 Yongzhe Chen1,2
Yongzhe Chen1,2 Zesong Wang3
Zesong Wang3 Yueren Wu3Wenbin He4,5
Yueren Wu3Wenbin He4,5 Juan Du1,2,4,5,6Sufen Cai1,2,4,5
Juan Du1,2,4,5,6Sufen Cai1,2,4,5 Fei Gong1,2,4,5,6Guangxiu Lu3,4,5,6Ge Lin1,2,4,5,6*Can Dai3,4,5*
Fei Gong1,2,4,5,6Guangxiu Lu3,4,5,6Ge Lin1,2,4,5,6*Can Dai3,4,5*- 1School of Basic Medical Science, Central South University, Changsha, China
- 2National Health Commission Key Laboratory of Human Stem Cell and Reproductive Engineering, Central South University, Changsha, China
- 3Department of Basic Medicine, School of Medicine, Hunan Normal University, Changsha, China
- 4Reproductive and Genetic Hospital of China International Trust Investment Corporation Xiangya, Changsha, China
- 5Clinical Research Center for Reproduction and Genetics in Hunan Province, Changsha, China
- 6National Engineering and Research Center of Human Stem Cell, Changsha, China
Background: Empty follicle syndrome (EFS) is defined as the complete failure to retrieve oocytes after ovarian stimulation. Although several mutations in ZP1, ZP2, ZP3, and LHCGR have been identified as genetic causes of EFS, its pathogenesis is still not well-understood.
Methods: Whole-exome sequencing (WES) was employed to identify the candidate pathogenic mutations, which were then verified by Sanger sequencing. A study in CHO-K1 cells was performed to analyze the effect of the mutation on protein expression. Additionally, immunohistochemistry (IHC) staining was used to examine follicular development and zona pellucida (ZP) assembly in the ovary of an EFS patient.
Results: A novel heterozygous deletion in ZP3 (c.565_579del[p.Thr189_Gly193del]) was identified in the EFS patient. It was inherited dominantly and resulted in significant degradation of the ZP3 protein. Oocytes with degenerated cytoplasm and abnormal ZP assembly were observed in follicles up to the secondary stage, and many empty follicle-like structures were present.
Conclusion: We identified a novel ZP3 mutation that expands the mutational spectrum associated with human EFS. We also showed the abnormal follicular development and ZP assembly of the EFS patient with the heterozygous ZP3 mutation, which provides new insights into the pathogenesis of EFS.
Introduction
Female infertility has become an increasingly prominent problem. Approximately 50 million women around the world suffer from infertility (Mascarenhas et al., 2012). Regular ovulation and healthy oocytes are the basis of human reproduction. With the development of in vitro fertilization (IVF) as a treatment for infertility, we have been able to retrieve oocytes and discover their defects in vitro.
Empty follicle syndrome (EFS), first reported in 1986 (Coulam et al., 1986), is commonly defined as the complete failure to retrieve oocytes after ovarian stimulation for IVF, despite normal follicular development and appropriate serum estradiol level (Revelli et al., 2017). Follicle atresia and oocyte degeneration due to ovarian aging may be possible causes (Awonuga et al., 1998). In recent years, mutations in four genes, including LHCGR, ZP1, ZP2, and ZP3, have been linked to EFS (Yuan et al., 2017; Chen et al., 2018; Dai et al., 2019a; Lu et al., 2019; Yang et al., 2020; Wang et al., 2021). Nevertheless, the pathogenesis of EFS, especially in patients with gene mutations, remains largely unknown.
During IVF treatment, cumulus–oocyte complexes (COCs), which consist of granulosa cells surrounding the centrally located oocyte, are recovered from the individual's follicular fluid (Veeck, 1988). Each oocyte is encapsulated in a zona pellucida (ZP), an extracellular matrix that plays important roles in the communication between oocyte and granulosa cells (GCs) (Fagbohun and Downs, 1990), fertilization (Pang et al., 2011; Avella et al., 2014), and protection of the early embryo (Hasegawa and Koyama, 2007).
