 Rulai Yang1†
Rulai Yang1† Pingping Jiang
Pingping Jiang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 12 August 2021
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.694683
This article is part of the Research Topic Genetics of Thyroid Glands View all 5 articles
Congenital hypothyroidism (CH) is the most common neonatal metabolic disorder. Although it has been understood to be a monogenic disease, some CH patients are reported to carry two or more variants at different genes. Here, ten permanent congenital hypothyroidism (PCH) patients were retrospectively reviewed, with elevated levels of serum thyroid-stimulating hormone and levothyroxine dependence during follow-up between 2015 and 2019. Each affected individual carried digenic variants, which were heterozygous at two of pathogenic genes. In total, five pathogenic genes, TSHR, TG, TPO, DUOX2 and DUOXA2, were simultaneously identified in subjects that were involved in the same metabolic pathway: thyroid hormone biosynthesis. There were digenic variants at TSHR and DUOX2 combined in three patients, DUOX2 and TG combined in two patients, DUOX2 and DUOXA2 combined in two patients, TG and DUOXA2 combined in two patients, and TG and TPO combined in one patient. Additionally, seven novel variants, TSHR c.679G>A, DUOX2 c.127A>T, c.608-619del, c.959T>C, TG c.2307G>A, and c.6759_6765del, and DUOXA2 c.93T>G, were identified in these PCH patients. Along with a literature review on digenic variants in patients with CH, our findings illustrated the complexity of genetic etiology in CH.
Congenital hypothyroidism (CH) is the most common neonatal metabolic disorder. It has an incidence ranging from 1:1,400 to 1:2,800 live births in many countries (Wassner and Brown, 2015), and it results in severe neurodevelopmental impairment if not treated early and effectively. Primary CH is usually classified into two categories by pathogenesis: thyroid dysgenesis, a defect in thyroid gland development in which a few cases were caused by FOXE1, NKX2-1, NKX2-5, and PAX8, and thyroid dyshormonogenesis (DH), an intrinsic defect of thyroid hormone biosynthesis caused by DUOX2, DUOXA2, IYD (DEHAL1), TG, TPO, SLC26A4 (PDS), SLC26A7, SLC5A5 (NIS), and TSHR (Cangul et al., 2018; Kwak, 2018). Based on the newborn screening (NBS) program and clinical diagnosis, thyroid dyshormonogenes dominate compared to thyroid dysgenesis in the Chinese population upon increased molecular diagnosis (Long et al., 2018; Sun et al., 2018). Whereas thyroid dysgenesis still accounts for more than 69% of primary CH worldwide (Wassner and Brown, 2015; Peters et al., 2018). The inheritance of CH is controversial. Although it has been understood to be autosomal recessive (biallelic) in most cases as a monogenic disorder, a few CH cases appear to be monoallelic in one gene (Nicholas et al., 2016, Fugazzola et al., 2003), or 2 or more variants in different genes (Sriphrapradang et al., 2011; Satoh et al., 2015; Makretskaya et al., 2018; Yamaguchi et al., 2020). Here, we report 10 permanent congenital hypothyroidism (PCH) cases carrying digenic variants in which each affected individual is heterozygous at two of pathogenic genes simultaneously as well as the identification of seven novel genetic variants.
During January 2015 and December 2019, the mean incidence of CH was 1:1,093 based on the NBS program in the Children's Hospital, Zhejiang University. CH screening strategies are designed to detect elevated levels of TSH and/or decreased concentrations of thyrocine (T4) (Group for Newborn Screening Society of Child Health Chinese Preventive Medicine Association, 2011). Total 2647 CH cases were diagnosed, of which 148 cases were offered genetic tests, and 66 cases (44.6%) had clear genetic confirmation, either carrying one P/LP variant in a dominate gene or two P/LP variants in a recessive gene. However, another 10 CH patients carrying digenic variants were retrospectively reviewed. They were clinically diagnosed to be PCH with a defect of thyroid hormone biosynthesis based on careful evaluation of clinical features and levothyroxine treatment during follow-up. As shown in Table 1, all patients had initially elevated TSH levels (≥9 μIU/mL), ranging from 9.15 to 25.5 μIU/mL, and were proven to be permanent by receiving a trail off levothyroxine (LT4) at 2–3 years of age. Additionally, the influences of preterm, low-birthweight, and autoimmune thyroid disease on these cases were excluded. The detailed clinical information of the patients was listed in Table 1. With LT4 treatment with a dose of 12.5–33.3 μg per day, all patients had normal ASQ (Ages & Stages Questionnaires) and maintained serum TSH levels ranging from 1 to 10 (mIU/L) with a normal level of free thyroxine (FT4) between 9.01 and 19.05 (pmol/L) cutoff during follow-up. Cases #5 has a goiter by ultrasound during NBS with dimensions 2.3 × 0.9 × 0.8 cm (Right) and 2.2 × 1.0 × 0.8 cm (Left) as previously reported (Wang et al., 2014). There was no compensatory goiter recorded in Case #5 after 1 year with the LT4 supplement.
