 Przemyslaw Kosinski1†
Przemyslaw Kosinski1† Milena Greczan
Milena Greczan Aleksandra Jezela-Stanek
Aleksandra Jezela-Stanek
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet., 07 June 2021
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.674722
This article is part of the Research TopicInherited Protein Glycosylation Defects in Human DiseasesView all 15 articles
Fryns syndrome is an autosomal recessive multiple congenital anomaly syndrome, with diaphragmatic defects and secondary lung hypoplasia as cardinal features. Despite it was reported first in 1979, its exact etiology has not been established to date. With this review, we would like to draw attention to the prenatal presentation of multiple congenital anomalies syndromes, resulting from defects in the synthesis of glycosylphosphatidylinositol anchors, to be considered in a prenatal assessment of fetuses with DH and Fryns-like phenotype.
Fryns syndrome (FRNS, %229850) is an autosomal recessive multiple congenital anomaly syndrome. It was reported first in 1979 in two siblings with numerous congenital anomaly, including coarse facies, diaphragmatic hernia, absence of lung lobulation, and distal limb deformities (Fryns and Van den Berghe, 1979). To date, the exact etiology of FRNS (FS) has not been established, and genetic heterogeneity remains highly probable. From the clinical perspective, diaphragmatic defects and secondary lung hypoplasia are cardinal features (Fryns, 1987).
Congenital diaphragmatic hernia (CDH) is a developmental discontinuity of the diaphragm. As a result, the abdominal viscera may herniate into the chest and leads to lung hypoplasia. It occurs in approximately 1 in 3,000 live births but results in high morbidity and mortality, in neonatal and infancy period (Wynn et al., 2014). The most common localization is the posterolateral left side of the diaphragm (75–90% of cases), but the defect can also be right-sided (10–15% of cases) or even bilateral (1–2% of cases) (Deprest et al., 2011). The pulmonary vasculature malformation are the main cause of CDH-related pulmonary hypertension (Sluiter et al., 2012). Moreover, increased vascular resistance and decreased surface area for gas exchange are observed (Hislop and Reid, 1976).
Unfortunately, the pathogenesis of CDH has not been established definitively, and encompass wide range of genetic disorders, from chromosomal defects, noted in at least 10–30% cases, including polyploidies, trisomy 18, partial trisomy 5, partial trisomy 20 or tetrasomy 12p (Pallister-Killian syndrome) (Graham and Devine, 2005; Pober, 2008; Kosinski and Wielgos, 2017) to monogenic syndromes (as Apert, CHARGE, Coffin-Siris, Goltz, Swyer, Brachmann-Cornelia De Lange, Simpson-Golabi-Behmel, Donnai-Barrow, Mathew-Wood, Jarcho-Levin, and Fraser). The influence of genetics in CDH was recently reviewed by Yu et al. (2020). This comprehensive review explains the genetic contributions to CDH are highly heterogeneous and suggests CDH genes are often transcription factors, genes involved in cell migration or the components of extracellular matrix. In cases of multiple abnormalities, malformations may occur in all major organ systems (Pober et al., 2005; Pober, 2007; Taylor et al., 2009). Since the etiology of CDH varies and is difficult to establish, with this review, we would like to draw attention to the prenatal presentation of multiple congenital anomalies syndromes, resulting from defects in the synthesis of glycosylphosphatidylinositol (GPI) anchors, as a possible cause, and to be considered in a prenatal assessment of fetuses with DH and Fryns-like phenotype.
In 1987 following criteria were suggested by Fryns (1987) to establish the diagnosis of FRNS: polyhdramnios, often occurring in the presence of normal fetal growth, in an infant with characteristic facial dysmorphism – a coarse face, a broad flat nasal bridge (but a large nose anteriorly), a short upper lip, a small jaw, a cleft lip and palate, and poorly shaped auricles with attached earlobes. The phalanges are distally hypoplastic with rudimentary and dysplastic nails. The cardinal features within internal organs are diaphragmatic defects with secondary lung hypoplasia. Moreover, malrotation of the intestine, duodenal or multiple atresias and a bicornuate uterus may be noted. Cloudy corneas, facial hirsutism and pulmonary segmentation defects all help in establishing the diagnosis. The eye findings in the syndrome were reviewed by Pierson et al. (2004), who found corneal clouding in nine out of 15 patients, anophthalmia in two and retinal dysplasia in two. Subtle skeletal abnormalities are also part of the condition (Kershisnik et al., 1991; Tsukahara et al., 1995).
