 Yangchun Zhang
Yangchun Zhang Ziqi Liu1
Ziqi Liu1 Yongmei Xiao
Yongmei Xiao Wen Chen
Wen Chen Qing Wang
Qing Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 23 July 2021
Sec. Toxicogenomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.696892
This article is part of the Research TopicGene-environment interactions: impact of inorganic and organic toxicants on redox status, DNA stability and epigenetic profileView all 5 articles
Although it is recognized that cadmium (Cd) causes renal tubular dysfunction, the mechanism of Cd-induced nephrotoxicity is not yet fully understood. Mode of action (MOA) is a developing tool for chemical risk assessment. To establish the mechanistic MOA of Cd-induced renal tubular dysfunction, the Comparative Toxicogenomics Database (CTD) was used to obtain genomics data of Cd-induced nephrotoxicity, and Ingenuity® Pathway Analysis (IPA) software was applied for bioinformatics analysis. Based on the perturbed toxicity pathways during the process of Cd-induced nephrotoxicity, we established the MOA of Cd-induced renal tubular dysfunction and assessed its confidence with the tailored Bradford Hill criteria. Bioinformatics analysis showed that oxidative stress, DNA damage, cell cycle arrest, and cell death were the probable key events (KEs). Assessment of the overall MOA of Cd-induced renal tubular dysfunction indicated a moderate confidence, and there are still some evidence gaps to be filled by rational experimental designs.
Cadmium (Cd) is one of the most toxic metals and naturally exists in the environment. However, anthropogenic activities, including agricultural and industrial activities, contribute greatly to the pollution by Cd. The exposure sources in the general population include food, drinking water, smoking, and air. Dietary exposure is the main source in the general population, whereas occupational exposure in individuals working in alloy production, battery production, pigment production and use, plastics production, and smelting and refining mostly involves the respiratory tract (Faroon et al., 2012). Cd can accumulate in the human body due to its long biological half-life (10–30 years) (Kjellström and Nordberg, 1978; Nawrot et al., 2006; Amzal et al., 2009). After being absorbed, Cd is distributed throughout the body via blood circulation and accumulates mainly in the kidneys and liver—the major target organs of Cd toxicity. Epidemiological studies have demonstrated the association of Cd exposure and the increased risk of renal tubular impairment in both occupationally and environmentally exposed populations (Jarup et al., 2000; Akesson et al., 2005; Satarug et al., 2005; Teeyakasem et al., 2007; Wang et al., 2016). In blood, Cd is mostly found in the form of complexes consisting of low-molecular weight protein [mainly metallothionein (MT)]. These complexes readily pass through the glomeruli and are reabsorbed at the brush broader of renal tubules, where they subsequently accumulate and generate toxicity (Fujishiro et al., 2012, 2019). Cd accumulation in renal tubules can lead to tubular dysfunction characterized by Fanconi syndrome, which includes polyuria, proteinuria, and glucosuria (Johri et al., 2010). Although a large number of studies have focused on the mechanism of Cd-induced nephrotoxicity, which includes oxidative stress, mitochondrial dysfunction, genotoxicity, cell cycle arrest, and apoptosis, the data are still fragmented, and a systematic mode of action (MOA) remains to be established and applied to health risk assessment (Fujiwara et al., 2012; Prozialeck and Edwards, 2012; Matović et al., 2015; Song et al., 2016; Wang et al., 2017; Ge et al., 2019). Cd exposure has been recognized as an important public health issue since the beginning of the last century. The World Health Organization (WHO) and other institutes have issued a series of guidelines establishing limits for Cd exposure to protect the population from Cd-caused damage. However, it has been shown that damage, especially kidney impairment, occurs even under a “safe exposure level” (Satarug et al., 2010). Therefore, we need to improve the risk assessment of Cd and establish safer guidelines for exposure limits. MOA is a useful tool recommended by collaborative groups, including the International Programme on Chemical Safety and US Environmental Protection Agency, to identify critical precursor key events (KEs) and set the stage for human health risk assessment (Boobis et al., 2008).
MOA represents biologically plausible causal relationships and series of KEs, including molecular initiating event (MIE), which are essential for the occurrence of an effect [usually an adverse outcome (AO)]. These KEs can be empirically observed steps or their markers. They are essential but not necessarily sufficient in their own right for the occurrence of AO, and can occur on different biological levels, such as molecular level, cellular level, tissue/organ level, and individual level (Meek and Klaunig, 2010; Meek et al., 2014). With the indication of the robust MOA based on mechanistic data, early KEs and lower dose of chemical exposure related to the apical end points, which are traditionally observed in animal tests, can be effectively identified; thus, in vitro studies in human cells for characterizing early toxicity pathway perturbation with the employment of computational biology techniques can be applied for chemical risk assessment with less use of animals and reduced uncertainty in species, thereby conforming to the vision of “Toxicity Testing in the 21st Century” (National Research Council, 2007). To date, MOAs for toxicants, such as benzo[a]pyrene (BaP) and multi-walled carbon nanotubes have been established based on transcriptional genomics and applied to derive the points of departure (PODs) (Moffat et al., 2015; Labib et al., 2016), providing a new cost-effective paradigm for chemical risk assessment. However, compared with the construction of a MOA framework, the assessment of its confidence plays an equally important role and is pivotal to determine whether the putative MOA is robust enough to be further applied in chemical risk assessment.
Therefore, to provide a theoretical basis for Cd risk assessment, we constructed a putative MOA for Cd-induced renal tubular dysfunction based on bioinformatics analysis of genomics data from the Comparative Toxicogenomics Database (CTD). Weight of evidence analysis of the MOA revealed the potential data gaps in the present evidence, and further studies are needed to fill them.
Comparative Toxicogenomics Database (CTD)1 is a public database containing the association information between chemical exposure, gene products, and diseases. It is manually organized by professional biocurators to promote the understanding of the effects of environmental chemicals on human health (Davis et al., 2011). The data of toxicogenomics, chemicals, diseases, and their interaction can help researchers to generate hypotheses about the molecular mechanisms of chemical-induced adverse health outcomes in support of experimental evidence (Davis et al., 2019). Therefore, CTD was employed to define the set of genes whose expression is influenced by Cd in the kidneys. The word “cadmium” was input into the chemical search box of CTD as a search term, and a total of 960 references were retrieved on July 14, 2020. Then, we manually screened the title or abstract of each reference. We excluded 149 references (review articles, studies irrelevant to Cd toxicity, assay development, in vitro enzyme activity tests, characterization of protein structure or function, and exposure characterization). The remaining references were then categorized according to Cd-affected organs/tissues. In addition, due to the lack of kidney specificity, we excluded epidemiological studies where human blood or urine samples had been used. The final set included 107 references (see Supplementary Sheet 1) related to Cd-induced nephrotoxicity and 869 genes whose expression or protein activity was increased/decreased/affected by Cd under the function model of chemical–gene interaction within each reference. The strategies of reference exclusion and differentially expressed gene selection are shown in Figure 1.
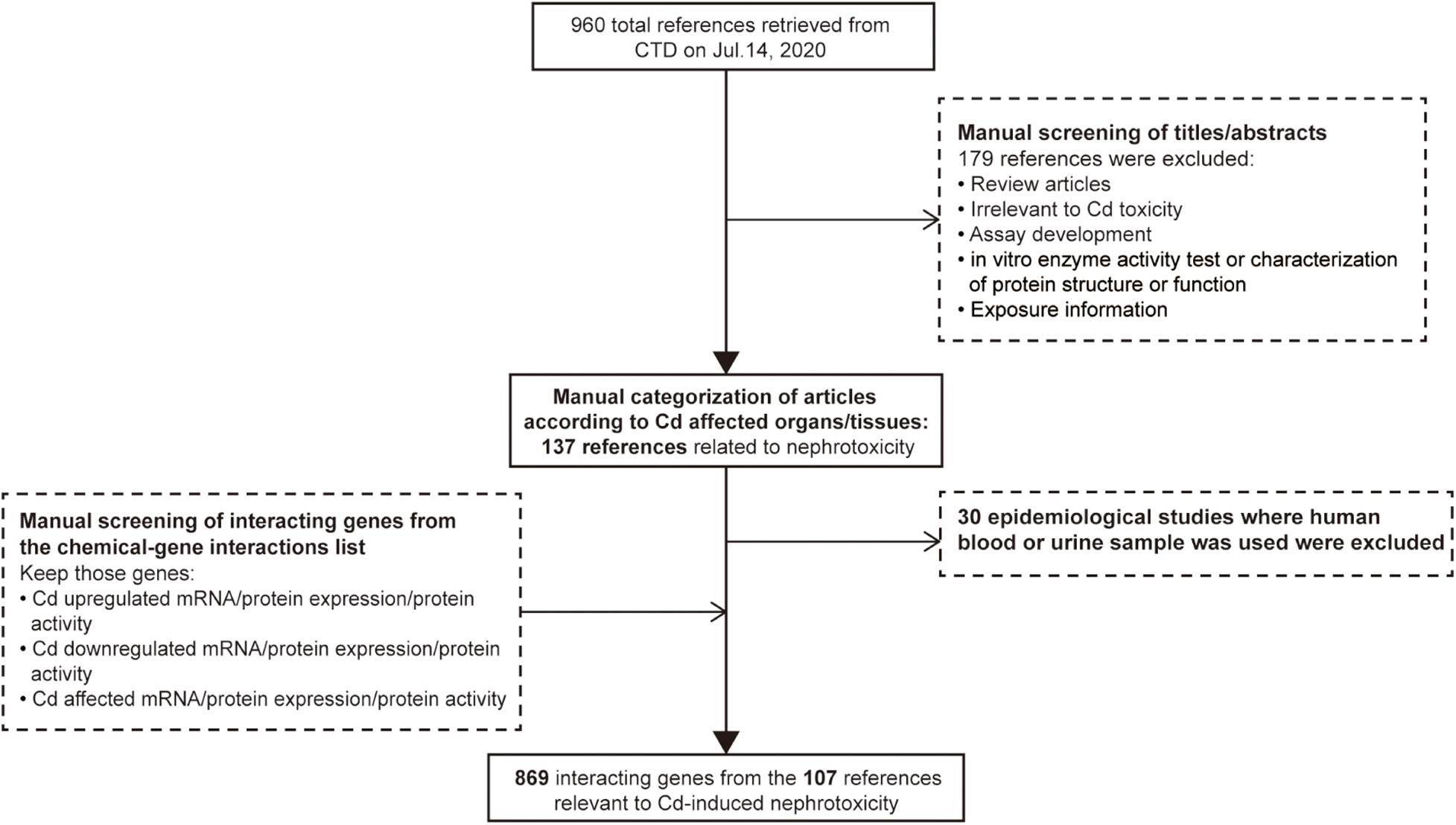
Figure 1. Workflow for selecting differentially expressed genes associated with cadmium-induced nephrotoxicity.