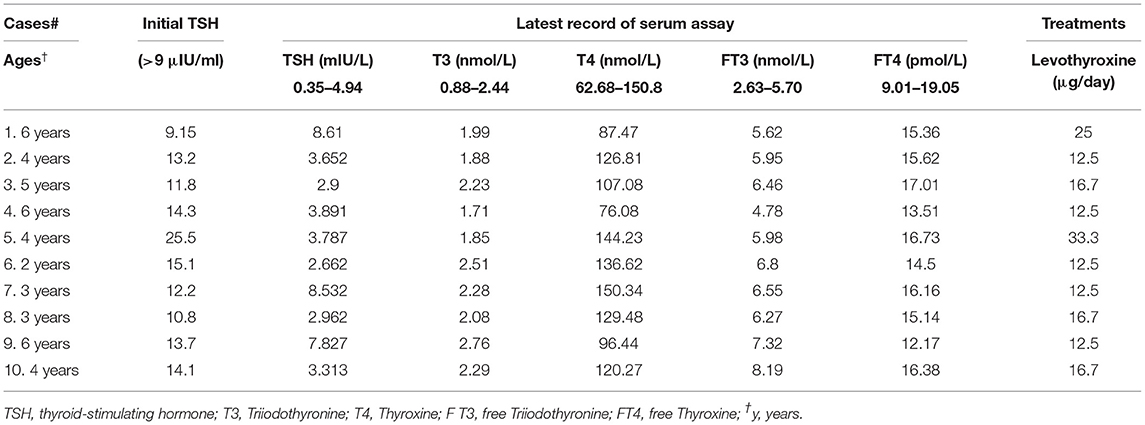
Table 1. The clinical data of 10 primary congenital hypothyroidism cases.
Identification of causative gene via whole-exome sequencing (WES) using peripheral blood was performed for 10 patients. The DNA library was prepared by an Agilent SureSelect Inherited Disease Capture Kit and sequenced using an Illumina HiSeq 2500 platform. All sequencing reads were mapped to the human reference genome (GRCh37) by BWA (Li and Durbin, 2010) and annotated by ANNOVAR (http://annovar.openbioinformatics.org). A series of automatic tools (SIFT, Polyphen, MutationTaster, etc.) were used to predict the functional significance of variants (Table 2). DNA samples from family #1, #3, #4, #6, #7, #8, and #10 were verified further by Sanger sequencing (Supplementary Figure 1).
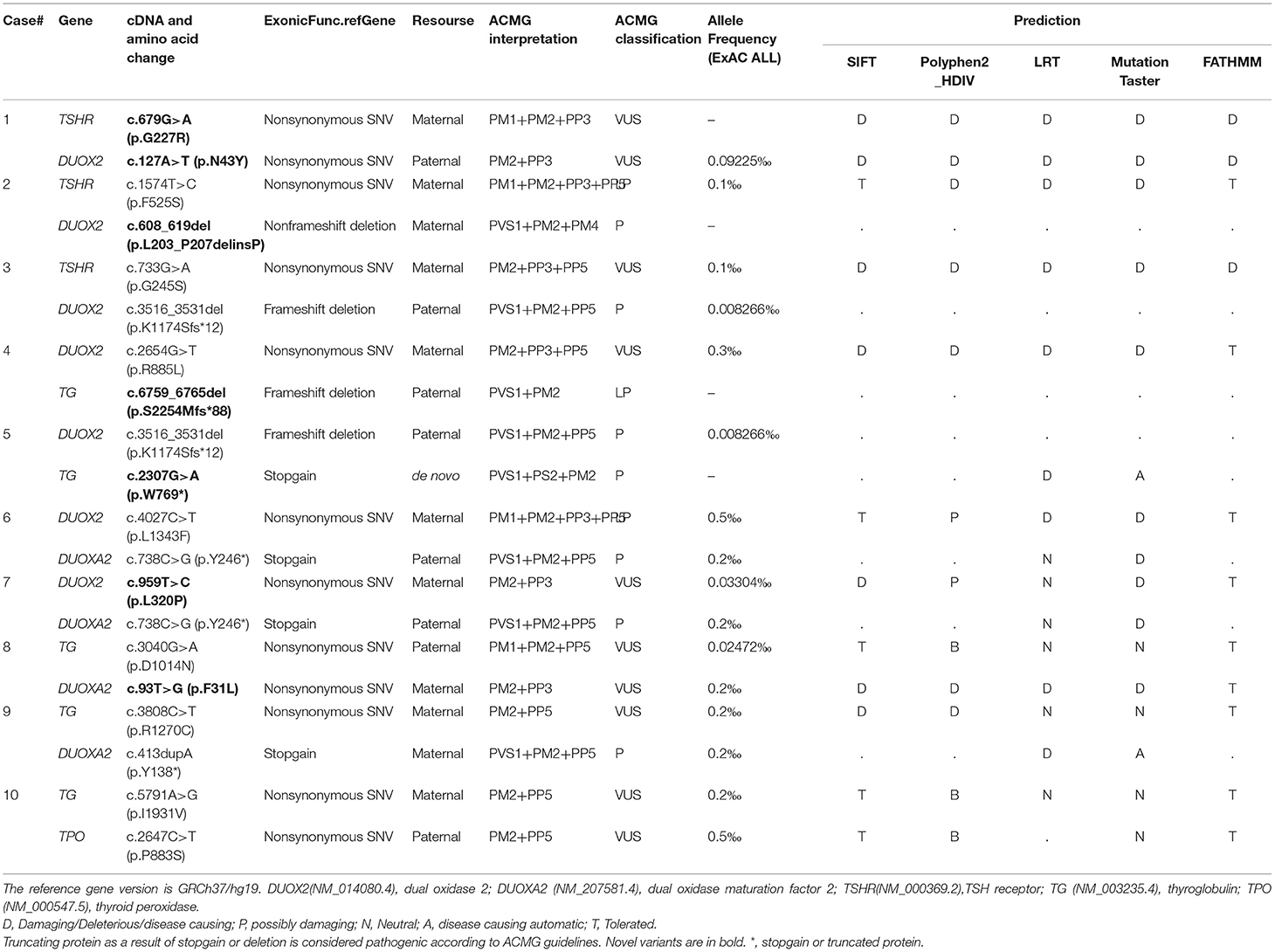
Table 2. Genetic variants and their prediction on protein function.
The majority of dyshormonogenesis has an identifiable genetic basis since there are more than 10 genes reported to be involved in thyroid hormone biosynthesis (Kwak, 2018). All identified genes and variants were summarized in Figure 1. Five causative genes, TSHR, TG, TPO, DUOX2, and DUOXA2, were identified among the 10 patients. DUOX2 was detected in seven patients, followed by TG in five patients, DUOXA2 in four patients, TSHR in three patients, and TPO in one patient. There were digenic variants involving TSHR and DUOX2 in Case #1 (TSHR c.679G>A and DUOX2 c.127A>T), #2 (TSHR c.1574T>A and DUOX2 c.608-619del), and #3 (TSHR c.733G>A and DUOX2 c.3516_3531del), DUOX2 and TG in Case #4 (DUOX2 c.2654G>T