 Xuefei Yuan
Xuefei Yuan Ian C. Scott
Ian C. Scott Michael D. Wilson
Michael D. Wilson- 1Program in Genetics and Genome Biology, The Hospital for Sick Children, Toronto, ON, Canada
- 2Program in Developmental and Stem Cell Biology, The Hospital for Sick Children, Toronto, ON, Canada
- 3Department of Molecular Genetics, University of Toronto, Toronto, ON, Canada
Bound by lineage-determining transcription factors and signaling effectors, enhancers play essential roles in controlling spatiotemporal gene expression profiles during development, homeostasis and disease. Recent synergistic advances in functional genomic technologies, combined with the developmental biology toolbox, have resulted in unprecedented genome-wide annotation of heart enhancers and their target genes. Starting with early studies of vertebrate heart enhancers and ending with state-of-the-art genome-wide enhancer discovery and testing, we will review how studying heart enhancers in metazoan species has helped inform our understanding of cardiac development and disease.
Introduction
The heart is a vital organ whose primary role is to pump blood through the circulatory system to reach different organs. Heart-like structures are ancient and observed across diverse metazoans, including arthropods (such as Drosophila), mollusks (such as octopus) and chordates. Heart structures vary widely across metazoans ranging from a single-layered tubular heart in arthropods and tunicates (including Ciona), three separate hearts in some cephalopods (including octopus), a two-chambered heart in jawed fish, a three-chambered heart in amphibians, to a four-chambered heart in other tetrapods (reviewed in Stephenson et al., 2017; Poelmann and Gittenberger-de Groot, 2019). This lineage-specific tuning of cardiac structures is accompanied by changes in the whole circulatory system and highly adapted to the specific physiological needs of different animals. Despite these differences in heart structure, which are mostly related to later-stage heart morphogenesis, many cellular events and molecular regulators involved in early heart development are broadly shared across metazoan species.
A core set of cardiac transcription factors (TFs), including NK2 (Drosophila homolog: Tinman), MEF2 (Drosophila homolog: Mef2), GATA (Drosophila homolog: Pannier), TBX (Drosophila homolog: Nmr1/2, Doc1/2/3, etc.), and HAND (Drosophila homolog: Hand) families, interact with enhancers to control cardiac gene expression and cell fates in Drosophila, fish, and tetrapods (reviewed in Olson, 2006; Tolkin and Christiaen, 2012; Waardenberg et al., 2014). Though specific usage of paralogs and dosage sensitivities may vary between different species, these core TFs form the “cardiac regulatory kernel” (Tolkin and Christiaen, 2012; Waardenberg et al., 2014) in metazoans by closely interacting with each other and extracellular signaling cues. The requirement of extracellular signaling pathways in cardiogenesis also shows a high degree of conservation. The core signaling pathways, such as WNT, FGF, NOTCH, and BMP, play essential cardiogenic roles in both Drosophila and vertebrates (reviewed in Noseda et al., 2011).
Early vertebrate heart development involves a conserved sequence of cellular events that are seen in most, if not all, classes of vertebrate species (reviewed in Miquerol and Kelly, 2013). These events include: the emergence of specified cardiac progenitors within the anterior lateral plate mesoderm; migration of the cardiac progenitors to the midline to form the linear heart tube; rightward looping and elongation of the primitive heart tube; ballooning of the atrial and ventricular chambers out from the looped tube; and cardiac cushion and valve formation at the atrioventricular canal and outflow tract. This conserved set of events involve the complex interplay of multiple cardiac cell types, including the first heart field progenitors (FHF) that give rise to the linear heart tube and second heart field progenitors (SHF) that provide later addition to both poles of the heart tube (Kelly, 2012). Although cardiomyocytes make up a significant portion of mature hearts, other cell types, such as endocardial cells, smooth muscle cells, and cardiac fibroblasts, are also involved in cardiac development and physiological function (Hu et al., 2018; Honkoop et al., 2019; Tucker et al., 2020).
Understanding the interplay between multiple cardiac TFs and signaling pathways, within and between the cell types involved in cardiogenesis, requires a detailed knowledge of the cis-regulatory elements (CREs) that comprise heart enhancers. The regulatory logic encoded within CREs is readily understood by the embryo and is sufficient to organize multiple cardiac TFs and signaling pathways that ultimately result in a fully formed and functioning heart. In contrast, it has taken decades of experimental advances and insights to develop systems and technologies where cardiac CREs can be discovered and tested.
In this review, we discuss the genetic control of heart development and disease from an enhancer-centric perspective. From early gene-centric enhancer dissection in the 1990s to genome-wide characterization of heart enhancers in development and disease today, the discovery of heart enhancers has substantially shaped our understanding of the principles in cardiac gene regulation. We begin with a brief overview of developmental enhancers followed by a discussion of regulatory principles gained from pre-genomics enhancer studies. We then discuss how rapid advances in genome-wide approaches have transformed our knowledge regarding the locations, interactions, temporal dynamics and functions of heart enhancers. Our review will incorporate evolutionary characteristics of heart enhancers and discuss how new methods for dissecting heart enhancer functions promises to improve our understanding of heart development and cardiovascular diseases.
Enhancer Structure and Function in Development: A Primer
Enhancers are traditionally defined as short non-coding DNA sequences with the ability to drive gene expression regardless of the genomic distance, position, and orientation relative to the cognate genes [i.e., (Blackwood and Kadonaga, 1998) recently reviewed by Field and Adelman, 2020]. Enhancers can influence gene expression over short (hundreds of base pairs, bp) or large (megabases) genomic distances. These distal enhancers form long-range chromatin interactions with their target genes, such as the well-studied ZRS enhancer that is 1 Mb away from its target Shh (Lettice et al., 2003). This flexibility allows a single gene to be regulated by multiple enhancers with different spatiotemporal activities, as well as a single enhancer to contribute to the regulation of multiple genes, which was shown in recent genome-wide enhancer interaction maps (Montefiori et al., 2018; Jung et al., 2019). Together this many-to-many relationship sets up a complex regulatory network to achieve the highly diverse tissue-specific expression patterns evident in development.
Spatial-temporal developmental gene expression is achieved through the combinatorial recruitment of a discrete set of TFs to enhancers (for a recent review of how TFs recognize CREs see Zeitlinger, 2020). TFs interact with enhancers through short degenerate DNA sequence motifs. Recent work investigating the regulatory logic of a typical developmental enhancer supports an overarching principle that specific developmental gene expression relies on sub-maximal TF recognition motifs (Farley et al., 2015). Layered on top of TF motif affinity is the motif syntax within an enhancer, where the spacing, orientation, and order of the motifs themselves can impact the ability of the enhancer to drive developmental gene expression (Farley et al., 2016). It is also important to recognize that developmental genes are commonly regulated by additional redundant enhancers and ascertaining the contributions of individual enhancers remains an outstanding challenge for the majority of developmentally expressed genes (Cannavò et al., 2016; Osterwalder et al., 2018).
Some lineage-determining TFs can bind to compact chromatin regions that are largely inaccessible to other factors. These pioneer factors recruit chromatin-remodeling complexes that promote nucleosome eviction, facilitating the subsequent binding of other collaborating TFs and signal effectors (McPherson et al., 1993; Cirillo et al., 2002; reviewed in Zaret, 2020). To impact gene expression, TFs recruit transcriptional cofactors to enhancers. Cofactors can in turn modify chromatin states by catalyzing post-translational histone modifications (e.g., P300/CBP, MLL3/4), initiate chromatin remodeling (e.g., BRG1), bridge the gap between promoters and enhancer-bound transcription machinery (e.g., Mediator), or affect the affinity of TF binding at enhancers (Malik and Roeder, 2010; Siggers et al., 2011; Slattery et al., 2011; Krasnov et al., 2016). Despite these advances (and many others), much remains to be learned about the mechanisms underlying the recruitment of pioneer factors to a small subset of genomic sites and the molecular events that follow.
Enhancer activation in development is accompanied by progressive changes at the chromatin level, which in turn can be used to annotate enhancer states. Repressed enhancers are located in nucleosome dense regions. Certain repressed regions are characterized by the post-translational histone modification H3K27me3 which is deposited by the Polycomb repressive complex 2 (PRC2). The binding of pioneer factors and chromatin-remodeling complexes may switch enhancers to a poised state, in which enhancers share many features with those in an active state. Poised enhancers show features of low nucleosome occupancy, limited TF binding, and post-translational histone modifications H3K4me1 and H3K4me2 without the presence of H3K27ac, a histone mark of active developmental enhancers (Creyghton et al., 2010; Rada-Iglesias et al., 2011; Zentner et al., 2011). These poised developmental enhancers may even retain the repressive mark H3K27me3 (Rada-Iglesias et al., 2011; Zentner et al., 2011). Upon full activation, transcription co-factor P300 and RNA polymerase II are recruited to enhancers, leading to bi-directional transcription of enhancer RNAs and active enhancer regions marked with H3K27ac (reviewed by Calo and Wysocka, 2013; Heinz et al., 2015).
Enhancer activities are influenced by both local chromatin interactions and higher-order chromatin architectures. Eukaryotic genomes are compartmentalized into large self-interacting chromatin domains, termed topologically associated domains (TADs) (Dixon et al., 2012; Rao et al., 2014). TADs largely constrain the chromatin span that enhancers search through and define the regulatory domains within which enhancer-promoter interactions most frequently occur (Long et al., 2016). For example, promoter capture Hi-C experiments have revealed that 60–80% of the detected promoter interactions occur within TADs (Javierre et al., 2016; Choy et al., 2018; Montefiori et al., 2018). Early studies have noticed that TAD boundaries are shared between different cell types and conserved between species (Dixon et al., 2012; Vietri Rudan et al., 2015), however, these two concepts have been revised more recently. An increasing number of studies reported dynamic loss and gain of TADs and changes of TAD sizes during differentiation (Bonev et al., 2017; Bertero et al., 2019; Zhang et al., 2019). While evolutionarily conserved TADs correspond to regions of conserved synteny harboring important developmental genes and enhancers (Harmston et al., 2017), new analyses have questioned the extent to which TAD boundaries themselves correspond to evolutionary breakpoints (Eres et al., 2019; Eres and Gilad, 2020; Torosin et al., 2020). The importance of understanding how TADs relate to gene regulation is underscored by the increasing number of experiments showing that the disruption of TAD boundaries and sub-TAD domains can rewire enhancer-promoter interactions and fundamentally change the regulatory environment (Guo et al., 2015; Lupiáñez et al., 2015; Flavahan et al., 2016; Franke et al., 2016; Liang et al., 2020).
In sum, the precise and robust transcriptional regulation that occurs during development is achieved by the complex interplay between enhancers, TFs, co-factors, and epigenetic modifications, which together are organized under higher orders of chromatin architectures.
Heart Enhancers: Fundamental Insights, One Cre at a Time
Studies of heart enhancers initiated from targeted searches around cardiac genes. Putative enhancer regions were screened by “promoter bashing,” in which regulatory regions near the TSS are narrowed down via a series of deletions/mutations to produce overlapping DNA segments that are tested in reporter assays (Table 1). One of the best-studied examples is the mouse Nkx2.5 locus. LacZ reporter assays identified enhancer elements that specifically drove Nkx2.5 expression in different chambers of the hearts, as well as in thyroid, pharynx, and stomach within a 14 kb window around the TSS, revealing previously unappreciated complex enhancer modules underlying the control of cardiac TFs. Similar complexities were seen at genes encoding other cardiac TFs, such as Hand2 (heart and pharyngeal specific enhancers) (McFadden et al., 2000; Charité et al., 2001; Iklé et al., 2012), Mef2c (anterior heart field and somite specific enhancers) (Wang et al., 2001; Dodou et al., 2004), and Gata4 (lateral mesoderm, endocardium, and endoderm specific) (Rojas et al., 2005, 2009; Schachterle et al., 2012). Although limited in number and biased toward proximal gene promoter regions, these studies (and many others) have revealed fundamental principles and mechanisms underlying cardiac gene regulation.
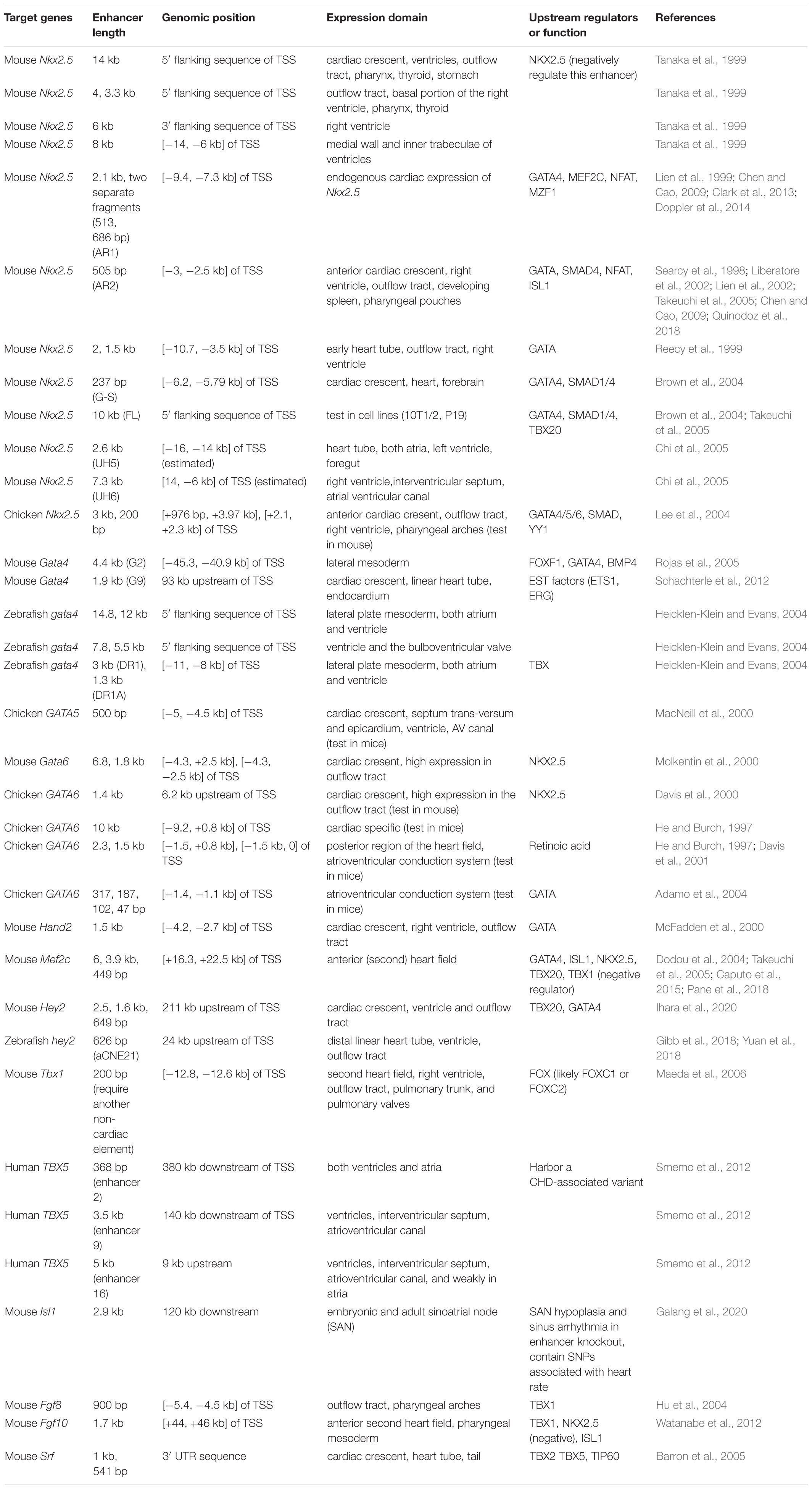
Table 1. Functionally characterized enhancer regions near cardiac genes.
Establishing Molecular Cascades Regulating Heart Development
Enhancers represent information hubs that integrate multiple upstream regulatory inputs such as lineage-determining master TFs and signaling effectors. Dissecting the transcription factors that bind to enhancers unveils these direct upstream regulators (Figure 1 and Table 1). By combining motif mutagenesis, gel shift, and transgenic assays, Nkx2.5 enhancer studies revealed that GATA4 and SMAD-mediated BMP signaling directly activated Nkx2.5 expression through multiple enhancer regions (Searcy et al., 1998; Lien et al., 1999, 2002; Liberatore et al., 2002; Brown et al., 2004) (Figure 1). Dissections of Nkx2.5 enhancers in the following years added ISL1, TBX20, MEF2C, and NFAT into direct upstream regulators that collectively drove Nkx2.5 expression in cardiac cells (Takeuchi et al., 2005; Chen and Cao, 2009; Clark et al., 2013). Furthermore, mining known heart enhancers can also lead to discoveries of novel cardiac regulators. For example, MZF1, previously known as a hematopoietic TF, was found to bind to an Nkx2.5 enhancer from in silico motif analysis and validated in embryonic stem cell (ESC) differentiation. Overexpression of MZF1 at different stages of cardiac differentiation revealed its novel, stage-dependent roles in cardiogenesis (Doppler et al., 2014).
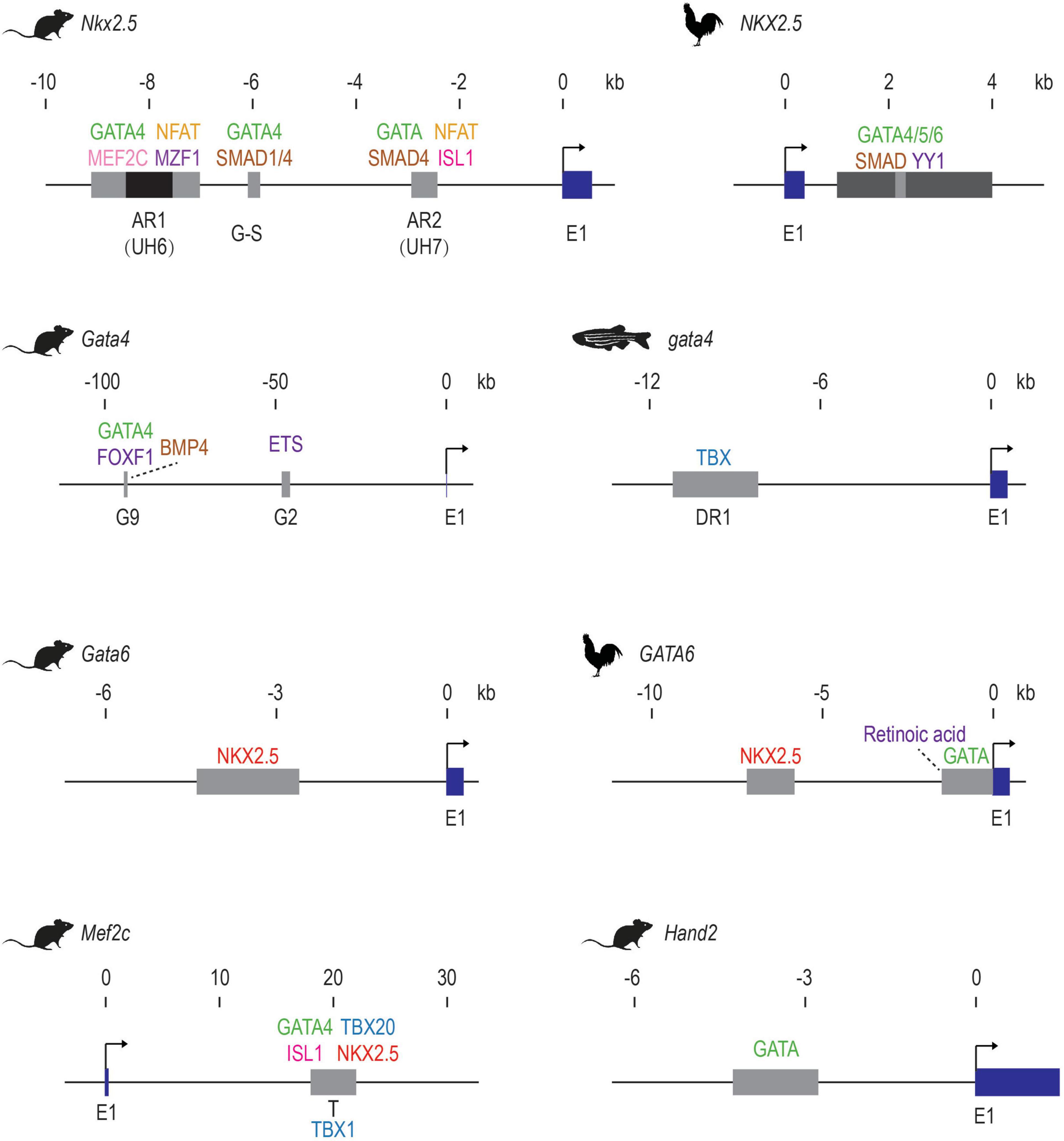
Figure 1. Early examples of validated of cardiac TF-enhancer interactions. The first exons of the cardiac genes are shown in dark blue. Enhancer elements are shown as gray boxes. The AR1 enhancer of mouse Nkx2.5 contains a repressive element in the middle, which is shown in black. Direct activators are listed above the enhancer elements while repressors are shown below. Upstream factors without direct binding evidence are indicated with dotted lines. E1: exon 1. These schematics are generated based on data from these publications: mouse Nkx2.5 (Searcy et al., 1998; Lien et al., 1999, 2002; Liberatore et al., 2002; Brown et al., 2004; Chi et al., 2005; Takeuchi et al., 2005; Chen and Cao, 2009; Clark et al., 2013; Doppler et al., 2014; Quinodoz et al., 2018); Chicken NKX2.5 (Lee et al., 2004); Mouse Gata4 (Rojas et al., 2005; Schachterle et al., 2012); zebrafish gata4 (Heicklen-Klein and Evans, 2004); mouse Gata6 (Molkentin et al., 2000); Chicken GATA6 (He and Burch, 1997; Davis et al., 2001; Adamo et al., 2004); mouse Mef2c (Dodou et al., 2004; Takeuchi et al., 2005; Pane et al., 2018); mouse Hand2 (McFadden et al., 2000).
Through similar enhancer dissection, the upstream signals of many other cardiac TFs have been identified (Table 1 and Figure 1). For example, the lateral mesoderm expression of mouse Gata4 relies on transcriptional inputs from FOXF1, BMP4, and its autoregulation (Rojas et al., 2005), while its expression in endocardia requires binding of ETS factors such as ETS1 and ERG (Schachterle et al., 2012). The anterior heart field (AHF) expression of Mef2c is positively regulated by GATA4, ISL1, and TBX20 and repressed by TBX1 through an intronic enhancer (Dodou et al., 2004; Takeuchi et al., 2005; Pane et al., 2018). Ventricular expression of Hey2 is dependent on TBX20 and GATA factor binding, but not NK-2 proteins. Summarizing the existing examples, it is clear that GATA factors, which regulate the expression of many other cardiac TFs (NKX2.5, HAND2, HEY2, MEF2C, etc.), sit among the top of the cardiac molecular cascade. Importantly, sustained cardiac expression of GATA itself requires the transcriptional inputs of other cardiac genes such as NKX2.5 and TBX factors, likely establishing a reciprocal feedback loop to maintain the robustness of the cardiac regulatory network.
