 Hong-Yan Shu1
Hong-Yan Shu1 Cong-Cong Zheng
Cong-Cong Zheng Yan-Gang Wang
Yan-Gang Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 23 August 2021
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.650077
We report a single-point variant of low-density lipoprotein receptor (LDLR) in a Chinese proband with a clinical diagnosis of familial hypercholesterolemia (FH) with a comprehensive functional analysis. Target exome capture-based next-generation sequencing was used for sequencing and identification of genomic variants in the LDLR gene. The expression, cellular location, and function of the mutant LDLR were analyzed. Sequencing of LDLR in FH patients indicated a point variant of single-base substitution (G < A) at a position of 2389 in the 16th exon, which led to a loss of the 16th exon in the LDLR messenger RNA. This genomic variant was found to cause exon 16 deletion in the mutant LDLR protein. Subsequent functional analyses showed that the mutant LDLR was retained in the Golgi apparatus and rarely expressed in the cellular membranes of HepG2 cells. Accordingly, the intake ability of HepG2 cells with the mutant LDLR was significantly reduced (P < 0.05). In conclusion, our results suggest that a mutant with a single-base substitution (c. 2389G > A) in the 16th exon of the LDLR gene was associated with miscleavage of messenger RNA and the retention of mutant LDLR in the Golgi apparatus, which revealed a pathogenic variant in LDLR underlying the pathogenesis of FH.
Family hypercholesterolemia (FH) is an inherited disease that mainly affects the metabolism of cholesterol (Watts et al., 2020). Clinically, FH is characterized by significantly raised serum levels of total cholesterol (TC) and low-density lipoprotein cholesterol (LDL-C), the presence of tendinous xanthomata, and high incidence of premature vascular disease, particularly coronary artery disease (Garg et al., 2019; Hamasaki and Kotani, 2020). The serum level of TC can be as high as 7–15 mmol/L in patients with heterozygous FH, whereas the level of TC can reach 20–25 mmol/L in patients with homozygous FH (Santos et al., 2016). The prevalence of heterozygous FH has been reported to be 1/500 (0.2%), whereas the prevalence of homozygous FH has been reported to be 1/1,000,000 (Reiner, 2015). However, this prevalence is thought to be underestimated. In a study of the general Danish population in 2012, the prevalence of FH was reported to be 1/200 (0.5%) (Benn et al., 2012), which is similar to the results of the National Health and Nutrition Examination Surveys conducted in the United States between 1999 and 2012 (de Ferranti et al., 2016). A study based on the Chinese adult population showed a prevalence of FH of 0.47% in 2014, which is similar to values in western populations (Shi et al., 2014; Zhou and Zhao, 2016). More importantly, comorbidities of FH have been associated with poor prognosis, particularly premature cardiovascular death. A previously published study in Norway showed that cardiovascular mortality in patients with FH was significantly higher as compared with that among patients without FH (standardized mortality ratio: 2–3, despite the use of statins; Mundal et al., 2014). Treatment strategies for FH include statins, which lower the LDL-C level in the short term, but the long-term efficacy of statin treatment in patients with FH, particularly related to cardiovascular outcomes, remains to be determined (Vuorio et al., 2017). Therefore, continuous efforts are needed to improve the diagnosis and treatment of FH.
Pathophysiologically, FH is caused by genomic variants of key genes that are involved in the metabolism of cholesterol, including the low-density lipoprotein receptor (LDLR), the proprotein convertase subtilisin/kexin type 9 (PCSK9), and the apolipoprotein B (APOB) genes (Henderson et al., 2016; Sharifi et al., 2017; Vrablik et al., 2020). Of note, genomic variants in LDLR are considered the most important pathogenic mechanism in FH. LDLR is a transmembrane protein that functions in the uptake and removal of LDL-C from circulation. Many pathogenic variants have been reported to be involved in the pathogenesis of FH. A recent meta-analysis including Chinese patients with FH reported 131 genomic variants in LDLR, mostly allocated in exon 4 as missense variants, which may participate in the pathogenesis of FH (Jiang et al., 2015). Overall, more than 2,000 genomic variants have been reported in LDLR (Chemello et al., 2021), and these are classified into five categories (Soutar and Naoumova, 2007; Usifo et al., 2012; Hendricks-Sturrup et al., 2020). Class 1 variants represent LDLR genomic variants that lead to the prevented synthesis of immunodetectable LDLR protein. Class 2 variants indicate LDLR variants that cause complete (2a) or partial (2b) retainment of LDLR protein in the endoplasmic reticulum (ER). Class 3 variants cause the membrane to incorporate LDLR, but they are unable to bind LDL. Class 4 variants of LDLR consist of mutant LDLR that are unable to concentrate in clathrin-coated pits. Class 5 variants of LDLR present mutant LDLR that are unable to recycle LDL into the endosome. However, it has been noted that not all of the genomic variants in LDLR are associated with FH, which highlights the importance of functional characterization of LDLR genomic variants (Henderson et al., 2016; Lee, 2017; Sharifi et al., 2017).
In this study, we reported a genomic variant of LDLR that contributes to the pathogenesis of FH. Subsequent functional analysis showed a single-point genomic variant led to a miscleavage of messenger RNA (mRNA) and the retaining of mutant LDLR in the Golgi apparatus.
The protocol for this study was reviewed and approved by the Ethics Committee of The Affiliated Hospital of Qingdao University before its performance. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and national research committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. We obtained written informed consent from the patient for publication of identifying information/images in an online, open-access publication.
The patient was diagnosed with FH at The Affiliated Hospital of Qingdao University. We used the Dutch Lipid Clinic Network diagnostic criteria for the diagnosis of FH (Catapano et al., 2016). Briefly, the Dutch Lipid Clinic Network criteria evaluate the possibility for the diagnosis of FH based on scores for the following four aspects: (1) family history of premature vascular disease, LDL-C level above the 95th percentile, presentation of tendinous xanthoma and/or arcus cornealis in a first-degree relative, or LDL-C level above the 95th percentile in a child < 18 years of age; (2) clinical presentation of the patient with premature vascular disease or LDL-C level above the 95th percentile; (3) physical examination of the patient showing tendinous xanthomata and/or arcus cornealis; (4) degree of LDL-C elevation of at least 4.0 mmol/L; and (5) DNA analyses showing functional genomic variants in the LDLR, APOB, or PCSK9 gene. The diagnosis of FH is considered definite if the total score is over 8.
