
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
DATA REPORT article
Front. Genet. , 02 July 2021
Sec. Genomics of Plants and the Phytoecosystem
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.651298
This article is part of the Research Topic Genetics and Genomics to Enhance Crop Production, Towards Food Security View all 36 articles
 Sin-Gi Park1†
Sin-Gi Park1† Eonji Noh1†SuRyun Choi2Boram Choi1In-Gang Shin1Seung-il Yoo1Dong Jin Lee1Sumin Ji1Hae-Suk Kim1
Eonji Noh1†SuRyun Choi2Boram Choi1In-Gang Shin1Seung-il Yoo1Dong Jin Lee1Sumin Ji1Hae-Suk Kim1 Yoon-Jung Hwang3
Yoon-Jung Hwang3 Jung Sun Kim4
Jung Sun Kim4 Jacqueline Batley5
Jacqueline Batley5 Yong Pyo Lim2David Edwards5*
Yong Pyo Lim2David Edwards5* Chang Pyo Hong1*
Chang Pyo Hong1*The Brassica genus consists of economically important oil and leafy vegetable crops, which are cultivated worldwide. The Brassica species represent as the “U's triangle” (Nagahara, 1935), which includes the three basic diploid species Brassica rapa (A genome), Brassica nigra (B genome), and Brassica oleracea (C genome), as well as the three amphidiploid species Brassica juncea (A and B genomes), Brassica napus (A and C genomes), and Brassica carinata (B and C genomes). Brassica rapa (2n = 2x = 20) has been cultivated for specific phenotypic characteristics such as heading (i.e., ssp. pekinensis; Chinese cabbage) and non-heading (i.e., ssp. chinesis; pak choi) leafy vegetables, tuberized hypocotyl/roots (i.e., ssp. rapifera; turnip), and oil rich seedpods (i.e., ssp. trilocularis; yellow sarson) in the oil crops. Although the genomes of its subspecies are very similar (Lin et al., 2014), B. rapa demonstrates extreme morphological diversity. Because of the economical value and scientific interest in phenotypic diversity, the genome sequence of mesopolyploid B. rapa ssp. pekinensis Chiifu, a Chinese cabbage, was the first published B. rapa reference genome, and revealed that B. rapa evolved via a two-step whole-genome triplication and, as a result, has three syntenic subgenomes (Wang et al., 2011). The polyploidization is hypothesized to have facilitated the diversification of genes as well as gene fractionation, and as a consequence led to the evolution of different morphotypes within and between related Brassica species. The study of intraspecific diversity in B. rapa offers opportunities to advance our understanding of plant growth, development, and phenotypic evolution (Paterson et al., 2001).
Over the last few years, the genome assembly of B. rapa (version 3.0) has been improved using single-molecule sequencing, optical mapping, and chromosome conformation capture technologies (Hi-C), resulting in an approximately 30-fold improvement with a contig N50 size of 1.45 Mb compared with that of previous references (Zhang et al., 2018). The assembly refined the syntenic relationship of genome blocks and centromere locations in the genome, and identified a greater number of annotated transposable elements (TEs) than in previous assemblies. In addition to the whole-genome sequence of Chinese cabbage, chromosome-level genome sequences of other B. rapa subspecies, including yellow sarson (Belser et al., 2018) and pak choi (Li P. et al., 2020; Li Y. et al., 2020), have been recently reported. The studies provide insight to the understanding of genetic drivers underlying the morphological variation among B. rapa subspecies. Moreover, a pangenome from Chinese cabbage, rapid-cycling Brassica, and Japanese vegetable turnip was constructed using Illumina short-read data (Lin et al., 2014), identifying genomic determinants of morphological variation, especially copy number differences in peroxidases associated with the phenylpropanoid biosynthetic pathway in turnip.
Turnip (B. rapa L. ssp. rapifera) represent one of the morphotypes in B. rapa that forms tubers (hypocotyl/taproot tubers), produces lobed leaves with long petioles, and can be used to study the genetics underlying storage organ formation (Zhang et al., 2014). Brassica species are susceptible to clubroot disease, caused by Plasmodiophora brassicae (Schwelm et al., 2015), and forms galls (clubs) on infected root tissues with abnormal proliferation, preventing water and nutrient uptake and retarding the normal growth and development of plants, resulting in significantly reduced yield and quality. Sources of known clubroot resistance genes are derived from European turnip, carrying strong resistance to this disease (Matsumoto et al., 1998; Hirani et al., 2018). The resistance genes of European turnip have been introduced into other Brassica crops including vegetables and oilseed rape. In particular, European clubroot differential (ECD) turnips, that exhibited high levels of resistance to clubroot and that consist of four accessions (ECD1–ECD4), were developed and have been helpful for discovering dominant loci conferring clubroot resistance genes including CRa, CRb, and CRc for marker-assisted selection in canola and other Brassica species (Hirani et al., 2018).