Cardiac TF Crosstalk
Enhancer activation requires the cooperative binding of multiple TFs, therefore studying heart enhancers reveals cooperation and competition between these upstream factors. By co-expressing different combinations of factors together with a specific enhancer, the synergistic effect of factors in activating the enhancer can be revealed by quantitative measures like luciferase assays. Using this type of approach, GATA4 and SMAD1/4 were found to work as mutual co-activators in activating Nkx2.5 expression through a distal enhancer (commonly referred to as the G-S enhancer) (Brown et al., 2004). At another Nkx2.5 enhancer (AR1), GATA binding is indispensable for the transcriptional activation mediated by NFAT, likely through cooperative binding (Chen and Cao, 2009). Besides cooperativity, competitive binding between different TFs at heart enhancers can also play an important role in cardiac lineage specification. For example, the two homeodomain TFs, NKX2.5 and ISL1 compete for the same binding sites within an anterior second heart field enhancer of Fgf10, reflecting the antagonism between NKX2.5 and ISL1 during the differentiation from SHF progenitors to cardiomyocytes (Watanabe et al., 2012).
Putting Enhancers to Work
Besides providing direct evidence for building cardiac transcriptional networks, validated cardiac enhancers also frequently serve as genetic tools to label a specific cardiac population of interest for developmental studies. Transgenic mice in which Cre recombinase expression is driven by the Mef2c AHF enhancer have been used to determine anterior heart field derived structures and conditionally knock-out many developmental genes (Mef2c, Tbx1, β-catenin, Ezh2) to reveal their specific roles in anterior heart field development and congenital heart disease (Verzi et al., 2005; Delgado-Olguín et al., 2012; Barnes et al., 2016; Racedo et al., 2017). A GFP line driven by the Nkx2.5 AR1 enhancer was used to discover an immature cardiomyoblast population in neonatal mice that was required for normal heart development (Serpooshan et al., 2017). Recently, this Nkx2.5 enhancer was found to be reactivated after myocardial infarction in the adult heart, suggesting the role of this enhancer in responses to heart injuries (Deutsch et al., 2018). A mouse Smarcd3 enhancer was found to label early cardiac progenitor cells before the expression of known cardiac markers (Nkx2.5, Isl1, Tbx5) in mice, indicating an early molecular distinction between cardiac progenitors and neighboring cells (Devine et al., 2014). This enhancer was later shown to function similarly in zebrafish and helped identify ∼160 putative cardiac enhancers conserved between zebrafish and mammals (Yuan et al., 2018). One of these deeply conserved heart enhancers recapitulated the cardiac expression of the nearby gene hey2 thus was subsequently used in dissecting how hey2 restricted cardiac progenitor proliferation (Gibb et al., 2018).
In sum, deeply dissecting cardiac enhancers reveals both molecular tools for visualizing, isolating, and manipulating cardiac populations as well as cis- and trans-regulatory mechanisms that control cardiac gene expression.
Unmasking Heart Enhancers With Comparative and Functional Genomics
Enhancer Hunting: Tools of the Trade
Comparative genomics has long been used to identify putative enhancer regions (Tagle et al., 1988; Aparicio et al., 1995). Such comparative approaches are based on the assumption that functionally relevant enhancer sequences will be under negative selection and will thus show higher sequence constraints than non-functional regions. This assumption is supported by the genome-wide identification of conserved non-coding elements (CNEs) and the following discoveries that many CNEs work as developmental enhancers (Nobrega et al., 2003; Bejerano et al., 2004; Johnson et al., 2004; de la Calle-Mustienes et al., 2005; Shin et al., 2005; Woolfe et al., 2005; Pennacchio et al., 2006). Substantial work using a variety of approaches including transitive alignment (Hiller et al., 2013; Braasch et al., 2016), ancestral reconstruction (Hiller et al., 2013), and conserved microsynteny (Irimia et al., 2012; Clément et al., 2020; Wong et al., 2020) have further enhanced our ability to detect more distantly related conserved non-coding elements.
Although CNEs are enriched for developmental enhancers, the vast majority of enhancers appear to evolve more rapidly, with many being lineage- or species-specific. This feature has been demonstrated in many different tissues or cell types and in both vertebrates and invertebrates (Odom et al., 2007; Kunarso et al., 2010; Schmidt et al., 2010b; Mikkelsen et al., 2010; Cotney et al., 2013; Paris et al., 2013; Arnold et al., 2014; Villar et al., 2015). Although enhancers in different tissues or at different developmental stages may be under varied selection pressures (Blow et al., 2010; Nord et al., 2013; Visel et al., 2013), rapid evolution is an overall feature of enhancer sequences, which suggests that many enhancers would be missed in detection approaches based on sequence conservation alone.
Over the past 15 years, large scale genomic assays have enabled enhancer discoveries at an unprecedented scale (Table 2). In particular, chromatin immunoprecipitation with high-throughput sequencing (ChIP-seq) can locate enhancers by profiling the co-occupancy of lineage-specific TFs, binding of co-factors, or post-transcriptional modifications that marks active enhancers (reviewed in Buecker and Wysocka, 2012). As ChIP-seq requires large numbers of input cells, which is often difficult to obtain from early embryonic tissues, many low input ChIP methods (O’Neill et al., 2006; Brind’Amour et al., 2015) and alternative strategies, such as enzyme-tethering based approaches have been established (e.g., CUT&RUN, CUT&Tag, and CUTAC) (Skene and Henikoff, 2017; Kaya-Okur et al., 2019; Meers et al., 2019; Henikoff et al., 2020).
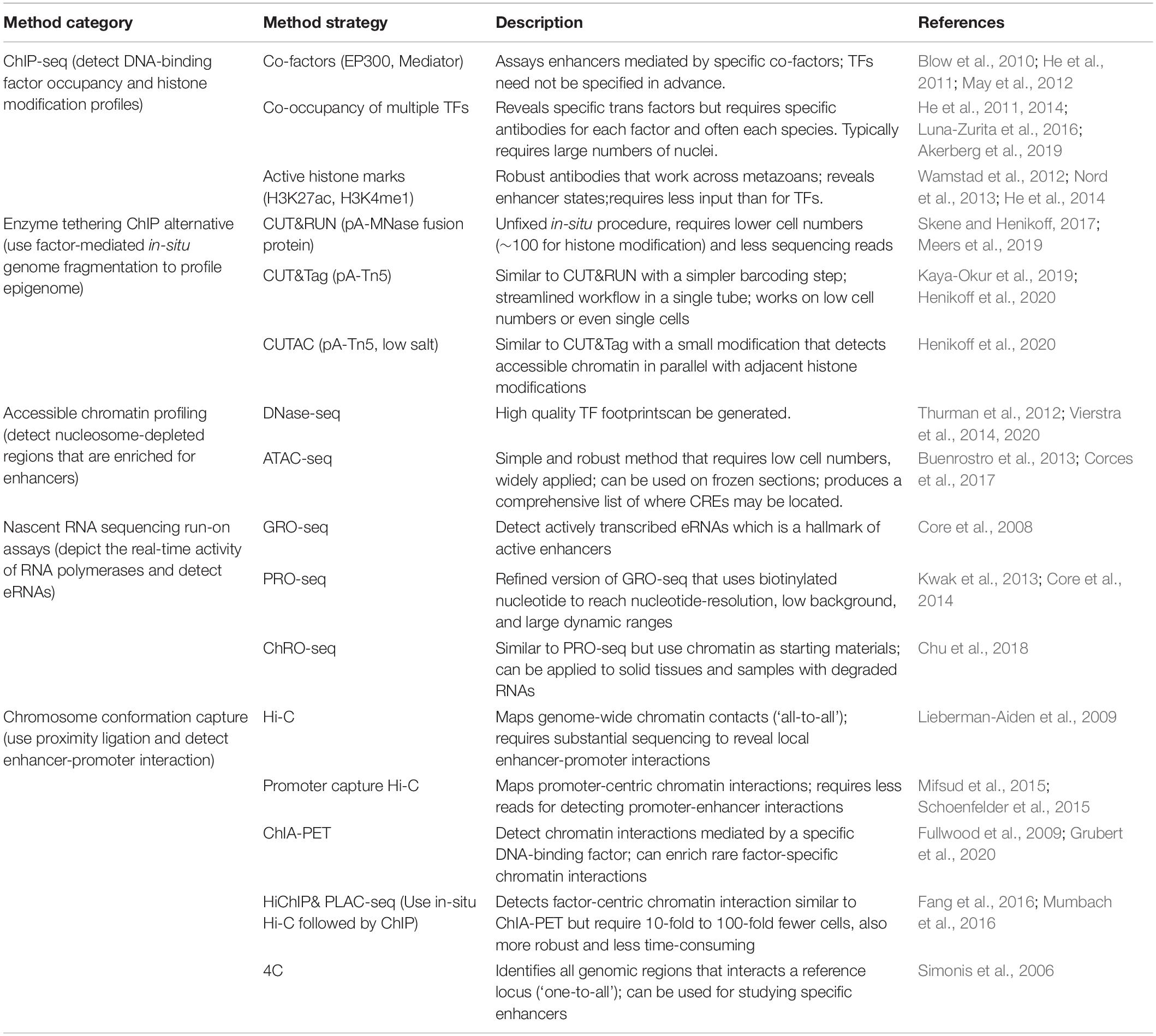
Table 2. Genomic approaches for enhancer mapping.
Chromatin accessibility profiling provides a comprehensive view of the candidate regions most likely to harbor CREs, making them arguably the most widely used assay to identify putative enhancers (Thurman et al., 2012; Buenrostro et al., 2013; Vierstra et al., 2014, 2020; Corces et al., 2017). Since active enhancers are transcribed bidirectionally to produce eRNA, nascent RNA sequencing technologies, specifically the run-on assays (GRO-seq, PRO-seq, ChRO-seq, etc.), can be used as a direct readout of enhancer activity. Furthermore, when coupled with chromatin accessibility assays (i.e., ATAC-seq), run-on assays can distinguish active enhancers (producing bi-directional RNAs) from other CREs such as CTCF bound insulators (reviewed in Wissink et al., 2019).
After discovering a distal putative enhancer, one of the most pressing questions is to discover what gene or genes it associates within a cell type and condition of interest. To address this, chromosome conformation capture (3C) based assays (including 4C, 5C, HiChIP, promoter capture Hi-C, and Hi-C) are commonly used to characterize enhancer-promoter interactions (Denker and De Laat, 2016; Fang et al., 2016; Mumbach et al., 2016). Capture Hi-C approaches, such as promoter-capture Hi-C and HiCap (Mifsud et al., 2015; Sahlén et al., 2015; Schoenfelder et al., 2015), are increasingly being used to reveal promoter-centric chromatin interactions at high resolution. Capture-based methods that target putative enhancer regions, such as those discovered by DNAse-seq (Sönmezer et al., 2020), could also be used for ‘enhancer-capture’ Hi-C. Naturally, the choice of 3C-based methods depends on the research question and practical considerations such as the quantity of sample material, genome size, capture probe availability, and sequencing costs.
These widely used genome-scale assays, each with their own strengths (Table 2), continue to reveal new insights into enhancer location, activity and function. The increasing number of high-quality datasets are also creating new opportunities and challenges for integrative data analysis that will further expand our understanding of metazoan heart development and human disease.
Heart Enhancers: From Genome-Wide Mapping to Metazoan Regulatory Logic
The development of ChIP-chip, ChIP-seq, and other genomic techniques has enabled genome-wide enhancer discoveries and analysis of distinct cardiac samples obtained from diverse model systems (Table 3). Pioneering studies in Drosophila using ChIP-chip against master regulators (Twi, Tin, Mef2, Bag, Bin, Doc, and Pnr) and signaling effectors (dTCF and pMad) required for the specification of cardiac mesoderm revealed fundamental principles of combinatorial TF binding dynamics and TF-signaling interactions at cardiac enhancers (Zinzen et al., 2009; Junion et al., 2012). These Drosophila cardiac TF mapping studies, together with a comparative analysis of Twi, Tin, Mef2, Bin, and Bap in two distant Drosophila species, underscore the conserved presence of combinatorial TF binding, even when the underlying DNA sequence has changed (Khoueiry et al., 2017). The Junion et al. (2012) study led to a “transcription factor collective” model of TF binding where TFs use both protein-DNA and protein–protein interactions to regulate gene expression (reviewed by Spitz and Furlong, 2012), which was later supported by the comparative Khoueiry et al. (2017) study.
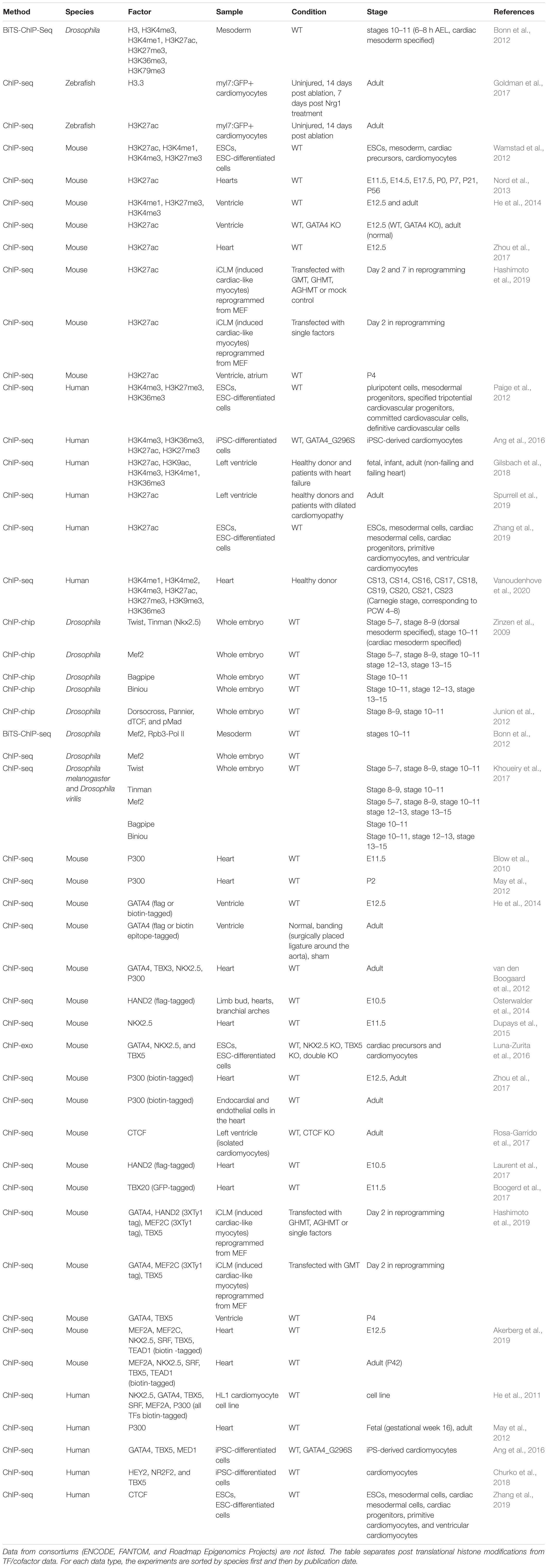
Table 3. Genome-wide metazoan heart enhancer profiling datasets generated using chromatin immunoprecipitation of post translational histone modifications, transcription factors, and cofactors.
To demarcate the location of putative enhancers active in embryonic and adult hearts, pioneering mammalian studies performed ChIP-seq for the histone acetyltransferase EP300 and the active post-translational histone modification H3K27ac (Blow et al., 2010; May et al., 2012). To overcome the challenge of having to obtain specific antibodies for each TF of interest, many ChIP-seq studies have used tagging methods to biotinylate DNA binding proteins including EP300 and cardiac TFs to define heart enhancers in Drosophila (Bonn et al., 2012), mouse embryos (He et al., 2014; Zhou et al., 2017; Akerberg et al., 2019) and human cardiomyocyte cell lines (He et al., 2011). These biotin tagging-based approaches achieve more sensitive and reliable identifications of heart enhancers and enable enhancers discoveries in specific cardiac cell types (Zhou et al., 2017).
Like in Drosophila, the combinatorial binding of cardiac TFs defines mammalian heart enhancers (He et al., 2011; Akerberg et al., 2019). However, it is still unclear whether the mammalian cardiac enhancers discovered by these and other studies fit the “TF collective model” proposed for Drosophila; the “billboard model (Arnosti and Kulkarni, 2005),” in which specific sets of TFs are recruited to enhancers with flexible motif grammar; or a mixture of models (Long et al., 2016). For example, the importance of heterotypic interactions between mouse TBX5 and NKX2-5 was demonstrated using co-crystal structure together with DNA, as well as ChIP-exo experiments (Luna-Zurita et al., 2016). Intriguingly, the genetic loss of either Tbx5 or Nkx2-5 led to ectopic interactions of the other remaining TF. Unlike the more flexible “TF collective” or “billboard” models, TBX5 and NKX2-5 co-occupancy highlighted in this study featured preferred motif arrangements. Most recently, a novel single molecule footprinting (SMF) method was used to ascertain TF co-occupancy in mouse embryonic stem cells (Sönmezer et al., 2020). In this study, simultaneous TF binding did not depend on the identity of the TFs involved, and the co-occupancy of TFs on chromatin lacked of strict motif organization, which the authors proposed agreed with the “billboard model” (Sönmezer et al., 2020). Indeed, comparative approaches using this SMF method to study enhancer logic during metazoan cardiac development will be insightful for both learning general principles governing enhancer regulation as well as the biologically important exceptions that define key physiological processes.
To study cardiac enhancer dynamics across multiple stages of in vitro cardiac differentiation or in vivo development, several studies from individual labs as well as consortiums, have utilized robust genome-wide assays that do not rely on mapping specific transcription factors, namely ChIP-seq for histone modifications (Paige et al., 2012; Wamstad et al., 2012; Nord et al., 2013; Vanoudenhove et al., 2020), and DNase-seq and ATAC-seq for chromatin accessibility (Bertero et al., 2019; Gorkin et al., 2020; Meuleman et al., 2020). These studies revealed highly dynamic chromatin states accompanying cardiac differentiation and development. Specifically, ATAC-seq is widely used on precious in vivo cardiac samples to identify genomic regions that are enriched for TF binding and functional enhancer elements (Jia et al., 2018; Yuan et al., 2018; Pawlak et al., 2019; Racioppi et al., 2019). Recently, accessible chromatin profiling has also enabled the discovery of enhancers specific to cardiac subpopulations, such as pacemaker cells (Galang et al., 2020; van Eif et al., 2020) and endocardial populations (Boogerd et al., 2017).
Functional insights into cardiac enhancer regions continue to be made by studying TF occupancy and chromatin states upon the perturbation of cardiac TFs or signaling pathways in multiple organisms (e.g., Gata4, gata5, Nkx2.5, Tbx5/tbx5, Tbx20, Hand2/hand2, Isl1, Foxf, Fgfr, Mek, and Ras) (He et al., 2014; Luna-Zurita et al., 2016; Boogerd et al., 2017; Jia et al., 2018; Pawlak et al., 2019; Racioppi et al., 2019), as well as in a human congenital heart disease (CHD) model (cardiomyocytes with a disease-associated missense mutation of GATA4) (Ang et al., 2016). These studies reveal the master regulatory roles of cardiac TFs at the chromatin level. For example, GATA4 is essential for establishing open chromatin, promoting active epigenetic modification (H3K27ac) and recruiting TBX5 to the proper cardiac enhancers (He et al., 2014; Ang et al., 2016). On the other hand, TBX5 and NKX2.5 are important for preventing ectopic binding of GATA4 during cardiac differentiation, highlighting the importance of interdependent co-occupancy of these cardiac TFs in precisely controlling cardiac gene expression (Luna-Zurita et al., 2016). This interdependent co-occupancy is also essential in cardiac reprogramming, as only co-expression of cardiac factor cocktails (GATA4, HAND2, TBX5, MEF2C, etc.), but not single-TF overexpression, can leads to robust cardiac TF occupancy to reprogramming enhancers (Hashimoto et al., 2019).
Heart Enhancers in Space: Chromatin Interactions and Architectures
Heart enhancer activity not only requires proper TF binding, but is under the control of local chromatin interactions and higher-order chromatin architectures. Several groups have conducted promoter capture Hi-C in ESC/iPSC-derived cardiomyocytes or adult hearts to map enhancer-promoter interactions (Choy et al., 2018; Montefiori et al., 2018; Jung et al., 2019). These promoter capture Hi-C studies identified potential target genes for a substantial fraction of candidate heart enhancers. Interestingly, on average 25–35 distal interacting regions per gene and 40–60% of distal regions interacting with more than one gene. Hi-C has also been recently used to profile high-order chromatin architectures such as TADs and compartments across closely sampled time points during the differentiation from stem cells to cardiomyocytes (Bertero et al., 2019; Zhang et al., 2019). These studies showed extensive rearrangement of chromatin architectures during cardiac cell differentiation, with 19% genome switching compartments and 20–40% of TADs being stage-specific. Integrated analyses based on these datasets also revealed important regulatory mechanisms and unknown regulators in heart development. For example, Bertero et al. (2019) detected spatial coalescence of multiple cardiac genes from different chromosomes. This coalescence formed a trans-interacting chromatin domain that recruited the muscle-specific splicing factor RBM20 for efficient pre-mRNA splicing (Bertero et al., 2019).
The importance of chromatin interactions and architecture in heart development and function is also revealed by the essentiality of genome organizing factors such as CTCF and the cohesin complex. CTCF knock-out in cardiac progenitor cells leads to severe defects in cardiac cell maturation due to the disruption of enhancer-promoter interaction and subsequent misregulation of cardiac genes (Gomez-Velazquez et al., 2017). In the adult heart, CTCF depletion is sufficient to induce pathological consequences that are very similar to heart failure (Rosa-Garrido et al., 2017). Knock-out of Stag2 (which encodes a cohesin subunit) in embryonic mice leads to lethality by E10.5 due to severe morphogenesis defects in SHF-derived structures (right ventricle, outflow tract and septation), however, loss of Stag2 in adults only moderately reduces their fitness, indicating a strong developmental role of Stag2 (De Koninck et al., 2020). Perturbation of the cohesin loading factor NIPBL in both mouse and zebrafish results in multi-organ defects (including heart abnormalities) reminiscent to the Cornelia de Lange Syndrome, a congenital disease linked to NIPBL mutation (Kawauchi et al., 2009; Muto et al., 2011; Santos et al., 2016). The different phenotypes observed upon the loss of cohesin complex members, cohesin associated loading proteins, and CTCF indicate that in addition to their roles in sister chromatid cohesion and chromatin organization (Merkenschlager and Nora, 2016; Hanssen et al., 2017; Pugacheva et al., 2020), there are likely more subtle and CTCF-independent roles (i.e., Schmidt et al., 2010a) for these proteins in cardiac gene regulation.
Enhancing Enhancers With Enhancer-Associated RNAs
Upon activation, many enhancers are transcribed into non-coding RNAs, which are broadly referred to as enhancer RNAs (eRNAs). The expression of eRNAs is well correlated with their putative target gene expression (Kim et al., 2010; Kaikkonen et al., 2013; Li et al., 2013; Andersson et al., 2014; Arner et al., 2015). eRNAs may not only serve as hallmarks of enhancer activation, but also exert important functions in driving target gene expression by promoting chromatin accessibility (Mousavi et al., 2013), mediating enhancer-promoter interaction (Lai et al., 2013; Li et al., 2013; Hsieh et al., 2014), regulating chromatin remodeling (Kaikkonen et al., 2013), and facilitating PolII pause-release at promoters (Schaukowitch et al., 2014; Shii et al., 2017). However, for the vast majority of eRNAs, it remains unclear whether they are simply by-products of enhancer transcription or whether they possess functional roles based on the transcriptional process itself, or through additional molecular interactions in cis or in trans (reviewed in Li et al., 2016; Arnold et al., 2020).