We obtained peripheral venous blood samples from the proband and her first-degree relatives in a fasting condition for the measurement of TC, LDL-C, triglycerides, and high-density lipoprotein cholesterol. Measurements of the lipid parameters mentioned earlier were performed with a chemiluminescence method with an automated biochemistry analyzer (Beckman AU 4500, United States) in The Affiliated Hospital of Qingdao University.
Genomic DNA of the proband, as well as the family members, was extracted from the peripheral venous blood sample with the phenol–chloroform centrifugation method as previously described (Schiebelhut et al., 2017). After treatment with ethylenediaminetetraacetic acid, genomic DNA samples of the proband were sent for customized target exome capture-based next-generation sequencing of the LDLR, PCSK9, and APOB genes at the Beijing Genomic Institution. The samples were sequenced simultaneously and analyzed for genomic variants of LDLR on the Illumina Platform, and a single-base variant within exon 16 was detected. Then, polymerase chain reaction (PCR) was performed targeting the mutated exome (exon 16) for the proband, her father, and paternal aunt and uncle (Table 1). Briefly, the primers were designed with Prime 5 as 5′-CCTTCCTTTAGACCTGGGCCT-3′ and 5′-CATAGCGGGAGGCTGTGACC-3′. We used 100 ng of genomic DNA as the template, which was mixed to obtain a total 50-μl reaction system using the HotStarTaq Plus DNA Polymerase Kit (Qiagen, Germany). The reactive conditions were as follows: Taq polymerase activation at 94±C for 5 min, 35 cycles of denaturing at 94±C for 30 s, annealing at 61–64±C for 30 s, extension at 72±C for 45 s, and final extension at 72±C for 5 min. We used agarose gel to separate and confirm the amplified products by electrophoresis. The PCR products were further purified using the DNA fragment purification kit (TaKaRa, Japan). Sequencing of the products was performed at the Beijing Genomic Institution with the Sanger sequencing method.
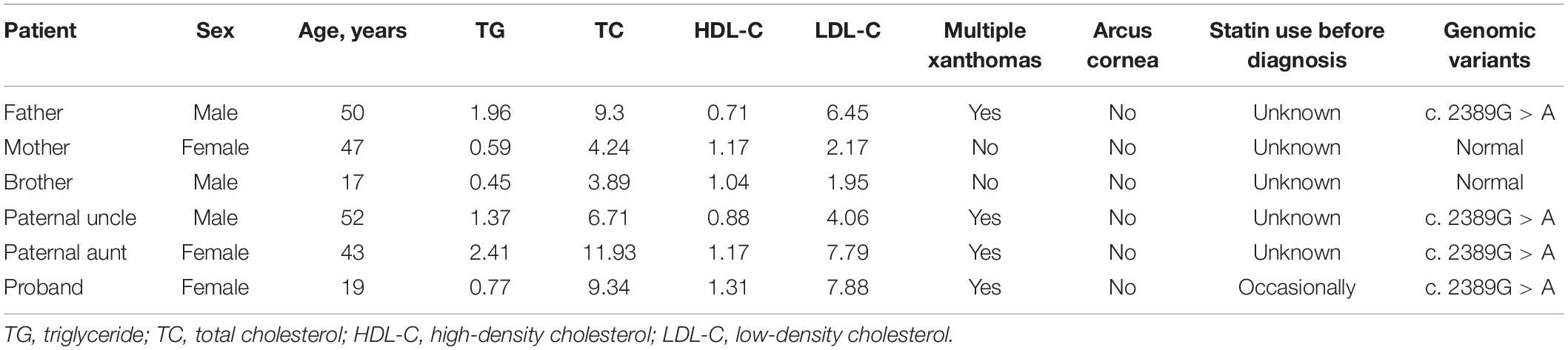
Table 1. Results of lipids analyses at admission for the proband and her relatives.
Total RNA from the peripheral venous blood samples obtained from the proband and an age- and sex-matched health control was extracted using the Trizol method as previously described (Kim et al., 2017). Then, reverse transcription PCR was performed to evaluate the transcription of exon 16 with the primers designed by Primer 5 software, forward: 5′-TGAA CTGGTGTGAGAGGACC-3′, and reverse 5′-CTGGTTGTGG CAAATGTGGA-3′. After electrophoresis with agarose gel, the targeted bands were confirmed and separated, and the purified PCR products were sequenced with the Sanger sequencing method at the Beijing Genomic Institution.
The construction of lentiviral vectors containing the coding regions of the mutant LDLR mentioned earlier at exon 16 and the wild-type (WT) LDLR genes, both with a merged FLAG protein on the C-terminus, was performed by the GeneChem Biotech Company (Shanghai, China). A GV416 plasmid with BamHI enzymatic sites was used when preparing the vector, and the vectors were labeled with a green fluorescent protein. 293T cells were used to measure the supernatant virus titer.
HepG2 cells were purchased from YRgene Biotech Company (Shanghai, China) and cultured with Roswell Park Memorial Institute 1640 medium (Gibco, United States) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37±C in a humidified atmosphere at 5% carbon dioxide. For vector transfection, HepG2 cells were plated in six-well culture plates and infected with the lentiviruses carrying the WT LDLR or mutant LDLR with Polybrene Reagent (Invitrogen Life Technologies, Spain) according to the manufacturer’s instructions. Infected cells were maintained in culture for 72 h to achieve the maximal LDLR expression, and then the cells were cultured in a medium with Puromycin (1 μg/ml) to exclude the non-transfected cells.
The membrane proteins of HepG2 cells were biotinylated and extracted as previously described (Whitaker et al., 2007). Briefly, HepG2 cells were washed with ice-cold phosphate-buffered saline (PBS) and incubated with buffer A [1 mg/ml Sulfo-NHS-(LC)-biotin in PBS, containing 1-mM MgCl2 and 1.3-mM CaCl2, pH 8.0] for 45 min at 4±C. Then, the cells were washed with cold PBS three times, scraped down, and centrifuged. After the cells were cultured in 300-μl buffer B (1% Triton X-100, 4-mM egtazic acid, 10-mM Tris, pH 8.0), they were lysed with ultrasonication, and the lysates were clarified by centrifugation for 15 min at 20,000 × g at 4±C. Then, 500-μg total protein was taken for each sample, and buffer B was added to a total volume of 280 μl before the protein sample was quantified. After the addition of 30 μl of a 50% slurry of High Capacity NeutrAvidin Agarose Resin (Pierce, United States) followed by mixing for 1 h at room temperature, the biotinylated proteins were precipitated from 280 μl of lysate. Then, the biotinylated proteins binding to the agarose were washed three times with ice-cold PBS and eluted from NeutrAvidin agarose after the addition of 20 μl of 2 × sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis sample buffer. Then, 20 μl of 50-mM dithiothreitol in SDS sample buffer was added in each sample and centrifuged at room temperature, and Bromophenol Blue was added and mixed before centrifugation for 1 min at 10,000 × g at 4±C. Then, the samples were used for Western blotting, with 500 μg of total protein loaded for each sample on 10% SDS-polyacrylamide gel electrophoresis gels, transferred to polyvinylidene difluoride membranes, and immunoblotted for FLAG proteins. All experiments for each sample were repeated at least three times.