and TG c.6759_6765del) and #5 (DUOX2 c.3516_3531del and TG c.2307G>A), DUOX2 and DUOXA2 in Case #6 (DUOX2 c.4027C>T and DUOXA2 c.738C>G) and #7 (DUOX2 c.959T>C and DUOXA2 c.738C>G), TG and DUOXA2 in Case #8 (TG c.3040G>A and DUOXA2 c.93T>G) and #9 (TG c.3808C>T and DUOXA2 c.413dupA), and TG and TPO in Case #10 (TG c.5791A>G and TPO c.2647C>T). In total, 18 variants were identified: 12 missense, 2 nonsense, and 4 frameshifts. These resulted from three deletions and one duplication. Five truncating proteins were observed in 7 cases, including DUOX2 p.K1174S fs*12 (c.3516_3531del) in Cases #3 and #5, TG p.S2254M fs*88 (c.6759_6765del) in Case #4, p.W769* (c.2307G>A) in Case #5, DUOXA2 p.Y246* (c.738C>G) in Cases #6 and #7, and p.Y138* (c.413dupA) in Case #9. Usually, truncating variants were pathogenic based on American College of Medical Genetics (ACMG) guidelines. Variants were mostly transmitted from both parents, although two heterozygous variants of Case #2 and #9 were solely from the mother (Figure 1). Moreover, a de novo variant resulting in a truncated protein, TG c.2307G>A (p. W769*), was detected in Case #5 (Supplementary Figure 1). Among these 18 variants, 7 were novel, identified as TSHR c.679G>A (p.G227R); DUOX2 c.127A>T (p.N43Y), c.608-619del (p.L203-P207delinsP) and c.959T>C (p.L320P); TG c.2307G>A (p.W769*) and c.6759_6765del (p.S2254Mfs*88); and DUOXA2 c.93T>G (p.F31L). All the novel variants were localized in highly conserved regions of each protein (Figure 1) and predicted to be potential pathogenic variants by functional consequences annotation through multiple software (Table 2).
