 Xiaoyuan Wang
Xiaoyuan Wang Huijie XiaoYong YaoKe XuXiaoyu LiuBaige SuHongwen Zhang
Huijie XiaoYong YaoKe XuXiaoyu LiuBaige SuHongwen Zhang Na Guan
Na Guan Xuhui Zhong
Xuhui Zhong Yanqin Zhang
Yanqin Zhang Jie Ding*
Jie Ding* Fang Wang*
Fang Wang*- Department of Pediatrics, Peking University First Hospital, Beijing, China
Renal hypodysplasia and cystic kidney diseases, the common non-glomerular causes of pediatric chronic kidney disease (CKD), are usually diagnosed by their clinical and imaging characteristics. The high degree of phenotypic heterogeneity, in both conditions, makes the correct final diagnosis dependent on genetic testing. It is not clear, however, whether the frequencies of damaged alleles vary among different ethnicities in children with non-glomerular CKD, and this will influence the strategy used for genetic testing. In this study, 69 unrelated children (40 boys, 29 girls) of predominantly Han Chinese ethnicity with stage 2–5 non-glomerular CKD caused by suspected renal hypodysplasia or cystic kidney diseases were enrolled and assessed by molecular analysis using proband-only targeted exome sequencing and array-comparative genomic hybridization. Targeted exome sequencing discovered genetic etiologies in 33 patients (47.8%) covering 10 distinct genetic disorders. The clinical diagnoses in 13/48 patients (27.1%) with suspected renal hypodysplasia were confirmed, and two patients were reclassified carrying mutations in nephronophthisis (NPHP) genes. The clinical diagnoses in 16/20 patients (80%) with suspected cystic kidney diseases were confirmed, and one patient was reclassified as carrying a deletion in the hepatocyte nuclear factor-1-beta gene (HNF1B). The diagnosis of one patient with unknown non-glomerular disease was elucidated. No copy number variations were identified in the 20 patients with negative targeted exome sequencing results. NPHP genes were the most common disease-causing genes in the patients with disease onsets above 6 years of age (14/45, 31.1%). The children with stage 2 and 3 CKD at onset were found to carry causative mutations in paired box gene 2 (PAX2) and HNF1B gene (11/24, 45.8%), whereas those with stage 4 and 5 CKD mostly carried causative mutations in NPHP genes (19/45, 42.2%). The causative genes were not suspected by the kidney imaging patterns at disease onset. Thus, our data show that in Chinese children with non-glomerular renal dysfunction caused by renal hypodysplasia and cystic kidney diseases, the common causative genes vary with age and CKD stage at disease onset. These findings have the potential to improve management and genetic counseling of these diseases in clinical practice.
Introduction
The presence of structural or functional abnormalities in the kidney over a 3-month period is defined as chronic kidney disease (CKD), and is classified into five stages based on the glomerular filtration rate (Andrassy, 2013). End-stage renal disease (ESRD), which is the most serious CKD stage, requires the use of renal replacement therapy. Pediatric CKDs are less common than in adults, but affected children are at increased risk of early mortality and disabling physical comorbidities, which highlights the need for appropriate management of the affected children.
Congenital anomalies of the kidney and urinary tract (CAKUT) are the most common non-glomerular presentations of pediatric ESRD followed by cystic kidney diseases (Smith et al., 2007; Wuhl et al., 2014). In fact, the most prevalent malformation is reported to be renal hypodysplasia, which includes renal aplasia, hypoplasia, and dysplasia (Smith et al., 2007). Renal ultrasound provides essential diagnostic information about renal hypodysplasia and cystic kidney diseases (Sanna-Cherchi et al., 2007; Vester et al., 2010; Gimpel et al., 2019). For example, hypodysplastic kidney is defined by renal ultrasonography findings as a reduced renal size of greater than two standard deviations from the mean size in terms of age and loss of corticomedullary differentiation, and the sonographic signs of parenchymal hyperechogenicity and renal enlargement in a child are highly suggestive of polycystic kidney disease. However, a clinically definitive diagnosis of hypodysplastic kidney disease or cystic kidney disease remains challenging to arrive at because the sonographic appearance of these two conditions is observed in a variety of renal diseases.
With advances in genomic DNA sequencing technologies, the genetic mechanisms leading to renal hypodysplasia and cystic kidney diseases have been more readily assessed (Vivante and Hildebrandt, 2016; Sanna-Cherchi et al., 2018; Armstrong and Thomas, 2019; Devlin and Sayer, 2019; Nigam et al., 2019), and this improves the diagnostic accuracy of genetic testing. Our previous study reported that the genetic test results for pediatric steroid-resistant nephrotic syndrome vary by ethnicity (Wang et al., 2017b). It is not clear, however, whether a similar phenomenon exists with the pediatric chronic renal dysfunction caused by renal hypodysplasia and cystic kidney diseases, which provides the impetus for the reasonable selection of genetic testing approaches. To address this question, index-only targeted exome sequencing and array-comparative genomic hybridization (CGH) were performed in a cohort of 69 unrelated children with non-glomerular stage 2–5 CKD who were clinically suspected of having renal hypodysplasia or cystic kidney diseases.
Materials and Methods
Patients
Patients were enrolled in the study between January 2011 and September 2018 by a group of trained pediatric nephrologists from the Department of Pediatrics, Peking University First Hospital based on fulfillment of the following criteria: (i) the presence of stage 2–5 CKD below the age of 18 years; (ii) clinical diagnosis or suspicion of renal hypoplasia/dysplasia, cystic kidney diseases, or unknown non-glomerular diseases. Patients with polycystic kidney disease, incomplete clinical data (especially the absence of kidney imaging results), and an unwillingness to participate in the study were excluded. The study was approved by the Ethics Committee of Peking University First Hospital and was performed in accordance with the Declaration of Helsinki.
Comprehensive clinical data [including age of onset, age of renal failure, urinalysis, examination of urinary protein, renal imaging, estimated glomerular filtration rate using 24-h endogenous creatinine clearance or the Schwartz formula (Schwartz et al., 1987), extrarenal manifestations, renal biopsy, information from the last follow-up, and family history] and demographics were extracted from the Chinese Registry Database of Hereditary Kidney Diseases and then analyzed. Sonographic measurements of the longitudinal sections of both kidneys in each patient were compared with those of age-matched controls (Loftus et al., 1998).
After receiving informed consent from the patients or their parents/legal guardians, blood samples and comprehensive clinical data were collected and analyzed.
Genetic Examination
Genomic DNA was extracted from peripheral white blood cells using the QIAamp DNA Blood Mini Kit (A1120, Qiagen, Germany). DNA quantity and quality were determined by NanoDrop (Thermo Fisher Scientific, United States). When available, DNA samples from the participants’ relatives were obtained.
Because targeted exome sequencing is a cost-effective diagnostic strategy for identifying the genetic causes of kidney disorders (Vivante and Hildebrandt, 2016; Groopman et al., 2018), we used it to simultaneously examine 30 genes that are known to be associated with renal hypodysplasia and 118 genes associated with cystic kidney diseases (Supplementary Table 1). These genes were selected from the relevant literature (Devuyst et al., 2014; Mann et al., 2019; Nigam et al., 2019). DNA library preparation, capture, enrichment, next-generation sequencing, and data analysis were performed at BGI-Shenzhen, China, as described previously (Wang et al., 2017b). Variants with minor allele frequencies <0.01 were selected based on the control database such as NCBI dbSNP (snp137), 1000 Genomes Project (phase I), Exome sequencing project (ESP6500), Exome Aggregation Consortium (ExAC), Genome Aggregation Database (gnomAD), and the BGI in-house database. The Human Gene Mutation Database (HGMD) and ClinVar were used to detect previously reported pathogenic variants. The prioritized variants were classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015).