Confocal laser scanning microscopy (CLSM) was used to evaluate the colocalization of LDLR-FLAG fusion proteins in the HepG2 cells. Briefly, the transfected cells with WT or mutant LDLR were seeded on coverslips, fixed with 4% paraformaldehyde for 15 min, and washed three times with ice-cold PBS. Then, cells were permeabilized with 1% Triton X-100 for 30 min at room temperature. The samples were washed and blocked with PBS-5% bovine serum albumin for 1 h. After washing three times, the samples were incubated with tetramethylrhodamine-conjugated concanavalin A (1:100, Molecular Probes, United States) at room temperature for 1 h. Then, the cells were washed three times with PBS and fixed with 4% paraformaldehyde for 15 min at room temperature for observation of the intracellular fluorescent dye by CLSM (Olympus IX 81, Tokyo, Japan). The intracellular fluorescent dye was observed with sequential excitation and capture image acquisition with a digital camera (Axiocam NRc5; Zeiss, Jena, Germany). We used the Fluoview v50 software (Olympus, Miami, FL, United States) to obtain the images, and quantitative analyses of the fluorescence intensities were performed with ImageJ software (NIH, Bethesda, MD, United States). Finally, the intracellular fluorescent dye was observed by CLSM.
To evaluate the LDL uptake ability of the cells, the HepG2 cells transfected with the mutant and WT LDLR were incubated with 20 mg/ml fluorescent 1,19-dioctadecyl-3,3,3939-tetramethylindocarbocyanine perchlorate (Dil)-conjugated LDL (Molecular Probes) in serum-free media and kept at 37±C for 4 h. The Golgi apparatus was labeled with anti-syntaxin 6. Then, the medium was removed, and the cells were washed with PBS three times. After fixation with 4% paraformaldehyde for 15 min at room temperature, the intracellular fluorescent dye, which is reflective of the uptake ability of LDL, was observed by CLSM.
We used the GraphPad Prism 6 (GraphPad Software, San Diego, CA) and SPSS 16.0 software programs (SPSS, Inc., Chicago, IL, United States) for statistical analyses. Data are presented as the mean ± standard error of the mean. The Student’s t-test was used for comparisons between two groups. A P < 0.05 was considered to be statistically significant.
The proband was a 19-year-old female who presented at our hospital with high levels of TC and LDL-C and multiple xanthomas on the buttocks that had been present she was 9 years old. Physical examination showed multiple xanthomas on the buttocks, tendon, and popliteal space of the proband (Figure 1A). As for the proband’s family history, her father had died at 48 years of age due to myocardial infarction, and he also had hypercholesterolemia and multiple tendon xanthomas. Moreover, one paternal aunt and one paternal uncle of the proband also had hypercholesterolemia and multiple tendon xanthomas. None of the family members mentioned earlier had arcus cornea. The paternal grandmother of the proband died at the age of 41 years from myocardial infarction, whereas the mother, brother, and grandfather of the proband had no hypercholesterolemia. The family tree of the proband is shown in Figure 1B, and the results of lipid analyses before treatment are shown in Table 1. Based on the proband’s family history, physical examination, lipids results, and subsequent DNA analyses, the proband was diagnosed with FH. According to the statement of the proband, statins were occasionally used before the diagnosis. After the diagnosis, atorvastatin (20 mg per night) and ezetimibe (10 mg per night) were prescribed, and the proband was followed in the clinic each month for 6 months. The serum TC and LDL-C varied between 6–7 and 4–5 mmol/L during follow-up.
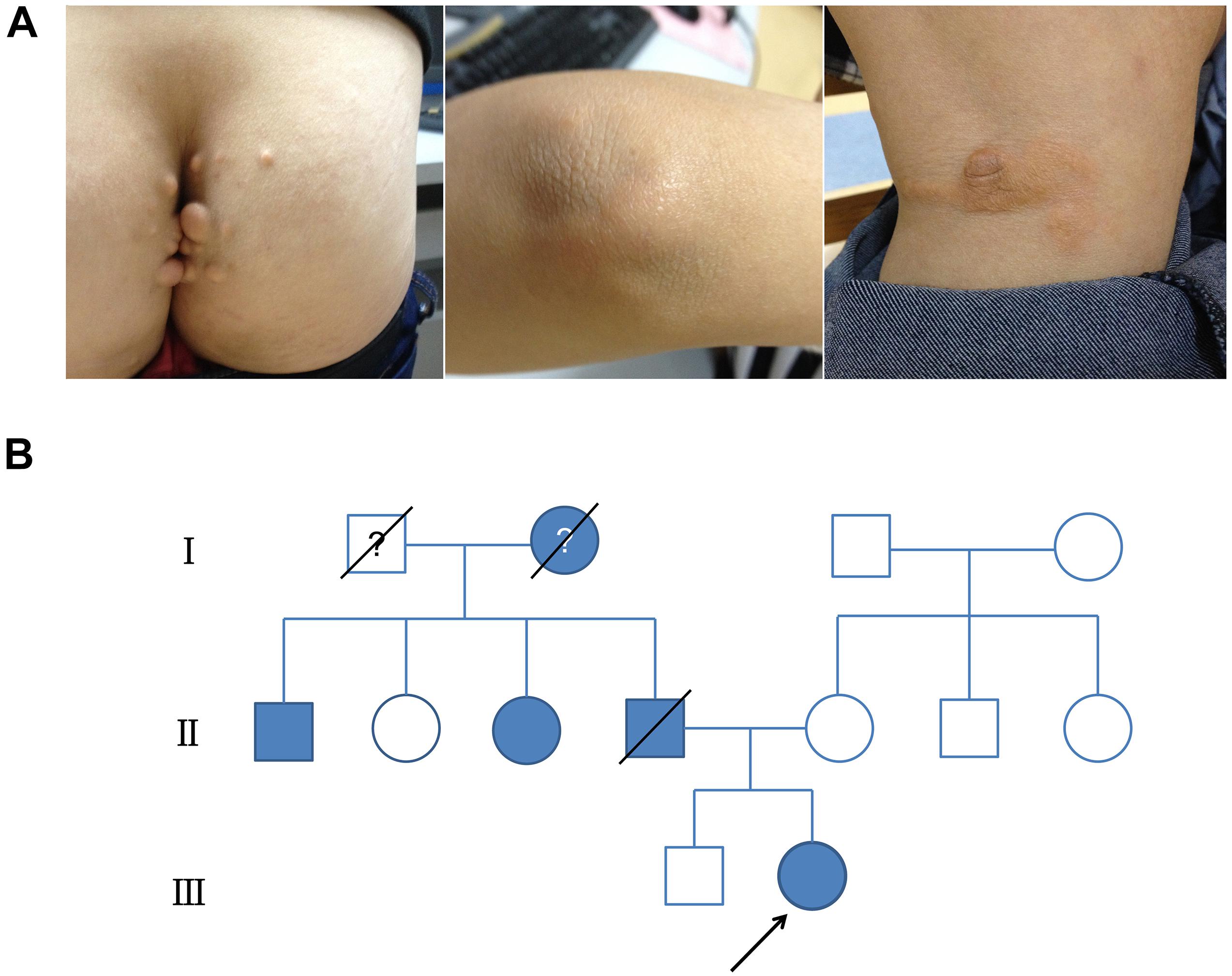
Figure 1. Clinical presentation and family tree of proband. (A) Physical examination revealed multiple xanthomas on buttocks, tendon, and popliteal space on proband. (B) Family tree showing confirmed hypercholesterolemia in family of proband (colored in gray).
