 Rongchun Wang1,2,3†
Rongchun Wang1,2,3† Danhui Yang1,2,3†
Danhui Yang1,2,3† Ting Guo1,2,3
Ting Guo1,2,3 Cheng Lei1,2,3Xu Chen1,2,3Xi Kang1,2,3Jie Qing1,2,3*
Cheng Lei1,2,3Xu Chen1,2,3Xi Kang1,2,3Jie Qing1,2,3* Hong Luo1,2,3*
Hong Luo1,2,3*- 1Department of Pulmonary and Critical Care Medicine, The Second Xiangya Hospital, Central South University, Changsha, China
- 2Research Unit of Respiratory Disease, Central South University, Changsha, China
- 3Hunan Diagnosis and Treatment Center of Respiratory Disease, Changsha, China
Background: ODAD3 encodes a protein of 595 amino acids and contain three highly conserved coiled-coil domains, which is essential for cilia axoneme dynein arm assembly and docking. Primary ciliary dyskinesia (PCD) of ODAD3 deficiency are rarely reported. Female infertility in PCD related to ODAD3 variants has not been reported.
Methods: Whole-exome and Sanger sequencing were used to identify the disease-related gene of the patient with PCD in a consanguineous Chinese family. Domain analysis was applied to predict the impact of the variant on ODAD3 protein.
Results: The 35 year-old female patient exhibited chronic sinusitis, diffuse bronchiectasis, dextrocardia and infertility. We identified a novel homozygous variant in ODAD3, c.1166_1169dupAGAC, p.(Leu391Aspfs*105) in the PCD patient by exome sequencing and Sanger sequencing. This frameshift variant was predicted to be disease causing by bioinformatics analysis and was also not presented in the current authorized large genetic databases.
Conclusions: Our study enriches the genetic spectrum and clinical phenotypes of ODAD3 variants in PCD and provide more evidence for future genetic counseling and gene-targeted therapy for this disease.
Introduction
Primary ciliary dyskinesia (PCD, MIM 244400) is a clinically and genetically heterogeneous motile ciliopathy characterized by recurrent upper and lower respiratory infections, subfertility and laterality defect (Mirra et al., 2017). The estimated prevalence of PCD is 1:10 000 to 1:20 000 live-born children (Afzelius and Stenram, 2006). So far, more than 47 genes associated with cilia structure or function dysfunction have been identified causative for PCD (Wallmeier et al., 2020).
ODAD3 (Outer dynein arm docking complex subunit 3; formerly known as CCDC151; coiled-coil domain containing 151) encodes a protein of 595 amino acids which contains three highly conserved coiled-coil domains. This protein was initially recognized as a potential ciliome gene due to its expression in human ciliary axonemes (Ostrowski et al., 2002) and it is essential for cilia axoneme dynein arm assembly and docking (Hjeij et al., 2014). Up to now, PCD caused by ODAD3 variants (MIM 616037, CILD30) are rarely reported and female infertility has not been identified in PCD caused by ODAD3 variants.
In the present study, a novel variant in ODAD3 was identified in a female PCD patient with infertility from a Chinese consanguineous family.
Case Presentation
The patient is a 35 year-old female. She was admitted to our hospital because of chronic cough, purulent sputum, nasal congestion for over 30 years and dyspnea for about 5 years. She has been inflicted by recurrent infections of the lower and upper airways since newborn. And he has been married for 10 years but without pregnancy and was diagnosed with infertility. She had no history of drowning, measles, polio, tuberculosis, immunodeficiency, connective tissue disease, etc. Her family members denied any obvious respiratory or other disease history. Physical examination indicated right-side heart sound. High resolution computed tomography of this patient revealed chronic sinusitis (Figure 1A), diffuse bronchiectasis (Figure 1B), situs inversus: heart dextroversion (Figure 1C) and abdominal organs in the reverse location (Figure 1D). Lung function test revealed a moderate to severe mixed (restrictive and obstructive) ventilation dysfunction. Her nasal nitric oxide concentration (nNO) (6 nl/min) was far below the reference nNO cutoff value of PCD (77 nl/min) (Leigh et al., 2013).