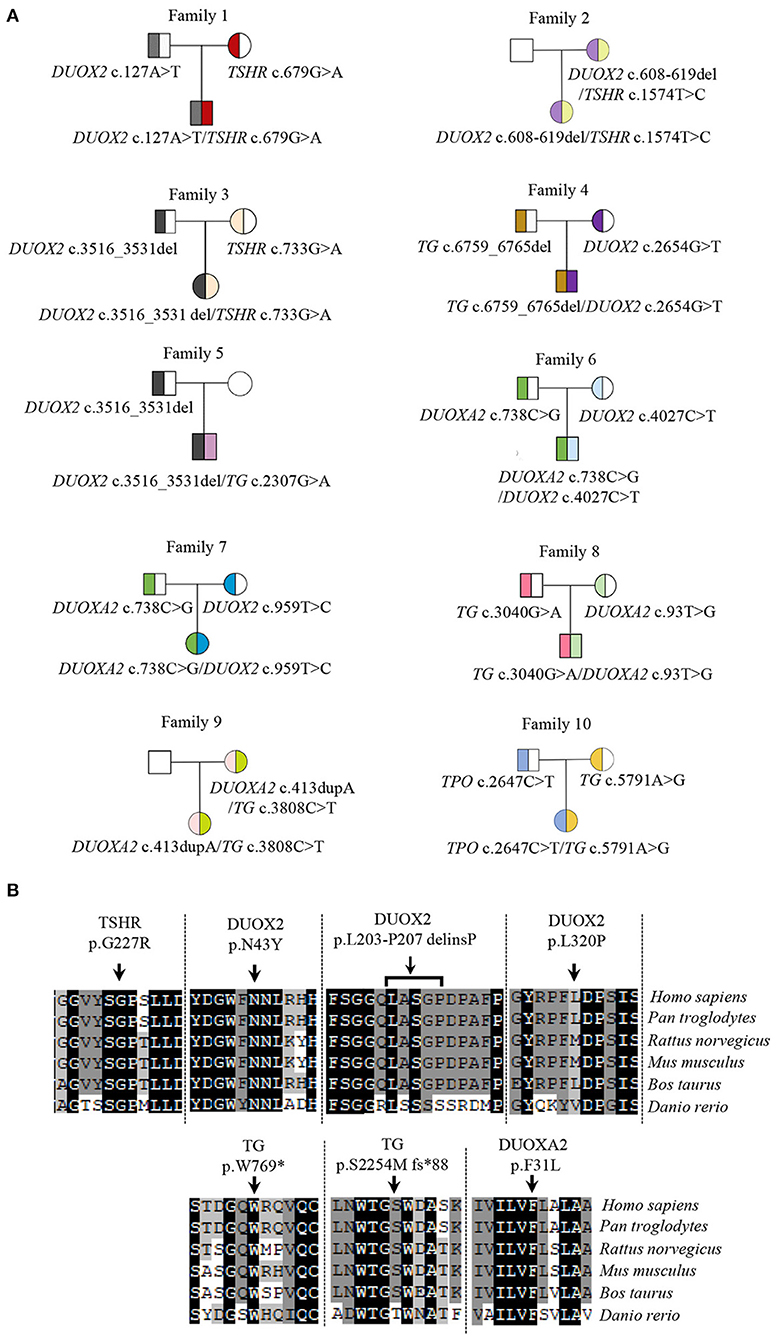
Figure 1. Variants in CH patients and their conservative analysis. (A) Genotypes of CH pedigrees; (B) Conservative analysis of seven novel variants in species. The reference sequences for TSHR, DUOX2, TG, and DUOXA2 in species are followed as: TSHR: Homo sapiens, NP_000360.2; Pan troglodytes, XP_009426511.1; Rattus norvegicus, NP_037020.2; Mus musculus, NP_035778.3; Bos taurus, NP_776631.1; Danio rerio, NP_001139235.1. DUOX2: Homo sapiens, NP_054799.4; Pan troglodytes, XP_009427327.1; Rattus norvegicus, NP_077055.2; Mus musculus, NP_001349684.1; Bos taurus, XP_005211958.1; Danio rerio, XP_002666953.2. TG: Homo sapiens, NP_003226.4; Pan troglodytes, XP_016815373.2; Rattus norvegicus, NP_112250.2; Mus musculus, NP_033401.2; Bos taurus, NP_776308.1; Danio rerio, NP_001316794.1. DUOXA2: Homo sapiens, NP_997464.2; Pan troglodytes, XP_001146826.2; Rattus norvegicus, NP_001178894.1; Mus musculus, NP_080053.1; Bos taurus, XP_002690989.1; Danio rerio, XP_017209762.1.