The human ZP comprises four glycoproteins encoded by ZP1–ZP4 genes (Lefievre et al., 2004). In recent years, mutations in ZP genes have been reported to be responsible for oocyte anomalies and female infertility (Yang et al., 2020). An autosomal recessive truncating variant in ZP1 was identified in a consanguineous Chinese family, and this was suggested to explain infertility in females with abnormal oocytes lacking a ZP (Huang et al., 2014). Chen et al. was the first to identify a heterozygous recurrent missense mutation in ZP3 as the cause of EFS. This mutation exerts its effect via a dominant negative effect on the interaction of ZP proteins, thereby possibly affecting ZP assembly, preventing communication between the cumulus cells and the oocyte, and eventually leading to oocyte degeneration (Chen et al., 2017). In the past 3 years, 25 ZP1 mutations, two ZP2 mutations, and two ZP3 mutations were identified in patients with EFS (Dai et al., 2019; Yang et al., 2020; Wang et al., 2021; Zhang et al., 2021).
In the present study, we identified a novel heterozygous ZP3 deletion associated with EFS and female infertility, which was inherited dominantly in a Chinese family. We also assessed the EFS patient's follicular development and ZP assembly by using ovarian sections and immunohistochemical staining (IHC).
Case Presentation
Clinical Characterization
A patient affected by EFS (Figures 1A,B) was recruited from the Reproductive and Genetic Hospital of CITIC–Xiangya. She was 37 years old, with a diagnosis of primary infertility for 7 years. She underwent three failed artificial insemination (AI) treatments and four cycles of ovarian stimulation and oocyte retrieval in another hospital, in which no oocytes but three empty COCs containing small ooplasm-like fragments without ZP structure were obtained (Table 1).
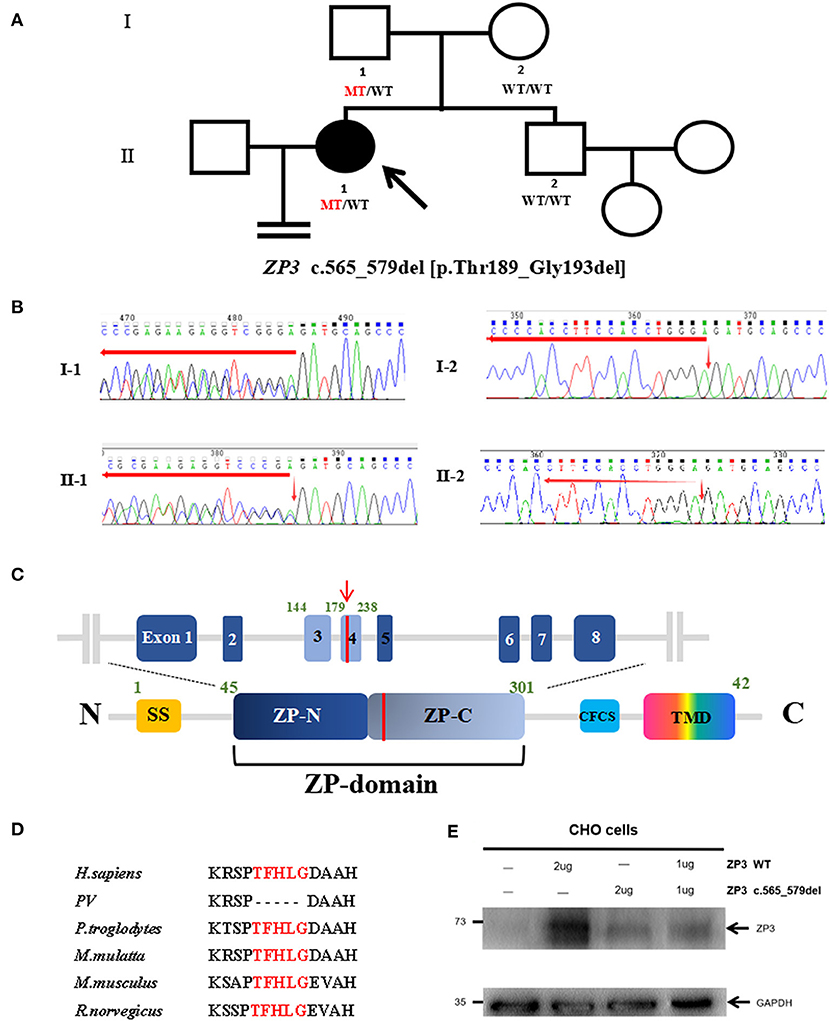
Figure 1. Genotypic features of the EFS patient. (A) Pedigrees of the family with EFS and female infertility. The filled circle indicates the EFS patient. The arrow indicates the proband. The ZP3 genotype for each subject detected is shown, with WT indicating a normal allele and MT indicating the mutant allele. (B) Sanger genotype of ZP3 for each individual. (C) Location of the mutation (red line) is indicated in the genomic structure (top) and domain organization (bottom) of ZP3. SS, signals sequence; CFCS, consensus furin cleavage site; TMD, transmembrane domain. (D) Thr189_Gly193 is highlighted in red and conserved in the selected species. (E) Expression of wild-type (WT) and mutant ZP3 proteins in Chinese hamster ovary (CHO) cells examined by immunoblot analysis using anti-ZP3 antibodies.
