
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 02 July 2021
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.637096
 Luhang Cai1†Ya Liu1†
Luhang Cai1†Ya Liu1† Yaping Xu1*Hang Yang2Lihui Lv3
Yaping Xu1*Hang Yang2Lihui Lv3 Yang Li4Qiongqiong Chen1Xiaojiang Lin5Yihui Yang6Guangwei Hu7Guofeng Zheng8Jing Zhou9Qiyong Qian10Mei-ai Xu11Jin Fang12Jianjun Ding13Wei Chen1Jiong Gao14
Yang Li4Qiongqiong Chen1Xiaojiang Lin5Yihui Yang6Guangwei Hu7Guofeng Zheng8Jing Zhou9Qiyong Qian10Mei-ai Xu11Jin Fang12Jianjun Ding13Wei Chen1Jiong Gao14Purpose: The conventional genetic screening for deafness involves 9–20 variants from four genes. This study expands screening to analyze the mutation types and frequency of hereditary deafness genes in Zhejiang, China, and explore the significance of in-depth deafness genetic screening in newborns.
Methods: This was a multi-centre study conducted in 5,120 newborns from 12 major hospitals in the East-West (including mountains and islands) of Zhejiang Province. Concurrent hearing and genetic screening was performed. For genetic testing, 159 variants of 22 genes were screened, including CDH23, COL11A1, DFNA5, DFNB59, DSPP, GJB2, GJB3, KCNJ10, MT-RNR1, MT-TL1, MT-TS1, MYO15A, MYO7A, OTOF, PCDH15, SLC26A4, SOX10, TCOF1, TMC1, USH1G, WFS1, and WHRN using next-generation sequencing. Newborns who failed to have genetic mutations or hearing screening were diagnosed audiologically at the age of 6 months.
Results: A total of 4,893 newborns (95.57%) have passed the initial hearing screening, and 7 (0.14%) have failed in repeated screening. Of these, 446 (8.71%) newborns carried at least one genetic deafness-associated variant. High-risk pathogenic variants were found in 11 newborns (0.21%) (nine homozygotes and two compound heterozygotes), and eight of these infants have passed the hearing screening. The frequency of mutations in GJB2, GJB3, SLC26A4, 12SrRNA, and TMC1 was 5.43%, 0.59%, 1.91%, 0.98%, and 0.02%, respectively. The positive rate of in-depth screening was significantly increased when compared with 20 variants in four genes of traditional testing, wherein GJB2 was increased by 97.2%, SLC26A4 by 21% and MT-RNR1 by 150%. The most common mutation variants were GJB2c.235delC and SLC26A4c.919-2A > G, followed by GJB2c.299_300delAT. Homoplasmic mutation in MT-RNR1 was the most common, including m.1555A > G, m.961T > C, m.1095T > C. All these infants have passed routine hearing screening. The positive rate of MT-RNR1 mutation was significantly higher in newborns with high-risk factors of maternal pregnancy.
Conclusion: The positive rate of deafness gene mutations in the Zhejiang region is higher than that of the database, mainly in GJB2c.235delC, SLC26A4 c.919-2A > G, and m.1555A > G variants. The expanded genetic screening in the detection rate of diseasecausing variants was significantly improved. It is helpful in identifying high-risk children for follow-up intervention.
Deafness is one of the most common birth defects in humans, often causing lifelong disability and seriously affecting the quality of life of individuals. According to WHO in 2018, more than 5% of the world’s total population (466 million) suffer from some form of disabling hearing loss and 34 million of these are children. 1.1 out of every 1,000 newborns suffer from hearing impairment (Butcher et al., 2019). It is estimated that over 900 million people will have disabling hearing loss by 2050 (World Health Organization, 2018). According to the 2012 China Birth Defect Prevention Report, hearing impairment has become the second largest birth defect in China, with an incidence rate of 1–3‰ (Qin and Zhu, 2013), and about 30,000 newborns with severe hearing impairment are born every year (Xin et al., 2013). Therefore, it is important to carry out genetic screening for deafness in newborns, as this assists in detecting it as soon as possible and provides suitable intervention in time.
As early as 2019, Shearer et al. (2019) recommended deafness gene testing and believed that in the next decade, genome sequencing will become a supplement to universal physiological neonatal hearing screening. Currently, the combined screening of newborn hearing and deafness genes aid in identifying hearing loss at birth, give early warning to delayed, progressive, and ototoxic-drug deafness, and effectively avoid the occurrence of hearing loss. However, the vast majority of deafness gene screening is aimed only at hot spot variants of few genes. Although these genes and loci cover most of the deafness gene pathogenic and likely pathogenic variants in individuals, there is still a high proportion of omissions. In some areas of China, large-scale neonatal gene screening has been conducted (Wang et al., 2011, 2019; Dai et al., 2019), while a large-scale multi-center neonatal genetic screening or exact epidemiological survey on the incidence of deaf children in Zhejiang Province, which has been a major economic province along the southeast coast of China, has not been carried out. This study conducted newborn hearing and genetic screening in 12 major hospitals and delivery institutions, including the mountainous areas and Zhoushan Archipelago of the Zhejiang region. A large number of previous studies have also focused on four genes (GJB2, GJB3, SLC26A, MT-RNR1) and 20 variants (GJB2:c.35delG, c.167delT, c.176_191del16, c.235delC, c.299_300delAT; GJB3:c.538C > T, c.547G > A; SLC26A:c.281C > T, c.589G > A, c.919-2A > G, c.1174A > T, c.1226G > A, c.1229C > T, c.1707 + 5G > A, c.1975G > C, c.2027T > A, c.2162C > T, c.2168A > G; MT-RNR1:m.1494C > T, m.1555A > G). This study conducted expanded screening of 159 variants of 22 deafness genes. We comprehensively analyzed the genotypes, carrying spectrum, and frequency of deafness pathogenic/likely pathogenic variants in newborns of Zhejiang province, hoping to establish a database of disease-causing variants with regional characteristics to lay a foundation for the establishment of a database of deafness genes in China.
The study was a prospective and multi-center study of 5,120 newborns from maternal and children’s health and general hospitals (including mountains and islands) in Zhejiang Province from January 2018 to December 2019. A total of 12 regional hospitals included Hangzhou, Fenghua, Jiaxing, Kaihua, Jiangshan, Linhai, Ningbo, Ruian, Sanmen, Shaoxing, Zhoushan, and Shengzhou. Medical history and physical examination of the participating subjects were collected, including detailed family history, drug exposure history, and potential deafness factors.
This study was carried out with the authorization of the Hospital Ethics Committee of The First Affiliated Hospital, Zhejiang University School of Medicine. The participating subjects signed informed consent forms before sample collection and agreed that the sequencing data could be used for research after anonymization.
At least three dried blood spots were collected from the heels of the newborns on the third day after birth. When the samples were sent to the laboratory, the appearance and information of the samples were checked, and then into the laboratory information management system for establishing a unified blood card, questionnaire, and information file.
Initial screening was performed using transiently evoked otoacoustic emission (TEOAE) testing. TEOAE was performed 48 h after birth. For those referred (hearing screening failed) after initial testing, a repeat TEOAE test was performed by the age of 42 days and diagnosed audiologically at the age of 3 and 6 months.
Deafness genetic screening was used to identify 159 variants in 22 genes, including CDH23, COL11A1, DFNA5, DFNB59, DSPP, GJB2, GJB3, KCNJ10, MT-RNR1, MT-TL1, MT-TS1, MYO15A, MYO7A, OTOF, PCDH15, SLC26A4, SOX10, TCOF1, TMC1, USH1G, WFS1, and WHRN.
Genomic DNA was extracted from the blood samples using the Nucleic Acid Purification kit (BGI, Shenzhen, China). Multiplex PCR was used to amplify the target sequence of enriched human genomic DNA and introduce the sample tag sequence for sample identification. After purification and physical interruption, the PCR products were flattened at the end and added “A” at the 3′ end to complete the connection of the special connector at the two ends of the PCR product under the action of DNA polymerase, ligase, and phosphatase, and the preparation of the library was completed by magnetic bead purification.
We then carried out quality inspection and pooling of the library. The DNA double strands were thermally denatured into single strands after pooling, followed by the addition of the cyclic buffer and ligase to prepare circular DNA according to the cyclization reaction. The circular DNA molecules were used to make DNA nanoballs (DNBs) by rolling-circle replication (RCR). The concentration of DNBs was determined on a Qubit® 2.0 fluorometer using the QubitTM ssDNA Assay kit (Thermo Fisher Scientific, Waltham, MA, United States), and a DNB concentration within the range of 8–40 ng/μL was considered ideal. The DNBs were loaded onto chips and sequenced on the MGISEQ-2000 sequencing platform (BGI, Shenzhen, China). Figure 1 indicates the overall technical route.