Here, we report the draft genome assembly of a European turnip, ECD4, which has strong clubroot resistance, that was generated by PacBio single-molecule long-read sequencing technology. This draft genome assembly coupled with transcriptome data derived from various leaf and root tissues will support the discovery of disease resistance genes (R genes) especially clubroot resistance genes, enabling the development of allele-specific markers for marker-assisted selection in Brassica breeding. Moreover, these data provide a valuable resource for studying the morphological diversity and evolution of turnips.
For the genome assembly of European turnip (ECD4), a total of 70.63 Gb of PacBio long reads (an average sequencing coverage of 136.36x) and 72.16 Gb of Illumina short reads (139.3x) were generated (Supplementary Table 1). Based on k-mer analysis (k = 21), the estimated genome size of European turnip is approximately 518 Mbp (Figure 1A). The genome assembly resulted in a 315.8 Mb draft genome with 655 contigs (contig N50 length of 1.45 Mb; the longest contig length was 21.92 Mb) (Table 1). Our assembly data covered about 61% of the genome of European turnip, being predicted as mostly euchromatins. The assembly coverage is slightly low compared with those reported in Chinese cabbage (353 Mb) (Zhang et al., 2018), pak choi (370 Mb) (Li P. et al., 2020; Li Y. et al., 2020), and yellow sarson (402 Mb) (Belser et al., 2018). Whole-genome sequences of Chinese cabbage, pak choi, and yellow sarson were assembled at the chromosome-level by using chromosome conformation capture (Hi-C) technology [Chinese cabbage (reference genome version 3.0) and pak choi] and/or BioNano optical mapping [Chinese cabbage (v3.0) and yellow sarson] besides long-read sequencing such as PacBio SMRT sequencing or Oxford Nanopore sequencing. Therefore, such long-range scaffolding technologies are further required to improve the quality of genome assembly in European turnip. While the remained 39% is expected to contain considerably repetitive DNAs, including tandem satellite repeats, rDNAs, and retrotransposons which can disturb assembly (Wang et al., 2011; Perumal et al., 2017; Zhang et al., 2018; Li P. et al., 2020). Of the contigs, 260 were anchored to 10 chromosomes of Chinese cabbage (cv. Chiifu-401) (version 3.0), with a total length of 260.47 Mb (Figure 1B; Supplementary Table 2).