Ingenuity® Pathway Analysis (IPA) software (QIAGEN Inc.)2 was used to analyze the biological effects during Cd-induced nephrotoxicity by canonical pathway analysis and biofunction and disease analysis. Genes identified to be differentially expressed from CTD were input into IPA. Since CTD does not provide the detailed expression values or fold change values of the corresponding genes, we manually assigned the fold change value for those genes for the analysis in IPA. For transforming the qualitative expression alteration into a quantitative fold change value, we manually assigned the expression level of genes to 2, −2, or 0 based on the overall change in all of the included references. Genes with consistent change on either expression or protein activity (upregulated/downregulated/affected) in all the concerning references were assigned 2, −2, or 0, while the assignment of genes with contradictory expression change depended on the majority of cases. For example, SQSTM1 was involved in five references; in three of them, SQSTM1 was upregulated, while in two references, it was downregulated. Therefore, the expression level of SQSTM1 was assigned to 2. The 869 genes relevant to Cd-induced nephrotoxicity from 107 references in CTD were summarized in Supplementary Sheet 3. When setting parameters, we chose “human” as the specific species and the kidney as the target organ. We chose p < 0.05 as the threshold for significant perturbed pathways and excluded those that lacked z-score used to predict whether the pathway was activated or inhibited and the corresponding degree. The top 10 enriched pathways were selected to recognize the main effects relevant to the perturbed pathways.
Mode of action (MOA) of a specific chemical consists of a series of events that are crucial to the occurrence of the AO; these KEs are measurable and sequential in chronological order (Meek and Klaunig, 2010). We recognized the biological effects of the top 10 significantly perturbed pathways as KEs. Their order was recognized according to the biological plausibility.
To initially confirm the reliability of the postulated MOA, the evolved Bradford Hill criteria were used to assess the evidence of the MOA, especially the causal relationship between the upstream KEs and downstream KEs. Five main aspects were considered in the evolved Bradford Hill criteria: biological plausibility, essentiality, empirical observation, consistency, and analogy. In the evaluation of essentiality, the focus was on the KEs, whereas in the evaluation of biological plausibility, empirical support, consistency, and analogy, the emphasis was on key event relationships (KERs). Biological plausibility required that the mechanistic relationships between KEs were consistent with the established biological knowledge. Essentiality was defined when the downstream KE would be prevented if the upstream KE was blocked. Empirical support included considerations about evidence for both dose–response and temporal concordance of KERs. Consistency referred to the consistency of empirical support for KERs across different species/strains/organs/test systems. Analogy support required that dependent alteration of KEs was observed after the exposure to the structurally related chemicals. Here, we put stress on the evaluation of KEs’ essentiality and empirical support for KERs because they have been weighted up to 80% in the assessment of MOA (Becker et al., 2017). The detailed defining questions and the relative confidence levels of evidence for the assessment of MOA were described in the published articles (Becker et al., 2015, 2017; OECD, 2018).
To identify the crucial toxicity pathways in Cd-induced nephrotoxicity, we entered the differentially expressed genes selected from CTD into IPA software to generate perturbed pathways. The summary of pathway perturbation and the corresponding enriched genes is presented in Supplementary Sheet 2. The top 10 toxicity pathways with the smallest p-value were as follows: nuclear factor erythroid 2-related factor 2 (Nrf2)-mediated oxidative stress response, aryl hydrocarbon receptor signaling, endoplasmic reticulum (ER) stress pathway, hypoxia signaling in the cardiovascular system, ferroptosis signaling pathway, unfolded protein response, mitotic roles of polo-like kinase, hypoxia-inducible factor-1-alpha (HIF1α) signaling, apoptosis signaling, and cyclins and cell cycle regulation (Figure 2A and Supplementary Figures 1–10). The positive value of z-score indicated the predicted activation of the pathway, while the negative value represented the predicted inhibition of the pathway, and the absolute value represented the degree of activation/inhibition. Hypoxia signaling in the cardiovascular system pathway, whose z score was 0, was neither activated nor inhibited (Figure 2B). Oxidative stress, DNA damage repair, cell cycle arrest, and apoptosis were the main biological effects triggered in the remaining nine pathways, which were either activated or inhibited (Table 1). The diseases and biofunctions analysis (Figure 2C) showed that cell death, especially apoptosis and necrosis, may be pivotal to the Cd-induced nephrotoxicity. This is concordant with the results from animal and cell experiments that showed that Cd could induce tubular necrosis in vivo and cell death in vitro (Fujiki et al., 2019; Hamidian et al., 2020).

Figure 2. Crucial perturbed pathways, diseases, and biological functions involved in cadmium (Cd)-induced nephrotoxicity. A total of 869 genes (selection flow is shown in Figure 1) were entered into Ingenuity® Pathway Analysis (IPA) together with the manually assigned fold change values to conduct core analysis. (A) Top 10 toxicity pathways involved in Cd-induced nephrotoxicity from canonical pathway analysis in IPA. Under the threshold p < 0.05 and rejecting the pathways without a z-score, we obtained the top 10 pathways in order of p-values from minimum to maximum. The red bars refer to the percentages of upregulated genes enriched in the corresponding pathways, the green bars refer to the percentage of downregulated genes enriched in the corresponding pathways, and the gray bars refer to the percentage of upregulated/downregulated genes enriched in the corresponding pathways. The overlap percentages were calculated as × 100%. (B) The activation z-score of the top 10 perturbed pathways. (C) Top 10 diseases and biological functions related to Cd from the module diseases and functions in IPA. After excluding the diseases and biological function terms without a predicted activation z-score, we ranked the diseases and biological functions in order of p-values. −log (p-value) and corresponding z-score of each entry are presented.
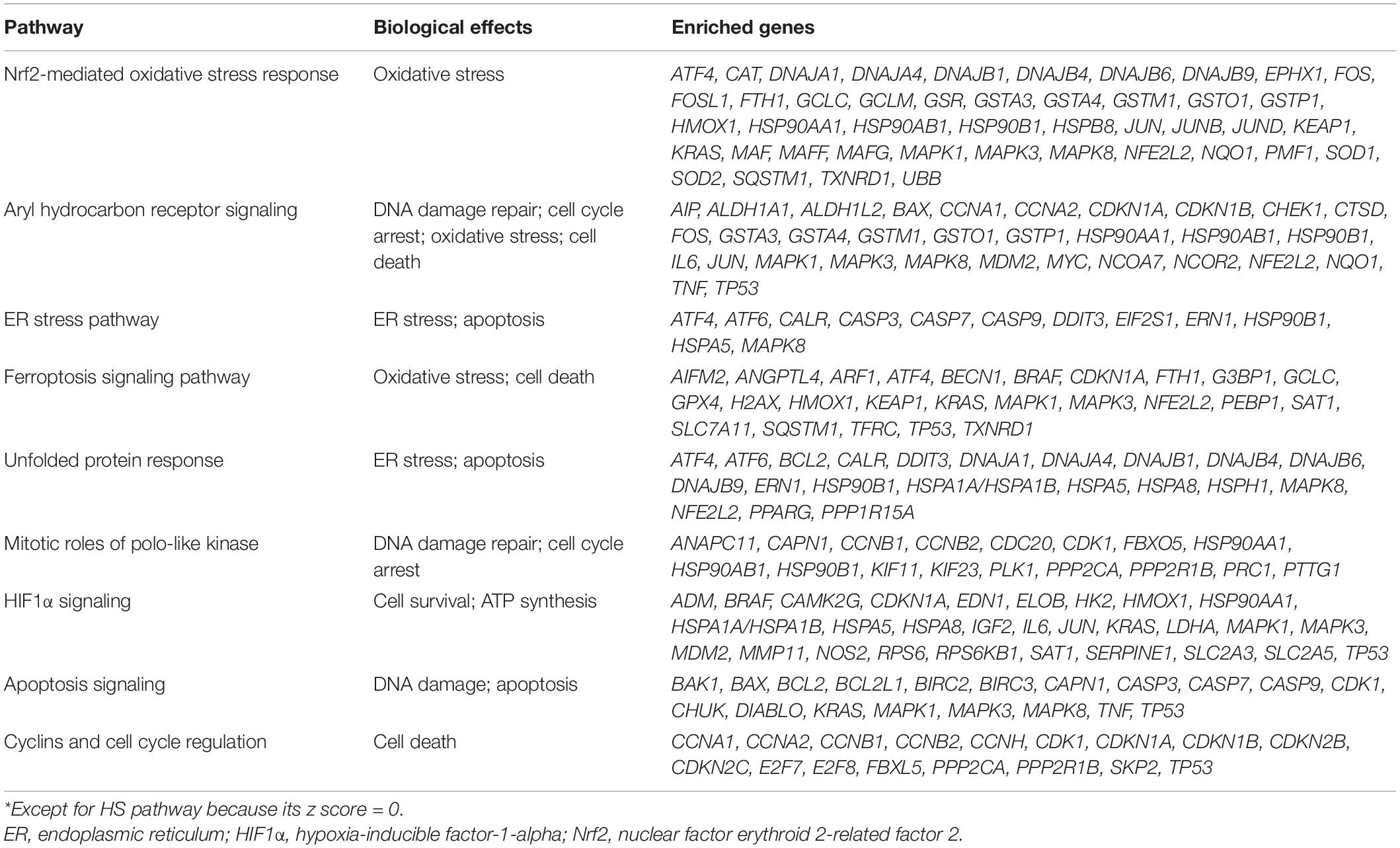
Table 1. Biological effects triggered by the top 10 pathways*.