Though early discoveries described eRNAs as short, non-polyadenylated, bidirectionally transcribed RNAs (Kim et al., 2010; Andersson et al., 2014), a diverse group of molecules with other structures (long, polyadenylated, or unidirectionally transcribed) have been attributed to eRNAs (Koch et al., 2011; Kaikkonen et al., 2013; Li et al., 2013; Alvarez-Dominguez et al., 2017). The structure and functional similarities between some eRNAs and cis-acting long non-coding RNAs (lncRNAs) have raised an emerging concept that they represent overlapping categories of regulatory non-coding RNAs (ncRNAs) (Espinosa, 2016; Paralkar et al., 2016; Arnold et al., 2020; Gil and Ulitsky, 2020).
The roles of eRNAs and lncRNAs in the contexts of heart development have been explored by many studies (Grote et al., 2013; Klattenhoff et al., 2013; Ounzain et al., 2014, 2015; Anderson et al., 2016; Alexanian et al., 2017; Yang et al., 2017; Turton et al., 2019; Nicole Ritter et al., 2019). Given the challenge in categorizing these ncRNAs, we consider all ncRNAs that are associated with heart enhancers and discuss the different ways through which they may regulate heart development using two examples: (1) The ncRNA transcript itself is involved in target gene regulation. For example, using anti-sense mediated RNA knockdown, Yang et al. (2017) showed that the expression of Ryr2, a TBX5 target that is critical for maintaining cardiac rhythm, depends on a novel TBX5-dependent eRNA, RACER; and (2) Instead of the ncRNA molecule, it is the transcriptional activity of the ncRNA locus that appears to be important for controlling the target genes. Two such ncRNAs come from the Hand2 locus, upperhand (Uph) (Anderson et al., 2016) and handsdown (Hdn) (Nicole Ritter et al., 2019). Particularly, the Hdn locus interacts with the Hand2 promoter and putative cardiac enhancers, suggesting it may regulate Hand2 expression via direct chromatin interaction, reminiscent of CREs (Nicole Ritter et al., 2019). The transcription of Uph over a cardiac enhancer upstream of Hand2 allows the binding of GATA4 and deposition of H3K27ac to this enhancer (Anderson et al., 2016). Together, these examples showcase a few models of the complex interactions between enhancers and the ncRNAs associated with them.
The field of enhancer-associated ncRNAs in heart development has many unanswered questions. Future studies that use chromatin run-on assays (GRO-seq, PRO-seq) or generate deeply sequenced RNA-seq datasets coupled with enhancer annotations should help to understand the dynamic changes of eRNAs in development. Functional experiments such as those use RNA targeting Cas protein (Cas13) (Abudayyeh et al., 2017), shRNA (Lambeth and Smith, 2013), or antisense oligonucleotide-mediated knockdown (Dias and Stein, 2002) will also be essential for teasing out the roles of enhancer-associated ncRNAs in gene-regulation independent of the enhancer elements themselves.
Heart Enhancers: Keeping Track of Time
As the activity of enhancers are not only tissue-specific but also stage-specific, it is important to obtain high-resolution temporal profiles of heart enhancers to truly understand their function. This is specifically highlighted by the in vitro cardiac differentiation study from Wamstad et al. (2012), which showed that enhancers active in ESC, mesoderm progenitors, cardiac progenitors, and cardiomyocytes were largely non-overlapping (Wamstad et al., 2012). Consistently, Luna-Zurita et al. (2016) discovered thousands of GATA4, NKX2.5, and TBX5 binding sites were specific to either cardiac progenitor cells or cardiomyocytes. Similar results have also been reported for in vivo development, for example, 80% of the GATA4 binding sites in fetal heart are not occupied by GATA4 in adult heart (He et al., 2014).
Since the heart is the first organ formed in embryogenesis, the embryonic stage that is required to capture the initial phase of cardiogenesis is especially early in development, and is likely during early gastrulation (Scott, 2012; Devine et al., 2014; Lescroart et al., 2014). Though heart enhancers have been extensively characterized across many developmental stages in various species [such as Nord et al. (2013) and Vanoudenhove et al. (2020) and many others in Tables 3–5], there is a paucity of datasets that characterize enhancers active at the initial stage of vertebrate heart development, such as the transition from mesoderm progenitors to cardiac lineages. The majority of in vivo studies in vertebrates used relatively mature cardiac samples, including embryonic hearts with defined chamber structures (e.g., E10.5 and onward in mice) or postnatal heart tissues (Tables 3–5). As these stages are later than when cardiac lineage commitment occurs, these studies may not capture the enhancers that specifically drive early cardiogenesis.
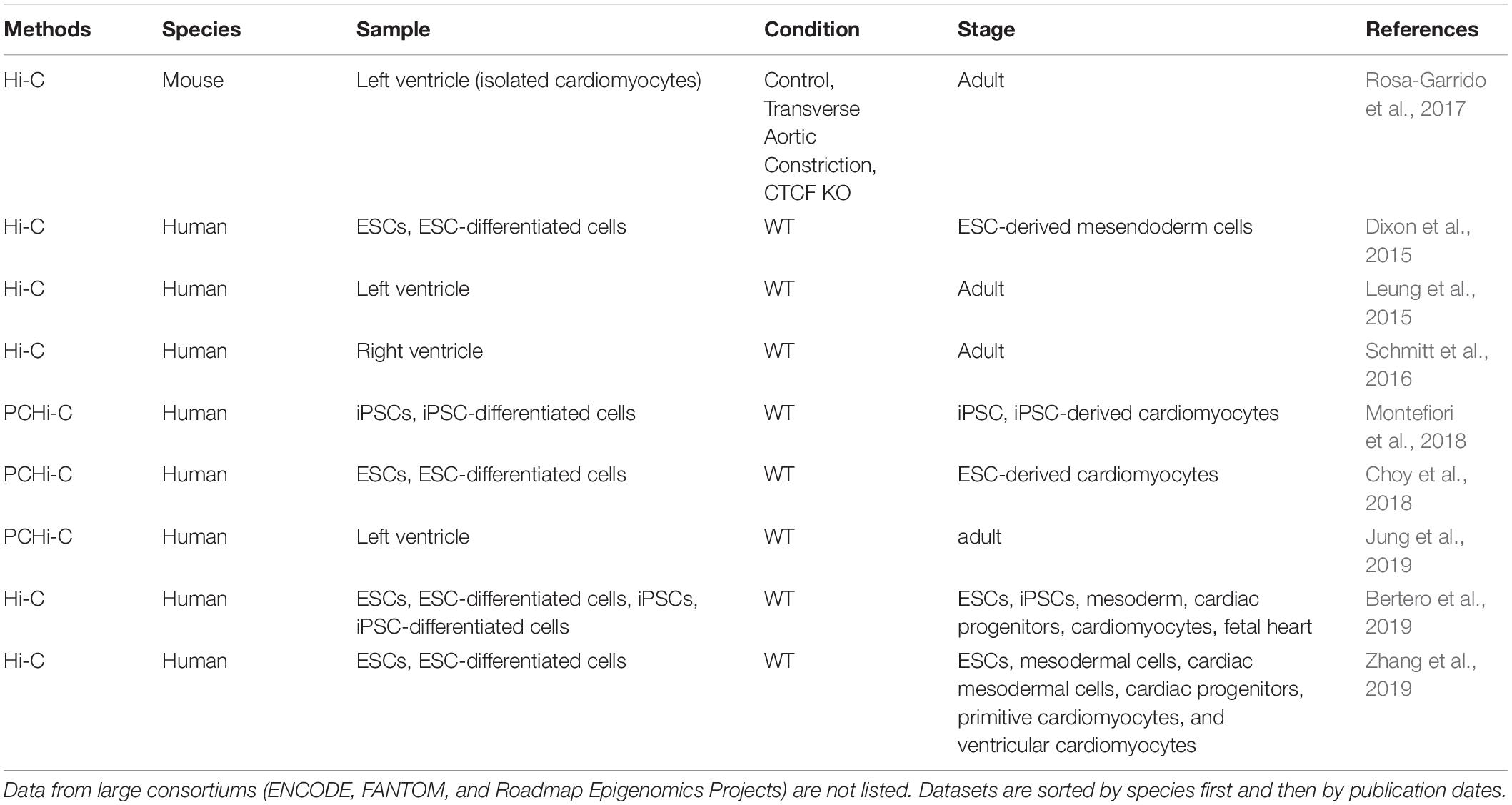
Table 4. Chromatin interaction datasets used for annotating heart enhancers.
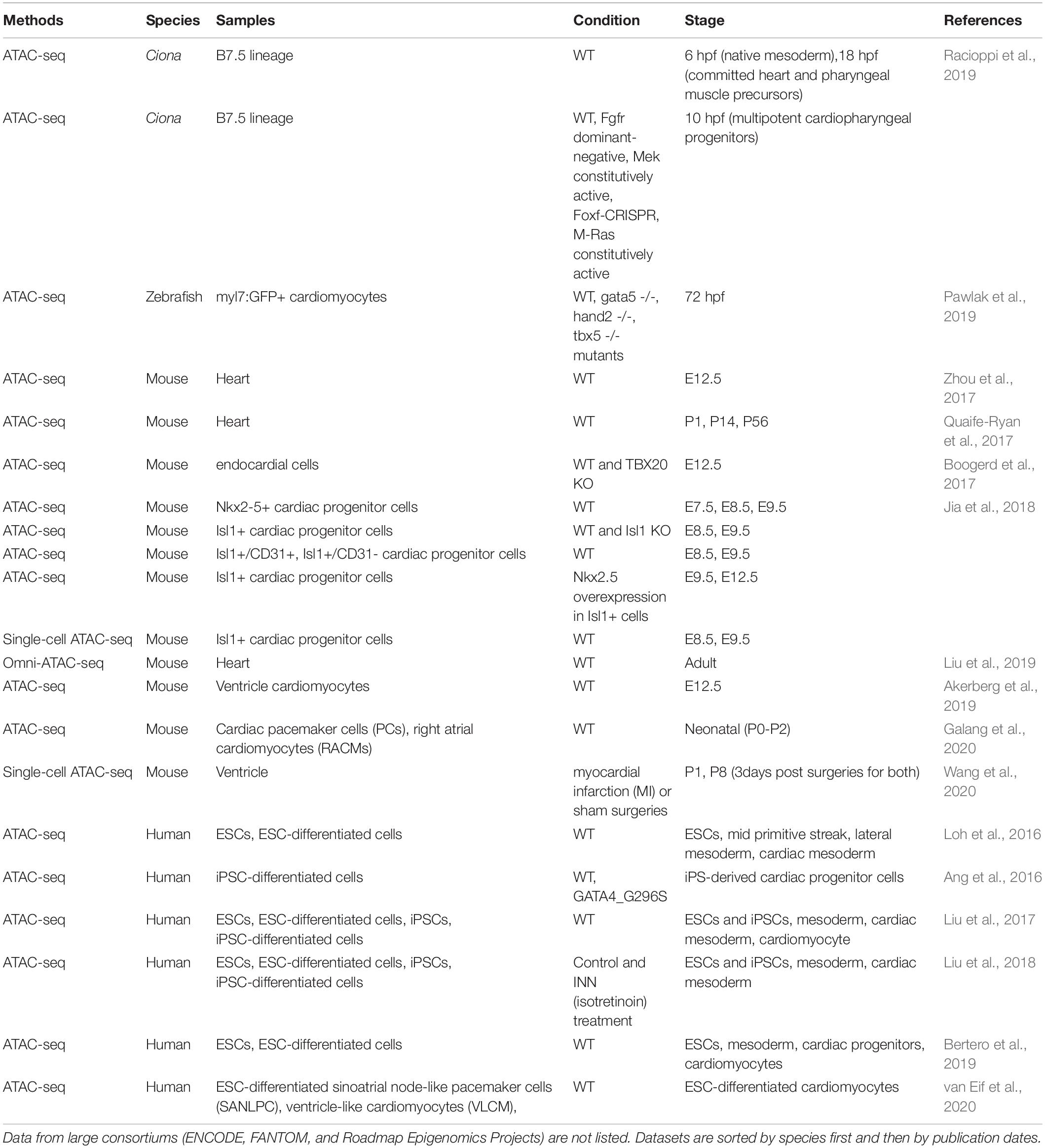
Table 5. Chromatin accessibility datasets used for annotating heart enhancers.
A few recent in vivo studies confirm the observations made from in vitro differentiation that enhancer-associated chromatin states are highly dynamic, especially during early cardiac lineage specification. A recent study that profiled mouse Nkx2.5+ cardiac progenitor cells revealed major changes in chromatin accessibility between E7.5 and E8.5 but only minor differences between E8.5 and E9.5 (Jia et al., 2018). This suggests that early lineage fate transitions may be accompanied by major changes of chromatin states, which become more stabilized in committed cell types. Similar trends are observed in cardiopharyngeal lineage specification in the tunicate Ciona, in which most significant chromatin changes occur between the transition from mesoderm progenitors to cardiopharyngeal progenitors compared to later stages (Racioppi et al., 2019). These examples reveal intriguing dynamics of the enhancers involved in early cardiac lineage decisions, however, much remains to be explored. Filling this knowledge gap, especially in the context of developing embryos, can bring valuable insights into key cellular events in early cardiogenesis.
Evolutionary Mysteries of Heart Enhancers
Intriguing results have emerged from evolutionary studies of heart enhancers. Although the TFs controlling heart enhancers are highly conserved, validated heart enhancers show weak DNA constraint compared to brain enhancers identified at the same developmental stage (E11.5) (Blow et al., 2010). For instance, only 6% of the candidate heart enhancers were deemed to possess high DNA constraint (phastCon score > 600) compared to 44% of forebrain, 39% of midbrain, and 30% of limb enhancers. This could be in part due to the fact that molecularly, the brain seems to be a more conserved organ in terms of the low proportion of positively select genes, old phylogenetic ages of the transcriptomes, and the low percentage of genes showing trajectory changes between different species (Cardoso-Moreira et al., 2019).
It remains an open and intriguing question how heart enhancers that lack evolutionary conservation work together with many conserved cardiac TFs to orchestrate the development of the heart. Several reasons may contribute to this phenomenon. First, it has been demonstrated by many studies that enhancers are rapidly evolving with pervasive turnovers of TF binding sites (TFBSs) (Kunarso et al., 2010; Mikkelsen et al., 2010; Schmidt et al., 2010b; Cotney et al., 2013; Paris et al., 2013; Arnold et al., 2014; Ballester et al., 2014; Villar et al., 2015; Khoueiry et al., 2017). The rapid changes in the sequence, orientation, spacing and numbers of TFBSs within enhancers may not necessarily alter the functional roles of enhancers but do make it hard to detect enhancer sequence homology via genomic sequence alignment. As a consequence, some functionally conserved enhancers will not share detectable sequence homology. A recent and striking example is a sponge Islet enhancer, which drives expression that overlaps endogenous islet gene (isl2a) expression in zebrafish, despite the absence of homologous sequence in the vertebrate genomes. Nevertheless, enhancers with similar TFBS compositions can be found in human and mouse ISLET/Islet regions and their activities resemble that of the sponge enhancer in zebrafish (Wong et al., 2020). A similar strategy based on motif composition also identified conserved brain enhancers between chordates and hemichordates, which would not have been detected by sequence alignment alone (Yao et al., 2016). These two examples and many others i.e. (Fisher et al., 2006; Hare et al., 2008; Friedli et al., 2010; Chatterjee et al., 2011) indicate that a grammar more flexible than strict sequence conservation is used in some enhancers to produce conserved transcriptional “output.” Overall, the discordance between sequence and functional conservation may account for a significant portion of the weakly conserved heart enhancers.
Second, an increasing number of studies indicate that the conservation of enhancers active in early embryonic development follows an hour-glass like pattern (Bogdanovic et al., 2012; Bogdanović et al., 2016; Liu et al., 2020) similar to that of transcriptomes (Irie and Sehara-Fujisawa, 2007; Domazet-Lošo and Tautz, 2010; Kalinka et al., 2010; Irie and Kuratani, 2011; Yanai et al., 2011). However, much less is known about “phylotypic enhancers” that presumably are established prior to organogenesis to set up conserved vertebrate gene expression patterns. A temporal study of developmental enhancers compared the H3K27ac (a mark of active enhancers) profiles across the development of three mouse tissues (heart, brain, and liver) from ESC to adults (Nord et al., 2013). They showed that both sequence constraints (PhastCon scores) and evolutionary ages of candidate active enhancers peak at different developmental stages in different tissues. Though enhancers active in the brain show the highest conservation at E11.5, heart enhancers active at mouse E11.5 are less conserved compared to those active during earlier cardiac lineage specification (Figure 2A). This suggests that although enhancer turnover is a typical property of heart enhancers, deeply conserved CREs are more likely to be active in early cardiogenesis or even prior to cardiac lineage commitment.
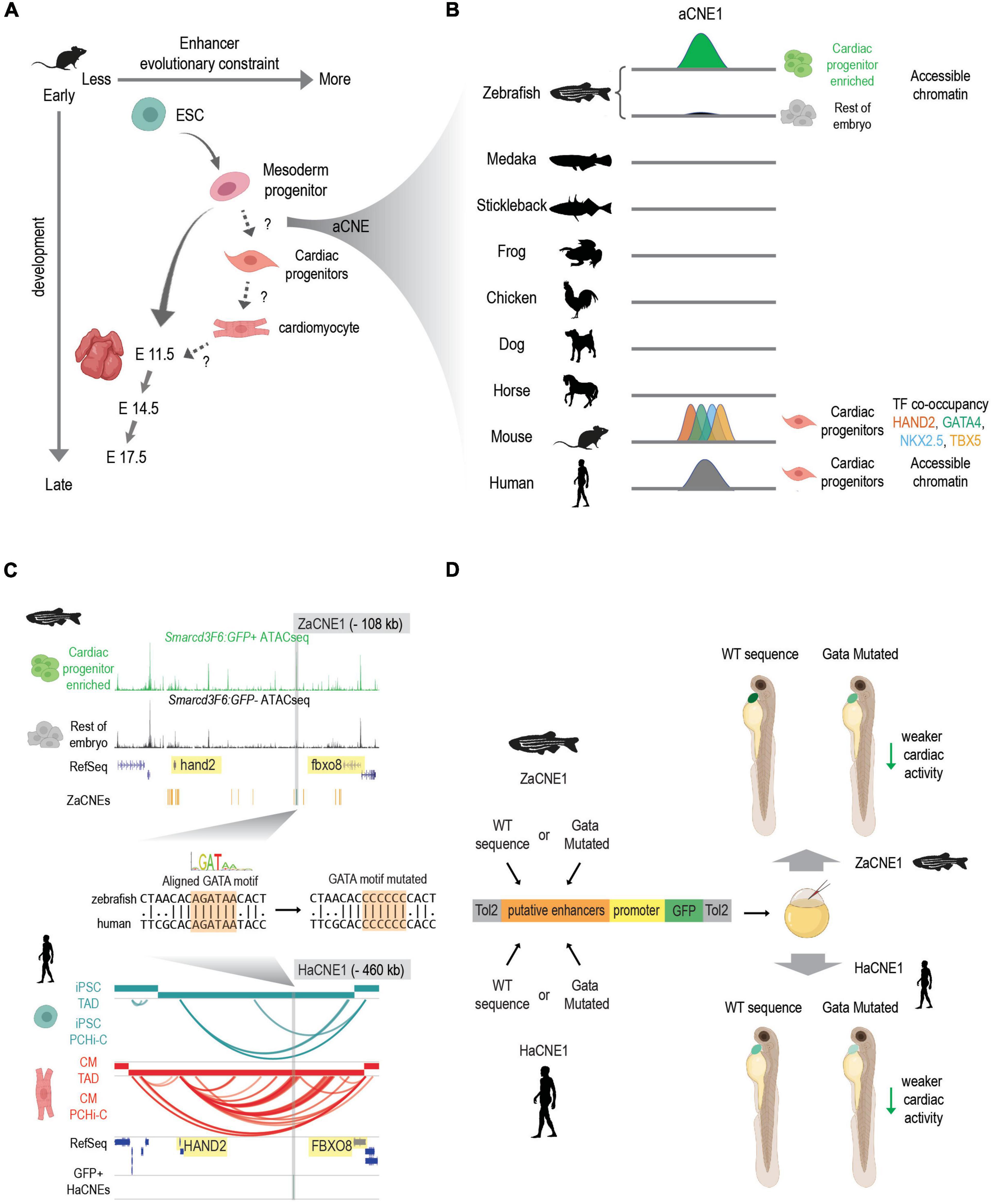
Figure 2. Discovering conserved heart enhancers during early heart development: a case study. (A) Enhancers that are active at different stages of heart development show different evolutionary constraints. In mouse, enhancers that are active in mesoderm progenitors show higher sequence conservation than enhancers active in ESC and E11.5 embryonic hearts. But conservation levels of enhancers that active during the transition of mesoderm progenitors to cardiac progenitors and cardiac progenitors to cardiomyocytes remain less characterized. aCNEs, the accessible chromatin shared between zebrafish and human (or zebrafish and mouse) were identified within the mesoderm to cardiac progenitor transition (Yuan et al., 2018). Schematics generated based on Figure 5 (Nord et al., 2013). (B) Schematics showing sequence homology and shared enhancer signatures for aCNE1 locus across multiple species. aCNE1 was first discovered as an accessible chromatin region specific for an early cardiac progenitor-enriched population in zebrafish. Gray lines indicate the existence of orthologous sequences to aCNE1 in the given species (based on CNEs identified in Hiller et al., 2013). In mouse, aCNE1 regions are co-occupied by multiple cardiac TFs in cardiac cells (based on data from Luna-Zurita et al., 2016; Laurent et al., 2017). Human aCNE1 region shows chromatin accessibility in cardiac progenitor cells (based on data from Paige et al., 2012). The stickleback and the frog icons were created by Milton Tan and Soledad Miranda-Rottmann, respectively, and shared through (http://phylopic.org/) under the following license (https://creativecommons.org/licenses/by/3.0/). (C) Genome browser view of aCNE1 in zebrafish (ZaCNE1) and human (HaCNE1) genome. aCNE1 is located 108 kb upstream of hand2 in the zebrafish genome and 406 kb upstream of HAND2 in the human genome. Yellow boxes highlight the genes flanking aCNE1, indicating the conserved synteny that aCNE1 resides in. ATAC-seq data from Yuan et al. (2018) is plotted for ZaCNE1 and promoter capture Hi-C data from Montefiori et al. (2018) is plotted for HaCNE1. Note that aCNE1 display conserved cardiac-specific activity in both zebrafish (accessibility) and human (interacting with cardiac gene HAND2). ZaCNE1 and HaCNE1 shares an aligned GATA motif, the mutation of which can be used to determine if the activity of aCNE1 depends on this GATA motif. (D) Functional enhancer assays of WT and GATA motif mutated zebrafish and human aCNE1 sequence in zebrafish embryos. Candidate sequences are cloned into an enhancer vector to drive GFP expression. The whole cassette will be chromatinized after injecting into zebrafish embryos. For both ZaCNE1 and HaCNE1, GATA motif mutation leads to decreased enhancer activity compared to the respective WT sequences. This example illustrates that human and zebrafish aCNE1 share conserved activity and regulation despite less than 60% sequence identity. Schematics generated based on data from Yuan et al. (2018). Parts of this figure were created with BioRender.com.