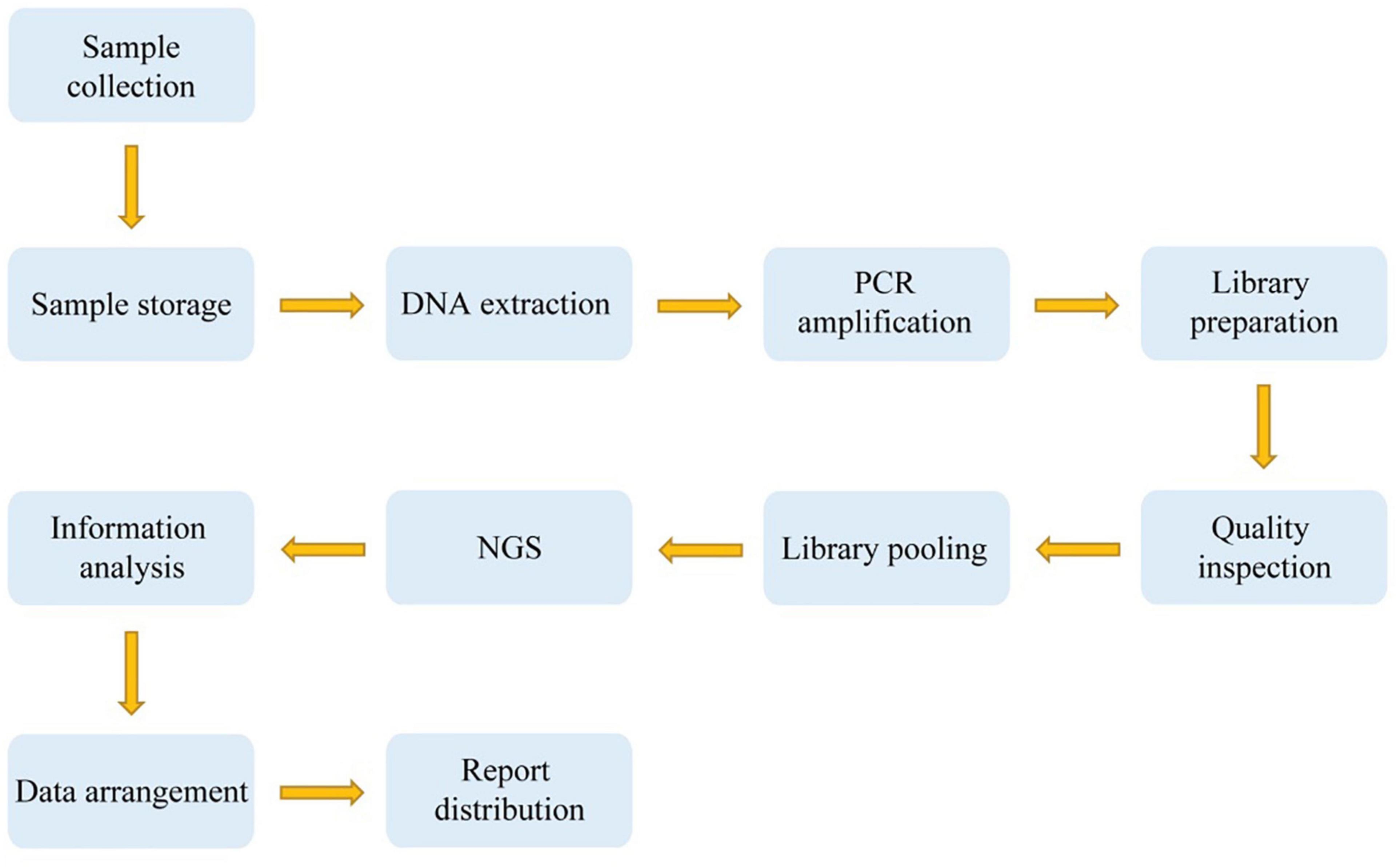
Figure 1. A technical route of deafness genetic screening.
The align sequence reading was compared with the human reference genome (hg19) and variants were called using the GATK software package and a localized database based on Human Gene Mutation Database, 2020. Further annotation was obtained using data from Human Gene Mutation Database,1 OMIM,2 PubMed,3 GeneReviews,4 dbSNP,5 1000 Genomes Project,6 the National Heart, Lung, and Blood Institute Exome Sequencing Project,7 the National Center for Biotechnology Information Reference Sequence Database,8 and the UCSC genome browser,9 as well hearing loss-specific databases such as the Hereditary Hearing Loss Homepage.10
For the positive samples detected by high-throughput sequencing, mass spectrometry or Sanger sequencing would be used for secondary verification, and the results of these two detection methods should be compared. If the results were consistent, these two detection results would be written into the reports. When the results of the two methods were inconsistent, we arranged high-throughput sequencing and Sanger sequencing for re-verification and wrote a report based on the results of these two rounds of testing (Figure 1).
All data is statistically analyzed using SPSS 20.0 software. Pearson chi-square test was used for comparison among the samples. The 95% confidence interval of frequency of genes was calculated. P < 0.05 was considered statistically significant.
A total of 5,120 neonatal babies were enrolled in the study, including 2,640 male (51.56%) and 2,480 female (48.44%) participants. In these cases, 2,611 mothers were primiparas (51%) and 2,509 mothers were nulliparous women (49%); 4,570 full term (89.26%), 264 premature delivery (5.16%), and 286 overdue delivery (5.58%); 2,974 natural delivery (58.09%) and 2,146 cesarean sections (41.91%). As Table 1 showed, 792 newborns (15.47%) were born with high-risk factors by neonatal screening. The highest proportion that occurred was hyperbilirubinemia, followed by premature delivery (Table 1).
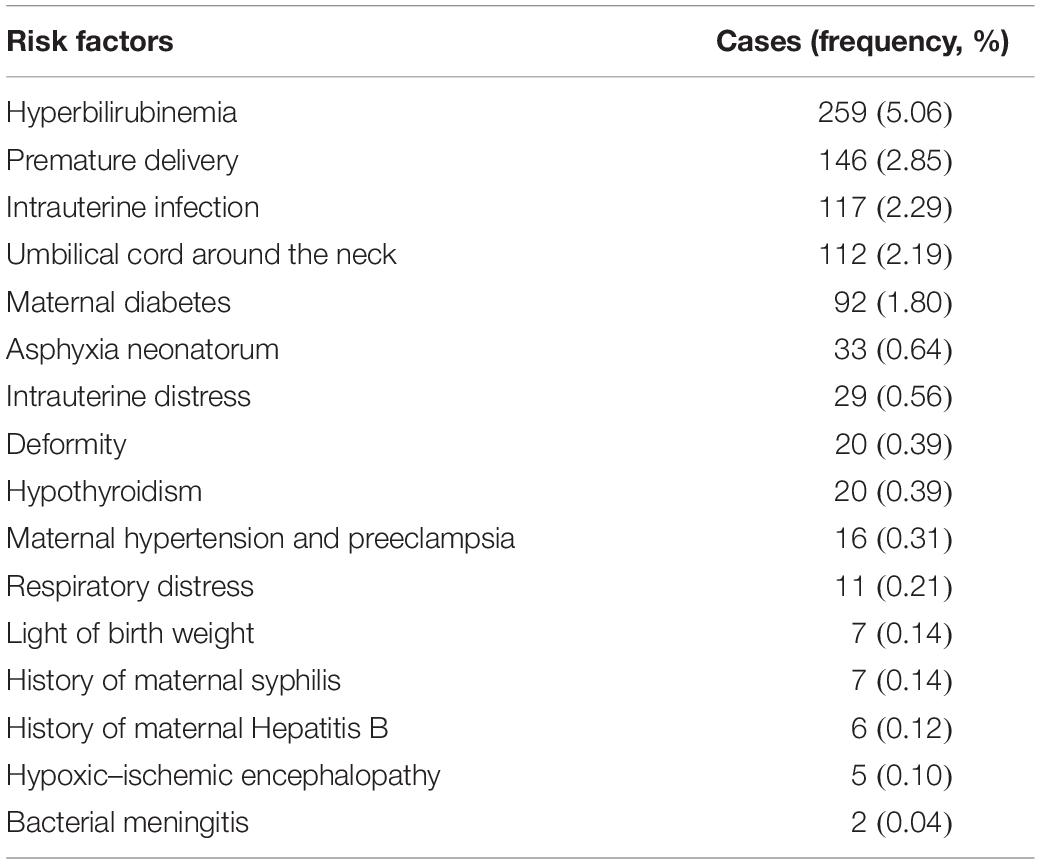
Table 1. Main risk factors on birth.