In this study, we used core analysis of IPA software through which we were able to simultaneously obtain the information of pathway enrichment, molecular interactions, and upstream prediction generated from the CTD gene set. CTD itself can also analyze pathway enrichment based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) and REACTOME databases; thus, we also analyzed pathway enrichment by CTD with the support of KEGG. The results (Supplementary Figure 11) were similar to those from IPA, showing that apoptosis, cell cycle, and ER stress-related pathways were significantly perturbed.
In addition to the bioinformatics analysis, we also retrieved review articles to understand the established cytotoxicity mechanisms of Cd. It has been shown that Cd could cause oxidative stress not by directly increasing free radicals due to its non-redox nature but by indirectly generating reactive oxygen species (ROS) by interaction with Fenton metal, such as Fe and affecting the antioxidative defense system (Matović et al., 2015). Cd is not directly genotoxic because it lacks the capacity to generate DNA adducts, but it could induce indirect DNA damage by the increased ROS levels (Valverde et al., 2001). To avoid the wrong DNA replication entering into the next cell generation, the unrepaired DNA damage subsequently blocks cell cycle progression and initiates cell death (Takahashi et al., 2019). Thus, according to the main biological effects of top significant pathways and the biological relationship among them, key cellular events and the chronological order among them were recognized. Oxidative stress was shown to be the KE1, followed by DNA damage (KE2), cell cycle arrest (KE3), and cell death (KE4). The order is biologically plausible.
Kidneys are the recognized target organs of chronic Cd exposure, especially the renal proximal tubules. Renal tubular dysfunction is an early clinical feature of renal damage caused by Cd, during which proteinuria (mainly low-molecular weight proteins, such as β2-microglobulin), glycosuria, amino aciduria, and polyuria have been observed in both humans and laboratory animals (Faroon et al., 2012). Furthermore, epidemiological studies have shown evidence of association between renal damage and Cd exposure (Akesson et al., 2005; Satarug et al., 2010). To date, researchers have explored various mechanisms of Cd-induced nephrotoxicity, but the MOA of Cd-induced renal tubular dysfunction has not been established yet. After integration of bioinformatics analysis and biological plausibility, we constructed the MOA of Cd-induced renal tubular dysfunction (Figure 3).
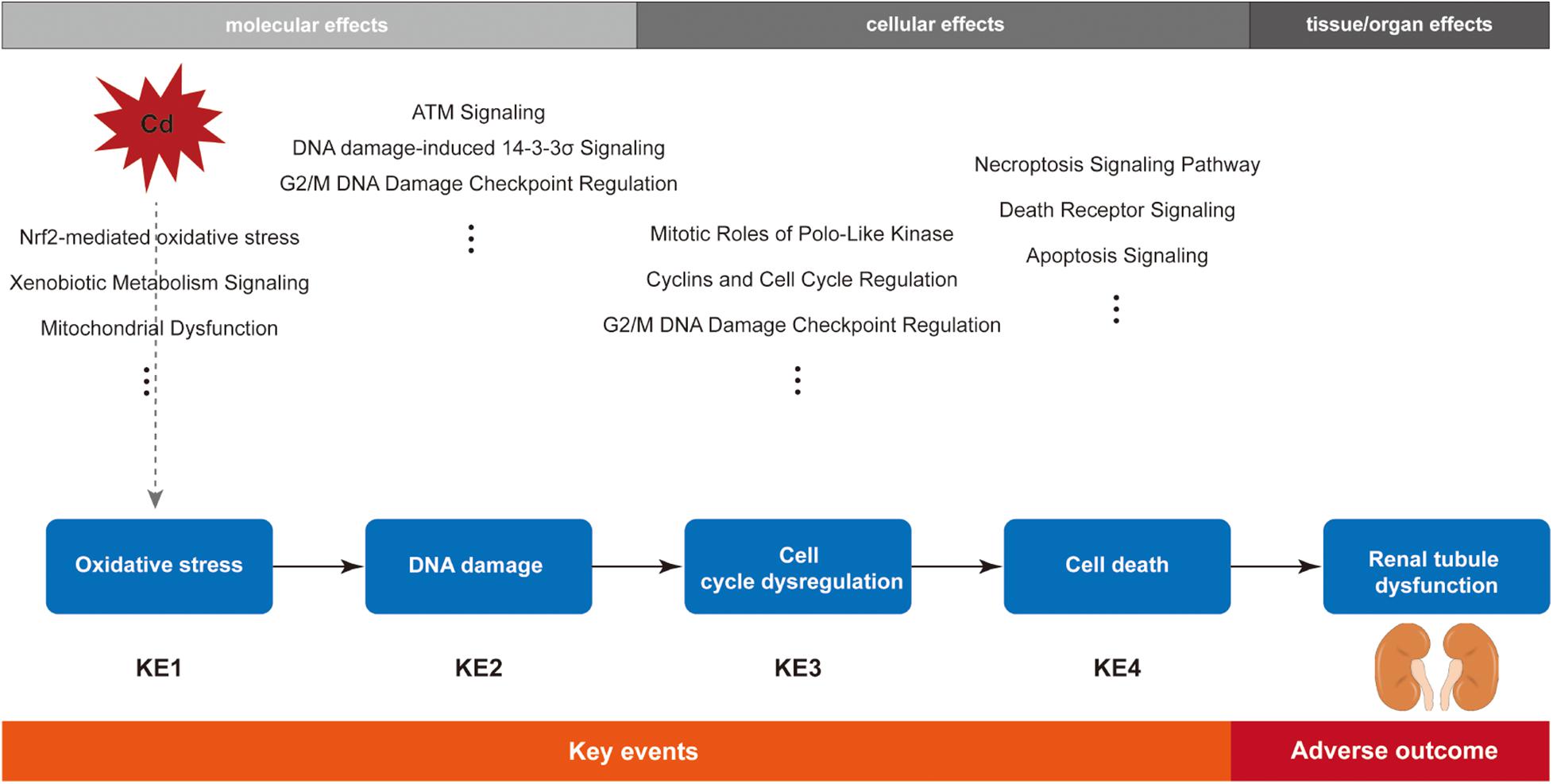
Figure 3. Schematic diagram of the postulated mode of action (MOA) for cadmium-induced renal tubular dysfunction.
In the results of canonical pathway analysis, in addition to the NRF2 pathway, which was 1 of the top 10 significant pathways, there are other pathways associated with oxidative stress, such as xenobiotic metabolism signaling pathway and mitochondrial dysfunction pathway (Supplementary Table 1), which may be involved in Cd-induced oxidative stress in the kidney.