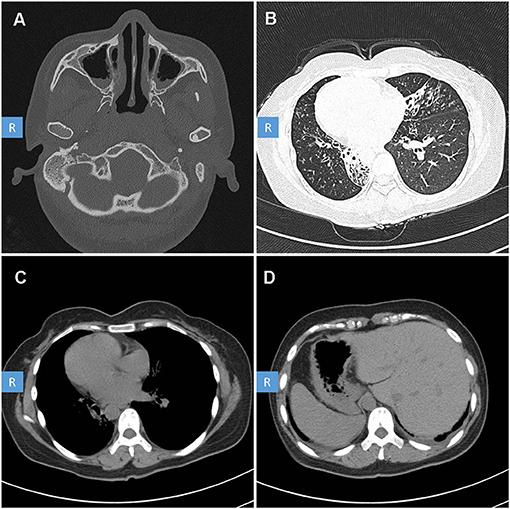
Figure 1. High-resolution computed tomography (HRCT) scan of the patient with PCD. CT scan showed: (A) Sinusitis. (B) Diffuse bronchiectasis. (C) Right-side heart. (D) Abdominal organ heterotaxy: liver in the left side, stomach and spleen in the right side.
Methods
Ethical Compliance
This study was approved by the Review Board of the Second Xiangya Hospital of Central South University in China. Written informed consent was obtained from the patient.
DNA Extraction and Variant Analysis
We extracted genomic DNA from peripheral blood of the proband and healthy control, using a QIAamp DNA Blood Mini Kit (250) (QIAGEN, Valencia, CA). Then we captured the DNA of the proband with the Agilent SureSelect Human All Exon V5 Kit (Agilent, California, USA) and sequenced on Illumina Hiseq 4000 (Illumina Inc., San Diego, USA).
The valid sequencing reads were aligned to the NCBI human reference genome (GRCh37/hg19) by the Burrows Wheeler Aligner software (Li and Durbin, 2010). ANNOVAR is used to do annotation for Variant Call Format file (Wang et al., 2010). We classified the variants as pathogenic, likely pathogenic and uncertain significance, likely benign, benign according to American College of Medical Genomics (ACMG) guidance (Richards et al., 2015).
We filtered single-nucleotide variants (SNVs) and short insertions and deletions (InDels) as follows (Figure 2): (1) High-frequency (minor allele frequency > 0.01) variants found in 1000 Genomes Project, Exome Sequencing Project (ESP), Genome Aggregation Database (gnomAD) and Novo inhouse Database, were excluded. (2) Variants within Exonic, splicing sites were included from subsequent analysis. (3) Bioinformatics programs: Mutation Taster, Polyphen2, SIFT, CADD, and LoFtool were used to predict the possible impacts of SNVs. (4) Homozygous variants were retained to be filtered by a PCD-related gene panel including 47 identified genes (Supplementary Table 1). Sanger sequencing was performed in the patient to validate the mutation. The location of the mutation was analyzed by SMART program (http://smart.embl-heidelberg.de). The DNA samples from the patient's parents were not available. The primer sequences were designed as follows: forward, 5′-ATTCTAAGACCGCTGTGTCCC-3′; reverse, 5′ TTGCACAGCAATGTATGGGG-3′.
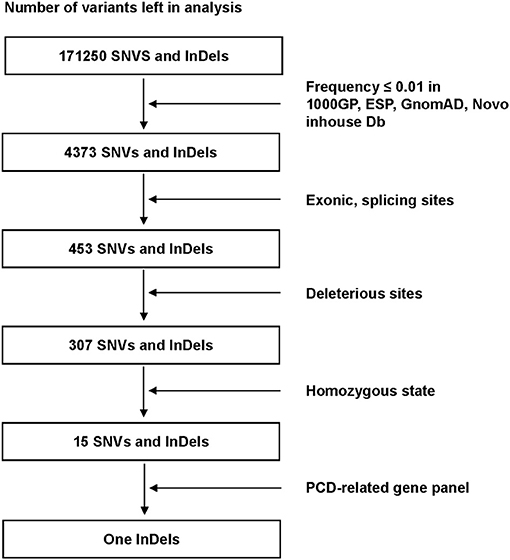
Figure 2. Filter process performed to all variants in the alignment SNVs and InDels. Only one InDels (ODAD3, NM_145045.4: c.1166_1169dup) left after all filter steps. SNVs, single-nucleotide variants: InDels, insertions and deletions; 1000GP, 1000 Genomes Project; ESP, Exome Sequencing Project; GnomAD, Genome Aggregation Database; PCD, Primary ciliary dyskinesia.