Fryns (1995) points out that growth parameters may be above the 75th centile at birth with apparent macrocephaly. The author suggested that the finding of diaphragmatic hernia with intrauterine growth retardation is against the diagnosis (Fryns, 1995). Vargas et al. (2000) described discordant phenotype for diaphragmatic hernia in monozygotic twins. A few cases have survived the neonatal period and are severely retarded (Bamforth et al., 1989; Cunniff et al., 1990; Hanssen et al., 1992; Van Hove et al., 1995). Dingens and Fryns (1999) reported that the girl first described by Hanssen et al. (1992) had died at the age of 10 years in status epilepticus. She was found to have hematometra.
The individual presented by Davis and Samarakkody (2002) (with no facial photographs available), was diagnosed with Fryns syndrome based on constellation of physical anomalies, as coarse facial feature, fifth digits’ hypoplasia and nail absence, tricuspid regurgitation, diaphragmatic defects, as well as Hirschsprung’s disease. The finding of a diaphragmatic defect and renal abnormalities might alert the astute investigator to the possibility of this syndrome, but note that Siebert et al. found that 13–27% of infants with a diaphragmatic defect had a urinary tract anomaly (Siebert et al., 1990). Willems et al. (1991) reported a possible case with the diaphragm reduced to a fibrous web without an overt hernia. In some cases CDH might be the only ultrasound abnormality detected prenatally.
In a review of 23 patients with a diaphragmatic hernia (Congenital Diaphragmatic Hernia Study Group, 2002), seven patients underwent surgical repair at an average age of 7.5 days (range, 6 h to 14 days); the mortality rate was as high as 83% in patients with both CDH and FS compared to 33% in patients with unilateral isolated CDH (Neville et al., 2002).
McPherson et al. reported a case of Pallister-Killian syndrome (resulted from mosaic isochromosome 12p) with some Fryns syndrome features (facial dysmorphism and digital anomalies) and pointed out the possibility of diagnostic challenge (McPherson et al., 1993). Stratton et al. (1994) and Rodriguez et al. (1994) noted similar cases with Pallister–Killian syndrome and re-emphasized the latter point.
Three cases have had osteochondrodysplasia (mostly delayed ossification) (Kershisnik et al., 1991; Slavotinek et al., 2005). Wilgenbus et al. (1994) reported two fetuses with features of the condition where there were no diaphragmatic hernias, but one of them had severe lung hypoplasia. Bartsch et al. (1995) reported similar two more cases with Fryns syndrome but without lateral diaphragmatic defects. Records of 1,833 liveborn patients with CDH from 83 hospitals revealed Fryns syndrome in 23 cases witch constitutes for 1.3% (Neville et al., 2002). On the other hand, CDH among Fryns syndrome patients is present in approximately 76% to 89% of reported cases (Cunniff et al., 1990; Van Hove et al., 1995).
While Ramsing et al. (2000) delineated three fetuses between 15 and 31 weeks of gestation with FS features (Cases 3–5). They also reported a family where two fetuses had a more severe form or a different condition (Cases 1 and 2). Several cases have been reported with features of the discussed condition and chromosome aberrations, including duplication of 1q24–31 (Clark and Fenner-Gonzales, 1989), terminal 6q deletion (Krassikoff and Sekhon, 1990), a submicroscopic deletion of 1q41–42 (Kantarci et al., 2006) and partial trisomy 22 (Dean et al., 1991). It should be noted that mosaic trisomy 9 also shares these features. Ladonne et al. (1996) reported a case of trisomy 22 with features of Fryns syndrome. de Beaufort et al. (2000) presented a patient with some features of the condition who had a partial trisomy 22. There is an excellent review by Slavotinek describing diagnostic features of Fryns syndrome (Slavotinek, 2004).
Based on previously published articles, Lin et al. (2005) suggested a definition of the Fryns phenotype, encompassing the combination of six criteria: diaphragmatic defects, pulmonary hypoplasia, recognizable facial features, distal digital hypoplasia, associated anomalies (polyhydramnios, cloudy corneas, cleft lip/palate and genitourinary, cardiovascular and cerebral malformations), and affected sibs. The presence of four among mentioned characteristics may provide a strict clinical definition of FS, while three constitute a broad definition of FS. To date, no gene had been clearly associated with FS. Thus the diagnosis typically remains clinical after the exclusion of chromosomal aberrations using classical karyotyping and/or array comparative genomic hybridization (aCGH) (Jezela-Stanek et al., 2020).