To explore the existence of pre-cardiac enhancers that could contribute to the initiation of cardiac gene regulatory networks, we recently characterized the open chromatin landscape of a cardiac-enriched population in zebrafish embryos before the expression of the canonical cardiac marker nkx2.5 (Yuan et al., 2018). This approach allowed us to detect cardiac CREs that were primed early in development prior to cardiac lineage commitment. We present this work in Figure 2 as a general example of how comparative genomic resources in combination with epigenomic profiling in two or more species can give insight into functionally conserved developmental enhancers. To determine to what extent deeply conserved CREs were involved in early heart development we exploited conserved non-coding element (CNE) datasets established using both direct alignment and indirect approaches (Hiller et al., 2013; Braasch et al., 2016) and found more than 160 human-zebrafish conserved candidate heart enhancers (referred to as aCNEs). Though most of these aCNEs remain to be tested in vivo, the majority of the aCNEs tested (15/18) drive robust cardiac expression in zebrafish. This example illustrates a comparative strategy for discovering early heart enhancers underscores that at least some of the regulatory logic driving vertebrate heart development can be found in orthologous sequences shared between humans and fish.
In sum, despite the overall rapid evolution of heart enhancers, a small fraction of deeply conserved heart enhancers likely contributes to the regulation of early cardiogenesis. The lack of overt sequence conservation in heart enhancers may be partially due to the rapid turnover of TFBSs. On the other hand, variants in heart enhancers that alter gene expression are likely to contribute to morphological differences of cardiac structures between species.
Heart Enhancers: One Cell at a Time
Currently, most of the data for annotating heart enhancers was generated at the bulk population level (Tables 3–5); however, both in vitro differentiated cardiac cells and animal hearts contain heterogeneous populations (reviewed in Paik et al., 2020). This was largely due to the challenges in isolating closely related developmental lineages and collecting enough material from early embryos for enhancer profiling. But as enhancer activity is highly context-specific, the existing data bias likely limits the discoveries of enhancers that are active only in specific subpopulations (e.g., SHF progenitors, endocardial cells, cardiac smooth muscle cells, etc.) or at certain stages.
Rapid advances in single-cell genomics techniques have brought unprecedented opportunities to circumvent the difficulties in cell type isolation. Specifically, single-cell ATAC-seq (scATAC-seq) has become more and more commonly used in delineating cell-type-specific CREs within diverse cellular populations (Buenrostro et al., 2015; Cusanovich et al., 2015). scATAC-seq of Isl1+ cells from E8.5 and E9.5 mouse embryonic hearts revealed the TF regulators involved in the different stages of two distinct developmental trajectories, the cardiomyocyte and endothelial trajectories (Jia et al., 2018). More recently, scATAC-seq of neonatal hearts post-injury uncovered previously uncharacterized TFs that potentially regulate specific cell types in mammalian heart regeneration and decoded the cis and trans regulators underlying regenerative and non-regenerative injury responses (Wang et al., 2020). Moreover, large single-cell atlases of chromatin accessibility have been generated for 13 adult mouse organs (∼100,000 nuclei) and 15 fetal human tissues (∼800,000 nuclei), illustrating the regulatory programs that define the cell repertoire for many mammalian organs including the heart (Cusanovich et al., 2018a; Domcke et al., 2020). Embryonic single-cell accessible chromatin landscapes have been profiled for E8.25 mouse embryos (∼19,000 nuclei) and Drosophila embryos (∼20,000 nuclei) spanning early blastoderm to terminally differentiated lineages (Cusanovich et al., 2018b; Pijuan-Sala et al., 2020). As all the above studies provide a variety of processed data and interactive web sessions for convenient exploration of the chromatin accessibility of one’s favorite genes or loci, they can be very useful resources for exploring cell type-specific cardiac enhancers.
Furthermore, with single-cell multimodal omics being selected as the Methods of the Year 2019 (Nature Methods, 2020), techniques for simultaneous measuring multiple modalities in the same single cells are blooming rapidly. Related to epigenomics, it has become possible to simultaneous profile accessible chromatin and transcriptome (Cao et al., 2018; Chen et al., 2019; Li et al., 2019; Moudgil et al., 2020), methylome and transcriptome (Angermueller et al., 2016), methylome and chromatin conformation (Li et al., 2019), or even three modalities altogether (Pott, 2017; Clark et al., 2018) within the same cells. The combinatorial use of single-cell epigenomics techniques on cardiac samples will potentially provide a holistic view of enhancer activities in all subtypes of cardiac cells across all stages in heart development. The multi-omics measurements not only enable a more comprehensive and accurate delineation of the state of the single cells but also provide unique opportunities in identifying the potential causal factors across multiple regulatory layers, by correlating changes from genetic, epigenetic, or chromatin conformation levels to the gene expression differences. Although technology and analytic challenges still lie ahead, the application of single-cell epigenomics, especially the multi-omics approaches, into heart development, will likely transform the way that we study and understand heart enhancers and cardiac gene regulatory networks.
Computing Heart Enhancers
With the rapid accumulation of hundreds of epigenomic and transcriptomic datasets from cardiac tissues, efforts have been made toward compiling them and extract sequence features from known cardiac enhancers to predict unknown ones. Dickel et al. (2016) conducted an integrative analysis of over 35 genome-wide H3K27ac or P300 profiles from mouse or human heart samples to compile a compendium of more than 80,000 heart enhancers, which serves as one of the most comprehensive putative heart enhancer lists available to date. The abundance of genomics datasets and the growing number of in vivo validated heart enhancers also provide ample input for building computational models for novel heart enhancer prediction. One kind of model is purely based on the sequence features of the gold standard heart enhancers experimentally validated in vivo. For example, Narlikar et al. (2010) combined motif discovery, Markov sequence feature characterization, and linear regression to build a heart enhancer classifier from ∼70 validated heart enhancers. They used this classifier to discover more than 40,000 putative heart enhancers within the conserved CNEs in the human genome, with an in vivo validation rate > 60% (Narlikar et al., 2010). By comparing validated cardiac and non-cardiac enhancer sequences from Drosophila, Jin et al. (2013) identified a novel motif as a classifier for heart enhancer prediction. They further showed that this motif was essential for driving cardiac activity in 3/8 enhancers tested. One widely used sequence-based machine learning method, gapped k-mer support-vector-machine (gkm-SVM) (Ghandi et al., 2014), has been applied to learn the sequence features from previously identified open chromatin regions. It predicted an addition of 80,000 putative cardiac CREs and the cognate TFs that bind to them (Lee et al., 2018).
Several studies have explored how including different genomics features in training models could affect their performance in enhancer prediction. A study in Drosophila added ChIP signals on top of sequence motifs into their classifiers and found this combined strategy significantly boosted the prediction accuracy of cell-type-specific cardiac enhancers than motif sequence alone (Ahmad et al., 2014). By further including ChIP data for a larger set of cardiac TFs and histone modifications, their updated model was able to distinguish enhancers active in distinct subpopulations of cardiac cells and pericardial cells in Drosophila embryos (Busser et al., 2015). Similarly, Akerberg et al. (2019) took advantage of the variety of ChIP-seq data that they generated for mouse hearts and compared the performance of different chromatin features (open chromatin, H3K27ac histone modification, cardiac TF occupancy) alone or combined in predicting heart enhancers. They found open chromatin had high sensitivity while TF binding profiles yielded high precision in enhancer prediction. Ultimately, the number of co-bound cardiac TFs turned out to be the most important classifier in heart enhancer prediction compared to signal intensities (Akerberg et al., 2019). With the rapid evolvement of the machine learning field, computational classification and predictions will become an important component that is complementary to experimental data in heart enhancer characterization. The two strategies will benefit from the advancement of each other and together expand our understanding of enhancer biology.
Heart Enhancers in Cardiovascular Disease
Heart diseases are a leading cause of death worldwide (Mozaffarian et al., 2015). As the most prevalent human birth defects, congenital heart disease (CHD) affects roughly 0.8% of newborns (Fahed et al., 2013). Though disruption of a set of developmental and structural genes have been recognized as the causes of a portion of CHD, the genetic factors underlying a large number of cases remain ambiguous (Fahed et al., 2013; Barnett and Postma, 2015; Postma et al., 2015; Richter et al., 2020). Genome-wide association studies (GWAS) have been carried out to identify the underlying genetic causes of a wide range of cardiovascular phenotypes and diseases, including CHD, cardiac arrest, coronary artery disease (CAD), cardiac arrhythmia, cardiomyopathy, and myocardial infarction (Arking et al., 2014; Nikpay et al., 2015; Eppinga et al., 2016; Nelson et al., 2017). Currently, thousands of variants have been implicated in heart-related disease risks (NHGRI GWAS catalog1).
Whole-genome sequencing (WGS) is becoming the method of choice for discovering de novo variants in CHD. Supporting the use of WGS for discovering molecular mechanisms underlying CHD, a recent study illustrated that the potential contribution from disruptive non-coding variants was at least as high as that from coding-variants (Richter et al., 2020). However, several factors complicate the functional annotation of disease-associated non-coding variants (Zhang and Lupski, 2015). In the case of common genetic variation associated with CHD-related phenotypes uncovered by GWAS, the tagged SNPs used will be in linkage disequilibrium (LD) with other SNPs that may represent the true causal variant. Even if a likely pathogenic non-coding mutation or copy number variation is nominated, one must then ascertain when and where this change impacts development and disease. In the following section, we briefly review insights into heart enhancer function revealed by human genetic studies.
Connecting Non-coding Variants to Cardiovascular Diseases
Only a handful of non-coding variants linked to cardiovascular diseases have been functionally dissected (Table 6). Compared to studying the function of a protein coding gene mutation, the functional characterization of non-coding disease associated variants is challenging. An early example of this was done for a genetic variant on human chromosome 9p21 harboring multiple SNPs associated with myocardial infarction and CAD (reviewed by Samani and Schunkert, 2008). A large 70 kb deletion of the whole orthologous sequence in the mouse genome severely reduced the expression of the nearby cardiac genes (Cdkn2a/b) and affected aortic smooth muscle cell proliferation and senescence. Allele-specific analysis of Cdkn2b transcripts in the heterozygous mice revealed a lack of cis-acting enhancers as the main mechanism underlying Cdkn2b downregulation, suggesting this genetic susceptibility interval contains enhancers that could be affected by the discovered sequence polymorphisms (Visel et al., 2010). However, disruption of cis-regulatory elements is not the only mechanism that contributes to diseases risk. Other studies revealed that expression of the long non-coding RNA (lncRNA) ANRII, which resides in chromosome 9q21, was affected by several SNPs within this region, and ANRII, in turn, could regulate other genes involved in vascular cell proliferation, adhesion, apoptosis, and remodeling (Holdt et al., 2010; Congrains et al., 2012a, b).
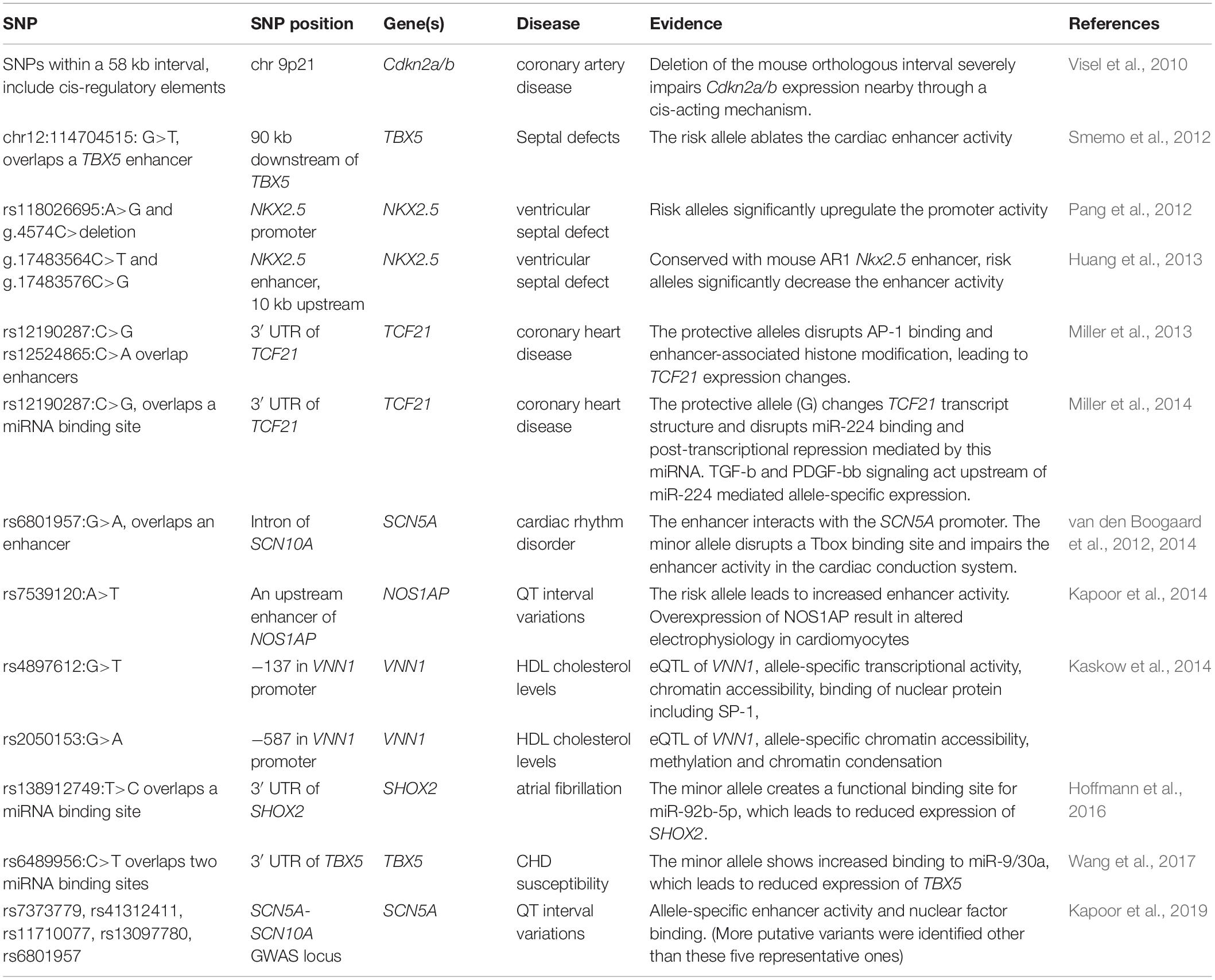
Table 6. Functionally characterized non-coding SNPs implicated in cardiovascular disease.
Another well-studied example is rs12190287, a CAD-associated variant located within the 3′ UTR of the TCF21 gene. Two continuous studies together revealed a dual mechanism of this SNP in modulating TCF21 expression at both transcriptional and post-transcriptional levels (Miller et al., 2013, 2014). Overlapping a TCF21 enhancer, this variant causes dysregulation of TCF21 through allele-specific histone modifications (H3K4me1, H3K27ac, H3K27me1) and AP-1 factor (c-Jun, JunD, ATF3) binding. These allele-specific chromatin effects are further augmented upon PDGFR-β stimulation, which indicates that the vascular growth factor signaling also acts differently on this variant (Miller et al., 2013). Moreover, the same minor allele disrupts a miR-224 binding site within the 3′ UTR of TCF21, therefore, prevents the post-transcriptional repression of TCF21 mediated by this miRNA (Miller et al., 2014).
The ion channel genes SCN5A/SCN10A locus is another hotspot heavily loaded with variants linked to cardiac arrhythmia and conduction system disorders (Veerman et al., 2015). One cardiac arrhythmia-associated SNP rs6801957 is located within the intron of SCN10A but is encompassed by a human-mouse conserved enhancer that interacts with the nearby gene SCN5A (van den Boogaard et al., 2014). This variant, but not other variants in LD disrupts the binding of TBX3/TBX5 in vitro and reduces the activity of this enhancer in the cardiac conduct system (van den Boogaard et al., 2012). Overall, these variant-oriented studies revealed the molecular mechanisms through which single nucleotide substitutions could alter enhancer activity and lead to pathological gene expression.
Discovering Disruptive Non-coding Variants Near Cardiac Genes
The CREs controlling the expression of TFs (i.e., the regulators of the regulators) are prime candidate regions for discovering damaging mutations that lead to gene dosage-related phenotypes (van der Lee et al., 2020). Indeed, hypothesis driven dissection of enhancers near cardiac genes have revealed several examples of disease causing non-coding mutations that control haploinsufficient cardiac genes TBX5, NKX2.5, and SHOX2 (reviewed in Chung and Rajakumar, 2016; Steimle and Moskowitz, 2017; Li et al., 2018).
It had been known for over a decade that heterozygous mutations within TBX5 lead to Holt-Oram syndrome in humans (Basson et al., 1997, 1999) when Smemo et al. (2012) went searching for disease-causing enhancer mutations around the TBX5 gene in families with septal defects, the predominant cardiac defect of Holt-Oram syndrome. This study, which involved scanning more than 700 kb for conserved non-coding sequences revealed three enhancer elements which together recapitulated the endogenous TBX5 heart expression in developing mouse embryos. Targeted sequencing revealed homozygous mutations in one of the enhancer elements in individuals with, but not in family members without, the disease. Another targeted sequencing of the NKX2.5 locus in ventricular septal defect patients revealed novel variants within the NKX2.5 promoter and a known distal enhancer (AR1). These novel variants significantly altered the transcriptional activity of the Nkx2.5 promoter and AR1 enhancer in luciferase assays (Pang et al., 2012; Huang et al., 2013). These tour de force experiments illustrate the lengths one must go to implicate regulatory mutations as a disease causing mechanism, and demonstrates how understanding the molecular mechanisms underlying human disease can reveal fundamental biological insights in cardiac enhancer elements.
In addition to enhancers and promoters, non-coding regulatory variation can impact miRNA binding sites, lncRNAs, or even several of these functional elements at the same time. In principle this could occur by disrupting or creating TF/miRNA binding sites, changing chromatin states, mediating different responses to extracellular signaling, or affecting lncRNA expression which in turn can affect gene regulation in trans (Table 6). For example a variant associated with increased CHD susceptibility was identified within the 3′ UTR of TBX5. This variant was shown to increase the binding of two miRNAs with the minor allele leading to a significant reduction in the expression of TBX5 through transcriptional and translational regulation (Wang et al., 2017). NKX2.5 mutations have also been implicated in diverse types of CHD, including ventricular septal defects (reviewed in Chung and Rajakumar, 2016). Similarly, target sequencing of the SHOX2 region in atrial fibrillation (AF) patients identified an AF-associated SNP within the 3′ UTR. The 3′ UTR allele created a binding site for an mRNA miR-92b-5p, which significantly reduced the SHOX2 3′UTR reporter activity in a luciferase assay (Hoffmann et al., 2016).
While there are relatively few hard-won examples of non-coding mutations that explain the molecular mechanism behind CHD, it is clear that a comprehensive annotation of heart enhancer location and function will accelerate molecular-based diagnoses and our understand of heart gene regulation.
Interpreting Non-coding Variants With Genome-Wide Enhancer Annotation
With the burst of cardiac epigenomic datasets in the past decade, the interpretation of heart disease-associated variants has developed from susceptible locus-centric to a genome-wide manner. Continuous efforts have been made to first establish a comprehensive enhancer annotation and then use for the fine-mapping non-coding variants (Dickel et al., 2016; Choy et al., 2018; Montefiori et al., 2018). For example, the heart enhancer list that they curated from ChIP-seq datasets, Dickel et al. (2016) found more than 2000 enhancer-overlapping variants that were associated with heart phenotypes. When deleting two of the variant-containing enhancers that were upstream of cardiac structure genes (Myl7 and Myl2), they showed that both enhancers are required for normal cardiac gene expression, cardiomyocyte morphology, and heart functions. On top of enhancer identification, chromatin conformation capture assays are especially helpful for linking cardiac GWAS SNPs to their targeted genes. The promoter capture Hi-C datasets generated in differentiated cardiomyocytes arguably pinpoint the true target genes of many GWAS and LD SNPs, some of which were different from the target genes proposed based on proximity (Choy et al., 2018; Montefiori et al., 2018). Remarkably, Montefiori et al. (2018) reported that 90% of the SNP-gene interactions skipped at least one gene promoter, arguing against the intuitive approach of assigning SNPs to their neighboring genes when interpreting possible causal mechanisms. In line with the cell-type-specificity of enhancer activities, the interaction networks identified using cardiomyocyte promoter capture Hi-C data turned out to be most informative to interpret cardiac arrhythmia phenotypes (which directly results from cardiomyocyte dysfunction) as compared to CHD, CAD, heart failure, and myocardial infarction (all of which involved cellular systems other than cardiomyocytes) (Choy et al., 2018; Montefiori et al., 2018). This indicates that generating chromatin maps for other cardiac cell types or at other differentiation stages could more effectively facilitate the mechanistic dissection of other types of cardiovascular diseases.
With the promising future of functional genomics in non-coding variants dissection, generation and curation of transcriptome and epigenome datasets have been tailed toward studying a specific type of heart disease to achieve higher precision. For example, to understand causal variants for atrial fibrillation (AF), RNA-seq data and ATAC-seq specifically from the left atria were generated to identify potential CREs and target genes that were likely to be affected by the genetic variants within 104 AF-associated loci (van Ouwerkerk et al., 2019). Following this study, a functional enhancer screening of these AF-associated loci using STARR-seq found 24/55 the variant-containing enhancers with allele-specific activities, demonstrating the robustness of this approach. Deletion of the orthologous region of one such enhancer near Hcn4 in the mouse genome caused a loss of Hcn4 expression and cardiac defects (van Ouwerkerk et al., 2020).
In addition to our growing understanding of the regulatory logic underlying developmental gene expression, it is also important to acknowledge the contribution of pro-inflammatory processes on heart enhancer usage and gene expression. For instance, the rapid pro-inflammatory gene expression by the NF-κB transcription factor complex, which across cell types utilizes clusters of strong enhancers (also known as “super enhancers”) to rapidly deploy pro-inflammatory gene expression (Brown et al., 2014; Schmidt et al., 2015). This mode of gene regulation can recruit transcriptional machinery from cell-lineage genes in a process known as cofactor squelching (Schmidt et al., 2015, 2016). Indeed a detailed knowledge of acute and chronic inflammatory enhancer biology during heart development and disease is essential and integrating this information with emerging compendiums of heart epigenomic data (such as Vanoudenhove et al., 2020) will be valuable.
Integrating enhancer information into the functional annotation of non-coding variants is no doubt a powerful approach; however, it should be noted that disrupting enhancer activities is not the only mechanism underlying the pathological consequences of non-coding variants. Even with extensive efforts in curating heart enhancers, nearly 90% of the heart disease-associated LD SNPs did not overlap any heart enhancers in the compendium (Dickel et al., 2016) and more than 80% of them could not be linked to gene promoters based on cardiomyocytes promoter capture Hi-C data (Montefiori et al., 2018). Apart from other possible technical reasons, this small overlap suggests regulatory mechanisms other than altering heart enhancers could account for a substantial portion of non-coding variants-mediated disease risk. In fact, unbiased examination of 98 amplicons (250–600 bp) containing 106 SNPs linked to QT interval phenotypes at the SCN5A locus found that 35% of the reference allele-containing amplicons showed enhancer activity while another 44% worked as silencers in luciferase assays (Kapoor et al., 2019), suggesting disease-associated SNPs likely fall into not only enhancers but also silencers. Besides CREs, functional non-coding variants have also been mapped to miRNA-binding sites and lncRNAs (Table 6). A recent CHD genomic analysis has demonstrated significant enrichment of RNA-binding-protein regulatory sites in de novo variants identified in CHD patients, indicating contribution from disrupted post-transcriptional regulation to CHD (Richter et al., 2020). Moreover, it has been shown that the same minor allele of a variant could regulate the target gene expression through both transcriptional and post-transcriptional mechanisms and, even more strikingly, in an opposite manner, highlighting the complexity of sequence polymorphisms in affecting gene expression (Miller et al., 2013, 2014). Therefore, a comprehensive annotation of different types of cardiac CREs that are not limited to enhancers, together with a good non-coding RNA annotation, will be necessary for truly understanding the mechanisms of the heart disease from the non-coding variant perspective. Additionally, it is likely that several coding and/or non-coding variants collectively explain a complex cardiovascular phenotype. Thus while it is important to dissect disease phenotype associated variants individually, more complex studies looking at genetic interactions and addictive effects may well be required.