Oxidative stress has been widely demonstrated to be a pivotal molecular mechanism in nephrotoxicity caused by acute or chronic Cd exposure (Renugadevi and Prabu, 2009; Wang et al., 2009; Nemmiche, 2017). Both in vivo and in vitro studies have provided plenty of evidence of oxidative stress playing an important role in Cd-induced nephrotoxicity, especially in tubular damage. Cd has been shown to enhance oxidative stress by triggering ROS production and inhibiting the antioxidant system in renal tubular epithelial cells; such oxidative damage and nephrotoxicity could be alleviated by various substances with antioxidant activity (Ognjanović et al., 2008; Wang et al., 2009, 2018, 2019; Nazima et al., 2015; Poontawee et al., 2016; Das et al., 2019; Joardar et al., 2019; Aqeel et al., 2020). Cd is a non-redox metal; thus, it is not able to directly produce free radicals, but it indirectly increases the generation of ROS and causes oxidative stress damage (Liu et al., 2008). Although Cd cannot directly generate free radicals, it can replace the iron component in some proteins, including ferritin and apoferritin, increasing free Fe2+ and ⋅OH generated from Fe-catalyzed Fenton action (Dewanjee et al., 2013; Matović et al., 2015). It has also been demonstrated that Cd could consume the antioxidant glutathione (GSH) and inhibit the activity of enzymes pivotal for maintaining GSH due to the high affinity between Cd2+ and -SH, such as glutathione reductase, glutathione peroxidase, and glutathione-S-transferase. Moreover, another potential mechanism by which Cd may decrease the activity of antioxidant enzymes is an interaction with divalent elements, such as manganese, zinc, and selenium; these elements function as cofactors and form the core active sites of antioxidant enzymes (e.g., superoxide dismutase, catalase, and glutathione reductase) (Cuypers et al., 2010; Matović et al., 2015). After chronic exposure to low-dose Cd, nephrotoxicity occurs with increased renal ROS level and decreased activity of antioxidant enzymes (Wang et al., 2013; Ansari et al., 2017; Liu et al., 2019). However, oxidative stress is a dynamic equilibrium process; there is an adaptive system to eliminate ROS, and oxidative damage would appear only when ROS was overloaded. Thus, we hypothesize that oxidative stress is an early event in Cd-induced nephrotoxicity, and it can be counterbalanced at least partly by the corresponding defense system.
As shown in the result of canonical pathway (Supplementary Table 1), ATM signaling, role of BRCA1 in DNA damage response, DNA damage-induced 14-3-3σ signaling, and cell cycle: G2/M DNA damage checkpoint regulation are potential pathways involved in the DNA damage and DNA damage repair induced by Cd other than the pathways in the top 10 significant pathways.
DNA is a common target of free radicals generated from oxidative stress induced by exogenous chemicals, including Cd. Cd probably induces DNA damage not by direct interaction but rather indirectly through oxidative stress. Cd2+ alone did not lead to DNA damage in plasmid DNA; however, it was able to cause DNA strand breaks in the presence of hydrogen peroxide in a dose-dependent manner due to the production of hydroxyl radicals (Badisa et al., 2007). Likewise, Valverde et al. (2001) reported that no direct induction of DNA damage by Cd was detected, and the genotoxicity probably involved an indirect interaction due to oxidative stress. The oxidative stress-associated DNA damage was found in both in vitro renal tubule cells and kidney tissues from in vivo experiments under Cd treatment (Saplakoğlu et al., 1997; Chater et al., 2008). Induction of DNA damage subsequently activates the DNA repair system to prevent the wrong DNA replication from entering the next cell generation. Cell cycle checkpoints and cell cycle arrest ensure adequate time for the DNA repair process (Nyberg et al., 2002). The cell fate depends on whether the DNA damage is repaired. If the DNA damage is successfully repaired, the cell cycle progression continues. If the repair fails, cell death is initiated (Harper and Elledge, 2007; Ciccia and Elledge, 2010).
The results of pathway analysis (Supplementary Table 1) showed that the significant pathways associated with cell cycle progression had negative z scores, while pathways associated with cell cycle arrest had positive z scores, indicating that the cell cycle may be arrested by Cd in the kidney. Pathways, such as ATM signaling and cell cycle: G2/M DNA damage checkpoint regulation were predicted to be activated (z score > 2), which indicated that these pathways may play critical roles in Cd-induced cell cycle dysregulation in the kidney.
Bork et al. (2010) found that the DNA damage caused by Cd could trigger the G2/M cell cycle arrest in kidney proximal tubule cells. Luo et al. (2020) reported that different concentrations of Cd led to different cell fates, depending on the extent of DNA damage in NRK-52E cells. At a low concentration (2.5 μM), Cd induced G0/G1 cell cycle arrest, and eventually the cells survived from slight damage; in contrast, 10 μM Cd caused severe DNA damage, leading to S phase cell cycle arrest and ultimately to cell death. These findings suggested that Cd could inhibit DNA repair and lead to cell death.
Previous studies showed that Cd could induce renal tubular cell death via oxidative stress and unrepaired DNA damage (Suliman Al-Gebaly, 2017; Zhuang et al., 2019). Different forms of cell death induced by Cd have been reported. Most studies have revealed that cell apoptosis and necrosis are the main forms of Cd-induced cell death, and a few studies indicated that pyroptosis might also be one of the forms of Cd-induced cell death (Fujiwara et al., 2012; Chou et al., 2019; Fujiki et al., 2019). It has been recognized that renal tubule necrosis occurs when animals are exposed to a high dose of Cd for a long time. However, before necrosis, renal tubular dysfunction with cell apoptosis has been observed (Hamada et al., 1991; Tanimoto et al., 1993; Yan et al., 1997). Therefore, cell apoptosis, rather than necrosis, is more likely to be the crucial event in the early stage of renal tubular dysfunction before obvious kidney injury. The following three pathways have been suggested to be the probable apoptotic pathways: ER stress mediated by unfolded protein reaction, caspase-dependent/independent pathway, and p53 signaling pathway (Fujiwara et al., 2012). These suggestions are consistent with the results of our bioinformatics analysis that showed that the ER stress-related pathways (ER stress pathway and unfolded protein in response pathway) were significantly activated and the apoptotic genes, such as CASPs, BAX/BCL2, and TP53, were involved in apoptosis-associated pathways. In addition, significant pathways, such as apoptosis signaling pathway, death receptor signaling, and MYC-mediated apoptosis signaling were predicted to be activated (z score > 2) (Supplementary Table 1).
Cd is a recognized toxicant that can generate toxicity in renal tubules and cause renal injury. In vivo studies have demonstrated that chronic Cd exposure leads to renal tubular dysfunction characterized as low-molecular weight proteinuria and histopathological changes of renal tubules (e.g., tubular degeneration, brush border atrophy, and epithelial cell fragmentation release into lumen) (Brzóska et al., 2003; Asar et al., 2004; Wu et al., 2012). Renal proximal tubule is the site where Cd tends to accumulate, and its dysfunction coincides with cell apoptosis prior to obvious tubular necrosis after Cd intoxication (Friberg, 1984; Prozialeck and Edwards, 2012). Therefore, dysfunction of reabsorption seems to be the early stage of Cd-induced renal tubular injury. However, in most experiments, renal tubular epithelial cell apoptosis and renal tubular dysfunction have been observed simultaneously; that is, there is a lack of evidence about dose/time-dependent response to explain the causal relationship between cell apoptosis and renal tubular dysfunction. In addition to in vivo experiments, epidemiological studies also showed associations between Cd exposure and renal tubular dysfunction among populations (Akesson et al., 2005; Satarug et al., 2010).
According to the guidance document released by the Organization for Economic Co-operation and Development (OECD, 2018), assessment of the essentiality of all KEs and overall assessment for KERs with tailored Bradford Hill criteria are required for the overall assessment of MOA. Herein, we focused on assessing essentiality of each KE and the empirical support for KERs using the defining questions described in previous studies (Becker et al., 2015, 2017; OECD, 2018). The latter included concordance in dose–response, temporal, and incidence relationships between KEs.
To explore the confidence of supporting data for the essentiality of KEs in the MOA, we assembled evidence and summarized it in Table 2.
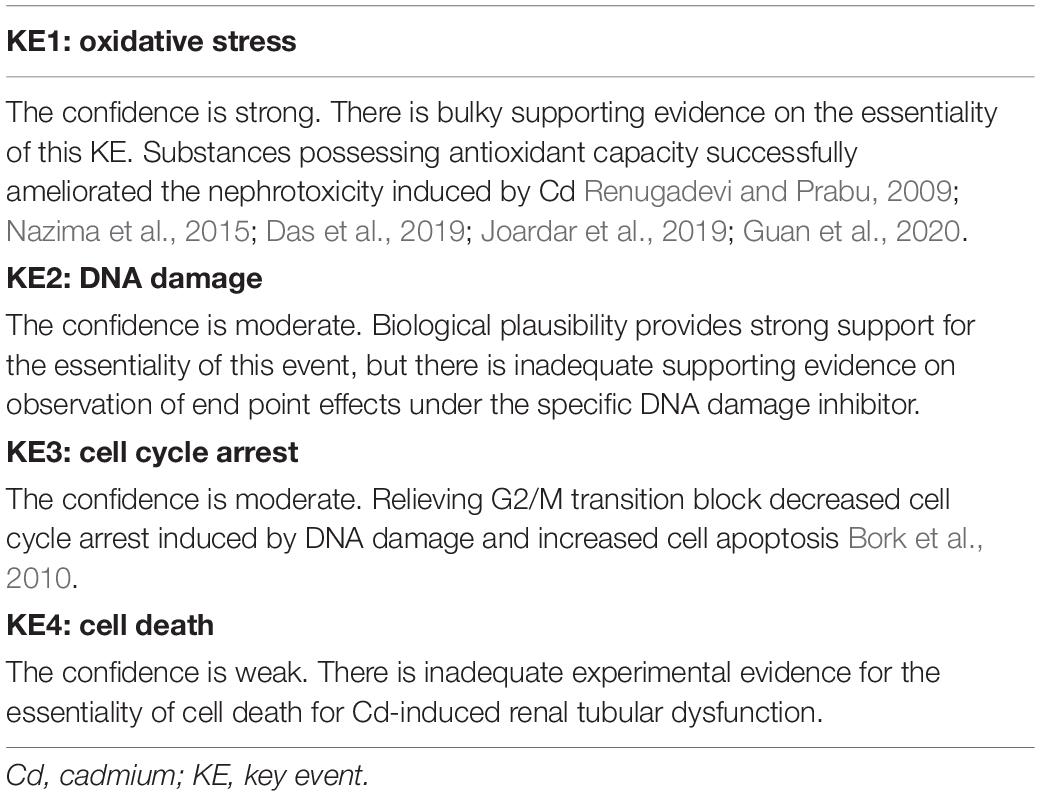
Table 2. Essentiality of KEs and the responding confidence.