Given that functional genomic variants in LDLR, PCSK9, and APOB are the main causes of FH (Henderson et al., 2016; Sharifi et al., 2017), we performed target exome capture-based next-generation sequencing of these genes to narrow down the potential genomic variants in this case of FH. Our genomic DNA sequencing found no genomic variants in the PCSK9 and APOB genes. However, there was a single-base substitution (G < A) at position 2389 in the 16th exon of the LDLR gene as shown in Figure 2A, which would lead to a change from valine (V) to methionine (M) at the protein sequence of 797 (V797M), if the protein is translated successfully. However, the genomic variants (c. 2389G > A) was located in the last base of exon 16, which led to the hypothesis that the variant in this case of FH may have a potential influence on mRNA cleavage progression. To test this hypothesis, we further performed quantitative reverse transcription PCR of peripheral venous blood samples from the proband and a healthy control to identify differences in the LDLR mRNA. With the primers for the 16th exon, the results showed that the proband had an additional PCR product of a different length from the one PCR product obtained for a healthy control subject (535 bp, Figure 2B). The sequencing results showed that the shorter PCR product exclusively observed in the proband had a deletion of the 16th exon of the mutant LDLR (Figure 2C), indicating that c. 2389G > A in LDLR disrupted normal mRNA transcription via disturbing mRNA cleavage. Genomic DNA sequencing by Sanger sequencing showed a similar variant (c. 2389G > A) in the family members with FH (father, paternal uncle, and aunt, Table 1) but not in the family members without FH (mother and brother).
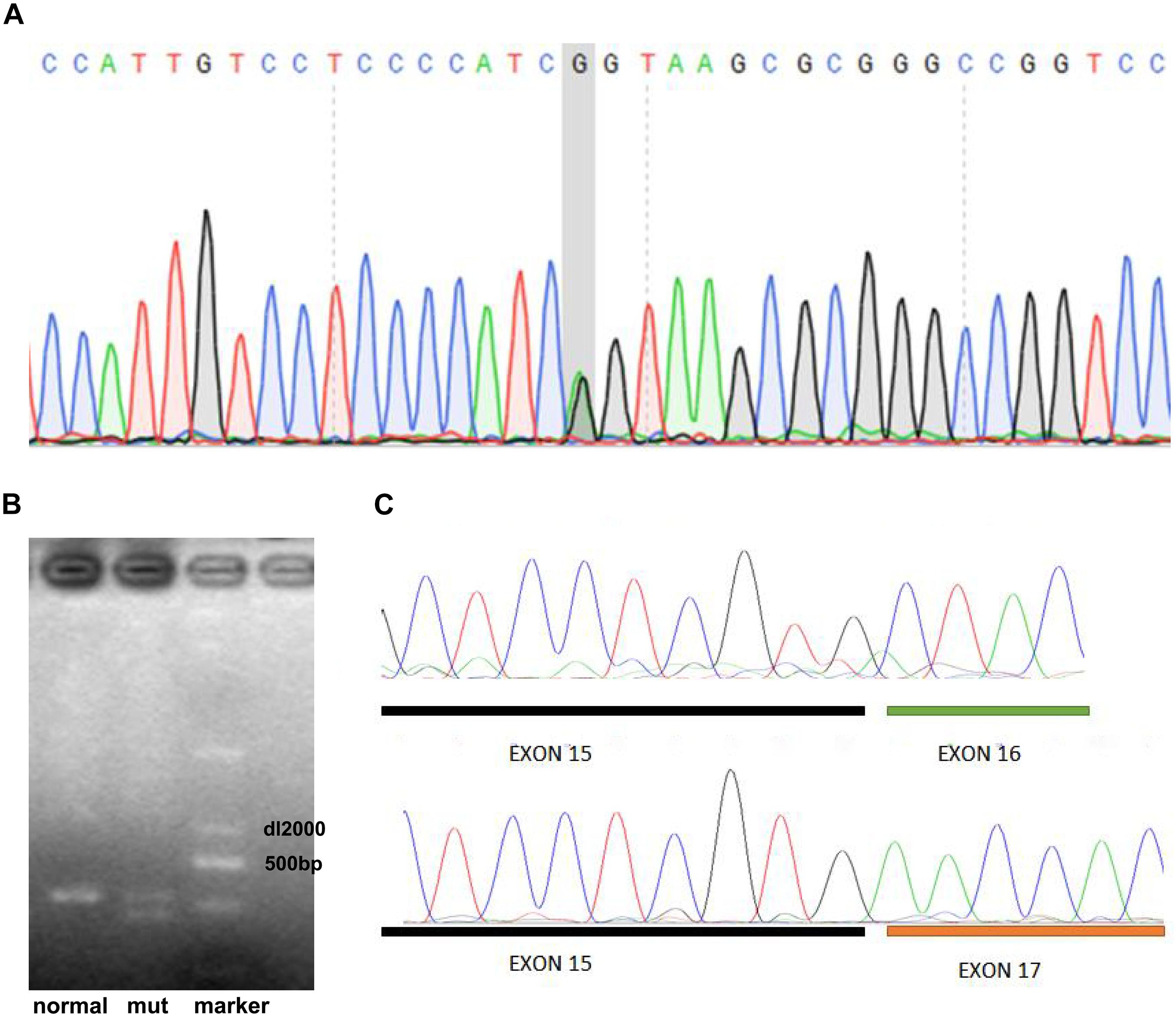
Figure 2. Identification of genomic variant in LDLR of proband. (A) DNA sequencing showed a single-base substitution (G < A) at position 2389 in 16th exon of LDLR gene. (B) RT-PCR including 16th exon showed that proband had an additional PCR product of a different length from one PCR product obtained for healthy control. (C) Sequencing results showed that shorter PCR product exclusively observed in proband had deletion of 16th exon of mutant LDLR.