Recently, several patients with syndromic CDH, harboring pathogenic variants in PIGN (Phosphatidyl Inositol Glycan Anchor Biosynthesis, Class N), PIGA (Phosphatidyl Inositol Glycan Anchor Biosynthesis, Class A), and PIGV (Phosphatidyl Inositol Glycan Anchor Biosynthesis, Class V) gene have been characterized (Longoni et al., 1993; Slavotinek, 1993; Brady et al., 2014; McInerney-Leo et al., 2016; Thompson and Cole, 2016; Reynolds et al., 2017; Alessandri et al., 2018; Witters et al., 2018; Bayat et al., 2020). These molecular diagnosed have expanded the genetic spectrum of Fryns-like phenotype to be part of congenital disorder of glycosylations (CDG), which encompass GPI-anchor biosynthesis defects (GPIBD).
Glycosylphosphatidylinositol is a phosphoglyceride that anchors proteins to the outer leaflet of the cellular membrane. GPIBDs form a group of heterogeneous diseases with a common underlying cause of inappropriate GPI-anchors biosynthesis or modification. Correct GPI biosynthesis and modification determine GPI-anchored proteins’ expression on the cell surface and thus determines the proper antigen presentation, cell adhesion, and signal transduction (Alessandri et al., 2018). According to the clinical and laboratory findings, GPIBDs have been historically divided into two main subgroups: hyperphosphatasia with mental retardation syndrome (HPMRS) – a group of GPIBDs with elevated serum alkaline phosphatase activity (ALP) (mutations in PIGV, PGAP2, PGAP3, PIGO, PIGW, and PIGY) and multiple congenital anomalies hypotonia seizures (MCAHS) – with normal serum ALP (mutations in PIGA, PIGN, PIGT). However, the number of GPIBSDs that cannot be divided into these two groups is growing (Knaus et al., 2018). Accordingly, the terminology is questioned and no longer recommended.
PIGN-CDG and PIGA-CDG are MCAHS, characterized by severe developmental delay, early-onset intractable seizures, global hypotonia, congenital cardiac, gastrointestinal, renal and central nervous system (CNS) anomalies, nystagmus, failure to thrive and facial dysmorphism. In some individuals, brachytelephalangy with nail hypoplasia, joint contractures and CDH have also been described (Alessandri et al., 2018; Knaus et al., 2018). Surprisingly, ALP levels are elevated in some individuals with PIGA-CDG and borderline in some individuals with PIGN-CDG (Knaus et al., 2018).
PIGV-CDG is HPMRS (also known as Mabry syndrome), characterized by the constantly elevated ALP, intellectual disability, seizures with facial dysmorphia, brachytelephalangy and nail hypoplasia. In some individuals, Hirschprung disease, renal and anorectal malformations and CDH have been described (Reynolds et al., 2017).
No specific treatment is available so far for these conditions, and early death may occur.
The summary of described cases is presented in Table 1. Please note that the diagnoses cited by us are the original ones described in the discussed publications (that is why the terminology is diverse).