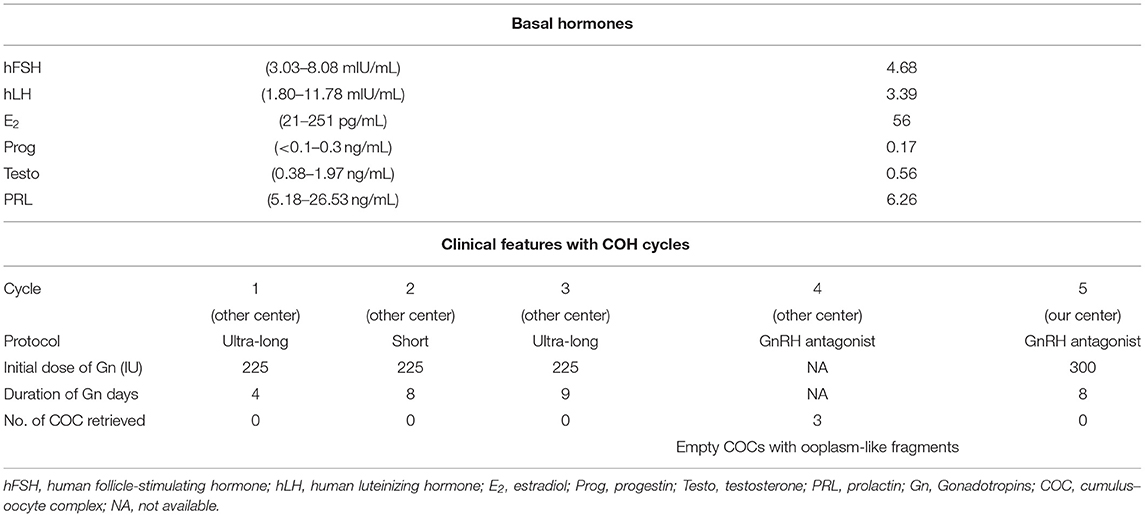
Table 1. Clinical features of the EFS patient with a heterozygous ZP3 deletion.
In our hospital, she received the fifth ovarian stimulation by the GnRH antagonist protocol. The rFSH/HMG was administered at a dose of 300 IU on day 2 of the menstrual cycle, and 300 IU per day for 8 days. Considering her history of recurrent failures and degenerated oocytes without ZP in the follicular aspirate, oocyte retrieval was performed by laparoscopy, upon identification of two follicles measuring 15.5 mm and 18.0 mm in diameters. Her serum estradiol level was 501 pg/mL. Unfortunately, no oocyte was obtained (Table 1).