Emerging Techniques for the Functional Dissection of Heart Enhancers
So far, numerous putative heart enhancers have been identified in different conditions and cell types from several model organisms. However, compared to enhancer mapping, the throughput of current approaches for enhancer functional dissection, especially in vivo, remains a major bottleneck. Traditionally, each candidate enhancer is accessed individually via being placed upstream of a reporter gene and introduced into cells or in vivo organisms. Collective efforts using this approach have led to the establishment of central resources of validated enhancers, such as the Vista Enhancer Browser2 (Visel et al., 2007). To measure enhancer activity in a more high throughput manner, several methods have been developed through the years, such as massively parallel reporter assays (MPRA) (Melnikov et al., 2012; Patwardhan et al., 2012; Sharon et al., 2012), and self-transcribing active regulatory region sequencing (STARR-seq) (Arnold et al., 2013). However, most of these approaches are typically carried out in vitro or in the absence of chromatin contexts, raising the question of how faithfully their results reflect the native activities of the candidate regions. Recently, the development of more robust and scalable in vivo enhancer assays, such as the site-directed enhancer-reporter assay (enSERT), has allowed systematic assessment of more than 100 variants in an essential limb enhancer (Kvon et al., 2020). For invertebrates like Drosophila, unbiased, automated enhancer mutational scanning has been established using robotic systems, which permits multi-stage quantitative measurement of enhancer activities in development (Fuqua et al., 2020). Developing similar systems for vertebrates will greatly improve our capacity in assessing vertebrate enhancer functions and advance our understanding of how regulatory information is encoded in developmental enhancers.
Compared to all enhancer reporter assays, which introduces an atypical distance between candidate enhancers and the reporter genes, a complementary perhaps preferred way to understand enhancer functions is to dissect their activity and function in their endogenous loci. The ever-growing CRISPR-Cas9 toolbox provides many options for in situ enhancer dissection (reviewed in Klein et al., 2018; Pickar-Oliver and Gersbach, 2019; Xu and Qi, 2019). Individual enhancer deletions or substitutions have been routinely used to characterize enhancer functions in specific developmental processes (Dickel et al., 2016, 2018; Kvon et al., 2016; Osterwalder et al., 2018; van Eif et al., 2020; van Ouwerkerk et al., 2020). To increase the throughput, a variety of CRISPR-based enhancer screens have been developed for in vitro systems, such as the saturated tilling arrays that can unbiased assess certain genomic loci for functional enhancers (Korkmaz et al., 2016; Diao et al., 2017; Gasperini et al., 2017) and epigenetic screens against candidate enhancers using deactivated Cas9 (dCas9) coupled with transcriptional activators or repressors (Klann et al., 2017; Simeonov et al., 2017; Fulco et al., 2019; Gasperini et al., 2019). Specifically, by using single-cell RNA-seq as readouts, CRISPR-mediated epigenetic screens have been successfully applied to perturb thousands of candidate enhancers in cell lines to determine their functional importance and target genes (Fulco et al., 2019; Gasperini et al., 2019). Though achieving the same throughput in vivo may still be challenging, increasing efforts have been made toward applying these powerful systems in animals. Very recently, a single-cell-based in vivo CRISPR/Cas9 screen (Perturb-seq) has been successfully used to screen 35 genes in the mouse developing neuronal cortex in utero (Jin et al., 2020). Though not large-scale yet, this study offers a very encouraging framework to achieve systematic assessment of genes or CREs in vivo. Moreover, dCas9-mediated epigenetic perturbation, which is likely more suitable for enhancer screens, has been continuously optimized over the years and showed a promising future of targeting enhancers in a more scalable manner in developing animals (Morita et al., 2016; Zhou et al., 2018; Li et al., 2020).
Discussion, Concluding Remarks, and Future Perspectives
The past decade has witnessed an exponential growth of the numbers of putative heart enhancer regions identified, largely owing to rapid advances in epigenomic profiling approaches. These techniques are still growing at an ever-increasing speed and will undoubtedly continue to revolutionize the way that researchers annotate and interpret enhancer activities. Single-cell epigenomic techniques, especially the multi-omics approaches, will likely become one of the main driving forces in expanding the horizon of cardiac enhancers and regulatory networks in the next decade. However, it should be noted that many analytical challenges are inherently associated with single-cell epigenomic datasets that currently remain sparse and noisy (reviewed in Schwartzman and Tanay, 2015; Verma and Kumar, 2019). Robust computational and statistical models are needed to extract biological information from other irrelevant signals (e.g., technical noises, batch effect) and for integrating the multimodal data of different characteristics, dimensionalities, and coverages to model them in a single space. Methods addressing these challenges are rapidly emerging (reviewed in Forcato et al., 2020; Hao et al., 2020) but still in the early stages in terms of accommodating all different data types and features. Both technical improvements of assay sensitivity and the development of analytic methods are essential for successfully applying these single-cell genomics techniques to understanding enhancer biology.
In vivo functional characterization of enhancers, especially developmental enhancers, is still one of the biggest challenges lying ahead. As developmental genes are usually regulated by multiple enhancers with overlapping activities, it is reasonable to assume that most enhancers may have redundant functions in normal development (Frankel et al., 2010; Perry et al., 2010; Cannavò et al., 2016; Dickel et al., 2018; Osterwalder et al., 2018). While these redundant enhancers may be seemingly dispensable in normal conditions, they could be required in stressed environments or sensitized genetic backgrounds (e.g., such as heterozygous deletion of developmental TFs) (Frankel et al., 2010; Perry et al., 2010; Osterwalder et al., 2018). It therefore becomes a very complicated task to determine the specific contexts in which a given developmental enhancer is required.
On the other hand, we are in an era with unprecedented opportunities to overcome these challenges. The combined use of CRISPR technologies and single-cell genomics is likely to make a substantial contribution to functional enhancer dissections in the near future. With the concurrent advancement of these two technologies, it probably will not be too far until we can conduct mid- to large-scale in vivo enhancer screening. Moreover, coupling CRISPR with other single-cell epigenomic assays (e.g., single-cell accessibility chromatin) to target TFs or chromatin modifiers (Rubin et al., 2019; Sanjana et al., 2020), can provide information complementary to enhancer screens and together build toward a comprehensive regulatory network.
From traditional approaches to the newest genomic assays, the rich history of heart enhancer studies has not only left us with a wealth of knowledge about the genomic locations, functional roles, evolutionary conservation, and disease implications of heart enhancers but also opened up many challenges and unanswered questions. What are the best experimental designs and analytic strategies of single-cell epigenomic assays? How can we increase the scalability of functional enhancer assays and efficiently adopt them into in vivo contexts? Could we develop more robust and transferable computational methods that can not only predict heart enhancers but also determine their chamber-, cell-type or developmental-stage specific activities and how the activity of enhancers can be affected by non-coding variants? We may not be sure when these questions will be fully answered, but we can confidently anticipate that efforts made in tackling these challenges will push our understanding of heart enhancers and cardiac regulatory network to an unprecedented level.
Author Contributions
XY researched, conceived the structure, created the figures and tables, and led the writing of the review. MW and IS developed the ideas and provided text for the review. All the authors read and edited the review.
Funding
This work was supported in part by The Hospital for Sick Children Restracomp Studentship and Connaught International Scholarship to XY and CIHR (FRN 156318 to MW and IS). MW was supported by the Canada Research Chairs Program and an Early Researcher Award from the Ontario Ministry of Research and Innovation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to the authors whose work could not be covered or thoroughly discussed in this review due to space limitations. We would like to thank Mengyi Song, Huayun Hou, and Anna Prentice for reading the manuscript and providing feedback.
Footnotes
References
Abudayyeh, O. O., Gootenberg, J. S., Essletzbichler, P., Han, S., Joung, J., Belanto, J. J., et al. (2017). RNA targeting with CRISPR-Cas13. Nature 550, 280–284.
Adamo, R. F., Guay, C. L., Edwards, A. V., Wessels, A., and Burch, J. B. E. (2004). GATA-6 gene enhancer contains nested regulatory modules for primary myocardium and the embedded nascent atrioventricular conduction system. Anat. Rec. 280A, 1062–1071. doi: 10.1002/ar.a.20105
Ahmad, S. M., Busser, B. W., Huang, D., Cozart, E. J., Michaud, S., Zhu, X., et al. (2014). Machine learning classification of cell-specific cardiac enhancers uncovers developmental subnetworks regulating progenitor cell division and cell fate specification. Dev. 141, 878–888. doi: 10.1242/dev.101709
Akerberg, B. N., Gu, F., VanDusen, N. J., Zhang, X., Dong, R., Li, K., et al. (2019). A reference map of murine cardiac transcription factor chromatin occupancy identifies dynamic and conserved enhancers. Nat. Commun. 10, 1–16.
Alexanian, M., Maric, D., Jenkinson, S. P., Mina, M., Friedman, C. E., Ting, C. C., et al. (2017). A transcribed enhancer dictates mesendoderm specification in pluripotency. Nat. Commun. 8, 1–19.
Alvarez-Dominguez, J. R., Knoll, M., Gromatzky, A. A., and Lodish, H. F. (2017). The super-enhancer-derived alncRNA-EC7/bloodlinc potentiates red blood cell development in trans. Cell Rep. 19, 2503–2514. doi: 10.1016/j.celrep.2017.05.082
Anderson, K. M., Anderson, D. M., McAnally, J. R., Shelton, J. M., Bassel-Duby, R., and Olson, E. N. (2016). Transcription of the non-coding RNA upperhand controls Hand2 expression and heart development. Nature 2, 1–13.
Andersson, R., Gebhard, C., Miguel-Escalada, I., Hoof, I., Bornholdt, J., Boyd, M., et al. (2014). An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461.
Ang, Y.-S., Rivas, R. N., Ribeiro, A. J. S., Srivas, R., Rivera, J., Stone, N. R., et al. (2016). Disease model of GATA4 mutation reveals transcription factor cooperativity in human cardiogenesis. Cell 167, 1734.e22–1749.e22.
Angermueller, C., Clark, S. J., Lee, H. J., Macaulay, I. C., Teng, M. J., Hu, T. X., et al. (2016). Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat. Methods 13, 229–232. doi: 10.1038/nmeth.3728
Aparicio, S., Morrison, A., Gould, A., Gilthorpe, J., Chaudhuri, C., Rigby, P., et al. (1995). Detecting conserved regulatory elements with the model genome of the Japanese puffer fish. Fugu rubripes. Proc. Natl. Acad. Sci. U.S.A. 92, 1684–1688. doi: 10.1073/pnas.92.5.1684
Arking, D. E., Pulit, S. L., Crotti, L., van der Harst, P., Munroe, P. B., Koopmann, T. T., et al. (2014). Genetic association study of QT interval highlights role for calcium signaling pathways in myocardial repolarization. Nat. Genet. 46, 826–836.
Arner, E., Daub, C. O., Vitting-Seerup, K., Andersson, R., Lilje, B., Drabløs, F., et al. (2015). Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 347, 1010–1014.
Arnold, C. D., Gerlach, D., Spies, D., Matts, J. A., Sytnikova, Y. A., Pagani, M., et al. (2014). Quantitative genome-wide enhancer activity maps for five Drosophila species show functional enhancer conservation and turnover during cis-regulatory evolution. Nat. Genet. 46, 685–692. doi: 10.1038/ng.3009
Arnold, C. D., Gerlach, D., Stelzer, C., Boryn, L. M., Rath, M., and Stark, A. (2013). Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339, 1074–1077. doi: 10.1126/science.1232542
Arnold, P. R., Wells, A. D., and Li, X. C. (2020). Diversity and emerging roles of enhancer RNA in regulation of gene expression and cell fate. Front. Cell Dev. Biol. 7:377. doi: 10.3389/fcell.2019.00377
Arnosti, D. N., and Kulkarni, M. M. (2005). Transcriptional enhancers: intelligent enhanceosomes or flexible billboards? J. Cell. Biochem. 94, 890–898. doi: 10.1002/jcb.20352
Ballester, B., Medina-Rivera, A., Schmidt, D., Gonzàlez-Porta, M., Carlucci, M., Chen, X., et al. (2014). Multi-species, multi-transcription factor binding highlights conserved control of tissue-specific biological pathways. eLife 3, 1–29. doi: 10.1515/bc.2003.001
Barnes, R. M., Harris, I. S., Jaehnig, E. J., Sauls, K., Sinha, T., Rojas, A., et al. (2016). MEF2C regulates outflow tract alignment and transcriptional control of Tdgf1. Development 143, 774–779. doi: 10.1242/dev.126383
Barnett, P., and Postma, A. V. (2015). Genetics of congenital heart disease: beyond half-measures. Trends Cardiovasc. Med. 25, 302–304. doi: 10.1016/j.tcm.2014.11.012
Barron, M. E., Belaguli, N. S., Shu, X. Z., Trinh, M., Iyer, D., Merlo, X., et al. (2005). Serum response factor, an enriched cardiac mesoderm obligatory factor, is a downstream gene target for Tbx genes. J. Biol. Chem. 280, 11816–11828. doi: 10.1074/jbc.m412408200
Basson, C. T., Bachinsky, D. R., Lin, R. C., Levi, T., Elkins, J. A., Soults, J., et al. (1997). Mutations in human cause limb and cardiac malformation in Holt-Oram syndrome. Nat. Genet. 15, 30–35. doi: 10.1038/ng0197-30
Basson, C. T., Huang, T., Lin, R. C., Bachinsky, D. R., Weremowicz, S., Vaglio, A., et al. (1999). Different TBX5 interactions in heart and limb defined by Holt-Oram syndrome mutations. Proc. Natl. Acad. Sci. U.S.A. 96, 2919–2924.
Bejerano, G., Pheasant, M., Makunin, I., Stephen, S., Kent, W. J., Mattick, J. S., et al. (2004). Ultraconserved elements in the human genome. Science 304, 1321–1325. doi: 10.1126/science.1098119
Bertero, A., Fields, P. A., Ramani, V., Bonora, G., Yardimci, G. G., Reinecke, H., et al. (2019). Dynamics of genome reorganization during human cardiogenesis reveal an RBM20-dependent splicing factory. Nat. Commun. 10, 1–19.
Blackwood, E. M., and Kadonaga, J. T. (1998). Going the distance: a current view of enhancer action. Science 281, 60–63. doi: 10.1126/science.281.5373.60
Blow, M. J., McCulley, D. J., Li, Z., Zhang, T., Akiyama, J. A., Holt, A., et al. (2010). ChIP-Seq identification of weakly conserved heart enhancers. Nat. Genet. 42, 806–810. doi: 10.1038/ng.650
Bogdanovic, O., Fernandez-Miñán, A., Tena, J. J., de la Calle-Mustienes, E., Hidalgo, C., van Kruysbergen, I., et al. (2012). Dynamics of enhancer chromatin signatures mark the transition from pluripotency to cell specification during embryogenesis. Genome Res. 22, 2043–2053. doi: 10.1101/gr.134833.111
Bogdanović, O., Smits, A. H., de la Calle Mustienes, E., Tena, J. J., Ford, E., Williams, R., et al. (2016). Active DNA demethylation at enhancers during the vertebrate phylotypic period. Nat. Genet. 48, 417–426. doi: 10.1038/ng.3522
Bonev, B., Mendelson Cohen, N., Szabo, Q., Fritsch, L., Papadopoulos, G. L., Lubling, Y., et al. (2017). Multiscale 3D genome rewiring during mouse neural development. Cell 171, 557.e24–572.e24.
Bonn, S., Zinzen, R. P., Girardot, C., Gustafson, E. H., Perez-Gonzalez, A., Delhomme, N., et al. (2012). Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat. Genet. 44, 148–156. doi: 10.1038/ng.1064
Boogerd, C. J., Aneas, I., Sakabe, N., Dirschinger, R. J., Cheng, Q. J., Zhou, B., et al. (2017). Probing chromatin landscape reveals roles of endocardial TBX20 in septation. J. Clin. Invest. 126, 3023–3035. doi: 10.1172/jci85350
Braasch, I., Gehrke, A. R., Smith, J. J., Kawasaki, K., Manousaki, T., Pasquier, J., et al. (2016). The spotted gar genome illuminates vertebrate evolution and facilitates human-teleost comparisons. Nat. Genet. 48, 427–437.
Brind’Amour, J., Liu, S., Hudson, M., Chen, C., Karimi, M. M., and Lorincz, M. C. (2015). An ultra-low-input native ChIP-seq protocol for genome-wide profiling of rare cell populations. Nat. Commun. 6, 1–8.
Brown, C. O., Chi, X., Garcia-Gras, E., Shirai, M., Feng, X. H., and Schwartz, R. J. (2004). The cardiac determination factor, Nkx2-5, is activated by mutual cofactors GATA-4 and Smad1/4 via a novel upstream enhancer. J. Biol. Chem. 279, 10659–10669. doi: 10.1074/jbc.m301648200
Brown, J. D., Lin, C. Y., Duan, Q., Griffin, G., Federation, A. J., Paranal, R. M., et al. (2014). Nf-kb directs dynamic super enhancer formation in inflammation and atherogenesis. Mol. Cell 56, 219–231. doi: 10.1016/j.molcel.2014.08.024
Buecker, C., and Wysocka, J. (2012). Enhancers as information integration hubs in development: lessons from genomics. Trends Genet. 28, 276–284. doi: 10.1016/j.tig.2012.02.008
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., and Greenleaf, W. J. (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. doi: 10.1038/nmeth.2688
Buenrostro, J. D., Wu, B., Litzenburger, U. M., Ruff, D., Gonzales, M. L., Snyder, M. P., et al. (2015). Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490. doi: 10.1038/nature14590
Busser, B. W., Haimovich, J., Huang, D., Ovcharenko, I., and Michelson, A. M. (2015). Enhancer modeling uncovers transcriptional signatures of individual cardiac cell states in Drosophila. Nucleic Acids Res. 43, 1726–1739. doi: 10.1093/nar/gkv011
Calo, E., and Wysocka, J. (2013). Modification of enhancer chromatin: what, how, and why? Mol. Cell 49, 825–837. doi: 10.1016/j.molcel.2013.01.038
Cannavò, E., Khoueiry, P., Garfield, D. A., Geeleher, P., Zichner, T., Gustafson, E. H., et al. (2016). Shadow enhancers are pervasive features of developmental regulatory networks. Curr. Biol. 26, 38–51. doi: 10.1016/j.cub.2015.11.034
Cao, J., Cusanovich, D. A., Ramani, V., Aghamirzaie, D., Pliner, H. A., Hill, A. J., et al. (2018). Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385. doi: 10.1126/science.aau0730
Caputo, L., Witzel, H. R., Kolovos, P., Cheedipudi, S., Looso, M., Mylona, A., et al. (2015). The Isl1/Ldb1 complex orchestrates genome-wide chromatin organization to instruct differentiation of multipotent cardiac progenitors. Cell Stem Cell 17, 287–299. doi: 10.1016/j.stem.2015.08.007
Cardoso-Moreira, M., Halbert, J., Valloton, D., Velten, B., Chen, C., Shao, Y., et al. (2019). Gene expression across mammalian organ development. Nature 571, 505–509.
Charité, J., McFadden, D. G., Merlo, G., Levi, G., Clouthier, D. E., Yanagisawa, M., et al. (2001). Role of Dlx6 in regulation of an endothelin-1-dependent, dHAND branchial arch enhancer. Genes Dev. 15, 3039–3049. doi: 10.1101/gad.931701
Chatterjee, S., Bourque, G., and Lufkin, T. (2011). Conserved and non-conserved enhancers direct tissue specific transcription in ancient germ layer specific developmental control genes. BMC Dev. Biol. 11:63. doi: 10.1186/1471-213X-11-63
Chen, S., Lake, B. B., and Zhang, K. (2019). High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat. Biotechnol. 37, 1452–1457. doi: 10.1038/s41587-019-0290-0
Chen, Y., and Cao, X. (2009). NFAT directly regulates Nkx2-5 transcription during cardiac cell differentiation. Biol. Cell 101, 335–350. doi: 10.1042/bc20080108
Chi, X., Chatterjee, P. K., Wilson, W., Zhang, S. X., DeMayo, F. J., and Schwartz, R. J. (2005). Complex cardiac Nkx2-5 gene expression activated by noggin-sensitive enhancers followed by chamber-specific modules. Proc. Natl. Acad. Sci. U.S.A. 102, 13490–13495. doi: 10.1073/pnas.0504295102
Choy, M.-K., Javierre, B. M., Williams, S. G., Baross, S. L., Liu, Y., Wingett, S. W., et al. (2018). Promoter interactome of human embryonic stem cell-derived cardiomyocytes connects GWAS regions to cardiac gene networks. Nat. Commun. 9:2526.
Chu, T., Rice, E. J., Booth, G. T., Salamanca, H. H., Wang, Z., Core, L. J., et al. (2018). Chromatin run-on and sequencing maps the transcriptional regulatory landscape of glioblastoma multiforme. Nat. Genet. 50, 1553–1564. doi: 10.1038/s41588-018-0244-3
Chung, I.-M., and Rajakumar, G. (2016). Genetics of congenital heart defects: the NKX2-5 Gene, a Key player. Genes 7: 6. doi: 10.3390/genes7020006
Churko, J. M., Garg, P., Treutlein, B., Venkatasubramanian, M., Wu, H., Lee, J., et al. (2018). Defining human cardiac transcription factor hierarchies using integrated single-cell heterogeneity analysis. Nat. Commun. 9:4906.
Cirillo, L. A., Lin, F. R., Cuesta, I., Friedman, D., Jarnik, M., and Zaret, K. S. (2002). Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell 9, 279–289. doi: 10.1016/s1097-2765(02)00459-8
Clark, C. D., Zhang, B., Lee, B., Evans, S. I., Lassar, A. B., and Lee, K. H. (2013). Evolutionary conservation of Nkx2.5 autoregulation in the second heart field. Dev. Biol. 374, 198–209. doi: 10.1016/j.ydbio.2012.11.007
Clark, S. J., Argelaguet, R., Kapourani, C. A., Stubbs, T. M., Lee, H. J., Alda-Catalinas, C., et al. (2018). ScNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells e. Nat. Commun. 9, 1–9.