Results
For the DNA sample of the patient, exome sequencing generated an average of 14.5 Gb data with an ~99% coverage and a depth of >50×. After alignment and frequency filter, 4,373 non-synonymous variants (SNVs and InDels) were further analyzed and 453 variants in exons or in canonical splice sites (splicing junction 10 bp) were further analyzed. Fifteen SNVs and InDels in homozygous state were left and then filtered by the PCD-related gene panel. Finally, only one homozygous variant, ODAD3, NC_000019.9:g.11533477_11533480dup, NM_145045.4: c.1166_1169dupAGAC, NP_659482.3: p.(Leu391Aspfs*105), which caused frameshift insertion, passed the filtration.
This variant was in accord with the hereditary mode from consanguineous family (Figure 3A) and was validated via Sanger sequencing (Figures 3B,C). According to ACMG guidelines (Richards et al., 2015), this variant was classified into pathogenic (meeting criteria of PVS1, PM2, PM3). The location of the variant is within the third coiled-coil domain of the 595-amino-acid ODAD3 protein (Refseq NP_659482.3) analyzed by SMART program (http://smart.embl-heidelberg.de) (Figure 3D).
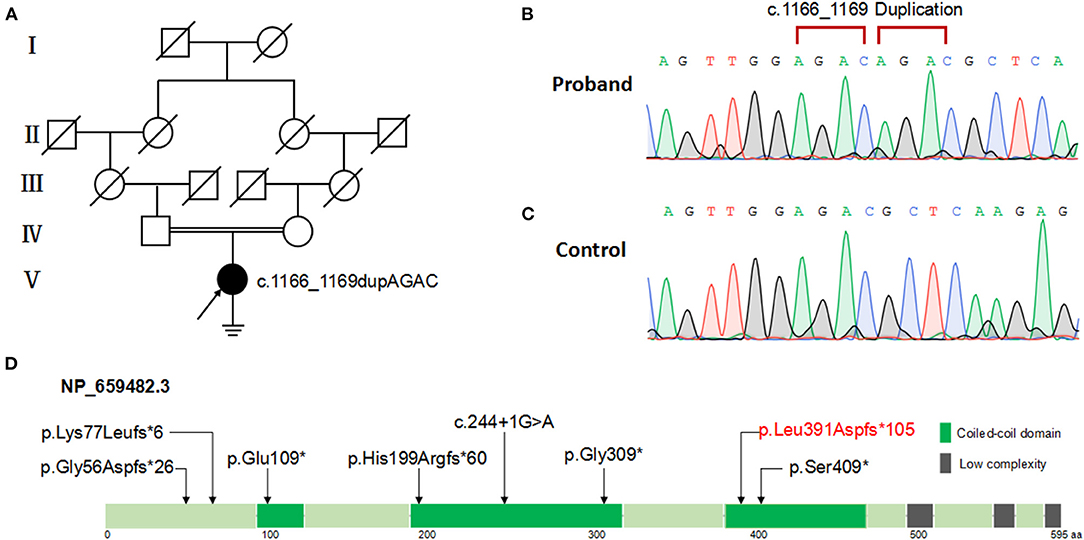
Figure 3. Pedigree and ODAD3 variants analysis of the patient with PCD. (A) Family pedigree of the patient with PCD. Filled symbol represents the affected member, empty symbol represents unaffected members, slashed symbols represent deceased members. Squares, males; circles, females. The patient (V:1) is indicated by a black arrow. (B) Patient with ODAD3 c.1166_1169dupAGAC alleles. (C) Normal control with wild-type alleles. (D) The location of the mutations is shown within the third coiled-coil domain of the 595-amino-acid ODAD3 protein (Refseq NP_659482.3) model. Green boxes indicate coiled-coil domain; gray boxes indicate low-complexity repeat regions.
Discussion and Conclusion
This study describes a female with PCD that carried a novel homozygous variant in the ODAD3 gene. This patient in our study exhibited chronic sinusitis, bronchiectasis, situs inversus, and infertility. Our case enriches the mutation spectrum and clinical phenotypes of ODAD3 variants in PCD.