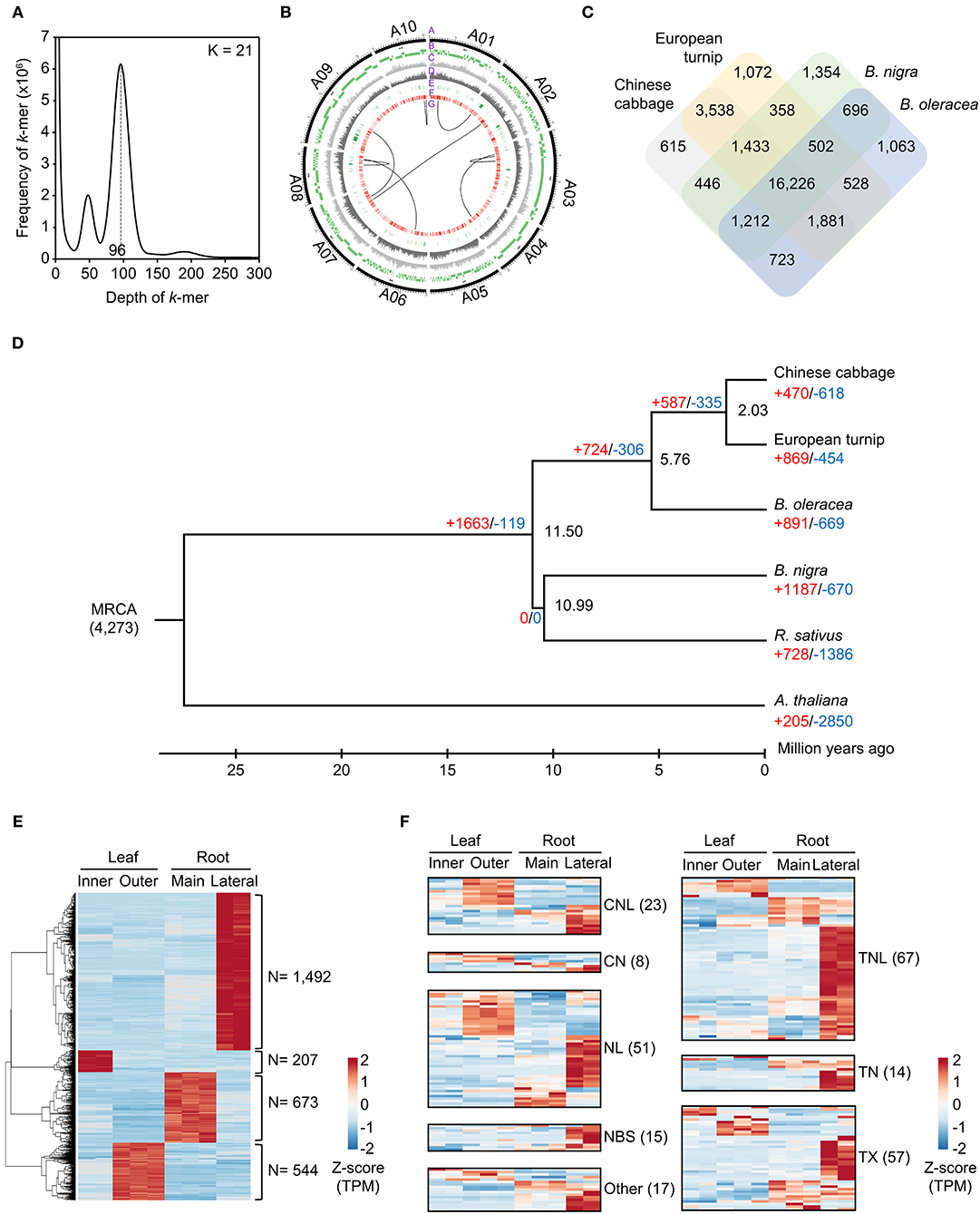
Figure 1. Characterization of the draft genome assembly for the European turnip ECD4. (A) Estimation of genome size on the basis of k-mer frequency analysis. (B) Anchoring genome assembly data of European turnip to 10 chromosomes of Chinese cabbage (A1–A10). In the Circos plot, A, B, C, D, E, F, and G indicate chromosomes of Chinese cabbage, alignment of genome assembly data of European turnip, SNPs, InDels, NBS-encoding R genes, membrane-associated R genes, and translocations, respectively. (C) Orthologous gene clusters among Chinese cabbage, European turnip, B. nigra, and B. oleracea. (D) Expansion and contraction of gene families based on a time-calibrated phylogeny of four Brassica species, R. sativus, and the outgroup A. thaliana. (E) Identification of genes showing tissue-specific expression in the inner and outer leaves and the main and lateral root tissues of European turnip. (F) Genome-wide expression of NBS-encoding R genes identified in the leaf and root tissues of European turnip.
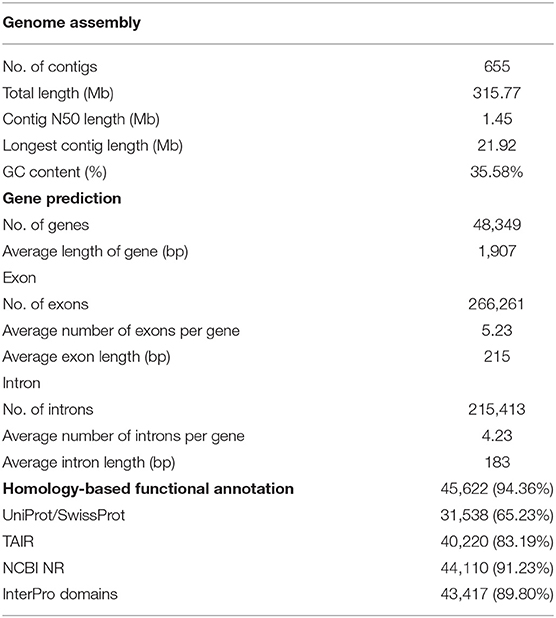
Table 1. Genome assembly and gene prediction of European turnip.
The quality of the genome assembly was assessed using BUSCO and paired-end read mapping. First, the draft genome assembly captured 99.6% of the complete Benchmarking Universal Single-Copy Orthologs (BUSCOs) with the Virdiplantae_odb10 database; 82.4, 17.2, 0.2, and 0.2% of the BUSCOs were predicted as complete and single-copy, complete and duplicated, fragmented, and missing, respectively. The high completeness of our assembly data seems to be resulted from error correction and curation of heterozygous assemblies by Pilon and Purge_haplotigs with high read depth and coverage (read depth: 139.3x of short reads for Pilon and 136.4x of long reads for Purge_haplotigs). In particular, the method led to a saturation of BUSCO scores with one round of error correction in Pilon (Supplementary Table 10). The genome completeness of European turnip is similar to that of Chinese cabbage (Supplementary Table 3). Second, 94.58% of the short-insert paired-end reads were successfully realigned to the assembly, with 99.84% assembly coverage. Moreover, the analysis showed that 0.32% of the nucleotides in the genome assembly were heterozygous.