Clément, Y., Torbey, P., Gilardi-Hebenstreit, P., and Crollius, H. R. (2020). Enhancer-gene maps in the human and zebrafish genomes using evolutionary linkage conservation. Nucleic Acids Res. 48, 2357–2371. doi: 10.1093/nar/gkz1199
Congrains, A., Kamide, K., Katsuya, T., Yasuda, O., Oguro, R., Yamamoto, K., et al. (2012a). CVD-associated non-coding RNA, ANRIL, modulates expression of atherogenic pathways in VSMC. Biochem. Biophys. Res. Commun. 419, 612–616. doi: 10.1016/j.bbrc.2012.02.050
Congrains, A., Kamide, K., Oguro, R., Yasuda, O., Miyata, K., Yamamoto, E., et al. (2012b). Genetic variants at the 9p21 locus contribute to atherosclerosis through modulation of ANRIL and CDKN2A/B. Atherosclerosis 220, 449–455. doi: 10.1016/j.atherosclerosis.2011.11.017
Corces, M. R., Trevino, A. E., Hamilton, E. G., Greenside, P. G., Sinnott-Armstrong, N. A., Vesuna, S., et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. doi: 10.1038/nmeth.4396
Core, L. J., Martins, A. L., Danko, C. G., Waters, C. T., Siepel, A., and Lis, J. T. (2014). Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat. Genet. 46, 1311–1320. doi: 10.1038/ng.3142
Core, L. J., Waterfall, J. J., and Lis, J. T. (2008). Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322, 1845–1848. doi: 10.1126/science.1162228
Cotney, J., Leng, J., Yin, J., Reilly, S. K., Demare, L. E., Emera, D., et al. (2013). The evolution of lineage-specific regulatory activities in the human embryonic limb. Cell 154, 185–196. doi: 10.1016/j.cell.2013.05.056
Creyghton, M. P., Cheng, A. W., Welstead, G. G., Kooistra, T., Carey, B. W., Steine, E. J., et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S A. 107, 21931–21936. doi: 10.1073/pnas.1016071107
Cusanovich, D. A., Daza, R., Adey, A., Pliner, H. A., Christiansen, L., Gunderson, K. L., et al. (2015). Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914. doi: 10.1126/science.aab1601
Cusanovich, D. A., Hill, A. J., Aghamirzaie, D., Daza, R. M., Pliner, H. A., Berletch, J. B., et al. (2018a). A single-cell atlas of in vivo mammalian chromatin accessibility. Cell 174, 1309.e18–1324.e18.
Cusanovich, D. A., Reddington, J. P., Garfield, D. A., Daza, R. M., Aghamirzaie, D., Marco-Ferreres, R., et al. (2018b). The cis-regulatory dynamics of embryonic development at single-cell resolution. Nature 555, 538–542. doi: 10.1038/nature25981
Davis, D. L., Edwards, A. V., Juraszek, A. L., Phelps, A., Wessels, A., and Burch, J. B. E. (2001). A GATA-6 gene heart-region-specific enhancer provides a novel means to mark and probe a discrete component of the mouse cardiac conduction system. Mech. Dev. 108, 105–119. doi: 10.1016/s0925-4773(01)00500-7
Davis, D. L., Wessels, A., and Burch, J. B. E. (2000). An Nkx-dependent enhancer regulates cGATA-6 gene expression during early stages of heart development. Dev. Biol. 217, 310–322. doi: 10.1006/dbio.1999.9561
De Koninck, M., Lapi, E., Badía-Careaga, C., Cossío, I., Giménez-Llorente, D., Rodríguez-Corsino, M., et al. (2020). Essential roles of cohesin STAG2 in Mouse embryonic development and adult tissue homeostasis. Cell Rep. 32:108014. doi: 10.1016/j.celrep.2020.108014
de la Calle-Mustienes, E., Feijóo, C. G., Manzanares, M., Tena, J. J., Rodríguez-Seguel, E., Letizia, A., et al. (2005). A functional survey of the enhancer activity of conserved non-coding sequences from vertebrate Iroquois cluster gene deserts. Genome Res. 15, 1061–1072. doi: 10.1101/gr.4004805
Delgado-Olguín, P., Huang, Y., Li, X., Christodoulou, D., Seidman, C. E., Seidman, J. G., et al. (2012). Epigenetic repression of cardiac progenitor gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nat. Genet. 44, 343–347. doi: 10.1038/ng.1068
Denker, A., and De Laat, W. (2016). The second decade of 3C technologies: detailed insights into nuclear organization. Genes Dev. 30, 1357–1382. doi: 10.1101/gad.281964.116
Deutsch, M. A., Doppler, S. A., Li, X., Lahm, H., Santamaria, G., Cuda, G., et al. (2018). Reactivation of the Nkx2.5 cardiac enhancer after myocardial infarction does not presage myogenesis. Cardiovasc. Res. 114, 1098–1114. doi: 10.1093/cvr/cvy069
Devine, W. P., Wythe, J. D., George, M., Koshiba-Takeuchi, K., and Bruneau, B. G. (2014). Early patterning and specification of cardiac progenitors in gastrulating mesoderm. eLife 3:e03848.
Diao, Y., Fang, R., Li, B., Meng, Z., Yu, J., Qiu, Y., et al. (2017). A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nat. Methods 14, 629–635. doi: 10.1038/nmeth.4264
Dias, N., and Stein, C. A. (2002). Antisense oligonucleotides: basic concepts and mechanisms. Mol. Cancer Ther. 1, 347–355.
Dickel, D. E., Barozzi, I., Zhu, Y., Fukuda-Yuzawa, Y., Osterwalder, M., Mannion, B. J., et al. (2016). Genome-wide compendium and functional assessment of in vivo heart enhancers. Nat. Commun. 7:12923.
Dickel, D. E., Ypsilanti, A. R., Rubenstein, J. L. R., Pennacchio, L. A., Correspondence, A. V., and Gov, D. (2018). Ultraconserved enhancers are required for normal development. Cell 172, 491–499. doi: 10.1016/j.cell.2017.12.017
Dixon, J. R., Jung, I., Selvaraj, S., Shen, Y., Antosiewicz-Bourget, J. E., Lee, A. Y., et al. (2015). Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336. doi: 10.1038/nature14222
Dixon, J. R., Selvaraj, S., Yue, F., Kim, A., Li, Y., Shen, Y., et al. (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. doi: 10.1038/nature11082
Dodou, E., Verzi, M. P., Anderson, J. P., Xu, S.-M., and Black, B. L. (2004). Mef2c is a direct transcriptional target of ISL1 and GATA factors in the anterior heart field during mouse embryonic development. Development 131, 3931–3942. doi: 10.1242/dev.01256
Domazet-Lošo, T., and Tautz, D. (2010). A phylogenetically based transcriptome age index mirrors ontogenetic divergence patterns. Nature 468, 815–819. doi: 10.1038/nature09632
Domcke, S., Hill, A. J., Daza, R. M., Cao, J., O’Day, D. R., Pliner, H. A., et al. (2020). A human cell atlas of fetal chromatin accessibility. Science 370:eaba7612. doi: 10.1126/science.aba7612
Doppler, S. A., Werner, A., Barz, M., Lahm, H., Deutsch, M. A., Dreßen, M., et al. (2014). Myeloid zinc finger 1 (Mzf1) differentially modulates murine cardiogenesis by interacting with an Nkx2.5 cardiac enhancer. PLoS One 9:e113775. doi: 10.1371/journal.pone.0113775
Dupays, L., Shang, C., Wilson, R., Kotecha, S., Wood, S., Towers, N., et al. (2015). Sequential binding of MEIS1 and NKX2-5 on the Popdc2 gene: a mechanism for spatiotemporal regulation of enhancers during cardiogenesis. Cell Rep. 13, 183–195. doi: 10.1016/j.celrep.2015.08.065
Eppinga, R. N., Hagemeijer, Y., Burgess, S., Hinds, D. A., Stefansson, K., Gudbjartsson, D. F., et al. (2016). Identification of genomic loci associated with resting heart rate and shared genetic predictors with all-cause mortality. Nat. Genet. 48, 1557–1563. doi: 10.1038/ng.3708
Eres, I. E., and Gilad, Y. (2020). A TAD Skeptic: is 3D genome topology conserved? Trends Genet. S0168-9525, 30298–30305.
Eres, I. E., Luo, K., Hsiao, C. J., Blake, L. E., and Gilad, Y. (2019). Reorganization of 3D genome structure may contribute to gene regulatory evolution in primates. PLoS Genet. 15:e1008278. doi: 10.1371/journal.pgen.1008278
Espinosa, J. M. (2016). Revisiting lncRNAs: how do you know yours is not an eRNA? Mol. Cell 62, 1–2. doi: 10.1016/j.molcel.2016.03.022
Fahed, A. C., Gelb, B. D., Seidman, J. G., and Seidman, C. E. (2013). Genetics of congenital heart disease: the glass half empty. Circ. Res. 112, 707–720. doi: 10.1161/circresaha.112.300853
Fang, R., Yu, M., Li, G., Chee, S., Liu, T., Schmitt, A. D., et al. (2016). Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Res. 26, 1345–1348. doi: 10.1038/cr.2016.137
Farley, E. K., Olson, K. M., Zhang, W., Brandt, A. J., Rokhsar, D. S., and Levine, M. S. (2015). Suboptimization of developmental enhancers. Science 350, 325–328. doi: 10.1126/science.aac6948
Farley, E. K., Olson, K. M., Zhang, W., Rokhsar, D. S., and Levine, M. S. (2016). Syntax compensates for poor binding sites to encode tissue specificity of developmental enhancers. Proc. Natl. Acad. Sci. U.S.A. 113, 6508–6513. doi: 10.1073/pnas.1605085113
Field, A., and Adelman, K. (2020). Evaluating enhancer function and transcription. Annu. Rev. Biochem. 89, 213–234. doi: 10.1146/annurev-biochem-011420-095916
Fisher, S., Grice, E. A., Vinton, R. M., Bessling, S. L., and McCallion, A. S. (2006). Conservation of RET regulatory function from human to zebrafish without sequence similarity. Science 312, 276–279. doi: 10.1126/science.1124070
Flavahan, W. A., Drier, Y., Liau, B. B., Gillespie, S. M., Venteicher, A. S., Stemmer-Rachamimov, A. O., et al. (2016). Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114. doi: 10.1038/nature16490
Forcato, M., Romano, O., and Bicciato, S. (2020). Computational methods for the integrative analysis of single-cell data. Brief. Bioinform. 2020, 1–10.
Franke, M., Ibrahim, D. M., Andrey, G., Schwarzer, W., Heinrich, V., Schöpflin, R., et al. (2016). Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538, 265–269. doi: 10.1038/nature19800
Frankel, N., Davis, G. K., Vargas, D., Wang, S., Payre, F., and Stern, D. L. (2010). Phenotypic robustness conferred by apparently redundant transcriptional enhancers. Nature 466, 490–493. doi: 10.1038/nature09158
Friedli, M., Barde, I., Arcangeli, M., Verp, S., Quazzola, A., Zakany, J., et al. (2010). A systematic enhancer screen using lentivector transgenesis identifies conserved and non-conserved functional elements at the Olig1 and Olig2 locus. PLoS One 5:e15741. doi: 10.1371/journal.pone.0015741
Fulco, C. P., Nasser, J., Jones, T. R., Munson, G., Bergman, D. T., Subramanian, V., et al. (2019). Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 51, 1664–1669. doi: 10.1038/s41588-019-0538-0
Fullwood, M. J., Liu, M. H., Pan, Y. F., Liu, J., Xu, H., Mohamed, Y. B., et al. (2009). An oestrogen-receptor-α-bound human chromatin interactome. Nature 462, 58–64.
Fuqua, T., Jordan, J., Elize van Breugel, M., Halavatyi, A., Tischer, C., Polidoro, P., et al. (2020). Dense and pleiotropic regulatory information in a developmental enhancer. Nature 587, 235–239. doi: 10.1038/s41586-020-2816-5
Galang, G., Mandla, R., Ruan, H., Jung, C., Sinha, T., Stone, N. R., et al. (2020). ATAC-seq reveals an Isl1 enhancer that regulates sinoatrial node development and function. Circ. Res. 127, 1502–1518. doi: 10.1161/circresaha.120.317145
Gasperini, M., Findlay, G. M., McKenna, A., Milbank, J. H., Lee, C., Zhang, M. D., et al. (2017). CRISPR/Cas9-mediated scanning for regulatory elements required for HPRT1 expression via thousands of large, programmed genomic deletions. Am. J. Hum. Genet. 101, 192–205. doi: 10.1016/j.ajhg.2017.06.010
Gasperini, M., Hill, A. J., McFaline-Figueroa, J. L., Martin, B., Kim, S., Zhang, M. D., et al. (2019). A genome-wide framework for mapping gene regulation via cellular genetic screens. Cell 176, 377.e19–390.e19.
Ghandi, M., Lee, D., Mohammad-Noori, M., and Beer, M. A. (2014). Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput. Biol. 10:e1003711. doi: 10.1371/journal.pcbi.1003711
Gibb, N., Lazic, S., Yuan, X., Deshwar, A. R., Leslie, M., Wilson, M. D., et al. (2018). Hey2 regulates the size of the cardiac progenitor pool during vertebrate heart development. Development 145:dev167510. doi: 10.1242/dev.167510
Gil, N., and Ulitsky, I. (2020). Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 21, 102–117. doi: 10.1038/s41576-019-0184-5
Gilsbach, R., Schwaderer, M., Preissl, S., Grüning, B. A., Kranzhöfer, D., Schneider, P., et al. (2018). Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat. Commun. 9:391.
Goldman, J. A., Kuzu, G., Lee, N., Karasik, J., Gemberling, M., Foglia, M. J., et al. (2017). Resolving heart regeneration by replacement histone profiling. Dev. Cell 40, 392.e6–404.e6.
Gomez-Velazquez, M., Badia-Careaga, C., Lechuga-Vieco, A. V., Nieto-Arellano, R., Tena, J. J., Rollan, I., et al. (2017). CTCF counter-regulates cardiomyocyte development and maturation programs in the embryonic heart. PLoS Genet. 13:e1006985. doi: 10.1371/journal.pgen.1006985
Gorkin, D. U., Barozzi, I., Zhao, Y., Zhang, Y., Huang, H., Lee, A. Y., et al. (2020). An atlas of dynamic chromatin landscapes in mouse fetal development. Nature 583, 744–751.
Grote, P., Wittler, L., Hendrix, D., Koch, F., Währisch, S., Beisaw, A., et al. (2013). The tissue-specific lncRNA fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell 24, 206–214. doi: 10.1016/j.devcel.2012.12.012
Grubert, F., Srivas, R., Spacek, D. V., Kasowski, M., Ruiz-Velasco, M., Sinnott-Armstrong, N., et al. (2020). Landscape of cohesin-mediated chromatin loops in the human genome. Nature 583, 737–743. doi: 10.1038/s41586-020-2151-x
Guo, Y., Xu, Q., Canzio, D., Shou, J., Li, J., Gorkin, D. U., et al. (2015). CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell 162, 900–910. doi: 10.1016/j.cell.2015.07.038
Hanssen, L. L. P., Kassouf, M. T., Oudelaar, A. M., Biggs, D., Preece, C., Downes, D. J., et al. (2017). Tissue-specific CTCF-cohesin-mediated chromatin architecture delimits enhancer interactions and function in vivo. Nat. Cell Biol. 19, 952–961. doi: 10.1038/ncb3573
Hao, Y., Hao, S., Andersen-Nissen, E., Mauck, W. M. III, Zheng, S., Butler, A., et al. (2020). Integrated analysis of multimodal single-cell data. bioRxiv [Preprint]. doi: 10.1101/2020.10.12.335331
Hare, E. E., Peterson, B. K., Iyer, V. N., Meier, R., and Eisen, M. B. (2008). Sepsid even-skipped enhancers are functionally conserved in Drosophila despite lack of sequence conservation. PLoS Genet. 4:e1000106. doi: 10.1371/journal.pgen.1000106
Harmston, N., Ing-Simmons, E., Tan, G., Perry, M., Merkenschlager, M., and Lenhard, B. (2017). Topologically associating domains are ancient features that coincide with Metazoan clusters of extreme noncoding conservation. Nat. Commun. 8:441.
Hashimoto, H., Wang, Z., Garry, G. A., Malladi, V. S., Botten, G. A., Ye, W., et al. (2019). Cardiac reprogramming factors synergistically activate genome-wide cardiogenic stage-specific enhancers. Cell Stem Cell 25, 69.e5–86.e5.
He, A., Gu, F., Hu, Y., Ma, Q., Ye, L. Y., Akiyama, J. A., et al. (2014). Dynamic GATA4 enhancers shape the chromatin landscape central to heart development and disease. Nat. Commun. 5:4907.
He, A., Kong, S. W., Ma, Q., and Pu, W. T. (2011). Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc. Natl. Acad. Sci. U.S.A. 108, 5632–5637. doi: 10.1073/pnas.1016959108
He, C. Z., and Burch, J. B. E. (1997). The chicken GATA-6 locus contains multiple control regions that confer distinct patterns of heart region-specific expression in transgenic mouse embryos. J. Biol. Chem. 272, 28550–28556. doi: 10.1074/jbc.272.45.28550
Heicklen-Klein, A., and Evans, T. (2004). T-box binding sites are required for activity of a cardiac GATA-4 enhancer. Dev. Biol. 267, 490–504. doi: 10.1016/j.ydbio.2003.09.042
Heinz, S., Romanoski, C. E., Benner, C., and Glass, C. K. (2015). The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 16, 144–154. doi: 10.1038/nrm3949
Henikoff, S., Henikoff, J., Kaya-Okur, H. S., and Ahmad, K. (2020). Efficient chromatin accessibility mapping in situ by nucleosome-tethered tagmentation. eLife 9:e63274. doi: 10.7554/eLife.63274
Hiller, M., Agarwal, S., Notwell, J. H., Parikh, R., Guturu, H., Wenger, A. M., et al. (2013). Computational methods to detect conserved non-genic elements in phylogenetically isolated genomes: application to zebrafish. Nucleic Acids Res. 41:e151. doi: 10.1093/nar/gkt557
Hoffmann, S., Clauss, S., Berger, I. M., Weiß, B., Montalbano, A., Röth, R., et al. (2016). Coding and non-coding variants in the SHOX2 gene in patients with early-onset atrial fibrillation. Basic Res. Cardiol. 111, 1–15.
Holdt, L. M., Beutner, F., Scholz, M., Gielen, S., Gäbel, G., Bergert, H., et al. (2010). ANRIL expression is associated with atherosclerosis risk at chromosome 9p21. Arterioscler. Thromb. Vasc. Biol. 30, 620–627.
Honkoop, H., de Bakker, D. E. M., Aharonov, A., Kruse, F., Shakked, A., Nguyen, P. D., et al. (2019). Single-cell analysis uncovers that metabolic reprogramming by ErbB2 signaling is essential for cardiomyocyte proliferation in the regenerating heart. eLife 8, 1–27.
Hsieh, C. L., Fei, T., Chen, Y., Li, T., Gao, Y., Wang, X., et al. (2014). Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc. Natl. Acad. Sci. U.S.A. 111, 7319–7324. doi: 10.1073/pnas.1324151111
Hu, P., Liu, J., Zhao, J., Wilkins, B. J., Lupino, K., Wu, H., et al. (2018). Single-nucleus transcriptomic survey of cell diversity and functional maturation in postnatal mammalian hearts. Genes Dev. 32, 1344–1357. doi: 10.1101/gad.316802.118
Hu, T., Yamagishi, H., Maeda, J., McAnally, J., Yamagishi, C., and Srivastava, D. (2004). Tbx1 regulates fibroblast growth factors in the anterior heart field through a reinforcing autoregulatory loop involving forkhead transcription factors. Development 131, 5491–5502. doi: 10.1242/dev.01399
Huang, W., Meng, H., Qiao, Y., Pang, S., Chen, D., and Yan, B. (2013). Two novel and functional DNA sequence variants within an upstream enhancer of the human NKX2-5 gene in ventricular septal defects. Gene 524, 152–155. doi: 10.1016/j.gene.2013.04.043
Ihara, D., Watanabe, Y., Seya, D., Arai, Y., Isomoto, Y., Nakano, A., et al. (2020). Expression of Hey2 transcription factor in the early embryonic ventricles is controlled through a distal enhancer by Tbx20 and Gata transcription factors. Dev. Biol. 461, 124–131. doi: 10.1016/j.ydbio.2020.02.001
Iklé, J. M., Artinger, K. B., and Clouthier, D. E. (2012). Identification and characterization of the zebrafish pharyngeal arch-specific enhancer for the basic helix-loop-helix transcription factor Hand2. Dev. Biol. 368, 118–126. doi: 10.1016/j.ydbio.2012.05.003
Irie, N., and Kuratani, S. (2011). Comparative transcriptome analysis reveals vertebrate phylotypic period during organogenesis. Nat. Commun. 2:248.
Irie, N., and Sehara-Fujisawa, A. (2007). The vertebrate phylotypic stage and an early bilaterian-related stage in mouse embryogenesis defined by genomic information. BMC Biol. 5:1. doi: 10.1186/1741-7007-5-1
Irimia, M., Tena, J. J., Alexis, M. S., Fernandez-Miñan, A., Maeso, I., Bogdanovic, O., et al. (2012). Extensive conservation of ancient microsynteny across metazoans due to cis-regulatory constraints. Genome Res 22, 2356–2367. doi: 10.1101/gr.139725.112
Javierre, B. M., Burren, O. S., Wilder, S. P., Kreuzhuber, R., Hill, S. M., Sewitz, S., et al. (2016). Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell 167, 1369.e19–1384.e19.
Jia, G., Preussner, J., Chen, X., Guenther, S., Yuan, X., Yekelchyk, M., et al. (2018). Single cell RNA-seq and ATAC-seq analysis of cardiac progenitor cell transition states and lineage settlement. Nat. Commun. 9:4877.
Jin, H., Stojnic, R., Adryan, B., Ozdemir, A., Stathopoulos, A., and Frasch, M. (2013). Genome-wide screens for in vivo tinman binding sites identify cardiac enhancers with diverse functional architectures. PLoS Genet. 9:e1003195. doi: 10.1371/journal.pgen.1003195
Jin, X., Simmons, S. K., Guo, A., Shetty, A. S., Ko, M., Nguyen, L., et al. (2020). In vivo Perturb-Seq reveals neuronal and glial abnormalities associated with autism risk genes. Science 370:eaaz6063. doi: 10.1126/science.aaz6063
Johnson, D. S., Davidson, B., Brown, C. D., Smith, W. C., and Sidow, A. (2004). Noncoding regulatory sequences of Ciona exhibit strong correspondence between evolutionary constraint and functional importance. Genome Res. 14, 2448–2456. doi: 10.1101/gr.2964504
Jung, I., Schmitt, A., Diao, Y., Lee, A. J., Liu, T., Yang, D., et al. (2019). A compendium of promoter-centered long-range chromatin interactions in the human genome. Nat. Genet. 51, 1442–1449. doi: 10.1038/s41588-019-0494-8
Junion, G., Spivakov, M., Girardot, C., Braun, M., Gustafson, E. H., Birney, E., et al. (2012). A transcription factor collective defines cardiac cell fate and reflects lineage history. Cell 148, 473–486. doi: 10.1016/j.cell.2012.01.030
Kaikkonen, M. U., Spann, N. J., Heinz, S., Romanoski, C. E., Allison, K. A., Stender, J. D., et al. (2013). Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol. Cell 51, 310–325. doi: 10.1016/j.molcel.2013.07.010
Kalinka, A. T., Varga, K. M., Gerrard, D. T., Preibisch, S., Corcoran, D. L., Jarrells, J., et al. (2010). Gene expression divergence recapitulates the developmental hourglass model. Nature 468, 811–816. doi: 10.1038/nature09634
Kapoor, A., Lee, D., Zhu, L., Soliman, E. Z., Grove, M. L., Boerwinkle, E., et al. (2019). Multiple SCN5A variant enhancers modulate its cardiac gene expression and the QT interval. Proc. Natl. Acad. Sci. U.S.A. 166, 10636–10645. doi: 10.1073/pnas.1808734116
Kapoor, A., Sekar, R. B., Hansen, N. F., Fox-Talbot, K., Morley, M., Pihur, V., et al. (2014). An enhancer polymorphism at the cardiomyocyte intercalated disc protein NOS1AP locus is a major regulator of the QT interval. Am. J. Hum. Genet. 94, 854–869. doi: 10.1016/j.ajhg.2014.05.001
Kaskow, B. J., Diepeveen, L. A., Michael Proffitt, J., Rea, A. J., Ulgiati, D., Blangero, J., et al. (2014). Molecular prioritization strategies to identify functional genetic variants in the cardiovascular disease-associated expression QTL Vanin-1. Eur. J. Hum. Genet. 22, 688–695. doi: 10.1038/ejhg.2013.208
Kawauchi, S., Calof, A. L., Santos, R., Lopez-Burks, M. E., Young, C. M., Hoang, M. P., et al. (2009). Multiple organ system defects and transcriptional dysregulation in the Nipbl+/- Mouse, a model of cornelia de lange syndrome. PLoS Genet. 5:e1000650. doi: 10.1371/journal.pgen.1000650
Kaya-Okur, H. S., Wu, S. J., Codomo, C. A., Pledger, E. S., Bryson, T. D., Henikoff, J. G., et al. (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10:1930.