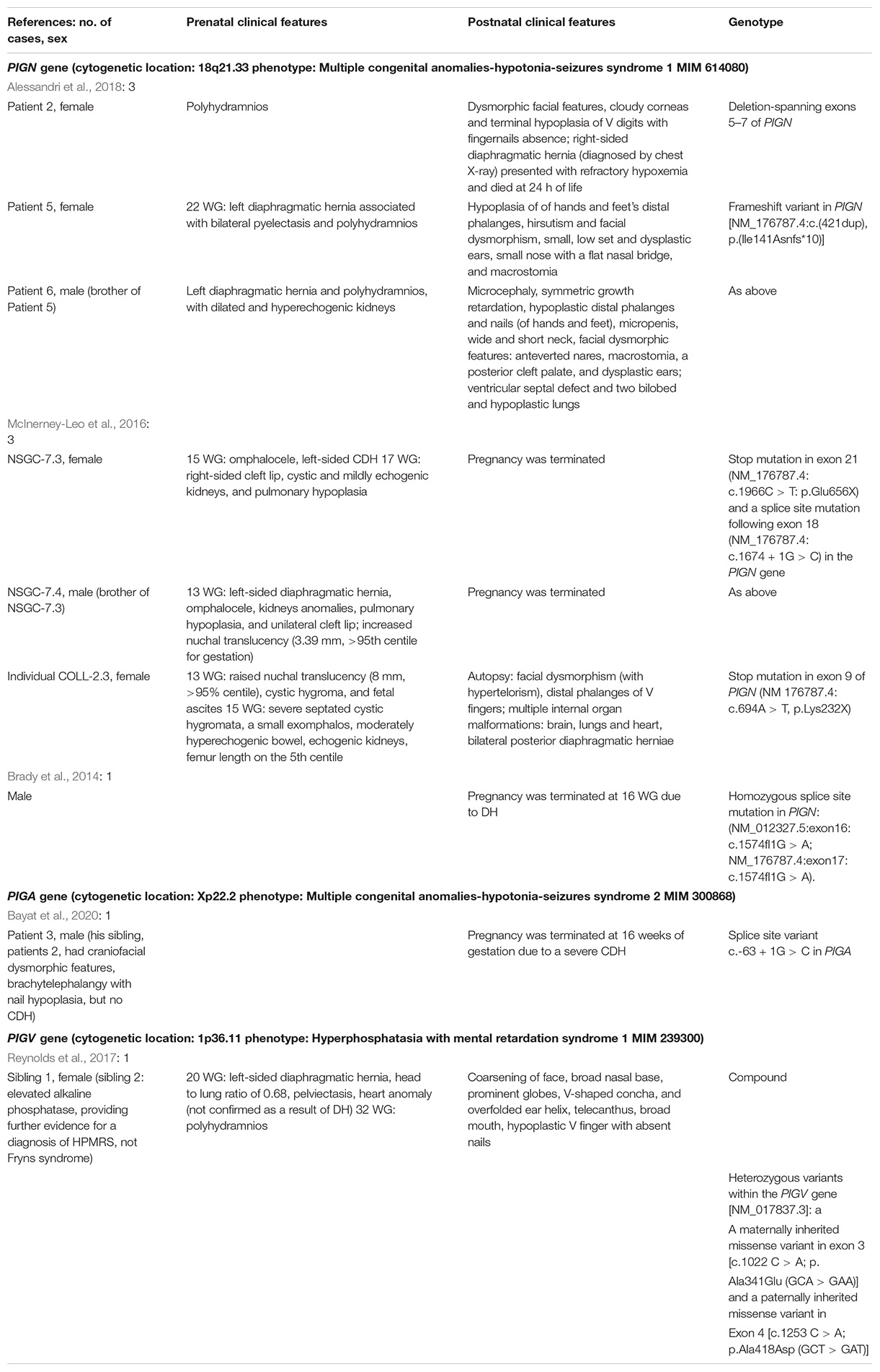
Table 1. Summary of reported to date cases with Fryns syndrome phenotype due to glycosylphosphatidylinositol biosynthesis defects (GPIBDs).
Due to the complex nature and not fully recognized etiology of “Fryns syndrome”, some confusion about the terminology exists. When it is a clinical entity, “Fryns-like phenotype” term should be used. Otherwise, Fryns syndrome is a disorder with presumed autosomal recessive inheritance. It is thus of primary importance to distinguish between a clinical or molecular diagnosis. In all individuals/fetuses within the spectrum of Fryns syndrome phenotype genetic diagnostics is rational.
It has to be underlined that a genetic abnormality must be suspected in each case of CDH diagnosed prenatally, when accompany with other malformations. Except for CDH, the clinical presentation of Fryns-like phenotype includes pulmonary hypoplasia, craniofacial dysmorphism, cleft lip/palate, brachytelephalangy accompany with nail hypoplasia, and various other internal organ malformations. Although no major genetic cause has been identified for FRNS, biallelic pathogenic variants in PIGN, PIGV and X-linked in PIGA, all encoding a component of the GPI-anchor biosynthesis pathway, have been identified in several probands with a phenotype fitting with FRNS.