The whole-genome comparison revealed eight genomic rearrangements between European turnip and Chinese cabbage (“G” layer in Figure 1B), with the identification of hotspots (Supplementary Table 4; Supplementary Figure 1), indicating genomic change since the divergence from a common ancestor of B. rapa. For example, the positions 629.5 to 774.9 kb (145.4 kb in length) and positions 11.5 to 579.4 kb (567.9 kb in length) on contig00007136 of European turnip were aligned to positions 18,057.7 to 18,217.2 kb (159.5 kb in length) and positions 18,966.7 to 19,568.7 kb (602 kb in length) on chromosome A10 of Chinese cabbage, respectively. These hotspots were validated by mapping Illumina paired-end reads of European turnip to the corresponding hotspot of Chinese cabbage and then identifying the split of paired-end read mapping in Chinese cabbage (Supplementary Figure 8). We further identified five and three genome rearrangements between pak choi and European turnip, and between yellow sarson and European turnip, respectively, showing large genome variations among B. rapa subspecies (Supplementary Figure 7). Additionally, 3,795,367 SNPs, with a transition/transversion ratio (Ts/Tv) of 1.35, and 954,051 InDels were identified (“C” and “D” layers in Figure 1B).
A total of 112.6 Mb of TEs were identified, accounting for 35.66% of the genome assembly (Supplementary Table 5). Of the class I elements, Ty3/Gypsy (29.5%) and Ty1/Copia (18.7%) for long terminal repeat (LTR) retrotransposons and LINE (L1) for non-LTR retrotransposons were abundant in the genome. The hAT family (11.7%) represented the most abundant DNA transposon (Supplementary Table 5). A total of 48,349 non-redundant protein-coding genes were predicted with an average number of exons per gene of 5.23 using evidence-driven gene prediction methods coupled with ab initio prediction (Table 1). The gene models were supported by 93.04% RNA-Seq data derived from two leaves and two root tissues of European turnip (Supplementary Table 6). Of the predicted genes, 45,622 (94.36%) were successfully annotated by at least one database, including UniProt/SwissProt, TAIR, NCBI NR, and InterPro. To investigate species-specific and shared genes in European turnip compared with other Brassica diploids, including Chinese cabbage (AA), B. nigra (BB), and B. oleracea (CC), we analyzed orthologous gene clusters. This analysis showed that 24,466 of 31,647 orthologous gene clusters were shared among European turnip and three Brassica diploids and that 1,072 orthologous gene clusters were species-specific in European turnip (Figure 1C). This highlighted the enrichment of genes with F-box domains that play various roles in developmental processes, including plant hormone signal transduction, floral development, secondary metabolism, senescence, circadian rhythm, and response to biotic and abiotic stress (Xu et al., 2009) (Supplementary Figure 2). Of the orthologous gene clusters, five R genes (three TIR- and two CC-NBS-LRR-containing domain proteins) were found. We also identified 709, 672, and 463 orthologous gene clusters specific to European turnip, yellow sarson, and Chinese cabbage, respectively (Supplementary Figure 9A). Similar to above finding, the enrichment of genes with F-box and leucine-rich repeat (LRR) domains was identified in orthologous gene clusters specific to European turnip (Supplementary Figure 9B), reflecting the characteristics of plant disease resistance in European turnip.
A phylogenetic tree of European turnip was constructed using a total of 3,139 single-copy orthologous genes (Figure 1D). Through orthologous gene cluster analysis, we identified that European turnip has 869 expanded and 454 contracted gene families (red- and blue-colored numbers in Figure 1D). Of the expanded gene families, protein domains found in genes associated with plant development (no apical meristem-associated C-terminal) and plant defense (haloacid dehalogenase-like hydrolase, xylanase inhibitor C-terminal, tetratricopeptide repeat, and oxidative-stress-responsive kinase 1 C-terminal) were enriched (Supplementary Figure 3).
Tissue-specific gene expression was identified in inner leaf (207 genes), outer leaf (544 genes), main root (673 genes), and lateral root (1,492 genes) tissues, with a statistical cut-off of q < 0.05, absolute 2-fold change, and at least one tissue with ≥10 FPKMs (Figure 1E). Functional annotation for these genes revealed the functions for each of tissues; for example, chloroplast organization (P = 1.1 × 10−12) for inner leaves, phosphorylation (P = 1.2 × 10−23), and defense response (P = 7.1 × 10−6) for outer leaves, cell wall organization (P = 6.7 × 10−9), response to salicylic acid (P = 1.7 × 10−5), and post-embryonic morphogenesis (P = 5.6 × 10−5) for the main root, and transcription (P = 2.2 × 10−19), response to salicylic acid (P = 3.9 × 10−8), wounding (P = 5.8 × 10−8), toxic substance (P = 4.4 × 10−7), abscisic acid (P = 5.7 × 10−8), and jasmonic acid (2.2 × 10−6) for lateral roots were significantly represented (Supplementary Figure 4).