Since MOA consists of a series of sequential, measurable KEs essential to AO, in addition to the assessment of essentiality of each KE, evidence concordance of all KERs is also required. Thus, we assessed the evidence concordance including the dose–response and the temporal relationship between KEs.
The existing knowledge has provided biological plausibility of KERs in the MOA that we constructed. ROS indirectly produced by Cd2+ could attack DNA and induce DNA damage by breaking DNA strands. DNA damage accumulation challenges DNA repair and subsequently blocks cell cycle progression and initiates cell death to avoid wrong replication entering into divided cells (Ciccia and Elledge, 2010). However, evidence supporting the concordance of KERs from either dose–response or temporal consistency is not as strong as biological plausibility. In most studies, these KEs were first observed at the same concentrations or time points as shown in Table 3. This may be attributed to the relatively high concentration of Cd, at which the tested cells or animals would undergo more severe damage and activate both the early and the late KEs. In addition, inability to select more intervals for the analysis of time-dependent effects increases the difficulty to figure out the temporal sequence of KEs. Noticeably, a study (Ge et al., 2018) of Cd-induced cytotoxicity on HK-2 cells under a low concentration of 1 μM showed that during the 12-day exposure period, oxidative stress (KE1) was first observed on day 2, while the early cell apoptosis (KE3) was significantly increased from day 10. The results indeed supported the temporal relationship of KE1 and KE3; however, the significant increase of ROS only occurred on days 2 and 4, suggesting that it is not the transient activation of oxidative stress, but rather another oxidative stress-related mechanism, that directly leads to apoptosis. Unfortunately, only a few studies showed obvious dose–response and temporal concordance between either adjacent or nonadjacent KEs (Fasanya-Odewumi et al., 1998; Nemmiche and Guiraud, 2016; Ge et al., 2018), suggesting that the supporting evidence is not strong and further verification needs to be designed.
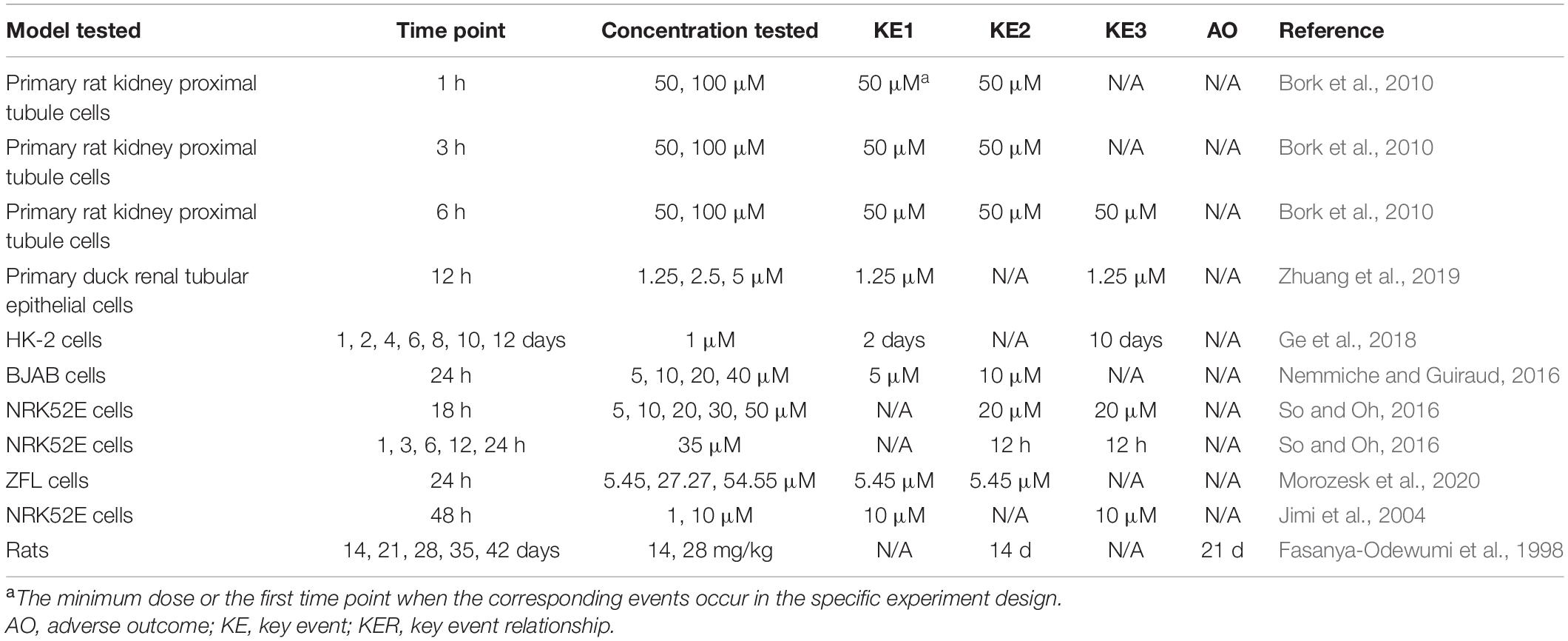
Table 3. Dose–response and temporal concordance of KERs.
In the present study, we used the toxicogenomics data from CTD to understand the perturbed pathways involved in Cd-induced nephrotoxicity, and we initially constructed the MOA of Cd-induced renal tubular dysfunction. Indeed, the genomics alteration induced by Cd treatment provided us with the bioinformatics indication of key cellular events, but the genomics data from CTD were not able to identify the dose–response relationship and temporal sequence between those KEs. Despite such flaws, this method could make use of the genomics information derived from the references collected by the frequently updated CTD, which would save us a lot of time on literature retrieval and review and reduce the reference omitting due to manual retrieval.
In the postulated MOA of Cd-induced renal tubular dysfunction, we could not recognize the MIE based on the bioinformatics analysis. Cd has a high affinity for the -SH-rich protein MT, which can respond to Cd stimulation. However, free Cd2+ instead of Cd–MT is the toxic form of Cd (Liu et al., 1998, 2000). Most Cd is absorbed at renal tubular epithelium in the form of Cd–MT complex by endocytosis, and the acidic environment of endosome or lysosome promotes the dissociation of Cd2+ from MT (Sabolić et al., 2010). Increased dissociation of Cd2+ from Cd–MT could be the MIE of Cd-induced renal tubular dysfunction. However, the MIE has not yet been experimentally verified, which requires detecting methods to distinguish the intracellular free Cd2+ from combined Cd. Methods, such as atomic absorption spectrometry (AAS) and inductively coupled plasma mass spectrometry (ICP-MS) have widely been used in the determination of Cd content in biological samples, but they failed to differentiate the intracellular ionic Cd (Jablonska et al., 2017; Wu et al., 2019). In vitro models need to be developed with the aim to measure the transition of Cd from the combined form to free ion and explore how the free Cd2+ affects the antioxidant system to trigger the downstream KEs.
To generate the postulated MOA of Cd-induced renal tubular dysfunction, we used a strategy of data mining from CTD to perform the pathway analysis with those genes whose mRNA and protein expression levels were affected by Cd. Actually, we also analyzed the perturbed pathways involved in Cd toxicity in liver and breast (see Supplementary Table 2) with the strategies similar to those depicted in the preceding text. In our MOA framework, KEs were not specific for nephrotoxicity. Oxidative stress, one of the reported mechanisms of Cd toxicity, was also involved in hepatotoxicity caused by Cd due to significant perturbation of Nrf2 signaling pathway. Similarly, cell cycle arrest-related pathway was activated by Cd in the breast, and apoptosis signaling was activated by Cd in the liver and breast. Thus, MIE is probably what determines the specificity of the MOA for Cd-induced nephrotoxicity. Regretfully, in our research, we failed to recognize the MIE for Cd-induced nephrotoxicity due to the lack of specific dose–effect and time–effect information from the CTD dataset to help determine the earliest event and no suggestive clue on structural information to predict the direct interaction between Cd and the potential initiating molecule.
We used the Hill Bradford criteria to assess the weight of evidence of the postulated MOA. The biological plausibility supporting the MOA was strong; however, for the essentiality and empirical support, the confidence was weaker. The essentiality of KE1 was well-supported by evidence considering that blockages of this event successfully reversed the downstream KEs. However, the essentiality of later KEs was insufficient, as there were data gaps for direct evidence (e.g., inhibitors of DNA damage/promoters for DNA repair block cell death induced by Cd in kidney tissue or in vitro renal tubular cells). Furthermore, not all KERs were well-supported by empirical evidence; indeed, in most cases, several KEs were observed simultaneously, especially the adjacent KEs. Thus, to increase the confidence of each KE, in vivo and in vitro studies involved in Cd-induced renal tubular dysfunction should set lower Cd concentrations and smaller time intervals to identify the dose–response and temporal sequence between each KE. Benchmark dose (BMD) analysis could help to verify the dose–response or temporal sequences of KEs.