To further characterize the localization of the mutant LDLR protein, we constructed viral vectors of LDLR with exon 16 deletion, and WT LDLR with FLAG merged on the C-terminus and infected the conventional cell line of human liver cells. We then used Western blot analyses to evaluate the amount of LDLR protein on the membrane and among total cellular proteins. As shown in Figure 3A, opposite to the considerable membrane expression of LDLR as detected by Western blotting on HepG2 cells transfected with WT LDLR, LDLR protein was barely detected on the membrane cells transfected with the mutant LDLR with exon 16 deletion. However, a considerable amount of LDLR at 120 kDa was detected when the total protein was used for Western blot analysis in the mutant cells, indicating the accumulation of an LDLR precursor-like protein in the plasma of the HepG2 cells with mutant LDLR. To further determine the cellular localization of the LDLR-FLAG fusion proteins, a CLSM examination was used. To our surprise, WT LDLR was mainly distributed at the cell surface. However, the mutant LDLR with exon 16 deletion was retained primarily in the Golgi apparatus (Figure 3B). Taken together, these results indicate that LDLR exon 16 deletion led to LDLR protein re-localization in the Golgi apparatus rather than at the cell surface.
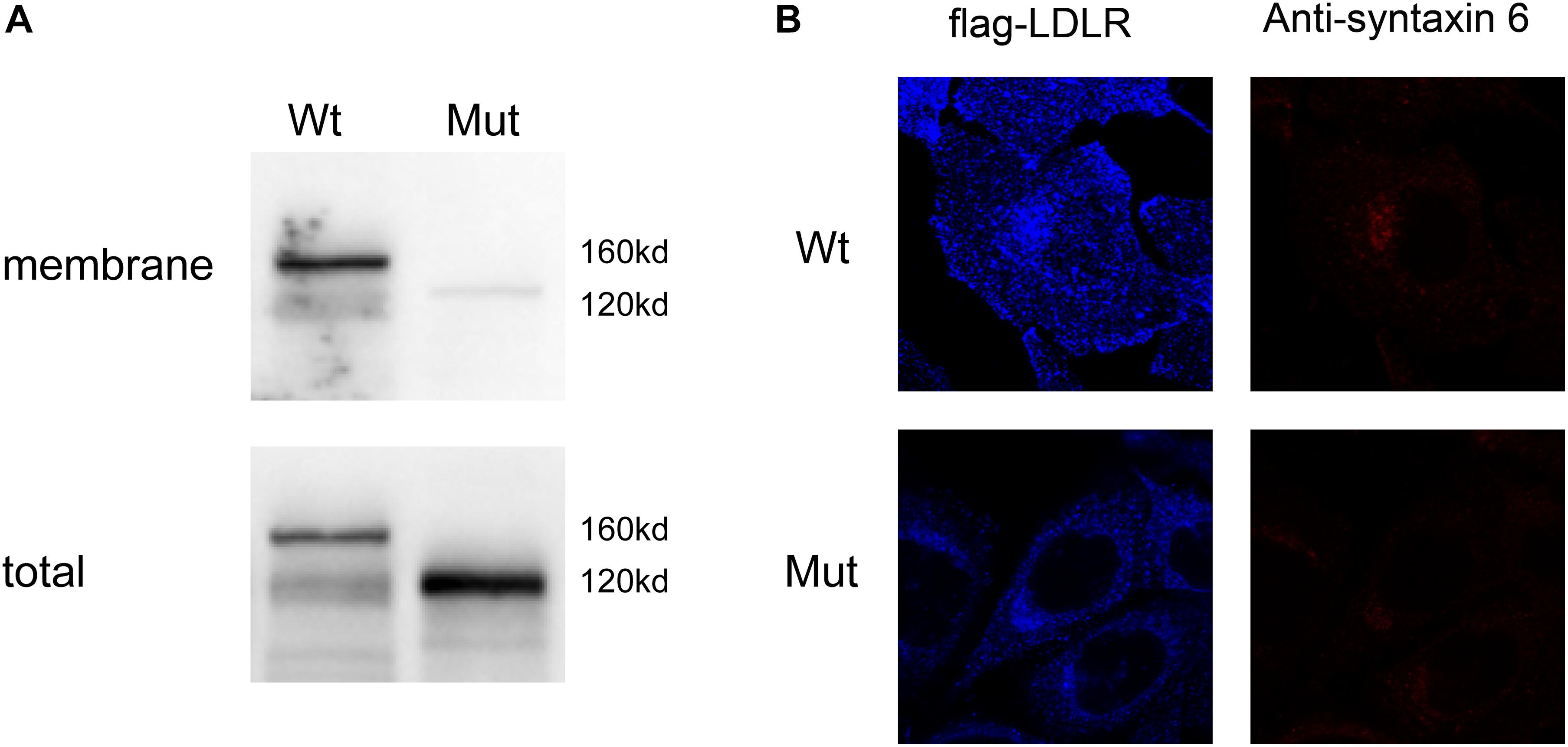
Figure 3. Expression of different LDLR variants on cell surface of HepG2-transfected cells. (A) Western blot analysis showed that mutant LDLR protein with exon 16 deletion was rarely expressed on membrane but accumulated in plasma of HepG2 cells with a molecular weight of 120 kDa. Membranous LDLR was analyzed by biotinylation as described in section “Analysis of Surface Biotinylated Proteins and Western Blotting.” (B) CLSM examination showed that WT LDLR was mainly distributed at cell surface, whereas mutant LDLR with exon 16 deletions was mainly retained in Golgi apparatus. Blue fluorescence represents distribution of Flag antibody. Because Flag gene and LDLR gene are fused, blue fluorescence distribution reflects distribution of LDLR protein in cell (LDL-R). Red fluorescence indicates the position of Golgi apparatus (marked with anti-syntaxin 6).