We also found a total of 252 plant defense-related R genes: 23 CC-NBS-LRRs (CNLs), 8 CC-NBSs (CNs), 67 TIR-NBS-LRRs (TNLs), 14 TIR-NBSs (TNs), 51 NBS-LRRs (NLs), 15 NBSs, 57 TIR-others (TXs), and 17 others (Supplementary Table 7). Interestingly, 103 R genes exhibited high expression in lateral root tissue (Figure 1F). In addition to these R genes, a total of 1,135 genes−119 receptor-like proteins (RLPs), 742 receptor-like kinases (RLKs), and 274 TM-CCs—categorized into membrane-associated R genes were identified (Figure 1F; Supplementary Table 7). Of those three gene families, 243 RLKs also showed high expression in the lateral roots (Supplementary Figure 5). Additionally, two contigs harboring microsatellite markers linked to clubroot resistance loci BraA.CR.a and BraA.CR.b (Hirani et al., 2018), which are located on chromosomes A3 and A8, respectively, were found (Supplementary Table 8; Supplementary Figure 6). Interestingly, the comparison of R genes between the linked regions of the two species showed the expansion of R genes, mostly R genes of TM-CC and TNL classes, in European turnip even though exhibiting their co-linearity in the linked regions (Supplementary Table 9). Moreover, three R genes homologous to Rcr6, which is a clubroot resistance gene of TNL class identified in B. nigra (Chang et al., 2019), were identified in European turnip, but only one was identified in Chinese cabbage (Supplementary Table 9).
We report the first draft genome of European turnip (ECD4) coupled with transcriptome data derived from various tissues. Our data can provide an invaluable genomic resource to study the morphological diversity and evolution of European turnip by applying pan-genomics to Brassica. Moreover, our data can help develop molecular markers as a genetic breeding tool to identify plant defense-related genes such as clubroot resistance genes (R genes).
Seeds of European turnip ECD4 (B. rapa ssp. rapifera) (accession no. 25026) were obtained from the Korea Brassica Genome Resource Bank (KBGRB, South Korea). The seeds were germinated in seedling trays containing autoclaved soil in a controlled chamber at 25°C, 60% humidity, and photoperiodic lighting (16 h of light:8 h of dark). From leaf tissue of 3-week-old seedlings, genomic DNA was extracted by a WizPrep Plant DNA Mini Kit (Wizbiosolutions, Seongnam, South Korea) according to the manufacturer's protocol for whole-genome sequencing. A long-read library with high-quality genomic DNA of ≥20 μg was prepared using the SMRTbell Express Template prep kit 2.0 (Pacific Biosciences, Menlo Park, CA, USA) and sequenced on a PacBio Sequel System using one SMRT cell. Additionally, DNA sequence libraries were prepared from 1 μg input DNA using a TruSeq Nano DNA Sample Prep Kit according to the manufacturer's instructions (Illumina, Inc., San Diego, CA, USA). The libraries were subjected to paired-end sequencing with a 150-bp read length using the Illumina NovaSeq 6000 platform.
Illumina short reads were processed by Jellyfish (version 2.2.0) (Marcais and Kingsford, 2011) to estimate the genome size of European turnip. The k-mer frequency with a k-mer size of 21 was counted and plotted as a k-mer frequency distribution. PacBio SMRT long reads were assembled using CANU (version 1.8) (Koren et al., 2017) with the following primary parameters; genomeSize 518M, minReadLength 1000, minOverlapLength 500, rawErrorRate 0.3, correctedErrorRate 0.045. For assembly polishing, short reads were aligned to the CANU assembly data using BWA-MEM (version 0.7.17) (Li and Durbin, 2009), and errors in nucleotides in the CANU assembly data were then corrected using Pilon (version 1.22) (Walker et al., 2014) with one round of polishing. The polished contigs were processed with Purge_haplotigs to curate heterozygous diploid genome assemblies (Roach et al., 2018). After filtering of ≤10 kb of contigs, a total of 655 assembled contigs were finally generated. This method was summarized in Supplementary Figure 10.
The genome completeness of the draft genome assembly of European turnip was assessed by using BUSCO (version 4.0.5) (Seppey et al., 2019) with 425 single-copy orthologs of the viridiplantae_odb10 database. For estimation of sequence coverage, Illumina short-insert paired-end reads were realigned to the draft genome assembly using BWA-MEM. Furthermore, heterozygous nucleotides were identified by using SAMtools mpileup (Li, 2011).