PCD is a motile ciliopathy involving multiple organs or system, especially in respiratory tract. Cilia can be classified into two types according their motility: motile cilia with 9+2 or 9+0 microtube structure and immotile cilia with 9+0 microtube (Satir and Christensen, 2007). Motile cilia has highly conserved cell structure among different species and extensively exists on airway epithelium, germinal center in fetal period, oviduct and sperm flagella (Bayless et al., 2019). The critical pathogenesis of PCD lies in the abnormality of motile cilia structure or function or ciliogenesis, which accounts for ineffective cilia clearance, abnormal left–right asymmetry and subfertility (Bustamante-Marin and Ostrowski, 2017).
To date, a total of seven likely loss-of-function variants in ODAD3 have been reported in PCD (Supplementary Table 2). The reported ODAD3 variants including three non-sense variants: p.(Gly309 *) (Hjeij et al., 2014); p.(Ser409*) (Hjeij et al., 2014); p.(Glu109*) (Zhang et al., 2019); three frame-shifting variants: p.(Gly56Aspfs*26) (Deng et al., 2020); p.(Lys77Leufs*6) (Mani et al., 2020); p.(His199Argfs*60) (Olm et al., 2019) and one splice site variant: c.244+1G>A (Monies et al., 2019). The only two hitherto reported female patients (10 week and 1 year-old, respectively) did not reach reproductive age. infertility has not been determined in the previously reported patients. However, it means that it was not reported (and probably not even investigated in adult patients) and not that it was not present. Therefore, assessing and reporting infertility in patients affected by PCD is essential, whenever possible. In our study, we detected a novel homozygous variant of ODAD3, p.(Leu391Aspfs*105) which was likely to be PCD disease-causing. The patient in our study exhibited typical PCD phenotypes including chronic sinusitis, bronchiectasis, situs inversus and infertility. The frame-shift variant results in an incorrect and premature termination of translation of ODAD3 protein which may undergo non-sense-mediated decay, indicating a loss of function of ODAD3 in outer dynein arm (ODA) assembly and docking.
Infertility is more common in males than females in PCD (Wallmeier et al., 2020). The prevalence of infertility among PCD females is unclear, partly due to insufficient awareness of clinicians. A cohort study (Vanaken et al., 2017) among PCD reported 61% (22/36) women are considered infertile related with 13 genes such as CCDC39, CCDC40, DNAH5, which affect motile cilia structure. CCDC39 and CCDC40, similar to ODAD3, also contain coiled-coil domains and their defects display disorganization of microtubule doublets and central pair in cilia (Becker-Heck et al., 2011; Merveille et al., 2011). DNAH5 is the ODA component of motile cilia and its defect lead to ciliary dyskinesia (Hornef et al., 2006). These data indicate that those genes impacting motile cilia structure, may related to female infertility. Our study is the first to report infertility in female PCD patient with ODAD3 deficiency. The frameshift ODAD3 variant may lead to ODA defects and then impede the oocytes transportation across the oviduct, which finally results in infertility. As there are currently no way for women to test their potentially PCD-related fertility before trying to conceive, it is necessary for clinicians to pay more attention to fertility status of PCD. Fertility counseling and appropriate assisted reproductive technologies should be included in the standard PCD patient care.
Animal models of deficient ODAD3 demonstrated phenotypes of ciliary dyskinesia and laterality defects similar to human. Odad3 in vertebrates is homologous to the Chlamydomonas ODA10 gene which is also associated with cilia outer ODA assembly (Dean and Mitchell, 2013). ODAD3 is involved in regulating intraflagellar transport (IFT)-dependent dynein arm assembly according to previous research in drosophila, zebrafish, and mice (Alsaadi et al., 2014; Hjeij et al., 2014; Jerber et al., 2014). ODAD3-knockdown zebrafish had motile ciliary dysfunction in the pronephros and Kupffer's vesicle, and showed abnormal left–right asymmetry (Hjeij et al., 2014). In mice, Odad3 is expressed in embryonic nodes and ependymal cells, and deficient Odad3 may result in ciliary dyskinesia and laterality defects (Jerber et al., 2014). Whether ODAD3 defect leads to infertility in animal remains unknown and need further research to recapture phenotype in human.