To evaluate the functional consequence of mutant LDLR with exon 16 deletion, we used DiI-LDL to evaluate the uptake ability of the LDLR in HepG2 cells. We observed that the internalization capacity of HepG2 cells with mutant LDLR was substantially lower than that of cells with WT LDLR (13.6 vs. 19.5%, P < 0.05; Figure 4). From their results taken together, this variant was classified as likely pathogenic with autosomal dominant inheritance by the American College of Medical Genetics and Genomics. Because this variant has been reported in individuals with FH in various studies1, this variant has been found in ≥ 10 unrelated FH cases (PS4). Moreover, our functional analysis fulfilled PS3_Moderate (1) and (3) criteria.
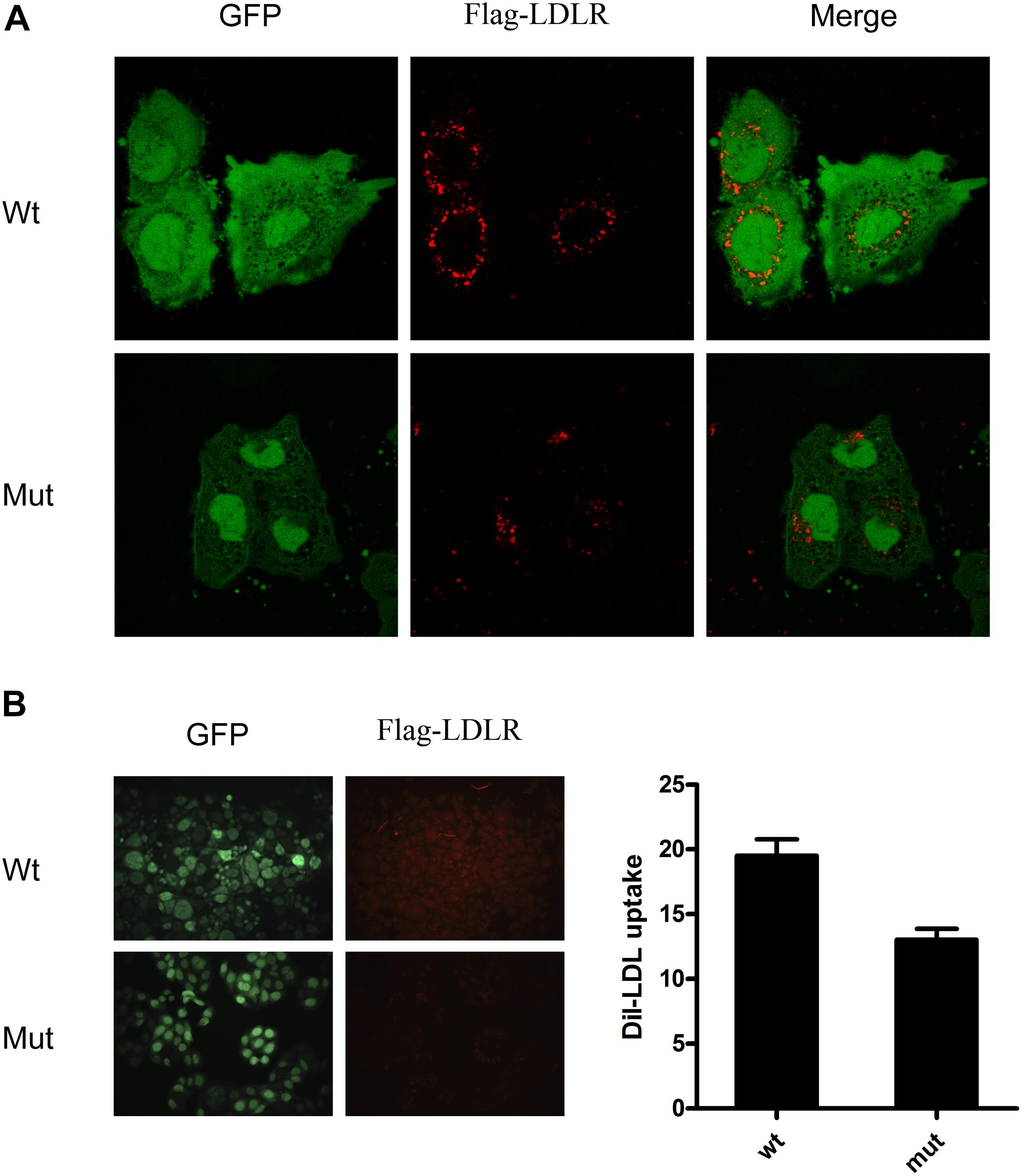
Figure 4. DiI-LDL uptake measurement showed that internalization capacity of HepG2 cells with mutant LDLR with exon 16 deletion was substantially lower than that observed in cells with WT LDLR. (A) Red fluorescence represents DiI-LDL, and green fluorescence represents HepG2-transfected cells. (B) Quantitative analysis showed that capacity of DiI-LDL uptake was significantly reduced in HepG2 cells transfected with c. G < A 2389 that resides in 16th exon of LDLR as compared with HepG2cells transfected with WT LDLR.
In this study, we identified and characterized a single-base substitution genomic variant (c. G < A 2389) in a Chinese proband with FH. Subsequent functional characterization of the variants showed that c. G < A 2389 resides in the 16th exon of LDLR and leads to the miscleavage of LDLR mRNA and loss of the 16th exon. Accordingly, exon 16 deletion occurred in the mutant LDLR protein, which resulted in the retention of the mutant LDLR in the Golgi apparatus, reduced expression of LDLR on the cellular membrane, and substantially reduced uptake ability of the LDLR protein for LDL-C.