In addition, in view of the weaker confidence for KE3 and KE4, we considered some alternative pathways in the MOA of Cd-induced kidney damage. ER stress, a probable mechanism of Cd toxicity, was also shown to be a potential KE based on the toxicity pathway analysis by IPA. In the top 10 perturbed pathways, ER stress pathway and unfolded protein response pathway were significantly enriched and were predicted to be activated. Furthermore, previous studies have reported the potential role of ER stress in Cd-induced nephrotoxicity. It has been reported that ER stress can regulate the apoptosis induced by Cd exposure (Ge et al., 2018; Li et al., 2020). However, the relationships between ER stress and the KEs (KE1, KE2, and KE3) in our postulated MOA were not clear. Thus, it is interesting to explore the crosstalk between the potential KEs and construct a comprehensive MOA.
MOA was recognized as a useful tool for chemical risk assessment. The use of quantitative transcriptomic data to determine BMDs based on a chemical’s MOA was demonstrated to be comparable to those derived from adverse apical effects (Hester et al., 2015; Webster et al., 2015; Labib et al., 2016). The KEs in the MOA could be alternative toxic end points for the traditional apical end points used in risk assessment, and more sensitive PODs may be obtained in this way. In addition, the pathway-oriented KE will help us to develop new strategies in future chemical toxicity tests. For example, key molecules in pathways associated with KEs can serve as biomarkers or new indications for toxicity assays. Thus, the MOA we constructed provides a new insight for Cd risk assessment.
In summary, based on the pathway-oriented approach, here we constructed the MOA framework for Cd-induced renal tubular dysfunction. Oxidative stress response was the KE1, followed by DNA damage, cell cycle arrest, and cell death as the later KEs, and eventually renal tubular dysfunction was the AO. Weight of evidence analysis for our MOA suggested that data gaps still exist, especially in terms of MIE. The MOA has to be verified in further studies before it can be used for chemical risk assessment.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
YZ wrote the main manuscript text, contributed to data collection and bioinformatics analysis, and prepared the figures. QW proposed and organized the study, contributed to data interpretation, and revised the manuscript. YJ, DY, YX, and WC contributed to the study design. ZL, QH, and FW contributed to the data analysis. All authors reviewed the manuscript.
This study was supported and funded by the National Key R&D Program of China (SQ2019YFC1604603 and 2017YFC1600201) and the National Natural Science Foundation of China (Grant No. 82073583).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.696892/full#supplementary-material
Akesson, A., Lundh, T., Vahter, M., Bjellerup, P., Lidfeldt, J., Nerbrand, C., et al. (2005). Tubular and glomerular kidney effects in Swedish women with low environmental cadmium exposure. Environ. Health Perspect. 113, 1627–1631. doi: 10.1289/ehp.8033
Amzal, B., Julin, B., Vahter, M., Wolk, A., Johanson, G., and Akesson, A. (2009). Population toxicokinetic modeling of cadmium for health risk assessment. Environ. Health Perspect. 117, 1293–1301. doi: 10.1289/ehp.0800317
Ansari, M. A., Raish, M., Ahmad, A., Alkharfy, K. M., Ahmad, S. F., Attia, S. M., et al. (2017). Sinapic acid ameliorate cadmium-induced nephrotoxicity: In vivo possible involvement of oxidative stress, apoptosis, and inflammation via NF-κB downregulation. Environ. Toxicol. Pharmacol. 51, 100–107. doi: 10.1016/j.etap.2017.02.014
Aqeel, T., Gurumallu, S. C., Bhaskar, A., Hashimi, S. M., and Javaraiah, R. (2020). Secoisolariciresinol diglucoside protects against cadmium-induced oxidative stress-mediated renal toxicity in rats. J. Trace Elem. Med. Biol. 61:126552. doi: 10.1016/j.jtemb.2020.126552
Asar, M., Kayisli, U. A., Izgüt-Uysal, V. N., and Akkoyunlu, G. (2004). Immunohistochemical and ultrastructural changes in the renal cortex of cadmium-treated rats. Biol. Trace Elem. Res. 97, 249–263.
Badisa, V. L., Latinwo, L. M., Odewumi, C. O., Ikediobi, C. O., Badisa, R. B., Ayuk-Takem, L. T., et al. (2007). Mechanism of DNA damage by cadmium and interplay of antioxidant enzymes and agents. Environ. Toxicol. 22, 144–151. doi: 10.1002/tox.20248
Becker, R. A., Ankley, G. T., Edwards, S. W., Kennedy, S. W., Linkov, I., Meek, B., et al. (2015). Increasing scientific confidence in adverse outcome pathways: application of tailored bradford-hill considerations for evaluating weight of evidence. Regul. Toxicol. Pharmacol. 72, 514–537. doi: 10.1016/j.yrtph.2015.04.004
Becker, R. A., Dellarco, V., Seed, J., Kronenberg, J. M., Meek, B., Foreman, J., et al. (2017). Quantitative weight of evidence to assess confidence in potential modes of action. Regul. Toxicol. Pharmacol. 86, 205–220. doi: 10.1016/j.yrtph.2017.02.017
Boobis, A. R., Doe, J. E., Heinrich-Hirsch, B., Meek, M. E., Munn, S., Ruchirawat, M., et al. (2008). IPCS framework for analyzing the relevance of a noncancer mode of action for humans. Crit. Rev. Toxicol. 38, 87–96. doi: 10.1080/10408440701749421
Bork, U., Lee, W. K., Kuchler, A., Dittmar, T., and Thévenod, F. (2010). Cadmium-induced DNA damage triggers G(2)/M arrest via chk1/2 and cdc2 in p53-deficient kidney proximal tubule cells. Am. J. Physiol. Renal. Physiol. 298, F255–F265. doi: 10.1152/ajprenal.00273.2009
Brzóska, M. M., Kamiński, M., Supernak-Bobko, D., Zwierz, K., and Moniuszko-Jakoniuk, J. (2003). Changes in the structure and function of the kidney of rats chronically exposed to cadmium. I. Biochemical and histopathological studies. Arch. Toxicol. 77, 344–352. doi: 10.1007/s00204-003-0451-1
Chater, S., Douki, T., Garrel, C., Favier, A., Sakly, M., and Abdelmelek, H. (2008). Cadmium-induced oxidative stress and DNA damage in kidney of pregnant female rats. C. R. Biol. 331, 426–432. doi: 10.1016/j.crvi.2008.03.009
Chou, X., Ding, F., Zhang, X., Ding, X., Gao, H., and Wu, Q. (2019). Sirtuin-1 ameliorates cadmium-induced endoplasmic reticulum stress and pyroptosis through XBP-1s deacetylation in human renal tubular epithelial cells. Arch. Toxicol. 93, 965–986. doi: 10.1007/s00204-019-02415-8
Ciccia, A., and Elledge, S. J. (2010). The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204. doi: 10.1016/j.molcel.2010.09.019
Cuypers, A., Plusquin, M., Remans, T., Jozefczak, M., Keunen, E., Gielen, H., et al. (2010). Cadmium stress: an oxidative challenge. Biometals 23, 927–940. doi: 10.1007/s10534-010-9329-x
Das, S., Dewanjee, S., Dua, T. K., Joardar, S., Chakraborty, P., Bhowmick, S., et al. (2019). Carnosic Acid attenuates cadmium induced nephrotoxicity by inhibiting oxidative stress, promoting Nrf2/HO-1 signalling and impairing TGF-β1/Smad/collagen IV signalling. Molecules 24:4176. doi: 10.3390/molecules24224176
Davis, A. P., Grondin, C. J., Johnson, R. J., Sciaky, D., McMorran, R., Wiegers, J., et al. (2019). The comparative toxicogenomics database: update 2019. Nucleic Acids Res. 47, D948–D954. doi: 10.1093/nar/gky868
Davis, A. P., Wiegers, T. C., Rosenstein, M. C., Murphy, C. G., and Mattingly, C. J. (2011). The curation paradigm and application tool used for manual curation of the scientific literature at the Comparative Toxicogenomics Database. Database (Oxford) 2011, bar034. doi: 10.1093/database/bar034
Dewanjee, S., Gangopadhyay, M., Sahu, R., and Karmakar, S. (2013). Cadmium induced pathophysiology: prophylactic role of edible jute (Corchorus olitorius) leaves with special emphasis on oxidative stress and mitochondrial involvement. Food Chem. Toxicol. 60, 188–198. doi: 10.1016/j.fct.2013.07.043
Faroon, O., Ashizawa, A., Wright, S., Tucker, P., Jenkins, K., Ingerman, L., et al. (2012). Toxicological Profile for Cadmium. Atlanta, GA: Agency for Toxic Substances and Disease Registry (US).