To detect copy number variations (CNVs), array CGH was performed using the Agilent SurePrint G3 Human 8 × 60 K CGH Microarray (Agilent Technologies, Technologies, Santa Clara, CA, United States). DNA labeling, array hybridization, scanning, and data analysis were conducted at the Department of Central Laboratory, Peking University First Hospital, Beijing, China, as described previously (Yi et al., 2016). Public CNV databases including DGV, NCBI, DECIPHER, ClinGen, OMIM, and ISCA were used to detect known CNVs. The prioritized CNVs were classified according to the ACMG guidelines (Riggs et al., 2020).
Validation of all candidate pathogenic or likely pathogenic variants was performed using Sanger sequencing or quantitative PCR (qPCR) on the genomic DNAs of the probands. Hepatocyte nuclear factor-1-beta gene (HNF1B) and the nephronophthisis type 1 (NPHP1) gene were used to normalize the gene dosage in qPCR, and they were analyzed in triplicate. Segregation analyses were performed for all the available first-degree relatives.
Results
Clinical Features
As shown in Figure 1 and Table 1, 69 unrelated patients (40 boys, 29 girls) were enrolled in this study. They were from 18 provinces, municipalities, and autonomous Chinese regions and were predominantly the Han Chinese ethnicity (61/69). Renal dysfunction was detected in most of these patients (36/69), either accidentally or for other reasons at disease onset, whereas complaints of fatigue or a sallow complexion were observed in 19 patients, edema in eight patients, short stature in four patients, polydipsia and polyuria in one patient, and enuresis in another patient. Their median age at disease onset was 8.5 years (range, 0 day–16.7 years). Renal hypoplasia/dysplasia, cystic kidney disease, and unknown non-glomerular disease were diagnosed or suspected in 48, 20, and 1 patient, respectively. There were seven patients with stage 2 CKD, 17 patients with stage 3 CKD, 16 patients with stage 4 CKD, and 29 patients with stage 5 CKD. Of the 17 patients undergoing renal biopsy, chronic tubulointerstitial nephropathy was the most common histopathological diagnosis. The patients’ extrarenal manifestations included short stature, ocular abnormalities (including ametropia, strabismus, microphthalmia, retinopathy, vitreous opacity, and nystagmus), auricle malformation, preauricular fistula, spina bifida, cryptorchidism, skeletal deformities (including polydactylism, tetradactylism, straw sandal-like feet, strephenopodia, and fourth metatarsal microsomia), elevated liver enzymes, ovarian teratoma, microcephaly, ventricular septal defect, and patent arterial duct in 23 patients. Parental consanguinity was reported in only one patient, whereas eight patients had positive family histories of ESRD, one patient had a positive family history of proteinuria, and one patient had a positive family history of renal cystic disease. Eight patients had received a renal transplant (median age, 12.9 years; range, 6–18 years), and no disease recurrence in their allografts was documented. Six patients died at a median age of 5.9 (2–12) years.
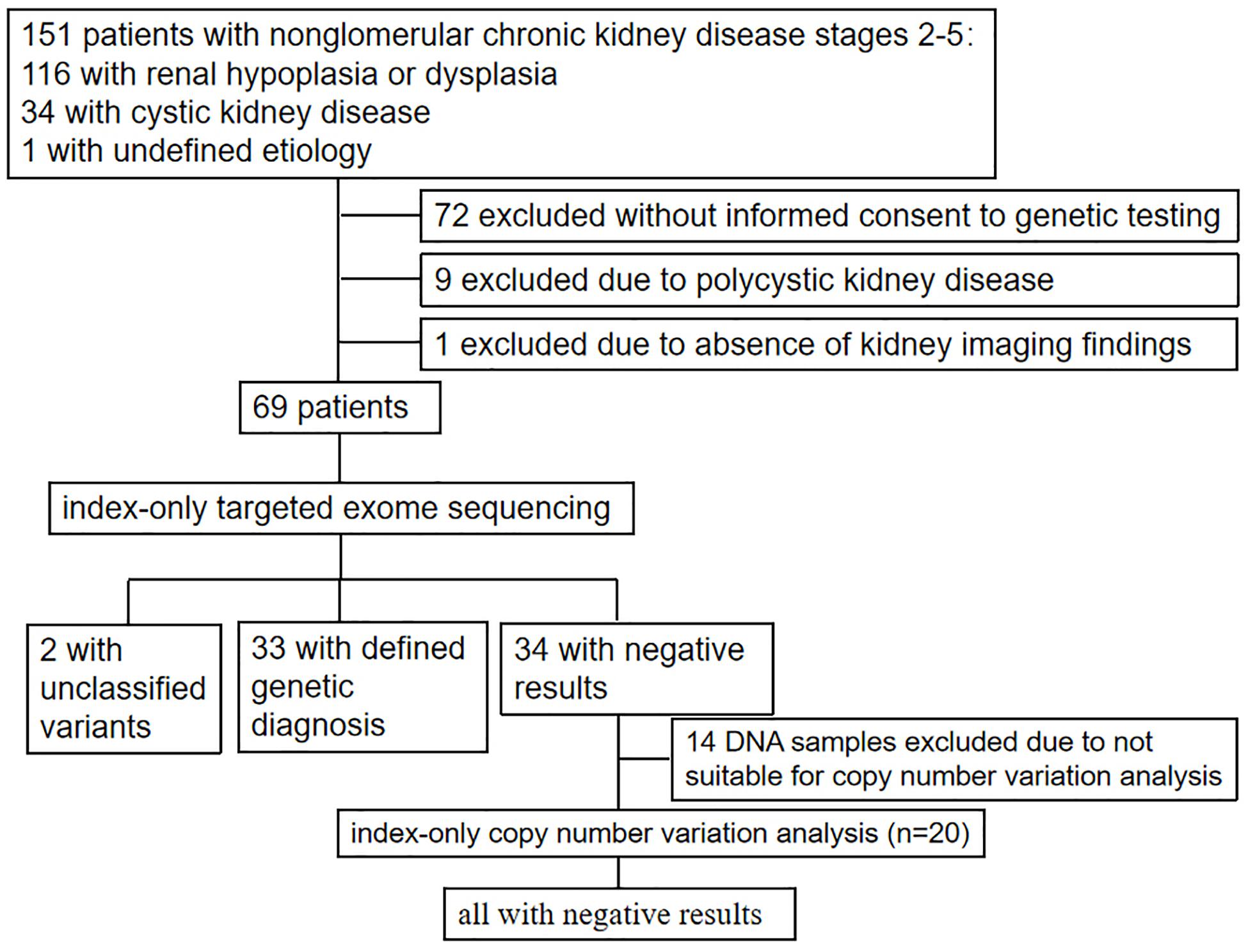
Figure 1. Flow diagram for patient selection and genetic testing.