A combination of ab initio and evidence-based approaches was employed for gene prediction of European turnip. The assembled genome was premasked for repetitive DNA sequences using RepeatMasker (version 4.0.6) (http://www.repeatmasker.org/). An unsupervised training gene structure was generated using GeneMark-ET (version 4.10) (Lomsadze et al., 2014) by incorporating RNA-Seq data. De novo prediction was performed using AUGUSTUS (version 3.3.1) (Keller et al., 2011) with the training gene set and with exon–intron boundary information predicted by RNA-Seq and protein sequence alignments. Here, STAR (version 2.7.1a) (Dobin et al., 2013) was used for RNA-Seq alignment of the species, and GenomeThreader (version 1.7.0) (Gremme et al., 2005) was used for protein sequence alignment with the B. rapa protein sequences (version 3.0). Functional annotation for predicted genes was performed on the basis of homology-based searches with TAIR11, UniProt/SwissProt, and NCBI non-redundant (NR) databases using BLASTP (version 2.3.0+) (Altschul et al., 1990) with a cutoff E-value of 1E-10. Protein domains were also searched by using InterProScan (version 5.19-58.0) (Mulder and Apweiler, 2007). Additionally, tRNA- and miRNA-like sequences were predicted using tRNAscan-SE (version 1.4 alpha) (Schattner et al., 2005) and Infernal (version 1.1.1) (Nawrocki and Eddy, 2013). Resistance gene analogs (RGAs), such as NBS-encoding proteins, RLKs and RLPs, were predicted using an RGAugury pipeline (Li et al., 2016).
The protein sequences from turnip (AA) and diploid Brassica species, including Chinese cabbage (AA; reference genome v3.0), B. nigra (BB; v1.1), B. oleracea (CC; v1.1), Raphanus sativus (v1.1), and the outgroup species Arabidopsis thaliana (version TAIR10) were used to predict orthologous gene clusters. The gene set of each species was filtered as follows: First, the genes encoding proteins of fewer than 30 amino acids were filtered out. Second, the similarity relation between the protein sequences of all of the species was obtained through BLASTP (i.e., all-vs.-all BLASTP searches) with a cutoff E-value of 1E-05. All of the protein datasets of the representative species were clustered into paralogues and orthologs by OrthoMCL (version 2.0.9) (Li et al., 2003), with an inflation parameter of 1.5.
The single-copy orthologs in these six species were aligned using MAFFT (version 7.123b) (Katoh and Standley, 2013) and then used to extract conserved regions with Gblocks (version 0.91b) (Talavera and Castresana, 2007). The alignment results were combined to create a superalignment matrix. A phylogenetic tree of these six species was constructed using MEGA X (Kumar et al., 2018) with the maximum likelihood method and 1,000 bootstrap values. The divergence time of European turnip was estimated using RelTime of MEGA X.
The expansion and contraction of the gene families were analyzed by comparing the cluster size differences between the ancestor and each species by using café (De Bie et al., 2006). A random birth and death model was used to study the changes in gene families along each lineage of the phylogenetic tree. A probabilistic graphical model (PGM) was introduced to calculate the probability of transitions in gene family size from parent to child nodes in the phylogeny. With conditional likelihoods as the test statistics, the corresponding p-value in each lineage was calculated, and a p-value of 0.05 was used to identify the families that were significantly expanded or contracted. The expanded and contracted genes were then subjected to PFAM functional annotation.
Three-week-old seedlings were transplanted to pots and grown for 4 weeks in a glasshouse. Total RNA from four tissues (the inner leaf, outer leaf, main root, and lateral root) was extracted using an RNeasy Plant Mini Kit (Qiagen, Hilden, Germany). RNA-Seq libraries from 1 μg of purified total RNA were prepared using the TruSeq Stranded mRNA Sample Prep Kit according to the manufacturer's manual (Illumina, Inc., San Diego, CA). cDNA was synthesized and then subjected to an end repair process, addition of a single “A” base, and ligation of the adaptors. Libraries that were purified and enriched with PCR amplification were subjected to paired-end sequencing with a 150 bp read length using an Illumina NovaSeq 6000 platform.
After removal of adaptor sequences and trimming of low-quality sequences (<Q20) using Cutadapt (version 2.8), clean reads were aligned to the draft genome assembly using STAR (version 2.7.1a). Gene expression quantification was performed using RSEM (version 1.3.1) (Li and Dewey, 2011) with TPM values (version 1.3.1). For analysis of tissue-specific expression in the inner/outer leaves and main/lateral roots, differential expression analysis was performed using DESeq2 (version 1.26.0) (Love et al., 2014) with a cutoff of q < 0.05 and absolute ≥2-fold change. Functional annotation for genes showing tissue-specific expression was performed by using DAVID (Huang et al., 2007), and relevant gene ontology (GO) terms were selected with a cutoff EASE score <1 × 10−4. The expression pattern of the analyzed genes was visualized as a heatmap by using ClustVis (Metsalu and Vilo, 2015) with the correlations and the average linkage method.
Syntenic blocks between the chromosomes of Chinese cabbage and contigs of European turnip were detected by using SyMAP (version 4.2) (Soderlund et al., 2006) and are shown in a Circos plot. Genome rearrangement events such translocation between the two genomes were verified by using MUMmer (version 3.9.4) (Delcher et al., 2003).