Most cases of CH are common endocrine disorders caused by biallelic or monoallelic variants in one gene. With the widespread use of newborn screening programs and the application of genetic testing, some cases were found to carry two or more variants at different genes (Satoh et al., 2015; Nicholas et al., 2016; Sun et al., 2018; Yamaguchi et al., 2020), indicating the complexity of genetic etiology in CH. Cases with two or more variants in different genes were usually understood to be oligogenic cases, compared to those in biallelic and monoallelic cases (Yamaguchi et al., 2020). Here, we present 10 PCH cases carrying digenic variants in genes involved in thyroid hormone biosynthesis. Similar to our findings, another 58 cases harboring digenic variants were reported elsewhere (Supplementary Table 1). A total of 24 cases had digenic variants in TSHR and DUOX2, including 5 cases out of 220 Chinese CH (Fang et al., 2019) and 6 cases in Japanese patients (Abe et al., 2018; Yamaguchi et al., 2020). The coexistence of heterozygous variants in TSHR and DUOX2 was also revealed in Caucasian cases (Makretskaya et al., 2018; Sasivari et al., 2019). More digenic variants were heterozygous in two causative genes, including combined DUOX2 and TG in 13 patients (Löf et al., 2016; Fan et al., 2017; Long et al., 2018; Sun et al., 2018; Yamaguchi et al., 2020), DUOX2 and DUOXA2 in 4 patients (Zheng et al., 2016; Yamaguchi et al., 2020), DUOX2 and TPO in 3 patients (Matsuo et al., 2016; Long et al., 2018; Makretskaya et al., 2018), TG and TPO in 6 patients (Nicholas et al., 2016; Makretskaya et al., 2018; Yamaguchi et al., 2020), and TG and SLC26A4 in 2 patients (Löf et al., 2016; Sun et al., 2018). Moreover, it was also demonstrated that 23% of Italian CH patients harbored pathogenic variants in more than one gene (Filippis et al., 2017), indicating that there was no ethnicity limiting the digenic form but rather a frequency of dyshormonogenesis-associated variants. As shown in Supplementary Table 1, eight genes (TSHR, TG, DUOX2, DUOX1, DUOXA2, TPO, IYD, and SLC26A4) were present in those oligogenic cases. The higher frequency genes were DUOX2 (35.3%), TSHR (22.8%), and TG (22.8%). This was consistent with prior studies showing that DUOX2 and TSHR variants were more prevalent in Chinese, Japanese, and Korean patients (Jin et al., 2014; Fu et al., 2016; Fang et al., 2019; Yamaguchi et al., 2020). Higher frequent TG variants were detected in the Sudanese population (Bruellman et al., 2020). However, only two cases harbored variants of IYD that one individual combined with TG (Makretskaya et al., 2018) and the other one with DUOX1(Sun et al., 2018). The digenic variants thereby seemed to be common in CH, but it is somewhat challenged in the variant interpretation by the dominant effect of some of these variants. For example, there were monoallelic variants reported in DUOX2 (Moreno et al., 2002), and later this turned out to be associated with transient hypothyroidism (Wang et al., 2014; Matsuo et al., 2016).
To date, all reported genes with digenic variants are involved in the same metabolic pathway: thyroid hormone biosynthesis. As shown in Figure 2, the thyroid hormone is synthesized at the apical surface of polarized thyroid follicular cells, where the initial step is the binding of TSH to its receptors (TSHR) in the basolateral membrane, activating TG expression. To date, only 5 oligogenic cases carried heterozygous TSHR and TG variants in the Chinese and Japanese population (Fu et al., 2016; Yamaguchi et al., 2020). However, most cases of heterozygous TSHR (77.4%, 24/31) were combined with heterozygous DUOX2 as shown in Supplementary Table 1 (involved in steps 1 and 2). Subsequently, TG, TPO, and the DUOXs (DUOX2 and DUOX1) and their accessory protein DUOXA2 are involved in iodide oxidation to form T4 and T3 (Kwak, 2018). Here, 7 out of 10 cases were caused by two of the five genes in this step. Additionally, overall 32 oligogenic cases (47%, 32/68) carried the combination of two heterozygous variants in two genes in this step, indicating that iodide organification defects may be more common in CH patients. Genes, IYD/DEHAL1, NIS/SLC5A5 and PDS/SLC26A4 involved in recycling of T4, T3, iodide and tyrosine (Spitzweg et al., 2000). Limited by the number of cases, there was no variant detected in NIS as elsewhere (Long et al., 2018). Only a few variants have been reported to date in IYD and PDS. As shown in Supplementary Table 1, there were only three cases carrying heterozygous PDS and two cases carrying heterozygous IYD. Theoretically, any defects of these eight proteins in substrates, enzymes, and transport molecules in the same metabolic pathway led to thyroid dyshormonogenesis. In fact, our data and recent evidence revealed that the combinations of two pathogenic genes predominantly happened in iodide organification and then in TG expression.