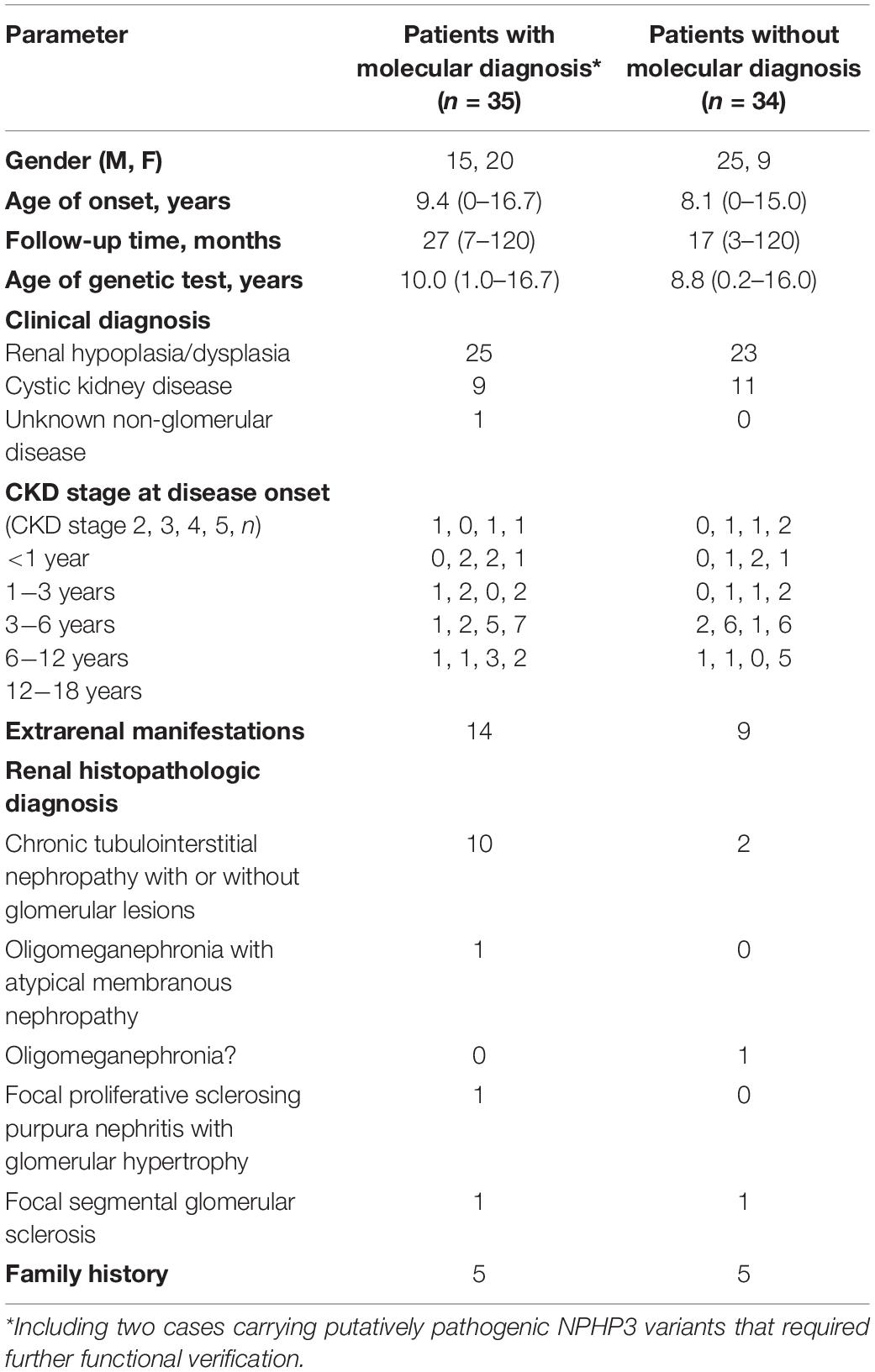
Table 1. Clinical features of the 69 patients from the present study.
Genetic Study
Twenty-two pathogenic variants and six likely pathogenic variants in 10/148 targeted genes, including nine non-sense, seven missense, six splice sites, three small deletions, two whole gene deletion, and one small insertion, were detected in 33/69 patients (47.8%), and these variants encompassed 10 distinct genetic disorders (Table 2 and Figure 2). Of these variants, the 14 (50.0%) novel ones included three variants that we reported on previously (Deng et al., 2019), whereas the remaining 14 variants were previously reported. Of the 18 patients harboring diagnostic variants in recessive genes, compound heterozygous variants were found in 10 patients and homozygous variants were found in eight patients. Of the 48 patients with suspected renal hypodysplasia, the targeted exome sequencing confirmed the clinical diagnoses of 13 patients (27.1%), and reclassified the clinical diagnoses of two patients carrying mutations in nephronophthisis (NPHP) genes (INVS and WDR19). Of the 20 patients with suspected cystic kidney diseases, the clinical diagnoses for 16 patients (80%) were confirmed, and that of one patient with a deletion in HNF1B was reclassified. The diagnosis of the remaining patient (patient 19) with renal dysfunction (serum creatinine, 122 μmol/L; evaluated glomerular filtration rate, 22.9 ml/min/1.73 m2), moderate anemia (70 g/L), short stature (height, 71 cm), and normal-sized, non-cystic kidneys combined with parenchymal hyperechogenicity and poor corticomedullary differentiation on renal ultrasonography (11 months of age) was classified by the compound heterozygous non-sense mutations present in the gene encoding the angiotensin-converting enzyme (ACE).
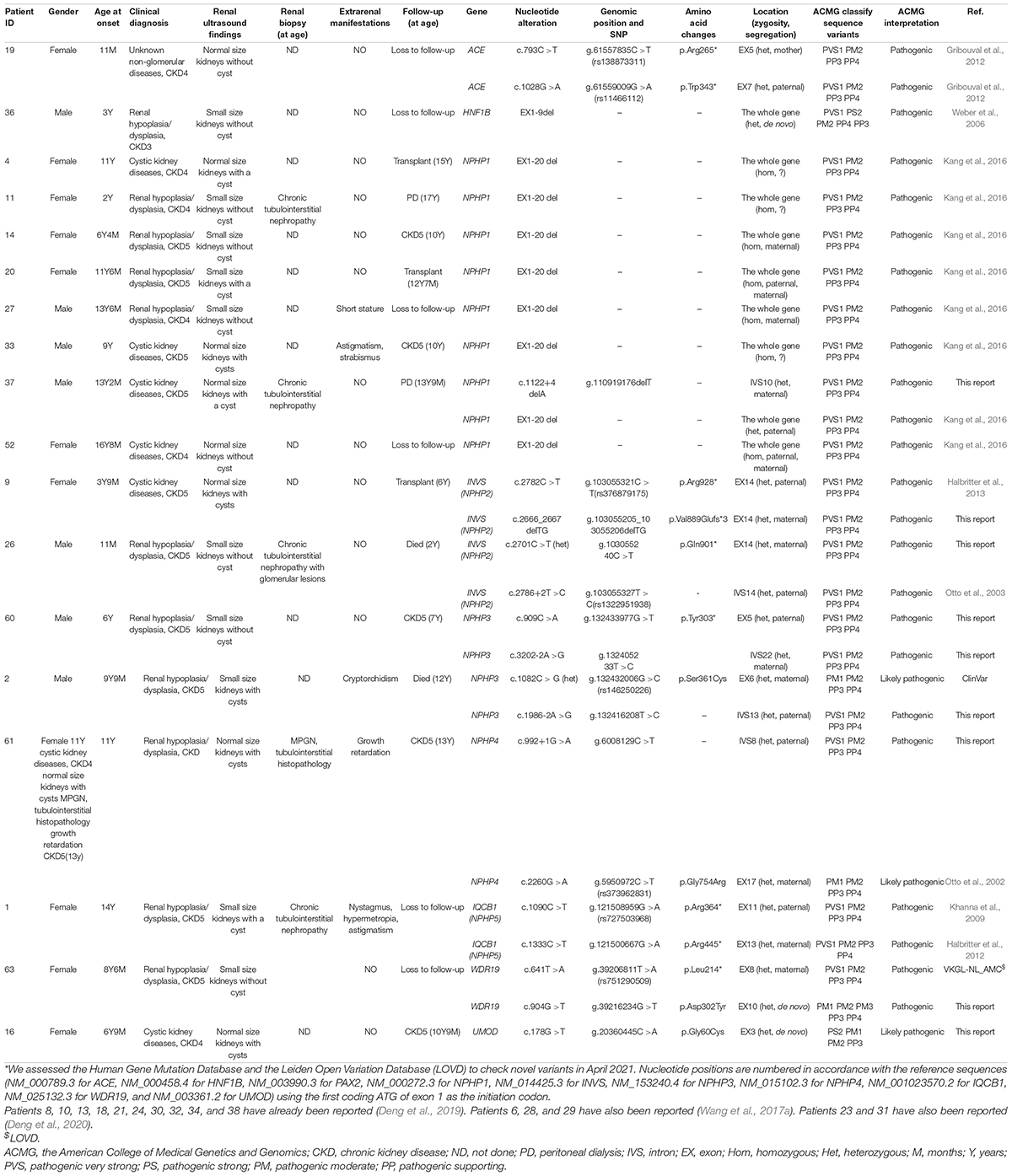
Table 2. Pathogenic or likely pathogenic variants detected by targeted exome sequencing*.