The datasets presented in this study can be found in online repositories . Sequencing data used in this study are available in the NCBI Sequence Read Archive (SRA) database under the following accession PRJNA690160; JAEOXI000000000 (whole genome assembly), SRR13375884 (PacBio SMRT long read sequencing data), SRR13375883 (Illumina DNA-Seq data), SRR13375896 - SRR13375905 (RNA-Seq data derived from four different tissues). In addition, datasets for functional annotation are available from our website http://turnip.genome.theragenbio.com/BRT_1.0.
CH and DE conceived this study. S-GP, BC, I-GS, S-iY, and DL performed the bioinformatics analyses. EN, SJ, H-SK, Y-JH, and JK performed the whole-genome and RNA sequencing. SC and YL prepared the samples and extracted genomic DNA and total RNA. CH, S-GP, JB, and DE wrote the manuscript. All authors read and approved the final manuscript, contributed to the article, and approved the submitted version.
This work was supported by the Cooperative Research Program for National Agricultural Genome Program (Grant No. PJ01338203), Rural Development Administration (RDA), South Korea.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.651298/full#supplementary-material
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Belser, C., Istace, B., Denis, E., Dubarry, M., Baurens, F. C., Falentin, C., et al. (2018). Chromosome-scale assemblies of plant genomes using nanopore long reads and optical maps. Nature Plants 4, 879–887. doi: 10.1038/s41477-018-0289-4
Chang, A., Lamara, M., Wei, Y., Hu, H., Parkin, I. A. P., Gossen, B. D., et al. (2019). Clubroot resistance gene Rcr6 in Brassica nigra resides in a genomic region homologous to chromosome A08 in B. rapa. BMC Plant Biol. 19:224. doi: 10.1186/s12870-019-1844-5
De Bie, T., Cristianini, N., Demuth, J. P., and Hahn, M. W. (2006). CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271. doi: 10.1093/bioinformatics/btl097
Delcher, A.L., Salzberg, S.L., and Phillippy, A.M. (2003). Using MUMmer to identify similar regions in large sequence sets. Curr. Protoc. Bioinformatics Chapter 10:Unit 10.3. doi: 10.1002/0471250953.bi1003s00
Dobin, A., Davis, C.A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Gremme, G., Brendel, V., Sparks, M.E., and Kurtz, S. (2005). Engineering a software tool for gene structure prediction in higher organisms. Inf. Softw. Technol. 47, 965–978. doi: 10.1016/j.infsof.2005.09.005
Hirani, A.H., Gao, F., Liu, J., Fu, G., Wu, C., McVetty, P.B.E., et al. (2018). Combinations of independent dominant loci conferring clubroot resistance in all four turnip accessions (Brassica rapa) from the European clubroot differential set. Front. Plant Sci. 9:1628. doi: 10.3389/fpls.2018.01628
Huang, D.W., Sherman, B.T., Tan, Q., Collins, J.R., Alvord, W.G., Roayaei, J., et al. (2007). The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 8:R183. doi: 10.1186/gb-2007-8-9-r183
Katoh, K., and Standley, D.M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Keller, O., Kollmar, M., Stanke, M., and Waack, S. (2011). A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 27, 757–763. doi: 10.1093/bioinformatics/btr010
Koren, S., Walenz, B.P., Berlin, K., Miller, J.R., Bergman, N.H., and Phillippy, A.M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Li, B., and Dewey, C.N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Li, H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. doi: 10.1093/bioinformatics/btr509
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, L., Stoeckert, C.J. Jr., and Roos, D.S. (2003). OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Li, P., Quan, X., Jia, G., Xiao, J., Cloutier, S., and You, F.M. (2016). RGAugury: a pipeline for genome-wide prediction of resistance gene analogs (RGAs) in plants. BMC Genomics 17:852. doi: 10.1186/s12864-016-3197-x
Li, P., Su, T., Zhao, X., Wang, W., Zhang, D., Yu, Y., et al. (2020). Assembly of the non-heading pak choi genome and comparison with the genomes of heading Chinese cabbage and the oilseed yellow sarson. Plant Biotechnol. J. 19, 966–976. doi: 10.1111/pbi.13522
Li, Y., Liu, G. F., Ma, L. M., Liu, T. K., Zhang, C. W., Xiao, D., et al. (2020). A chromosome-level reference genome of non-heading Chinese cabbage [Brassica campestris (syn. Brassica rapa) ssp. chinensis]. Hortic. Res. 7:212. doi: 10.1038/s41438-020-00449-z
Lin, K., Zhang, N., Severing, E.I., Nijveen, H., Cheng, F., Visser, R.G., et al. (2014). Beyond genomic variation–comparison and functional annotation of three Brassica rapa genomes: a turnip, a rapid cycling and a Chinese cabbage. BMC Genomics 15:250. doi: 10.1186/1471-2164-15-250
Lomsadze, A., Burns, P.D., and Borodovsky, M. (2014). Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res. 42:e119. doi: 10.1093/nar/gku557
Love, M.I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Marcais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Matsumoto, E., Yasui, C., Ohi, M., and Tsukada, M. (1998). Linkage analysis of RFLP markers for clubroot resistance and pigmentation in Chinese cabbage (Brassica rapa ssp. pekinensis). Euphytica 104, 79–86. doi: 10.1023/A:1018370418201
Metsalu, T., and Vilo, J. (2015). ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 43, W566–W570. doi: 10.1093/nar/gkv468
Mulder, N., and Apweiler, R. (2007). InterPro and InterProScan: tools for protein sequence classification and comparison. Methods Mol. Biol. 396, 59–70. doi: 10.1007/978-1-59745-515-2_5
Nagahara, U. (1935). Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Jpn. J. Bot. 7, 389–452.