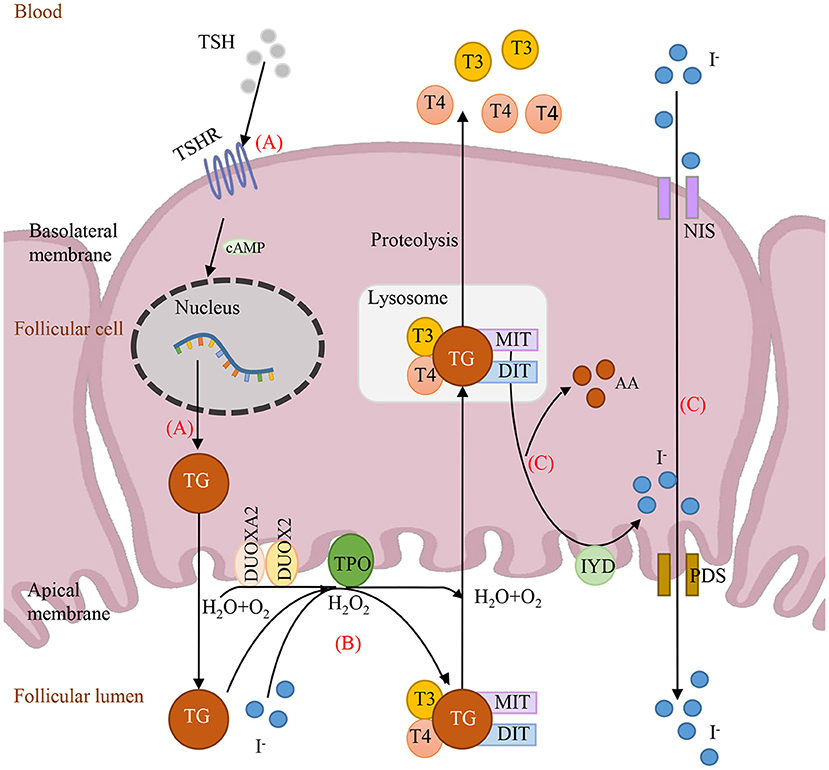
Figure 2. Schematic depiction of the causative genes involving in thyroid hormone synthesis. (A) TSH binds to TSHR and then activates TG expression. (B) The iodide organification, with substrates TG and H2O2, which is completed by enzymes TPO, DUOX2 and DUOXA2. (C) Iodide recycled by IYD and transported by NIS and PDS. TSH, thyroid-stimulating hormone; T3, Triiodothyronine; T4, Thyroxine; MIT, monoiodotyrosine; DIT, diiodotyrosine; DUOX2, dual oxidase 2; DUOXA2, dual oxidase maturation factor 2; TSHR, TSH receptor; TG, thyroglobulin; TPO, thyroid peroxidase; AA, amino acid.
The limitation here is the lack of a parental phenotype, and all our cases are simplex cases. Especially in Case#2 and #9, the parental phenotype is critical in understanding the functional effects of digenic variants. Unfortunately, the mothers carrying the same two variants refused to test their TSH levels. Moreover, the digenic variants in Case#1, #8, and #10 were classified to be VUS according to the ACMG guidelines, which need more cases or further functional experiments to evaluate their damage prediction.
Summarily, we reported here 10 PCH cases with digenic variants involved in the same metabolic pathway: thyroid hormone biosynthesis. To date, 68 CH patients have been reported harboring digenic variants in this metabolic pathway, including genes TSHR, TG, DUOX2, DUOX1, DUOXA2, TPO, IYD, and SLC26A4 with a high frequency of DUOX2, TSHR, and TG. The data present here will extend our awareness of the complexity of genetic etiology in CH.
The original contributions presented in the study are publicly available in NCBI using accession number PRJNA734721.
The studies involving human participants were reviewed and approved by the Research Ethics Committees, Children's Hospital of Zhejiang University School of Medicine. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
PJ and QS performed the conception, analysis, and interpretation of data. RY and LZ recruited patients and performed clinical evaluation. YL, CY, XW, and JF conducted the mutational sequencing and data analysis. PJ, YL, RY, and QS drafted and revised the article. All authors contributed to the article and approved the submitted version.
This work was supported by Grant 2018YFC1002700 to RY from the National Key R&D Program of China, and Grant 81870314 to PJ from the National Natural Science Foundation of China, and by Zhejiang Provincial Program for the Cultivation of High-Level Innovative Health Talents.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the patients and their families for their participation in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.694683/full#supplementary-material
CH, congenital hypothyroidism; PCH, permanent congenital hypothyroidism; TSH, thyroid-stimulating hormone; TSHR, thyroid-stimulating hormone receptor; T3, triiodothyronine; T4, thyroxine; MIT, monoiodotyrosine; DIT, diiodotyrosine; DUOX2, dual oxidase 2; DUOX1, dual oxidase 1; DUOXA2, dual oxidase maturation factor 2; TG, thyroglobulin; TPO, thyroid peroxidase; SLC26A4 (PDS), solute carrier family 26 member 4; SLC26A7, solute carrier family 26 member 7; SLC5A5 (NIS), solute carrier family 5 member 5 (sodium iodide symporter).