During the genetic evaluation of fetuses that manifest developmental anomalies, it is mandatory to check for chromosomal aberrations (and perform karyotype and/or chromosomal microarray) and also to consider monogenic disorders (Committee on Genetics and the Society for Maternal-Fetal Medicine, 2016; Hay et al., 2018). All of the GPIBDs presented above, means caused by pathogenic variants in the PIGN, PIGV, and PIGA genes, are clinically very heterogenic and congenital malformations are not among their primary characteristics in many other affected individuals. All these syndromes belong to neurodevelopmental disorders and manifest predominantly with facial dysmorphism, global developmental disorders (especially speech and language), muscular hypotonia and seizures, as well as cerebral or cerebellar atrophy in PIGN-CDG, minor CNS anomalies in PIGA- and PIGV-CDG (Siebert et al., 1990; Knaus et al., 2018; Bayat et al., 2020).
Given the high mortality rate resulting from severe congenital malformations, reliable and early genetic diagnosis is crucial. It only allows for determining the recurrence risk (which is 25% for autosomal recessive disorder and even up to 50% for X-linked disorders, if the genetic status of mothers is unknown) and perform genetic testing in subsequent pregnancies. Thus, we propose to include GPI biosynthesis defects in the differential diagnosis during the prenatal evaluation of fetuses with diaphragmatic defects, especially in families with positive family history. When isolated cases are evaluated, the additional ultrasound features that may direct the diagnosis are large for gestational age, hypertelorism, cleft lip/palate, atrial/ventricular septal defects, urogenital anomalies and hand/feet abnormal positioning. However, we wonder how much safer it would be, keeping in mind the complexity of Fryns syndrome, to utilize the term “Fryns syndrome – like phenotype”.
PK: manuscript writing and literature review. MG: manuscript writing. AJ-S: concept of the manuscript, writing, and literature review. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Alessandri, J. L., Gordon, C. T., Jacquemont, M. L., Gruchy, N., Ajeawung, N. F., Benoist, G., et al. (2018). Recessive loss of function PIGN alleles, including an intragenic deletion with founder effect in La Reunion Island, in patients with Fryns syndrome. Eur. J. Hum. Genet. 26, 340–349. doi: 10.1038/s41431-017-0087-x
Bamforth, J. S., Leonard, C. O., Chodirker, B. N., Chitayat, D., Gritter, H. L., Evans, J. A., et al. (1989). Congenital diaphragmatic hernia, coarse facies, and acral hypoplasia: Fryns syndrome. Am. J. Med. Genet. 32, 93–99. doi: 10.1002/ajmg.1320320120
Bartsch, O., Meinecke, P., and Kamin, G. (1995). Fryns syndrome: two further cases without lateral diaphragmatic defects. Clin. Dysmorphol. 4, 352–358. doi: 10.1097/00019605-199510000-00012
Bayat, A., Knaus, A., Pendziwiat, M., Afenjar, A., Barakat, T. S., Bosch, F., et al. (2020). Lessons learned from 40 novel PIGA patients and a review of the literature. Epilepsia 61, 1142–1155.
Brady, P. D., Moerman, P., De Catte, L., Deprest, J., Devriendt, K., and Vermeesch, J. R. (2014). Exome sequencing identifies a recessive PIGN splice site mutation as a cause of syndromic congenital diaphragmatic hernia. Eur. J. Med. Genet. 57, 487–493. doi: 10.1016/j.ejmg.2014.05.001
Clark, R. D., and Fenner-Gonzales, M. (1989). Apparent Fryns syndrome in a boy with a tandem duplication of 1q24-31.2. Am. J. Med. Genet. 34, 422–426. doi: 10.1002/ajmg.1320340319
Committee on Genetics and the Society for Maternal-Fetal Medicine (2016). Committee opinion No. 682 summary: microarrays and next-generation sequencing technology: the use of advanced genetic diagnostic tools in obstetrics and gynecology. Obstet. Gynecol. 128, 1462–1463. doi: 10.1097/aog.0000000000001814
Congenital Diaphragmatic Hernia Study Group (2002). Available online at: https://med.uth.edu/pediatricsurgery/research/research-centers-and-programs/cdhsg/
Cunniff, C., Jones, K. L., Saal, H. M., and Stern, H. J. (1990). Fryns syndrome: an autosomal recessive disorder associated with craniofacial anomalies, diaphragmatic hernia, and distal digital hypoplasia. Pediatrics 85, 499–504.