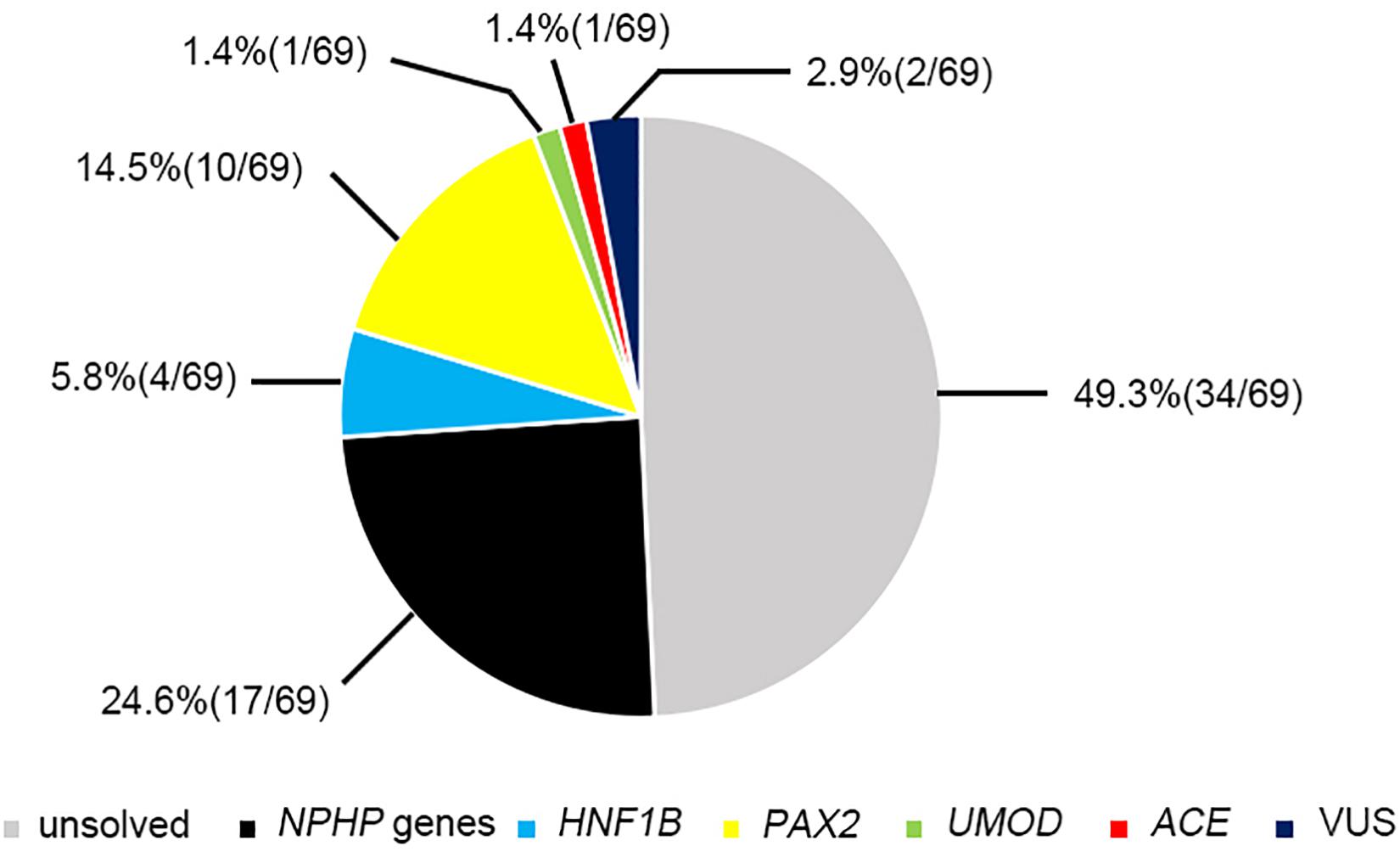
Figure 2. The results of genetic examination in 69 patients.
Patients 22 and 69 were strongly suspected of having NPHP based on the combination of ESRD before the age of 7 years without proteinuria or hematuria, normal-sized kidneys with hyperechogenicity and the absence of corticomedullary differentiation, and chronic tubulointerstitial nephropathy (in patient 22) or elevated liver enzymes of unknown cause (in patient 69), whereas targeted exome sequencing revealed three rare and predicted deleterious variants in NPHP3 that were classified as having unknown significance using ACMG criteria (Table 3). We assumed that these variants are pathogenic, although functional analyses on them are required.
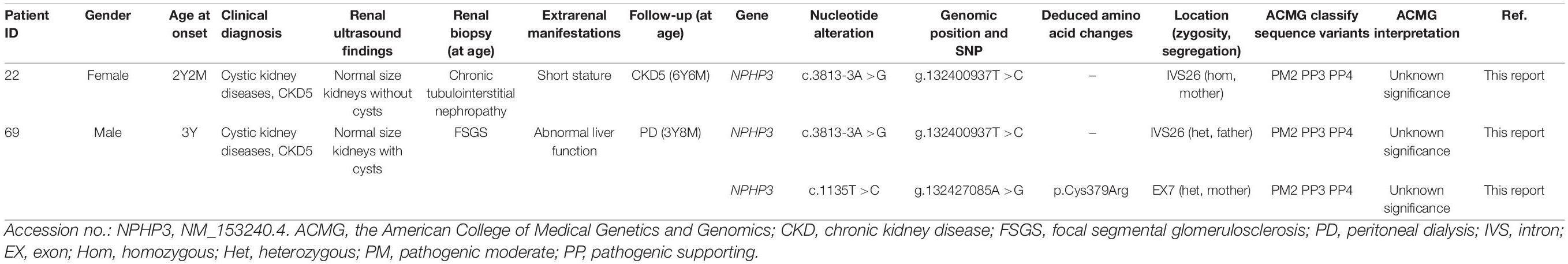
Table 3. Variants of unknown significance that were detected by targeted exome sequencing.
No CNVs were identified in the 20 patients with negative targeted exome sequencing results.
The likelihood of establishing an accurate molecular diagnosis of non-glomerular CKD did not improve with increasing age and remained roughly the same (at about 50%) (Figure 3A). We detected diagnostic PAX2 and NPHP gene variants in all four age groups, and the NPHP genes were the most common disease-causing ones in the patients whose disease onset was above 6 years of age (14/45, 31.1%).