Abe, K., Narumi, S., Suwanai, A. S., Adachi, M., Muroya, K., Asakura, Y., et al. (2018). Association between monoallelic TSHR mutations and congenital hypothyroidism: a statistical approach. Eur. J. Endocrinol. 178, 137–144. doi: 10.1530/EJE-16-1049
Bruellman, R. J., Watanabe, Y., Ebrhim, R. S., Creech, M. K., Abdullah, M. A., Dumitrescu, A. M., et al. (2020). Increased prevalence of TG and TPO mutations in sudanese children with congenital hypothyroidism. J. Clin. Endocrinol. Metab. 105, 1564–1572. doi: 10.1210/clinem/dgz297
Cangul, H., Liao, X. H., Schoenmakers, E., Kero, J., Barone, S., Srichomkwun, P., et al. (2018). Homozygous loss-of-function mutations in SLC26A7 cause goitrous congenital hypothyroidism. JCI Insight. 3:e99631. doi: 10.1172/jci.insight.99631
Fan, X., Fu, C., Shen, Y., Li, C., Luo, S., Li, Q., et al. (2017). Next-generation sequencing analysis of twelve known causative genes in congenital hypothyroidism. Clin. Chim. Acta 468, 76–80. doi: 10.1016/j.cca.2017.02.009
Fang, Y., Sun, F., Zhang, R. J., Zhang, C. R., Yan, C. Y., Zhou, Z., et al. (2019). Mutation screening of the TSHR gene in 220 Chinese patients with congenital hypothyroidism. Clin. Chim. Acta 497, 147–152. doi: 10.1016/j.cca.2019.07.031
Filippis, T. D., Gelmini, G., Paraboschi, E., Vigone, M. C., Di Frenna, M., Marelli, F., et al. (2017). A frequent oligogenic involvement in congenital hypothyroidism. Hum. Mol. Genet. 26, 2507–2514. doi: 10.1093/hmg/ddx145
Fu, C., Wang, J., Luo, S., Yang, Q., Li, Q., Zheng, H., et al. (2016). Next-generation sequencing analysis of TSHR in 384 Chinese subclinical congenital hypothyroidism (CH) and CH patients. Clin. Chim. Acta 462, 127–132. doi: 10.1016/j.cca.2016.09.007
Fugazzola, L., Cerutti, N., Mannavola, D., Vannucchi, G., Fallini, C., Persani, L., et al. (2003). Monoallelic expression of mutant thyroid peroxidase allele causing total iodide organification defect. J. Clin. Endocrinol. Metab. 88, 3264–3271. doi: 10.1210/jc.2002-021377
Group for Newborn Screening Society of Child Health and Chinese Preventive Medicine Association (2011). Consensus statement on the diagnosis and management of congenital hypothyroidism. Chin. J. Pediatr. 49, 421–424. doi: 10.3760/cma.j.issn.0578-1310.2011.06.006
Jin, H. Y., Heo, S. H., Kim, Y. M., Kim, G. H., Choi, J. H., Lee, B. H., et al. (2014). High frequency of DUOX2 mutations in transient or permanent congenital hypothyroidism with eutopic thyroid glands. Horm. Res. Paediatr. 82, 252–260. doi: 10.1159/000362235
Kwak, M. J. (2018). Clinical genetics of defects in thyroid hormone synthesis. Ann. Pediatr. Endocrinol. Metab. 23, 169–175. doi: 10.6065/apem.2018.23.4.169
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Löf, C., Patyra, K., Kuulasmaa, T., Vangipurapu, J., Undeutsch, H., Jaeschke, H., et al. (2016). Detection of novel gene variants associated with congenital hypothyroidism in a Finnish patient cohort. Thyroid 26, 1215–1224. doi: 10.1089/thy.2016.0016
Long, W., Lu, G., Zhou, W., Yang, Y., Zhang, B., Zhou, H., et al. (2018). Targeted next-generation sequencing of thirteen causative genes in Chinese patients with congenital hypothyroidism. Endocr. J. 65, 1019–1028. doi: 10.1507/endocrj.EJ18-0156
Makretskaya, N., Bezlepkina, O., Kolodkina, A., Kiyaev, A., Vasilyev, E. V., Petrov, V., et al. (2018). High frequency of mutations in 'dyshormonogenesis genes' in severe congenital hypothyroidism. PLoS ONE 13:e0204323. doi: 10.1371/journal.pone.0204323