Davis, C., and Samarakkody, U. (2002). Fryns syndrome: a surviving case with associated Hirschsprung’s disease and hemidiaphragmatic agenesis. J. Paediatr. Child Health 38, 318–320. doi: 10.1046/j.1440-1754.2002.00820.x
de Beaufort, C., Schneider, F., Chafai, R., Colette, J. M., Delneste, D., and Pierquin, G. (2000). Diaphragmatic hernia and Fryns syndrome phenotype in partial trisomy 22. Genet. Couns. 11, 181–182.
Dean, J. C., Couzin, D. A., Gray, E. S., Lloyd, D. J., and Stephen, G. S. (1991). Apparent Fryns’ syndrome and aneuploidy: evidence for a disturbance of the midline developmental field. Clin. Genet. 40, 349–352. doi: 10.1111/j.1399-0004.1991.tb03108.x
Deprest, J. A., Nicolaides, K., and Gratacos, E. (2011). Fetal surgery for congenital diaphragmatic hernia is back from never gone. Fetal Diagn. Ther. 29, 6–17. doi: 10.1159/000322844
Dingens, M., and Fryns, J. P. (1999). Hematometra and sudden death after status epilepticus in the adolescent female with Fryns syndrome. Genet. Couns. 10, 329–330.
Fryns, J. P. (1987). Fryns syndrome: a variable MCA syndrome with diaphragmatic defects, coarse face, and distal limb hypoplasia. J. Med. Genet. 24, 271–274. doi: 10.1136/jmg.24.5.271
Fryns, J. P. (1995). Prenatal diagnosis and long survival of Fryns syndrome. Prenat. Diagn. 15, 97–98. doi: 10.1002/pd.1970150125
Fryns, J. P., and Van den Berghe, H. (1979). Corneal clouding, subvalvular aortic stenosis, and midfacial hypoplasia associated with mental deficiency and growth retardation–a new syndrome? Eur. J. Pediatr. 131, 179–183. doi: 10.1007/bf00538941
Graham, G., and Devine, P. C. (2005). Antenatal diagnosis of congenital diaphragmatic hernia. Semin. Perinatol. 29, 69–76. doi: 10.1053/j.semperi.2005.04.002
Hanssen, A. M., Schrander-Stumpel, C. T., Thiry, P. A., and Fryns, J. P. (1992). Fryns syndrome: another example of non-lethal outcome with severe mental handicap. Genet. Couns. 3, 187–193.
Hay, S. B., Sahoo, T., Travis, M. K., Hovanes, K., Dzidic, N., Doherty, C., et al. (2018). ACOG and SMFM guidelines for prenatal diagnosis: is karyotyping really sufficient? Prenat. Diagn. 38, 184–189. doi: 10.1002/pd.5212
Hislop, A., and Reid, L. (1976). Persistent hypoplasia of the lung after repair of congenital diaphragmatic hernia. Thorax 31, 450–455. doi: 10.1136/thx.31.4.450
Jezela-Stanek, A., Mierzewska, H., and Szczepanik, E. (2020). Vertical nystagmus as a feature of PIGN-related glycosylphosphatidylinositol biosynthesis defects. Clin. Neurol. Neurosurg. 196:106033. doi: 10.1016/j.clineuro.2020.106033
Kantarci, S., Casavant, D., Prada, C., Russell, M., Byrne, J., Haug, L. W., et al. (2006). Findings from aCGH in patients with congenital diaphragmatic hernia (CDH): a possible locus for Fryns syndrome. Am. J. Med. Genet. A 140, 17–23. doi: 10.1002/ajmg.a.31025
Kershisnik, M. M., Craven, C. M., Jung, A. L., Carey, J. C., and Knisely, A. S. (1991). Osteochondrodysplasia in Fryns syndrome. Am. J. Dis. Child 145, 656–660. doi: 10.1001/archpedi.1991.02160060074024
Knaus, A., Pantel, J. T., Pendziwiat, M., Hajjir, N., Zhao, M., Hsieh, T. C., et al. (2018). Characterization of glycosylphosphatidylinositol biosynthesis defects by clinical features, flow cytometry, and automated image analysis. Genome Med. 10:3.