Fasanya-Odewumi, C., Latinwo, L. M., Ikediobi, C. O., Gilliard, L., Sponholtz, G., Nwoga, J., et al. (1998). The genotoxicity and cytotoxicity of dermally-administered cadmium: effects of dermal cadmium administration. Int. J. Mol. Med. 1, 1001–1006. doi: 10.3892/ijmm.1.6.1001
Fujiki, K., Inamura, H., Sugaya, T., and Matsuoka, M. (2019). Blockade of ALK4/5 signaling suppresses cadmium- and erastin-induced cell death in renal proximal tubular epithelial cells via distinct signaling mechanisms. Cell Death Differ. 26, 2371–2385. doi: 10.1038/s41418-019-0307-8
Fujishiro, H., Hamao, S., Isawa, M., and Himeno, S. (2019). Segment-specific and direction-dependent transport of cadmium and manganese in immortalized S1, S2, and S3 cells derived from mouse kidney proximal tubules. J. Toxicol. Sci. 44, 611–619. doi: 10.2131/jts.44.611
Fujishiro, H., Yano, Y., Takada, Y., Tanihara, M., and Himeno, S. (2012). Roles of ZIP8, ZIP14, and DMT1 in transport of cadmium and manganese in mouse kidney proximal tubule cells. Metallomics 4, 700–708. doi: 10.1039/c2mt20024d
Fujiwara, Y., Lee, J. Y., Tokumoto, M., and Satoh, M. (2012). Cadmium renal toxicity via apoptotic pathways. Biol. Pharm. Bull. 35, 1892–1897. doi: 10.1248/bpb.b212014
Ge, J., Zhang, C., Sun, Y. C., Zhang, Q., Lv, M. W., Guo, K., et al. (2019). Cadmium exposure triggers mitochondrial dysfunction and oxidative stress in chicken (Gallus gallus) kidney via mitochondrial UPR inhibition and Nrf2-mediated antioxidant defense activation. Sci. Total Environ. 689, 1160–1171. doi: 10.1016/j.scitotenv.2019.06.405
Ge, Z., Diao, H., Ji, X., Liu, Q., Zhang, X., and Wu, Q. (2018). Gap junctional intercellular communication and endoplasmic reticulum stress regulate chronic cadmium exposure induced apoptosis in HK-2 cells. Toxicol. Lett. 288, 35–43. doi: 10.1016/j.toxlet.2018.02.013
Guan, T., Xin, Y., Zheng, K., Wang, R., Zhang, X., Jia, S., et al. (2020). Metabolomics analysis of the effects of quercetin on renal toxicity induced by cadmium exposure in rats. Biometals 34, 33–48. doi: 10.1007/s10534-020-00260-2
Hamada, T., Nakano, S., Iwai, S., Tanimoto, A., Ariyoshi, K., and Koide, O. (1991). Pathological study on beagles after long-term oral administration of cadmium. Toxicol. Pathol. 19, 138–147. doi: 10.1177/019262339101900208
Hamidian, G., Mirdar, S., Raee, P., Asghari, K., and Jarrahi, M. (2020). Silymarin protects the structure of kidney in the neonatal rats exposed to maternal cadmium toxicity: a stereological study. Vet. Res. Forum. 11, 143–152. doi: 10.30466/vrf.2019.84202.2108
Harper, J. W., and Elledge, S. J. (2007). The DNA damage response: ten years after. Mol. Cell 28, 739–745. doi: 10.1016/j.molcel.2007.11.015
Hester, S., Eastmond, D. A., and Bhat, V. S. (2015). Developing toxicogenomics as a research tool by applying benchmark dose-response modelling to inform chemical mode of action and tumorigenic potency. Int. J. Biotechnol. 14, 28–46.
Jablonska, E., Socha, K., Reszka, E., Wieczorek, E., Skokowski, J., Kalinowski, L., et al. (2017). Cadmium, arsenic, selenium and iron- Implications for tumor progression in breast cancer. Environ. Toxicol. Pharmacol. 53, 151–157. doi: 10.1016/j.etap.2017.05.014
Jarup, L., Hellstrom, L., Alfven, T., Carlsson, M. D., Grubb, A., Persson, B., et al. (2000). Low level exposure to cadmium and early kidney damage: the OSCAR study. Occup. Environ. Med. 57, 668–672. doi: 10.1136/oem.57.10.668
Jimi, S., Uchiyama, M., Takaki, A., Suzumiya, J., and Hara, S. (2004). Mechanisms of cell death induced by cadmium and arsenic. Ann. N. Y. Acad. Sci. 1011, 325–331. doi: 10.1007/978-3-662-41088-2_32
Joardar, S., Dewanjee, S., Bhowmick, S., Dua, T. K., Das, S., Saha, A., et al. (2019). Rosmarinic acid attenuates cadmium-induced nephrotoxicity via inhibition of oxidative stress, apoptosis, inflammation and fibrosis. Int. J. Mol. Sci. 20:2027. doi: 10.3390/ijms20082027
Johri, N., Jacquillet, G., and Unwin, R. (2010). Heavy metal poisoning: the effects of cadmium on the kidney. Biometals 23, 783–792. doi: 10.1007/s10534-010-9328-y
Kjellström, T., and Nordberg, G. F. (1978). A kinetic model of cadmium metabolism in the human being. Environ. Res. 16, 248–269. doi: 10.1016/0013-9351(78)90160-3
Labib, S., Williams, A., Yauk, C. L., Nikota, J. K., Wallin, H., Vogel, U., et al. (2016). Nano-risk Science: application of toxicogenomics in an adverse outcome pathway framework for risk assessment of multi-walled carbon nanotubes. Part Fibre Toxicol. 13:15. doi: 10.1186/s12989-016-0125-9
Li, J. R., Ou, Y. C., Wu, C. C., Wang, J. D., Lin, S. Y., Wang, Y. Y., et al. (2020). Endoplasmic reticulum stress and autophagy contributed to cadmium nephrotoxicity in HK-2 cells and Sprague-Dawley rats. Food Chem. Toxicol. 146:111828. doi: 10.1016/j.fct.2020.111828
Liu, J., Liu, Y., Habeebu, S. S., and Klaassen, C. D. (1998). Susceptibility of MT-null mice to chronic CdCl2-induced nephrotoxicity indicates that renal injury is not mediated by the CdMT complex. Toxicol. Sci. 46, 197–203. doi: 10.1006/toxs.1998.2541
Liu, J., Qian, S. Y., Guo, Q., Jiang, J., Waalkes, M. P., Mason, R. P., et al. (2008). Cadmium generates reactive oxygen- and carbon-centered radical species in rats: insights from in vivo spin-trapping studies. Free Radic. Biol. Med. 45, 475–481. doi: 10.1016/j.freeradbiomed.2008.04.041
Liu, Q., Zhang, R., Wang, X., Shen, X., Wang, P., Sun, N., et al. (2019). Effects of sub-chronic, low-dose cadmium exposure on kidney damage and potential mechanisms. Ann. Trans. Med. 7:17.
Liu, Y., Liu, J., Habeebu, S. M., Waalkes, M. P., and Klaassen, C. D. (2000). Metallothionein-I/II null mice are sensitive to chronic oral cadmium-induced nephrotoxicity. Toxicol. Sci. 57, 167–176. doi: 10.1093/toxsci/57.1.167
Luo, T., Yu, Q., Zou, H., Zhao, H., Gu, J., Yuan, Y., et al. (2020). Role of poly (ADP-ribose) polymerase-1 in cadmium-induced cellular DNA damage and cell cycle arrest in rat renal tubular epithelial cell line NRK-52E. Environ. Pollut. 261:114149. doi: 10.1016/j.envpol.2020.114149
Matović, V., Buha, A., Ðukić-Ćosić, D., and Bulat, Z. (2015). Insight into the oxidative stress induced by lead and/or cadmium in blood, liver and kidneys. Food Chem. Toxicol. 78, 130–140. doi: 10.1016/j.fct.2015.02.011
Meek, M. E., Boobis, A., Cote, I., Dellarco, V., Fotakis, G., Munn, S., et al. (2014). New developments in the evolution and application of the WHO/IPCS framework on mode of action/species concordance analysis. J. Appl. Toxicol. 34, 1–18. doi: 10.1002/jat.2949
Meek, M. E., and Klaunig, J. E. (2010). Proposed mode of action of benzene-induced leukemia: Interpreting available data and identifying critical data gaps for risk assessment. Chem. Biol. Interact. 184, 279–285. doi: 10.1016/j.cbi.2010.02.006
Moffat, I., Chepelev, N., Labib, S., Bourdon-Lacombe, J., Kuo, B., Buick, J. K., et al. (2015). Comparison of toxicogenomics and traditional approaches to inform mode of action and points of departure in human health risk assessment of benzo[a]pyrene in drinking water. Crit. Rev. Toxicol. 45, 1–43. doi: 10.3109/10408444.2014.973934
Morozesk, M., Franqui, L. S., Pinheiro, F. C., Nóbrega, J. A., Martinez, D. S. T., and Fernandes, M. N. (2020). Effects of multiwalled carbon nanotubes co-exposure with cadmium on zebrafish cell line: metal uptake and accumulation, oxidative stress, genotoxicity and cell cycle. Ecotoxicol. Environ. Saf. 202:110892. doi: 10.1016/j.ecoenv.2020.110892
National Research Council (2007). Toxicity Testing In The 21st Century: A Vision And A Strategy. Washington, DC: National Academies Press.