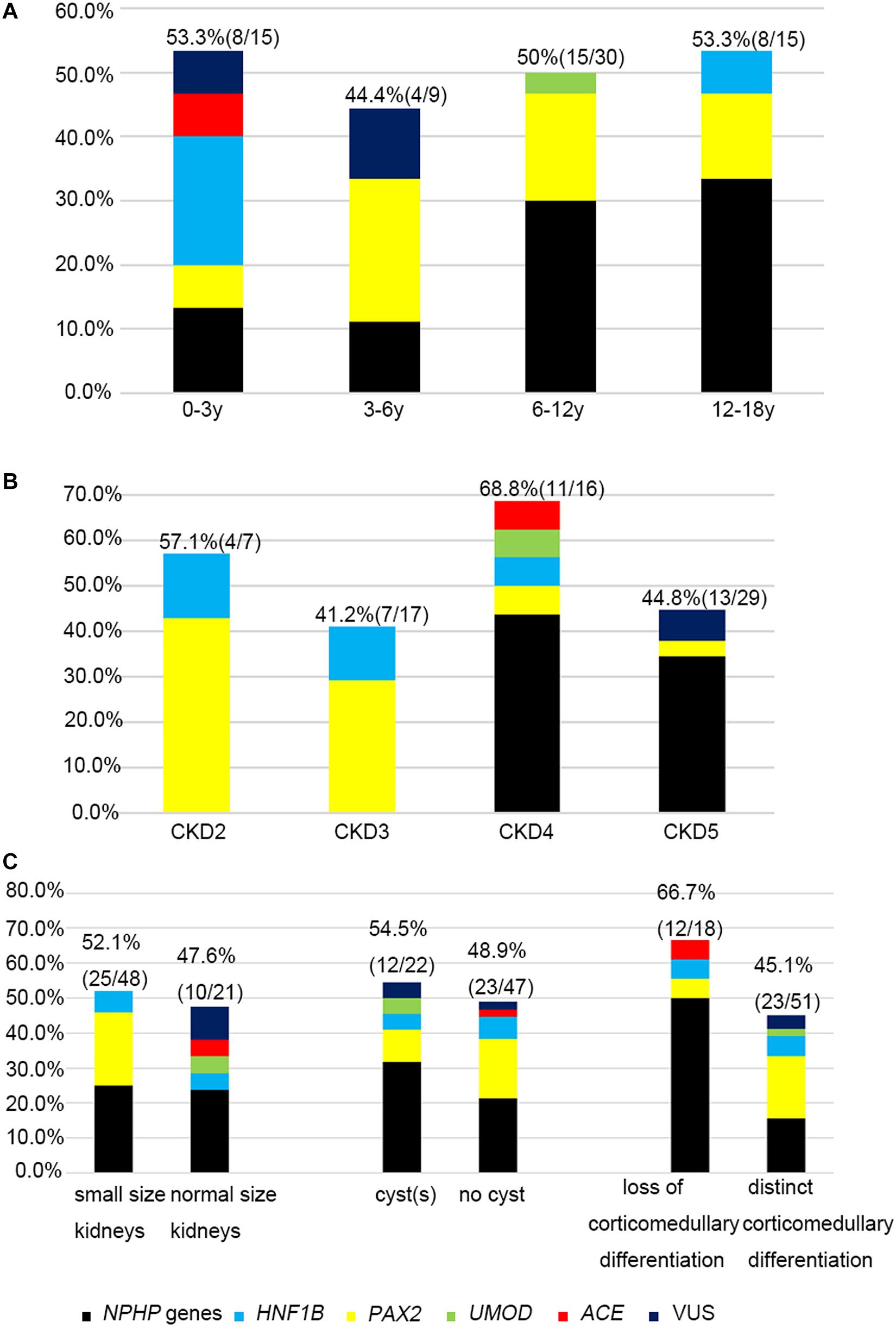
Figure 3. The relationship between causative genes and clinical features. Histograms indicate fractions (in percentage) of patients with disease gene detected per group. (A) Genetic diagnosis and the age at onset for non-glomerular CKD. (B) Genetic diagnosis and CKD stages. (C) Genetic diagnosis and renal ultrasound findings.
The molecular diagnostic performance and common mutated genes differed in line with the increased CKD stage at disease onset (Figure 3B). The genetic diagnostic yield was highest in the patients with stage 4 CKD at onset (11/16, 68.7%). The children whose CKD onset was stage 2 and 3 carried mutations in PAX2 and HNF1B genes (11/24, 45.8%), whereas those whose CKD onset was stage 4 and 5 mostly carried mutations in NPHP genes (19/45, 42.2%).
Because renal ultrasonography is used in the first instance to diagnose renal hypodysplasia and cystic nephropathies, we analyzed the renal imaging patterns at disease onset and the mutation detection rates in the patients (Figure 3C). An etiological diagnosis was found in 52.1% of the children with small kidneys (25/48), 47.6% with normal-sized kidneys (10/21), 48.9% without cysts (23/47), 54.5% with a single cyst or multiple cysts (12/22), 66.7% without corticomedullary differentiation (12/18), and 45.1% with distinct corticomedullary differentiation (23/51). Renal parenchymal hyperechogenicity was observed in all 69 patients.
The most prevalent genetic diagnosis in our study was NPHP. Chaki et al. (2011) reported that 100% of 440 patients with NPHP-related ciliopathies carried biallelic pathogenic variants in NPHP genes. We therefore analyzed the genetic test results from the patients who met at least one of the four criteria for NPHP used by Chaki et al. (2011). Hence, we were able to clinically diagnose 52 patients as having NPHP and found that 26 patients (50%) had pathogenic or likely pathogenic variants in the causative genes. Of these patients, 17 had NPHP, whereas we identified mutations in PAX2, HNF1B, ACE, and UMOD genes in the remaining patients (Figure 4).
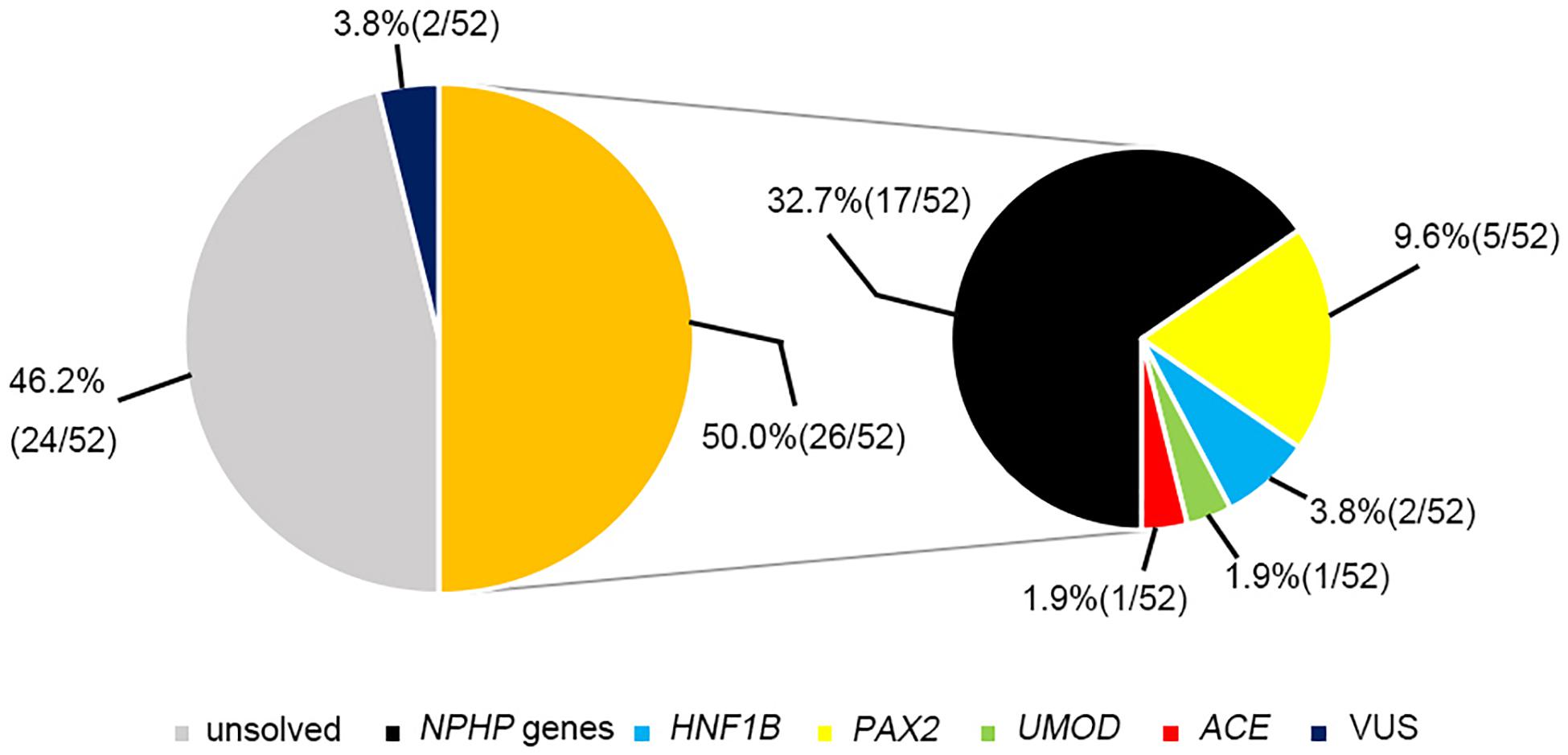
Figure 4. The genetic examination results for 52 patients who were highly suspected of having nephronophthisis.