Of the 5,120 newborns, 227 neonates (4.43%) were referred bilaterally or unilaterally for the initial screening, and seven subsequent neonates (0.14%) failed in repeat screening at the age of 3 and 6 months. Three neonates (4.51%) were positive for genetic screening. At follow-up at 32 months, one case received a cochlear implant with profound hearing loss at one year, but he had not carried the common deafness genes. Three cases had normal hearing speech (one case had speech retardation). one case with homozygous variant c.[235del] in GJB2 was referred bilaterally for hearing screening and developed moderate hearing loss, but her linguistic competence was normal. Two cases who were referred for hearing screening for two steps had normal linguistic competence, carried with c.[235del] heterozygote in GJB2 and c.920C > T heterozygote in SLC26A4.
In total, 446 (8.71%) of the 5,120 newborns carried at least one genetic deafness-associated variant. Table 2 shows the 95% confidence interval of frequency of genes (Table 2). The probability of the genetic variants occurring in these newborns was as follows.
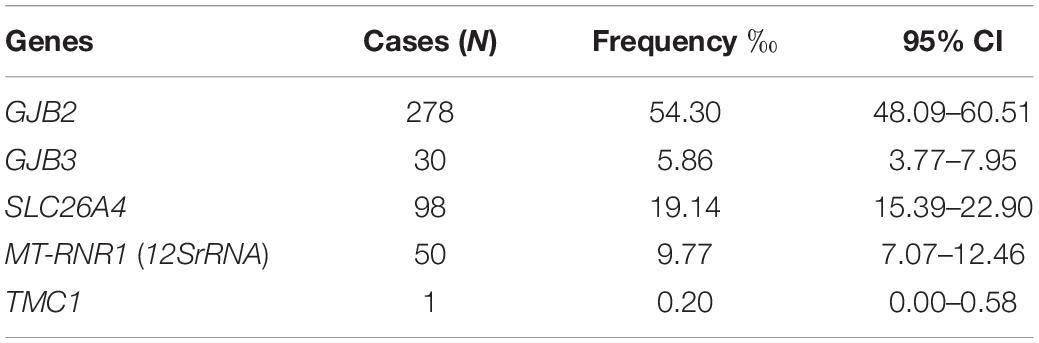
Table 2. Results of genetic screening.
1. For GJB2 gene: 271 heterozygotes (5.29%) and 7 homozygotes, among which nine cases (3.24%, 9/278) carried pathogenic deafness-associated variants, including seven homozygotes and another two cases with two variants identified (phase unknown) (c.[109G > A] + [299_300delAT] and c.[235delC] + [257C > G]) (Table 3).
2. For GJB3 gene: 30 heterozygotes (0.59%), including 16 c.[538C > T] and 14 c.[547G > A]. No cases carried pathogenic variants.
3. For SLC26A4 gene: 96 heterozygotes (1.88%) and 2 homozygotes (0.04%). The 2 homozygotes were identified to carry pathogenic alleles (homozygotes, c.[1229C > T] + [1229C > T], c.[2168A > G] + [2168A > G]) (Table 4).
4. For MT-RNR1: 26 heteroplasmic variants (0.51%) and 24 homoplasmic variants (0.47%) including m.[1555A > G], m.[961T > C], and m.[1095T > C] (Table 5).
5. For TMC1: only one case was c.100C > T heterozygotes carrier. Followed-up by 30 months, the baby had normal hearing and speech.
6. There were 11 multilocus heterozygous variants of more than one gene. (0.22%, 11/5,120). All children with these two genes have passed hearing screening (Table 6).

Table 3. 278 GJB2 variants and genotypes.
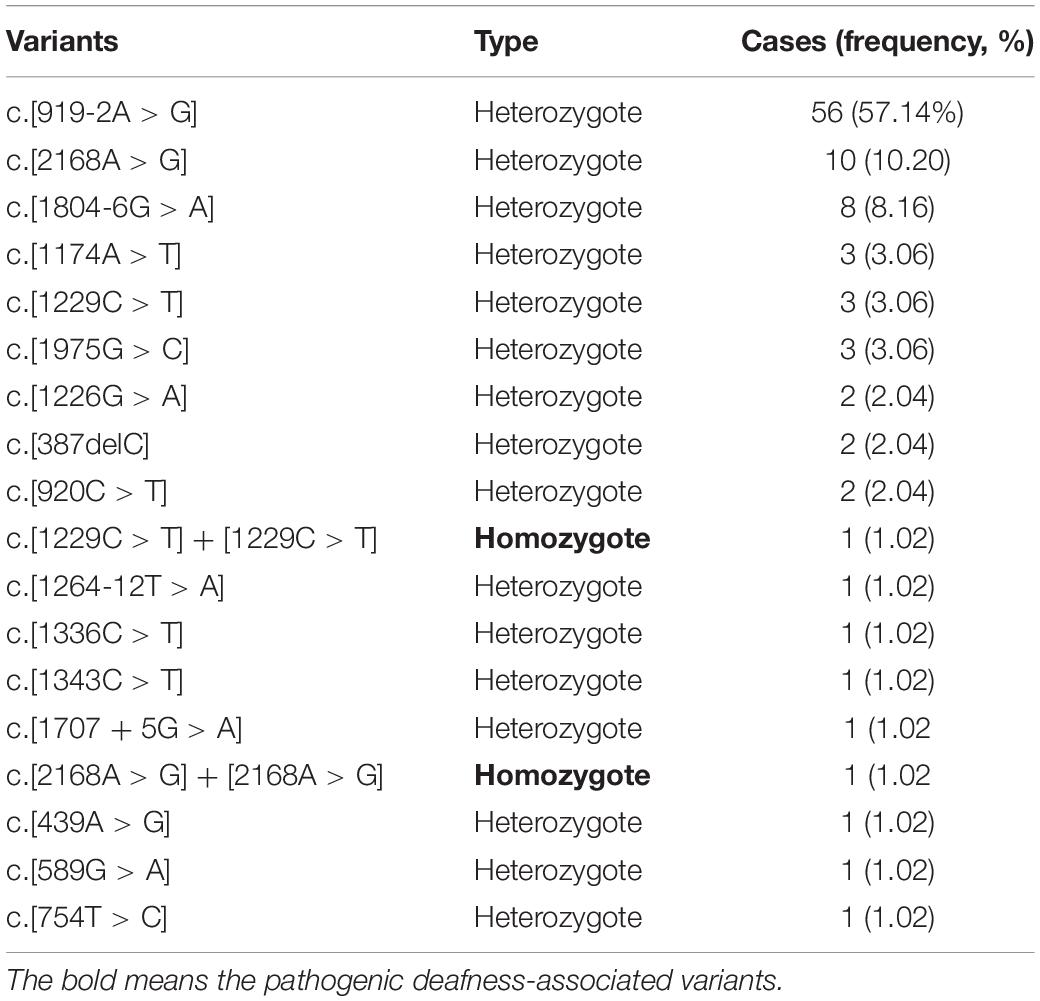
Table 4. 98 SLC26A4 variants and genotypes.

Table 5. Fifty MT-RNR1 variants and genotypes.

Table 6. Multilocus variation.
Compared with 20 variants in the four genes of traditional testing, the positive rate of expanded screening in 159 variants of 22 deafness related genes increased significantly (65.2% on average), including GJB2 increasing by 97.2%, SLC26A4 by 21%, and MT-RNR1 by 150%. It is suggesting that it was helpful to improve the clinical efficacy of deafness-related gene screening by increasing the scope of screening (Table 7).
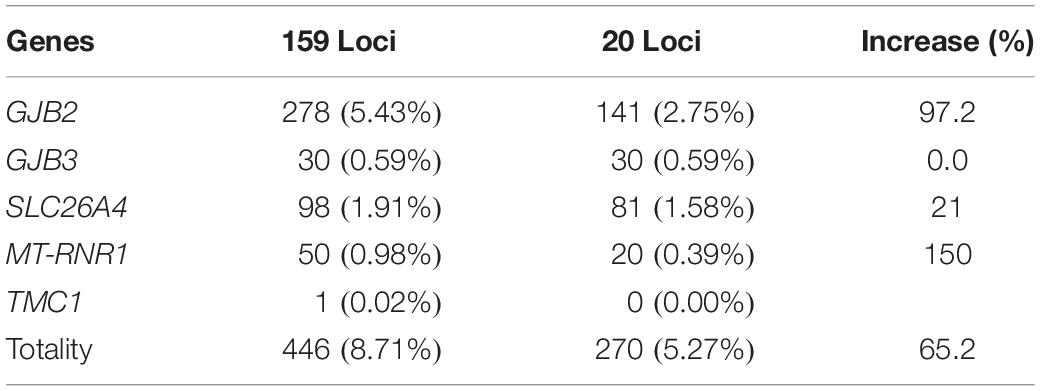
Table 7. Comparison of genetic variants by expanded and conventional screening.