Physically, LDLR is known as a type 1 transmembrane protein that functions by binding and internalizing LDL-C and mediating its subsequent release and degradation in the endosomes and lysosomes (Willnow, 1999; Defesche, 2004). LDLR is initially synthesized as a precursor of 860 amino acids (AAs), including a signaling peptide of 21 AAs. After cleavage of the signaling peptide, the mature LDLR protein with 839 AAs is transported into the ER, where it undergoes folding and glycosylation. Post-translational modification of LDLR also occurs in the Golgi apparatus, likely because LDLR requires O-glycosylation for its stable expression and incorporation into the cellular membrane, where clathrin-coated pits form to confer the internalization and subsequent intracellular metabolism of LDL-C. Five functional domains have been identified in the LDLR protein, and the transmembrane domain that consists of AAs 789–810 is considered to be evolutionarily preserved and to play a fundamental role in the incorporation of the protein into the cellular membrane (Willnow, 1999; Defesche, 2004; Mahdieh et al., 2020). The results of our sequencing analyses for the LDLR gene and its mRNA indicated the loss of exon 16 caused by the single-base substation genomic variant (G < A 2389), which led to coding of the last base of the 15th exon (2311) and the first two bases of the 17th exon (2390 and 2391). Accordingly, mutant LDLR with exon 16 deletion was formed, and the original 789–769 AAs of the transmembrane domain of the LDLR protein were missing, which finally led to the failure of the mutant LDLR to incorporate into the cellular membrane. From a search of the clinical variation database, this variant has been reported in individuals with FH in various studies (see text footnote 1). This variant has been found in several FH patients, along with evidence of co-segregation with the disease. Multiple clinical diagnostic laboratories/reputable databases have classified this variant as likely pathogenic/pathogenic. The variant was first reported by Mak et al. (1998) in a Chinese patient with FH. They found that G < A 2389 occurs at the last base of exon 16, which may be the −1 position of the 5′ donor splice site. Reverse transcription PCR investigated its effect on splicing. Only the normal G allele was found. This mutation thus appears to cause a donor site splicing error for the LDL receptor gene (Mak et al., 1998). However, functional analysis has not been reported, making the present analysis valuable in understanding this genomic variant. Our functional studies, as complementation, showed that c. G < A 2389 resides in the 16th exon of LDLR and leads to the miscleavage of LDLR mRNA and loss of the 16th exon. Accordingly, exon 16 deletion occurred in the mutant LDLR protein, which resulted in the retention of the mutant LDLR in the Golgi apparatus, reduced expression of LDLR on the cellular membrane, and substantially reduced uptake ability of the LDLR protein for LDL-C.
The mechanisms underlying the retention of the mutant LDLR in the Golgi apparatus are not fully understood. Considering the importance of O-glycosylation of the LDLR protein for its stable expression and incorporation into the cellular membrane and the fact the molecular weight of the mutant LDLR protein is similar to that of the precursor of LDLR (120 kDa), we hypothesized that the glycosylation process might also be impaired due to the genomic variants, causing retention of the mutant LDLR in the Golgi apparatus. Interestingly, no previous study has reported any genomic variants of LDLR that cause retention of the mutant LDLR protein in the Golgi apparatus. In fact, a recent study reported a p.L799R variant in LDLR that leads to the translocation of the entire L799R-LDLR into the lumen of the ER (Strom et al., 2015).
In conclusion, we identified a single-base substitution variant (g. G < A 2389) in a Chinese proband with FH, which is associated with miscleavage of LDLR mRNA and subsequent retention of the protein in the Golgi apparatus. Further studies are needed to determine the exact mechanisms underlying the pathogenic effects of this genomic variant in FH, which may be important for better understanding the pathogenesis of FH.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Ethic Committee of The Affiliated Hospital of Qingdao University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
H-YS and Y-GW designed the study. Y-CL and C-CZ were involved with patient care. H-YS analyzed patient samples. WZ advised on histological staining and analysis. Y-CL and C-CZ assisted with sample analysis. H-YS and WZ drafted and wrote the manuscript. Y-GW, Y-CL, and C-CZ revised the manuscript critically for intellectual content. Y-GW had primary responsibility for the final content. All authors read and approved the final manuscript.
This work was support by the National Natural Science Foundation of China (81600684) and the Natural Science Foundation of Shandong Province (ZR2017MH069).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.650077/full#supplementary-material
Supplementary Figure 1 | Full gel image from RT-PCR including the 16th exon showing that the proband had an additional PCR product of a different length from the single PCR product obtained for the healthy control.
Supplementary Figure 2 | Full gel image of western blot analysis showing that the mutant LDLR protein with exon 16 deletion was rarely expressed on the membrane, but accumulated in the plasma of the HepG2 cells with a molecular weight of 120 kDa.
Benn, M., Watts, G. F., Tybjaerg-Hansen, A., and Nordestgaard, B. G. (2012). Familial hypercholesterolemia in the danish general population: prevalence, coronary artery disease, and cholesterol-lowering medication. J. Clin. Endocrinol. Metab. 97, 3956–3964. doi: 10.1210/jc.2012-1563
Catapano, A. L., Graham, I., De Backer, G., Wiklund, O., Chapman, M. J., Drexel, H., et al. (2016). 2016 ESC/EAS Guidelines for the Management of Dyslipidaemias: The Task Force for the Management of Dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS) Developed with the special contribution of the European Assocciation for Cardiovascular Prevention & Rehabilitation (EACPR). Atherosclerosis 253, 281–344.
Chemello, K., Garcia-Nafria, J., Gallo, A., Martin, C., Lambert, G., and Blom, D. (2021). Lipoprotein Metabolism in Familial Hypercholesterolemia. J. Lipid Res. 2021:100062.
de Ferranti, S. D., Rodday, A. M., Mendelson, M. M., Wong, J. B., Leslie, L. K., and Sheldrick, R. C. (2016). Prevalence of Familial Hypercholesterolemia in the 1999 to 2012 United States National Health and Nutrition Examination Surveys (NHANES). Circulation 133, 1067–1072. doi: 10.1161/circulationaha.115.018791
Defesche, J. C. (2004). Low-density lipoprotein receptor–its structure, function, and mutations. Semin. Vasc. Med. 4, 5–11. doi: 10.1055/s-2004-822993
Garg, A., Garg, V., Hegele, R. A., and Lewis, G. F. (2019). Practical definitions of severe versus familial hypercholesterolaemia and hypertriglyceridaemia for adult clinical practice. Lancet Diabetes Endocrinol. 7, 880–886. doi: 10.1016/s2213-8587(19)30156-1
Hamasaki, M., and Kotani, K. (2020). Lipoprotein(a) and Familial Hypercholesterolemia: A Short Review Including the Laboratory Viewpoint. Cardiol. Res. 11, 356–359. doi: 10.14740/cr1145
Henderson, R., O’Kane, M., McGilligan, V., and Watterson, S. (2016). The genetics and screening of familial hypercholesterolaemia. J. Biomed. Sci. 23:39.