Discussion
In the present study, we used targeted exome sequencing and array CGH to depict the genetic features of 69 unrelated children with non-glomerular stage 2–5 CKD caused by suspected renal hypodysplasia or cystic kidney diseases. First, 27.1% of our patients with suspected renal hypodysplasia obtained a molecular diagnosis, and PAX2 was the most common mutated gene (found in ten patients). In contrast, in one cohort of 159 Chinese CAKUT children (Rao et al., 2019), only four carried PAX2 mutations. Ishiwa et al. (2019) performed genetic analysis on 66 Japanese patients with CAKUT (the number of patients with renal hypodysplasia is not available) and identified the etiologies in 14 individuals with renal hypodysplasia (21.2%). Ahn et al. (2020) identified the causative genes responsible for renal hypodysplasia in 12/76 Korean children (15.8%), and predominance of HNF1B mutations was seen in these patients. The mutation detection rates range from 7 to 17% in patients from Europe and the United States with renal hypodysplasia combined with or without renal failure (Weber et al., 2006; Thomas et al., 2011; Hwang et al., 2014; Nicolaou et al., 2016). The SALL1 (spalt-like transcription factor 1) gene was detected more frequently in one cohort of patients (Hwang et al., 2014), whereas PAX2 and HNF1B were detected in another cohort (Weber et al., 2006; Thomas et al., 2011; Nicolaou et al., 2016). One possible explanation for this discrepancy relates to the criteria used for selecting patients: our cohort contained patients with bilateral renal lesions, whereas other studies have contained patients with bilateral and unilateral renal hypodysplasia. Another possible explanation is the high genetic heterogeneity in this condition. Second, causative genes were identified in 85% of our patients with suspected cystic kidney diseases, and 10 patients carried NPHP1 mutations, making it the most prevalent mutated gene. However, in other patient cohorts, about 70% of the children with cystic kidney diseases had monogenic disease, and the most frequent molecular diagnosis was autosomal recessive polycystic kidney disease or polycystic kidney disease (Bullich et al., 2018; Rao et al., 2019; Obeidova et al., 2020). Excluding polycystic kidney disease from our study may in part explain this discrepancy. Finally, a genetic diagnosis was obtained in three children where phenotypic overlapping caused the initial disease to be clinically misdiagnosed, one case of which had undiagnosed stage 4 CKD, which stresses the importance of genetic testing as one of the diagnostic workups in the pediatric CKD population.
It is worth noting the difference we observed for the common causative genes in relation to the age and CKD stage at disease onset. NPHP genes were the most frequently mutated genes in the patients whose onset exceeded 6 years of age with stage 4–5 CKD, whereas mutations in HNF1B and PAX2 together were more prevalent in patients whose onset was less than 6 years of age and had become stage 2–3 CKD. These findings suggest that performing genetic testing in accordance with the age and CKD stage at disease onset may be an efficient strategy for the molecular diagnosis of children with non-glomerular CKD. In contrast, Weber’s study showed that HNF1B and PAX2 mutations caused CKDs with an age of onset between 10 and 23 years (Weber et al., 2006), and autosomal recessive polycystic kidney disease was reported to be the most prevalent etiology in neonatal-onset cystic kidney diseases (Obeidova et al., 2020). The difference is likely impacted by the use of small populations of patients and the patients’ ethnic origins. Because early stage renal hypodysplasia and cystic kidney diseases are often clinically silent, patients with renal insufficiency who are usually detected accidentally may be referred for clinical diagnosis. Renal ultrasonography is currently the diagnostic mainstay. The presence of small-sized kidneys in a child always leads clinicians to make a diagnosis of renal hypodysplasia, and the presence of renal cysts support the diagnosis of cystic kidney disease. However, the phenotypic and genetic variability of these two conditions makes establishing the final clinical diagnosis challenging. As our study has shown, small- to normal-sized kidneys with or without cyst formation or changes in corticomedullary differentiation can be caused by mutant NPHP and HNF1B genes (Chaki et al., 2011; Avni et al., 2015), whereas mutations in PAX2 lead to small-sized kidneys that often show distinct corticomedullary differentiation and no cysts (Bower et al., 2012).
NPHP is one of the most common inherited diseases leading to pediatric ESRD, and the phenotypes and genotypes in Chinese children with NPHP have been described (Tang et al., 2020; Yue et al., 2020). However, our finding shows that non-NPHP genes can also cause NPHP-like phenotypes, which emphasizes the difficulty in diagnosing NPHP in clinical settings. Similar phenomena have been reported elsewhere (Bullich et al., 2018; Mann et al., 2019).
Pathogenic CNVs, recognizably important etiological factors underlying renal hypodysplasia (Sanna-Cherchi et al., 2012; Verbitsky et al., 2019), are recommended to be detected by array-based technologies. However, consistent with prior reports on the use of targeted exome sequencing as a tool for identifying CNVs (Roberts et al., 2017; Ahn et al., 2020), our targeted exome sequencing and qPCR, we detected the deletion of the whole HNF1B and NPHP1 genes in 4 and 10 patients, respectively, but no additional CNVs were identified using array CGH.
Obtaining a definite molecular diagnosis is very important for patients and their families and for facilitating genetic counseling. For example, HNF1B mutations are associated with diabetes mellitus (Clissold et al., 2015), and NPHP1 genetic variants may cause multisystemic diseases and Joubert syndrome, among others (Soliman et al., 2012). Early discovery of related hidden symptoms and timely treatments are very important for patients.
We are conscious of some limitations in our study, which include the comparatively small patient cohort, the stringent clinical criteria for selecting patients, the absence of whole-exome sequencing in patients lacking a genetic diagnosis, and the lack of functional verification of novel unknown significance variants. False-negative results from targeted exome sequencing were possible in the patients with no detectable variants. Nonetheless, our study was performed in one of the largest referral centers on mainland China, and the patients were from 18 out of 34 provincial administrative Chinese regions, indicating that our study is somewhat representative of Chinese children with renal dysfunction caused by renal hypodysplasia and cystic kidney diseases, and our findings provide the genotypic features seen in them. To the best of our knowledge, this is the first cohort study to provide evidence about the association between causative mutations and the stage of CKD onset.