By comparing present literature and gnomAD available data we found that: (1) Among the GJB2 variants, the incidence rate of c.[235delC] was the highest, followed by c.[299_300delAT]. There were 115 cases with c.[109G > A], accounting for 2.25%, which was significantly lower than 8.35% in the East Asian population. (2) Among the SLC26A4 variants, the incidence rate of c.[919-2A > G] was the highest, which was significantly higher than that in other regions. There were 7 cases with c.[1804-6G > A], accounting for 0.16%, which was lower than 0.29% of the East Asian population. (3) Among the GJB3 variants, the incidence rate of the two hot spot variants (c.[538C > T] and c.[547G > A]) was higher than that of the literature reports. (4) Among the MT-RNR1 variants, the incidence rate of m.[1555A > G] was more than data in China (Wang et al., 2019). All of them had statistical differences by one-way ANOVA (P < 0.05) (Table 8).
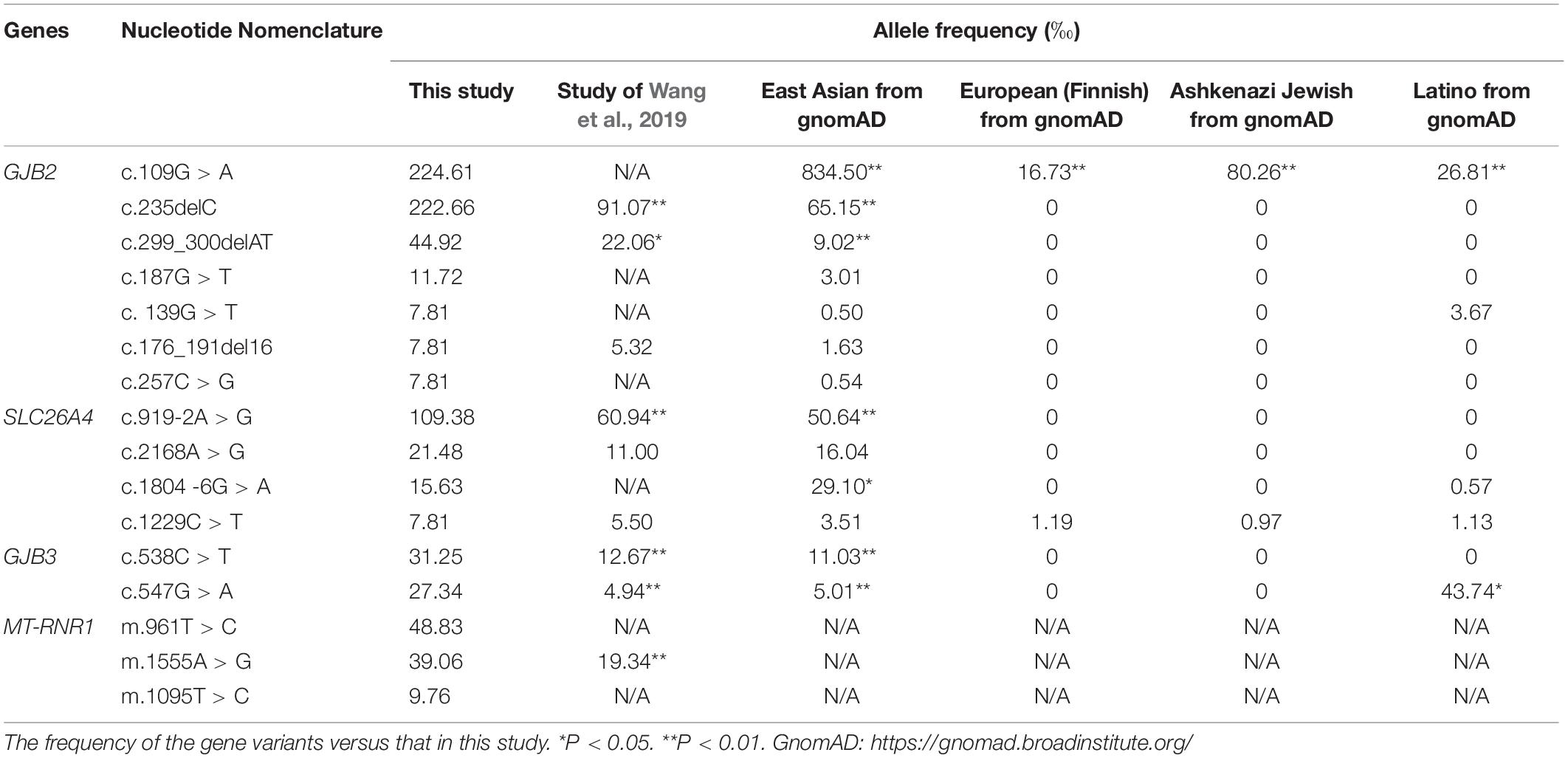
Table 8. The frequency of the pathogenic/likely pathogenic variants (more than four loci) in this study.
Subsequently, we focused on the high-risk pathogenic variants in 11 newborns (0.21%, 11/5,120). Among these genes, nine cases (homozygotes and complex heterozygotes) in GJB2 and 2 homozygotes in SLC26A4 were identified. Though most of them (72.72%, 8/11) have passed hearing screening, further follow-up and a diagnostic audiological test are crucial methods for delayed hearing disorder. Twenty-four cases were homoplasmic variants in MT-RNR1 who had a risk of deafness induced by ototoxic drugs, also had high-risk factors of pregnancy (Figure 2).
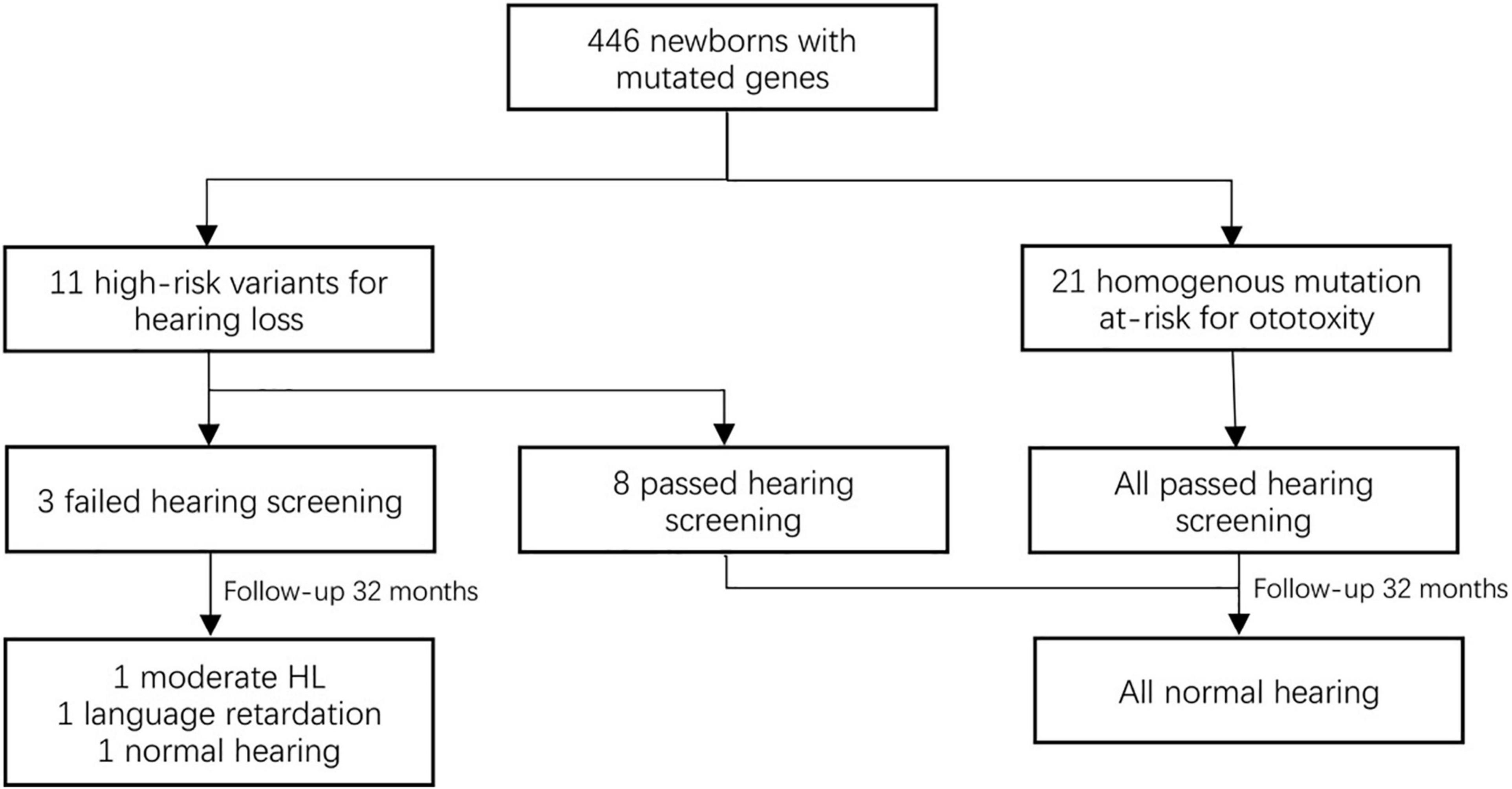
Figure 2. The hearing outcomes of newborns with positive genotypes.