Matsuo, K., Tanahashi, Y., Mukai, T., Suzuki, S., Tajima, T., Azuma, H., et al. (2016). High prevalence of DUOX2 mutations in Japanese patients with permanent congenital hypothyroidism or transient hypothyroidism. J. Pediatr. Endocrinol. Metab. 29, 807–812. doi: 10.1515/jpem-2015-0400
Moreno, J. C., Bikker, H., Kempers, M. J., van Trotsenburg, A. S., Baas, F., de Vijlder, J. J., et al. (2002). Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. N. Engl. J. Med. 347, 95–102. doi: 10.1056/NEJMoa012752
Nicholas, A. K., Serra, E. G., Cangul, H., Alyaarubi, S., Ullah, I., Schoenmakers, E., et al. (2016). Comprehensive Screening of Eight Known Causative Genes in Congenital Hypothyroidism with Gland-in-Situ. J. Clin. Endocrinol. Metab. 101, 4521–4531. doi: 10.1210/jc.2016-1879
Peters, C., van Trotsenburg, A. S. P., and Schoenmakers, N. (2018). Diagnosis of endocrine disease: congenital hypothyroidism: update and perspectives. Eur. J. Endocrinol. 179, R297–R317. doi: 10.1530/EJE-18-0383
Sasivari, Z., Szinnai, G., Seebauer, B., Konrad, D., and Lang-Muritano, M. (2019). Double variants in TSHR and DUOX2 in a patient with hypothyroidism: case report. J. Pediatr. Endocrinol. Metab. 32, 1299–1303. doi: 10.1515/jpem-2019-0051
Satoh, M., Aso, K., Ogikubo, S., Yoshizawa-Ogasawara, A., and Saji, T. (2015). Hypothyroidism caused by the combination of two heterozygous mutations: one in the TSH receptor gene the other in the DUOX2 gene. J. Pediatr. Endocrinol. Metab. 28, 657–661. doi: 10.1515/jpem-2014-0078
Spitzweg, C., Heufelder, A. E., and Morris, J. C. (2000). Thyroid iodine transport. Thyroid 10, 321–330. doi: 10.1089/thy.2000.10.321
Sriphrapradang, C., Tenenbaum-Rakover, Y., Weiss, M., Barkoff, M. S., Admoni, O., Kawthar, D., et al. (2011). The coexistence of a novel inactivating mutant thyrotropin receptor allele with two thyroid peroxidase mutations: a genotype-phenotype correlation. J. Clin. Endocrinol. Metab. 96, E1001–1006. doi: 10.1210/jc.2011-0127
Sun, F., Zhang, J. X., Yang, C. Y., Gao, G. Q., Zhu, W. B., Han, B., et al. (2018). The genetic characteristics of congenital hypothyroidism in China by comprehensive screening of 21 candidate genes. Eur. J. Endocrinol. 178, 623–633. doi: 10.1530/EJE-17-1017
Wang, F., Lu, K., Yang, Z., Zhang, S., Lu, W., Zhang, L., et al. (2014). Genotypes and phenotypes of congenital goitre and hypothyroidism caused by mutations in dual oxidase 2 genes. Clin. Endocrinol. 81, 452–457. doi: 10.1111/cen.12469
Wassner, A. J., and Brown, R. S. (2015). Congenital hypothyroidism: recent advances. Curr Opin. Endocrinol. Diabetes. Obes. 22, 407–412. doi: 10.1097/MED.0000000000000181
Yamaguchi, T., Nakamura, A., Nakayama, K., Hishimura, N., Morikawa, S., Ishizu, K., et al. (2020). Targeted next-generation sequencing for congenital hypothyroidism with positive neonatal TSH screening. J. Clin. Endocrinol. Metab. 105:dgaa308. doi: 10.1210/clinem/dgaa308
Keywords: digenic variants, thyroid hormone synthesis, congenital hypothyroidism, genetic counseling, oligogenic cases
Citation: Yang R, Lu Y, Yang C, Wu X, Feng J, Zhu L, Shu Q and Jiang P (2021) Case Report: Expanding the Digenic Variants Involved in Thyroid Hormone Synthesis−10 New Cases of Congenital Hypothyroidism and a Literature Review. Front. Genet. 12:694683. doi: 10.3389/fgene.2021.694683
Received: 13 April 2021; Accepted: 14 June 2021;
Published: 12 August 2021.
Edited by:
Desheng Liang, Central South University, ChinaCopyright © 2021 Yang, Lu, Yang, Wu, Feng, Zhu, Shu and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pingping Jiang, cHBqaWFuZ0B6anUuZWR1LmNu; Qiang Shu, c2h1cWlhbmdAemp1LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.