Nawrocki, E.P., and Eddy, S.R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Paterson, A.H., Lan, T.H., Amasino, R., Osborn, T.C., and Quiros, C. (2001). Brassica genomics: a complement to, and early beneficiary of, the Arabidopsis sequence. Genome Biol. 2:REVIEWS1011. doi: 10.1186/gb-2001-2-3-reviews1011
Perumal, S., Waminal, N.E., Lee, J., Lee, J., Choi, B. S., Kim, H. H., et al. (2017). Elucidating the major hidden genomic components of the A, C, and AC genomes and their influence on Brassica evolution. Sci. Rep. 7:17986. doi: 10.1038/s41598-017-18048-9
Roach, M.J., Schmidt, S.A., and Borneman, A.R. (2018). Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics 19:460. doi: 10.1186/s12859-018-2485-7
Schattner, P., Brooks, A.N., and Lowe, T.M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Schwelm, A., Fogelqvist, J., Knaust, A., Julke, S., Lilja, T., Bonilla-Rosso, G., et al. (2015). The Plasmodiophora brassicae genome reveals insights in its life cycle and ancestry of chitin synthases. Sci. Rep, 5:11153. doi: 10.1038/srep11153
Seppey, M., Manni, M., and Zdobnov, E.M. (2019). BUSCO: Assessing genome assembly and annotation completeness. Methods Mol. Biol. 1962, 227–245. doi: 10.1007/978-1-4939-9173-0_14
Soderlund, C., Nelson, W., Shoemaker, A., and Paterson, A. (2006). SyMAP: a system for discovering and viewing syntenic regions of FPC maps. Genome Res. 16, 1159–1168. doi: 10.1101/gr.5396706
Talavera, G., and Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Walker, B.J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, X., Wang, H., Wang, J., Sun, R., Wu, J., Liu, S., et al. (2011). The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1039. doi: 10.1038/ng.919
Xu, G., Ma, H., Nei, M., and Kong, H. (2009). Evolution of F-box genes in plants: different modes of sequence divergence and their relationships with functional diversification. Proc. Natl. Acad. Sci. U. S. A. 106, 835–840. doi: 10.1073/pnas.0812043106
Zhang, L., Cai, X., Wu, J., Liu, M., Grob, S., Cheng, F., et al. (2018). Improved Brassica rapa reference genome by single-molecule sequencing and chromosome conformation capture technologies. Hortic. Res. 5:50. doi: 10.1038/s41438-018-0071-9
Keywords: Brassica rapa L. ssp. rapifera, clubroot resistance, draft genome, European turnip, genome assembly, orthologous gene cluster, R gene
Citation: Park S-G, Noh E, Choi S, Choi B, Shin I-G, Yoo S-i, Lee DJ, Ji S, Kim H-S, Hwang Y-J, Kim JS, Batley J, Lim YP, Edwards D and Hong CP (2021) Draft Genome Assembly and Transcriptome Dataset for European Turnip (Brassica rapa L. ssp. rapifera), ECD4 Carrying Clubroot Resistance. Front. Genet. 12:651298. doi: 10.3389/fgene.2021.651298
Received: 09 January 2021; Accepted: 01 June 2021;
Published: 02 July 2021.
Edited by:
Ajay Kumar, North Dakota State University, United StatesReviewed by:
Chenggen Chu, Edward T. Schafer Agricultural Research Center (USDA-ARS), United StatesCopyright © 2021 Park, Noh, Choi, Choi, Shin, Yoo, Lee, Ji, Kim, Hwang, Kim, Batley, Lim, Edwards and Hong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Edwards, RGF2ZS5FZHdhcmRzQHV3YS5lZHUuYXU=; Chang Pyo Hong, Y2hhbmdweW8uaG9uZ0B0aGVyYWdlbmJpby5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.