In order to comprehensively evaluate the regional characteristics of deafness-related variants in different regions of Zhejiang province, we analyzed genetic variants between the remote mountain areas (971 cases) and island areas (500 cases) and found no statistical differences in positive rates of GJB2, GJB3, SLC26A4, and MT-RNR1 genes, P > 0.05.
The positive rates of GJB3, SLC26A4, and MT-RNR1 were not statistically different in every region. However, the frequency of GJB2 showed a statistical difference in every region, P < 0.05. Interestingly, it was significantly higher in Riuan and Fenghua (Figure 3).
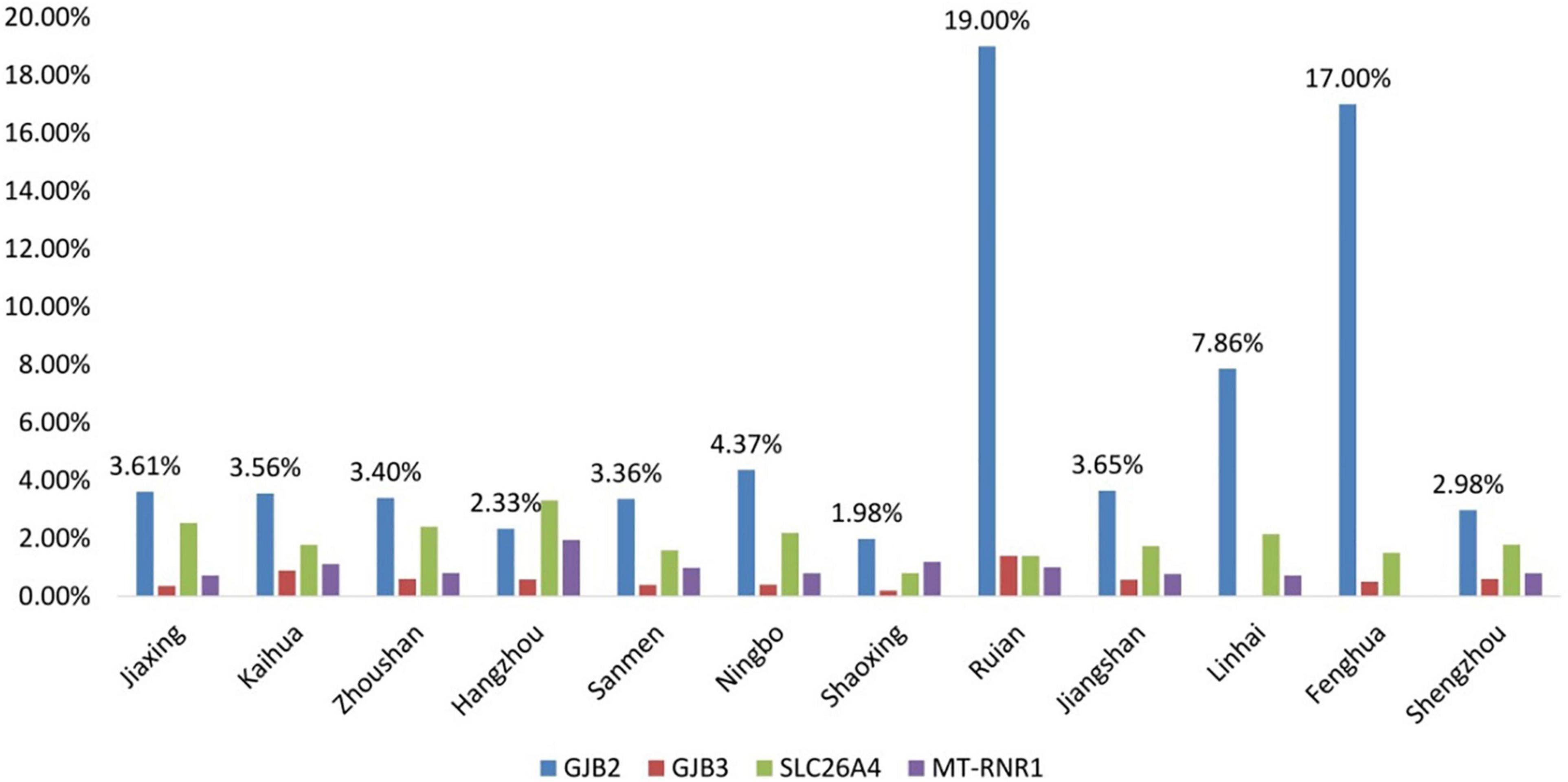
Figure 3. Comparison of the positive rates of genetic screening in 12 regional hospitals of Zhejiang Province.
The criteria for high-risk pregnancies are: (1) Pregnancy-related complications during pregnancy, or complications of chronic diseases such as hypertension and diabetes, (2) Mother-related complications during childbirth, (3) Premature expired or twin (multiple) pregnancies, and (4) Perinatal complications or malformations in newborns.
There were high-risk factors in 792 newborns (15.5%). The positive rate of the variant in MT-RNR1 was significantly higher in neonates with maternal pregnancy risk factors, P < 0.05. There was no significant difference in no high-risk factors, hyperbilirubinemia, and non-hyperbilirubinemia.
The positive rate of the variant in GJB2 was significantly lower in newborns with high-risk factors for maternal pregnancy, P < 0.001, especially those with non-hyperbilirubinemia (x2 = 15.416, P < 0.001) (Figure 4).
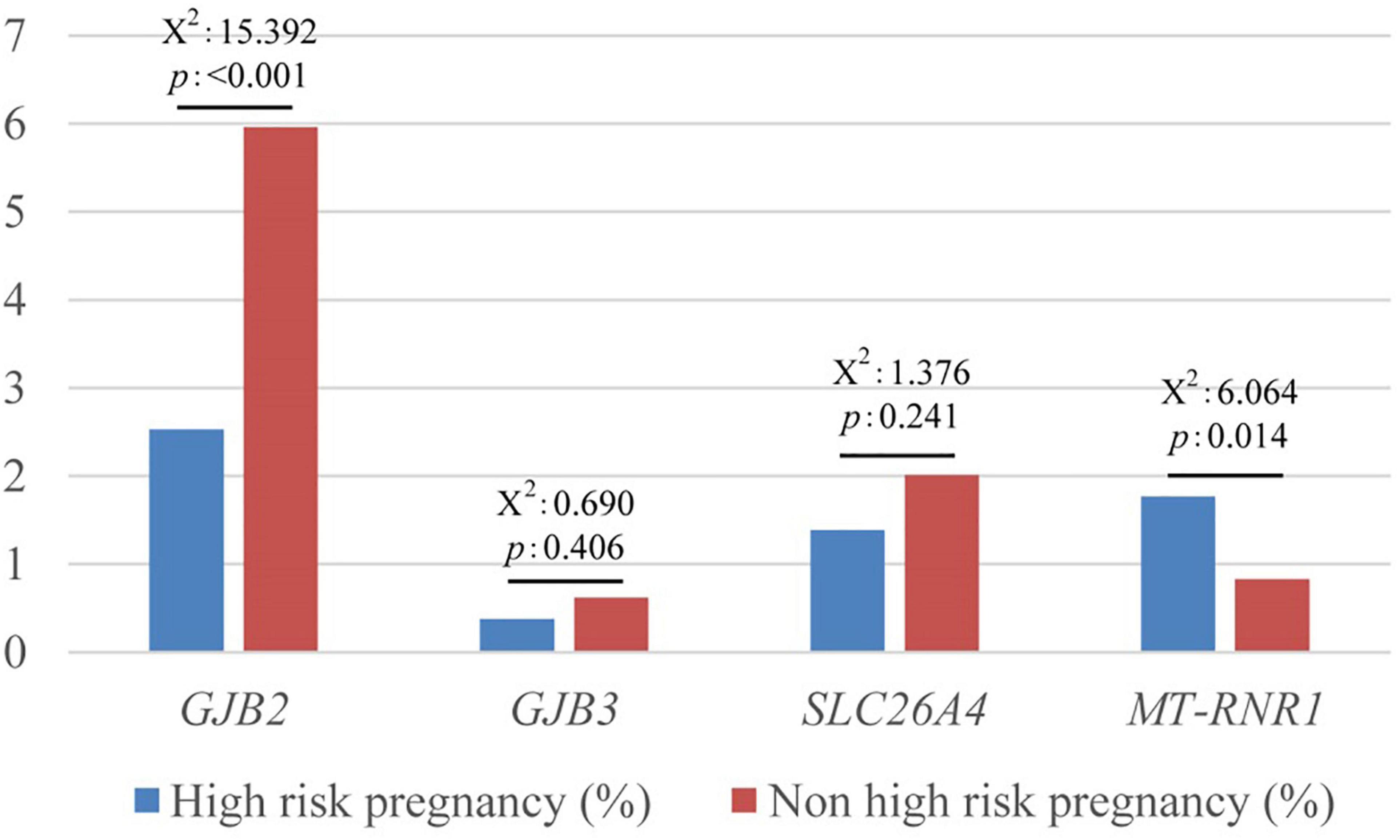
Figure 4. Comparison of high-risk and non-high-risk pregnancy in deafness gene mutations.