Identification of ZP3 Mutation
The recurrent EFS in the patient led us to consider a genetic cause. To identify the causative mutation, we initially performed whole-exome sequencing (WES) and variant analysis in the proband and her mother. The following mutations were considered as candidate mutations: (1) mutations detected in the proband but absent in her mother; (2) mutations with a minor allele frequency below 1% in three public databases (1000 Genomes, the ExAC, and gnomAD Browsers); (3) exonic non-synonymous or splice site variants, or coding INDELs; and (4) mutations with high gene expression (reads per kilobase million ≥ 30) in human oocytes in both the previously described (Yan et al., 2013) and in-house RNA sequencing data. These criteria led us to identify a heterozygous deletion c.565_579del [p.Thr189_Gly193del] in ZP3. This mutation is highly conserved among species (Figure 1D) and is located within the ZP domain (ZPD) of the ZP3 protein, a structural module essential for the polymerization and assembly of ZP proteins (Figure 1C) (Jovine et al., 2002). To confirm the c.565-579del and exclude the possibility of a heterozygous deletion of a large fragment in ZP3, the full-length ZP3 cDNA was amplified for Sanger sequencing using total RNA from the peripheral blood and primers: sense 5′- CCTCCTGCTCTGGGGTAGTA-3′ and antisense 5′- CTTTTATTCGGAAGCAGACACAG-3′. As a result, the proband and her father harbored the same heterozygous deletion c.565-579del, while both her mother and brother had the wild-type, suggesting that the deletion had been paternally transmitted (Figures 1A,B) and had not affected male fertility. The karyotype of the proband was 46, XX.
Effects of ZP3 Mutation on Protein Expression
To test the effect of this ZP3 mutation, expression plasmids carrying wild-type or mutant ZP3 cDNA were transfected into Chinese hamster ovary (CHO-K1) cells and immunoblot analyses were performed by using primary antibodies ZP3 (1:500; Santa Cruz Biotechnology, sc-25802) and GAPDH (1:3000; Beyotime, AF0006), respectively. Antibody binding was detected with HRP-conjugated goat anti-rabbit (1:2000; CWBIO, CW0103S, for primary antibody of ZP3) and goat anti-mouse (1:2000; CWBIO, CW0102S, for primary antibody of GAPDH) secondary antibodies. The expression level of the mutant ZP3 protein was significantly lower than that of the normal protein (Figure 1E), suggesting that the mutant protein was unstable and degraded. Moreover, in the experiment of co-transfection of wild-type and mutant plasmids, simulating heterozygous mutation in the patient, the total level of ZP3 protein was as low as in the cells transfected with the mutant plasmid (Figure 1E), indicating a dominant negative effect of the mutant ZP3.
Follicular Development in the EFS Patient With ZP3 Mutation
To examine follicular development and ZP assembly in the EFS patient carrying ZP3 mutation, IHC staining on the ovarian sections was performed. Control ovarian tissue was obtained from the ovarian wedge resection of an individual with polycystic ovary syndrome (PCOS). Antigen retrieval was performed using 0.01 M sodium citrate buffer for 10 min, then endogenous peroxidase activity was blocked with 0.3% H2O2 in PBS for 15 min, and non-specific binding sites were blocked with 5% BSA in PBS at room temperature for 1 h. Sections from the same follicle were then incubated overnight at 4°C with primary antibodies to VASA (1:100, Abcam, ab13840), ZP1 (1:100; Santa Cruz Biotechnology, sc-365435), and ZP3 (1:100; Santa Cruz Biotechnology, sc-398359), respectively. Antibody binding was detected with HRP-conjugated goat anti-rabbit (1:1000; Abcam, ab97051, for primary antibody of VASA) and goat anti-mouse (1:1000; Abcam, ab97245, for primary antibodies of ZP1 and ZP3) secondary antibodies. The sections were then developed using DAB kit and counterstained with hematoxylin. Using VASA staining, intact follicles containing an oocyte at the primordial to secondary stages were observed in the patient's ovary (Figures 2A–D). It is worth noting that, compared with homogeneous cytoplasm in normal oocytes (Figures 2P–AA), the patient's oocytes showed meshy cytoplasm (Figures 2D,I,N), which suggested that the oocytes might be undergoing degeneration. Using ZP staining, a continuous ZP structure increasing in width along with growing oocyte was observed in the normal control (Figures 2T–AA). In contrast, no clear and intact ZP structure was detected in the patient's secondary follicles (Figure 2N). Moreover, no follicles beyond secondary stage were found, but many empty follicle-like structures were detected (Figures 2E,J,O, 3). These results hint the abnormal ZP assembly and follicular development beyond the secondary stage in the EFS patient with ZP3 mutation.