Khoueiry, P., Girardot, C., Ciglar, L., Peng, P. C., Hilary Gustafson, E., Sinha, S., et al. (2017). Uncoupling evolutionary changes in DNA sequence, transcription factor occupancy and enhancer activity. eLife 6, 1–29.
Kim, T.-K., Hemberg, M., Gray, J. M., Costa, A. M., Bear, D. M., Wu, J., et al. (2010). Widespread transcription at neuronal activity-regulated enhancers. Nature 465, 182–187. doi: 10.1038/nature09033
Klann, T. S., Black, J. B., Chellappan, M., Safi, A., Song, L., Hilton, I. B., et al. (2017). CRISPR-Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat. Biotechnol. 35, 561–568. doi: 10.1038/nbt.3853
Klattenhoff, C. A., Scheuermann, J. C., Surface, L. E., Bradley, R. K., Fields, P. A., Steinhauser, M. L., et al. (2013). Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell 152, 570–583. doi: 10.1016/j.cell.2013.01.003
Klein, J. C., Chen, W., Gasperini, M., and Shendure, J. (2018). Identifying novel enhancer elements with CRISPR-Based screens. ACS Chem. Biol. 13, 326–332. doi: 10.1021/acschembio.7b00778
Koch, F., Fenouil, R., Gut, M., Cauchy, P., Albert, T. K., Zacarias-Cabeza, J., et al. (2011). Transcription initiation platforms and GTF recruitment at tissue-specific enhancers and promoters. Nat. Struct. Mol. Biol. 18, 956–963. doi: 10.1038/nsmb.2085
Korkmaz, G., Lopes, R., Ugalde, A. P., Nevedomskaya, E., Han, R., Myacheva, K., et al. (2016). Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat. Biotechnol. 34, 192–198. doi: 10.1038/nbt.3450
Krasnov, A. N., Mazina, M. Y., Nikolenko, J. V., and Vorobyeva, N. E. (2016). On the way of revealing coactivator complexes cross-talk during transcriptional activation. Cell Biosci. 6:15.
Kunarso, G., Chia, N.-Y., Jeyakani, J., Hwang, C., Lu, X., Chan, Y.-S., et al. (2010). Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat. Genet. 42, 631–634. doi: 10.1038/ng.600
Kvon, E. Z., Kamneva, O. K., Melo, U. S., Barozzi, I., Osterwalder, M., Mannion, B. J., et al. (2016). Progressive loss of function in a limb enhancer during snake evolution. Cell 167, 633.e11–642.e11.
Kvon, E. Z., Zhu, Y., Kelman, G., Visel, A., Dickel, D. E., and Pennacchio, L. A. (2020). Comprehensive in vivo interrogation reveals phenotypic impact of human enhancer variants. Cell 180, 1262.e15–1271.e15.
Kwak, H., Fuda, N. J., Core, L. J., and Lis, J. T. (2013). Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339, 950–953. doi: 10.1126/science.1229386
Lai, F., Orom, U. A., Cesaroni, M., Beringer, M., Taatjes, D. J., Blobel, G. A., et al. (2013). Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature 494, 497–501. doi: 10.1038/nature11884
Lambeth, L. S., and Smith, C. A. (2013). Short hairpin RNA-mediated gene silencing. Methods Mol. Biol. 942, 205–232. doi: 10.1007/978-1-62703-119-6_12
Laurent, F., Girdziusaite, A., Gamart, J., Barozzi, I., Osterwalder, M., Akiyama, J. A., et al. (2017). HAND2 target gene regulatory networks control atrioventricular canal and cardiac valve development. Cell Rep. 19, 1602–1613. doi: 10.1016/j.celrep.2017.05.004
Lee, D., Kapoor, A., Safi, A., Song, L., Halushka, M. K., Crawford, G. E., et al. (2018). Human cardiac cis-regulatory elements, their cognate transcription factors, and regulatory DNA sequence variants. Genome Res. 28, 1577–1588. doi: 10.1101/gr.234633.118
Lee, K. H., Evans, S., Ruan, T. Y., and Lassar, A. B. (2004). SMAD-mediated modulation of YY1 activity regulates the BMP response and cardiac-specific expression of a GATA4/5/6-dependent chick Nkx2.5 enhancer. Development 131, 4709–4723. doi: 10.1242/dev.01344
Lescroart, F., Chabab, S., Lin, X., Rulands, S., Paulissen, C., Rodolosse, A., et al. (2014). Early lineage restriction in temporally distinct populations of Mesp1 progenitors during mammalian heart development. Nat. Cell Biol. 16, 829–840. doi: 10.1038/ncb3024
Lettice, L. A., Heaney, S. J. H., Purdie, L. A., Li, L., de Beer, P., Oostra, B. A., et al. (2003). A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum. Mol. Genet. 12, 1725–1735. doi: 10.1093/hmg/ddg180
Leung, D., Jung, I., Rajagopal, N., Schmitt, A., Selvaraj, S., Lee, A. Y., et al. (2015). Integrative analysis of haplotype-resolved epigenomes across human tissues. Nature 518, 350–354. doi: 10.1038/nature14217
Li, G., Liu, Y., Zhang, Y., Kubo, N., Yu, M., Fang, R., et al. (2019). Joint profiling of DNA methylation and chromatin architecture in single cells. Nat. Methods 16, 991–993. doi: 10.1038/s41592-019-0502-z
Li, K., Liu, Y., Cao, H., Zhang, Y., Gu, Z., Liu, X., et al. (2020). Interrogation of enhancer function by enhancer-targeting CRISPR epigenetic editing. Nat. Commun. 11, 1–16.
Li, N., Wang, Z. S., Wang, X. H., Xu, Y. J., Qiao, Q., Li, X. M., et al. (2018). A SHOX2 loss-of-function mutation underlying familial atrial fibrillation. Int. J. Med. Sci. 15, 1564–1572. doi: 10.7150/ijms.27424
Li, W., Notani, D., Ma, Q., Tanasa, B., Nunez, E., Chen, A. Y., et al. (2013). Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 498, 516–520. doi: 10.1038/nature12210
Li, W., Notani, D., and Rosenfeld, M. G. (2016). Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat. Rev. Genet. 17, 207–223. doi: 10.1038/nrg.2016.4
Liang, M., Soomro, A. U., Tasneem, S., Abatti, L. E., Alizada, A., Yuan, X., et al. (2020). Enhancer-gene rewiring in the pathogenesis of quebec platelet disorder. Blood 136, 2679–2690.
Liberatore, C. M., Searcy-Schrick, R. D., Vincent, E. B., and Yutzey, K. E. (2002). Nkx-2.5 gene induction in mice is mediated by a Smad consensus regulatory region. Dev. Biol. 244, 243–256. doi: 10.1006/dbio.2002.0604
Lieberman-Aiden, E., Van Berkum, N. L., Williams, L., Imakaev, M., Ragoczy, T., Telling, A., et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. doi: 10.1126/science.1181369
Lien, C. L., McAnally, J., Richardson, J. A., and Olson, E. N. (2002). Cardiac-specific activity of an Nkx2-5 enhancer requires an evolutionarily conserved Smad binding site. Dev. Biol. 244, 257–266. doi: 10.1006/dbio.2002.0603
Lien, C. L., Wu, C., Mercer, B., Webb, R., Richardson, J. A., and Olson, E. N. (1999). Control of early cardiac-specific transcription of Nkx2-5 by a GATA-dependent enhancer. Development 126, 75–84.
Liu, C., Wang, M., Wei, X., Wu, L., Xu, J., Dai, X., et al. (2019). An ATAC-seq atlas of chromatin accessibility in mouse tissues. Sci. Data 6:65.
Liu, J., Viales, R. R., Khoueiry, P., Reddington, J. P., Girardot, C., Furlong, E. E. M., et al. (2020). The hourglass model of evolutionary conservation during embryogenesis extends to developmental enhancers with signatures of positive selection. bioRxiv [Preprint]. doi: 10.1101/2020.11.02.364505
Liu, Q., Jiang, C., Xu, J., Zhao, M. T., Van Bortle, K., Cheng, X., et al. (2017). Genome-wide temporal profiling of transcriptome and open chromatin of early cardiomyocyte differentiation derived from hiPSCs and hESCs. Circ. Res. 121, 376–391. doi: 10.1161/circresaha.116.310456
Liu, Q., Van Bortle, K., Zhang, Y., Zhao, M. T., Zhang, J. Z., Geller, B. S., et al. (2018). Disruption of mesoderm formation during cardiac differentiation due to developmental exposure to 13-cis-retinoic acid. Sci. Rep. 8, 1–11.
Loh, K. M., Chen, A., Koh, P. W., Deng, T. Z., Sinha, R., Tsai, J. M., et al. (2016). Mapping the pairwise choices leading from pluripotency to human bone. Heart, and other mesoderm cell types. Cell 166, 451–467. doi: 10.1016/j.cell.2016.06.011
Long, H. K., Prescott, S. L., and Wysocka, J. (2016). Ever-changing landscapes: transcriptional enhancers in development and evolution. Cell 167, 1170–1187. doi: 10.1016/j.cell.2016.09.018
Luna-Zurita, L., Stirnimann, C. U., Glatt, S., Kaynak, B. L., Thomas, S., Baudin, F., et al. (2016). Complex interdependence regulates heterotypic transcription factor distribution and coordinates cardiogenesis. Cell 164, 999–1014. doi: 10.1016/j.cell.2016.01.004
Lupiáñez, D. G., Kraft, K., Heinrich, V., Krawitz, P., Brancati, F., Klopocki, E., et al. (2015). Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012–1025. doi: 10.1016/j.cell.2015.04.004
MacNeill, C., French, R., Evans, T., Wessels, A., and Burch, J. B. E. (2000). Modular regulation of cGATA-5 gene expression in the developing heart and gut. Dev. Biol. 217, 62–76. doi: 10.1006/dbio.1999.9539
Maeda, J., Yamagishi, H., McAnally, J., Yamagishi, C., and Srivastava, D. (2006). Tbx1 is regulated by forkhead proteins in the secondary heart field. Dev. Dyn. 235, 701–710. doi: 10.1002/dvdy.20686
Malik, S., and Roeder, R. G. (2010). The metazoan mediator co-activator complex as an integrative hub for transcriptional regulation. Nat. Rev. Genet. 11, 761–772. doi: 10.1038/nrg2901
May, D., Blow, M. J., Kaplan, T., McCulley, D. J., Jensen, B. C., Akiyama, J. A., et al. (2012). Large-scale discovery of enhancers from human heart tissue. Nat. Genet. 44, 89–93. doi: 10.1038/ng.1006
McFadden, D. G., Charité, J., Richardson, J. A., Srivastava, D., Firulli, A. B., and Olson, E. N. (2000). A GATA-dependent right ventricular enhancer controls dHAND transcription in the developing heart. Development 127, 5331–5341.
McPherson, C. E., Shim, E. Y., Friedman, D. S., and Zaret, K. S. (1993). An active tissue-specific enhancer and bound transcription factors existing in a precisely positioned nucleosomal array. Cell 75, 387–398. doi: 10.1016/0092-8674(93)80079-t
Meers, M. P., Bryson, T. D., Henikoff, J. G., and Henikoff, S. (2019). Improved CUT&RUN chromatin profiling tools. eLife 8:e46314.
Melnikov, A., Murugan, A., Zhang, X., Tesileanu, T., Wang, L., Rogov, P., et al. (2012). Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol. 30, 271–277. doi: 10.1038/nbt.2137
Merkenschlager, M., and Nora, E. P. (2016). CTCF and cohesin in genome folding and transcriptional gene regulation. Annu. Rev. Genomics Hum. Genet. 17, 17–43. doi: 10.1146/annurev-genom-083115-022339
Meuleman, W., Muratov, A., Rynes, E., Halow, J., Lee, K., Bates, D., et al. (2020). Index and biological spectrum of human DNase I hypersensitive sites. Nature 584, 244–251. doi: 10.1038/s41586-020-2559-3
Mifsud, B., Tavares-Cadete, F., Young, A. N., Sugar, R., Schoenfelder, S., Ferreira, L., et al. (2015). Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet. 47, 598–606. doi: 10.1038/ng.3286
Mikkelsen, T. S., Xu, Z., Zhang, X., Wang, L., Gimble, J. M., Lander, E. S., et al. (2010). Comparative epigenomic analysis of murine and human adipogenesis. Cell 143, 156–169. doi: 10.1016/j.cell.2010.09.006
Miller, C. L., Anderson, D. R., Kundu, R. K., Raiesdana, A., Nürnberg, S. T., Diaz, R., et al. (2013). Disease-related growth factor and embryonic signaling pathways modulate an enhancer of TCF21 Expression at the 6q23.2 coronary heart disease locus. PLoS Genet. 9:e1003652. doi: 10.1371/journal.pgen.1003652
Miller, C. L., Haas, U., Diaz, R., Leeper, N. J., Kundu, R. K., Patlolla, B., et al. (2014). Coronary heart disease-associated variation in TCF21 Disrupts a miR-224 binding site and miRNA-Mediated regulation. PLoS Genet. 10:e1004263. doi: 10.1371/journal.pgen.1004263
Miquerol, L., and Kelly, R. G. (2013). Organogenesis of the vertebrate heart. Wiley Interdiscip. Rev. Dev. Biol. 2, 17–29. doi: 10.1002/wdev.68
Molkentin, J. D., Antos, C., Mercer, B., Taigen, T., Miano, J. M., and Olson, E. N. (2000). Direct activation of a GATA6 cardiac enhancer by Nkx2.5: evidence for a reinforcing regulatory network of Nkx2.5 and GATA transcription factors in the developing heart. Dev. Biol. 217, 301–309. doi: 10.1006/dbio.1999.9544
Montefiori, L. E., Sobreira, D. R., Sakabe, N. J., Aneas, I., Joslin, A. C., Hansen, G. T., et al. (2018). A promoter interaction map for cardiovascular disease genetics. eLife 7:e35788.
Morita, S., Noguchi, H., Horii, T., Nakabayashi, K., Kimura, M., Okamura, K., et al. (2016). Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat. Biotechnol. 34, 1060–1065. doi: 10.1038/nbt.3658
Moudgil, A., Wilkinson, M. N., Chen, X., He, J., Cammack, A. J., Vasek, M. J., et al. (2020). Self-reporting transposons enable simultaneous readout of gene expression and transcription factor binding in single cells. Cell 182, 992.e21–1008.e21.
Mousavi, K., Zare, H., Dell’Orso, S., Grontved, L., Gutierrez-Cruz, G., Derfoul, A., et al. (2013). ERNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol. Cell 51, 606–617. doi: 10.1016/j.molcel.2013.07.022
Mozaffarian, D., Benjamin, E. J., Go, A. S., Arnett, D. K., Blaha, M. J., Cushman, M., et al. (2015). Heart disease and stroke statistics—2015 update. Circulation 131, e29–e322.
Mumbach, M. R., Rubin, A. J., Flynn, R. A., Dai, C., Khavari, P. A., Greenleaf, W. J., et al. (2016). HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 13, 919–922. doi: 10.1038/nmeth.3999
Muto, A., Calof, A. L., Lander, A. D., and Schilling, T. F. (2011). Multifactorial origins of heart and gut defects in nipbl-deficient zebrafish, a model of cornelia de lange syndrome. PLoS Biol. 9:e1001181. doi: 10.1371/journal.pbio.1001181
Narlikar, L., Sakabe, N. J., and Blanski, A. A. (2010). Genome-wide discovery of human heart enhancers. Genome Res. 20, 381–392. doi: 10.1101/gr.098657.109
Nature Methods. (2020). Method of the Year 2019: single-cell multimodal omics. Nat. Methods 17:1. doi: 10.1038/s41592-019-0703-5
Nelson, C. P., Goel, A., Butterworth, A. S., Kanoni, S., Webb, T. R., Marouli, E., et al. (2017). Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat. Genet. 49, 1385–1391.
Nicole Ritter, A., Ali, T., Kopitchinski, N., Dimmeler, S., and Grote Correspondence, P. (2019). The lncRNA locus handsdown regulates cardiac gene programs and is essential for early mouse development. Dev. Cell 50, 644–657. doi: 10.1016/j.devcel.2019.07.013
Nikpay, M., Goel, A., Won, H.-H., Hall, L. M., Willenborg, C., Kanoni, S., et al. (2015). A comprehensive 1000 genomes–based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 47, 1121–1130. doi: 10.1038/ng.3396
Nobrega, M. A., Ovcharenko, I., Afzal, V., and Rubin, E. M. (2003). Scanning human gene deserts for long-range enhancers. Science 302, 413–413. doi: 10.1126/science.1088328
Nord, A. S., Blow, M. J., Attanasio, C., Akiyama, J. A., Holt, A., Hosseini, R., et al. (2013). Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell 155, 1521–1531. doi: 10.1016/j.cell.2013.11.033
Noseda, M., Peterkin, T., Simões, F. C., Patient, R., and Schneider, M. D. (2011). Cardiopoietic factors: extracellular signals for cardiac lineage commitment. Circ. Res. 108, 129–152. doi: 10.1161/circresaha.110.223792
Odom, D. T., Dowell, R. D., Jacobsen, E. S., Gordon, W., Danford, T. W., MacIsaac, K. D., et al. (2007). Tissue-specific transcriptional regulation has diverged significantly between human and mouse. Nat. Genet. 39, 730–732. doi: 10.1038/ng2047
Olson, E. N. (2006). Gene regulatory networks in the evolution and development of the heart. Science 313, 1922–1927. doi: 10.1126/science.1132292
O’Neill, L. P., VerMilyea, M. D., and Turner, B. M. (2006). Epigenetic characterization of the early embryo with a chromatin immunoprecipitation protocol applicable to small cell populations. Nat. Genet. 38, 835–841. doi: 10.1038/ng1820
Osterwalder, M., Barozzi, I., Tissières, V., Fukuda-Yuzawa, Y., Mannion, B. J., Afzal, S. Y., et al. (2018). Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 554, 239–243. doi: 10.1038/nature25461
Osterwalder, M., Speziale, D., Shoukry, M., Mohan, R., Ivanek, R., Kohler, M., et al. (2014). HAND2 targets define a network of transcriptional regulators that compartmentalize the early limb bud mesenchyme. Dev. Cell 31, 345–357. doi: 10.1016/j.devcel.2014.09.018
Ounzain, S., Micheletti, R., Arnan, C., Plaisance, I., Cecchi, D., Schroen, B., et al. (2015). CARMEN, a human super enhancer-associated long noncoding RNA controlling cardiac specification, differentiation and homeostasis. J. Mol. Cell. Cardiol. 89, 98–112. doi: 10.1016/j.yjmcc.2015.09.016
Ounzain, S., Pezzuto, I., Micheletti, R., Burdet, F., Sheta, R., Nemir, M., et al. (2014). Functional importance of cardiac enhancer-associated noncoding RNAs in heart development and disease. Curr. Ther. Res. Clin. Exp. 76, 55–70. doi: 10.1016/j.yjmcc.2014.08.009
Paige, S. L., Thomas, S., Stoick-Cooper, C. L., Wang, H., Maves, L., Sandstrom, R., et al. (2012). A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell 151, 221–232. doi: 10.1016/j.cell.2012.08.027
Paik, D. T., Cho, S., Tian, L., Chang, H. Y., and Wu, J. C. (2020). Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nat. Rev. Cardiol. 17, 457–473. doi: 10.1038/s41569-020-0359-y
Pane, L. S., Fulcoli, F. G., Cirino, A., Altomonte, A., Ferrentino, R., Bilio, M., et al. (2018). Tbx1 represses Mef2c gene expression and is correlated with histone 3 deacetylation of the anterior heart field enhancer. DMM Dis. Model. Mech. 11:dmm029967. doi: 10.1242/dmm.029967
Pang, S., Shan, J., Qiao, Y., Ma, L., Qin, X., Wanyan, H., et al. (2012). Genetic and functional analysis of the NKX2-5 gene promoter in patients with ventricular septal defects. Pediatr. Cardiol. 33, 1355–1361. doi: 10.1007/s00246-012-0346-0
Paralkar, V. R., Taborda, C. C., Huang, P., Yao, Y., Kossenkov, A. V., Prasad, R., et al. (2016). Unlinking an lncRNA from its associated cis element. Mol. Cell 62, 104–110. doi: 10.1016/j.molcel.2016.02.029
Paris, M., Kaplan, T., Li, X. Y., Villalta, J. E., Lott, S. E., and Eisen, M. B. (2013). Extensive divergence of transcription factor binding in Drosophila embryos with highly conserved gene expression. PLoS Genet. 9:e1003748. doi: 10.1371/journal.pgen.1003748
Patwardhan, R. P., Hiatt, J. B., Witten, D. M., Kim, M. J., Smith, R. P., May, D., et al. (2012). Massively parallel functional dissection of mammalian enhancers in vivo. Nat. Biotechnol. 30, 265–270. doi: 10.1038/nbt.2136
Pawlak, M., Kedzierska, K. Z., Migdal, M., Nahia, K. A., Ramilowski, J. A., Bugajski, L., et al. (2019). Dynamics of cardiomyocyte transcriptome and chromatin landscape demarcates key events of heart development. Genome Res. 29, 506–519. doi: 10.1101/gr.244491.118
Pennacchio, L. A., Ahituv, N., Moses, A. M., Prabhakar, S., Nobrega, M. A., Shoukry, M., et al. (2006). In vivo enhancer analysis of human conserved non-coding sequences. Nature 444, 499–502. doi: 10.1038/nature05295
Perry, M. W., Boettiger, A. N., Bothma, J. P., and Levine, M. (2010). Shadow enhancers foster robustness of Drosophila gastrulation. Curr. Biol. 20, 1562–1567. doi: 10.1016/j.cub.2010.07.043
Pickar-Oliver, A., and Gersbach, C. A. (2019). The next generation of CRISPR–Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 20, 490–507. doi: 10.1038/s41580-019-0131-5
Pijuan-Sala, B., Wilson, N. K., Xia, J., Hou, X., Hannah, R. L., Kinston, S., et al. (2020). Single-cell chromatin accessibility maps reveal regulatory programs driving early mouse organogenesis. Nat. Cell Biol. 22, 487–497. doi: 10.1038/s41556-020-0489-9
Poelmann, R. E., and Gittenberger-de Groot, A. C. (2019). Development and evolution of the metazoan heart. Dev. Dyn. 248, 634–656. doi: 10.1002/dvdy.45
Postma, A. V., Bezzina, C. R., and Christoffels, V. M. (2015). Genetics of congenital heart disease: the contribution of the noncoding regulatory genome. J. Hum. Genet. 61, 13–19. doi: 10.1038/jhg.2015.98
Pott, S. (2017). Simultaneous measurement of chromatin accessibility. DNA methylation, and nucleosome phasing in single cells. eLife 6:e23203.