Since Wang et al. (2011) first conducted multi-center large-scale neonatal hearing and deafness genetic screening in China in 2011, a large number of related studies have also been carried out in South China, Southwest China, North China, Taiwan, and other regions of China to provide abundant epidemiological data concerning the distribution and frequency of major genetic variants in China (Chu et al., 2015; Chen et al., 2019; Dai et al., 2019; Guo et al., 2020).
According to our study, of all the 446 newborns with positive deafness genes, 176 cases carried 20 loci in four genes, and more disease-causing variants have been detected. The overall positive rate increased by 65.2%. The positive rates of GJB2, SLC26A4, and MT-RNR1 variants were increased by 97.2, 21, and 150%, respectively. Based on a meta-analysis conducted in China in 2019, there was a positive rate of 4.3% for 9 loci and 4.7% for 20 loci (Chen et al., 2019). Through expanded screening of deafness genes at 159 loci of 22 genes, the positive screening rate could be significantly increased.
Based on the study, high-risk pathogenic variants were found in 11 newborns (0.21%), most of them (90.91%) passed hearing screening. Further follow-up and diagnostic audiological tests are very important for a delayed hearing disorder.
GJB2 encodes gap junction protein Cx26 and has been shown to be the main cause of congenitally severe or profound sensorineural deafness. There are significant differences in frequency and variants among different races and populations (Jiang et al., 2016). In Caucasian people, more than 50% of GJB2 pathogenic/likely pathogenic variants were found at c.35delG, about 70% were found in humans of Northern and Southern Europe, and at 167delT in German Jewish populations (Gasparini et al., 2000; Sugata et al., 2002). However, c.235delC is the most common variant in Asian populations (Kudo et al., 2001; Liu et al., 2002). The c.235delC could lead to frameshift variants and premature termination of Cx26 protein (Martínez et al., 2009). This study revealed c.235delC as the first mutant in GJB2, accounting for 40.74%, followed by c.299_300delAT, and the positive rate was higher than that in the East Asian population and gene pool data (Van Camp and Smith, 2020).
The results found that GJB2 c.[109G > A] accounted for 2.25% of the total population, which was significantly lower than 8.35% in an East Asian population (Van Camp and Smith, 2020). This locus showed a significant difference in different regions and is rarely reported in the southeast coastal areas of China; while c.109G > A is frequently reported in patients with sensorineural deafness in the Hainan region of China (Huang et al., 2015). Homozygote or compound heterozygote in c.109G > A have been reported in a normal population, and whether this locus is pathogenic remains controversial (Chen et al., 2014). Xiao et al. (2017) have believed that c.109G > A might be related to delayed deafness. Previous cell experiments have proved that homozygote in c.109G > A can affect the adhesion between the expression product connexin, making it unable to form gap junction channels, leading to deafness. Thus, the variants might be related to mild to moderate deafness (Chen et al., 2016).
The positive rate of GJB2 variants is found to be the highest in Ruian and Fenghua regions, suggesting that the rate of GJB2 is high in certain regions.
In China, GJB2 c.[235delC] + [235delC] homozygotes were the most common, which accounted for 55.10% of GJB2 gene-related deafness, followed by 235delC heterozygote (20.40%), and 235delC/299-300delAT compound heterozygote (10.2%) (Zheng et al., 2000). The proportion of deafness caused by GJB2 and common loci is different due to ethnic compositions and intermarriage within the ethnic groups in the northern and southern regions of Iran (Koohiyan et al., 2020). Some heterozygotes of GJB2 can also cause deafness (Dai et al., 2019). GJB2c.551G > A (p. Arg184Gln) heterozygote is found in patients with deafness (Liu et al., 2020).
This study revealed two genetic patterns in GJB2, including GJB3 c.[538C > T] + GJB2 c.[109G > A] and GJB3 c.[547G > A] + GJB2 c.[176_191delGCAAGAACGTGTG]. Similar reports have shown GJB2/GJB6 (Cheng et al., 2005) and GJB2/GJB3 (Yuan et al., 2010) variants in 11% of deaf children, and genetic interaction between GJB2 and GJB3 in three unrelated families with autosomal recessive hereditary diseases (Liu et al., 2009). Therefore, the variant pattern with regard to hearing needs further follow-up study.
The SLC26A4 is located on chromosome 7q22.3 and is mainly expressed in the thyroid gland, inner ear, and kidney. The gene encodes Pendrin and is related to chlorine-iodine ion transportation (Scott et al., 1999). The variant leads to Pendred syndrome (PDS, congenital sensorineural deafness, cochlear abnormality, and goiter) and non-syndromic deafness with enlargement of the vestibular aqueduct (EVA). The main phenotype reported is prelingually or post-lingually progressive fluctuating hearing loss (Campbell et al., 2001).
Approximately 4–17% of deafness in patients in China is caused by SLC26A4 variants, followed by the GJB2 gene. The majority of the subjects (i.e., 81.81%) with EVA were detected with 919-2A > G (Qing et al., 2015). Up to now, 587 variants of SLC26A4 have been found.11 c.919-2A > G is shown to be the most common variant of SLC26A4 in the Chinese population, accounting for 57.63% of the total variants (Dai et al., 2020). This study found that c.919-2A > G has been the first to be observed, accounting for 56.99%, and the positive rate was higher than reported in Wang’s study and Southeast Asian data (Wang et al., 2019; Van Camp and Smith, 2020). SLC26A4 c.[1804-6G > A] accounted for 0.16% of the total population, which is significantly lower than 0.29% (Van Camp and Smith, 2020) in East Asian populations. This reflects the regional characteristics of the SLC26A4 variant spectrum in this region.
The two pathogenic sites of SLC26A4 are c.[1229C > T] + [1229C > T] and c.[2168A > G] + [2168A > G]. The positive rate of these is similar to that of the gene pool (Van Camp and Smith, 2020). It has been found that SLC26A4, GJB2, and GJB3 have double gene inheritance (Van Camp and Smith, 2020). No studies have reported this information, and it should be explored further in future studies. It has been reported that EVA might also be caused by the heterozygote of SLC26A4 and FOXI1, or double gene inheritance of SLC26A4 and KCNJ10, which is the first example where double gene inheritance has been proven to be the cause of deafness in human beings (Naseri et al., 2018).
Variants in the mitochondrial gene MT-RNR1 can lead to genetic susceptibility to aminoglycoside-induced hearing loss. At present, it is believed that a homoplasmic variant of the mtDNA12SrRNA gene is closely related to aminoglycoside antibiotic-induced deafness. Aminoglycoside drugs can accumulate in the cochlea and the vestibule and combine with 12SrRNA, resulting in cell damage, apoptosis, and hearing loss. Previous studies have found that the MT-RNR1genotype demonstrated extensive heterogeneity, and MT-RNR1 variants in different members of the same family can also have different penetrance rates. Some mutants carriers were also associated with mild to moderate sensorineural deafness without using aminoglycoside antibiotics (Barbarino et al., 2016). The variant modes are homozygotes and heterozygotes. The main genetic types include A155G, C1494T, T1095C, 961delC, A827G in 12SrRNA and A7445G, G7444A, 7472insG, T7510C, and T7511C variants in COI/tRNASer (UCN) gene (Jacobs et al., 2005). According to previous Japanese studies, the carrying rate of m.1555A > G in sporadic neurosensorial hearing loss cases is about 3% (Mutai et al., 2017). The frequency in Africa is 2.4% (Tingang et al., 2019), and in eastern China it is 5.95%, which is significantly higher than that in other regions (Sun et al., 2019).