In conclusion, combined clinical profile and sequencing data, we suggest that novel homozygous variant of ODAD3, p.(Leu391Aspfs*105), is one of the pathogenic cause of PCD. This is also the first report about female infertility in PCD of ODAD3 deficiency. Our study enriches the genetic spectrum and clinical phenotypes of ODAD3 variants in PCD and provides more evidence for future genetic counseling and gene-targeted therapy for this disease.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Review Board of the Second Xiangya Hospital of Central South University in China. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
HL and JQ designed the study. RW, DY, TG, and CL performed the genetic analysis and bioinformatics evaluations. RW and DY drafted the manuscript. HL, JQ, XC, and XK conducted the clinical evaluations. All authors analyzed the data and approved the final manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (81470202, 81770002, 82070003, 81900002), Natural Science Foundation of Hunan Province, China (2020JJ5805), Fundamental Research Funds for Central Universities of Central South University (2020zzts292, 2020zzts281), the Hunan Provincial Innovation Foundation For Postgraduate (CX20200289), the Hunan Province Health Commission Scientific Research Project (B2019159), the Science and Technology Program of Changsha, China (kq1901120), Xiangya Clinical Big Data System Construction Project in Pulmonary Inflammatory Disease of Central South University, and the National Key Clinical Specialty Construction Projects of China.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to the patient and the healthy control for their collaboration. We acknowledge the State Key Laboratory of Medical Genetics of China for technical assistance. We acknowledge Shuizi Ding in Yale University for language editing advice of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.652381/full#supplementary-material
References
Afzelius, B. A., and Stenram, U. (2006). Prevalence and genetics of immotile-cilia syndrome and left-handedness. Int. J. Dev. Biol. 50, 571–573. doi: 10.1387/ijdb.052132ba
Alsaadi, M. M., Erzurumluoglu, A. M., Rodriguez, S., Guthrie, P. A., Gaunt, T. R., Omar, H. Z., et al. (2014). Nonsense mutation in coiled-coil domain containing 151 gene (CCDC151) causes primary ciliary dyskinesia. Hum. Mutat. 35, 1446–1448. doi: 10.1002/humu.22698
Bayless, B. A., Navarro, F. M., and Winey, M. (2019). Motile cilia: innovation and insight from ciliate model organisms. Front. Cell Dev. Biol. 7:265. doi: 10.3389/fcell.2019.00265
Becker-Heck, A., Zohn, I. E., Okabe, N., Pollock, A., Lenhart, K. B., Sullivan-Brown, J., et al. (2011). The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat. Genet. 43, 79–84. doi: 10.1038/ng.727
Bustamante-Marin, X. M., and Ostrowski, L. E. (2017). Cilia and mucociliary clearance. Cold Spring Harb. Perspect. Biol. 9:a028241. doi: 10.1101/cshperspect.a028241
Dean, A. B., and Mitchell, D. R. (2013). Chlamydomonas ODA10 is a conserved axonemal protein that plays a unique role in outer dynein arm assembly. Mol. Biol. Cell 24, 3689–3696. doi: 10.1091/mbc.e13-06-0310
Deng, S., Wu, S., Xia, H., Xiong, W., Deng, X., Liao, J., et al. (2020). Identification of a frame shift mutation in the CCDC151 gene in a Han-Chinese family with Kartagener syndrome. Biosci. Rep. 40:BSR20192510. doi: 10.1042/BSR20192510
Hjeij, R., Onoufriadis, A., Watson, C. M., Slagle, C. E., Klena, N. T., Dougherty, G. W., et al. (2014). CCDC151 mutations cause primary ciliary dyskinesia by disruption of the outer dynein arm docking complex formation. Am. J. Hum. Genet. 95, 257–274. doi: 10.1016/j.ajhg.2014.08.005