Pugacheva, E. M., Kubo, N., Loukinov, D., Tajmul, M., Kang, S., Kovalchuk, A. L., et al. (2020). CTCF mediates chromatin looping via N-terminal domain-dependent cohesin retention. Proc. Natl. Acad. Sci. U.S.A. 117, 2020–2031. doi: 10.1073/pnas.1911708117
Quaife-Ryan, G. A., Sim, C. B., Ziemann, M., Kaspi, A., Rafehi, H., Ramialison, M., et al. (2017). Multicellular transcriptional analysis of mammalian heart regeneration. Circulation 136, 1123–1139. doi: 10.1161/circulationaha.117.028252
Quinodoz, S. A., Ollikainen, N., Tabak, B., Palla, A., Schmidt, J. M., Detmar, E., et al. (2018). Higher-Order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell 174, 744.e24–757.e24.
Racedo, S. E., Hasten, E., Lin, M., Devakanmalai, G. S., Guo, T., Ozbudak, E. M., et al. (2017). Reduced dosage of β-catenin provides significant rescue of cardiac outflow tract anomalies in a Tbx1 conditional null mouse model of 22q11.2 deletion syndrome. PLoS Genet. 13:e1006687. doi: 10.1371/journal.pgen.1006687
Racioppi, C., Wiechecki, K. A., and Christiaen, L. (2019). Combinatorial chromatin dynamics foster accurate cardiopharyngeal fate choices. eLife 8:e49921.
Rada-Iglesias, A., Bajpai, R., Swigut, T., Brugmann, S. A., Flynn, R. A., and Wysocka, J. (2011). A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283. doi: 10.1038/nature09692
Rao, S. S. P., Huntley, M. H., Durand, N. C., Stamenova, E. K., Bochkov, I. D., Robinson, J. T., et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. doi: 10.1016/j.cell.2014.11.021
Reecy, J. M., Li, X., Yamada, M., DeMayo, F. J., Newman, C. S., Harvey, R. P., et al. (1999). Identification of upstream regulatory regions in the heart-expressed homeobox gene Nkx2-5. Development 126, 839–849.
Richter, F., Morton, S. U., Kim, S. W., Kitaygorodsky, A., Wasson, L. K., Chen, K. M., et al. (2020). Genomic analyses implicate noncoding de novo variants in congenital heart disease. Nat. Genet. 52, 769–777. doi: 10.1038/s41588-020-0652-z
Rojas, A., De Val, S., Heidt, A. B., Xu, S. M., Bristow, J., and Black, B. L. (2005). Gata4 expression in lateral mesoderm is downstream of BMP4 and is activated directly by Forkhead and GATA transcription factors through a distal enhancer element. Development 132, 3405–3417. doi: 10.1242/dev.01913
Rojas, A., Schachterle, W., Xu, S. M., and Black, B. L. (2009). An endoderm-specific transcriptional enhancer from the mouse Gata4 gene requires GATA and homeodomain protein-binding sites for function in vivo. Dev. Dyn. 238, 2588–2598. doi: 10.1002/dvdy.22091
Rosa-Garrido, M., Chapski, D. J., Schmitt, A. D., Kimball, T. H., Karbassi, E., Monte, E., et al. (2017). High-resolution mapping of chromatin conformation in cardiac myocytes reveals structural remodeling of the epigenome in heart failure. Circulation 136, 1613–1625. doi: 10.1161/circulationaha.117.029430
Rubin, A. J., Parker, K. R., Satpathy, A. T., Qi, Y., Wu, B., Ong, A. J., et al. (2019). Coupled single-Cell CRISPR screening and epigenomic profiling reveals causal gene regulatory networks. Cell 176, 361.e17–376.e17.
Sahlén, P., Abdullayev, I., Ramsköld, D., Matskova, L., Rilakovic, N., Lötstedt, B., et al. (2015). Genome-wide mapping of promoter-anchored interactions with close to single-enhancer resolution. Genome Biol. 16:156.
Samani, N. J., and Schunkert, H. (2008). Chromosome 9p21 and CARDIOVASCULAR DISEase. Circ. Cardiovasc. Genet. 1, 81–84. doi: 10.1161/circgenetics.108.832527
Sanjana, N., Montalbano, A., Deng, J., MéndezMancilla, A., Wessels, H.-H., Moss, N. G., et al. (2020). Scalable pooled CRISPR screens with single-cell chromatin accessibility profiling. bioRxiv [Preprint]. doi: 10.1101/2020.11.20.390971
Santos, R., Kawauchi, S., Jacobs, R. E., Lopez-Burks, M. E., Choi, H., Wikenheiser, J., et al. (2016). Conditional creation and rescue of nipbl-deficiency in mice reveals multiple determinants of risk for congenital heart defects. PLoS Biol. 14:e2000197. doi: 10.1371/journal.pbio.2000197
Schachterle, W., Rojas, A., Xu, S. M., and Black, B. L. (2012). ETS-dependent regulation of a distal Gata4 cardiac enhancer. Dev. Biol. 361, 439–449. doi: 10.1016/j.ydbio.2011.10.023
Schaukowitch, K., Joo, J. Y., Liu, X., Watts, J. K., Martinez, C., and Kim, T. K. (2014). Enhancer RNA facilitates NELF release from immediate early genes. Mol. Cell 56, 29–42. doi: 10.1016/j.molcel.2014.08.023
Schmidt, D., Schwalie, P. C., Ross-Innes, C. S., Hurtado, A., Brown, G. D., Carroll, J. S., et al. (2010a). A CTCF-independent role for cohesin in tissue-specific transcription. Genome Res. 20, 578–588. doi: 10.1101/gr.100479.109
Schmidt, D., Wilson, M. D., Ballester, B., Schwalie, P. C., Brown, G. D., Marshall, A., et al. (2010b). Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science 328, 1036–1040. doi: 10.1126/science.1186176
Schmidt, S. F., Larsen, B. D., Loft, A., and Mandrup, S. (2016). Cofactor squelching: artifact or fact? BioEssays 38, 618–626. doi: 10.1002/bies.201600034
Schmidt, S. F., Larsen, B. D., Loft, A., Nielsen, R., Madsen, J. G. S., and Mandrup, S. (2015). Acute TNF-induced repression of cell identity genes is mediated by NFκB-directed redistribution of cofactors from super-enhancers. Genome Res. 25, 1281–1294. doi: 10.1101/gr.188300.114
Schmitt, A. D., Hu, M., Jung, I., Xu, Z., Qiu, Y., Tan, C. L., et al. (2016). A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep. 17, 2042–2059. doi: 10.1016/j.celrep.2016.10.061
Schoenfelder, S., Furlan-Magaril, M., Mifsud, B., Tavares-Cadete, F., Sugar, R., Javierre, B. M., et al. (2015). The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res. 25, 582–597. doi: 10.1101/gr.185272.114
Schwartzman, O., and Tanay, A. (2015). Single-cell epigenomics: techniques and emerging applications. Nat. Rev. Genet. 16, 716–726. doi: 10.1038/nrg3980
Scott, I. C. (2012). Life Before Nkx2.5: Cardiovascular Progenitor Cells: Embryonic Origins and Development. Amsterdam: Elsevier Inc.
Searcy, R. D., Vincent, E. B., Liberatore, C. M., and Yutzey, K. E. (1998). A GATA-dependent nkx-2.5 regulatory element activates early cardiac gene expression in transgenic mice. Development 125, 4461–4470.
Serpooshan, V., Liu, Y. H., Buikema, J. W., Galdos, F. X., Chirikian, O., Paige, S., et al. (2017). Nkx2.5+ cardiomyoblasts contribute to cardiomyogenesis in the neonatal heart. Sci. Rep. 7, 1–13.
Sharon, E., Kalma, Y., Sharp, A., Raveh-Sadka, T., Levo, M., Zeevi, D., et al. (2012). Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat. Biotechnol. 30, 521–530. doi: 10.1038/nbt.2205
Shii, L., Song, L., Maurer, K., Zhang, Z., and Sullivan, K. E. (2017). SERPINB2 is regulated by dynamic interactions with pause-release proteins and enhancer RNAs. Mol. Immunol. 88, 20–31. doi: 10.1016/j.molimm.2017.05.005
Shin, J. T., Priest, J. R., Ovcharenko, I., Ronco, A., Moore, R. K., Burns, C. G., et al. (2005). Human-zebrafish non-coding conserved elements act in vivo to regulate transcription. Nucleic Acids Res. 33, 5437–5445. doi: 10.1093/nar/gki853
Siggers, T., Duyzend, M. H., Reddy, J., Khan, S., and Bulyk, M. L. (2011). Non-DNA-binding cofactors enhance DNA-binding specificity of a transcriptional regulatory complex. Mol. Syst. Biol. 7:555. doi: 10.1038/msb.2011.89
Simeonov, D. R., Gowen, B. G., Boontanrart, M., Roth, T. L., Gagnon, J. D., Mumbach, M. R., et al. (2017). Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature 549, 111–115.
Simonis, M., Klous, P., Splinter, E., Moshkin, Y., Willemsen, R., de Wit, E., et al. (2006). Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture–on-chip (4C). Nat. Genet. 38, 1348–1354. doi: 10.1038/ng1896
Skene, P. J., and Henikoff, S. (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 6, 1–35.
Slattery, M., Riley, T., Liu, P., Abe, N., Gomez-Alcala, P., Dror, I., et al. (2011). Cofactor binding evokes latent differences in DNA binding specificity between hox proteins. Cell 147, 1270–1282. doi: 10.1016/j.cell.2011.10.053
Smemo, S., Campos, L. C., Moskowitz, I. P., Krieger, J. E., Pereira, A. C., and Nobrega, M. A. (2012). Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Hum. Mol. Genet. 21, 3255–3263. doi: 10.1093/hmg/dds165
Sönmezer, C., Kleinendorst, R., Imanci, D., Barzaghi, G., Villacorta, L., Schübeler, D., et al. (2020). Molecular Co-occupancy identifies transcription factor binding cooperativity in vivo. Mol. Cell 81, 1–13. doi: 10.1016/s0022-2836(02)00894-x
Spitz, F., and Furlong, E. E. M. (2012). Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 13, 613–626. doi: 10.1038/nrg3207
Spurrell, C. H., Barozzi, I., Mannion, B. J., Blow, M. J., Fukuda-Yuzawa, Y., Afzal, S. Y., et al. (2019). Genome-wide fetalization of enhancer architecture in heart disease. bioRxiv [Preprint]. doi: 10.1101/591362
Steimle, J. D., and Moskowitz, I. P. (2017). TBX5: a key regulator of heart development. Curr. Top. Dev. Biol. 122, 195–221.
Stephenson, A., Adams, J. W., and Vaccarezza, M. (2017). The vertebrate heart: an evolutionary perspective. J. Anat. 231, 787–797. doi: 10.1111/joa.12687
Tagle, D. A., Koop, B. F., Goodman, M., Slightom, J. L., Hess, D. L., and Jones, R. T. (1988). Embryonic ε and γ globin genes of a prosimian primate (Galago crassicaudatus). Nucleotide and amino acid sequences, developmental regulation and phylogenetic footprints. J. Mol. Biol. 203, 439–455. doi: 10.1016/0022-2836(88)90011-3
Takeuchi, J. K., Mileikovskaia, M., Koshiba-Takeuchi, K., Heidt, A. B., Mori, A. D., Arruda, E. P., et al. (2005). Tbx20 dose-dependently regulates transcription factor networks required for mouse heart and motoneuron development. Development 132, 2463–2474. doi: 10.1242/dev.01827
Tanaka, M., Chen, Z., Bartunkova, S., Yamasaki, N., and Izumo, S. (1999). The cardiac homeobox gene Csx/Nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development 126, 1269–1280.
Thurman, R. E., Rynes, E., Humbert, R., Vierstra, J., Maurano, M. T., Haugen, E., et al. (2012). The accessible chromatin landscape of the human genome. Nature 489, 75–82.
Tolkin, T., and Christiaen, L. (2012). Development and Evolution of the Ascidian Cardiogenic Mesoderm. Amsterdam: Elsevier Inc.
Torosin, N. S., Anand, A., Golla, T. R., Cao, W., and Ellison, C. E. (2020). 3D genome evolution and reorganization in the Drosophila melanogaster species group. PLoS Genet. 16:e1009229. doi: 10.1371/journal.pgen.1009229
Tucker, N. R., Chaffin, M., Fleming, S. J., Hall, A. W., Parsons, V. A., Bedi, K. C., et al. (2020). Transcriptional and cellular diversity of the human heart. Circulation 142, 466–482.
Turton, N., Swan, R., Mahenthiralingam, T., Pitts, D., and Dykes, I. M. (2019). The functions of long non-coding RNA during embryonic cardiovascular development and its potential for diagnosis and treatment of congenital heart disease. J. Cardiovasc. Dev. Dis. 6:21. doi: 10.3390/jcdd6020021
van den Boogaard, M., Smemo, S., Burnicka-Turek, O., Arnolds, D. E., van de Werken, H. J. G., Klous, P., et al. (2014). A common genetic variant within SCN10A modulates cardiac SCN5A expression. J. Clin. Invest. 124, 1844–1852. doi: 10.1172/jci73140
van den Boogaard, M., Wong, L. Y. E., Tessadori, F., Bakker, M. L., Dreizehnter, L. K., and Wakker, et al. (2012). Genetic variation in T-box binding element functionally affects SCN5A / SCN10A enhancer Find the latest version. J. Clin. Invest. 122, 2519–2530. doi: 10.1172/jci62613
van der Lee, R., Correard, S., and Wasserman, W. W. (2020). Deregulated regulators: disease-causing cis variants in transcription factor genes. Trends Genet. 36, 523–539. doi: 10.1016/j.tig.2020.04.006
van Eif, V. W., Protze, S., Bosada, F. M., Yuan, X., Sinha, T., van Duijvenboden, K., et al. (2020). Genome-wide analysis identifies an essential human tbx3 pacemaker enhancer. Circ. Res. 127, 1522–1535. doi: 10.1161/circresaha.120.317054
van Ouwerkerk, A. F., Bosada, F., Liu, J., Zhang, J., van Duijvenboden, K., Chaffin, M., et al. (2020). Identification of functional variant enhancers associated with atrial fibrillation. Circ. Res. 127, 229–243. doi: 10.1161/circresaha.119.316006
van Ouwerkerk, A. F., Bosada, F. M., van Duijvenboden, K., Hill, M. C., Montefiori, L. E., Scholman, K. T., et al. (2019). Identification of atrial fibrillation associated genes and functional non-coding variants. Nat. Commun. 10, 1–14.
Vanoudenhove, J., Yankee, T. N., Wilderman, A., and Cotney, J. (2020). Epigenomic and transcriptomic dynamics during human heart organogenesis. Circ. Res. 127, E184–E209.
Veerman, C. C., Wilde, A. A. M., and Lodder, E. M. (2015). The cardiac sodium channel gene SCN5A and its gene product NaV1.5: role in physiology and pathophysiology. Gene 573, 177–187. doi: 10.1016/j.gene.2015.08.062
Verma, M., and Kumar, V. (2019). Single-Cell Epigenomics: Technology and Applications. Amsterdam: Elsevier Inc.
Verzi, M. P., McCulley, D. J., De Val, S., Dodou, E., and Black, B. L. (2005). The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol. 287, 134–145. doi: 10.1016/j.ydbio.2005.08.041
Vierstra, J., Lazar, J., Sandstrom, R., Halow, J., Lee, K., Bates, D., et al. (2020). Global reference mapping of human transcription factor footprints. Nature 583, 729–736. doi: 10.1038/s41586-020-2528-x
Vierstra, J., Rynes, E., Sandstrom, R., Zhang, M., Canfield, T., Hansen, R. S., et al. (2014). Mouse regulatory DNA landscapes reveal global principles of cis-regulatory evolution. Science 346, 1007–1012. doi: 10.1126/science.1246426
Vietri Rudan, M., Barrington, C., Henderson, S., Ernst, C., Odom, D. T., Tanay, A., et al. (2015). Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep. 10, 1297–1309. doi: 10.1016/j.celrep.2015.02.004
Villar, D., Berthelot, C., Aldridge, S., Rayner, T. F., Lukk, M., Pignatelli, M., et al. (2015). Enhancer evolution across 20 mammalian species. Cell 160, 554–566. doi: 10.1016/j.cell.2015.01.006
Visel, A., Minovitsky, S., Dubchak, I., and Pennacchio, L. A. (2007). VISTA Enhancer Browser–a database of tissue-specific human enhancers. Nucleic Acids Res. 35, D88–D92.
Visel, A., Taher, L., Girgis, H., May, D., Golonzhka, O., Hoch, R. V., et al. (2013). A high-resolution enhancer atlas of the developing telencephalon. Cell 152, 895–908. doi: 10.1016/j.cell.2012.12.041
Visel, A., Zhu, Y., May, D., Afzal, V., Gong, E., Attanasio, C., et al. (2010). Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature 464, 409–412. doi: 10.1038/nature08801
Waardenberg, A. J., Ramialison, M., Bouvere, R., and Harvey, R. P. (2014). Genetic networks governing heart development. Cold Spring Harb. Perspect. Med. 4, 1–24. doi: 10.1155/2017/4135956
Wamstad, J. A., Alexander, J. M., Truty, R. M., Shrikumar, A., Li, F., Eilertson, K. E., et al. (2012). Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell 151, 206–220. doi: 10.1016/j.cell.2012.07.035
Wang, F., Liu, D., Zhang, R.-R., Yu, L.-W., Zhao, J.-Y., Yang, X.-Y., et al. (2017). A TBX5 3′UTR variant increases the risk of congenital heart disease in the Han Chinese population. Cell Discov. 3, 1–13. doi: 10.1161/circulationaha.111.circulationaha.111.050245
Wang, Z., Cui, M., Shah, A. M., Tan, W., Liu, N., Bassel-Duby, R., et al. (2020). Cell-type-specific gene regulatory networks underlying murine neonatal heart regeneration at single-cell resolution. CellReports 33:108472. doi: 10.1016/j.celrep.2020.108472
Wang, D. Z., Valdez, M. R., McAnally, J., Richardson, J., Olson, E. N., and Da-Zhi, W. (2001). Myogenic bHLH and MEF2 Proteins Directly Regulate the Mef2c gene - 4623.Full.pdf. Development. Available online at: https://dev.biologists.org/content/128/22/4623.short
Watanabe, Y., Zaffran, S., Kuroiwa, A., Higuchi, H., Ogura, T., Harvey, R. P., et al. (2012). Fibroblast growth factor 10 gene regulation in the second heart field by Tbx1, Nkx2-5, and Islet1 reveals a genetic switch for down-regulation in the myocardium. Proc. Natl. Acad. Sci. U.S.A. 109, 18273–18280. doi: 10.1073/pnas.1215360109
Wissink, E. M., Vihervaara, A., Tippens, N. D., and Lis, J. T. (2019). Nascent RNA analyses: tracking transcription and its regulation. Nat. Rev. Genet. 20, 705–723. doi: 10.1038/s41576-019-0159-6
Wong, E. S., Zheng, D., Tan, S. Z., Bower, N. L., Garside, V., Vanwalleghem, G., et al. (2020). Deep conservation of the enhancer regulatory code in animals. Science 370:eaax8137. doi: 10.1126/science.aax8137
Woolfe, A., Goodson, M., Goode, D. K., Snell, P., McEwen, G. K., Vavouri, T., et al. (2005). Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 3:e7. doi: 10.1371/journal.pbio.0030007
Xu, X., and Qi, L. S. (2019). A CRISPR–dCas toolbox for genetic engineering and synthetic biology. J. Mol. Biol. 431, 34–47. doi: 10.1016/j.jmb.2018.06.037
Yanai, I., Peshkin, L., Jorgensen, P., and Kirschner, M. W. (2011). Mapping gene expression in two Xenopus species: evolutionary constraints and developmental flexibility. Dev. Cell 20, 483–496. doi: 10.1016/j.devcel.2011.03.015
Yang, X. H., Nadadur, R. D., Hilvering, C. R., Bianchi, V., Werner, M., Mazurek, S. R., et al. (2017). Transcription-factor-dependent enhancer transcription defines a gene regulatory network for cardiac rhythm. eLife 6:e31683.
Yao, Y., Minor, P. J., Zhao, Y.-T., Jeong, Y., Pani, A. M., King, A. N., et al. (2016). Cis-regulatory architecture of a brain signaling center predates the origin of chordates. Nat. Genet. 48, 575–580. doi: 10.1038/ng.3542
Yuan, X., Song, M., Devine, P., Bruneau, B. G., Scott, I. C., and Wilson, M. D. (2018). Heart enhancers with deeply conserved regulatory activity are established early in zebrafish development. Nat. Commun. 9:4977.
Zaret, K. S. (2020). Pioneer transcription factors initiating gene network changes. Annu. Rev. Genet. 54, 367–385. doi: 10.1146/annurev-genet-030220-015007
Zeitlinger, J. (2020). Seven myths of how transcription factors read the cis-regulatory code. Curr. Opin. Syst. Biol. 23, 22–31. doi: 10.1016/j.coisb.2020.08.002
Zentner, G. E., Tesar, P. J., and Scacheri, P. C. (2011). Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 21, 1273–1283. doi: 10.1101/gr.122382.111
Zhang, F., and Lupski, J. R. (2015). Non-coding genetic variants in human disease. Hum. Mol. Genet. 24, R102–R110.
Zhang, Y., Li, T., Preissl, S., Amaral, M. L., Grinstein, J. D., Farah, E. N., et al. (2019). Transcriptionally active HERV-H retrotransposons demarcate topologically associating domains in human pluripotent stem cells. Nat. Genet. 51, 1380–1388. doi: 10.1038/s41588-019-0479-7
Zhou, H., Liu, J., Zhou, C., Gao, N., Rao, Z., Li, H., et al. (2018). In vivo simultaneous transcriptional activation of multiple genes in the brain using CRISPR-dCas9-activator transgenic mice. Nat. Neurosci. 21, 440–446. doi: 10.1038/s41593-017-0060-6
Zhou, P., Gu, F., Zhang, L., Akerberg, B. N., Ma, Q., Li, K., et al. (2017). Mapping cell type-specific transcriptional enhancers using high affinity, lineage-specific Ep300 bioChIP-seq. eLife 6:e22039.
Keywords: gene regulation, cardiac gene expression, transcription factor (TF), epigenomics and epigenetics, comparative genomics, enhancer
Citation: Yuan X, Scott IC and Wilson MD (2021) Heart Enhancers: Development and Disease Control at a Distance. Front. Genet. 12:642975. doi: 10.3389/fgene.2021.642975
Received: 17 December 2020; Accepted: 29 January 2021;
Published: 10 March 2021.
Edited by:
Mayra Furlan-Magaril, National Autonomous University of Mexico, MexicoReviewed by:
Mikhail Spivakov, Babraham Institute (BBSRC), United KingdomKoen Prange, University of Amsterdam, Netherlands
Copyright © 2021 Yuan, Scott and Wilson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuefei Yuan, eHVlZmVpLnl1YW5AdW5pLWhlaWRlbGJlcmcuZGU=; Ian C. Scott, aWFuLnNjb3R0QHNpY2traWRzLmNh; Michael D. Wilson, bWljaGFlbHdpbHNvbkBzaWNra2lkcy5jYQ==
†Present address: Xuefei Yuan, Center for Molecular Biology, Heidelberg University (ZMBH), Heidelberg, Germany