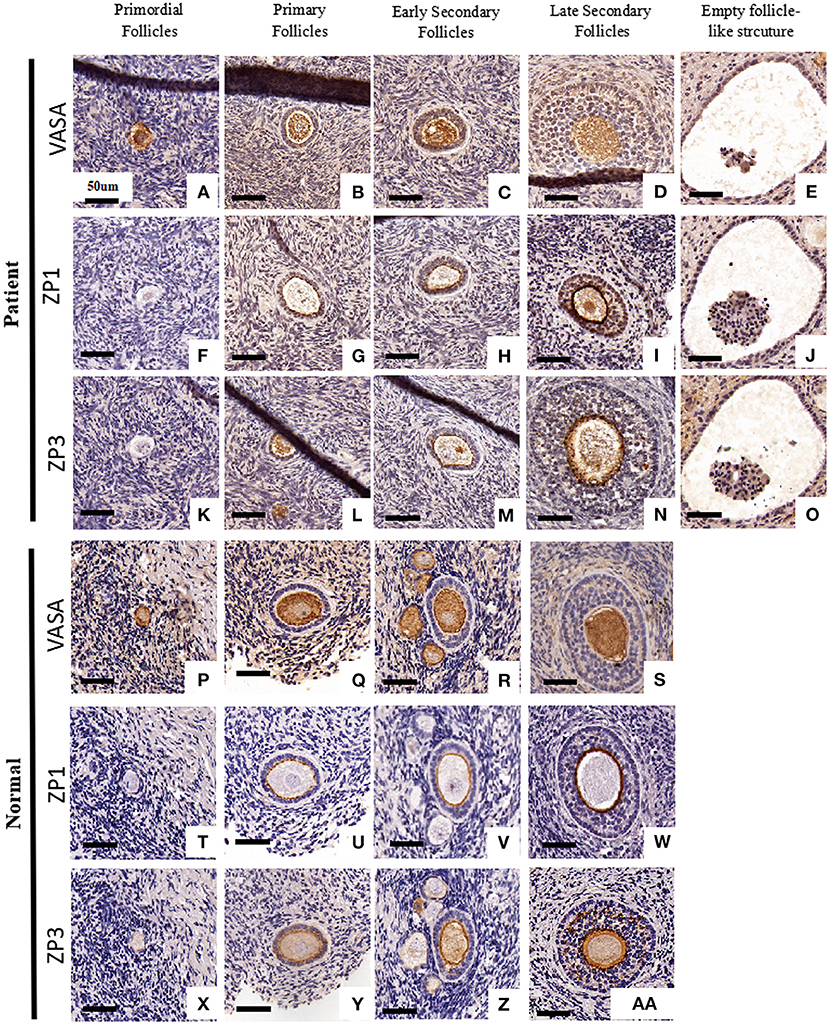
Figure 2. Ovarian histology and immunohistochemistry staining of the EFS patient with a heterozygous ZP3 deletion. Ovaries isolated from the EFS patient carrying a ZP3 mutation (A–O) and a normal control (P–Z, AA) were sectioned and stained with antibodies against VASA (A–E, P–S), ZP1 (F–J, T–W), or ZP3 (K–O, X–Z, AA). The scale bars represent 50 μm.
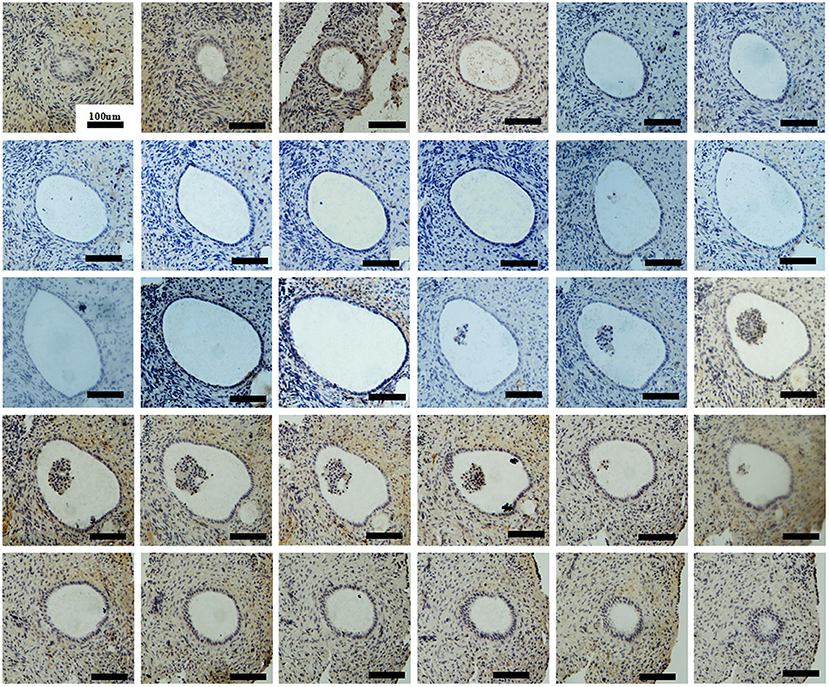
Figure 3. Ovarian histology and immunohistochemistry staining of serial sections showing an intact empty follicle-like structure of the EFS patient with a heterozygous ZP3 deletion. There are a total of 30 sections with 5 μm per section and 223 μm in its widest part.