Hornef, N., Olbrich, H., Horvath, J., Zariwala, M. A., Fliegauf, M., Loges, N. T., et al. (2006). DNAH5 mutations are a common cause of primary ciliary dyskinesia with outer dynein arm defects. Am. J. Respir. Crit. Care Med. 174, 120–126. doi: 10.1164/rccm.200601-084OC
Jerber, J., Baas, D., Soulavie, F., Chhin, B., Cortier, E., Vesque, C., et al. (2014). The coiled-coil domain containing protein CCDC151 is required for the function of IFT-dependent motile cilia in animals. Hum. Mol. Genet. 23, 563–577. doi: 10.1093/hmg/ddt445
Leigh, M. W., Hazucha, M. J., Chawla, K. K., Baker, B. R., Shapiro, A. J., Brown, D. E., et al. (2013). Standardizing nasal nitric oxide measurement as a test for primary ciliary dyskinesia. Ann. Am. Thorac. Soc. 10, 574–581. doi: 10.1513/AnnalsATS.201305-110OC
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Mani, R., Belkacem, S., Soua, Z., Chantot, S., Montantin, G., Tissier, S., et al. (2020). Primary ciliary dyskinesia gene contribution in Tunisia: identification of a major Mediterranean allele. Hum. Mutat. 41, 115–121. doi: 10.1002/humu.23905
Merveille, A. C., Davis, E. E., Becker-Heck, A., Legendre, M., Amirav, I., Bataille, G., et al. (2011). CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 43, 72–78. doi: 10.1038/ng.726
Mirra, V., Werner, C., and Santamaria, F. (2017). Primary ciliary dyskinesia: an update on clinical aspects, genetics, diagnosis, and future treatment strategies. Front. Pediatr. 5:135. doi: 10.3389/fped.2017.00135
Monies, D., Abouelhoda, M., Assoum, M., Moghrabi, N., Rafiullah, R., Almontashiri, N., et al. (2019). Lessons learned from large-scale, first-tier clinical exome sequencing in a highly consanguineous population. Am. J. Hum. Genet. 105:879. doi: 10.1016/j.ajhg.2019.09.019
Olm, M. A. K., Marson, F. A. L., Athanazio, R. A., Nakagawa, N. K., Macchione, M., Loges, N. T., et al. (2019). Severe pulmonary disease in an adult primary ciliary dyskinesia population in Brazil. Sci. Rep. 9:8693. doi: 10.1038/s41598-019-45017-1
Ostrowski, L. E., Blackburn, K., Radde, K. M., Moyer, M. B., Schlatzer, D. M., Moseley, A., et al. (2002). A proteomic analysis of human cilia: identification of novel components. Mol. Cell. Proteomics 1, 451–465. doi: 10.1074/mcp.M200037-MCP200
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Satir, P., and Christensen, S. T. (2007). Overview of structure and function of mammalian cilia. Annu. Rev. Physiol. 69, 377–400. doi: 10.1146/annurev.physiol.69.040705.141236
Vanaken, G. J., Bassinet, L., Boon, M., Mani, R., Honore, I., Papon, J. F., et al. (2017). Infertility in an adult cohort with primary ciliary dyskinesia: phenotype-gene association. Eur. Respir. J. 50:1700314. doi: 10.1183/13993003.00314-2017
Wallmeier, J., Nielsen, K. G., Kuehni, C. E., Lucas, J. S., Leigh, M. W., Zariwala, M. A., et al. (2020). Motile ciliopathies. Nat. Rev. Dis. Primers 6:77. doi: 10.1038/s41572-020-0209-6
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
Keywords: primary ciliary dyskinesia, ODAD3, CCDC151, sinusitis, bronchiectasis, dextrocardia, infertility
Citation: Wang R, Yang D, Guo T, Lei C, Chen X, Kang X, Qing J and Luo H (2021) Case Report: Identification of a Novel ODAD3 Variant in a Patient With Primary Ciliary Dyskinesia. Front. Genet. 12:652381. doi: 10.3389/fgene.2021.652381
Received: 12 January 2021; Accepted: 09 February 2021;
Published: 26 February 2021.
Edited by:
Emiliano González Vioque, University Clinical Hospital of Santiago, SpainReviewed by:
Fulya Taylan, Karolinska Institutet (KI), SwedenAna Westenberger, University of Lübeck, Germany
Copyright © 2021 Wang, Yang, Guo, Lei, Chen, Kang, Qing and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Luo, ZHJsdW9ob25nQGNzdS5lZHUuY24=; Jie Qing, cWluZ2ppZTAwQGNzdS5lZHUuY24=
†These authors have contributed equally to this work