Kosinski, P., and Wielgos, M. (2017). Congenital diaphragmatic hernia: pathogenesis, prenatal diagnosis and management - literature review. Ginekol. Pol. 88, 24–30. doi: 10.5603/gp.a2017.0005
Krassikoff, N., and Sekhon, G. S. (1990). Terminal deletion of 6q and Fryns syndrome: a microdeletion/syndrome pair? Am. J. Med. Genet. 36, 363–364. doi: 10.1002/ajmg.1320360327
Ladonne, J. M., Gaillard, D., Carre-Pigeon, F., and Gabriel, R. (1996). Fryns syndrome phenotype and trisomy 22. Am. J. Med. Genet. 61, 68–70. doi: 10.1002/(sici)1096-8628(19960102)61:1<68::aid-ajmg13>3.0.co;2-u
Lin, A. E., Pober, B. R., Mullen, M. P., and Slavotinek, A. M. (2005). Cardiovascular malformations in Fryns syndrome: is there a pathogenic role for neural crest cells? Am. J. Med. Genet. A 139, 186–193. doi: 10.1002/ajmg.a.31023
Longoni, M., Pober, B. R., and High, F. A. (1993). “Congenital diaphragmatic hernia overview,” in GeneReviews((R)), eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, G. Mirzaa, et al. (Seattle, WA: Wiely).
McInerney-Leo, A. M., Harris, J. E., Gattas, M., Peach, E. E., Sinnott, S., Dudding-Byth, T., et al. (2016). Fryns syndrome associated with recessive mutations in PIGN in two separate families. Hum. Mutat. 37, 695–702. doi: 10.1002/humu.22994
McPherson, E. W., Ketterer, D. M., and Salsburey, D. J. (1993). Pallister-killian and fryns syndromes: nosology. Am. J. Med. Genet. 47, 241–245. doi: 10.1002/ajmg.1320470219
Neville, H. L., Jaksic, T., Wilson, J. M., Lally, P. A., Hardin, W. D. Jr., Hirschl, R. B., et al. (2002). Fryns syndrome in children with congenital diaphragmatic hernia. J. Pediatr. Surg. 37, 1685–1687. doi: 10.1053/jpsu.2002.36695
Pierson, D. M., Taboada, E., and Butler, M. G. (2004). Eye abnormalities in fryns syndrome. Am. J. Med. Genet. A 125A, 273–277. doi: 10.1002/ajmg.a.20520
Pober, B. R. (2007). Overview of epidemiology, genetics, birth defects, and chromosome abnormalities associated with CDH. Am. J. Med. Genet. C Semin. Med. Genet. 145C, 158–171. doi: 10.1002/ajmg.c.30126
Pober, B. R. (2008). Genetic aspects of human congenital diaphragmatic hernia. Clin. Genet. 74, 1–15. doi: 10.1111/j.1399-0004.2008.01031.x
Pober, B. R., Lin, A., Russell, M., Ackerman, K. G., Chakravorty, S., Strauss, B., et al. (2005). Infants with Bochdalek diaphragmatic hernia: sibling precurrence and monozygotic twin discordance in a hospital-based malformation surveillance program. Am. J. Med. Genet. Part A 138A, 81–88. doi: 10.1002/ajmg.a.30904
Ramsing, M., Gillessen-Kaesbach, G., Holzgreve, W., Fritz, B., and Rehder, H. (2000). Variability in the phenotypic expression of fryns syndrome: a report of two sibships. Am. J. Med. Genet. 95, 415–424. doi: 10.1002/1096-8628(20001218)95:5<415::aid-ajmg2>3.0.co;2-j
Reynolds, K. K., Juusola, J., Rice, G. M., and Giampietro, P. F. (2017). Prenatal presentation of Mabry syndrome with congenital diaphragmatic hernia and phenotypic overlap with Fryns syndrome. Am. J. Med. Genet. A 173, 2776–2781. doi: 10.1002/ajmg.a.38379
Rodriguez, J. I., Garcia, I., Alvarez, J., Delicado, A., and Palacios, J. (1994). Lethal Pallister-Killian syndrome: phenotypic similarity with Fryns syndrome. Am. J. Med. Genet. 53, 176–181. doi: 10.1002/ajmg.1320530211
Siebert, J. R., Benjamin, D. R., Juul, S., and Glick, P. L. (1990). Urinary tract anomalies associated with congenital diaphragmatic defects. Am. J. Med. Genet. 37, 1–5. doi: 10.1002/ajmg.1320370102
Slavotinek, A. (1993). “Fryns syndrome,” in GeneReviews((R)), eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, G. Mirzaa, et al. (Seattle, WA: Wiely).