Discussion
Since 2014, dozens of mutations in human ZP genes have been identified as the cause of oocyte anomalies and female infertility. The mutations in ZP gene are the predominant causes of EFS (Yang et al., 2020). Most ZP mutations in EFS patients have been identified in ZP1, while only two were found in ZP2 and ZP3. Here, we performed genetic analysis on a Chinese woman who was diagnosed with primary infertility and EFS, considering that oocytes could not be obtained in five cycles of oocyte retrieval following ovarian stimulation. By using WES and Sanger sequencing, we identified a novel heterozygous deletion of ZP3 inherited in an autosomal dominant pattern. This is in agreement with the earlier studies that identified two heterozygous missense mutations in ZP3 inherited dominantly in four independent EFS families. Our functional studies showed that this heterozygous ZP3 deletion led to the degradation of both the mutant and wild-type ZP3 proteins.
The human ZP is composed of four glycoproteins, called ZP1–4 (Lefievre et al., 2004). During folliculogenesis, ZP2 and ZP3, acting as building blocks, polymerize and incorporate into fibrils that form an extracellular matrix around the growing oocytes, while ZP1 acts as a cross-linker of the individual fibrils (Wassarman, 2008). ZPD is a common element of ZP proteins that consists of two separated domains marked as ZP-N and ZP-C. It is responsible for mediating homodimerization of intracellular ZP3 (dormant status) as well as the heteropolymerization of extracellular ZP2 and ZP3 and their incorporation into ZP fibrils (Bokhove and Jovine, 2018). Thr189_Gly193 is located within the ZP-C subdomain, and its deletion might affect formation and stabilization of dimeric ZP3, thereby leading to the degradation of ZP3 proteins within cells.
The zona matrix acts as a structural basis supporting the formation of gap junctions, a key structure for signal transduction between the growing oocyte and the surrounding cumulus cells (Wassarman, 2008). Our recent study presented the morphological evidence showing an abnormal ZP assembly and antral-follicular development in EFS patients with biallelic ZP1 mutations (Dai et al., 2019). Here, similar anomalies in ZP assembly were observed in the EFS patient carrying a heterozygous ZP3 mutation, but degenerated oocytes were observed in the earlier stages and pre-antral follicles. However, dispersive ZP3 signals were detected surrounding the patient's oocyte within a secondary follicle (Figure 2N), which might result from secretion and accumulation of the relict ZP3 protein during follicle growth. We speculated that the shortage of secreted ZP3 protein and the defective ZPD in mutant ZP3 affect the ZP assembly, which further leads to a defective gap junction and cumulus–oocyte complex organization, eventually resulting in oocyte degeneration and empty follicle-like structures.
Interestingly, our earlier study showed that patients carrying homozygous ZP2-truncating mutations produced mature oocytes with a thin ZP that lacks ZP2, and this ZP2-null matrix sustained until the blastocyst stage (Dai et al., 2019b). ZP4 is a pseudogene in mice, while it is expressed in humans and may substitute ZP2 in forming a zona matrix as reported in mice (Avella et al., 2014). Collectively, these observations suggest a key role of ZP3 in ZP assembly and support the opinion that the absence of ZP3 cannot be overcome (Fahrenkamp et al., 2020).
