi: 10.1007/s11103-005-2910-y
Yu, S., Kakar, K. U., Yang, Z., Nawaz, Z., Lin, S., Guo, Y., et al. (2020). Systematic study of the stress-responsive Rboh gene family in Nicotiana tabacum: genome-wide identification, evolution and role in disease resistance. Genomics 112, 1404–1418. doi: 10.1016/j.ygeno.2019.08.010
Keywords: protein annotation, gene prediction, gene family, mis-prediction, mis-annotation
Citation: Mathé C and Dunand C (2021) Automatic Prediction and Annotation: There Are Strong Biases for Multigenic Families. Front. Genet. 12:697477. doi: 10.3389/fgene.2021.697477
Received: 21 April 2021; Accepted: 05 August 2021;
Published: 16 September 2021.
Edited by:
Ajay Kumar, North Dakota State University, United StatesReviewed by:
Claudio Casola, Texas A&M University, United StatesVirag Sharma, Dresden University of Technology, Germany
Copyright © 2021 Mathé and Dunand. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christophe Dunand, dunand@lrsv.ups-tlse.fr