In this study, 21 cases are homoplasmic variants and are potentially sensitive to aminoglycoside antibiotics-induced deafness. The main locus is m.1555A > G accounted for 3.906‰ in the total sample, and the frequency is significantly higher than that reported in China, Europe, and Southeast Asia (Van Camp and Smith, 2020). The second is m.961T > C and m.1095T > C. Due to maternal inheritance of MT-RNR1, the variants are relatively stable in the population, suggesting a high incidence of MT-RNR1 variants in this region. The positive rate of MT-RNR1 variants is found to be significantly higher in newborns with high-risk pregnancy factors (P < 0.05). No relevant literature has reported its correlation.
GJB3 is located on the human chromosome lp33-35 and encodes connexin 31 with 270 amino acids. Its variant is closely related to the gap connexin 31 (eonnexin31, Cx31) variant encoded by GJB3 (Xia et al., 1998). GJB3 was first cloned by Chinese scientist Xia Jiahui in 1998. The variants at c.538C > T and c.547G > A sites lead to autosomal dominant non-syndromic deafness, mainly resulting in high-frequency hearing loss (Xia et al., 1998). More than 10 different variants were found in deaf patients of Spain (López-Bigas et al., 2001), Turkey (Uyguner et al., 2003), and China (Xia et al., 1998; Liu et al., 2000, 2009). However, the pathogenicity of GJB3 is based only on reports with some cases and there is no available epidemiological incidence. Therefore, its pathogenicity is still controversial, and it is believed that it can lead to delayed deafness (Li et al., 2014). Two common disease-causing variants c.538C > T and c.547G > A were detected in this study, which is higher in proportion than that reported in the previous studies conducted in China and the data of the East Asian population (Van Camp and Smith, 2020). Under the condition that the pathogenicity of this gene cannot be excluded, it is necessary to strengthen the follow-up of this population.
In conclusion, the number of genes and loci for deafness screening was increased and the positive rate of screening is greatly improved in this study. Patients with genetic variants and normal hearing need to prolong the follow-up time for observing changes in hearing. Moreover, this study has defined the distribution range, genotypes, and pathogenic variants in Zhejiang province. It is expected to further expand the target range, more accurately and comprehensively analyze the spectrum and frequency of disease-causing variants, and establish a database of deafness gene variants carried with regional characteristics.
Even though this study involved in-depth screening of 159 variants in 22 deafness genes, it is impossible to cover all deafness-related variants or some new and rare loci. Genetic screening is not undertaken in all newborns in Zhejiang province. We will expand the screening population in the future and search for more regional characteristics of the pathogenic and likely pathogenic variants spectrum. For cases with high-risk deafness genes, it is necessary to further extend the follow-up time as later-onset hearing disorders may occur in.
The data presented in the study are deposited in the CNGB (db.cngb.org) repository, accession number CNP0001814.
The studies involving human participants were reviewed and approved by Research Ethics Committee of The First Affliated Hospital, College of Medcine, Zhejiang University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
YL and LC were responsible for the collection, collation, and statistics of the data, the drawing of the figures and forms, and retrieving of the references. YL was responsible for the gene detection, analyzing, and discussion. YX was corresponding author and responsible for designing, organization, analyzing, and discussion. WC was responsible for part of audiological examinations. The other authors were responsible for the collection and collation of samples and data. All authors contributed to the article and approved the submitted version.
This work was supported by a grant from Science and Technology Department of Zhejiang Province Government in China (Grant number: 2018C03027).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Zhang Jinglan and Jia Junling for their guidance on the research design and experimental process. We sincerely thank all of the patients and families who agreed to participate in this study. In addition, we would like to thank BGI-Shanghai for their technical support and the staff at all the hospitals listed in the article for their assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.637096/full#supplementary-material
Barbarino, J. M., McGregor, T. L., Altman, R. B., and Klein, T. E. (2016). PharmGKB summary: very important pharmacogene information for MT-RNR1. Pharmacogenet. Genomics 26, 558–567. doi: 10.1097/FPC.0000000000000247
Butcher, E., Dezateux, C., Cortina-Borja, M., and Knowles, R. L. (2019). Prevalence of permanent childhood hearing loss detected at the universal newborn hearing screen: systematic review and meta-analysis. PLoS One 14:e0219600. doi: 10.1371/journal.pone.0219600
Campbell, C., Cucci, R. A., Prasad, S., Green, G. E., Edeal, J. B., Galer, C. E., et al. (2001). Pendred syndrome, DFNB4, and PDS/SLC26A4 identification of eight novel mutations and possible genotype-phenotype correlations. Hum. Mutat. 17, 403–411. doi: 10.1002/humu.1116
Chen, S., Liang, Z., Chen, B., Yao, F., Huang, G., Zhu, J., et al. (2019). The prevalence of deafness-associated mutations in neonates: a meta-analysis of clinical trials. Int. J. Pediatr. Otorhinolaryngol. 121, 99–108. doi: 10.1016/j.ijporl
Chen, T., Jiang, L., Liu, C., Shan, H., Chen, J., Yang, B., et al. (2014). Update of the spectrum of GJB2 mutations in 107 patients with nonsyndromic hearing loss in the Fujian population of China. Ann. Hum. Genet. 78, 235–242. doi: 10.1111/ahg.12062
Chen, Y., Hu, L., Wang, X., Sun, C., Lin, X., Li, L., et al. (2016). Characterization of a knock-in mouse model of the homozygous p.V37I variant in Gjb2. Sci. Rep. 6:33279. doi: 10.1038/srep33279
Cheng, X., Li, L., Brashears, S., Morlet, T., Ng, S. S., Berlin, C., et al. (2005). Connexin 26 variants and auditory neuropathy/dys-synchrony among children in schools for the deaf. Am. J. Med. Genet. A. 139, 13–18. doi: 10.1002/ajmg.a.30929
Chu, C., Chen, Y., Lee, Y., Jaung, S., Lee, F., and Huang, H. (2015). Government-funded universal newborn hearing screening and genetic analyses of deafness predisposing genes in Taiwan. Int. J. Pediatr. Otorhinolaryngol. 79, 584–590. doi: 10.1016/j.ijporl.2015.01.033
Dai, P., Huang, L., Wang, G., Gao, X., Qu, C., Chen, X., et al. (2019). Concurrent hearing and genetic screening of 180,469 neonates with follow-up in Beijing, China. Am. J. Hum. Genet. 105, 803–812. doi: 10.1016/j.ajhg.2019.09.003
Dai, S., Wang, H., and Lin, Z. (2020). Novel and recurrent mutations in GJB3 and GJB4 cause erythrokeratodermia variabilis et progressiva. Indian J. Dermatol. Venereol. Leprol. 86, 87–90. doi: 10.4103/ijdvl.IJDVL_926_18
Gasparini, P., Rabionet, R., Barbujani, G., Melçhionda, S., Petersen, M., Brøndum-Nielsen, K., et al. (2000). High carrier frequency of the 35delG deafness mutation in European populations. Genetic analysis consortium of GJB2 35delG. Eur. J. Hum. Genet. 8, 19–23. doi: 10.1038/sj.ejhg.5200406
Guo, L., Xiang, J., Sun, L., Yan, X., Yang, J., Wu, H., et al. (2020). Concurrent hearing and genetic screening in a general newborn population. Hum. Genet. 139, 521–530. doi: 10.1007/s00439-020-02118-6
Human Gene Mutation Database (2020). Mutations of Solute Carrier Family 26 Member 4. Available online at: http://www.hgmd.cf.ac.uk/ac/gene.php?gene=SLC26A4 (accessed December 1, 2020).
Huang, Y., Yang, X., Chen, W., Duan, B., Lu, P., and Wang, Y. (2015). Prevalence of p.V37I variant of GJB2 among Chinese infants with mild or moderate hearing loss. Int. J. Clin. Exp. Med. 8, 21674–21678.