In conclusion, we reported a novel heterozygous ZP3 deletion associated with EFS and female infertility. We also showed the abnormal ZP assembly and pre-antral follicular development. Our findings expand the mutational spectrum associated with human EFS, and provide new insights into its pathogenesis.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of the Reproductive and Genetic Hospital of CITIC-Xiangya. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
CD, GLu, and GLin conceived and designed the study. YC, ZW, WH, and YW carried out the experiments. JD, SC, and FG provided the clinical samples. YC wrote the manuscript. CD and GLin critically commented on and edited the manuscript. All authors read and approved the final version of the manuscript.
Funding
This work was supported by grants from the National Key Research and Development Program of China (2016YFC1000200), the National Natural Science Foundation of China (81901553), and the Scientific Research Foundation of Reproductive and Genetic Hospital of China International Trust Investment Corporation (CITIC) Xiangya (YNXM-201911).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
References
Avella, M. A., Baibakov, B., and Dean, J. (2014). A single domain of the ZP2 zona pellucida protein mediates gamete recognition in mice and humans. J. Cell Biol. 205, 801–809. doi: 10.1083/jcb.201404025
Awonuga, A., Govindbhai, J., Zierke, S., and Schnauffer, K. (1998). Continuing the debate on empty follicle syndrome: can it be associated with normal bioavailability of beta-human chorionic gonadotrophin on the day of oocyte recovery? Hum. Reprod. 13, 1281–1284. doi: 10.1093/humrep/13.5.1281
Bokhove, M., and Jovine, L. (2018). Structure of zona pellucida module proteins. Curr. Top. Dev. Biol. 130, 413–442. doi: 10.1016/bs.ctdb.2018.02.007
Chen, C., Xu, X., Kong, L., Li, P., Zhou, F., Zhao, S., et al. (2018). Novel homozygous nonsense mutations in LHCGR lead to empty follicle syndrome and 46, XY disorder of sex development. Hum. Reprod. 33, 1364–1369. doi: 10.1093/humrep/dey215
Chen, T., Bian, Y., Liu, X., Zhao, S., Wu, K., Yan, L., et al. (2017). A recurrent missense mutation in ZP3 causes empty follicle syndrome and female infertility. Am. J. Hum. Genet. 101, 459–465. doi: 10.1016/j.ajhg.2017.08.001
Coulam, C. B., Bustillo, M., and Schulman, J. D. (1986). Empty follicle syndrome. Fertil. Steril. 46, 1153–1155. doi: 10.1016/S0015-0282(16)49898-5
Dai, C., Chen, Y., Hu, L., Du, J., Gong, F., Dai, J., et al. (2019a). ZP1 mutations are associated with empty follicle syndrome: evidence for the existence of an intact oocyte and a zona pellucida in follicles up to the early antral stage. A case report. Hum. Reprod. 34, 2201–2207. doi: 10.1093/humrep/dez174
Dai, C., Hu, L., Gong, F., Tan, Y., Cai, S., Zhang, S., et al. (2019b). ZP2 pathogenic variants cause in vitro fertilization failure and female infertility. Genet. Med. 21, 431–440. doi: 10.1038/s41436-018-0064-y
Fagbohun, C. F., and Downs, S. M. (1990). Maturation of the mouse oocyte-cumulus cell complex: stimulation by lectins. Biol. Reprod. 42, 413–423. doi: 10.1095/biolreprod42.3.413
Fahrenkamp, E., Algarra, B., and Jovine, L. (2020). Mammalian egg coat modifications and the block to polyspermy. Mol. Reprod. Dev. 87, 326–340. doi: 10.1002/mrd.23320
Hasegawa, A., and Koyama, K. (2007). Contribution of zona proteins to oocyte growth. Soc. Reprod. Fertil. Suppl. 63, 229–235. doi: 10.3192/jsirib.22.1