In summary, the Chinese children with non-glomerular renal dysfunction caused by renal hypodysplasia and cystic kidney diseases in this study that carried the common causative genes varied in the age and CKD stage at disease onset. This new knowledge should help with improving the management and genetic counseling of the abovementioned diseases in clinical practice.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethical Committee of Peking University First Hospital approved the procedures in this study. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
FW and JD: conceptualization, formal analysis, writing – review and editing, visualization, supervision, project Administration, and funding acquisition. XW: methodology, software, investigation, and resources. HX, YY, KX, HZ, XL, BS, NG, XZ, and YZ: data curation. XW and FW: writing – original draft preparation. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the National Key Research and Development Program of China (No. 2016YFC0901505) (the registry study of rare diseases in children) and the Beijing Key Laboratory of Molecular Diagnosis and Study on Pediatric Genetic Diseases (BZ0317).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the patients, families, and physicians who contributed to this project. We are very grateful to the NCBI, LOVD, and UCSC databases for their free use.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.697085/full#supplementary-material
Supplementary Table 1 | List of genes list associated with renal hypodysplasia and cystic kidney diseases.
References
Ahn, Y. H., Lee, C., Kim, N. K. D., Park, E., Kang, H. G., Ha, I. S., et al. (2020). Targeted exome sequencing provided comprehensive genetic diagnosis of congenital anomalies of the kidney and urinary tract. J. Clin. Med. 9:751. doi: 10.3390/jcm9030751
Andrassy, K. M. (2013). Comments on ‘KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease’. Kidney Int. 84, 622–623. doi: 10.1038/ki.2013.243
Armstrong, M. E., and Thomas, C. P. (2019). Diagnosis of monogenic chronic kidney diseases. Curr. Opin. Nephrol. Hypertens 28, 183–194. doi: 10.1097/MNH.0000000000000486
Avni, F. E., Lahoche, A., Langlois, C., Garel, C., Hall, M., and Vivier, P. H. (2015). Renal involvement in children with HNF1beta mutation: early sonographic appearances and long-term follow-up. Eur. Radiol. 25, 1479–1486. doi: 10.1007/s00330-014-3550-x
Bower, M., Salomon, R., Allanson, J., Antignac, C., Benedicenti, F., Benetti, E., et al. (2012). Update of PAX2 mutations in renal coloboma syndrome and establishment of a locus-specific database. Hum. Mutat. 33, 457–466. doi: 10.1002/humu.22020
Bullich, G., Domingo-Gallego, A., Vargas, I., Ruiz, P., Lorente-Grandoso, L., Furlano, M., et al. (2018). A kidney-disease gene panel allows a comprehensive genetic diagnosis of cystic and glomerular inherited kidney diseases. Kidney Int. 94, 363–371. doi: 10.1016/j.kint.2018.02.027
Chaki, M., Hoefele, J., Allen, S. J., Ramaswami, G., Janssen, S., Bergmann, C., et al. (2011). Genotype-phenotype correlation in 440 patients with NPHP-related ciliopathies. Kidney Int. 80, 1239–1245. doi: 10.1038/ki.2011.284
Clissold, R. L., Hamilton, A. J., Hattersley, A. T., Ellard, S., and Bingham, C. (2015). HNF1B-associated renal and extra-renal disease-an expanding clinical spectrum. Nat. Rev. Nephrol. 11, 102–112. doi: 10.1038/nrneph.2014.232
Deng, H., Zhang, Y., Xiao, H., Yao, Y., Liu, X., Su, B., et al. (2019). Diverse phenotypes in children with PAX2-related disorder. Mol. Genet. Genomic Med. 7:e701. doi: 10.1002/mgg3.701
Deng, H., Zhang, Y., Yao, Y., Xiao, H., Su, B., Xu, K., et al. (2020). Interpretation of autosomal recessive kidney diseases with “presumed homozygous” pathogenic variants should consider technical pitfalls. Front. Pediatr. 8:165. doi: 10.3389/fped.2020.00165
Devlin, L. A., and Sayer, J. A. (2019). Renal ciliopathies. Curr. Opin. Genet. Dev. 56, 49–60. doi: 10.1016/j.gde.2019.07.005
Devuyst, O., Knoers, N. V., Remuzzi, G., Schaefer, F., Board of the Working Group for Inherited Kidney Diseases of the European Renal A., European, D., et al. (2014). Rare inherited kidney diseases: challenges, opportunities, and perspectives. Lancet 383, 1844–1859. doi: 10.1016/S0140-6736(14)60659-0
Gimpel, C., Avni, E. F., Breysem, L., Burgmaier, K., Caroli, A., Cetiner, M., et al. (2019). Imaging of kidney cysts and cystic kidney diseases in children: an international working group consensus statement. Radiology 290, 769–782. doi: 10.1148/radiol.2018181243
Gribouval, O., Moriniere, V., Pawtowski, A., Arrondel, C., Sallinen, S. L., Saloranta, C., et al. (2012). Spectrum of mutations in the renin-angiotensin system genes in autosomal recessive renal tubular dysgenesis. Hum. Mutat. 33, 316–326. doi: 10.1002/humu.21661
Groopman, E. E., Rasouly, H. M., and Gharavi, A. G. (2018). Genomic medicine for kidney disease. Nat. Rev. Nephrol. 14, 83–104. doi: 10.1038/nrneph.2017.167
Halbritter, J., Diaz, K., Chaki, M., Porath, J. D., Tarrier, B., Fu, C., et al. (2012). High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J. Med. Genet. 49, 756–767. doi: 10.1136/jmedgenet-2012-100973
Halbritter, J., Porath, J. D., Diaz, K. A., Braun, D. A., Kohl, S., Chaki, M., et al. (2013). Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum. Genet. 132, 865–884. doi: 10.1007/s00439-013-1297-0
Hwang, D. Y., Dworschak, G. C., Kohl, S., Saisawat, P., Vivante, A., Hilger, A. C., et al. (2014). Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int. 85, 1429–1433. doi: 10.1038/ki.2013.508
Ishiwa, S., Sato, M., Morisada, N., Nishi, K., Kanamori, T., Okutsu, M., et al. (2019). Association between the clinical presentation of congenital anomalies of the kidney and urinary tract (CAKUT) and gene mutations: an analysis of 66 patients at a single institution. Pediatr. Nephrol. 34, 1457–1464. doi: 10.1007/s00467-019-04230-w
Kang, H. G., Lee, H. K., Ahn, Y. H., Joung, J. G., Nam, J., Kim, N. K., et al. (2016). Targeted exome sequencing resolves allelic and the genetic heterogeneity in the genetic diagnosis of nephronophthisis-related ciliopathy. Exp. Mol. Med. 48:e251. doi: 10.1038/emm.2016.63
Khanna, H., Davis, E. E., Murga-Zamalloa, C. A., Estrada-Cuzcano, A., Lopez, I., den Hollander, A. I., et al. (2009). A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat. Genet. 41, 739–745. doi: 10.1038/ng.366
Loftus, W. K., Gent, R. J., LeQuesne, G. W., and Metreweli, C. (1998). Renal length in Chinese children: sonographic measurement and comparison with western data. J. Clin. Ultrasound 26, 349–352. doi: 10.1002/(sici)1097-0096(199809)26:7<349::aid-jcu4<3.0.co;2-9
Mann, N., Braun, D. A., Amann, K., Tan, W., Shril, S., Connaughton, D. M., et al. (2019). Whole-Exome sequencing enables a precision medicine approach for kidney transplant recipients. J. Am. Soc. Nephrol. 30, 201–215. doi: 10.1681/ASN.2018060575