Jacobs, H. T., Hutchin, T. P., Käppi, T., Gillies, G., Minkkinen, K., Walker, J., et al. (2005). Mitochondrial DNA mutations in patients with postlingual, nonsyndromic hearing impairment. Eur. J. Hum. Genet. 13, 26–33. doi: 10.1038/sj.ejhg.5201250
Jiang, H., Shi, X., Qiu, S., Dong, Y., Qiao, Y., and Wei, D. (2016). A screening analysis of the GJB2 c.176 del 16 mutation responsible for hereditary deafness in a Chinese family. J. Otol. 11, 134–137. doi: 10.1016/j.joto.2016.09.002
Koohiyan, M., Koohian, F., and Azadegan-Dehkordi, F. (2020). GJB2-related hearing loss in central Iran: review of the spectrum and frequency of gene mutations. Ann. Hum. Genet. 84, 107–113. doi: 10.1111/ahg.12354
Kudo, T., Ikeda, K., Oshima, T., Kure, S., Tammasaeng, M., Prasansuk, S., et al. (2001). GJB2 (connexin 26) mutations and childhood deafness in Thailand. Otol Neurotol. 22, 858–861. doi: 10.1097/00129492-200111000-00025
Li, T., Kuan, Y., Ko, T., Li, C., and Yang, J. (2014). Mechanism of a novel missense mutation, p.V174M, of the human connexin31 (GJB3) in causing nonsyndromic hearing loss. Biochem. Cell Biol. 92, 251–257. doi: 10.1139/bcb-2013-0126
Liu, X., Wang, J., Wang, S., Li, S., Zhu, Y., Ding, W., et al. (2020). The mutation frequencies of GJB2, GJB3, SLC26A4 and MT-RNR1 of patients with severe to profound sensorineural hearing loss in northwest China. Int. J. Pediatr. Otorhinolaryngol. 136:110143. doi: 10.1016/j.ijporl.2020.110143
Liu, X., Xia, X., Ke, X., Ouyang, X., Du, L., Liu, Y., et al. (2002). The prevalence of connexin 26 (GJB2) mutations in the Chinese population. Hum. Genet. 111, 394–397. doi: 10.1007/s00439-002-0811-6
Liu, X., Xia, X., Xu, L., Pandya, A., Liang, C., Blanton, S., et al. (2000). Mutations in connexin31 underlie recessive as well as dominant non-syndromic hearing loss. Hum. Mol. Genet. 9, 63–67. doi: 10.1093/hmg/9.1.63
Liu, X., Yuan, Y., Yan, D., Ding, E., Ouyang, X., Fei, Y., et al. (2009). Digenic inheritance of non-syndromic deafness caused by mutations at the gap junction proteins Cx26 and Cx31. Hum. Genet. 125, 53–62. doi: 10.1007/s00439-008-0602-9
López-Bigas, N., Olivé, M., Rabionet, R., Ben-David, O., Martínez-Matos, J. A., Bravo, O., et al. (2001). Connexin 31 (GJB3) is expressed in the peripheral and auditory nerves and causes neuropathy and hearing impairment. Hum. Mol. Genet. 10, 947–952. doi: 10.1093/hmg/10.9.947
Martínez, A. D., Acuña, R., Figueroa, V., Maripillan, J., and Nicholson, B. (2009). Gap-junction channels dysfunction in deafness and hearing loss. Antioxid. Redox Signal. 11, 309–322. doi: 10.1089/ars.2008.2138
Mutai, H., Watabe, T., Kosaki, K., Ogawa, K., and Matsunaga, T. (2017). Mitochondrial mutations in maternally inherited hearing loss. BMC Med. Genet. 18:32. doi: 10.1186/s12881-017-0389-4
Naseri, M., Akbarzadehlaleh, M., Masoudi, M., Ahangari, N., Zonouzi, A. A. P., Zonouzi, A. P., et al. (2018). Genetic linkage analysis of DFNB4, DFNB28, DFNB93 loci in autosomal recessive non-syndromic hearing loss: evidence for digenic inheritance in GJB2 and GJB3 mutations. Iran J. Public Health 47, 95–102.
Qin, H., and Zhu, J. (2013). [Report on Prevention and Treatment of Birth Defects in China]. Zhong guo chu sheng que xian fang zhi bao gao. Chinese. Available online at: http://www.nhc.gov.cn/wsb/pxwfb/201209/55840/files/0af7007b1a68469397531b154d9425f9.pdf (accessed December 1, 2020).
Qing, J., Zhou, Y., Lai, R., Hu, P., Ding, Y., Wu, W., et al. (2015). Prevalence of mutations in GJB2, SLC26A4, and mtDNA in children with severe or profound sensorineural hearing loss in southwestern China. Genet. Test Mol. Biomarkers 19, 52–58. doi: 10.1089/gtmb.2014.0241
Scott, D. A., Wang, R., Kreman, T. M., Sheffield, V. C., and Karniski, L. P. (1999). The pendred syndrome gene encodes a chloride-iodide transport protein. Nat. Genet. 21, 440–443. doi: 10.1038/7783
Shearer, A. E., Shen, J., Amr, S., Morton, C. C., and Smith, R. J. Newborn Hearing Screening Working Group of the National Coordinating Center for the Regional Genetics Networks (2019). A proposal for comprehensive newborn hearing screening to improveidentification of deaf and hard-of-hearing children. Genet. Med. 21, 2614–2630.
Sugata, A., Fukushima, K., Sugata, K., Fukuda, S., Kimura, N., Gunduz, M., et al. (2002). High-throughput screening for GJB2 mutations–its clinical application to genetic testing in prelingual deafness screening for GJB2 mutations. Auris Nasus Larynx 29, 231–239. doi: 10.1016/s0385-8146(02)00014-7
Sun, S., Niu, L., Tian, J., Chen, W., Li, Y., Xia, N., et al. (2019). [Analysis of GJB2, SLC26A4, GJB3 and 12S rRNA gene mutations among patients with nonsyndromic hearing loss from eastern Shandong]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 36, 433–438. doi: 10.3760/cma.j.issn.1003-9406.2019.05.005 Chinese,
Tingang, E., Wonkam, E., Chimusa, E., Noubiap, J. J., Adadey, S. M., Fokouo, J. V. F., et al. (2019). GJB2 and GJB6 mutations in hereditary recessive non-syndromic hearing impairment in cameroon. Genes (Basel) 10:844. doi: 10.3390/genes10110844
Uyguner, O., Emiroglu, M., Uzumcu, A., Hafiz, G., Ghanbari, A., Baserer, N., et al. (2003). Frequencies of gap- and tight-junction mutations in Turkish families with autosomal-recessive non-syndromic hearing loss. Clin. Genet. 64, 65–69. doi: 10.1034/j.1399-0004.2003.00101.x
Van Camp, G., and Smith, R. J. H. (2020). Hereditary Hearing Loss Homepage. Available online at: https://hereditaryhearingloss.org (accessed December 1, 2020).
Wang, Q., Xiang, J., Sun, J., Yang, Y., Guan, J., Wang, D., et al. (2019). Nationwide population genetic screening improves outcomes of newborn screening for hearing loss in China. Genet. Med. 21, 2231–2238. doi: 10.1038/s41436-019-0481-6
Wang, Q., Zhao, Y., Rao, S., Guo, Y., He, Y., Lan, L., et al. (2011). Newborn hearing concurrent gene screening can improve care for hearing loss: a study on 14,913 Chinese newborns. Int. J. Pediatr. Otorhinolaryngol. 75, 535–542. doi: 10.1016/j.ijporl.2011.01.016
World Health Organization (2018). Deafness and Hearing Loss. Available online at: https://www.who.int/health-topics/hearing-loss#tab=tab_2 (accessed December 1, 2020).
Xia, J., Liu, C., Tang, B., Pan, Q., Huang, L., Dai, H., et al. (1998). Mutations in the gene encoding gap junction protein beta-3 associated with autosomal dominant hearing impairment. Nat. Genet. 20, 370–373. doi: 10.1038/3845
Xiao, Y., Bai, W., Chen, C., Li, H., Tang, S., and Xiang, Y. (2017). [Pathogenicity and clinical phenotypes analysis for hearing loss pedigrees with variant of p.V37I in GJB2]. Chin. J. Birth Health 25, 14–16. Chinese,
Xin, F., Yuan, Y., Deng, X., Han, M., Wang, G., Zhao, J., et al. (2013). Genetic mutations in nonsyndromic deafness patients of Chinese minority and Han ethnicities in Yunnan, China. J. Transl. Med. 17:312. doi: 10.1186/1479-5876-11-312
Yuan, Y., Huang, D., Yu, F., Han, B., Wang, G., and Han, D. (2010). [Sequence analysis of GJB3 in Chinese deafness population who carry one heterozygous GJB2 pathogenic mutation]. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 45, 287–290. Chinese,
Keywords: deafness, hearing screening, genetic screening, genetic deafness, newborn deafness
Citation: Cai L, Liu Y, Xu Y, Yang H, Lv L, Li Y, Chen Q, Lin X, Yang Y, Hu G, Zheng G, Zhou J, Qian Q, Xu M-a, Fang J, Ding J, Chen W and Gao J (2021) Multi-Center in-Depth Screening of Neonatal Deafness Genes: Zhejiang, China. Front. Genet. 12:637096. doi: 10.3389/fgene.2021.637096
Received: 03 December 2020; Accepted: 07 May 2021;
Published: 02 July 2021.
Edited by:
Nelson L. S. Tang, The Chinese University of Hong Kong, ChinaReviewed by:
Jin Xu, Ningbo University, ChinaCopyright © 2021 Cai, Liu, Xu, Yang, Lv, Li, Chen, Lin, Yang, Hu, Zheng, Zhou, Qian, Xu, Fang, Ding, Chen and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaping Xu, MjE4NTAzM0B6anUuZWR1LmNu
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.