Huang, H. L., Lv, C., Zhao, Y. C., Li, W., He, X. M., Li, P., et al. (2014). Mutant ZP1 in familial infertility. N. Engl. J. Med. 370, 1220–1226. doi: 10.1056/NEJMoa1308851
Jovine, L., Qi, H., Williams, Z., Litscher, E., and Wassarman, P. M. (2002). The ZP domain is a conserved module for polymerization of extracellular proteins. Nat. Cell Biol. 4, 457–461. doi: 10.1038/ncb802
Lefievre, L., Conner, S. J., Salpekar, A., Olufowobi, O., Ashton, P., Pavlovic, B., et al. (2004). Four zona pellucida glycoproteins are expressed in the human. Hum. Reprod. 19, 1580–1586. doi: 10.1093/humrep/deh301
Lu, X., Yan, Z., Cai, R., Khor, S., Wu, L., Sun, L., et al. (2019). Pregnancy and live birth in women with pathogenic LHCGR variants using their own oocytes. J. Clin. Endocrinol. Metab. 104, 5877–5892. doi: 10.1210/jc.2019-01276
Mascarenhas, M. N., Flaxman, S. R., Boerma, T., Vanderpoel, S., and Stevens, G. A. (2012). National, regional, and global trends in infertility prevalence since 1990: a systematic analysis of 277 health surveys. PLoS Med. 9:e1001356. doi: 10.1371/journal.pmed.1001356
Pang, P. C., Chiu, P. C., Lee, C. L., Chang, L. Y., Panico, M., Morris, H. R., et al. (2011). Human sperm binding is mediated by the sialyl-Lewis(x) oligosaccharide on the zona pellucida. Science 333, 1761–1764. doi: 10.1126/science.1207438
Revelli, A., Carosso, A., Grassi, G., Gennarelli, G., Canosa, S., and Benedetto, C. (2017). Empty follicle syndrome revisited: definition, incidence, aetiology, early diagnosis and treatment. Reprod. Biomed. Online 35, 132–138. doi: 10.1016/j.rbmo.2017.04.012
Veeck, L. L. (1988). Oocyte assessment and biological performance. Ann. N. Y. Acad. Sci. 541, 259–274. doi: 10.1111/j.1749-6632.1988.tb22263.x
Wang, J., Yang, X., Sun, X., Ma, L., Yin, Y., He, G., et al. (2021). A novel homozygous nonsense mutation in zona pellucida 1 (ZP1) causes human female empty follicle syndrome. J. Assist. Reprod. Genet. doi: 10.1007/s10815-021-02136-x. [Epub ahead of print].
Wassarman, P. M. (2008). Zona pellucida glycoproteins. J. Biol. Chem. 283, 24285–24289. doi: 10.1074/jbc.R800027200
Yan, L., Yang, M., Guo, H., Yang, L., Wu, J., Li, R., et al. (2013). Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol. 20, 1131–1139. doi: 10.1038/nsmb.2660
Yang, P., Chen, T., Liu, Y., Hou, Z., Wu, K., Cao, Y., et al. (2020). The critical role of ZP genes in female infertility characterized by empty follicle syndrome and oocyte degeneration. Fertil. Steril. doi: 10.1016/j.fertnstert.2020.11.003. [Epub ahead of print].
Yuan, P., He, Z., Zheng, L., Wang, W., Li, Y., Zhao, H., et al. (2017). Genetic evidence of “genuine” empty follicle syndrome: a novel effective mutation in the LHCGR gene and review of the literature. Hum. Reprod. 32, 944–953. doi: 10.1093/humrep/dex015
Keywords: ZP3 mutation, empty follicle syndrome, zona pellucida, female infertility, follicular development
Citation: Chen Y, Wang Z, Wu Y, He W, Du J, Cai S, Gong F, Lu G, Lin G and Dai C (2021) Case Report: A Novel Heterozygous ZP3 Deletion Associated With Empty Follicle Syndrome and Abnormal Follicular Development. Front. Genet. 12:690070. doi: 10.3389/fgene.2021.690070
Received: 02 April 2021; Accepted: 20 April 2021;
Published: 19 May 2021.
Edited by:
Fan Jin, Zhejiang University, ChinaReviewed by:
Tomoko Kawai, Hiroshima University, JapanQingxue Zhang, Sun Yat-Sen Memorial Hospital, China
Copyright © 2021 Chen, Wang, Wu, He, Du, Cai, Gong, Lu, Lin and Dai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Can Dai, ZGFpY2FuQGxpdmUuY24=; Ge Lin, bGluZ2dmQGhvdG1haWwuY29t