Nicolaou, N., Pulit, S. L., Nijman, I. J., Monroe, G. R., Feitz, W. F., Schreuder, M. F., et al. (2016). Prioritization and burden analysis of rare variants in 208 candidate genes suggest they do not play a major role in CAKUT. Kidney Int. 89, 476–486. doi: 10.1038/ki.2015.319
Nigam, A., Knoers, N., and Renkema, K. Y. (2019). Impact of next generation sequencing on our understanding of CAKUT. Semin. Cell Dev. Biol. 91, 104–110. doi: 10.1016/j.semcdb.2018.08.013
Obeidova, L., Seeman, T., Fencl, F., Blahova, K., Hojny, J., Elisakova, V., et al. (2020). Results of targeted next-generation sequencing in children with cystic kidney diseases often change the clinical diagnosis. PLoS One 15:e0235071. doi: 10.1371/journal.pone.0235071
Otto, E., Hoefele, J., Ruf, R., Mueller, A. M., Hiller, K. S., Wolf, M. T., et al. (2002). A gene mutated in nephronophthisis and retinitis pigmentosa encodes a novel protein, nephroretinin, conserved in evolution. Am. J. Hum. Genet. 71, 1161–1167. doi: 10.1086/344395
Otto, E. A., Schermer, B., Obara, T., O’Toole, J. F., Hiller, K. S., Mueller, A. M., et al. (2003). Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat. Genet. 34, 413–420. doi: 10.1038/ng1217
Rao, J., Liu, X., Mao, J., Tang, X., Shen, Q., Li, G., et al. (2019). Genetic spectrum of renal disease for 1001 Chinese children based on a multicentre registration system. Clin. Genet. 96, 402–410. doi: 10.1111/cge.13606
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Riggs, E. R., Andersen, E. F., Cherry, A. M., Kantarci, S., Kearney, H., Patel, A., et al. (2020). Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 22, 245–257. doi: 10.1038/s41436-019-0686-8
Roberts, S. C. M., Fuentes, L., Kriz, R., Williams, V., and Upadhyay, U. D. (2017). Corrigendum to “Implications for women of Louisiana’s law requiring abortion providers to have hospital admitting privileges” [Contraception (2015) 91:368-372. 10.1016/j.contraception.2015.02.001] Contraception 95, 221–222. doi: 10.1016/j.contraception.2016.08.015
Sanna-Cherchi, S., Caridi, G., Weng, P. L., Scolari, F., Perfumo, F., Gharavi, A. G., et al. (2007). Genetic approaches to human renal agenesis/hypoplasia and dysplasia. Pediatr. Nephrol. 22, 1675–1684. doi: 10.1007/s00467-007-0479-1
Sanna-Cherchi, S., Kiryluk, K., Burgess, K. E., Bodria, M., Sampson, M. G., Hadley, D., et al. (2012). Copy-number disorders are a common cause of congenital kidney malformations. Am. J. Hum. Genet. 91, 987–997. doi: 10.1016/j.ajhg.2012.10.007
Sanna-Cherchi, S., Westland, R., Ghiggeri, G. M., and Gharavi, A. G. (2018). Genetic basis of human congenital anomalies of the kidney and urinary tract. J. Clin. Invest. 128, 4–15. doi: 10.1172/JCI95300
Schwartz, G. J., Brion, L. P., and Spitzer, A. (1987). The use of plasma creatinine concentration for estimating glomerular filtration rate in infants, children, and adolescents. Pediatr. Clin. North Am. 34, 571–590. doi: 10.1016/s0031-3955(16)36251-4
Smith, J. M., Stablein, D. M., Munoz, R., Hebert, D., and McDonald, R. A. (2007). Contributions of the transplant registry: the 2006 annual report of the North American Pediatric renal trials and collaborative studies (NAPRTCS). Pediatr. Transpl. 11, 366–373. doi: 10.1111/j.1399-3046.2007.00704.x
Soliman, N. A., Hildebrandt, F., Otto, E. A., Nabhan, M. M., Allen, S. J., Badr, A. M., et al. (2012). Clinical characterization and NPHP1 mutations in nephronophthisis and associated ciliopathies: a single center experience. Saudi J. Kidney Dis. Transpl. 23, 1090–1098. doi: 10.4103/1319-2442.100968
Tang, X., Liu, C., Liu, X., Chen, J., Fan, X., Liu, J., et al. (2020). Phenotype and genotype spectra of a Chinese cohort with nephronophthisis-related ciliopathy. J. Med. Genet doi: 10.1136/jmedgenet-2020-107184 [Epub ahead of print],
Thomas, R., Sanna-Cherchi, S., Warady, B. A., Furth, S. L., Kaskel, F. J., and Gharavi, A. G. (2011). HNF1B and PAX2 mutations are a common cause of renal hypodysplasia in the CKiD cohort. Pediatr. Nephrol. 26, 897–903. doi: 10.1007/s00467-011-1826-9
Verbitsky, M., Westland, R., Perez, A., Kiryluk, K., Liu, Q., Krithivasan, P., et al. (2019). The copy number variation landscape of congenital anomalies of the kidney and urinary tract. Nat. Genet. 51, 117–127. doi: 10.1038/s41588-018-0281-y
Vester, U., Kranz, B., and Hoyer, P. F. (2010). The diagnostic value of ultrasound in cystic kidney diseases. Pediatr. Nephrol. 25, 231–240. doi: 10.1007/s00467-008-0981-0
Vivante, A., and Hildebrandt, F. (2016). Exploring the genetic basis of early-onset chronic kidney disease. Nat. Rev. Nephrol. 12, 133–146. doi: 10.1038/nrneph.2015.205
Wang, F., Yao, Y., Yang, H. X., Shi, C. Y., Zhang, X. X., Xiao, H. J., et al. (2017a). [Clinical phenotypes of hepatocyte nuclear factor 1 homeobox b-associated disease]. Zhonghua Er Ke Za Zhi 55, 658–662. doi: 10.3760/cma.j.issn.0578-1310.2017.09.006
Wang, F., Zhang, Y., Mao, J., Yu, Z., Yi, Z., Yu, L., et al. (2017b). Spectrum of mutations in Chinese children with steroid-resistant nephrotic syndrome. Pediatr. Nephrol. 32, 1181–1192. doi: 10.1007/s00467-017-3590-y
Weber, S., Moriniere, V., Knuppel, T., Charbit, M., Dusek, J., Ghiggeri, G. M., et al. (2006). Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J. Am. Soc. Nephrol. 17, 2864–2870. doi: 10.1681/ASN.2006030277
Wuhl, E., van Stralen, K. J., Wanner, C., Ariceta, G., Heaf, J. G., Bjerre, A. K., et al. (2014). Renal replacement therapy for rare diseases affecting the kidney: an analysis of the ERA-EDTA Registry. Nephrol. Dial. Transplant 29 Suppl 4, iv1–iv8. doi: 10.1093/ndt/gfu030
Yi, Z., Pan, H., Li, L., Wu, H., Wang, S., Ma, Y., et al. (2016). Chromosome Xq28 duplication encompassing MECP2: clinical and molecular analysis of 16 new patients from 10 families in China. Eur. J. Med. Genet. 59, 347–353. doi: 10.1016/j.ejmg.2016.05.004
Keywords: non-glomerular, chronic kidney disease, renal hypodysplasia, cystic kidney diseases, targeted exome sequencing, genetic diagnosis
Citation: Wang X, Xiao H, Yao Y, Xu K, Liu X, Su B, Zhang H, Guan N, Zhong X, Zhang Y, Ding J and Wang F (2021) Spectrum of Mutations in Pediatric Non-glomerular Chronic Kidney Disease Stages 2–5. Front. Genet. 12:697085. doi: 10.3389/fgene.2021.697085
Received: 18 April 2021; Accepted: 07 June 2021;
Published: 06 July 2021.
Edited by:
Xiu-An Yang, Chengde Medical College, ChinaReviewed by:
Nancy Monroy-Jaramillo, National Institute of Neurology and Neurosurgery, MexicoDesheng Liang, Central South University, China
Copyright © 2021 Wang, Xiao, Yao, Xu, Liu, Su, Zhang, Guan, Zhong, Zhang, Ding and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fang Wang, d2FuZ2ZhbmdwZWRAMTYzLmNvbQ==; Jie Ding, ZGpuY181ODU1QDEyNi5jb20=