Hendricks-Sturrup, R. M., Clark-LoCascio, J., and Lu, C. Y. (2020). A Global Review on the Utility of Genetic Testing for Familial Hypercholesterolemia. J. Pers. Med. 2020:10.
Jiang, L., Sun, L. Y., Dai, Y. F., Yang, S. W., Zhang, F., and Wang, L. Y. (2015). The distribution and characteristics of LDL receptor mutations in China: A systematic review. Sci. Rep. 5:17272.
Kim, S., Lee, H. S., Park, H. K., Linton, J. A., Lee, J. W., and Lee, H. (2017). Visceral adiposity and expression of clock genes in peripheral blood mononuclear cells: A pilot study. Chronobiol. Int. 2017, 1–10.
Lee, S. H. (2017). Update on Familial Hypercholesterolemia: Diagnosis, Cardiovascular Risk, and Novel Therapeutics. Endocrinol. Metab. 32, 36–40. doi: 10.3803/enm.2017.32.1.36
Mahdieh, N., Heshmatzad, K., and Rabbani, B. (2020). A systematic review of LDLR, PCSK9, and APOB variants in Asia. Atherosclerosis 305, 50–57. doi: 10.1016/j.atherosclerosis.2020.05.004
Mak, Y. T., Pang, C. P., Tomlinson, B., Zhang, J., Chan, Y. S., Mak, T. W., et al. (1998). Mutations in the low-density lipoprotein receptor gene in Chinese familial hypercholesterolemia patients. Arterioscler. Thromb. Vasc. Biol. 18, 1600–1605.
Mundal, L., Sarancic, M., Ose, L., Iversen, P. O., Borgan, J. K., Veierod, M. B., et al. (2014). Mortality among patients with familial hypercholesterolemia: a registry-based study in Norway, 1992-2010. J. Am. Heart. Assoc. 3:e001236.
Reiner, Z. (2015). Management of patients with familial hypercholesterolaemia. Nat. Rev. Cardiol. 12, 565–575. doi: 10.1038/nrcardio.2015.92
Santos, R. D., Gidding, S. S., Hegele, R. A., Cuchel, M. A., Barter, P. J., Watts, G. F., et al. (2016). Defining severe familial hypercholesterolaemia and the implications for clinical management: a consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Lancet Diabetes Endocrinol. 4, 850–861. doi: 10.1016/s2213-8587(16)30041-9
Schiebelhut, L. M., Abboud, S. S., Gomez Daglio, L. E., Swift, H. F., and Dawson, M. N. (2017). A comparison of DNA extraction methods for high-throughput DNA analyses. Mol. Ecol. Resour. 17, 721–729. doi: 10.1111/1755-0998.12620
Sharifi, M., Futema, M., Nair, D., and Humphries, S. E. (2017). Genetic Architecture of Familial Hypercholesterolaemia. Curr. Cardiol. Rep. 19:44.
Shi, Z., Yuan, B., Zhao, D., Taylor, A. W., Lin, J., and Watts, G. F. (2014). Familial hypercholesterolemia in China: prevalence and evidence of underdetection and undertreatment in a community population. Int. J. Cardiol. 174, 834–836. doi: 10.1016/j.ijcard.2014.04.165
Soutar, A. K., and Naoumova, R. P. (2007). Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat. Clin. Pract. Cardiovasc. Med. 4, 214–225. doi: 10.1038/ncpcardio0836
Strom, T. B., Laerdahl, J. K., and Leren, T. P. (2015). Mutation p.L799R in the LDLR, which affects the transmembrane domain of the LDLR, prevents membrane insertion and causes secretion of the mutant LDLR. Hum. Mol. Genet. 24, 5836–5844. doi: 10.1093/hmg/ddv304
Usifo, E., Leigh, S. E., Whittall, R. A., Lench, N., Taylor, A., Yeats, C., et al. (2012). Low-density lipoprotein receptor gene familial hypercholesterolemia variant database: update and pathological assessment. Ann. Hum. Genet. 76, 387–401. doi: 10.1111/j.1469-1809.2012.00724.x
Vrablik, M., Tichy, L., Freiberger, T., Blaha, V., Satny, M., and Hubacek, J. A. (2020). Genetics of Familial Hypercholesterolemia: New Insights. Front. Genet. 11:574474. doi: 10.3389/fgene.2020.574474
Vuorio, A., Kuoppala, J., Kovanen, P. T., Humphries, S. E., Tonstad, S., Wiegman, A., et al. (2017). Statins for children with familial hypercholesterolemia. Cochrane Database Syst. Rev. 7:CD006401.
Watts, G. F., Gidding, S. S., Mata, P., Pang, J., Sullivan, D. R., Yamashita, S., et al. (2020). Familial hypercholesterolaemia: evolving knowledge for designing adaptive models of care. Nat. Rev. Cardiol. 17, 360–377. doi: 10.1038/s41569-019-0325-8
Whitaker, H. C., Stanbury, D. P., Brinham, C., Girling, J., Hanrahan, S., Totty, N., et al. (2007). Labeling and identification of LNCaP cell surface proteins: a pilot study. Prostate 67, 943–954. doi: 10.1002/pros.20580
Willnow, T. E. (1999). The low-density lipoprotein receptor gene family: multiple roles in lipid metabolism. J. Mol. Med. 77, 306–315. doi: 10.1007/s001090050356
Keywords: low-density lipoprotein receptor gene, genomic variant, familial hypercholesterolemia, functional analyses, Golgi apparatus
Citation: Shu H-Y, Zhang W, Zheng C-C, Gao M-Y, Li Y-C and Wang Y-G (2021) Identification and Functional Characterization of a Low-Density Lipoprotein Receptor Gene Pathogenic Variant in Familial Hypercholesterolemia. Front. Genet. 12:650077. doi: 10.3389/fgene.2021.650077
Received: 06 January 2021; Accepted: 01 June 2021;
Published: 23 August 2021.
Edited by:
Jordi Pérez-Tur, Instituto de Biomedicina de Valencia, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Hayato Tada, Kanazawa University, JapanCopyright © 2021 Shu, Zhang, Zheng, Gao, Li and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan-Gang Wang, V2FuZ3lhbmdhbmcwNTMyQDEyNi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.