Nawrot, T., Plusquin, M., Hogervorst, J., Roels, H. A., Celis, H., Thijs, L., et al. (2006). Environmental exposure to cadmium and risk of cancer: a prospective population-based study. Lancet Oncol. 7, 119–126. doi: 10.1016/s1470-2045(06)70545-9
Nazima, B., Manoharan, V., and Miltonprabu, S. (2015). Grape seed proanthocyanidins ameliorates cadmium-induced renal injury and oxidative stress in experimental rats through the up-regulation of nuclear related factor 2 and antioxidant responsive elements. Biochem. Cell Biol. 93, 210–226. doi: 10.1139/bcb-2014-0114
Nemmiche, S. (2017). Oxidative signaling response to cadmium exposure. Toxicol. Sci. 156, 4–10. doi: 10.1093/toxsci/kfw222
Nemmiche, S., and Guiraud, P. (2016). Cadmium-induced oxidative damages in the human BJAB cells correlate with changes in intracellular trace elements levels and zinc transporters expression. Toxicol. Vitro 37, 169–177. doi: 10.1016/j.tiv.2016.09.014
Nyberg, K. A., Michelson, R. J., Putnam, C. W., and Weinert, T. A. (2002). Toward maintaining the genome: DNA damage and replication checkpoints. Annu. Rev. Genet. 36, 617–656. doi: 10.1146/annurev.genet.36.060402.113540
OECD (2018). Users’ Handbook Supplement to the Guidance Document for Developing and Assessing Adverse Outcome Pathways”: OECD Series on Adverse Outcome Pathways. Paris: OECD Publishing.
Ognjanović, B. I., Marković, S. D., Pavlović, S. Z., Zikić, R. V., Stajn, A. S., and Saicić, Z. S. (2008). Effect of chronic cadmium exposure on antioxidant defense system in some tissues of rats: protective effect of selenium. Physiol. Res. 57, 403–411.
Poontawee, W., Natakankitkul, S., and Wongmekiat, O. (2016). Protective effect of cleistocalyx nervosum var. paniala fruit extract against oxidative renal damage caused by cadmium. Molecules 21:133. doi: 10.3390/molecules21020133
Prozialeck, W. C., and Edwards, J. R. (2012). Mechanisms of cadmium-induced proximal tubule injury: new insights with implications for biomonitoring and therapeutic interventions. J. Pharmacol. Exp. Ther. 343, 2–12. doi: 10.1124/jpet.110.166769
Renugadevi, J., and Prabu, S. M. (2009). Naringenin protects against cadmium-induced oxidative renal dysfunction in rats. Toxicology 256, 128–134. doi: 10.1016/j.tox.2008.11.012
Sabolić, I., Breljak, D., Skarica, M., and Herak-Kramberger, C. M. (2010). Role of metallothionein in cadmium traffic and toxicity in kidneys and other mammalian organs. Biometals 23, 897–926. doi: 10.1007/s10534-010-9351-z
Saplakoğlu, U., Işcan, M., and Işcan, M. (1997). DNA single-strand breakage in rat lung, liver and kidney after single and combined treatments of nickel and cadmium. Mutat. Res. 394, 133–140.
Satarug, S., Garrett, S. H., Sens, M. A., and Sens, D. A. (2010). Cadmium, environmental exposure, and health outcomes. Environ. Health Perspect. 118, 182–190. doi: 10.1289/ehp.0901234
Satarug, S., Nishijo, M., Ujjin, P., Vanavanitkun, Y., and Moore, M. R. (2005). Cadmium-induced nephropathy in the development of high blood pressure. Toxicol. Lett. 157, 57–68. doi: 10.1016/j.toxlet.2005.01.004
So, K. Y., and Oh, S. H. (2016). Cadmium-induced heme-oxygenase-1 expression plays dual roles in autophagy and apoptosis and is regulated by both PKC-δ and PKB/Akt activation in NRK52E kidney cells. Toxicology 370, 49–59. doi: 10.1016/j.tox.2016.09.010
Song, X. B., Liu, G., Wang, Z. Y., and Wang, L. (2016). Puerarin protects against cadmium-induced proximal tubular cell apoptosis by restoring mitochondrial function. Chem. Biol. Interact. 260, 219–231. doi: 10.1016/j.cbi.2016.10.006
Suliman Al-Gebaly, A. (2017). Ameliorative effect of arctium lappa against cadmium genotoxicity and histopathology in kidney of wistar rat. Pak. J. Biol. Sci. 20, 314–319. doi: 10.3923/pjbs.2017.314.319
Takahashi, N., Ogita, N., Takahashi, T., Taniguchi, S., Tanaka, M., Seki, M., et al. (2019). A regulatory module controlling stress-induced cell cycle arrest in Arabidopsis. Elife 8:e43944. doi: 10.7554/eLife.43944
Tanimoto, A., Hamada, T., and Koide, O. (1993). Cell death and regeneration of renal proximal tubular cells in rats with subchronic cadmium intoxication. Toxicol. Pathol. 21, 341–352. doi: 10.1177/019262339302100401
Teeyakasem, W., Nishijo, M., Honda, R., Satarug, S., Swaddiwudhipong, W., and Ruangyuttikarn, W. (2007). Monitoring of cadmium toxicity in a Thai population with high-level environmental exposure. Toxicol. Lett. 169, 185–195. doi: 10.1016/j.toxlet.2007.01.004
Valverde, M., Trejo, C., and Rojas, E. (2001). Is the capacity of lead acetate and cadmium chloride to induce genotoxic damage due to direct DNA-metal interaction? Mutagenesis 16, 265–270. doi: 10.1093/mutage/16.3.265
Wang, D., Sun, H., Wu, Y., Zhou, Z., Ding, Z., Chen, X., et al. (2016). Tubular and glomerular kidney effects in the Chinese general population with low environmental cadmium exposure. Chemosphere 147, 3–8. doi: 10.1016/j.chemosphere.2015.11.069
Wang, L., Cao, J., Chen, D., Liu, X., Lu, H., and Liu, Z. (2009). Role of oxidative stress, apoptosis, and intracellular homeostasis in primary cultures of rat proximal tubular cells exposed to cadmium. Biol. Trace Elem. Res. 127, 53–68. doi: 10.1007/s12011-008-8223-7
Wang, L. Y., Fan, R. F., Yang, D. B., Zhang, D., and Wang, L. (2019). Puerarin reverses cadmium-induced lysosomal dysfunction in primary rat proximal tubular cells via inhibiting Nrf2 pathway. Biochem. Pharmacol. 162, 132–141. doi: 10.1016/j.bcp.2018.10.016
Wang, X. Y., Wang, Z. Y., Zhu, Y. S., Zhu, S. M., Fan, R. F., and Wang, L. (2018). Alleviation of cadmium-induced oxidative stress by trehalose via inhibiting the Nrf2-Keap1 signaling pathway in primary rat proximal tubular cells. J. Biochem. Mol. Toxicol. 32:e22011. doi: 10.1002/jbt.22011
Wang, X. Y., Yang, H., Wang, M. G., Yang, D. B., Wang, Z. Y., and Wang, L. (2017). Trehalose protects against cadmium-induced cytotoxicity in primary rat proximal tubular cells via inhibiting apoptosis and restoring autophagic flux. Cell Death Dis. 8:e3099. doi: 10.1038/cddis.2017.475
Wang, Y., Wu, Y., Luo, K., Liu, Y., Zhou, M., Yan, S., et al. (2013). The protective effects of selenium on cadmium-induced oxidative stress and apoptosis via mitochondria pathway in mice kidney. Food Chem. Toxicol. 58, 61–67. doi: 10.1016/j.fct.2013.04.013
Webster, A. F., Chepelev, N., Gagné, R., Kuo, B., Recio, L., Williams, A., et al. (2015). Impact of genomics platform and statistical filtering on transcriptional benchmark doses (BMD) and multiple approaches for selection of chemical point of departure (PoD). PLoS One 10:e0136764. doi: 10.1371/journal.pone.0136764
Wu, C. Y., Wong, C. S., Chung, C. J., Wu, M. Y., Huang, Y. L., Ao, P. L., et al. (2019). The association between plasma selenium and chronic kidney disease related to lead, cadmium and arsenic exposure in a Taiwanese population. J. Hazard Mater. 375, 224–232. doi: 10.1016/j.jhazmat.2019.04.082
Wu, X., Wei, S., Wei, Y., Guo, B., Yang, M., Zhao, D., et al. (2012). The reference dose for subchronic exposure of pigs to cadmium leading to early renal damage by benchmark dose method. Toxicol. Sci. 128, 524–531. doi: 10.1093/toxsci/kfs173
Yan, H., Carter, C. E., Xu, C., Singh, P. K., Jones, M. M., Johnson, J. E., et al. (1997). Cadmium-induced apoptosis in the urogenital organs of the male rat and its suppression by chelation. J. Toxicol. Environ. Health 52, 149–168. doi: 10.1080/00984109708984058
Keywords: cadmium, mode of action, renal tubular dysfunction, pathway analysis, weight of evidence
Citation: Zhang Y, Liu Z, He Q, Wu F, Xiao Y, Chen W, Jin Y, Yu D and Wang Q (2021) Construction of Mode of Action for Cadmium-Induced Renal Tubular Dysfunction Based on a Toxicity Pathway-Oriented Approach. Front. Genet. 12:696892. doi: 10.3389/fgene.2021.696892
Received: 18 April 2021; Accepted: 14 June 2021;
Published: 23 July 2021.
Edited by:
Gustavo Rafael Mazzaron Barcelos, Federal University of São Paulo, BrazilReviewed by:
Cynthia Grondin, North Carolina State University, United StatesCopyright © 2021 Zhang, Liu, He, Wu, Xiao, Chen, Jin, Yu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Wang, d2FuZ3EyN0BtYWlsLnN5c3UuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.