Slavotinek, A. M. (2004). Fryns syndrome: a review of the phenotype and diagnostic guidelines. Am. J. Med. Genet. A 124A, 427–433. doi: 10.1002/ajmg.a.20381
Slavotinek, A. M., Robinson, H., and Steele, M. A. (2005). Fryns syndrome with osteochondrodysplasia. Am. J. Med. Genet. A 134, 454–456. doi: 10.1002/ajmg.a.30351
Sluiter, I., Veenma, D., van Loenhout, R., Rottier, R., de Klein, A., Keijzer, R., et al. (2012). Etiological and pathogenic factors in congenital diaphragmatic hernia. Eur. J. Pediatr. Surg. 22, 345–354.
Stratton, R. F., Moore, C. M., Popham, C. S., DuPont, B. R., and Mattern, V. L. (1994). Pallister-Killian and Fryns syndromes. Am. J. Med. Genet. 51:90. doi: 10.1002/ajmg.1320510124
Taylor, G. A., Atalabi, O. M., and Estroff, J. A. (2009). Imaging of congenital diaphragmatic hernias. Pediatr. Radiol. 39, 1–16. doi: 10.1007/s00247-008-0917-7
Thompson, M. D., and Cole, D. E. (2016). Recessive PIGN mutations in fryns syndrome: evidence for genetic heterogeneity. Hum. Mutat. 37:621. doi: 10.1002/humu.23016
Tsukahara, M., Sase, M., Tateishi, H., Saito, T., Kato, H., and Furukawa, S. (1995). Skeletal manifestations in Fryns syndrome. Am. J. Med. Genet. 55, 217–220. doi: 10.1002/ajmg.1320550213
Van Hove, J. L., Spiridigliozzi, G. A., Heinz, R., McConkie-Rosell, A., Iafolla, A. K., and Kahler, S. G. (1995). Fryns syndrome survivors and neurologic outcome. Am. J. Med. Genet. 59, 334–340. doi: 10.1002/ajmg.1320590311
Vargas, J. E., Cox, G. F., and Korf, B. R. (2000). Discordant phenotype in monozygotic twins with Fryns syndrome. Am. J. Med. Genet. 94, 42–45. doi: 10.1002/1096-8628(20000904)94:1<42::aid-ajmg9>3.0.co;2-6
Wilgenbus, K. K., Engers, R., Crombach, G., and Majewski, F. (1994). Two fetuses with Fryns syndrome without diaphragmatic defects. J. Med. Genet. 31, 962–964. doi: 10.1136/jmg.31.12.962
Willems, P. J., Keersmaekers, G. H., Dom, K. E., Colpaert, C., Schatteman, E., Vergote, I. B., et al. (1991). Fryns syndrome without diaphragmatic hernia? Am. J. Med. Genet. 41, 255–257.
Witters, P., Breckpot, J., Foulquier, F., Preston, G., Jaeken, J., and Morava, E. (2018). Expanding the phenotype of metabolic cutis laxa with an additional disorder of N-linked protein glycosylation. Eur. J. Hum. Genet. 26, 618–621. doi: 10.1038/s41431-017-0044-8
Wynn, J., Yu, L., and Chung, W. K. (2014). Genetic causes of congenital diaphragmatic hernia. Semin. Fetal Neonatal Med. 19, 324–330.
Keywords: glycosylphosphatidylinositol biosynthesis defects, GPIBDs, congenital diaphragmatic hernia, CDH, Fryns syndrome
Citation: Kosinski P, Greczan M and Jezela-Stanek A (2021) Diaphragmatic Hernia as a Prenatal Feature of Glycosylphosphatidylinositol Biosynthesis Defects and the Overlap With Fryns Syndrome – Literature Review. Front. Genet. 12:674722. doi: 10.3389/fgene.2021.674722
Received: 01 March 2021; Accepted: 14 May 2021;
Published: 07 June 2021.
Edited by:
Thomas Schaible, University of Heidelberg, GermanyReviewed by:
Magdalena Sandu, Spitalul Clinic de Copii Doctor Victor Gomoiu, RomaniaCopyright © 2021 Kosinski, Greczan and Jezela-Stanek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aleksandra Jezela-Stanek, amV6ZWxhQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.