 Fengchang Qiao†
Fengchang Qiao† Cuiping Zhang
Cuiping Zhang Zhengfeng Xu
Zhengfeng Xu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 09 February 2021
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.628890
This article is part of the Research Topic Improving Medical Diagnosis in Rare Diseases View all 24 articles
Cornelia de Lange syndrome (CdLS) is a genetically heterogeneous disorder characterized by a wide spectrum of abnormalities, including craniofacial dysmorphism, upper limb anomalies, pre- and post-natal growth restrictions, hirsutism and intellectual disability. Approximately 60% of cases are caused by NIPBL variants. Herein we report on a prenatal case presented with bilateral upper-extremity malformations and cardiac defects. Whole-exome sequencing (WES) was performed on the fetus–parental trio and a de novo heterozygous synonymous variant in NIPBL [chr5:37020979; NM_133433.4: c.5328G>A, p. (Gln1776=)] was identified. Reverse transcriptase–polymerase chain reaction (RT–PCR) was conducted to evaluate the potential splicing effect of this variant, which confirmed that the variant caused a deletion of exon 27 (103 bp) by disrupting the splice-donor site and changed the reading frame with the insertion of at least three stop codons. Our finding not only expands the mutation spectrum of NIPBL gene but also establishes the crucial role of WES in searching for underlying genetic variants. In addition, our research raises the important issue that synonymous mutations may be potential pathogenic variants and should not be neglected in clinical diagnoses.
Cornelia de Lange syndrome (CdLS, MIM #122470, #300590, #610759, #614701, and #300882), a congenital developmental disorder inherited in an autosomal dominant (NIPBL, SMC3, and RAD21) or X-linked (SMC1A and HDAC8) manner, was initially discovered by Brachmann (1916) and was further summarized by de Lange (1933) (Jang et al., 2015; Li et al., 2020). CdLS is complicated by a wide spectrum of abnormalities, including craniofacial dysmorphism, upper limb anomalies, pre- and post-natal growth restrictions, hirsutism, and intellectual disability (Lalatta et al., 2007). Facial dysmorphisms include micrognathia, synophrys, long eyelashes, depressed nasal bridge, low-set ears, small head, small and widely spaced teeth, long philtrum and thin lips (Ireland and Burn, 1993). CdLS occurs sporadically with a prevalence of 1 in 10,000 to 1 in 30,000 live births (Kline et al., 2018); however, the actual number is far more than that as some mild cases with atypical symptoms are not clinically diagnosed.
CdLS is typically diagnosed after birth based on some abnormal phenotypes (Kinjo et al., 2019). Although several cases have been reported before birth, prenatal diagnosis is still a challenge for clinicians and geneticists, with only 23% of all cases being detected prenatally (Avagliano et al., 2017). It has been reported that maternal serum pregnancy-associated placental protein-A (PAPP-A) produced by the placenta in the first and second trimester of pregnancy is an early indicator of CdLS, but it is not specific as only a very low level can support this diagnosis (Kinjo et al., 2019). At present, prenatal diagnosis of CdLS primarily relies on sonographic findings during the second and third trimester of pregnancy, such as characteristic facial features and congenital malformations. However, some such prenatal phenotypes that are detected by ultrasound are not unique to CdLS. Undoubtedly, the complexity and variability of CdLS phenotypes increase the difficulty of prenatal diagnosis. Therefore, considerable clinical experience and professional knowledge about the dysmorphic features of CdLS are still needed for clinicians.
With the rapid development of molecular diagnostic technology, the variants of relative pathogenic genes can be confirmed by molecular testing, which is the key to diagnosis. To date, it has been acknowledged that pathogenic variants of five genes encoding the structural components (SMC1A, SMC3, and RAD21) or function-related factors (NIPBL and HDAC8) of the cohesin complex are associated with CdLS. In particular, the heterozygous mutations of NIPBL account for ~60–70% of all CdLS cases (Teresa-Rodrigo et al., 2014; Kline et al., 2018). NIPBL localized on 5p13.2 facilitates cohesin loading on chromatin, whereas the cohesin complex is involved in sister chromatid cohesion and ensures accurate chromosome segregation, recombination-mediated DNA repair, and genomic stability (Baquero-Montoya et al., 2014; Teresa-Rodrigo et al., 2014). It has been reported that the variants in NIPBL also manifest as severe CdLS phenotypes with a high frequency of limb anomalies (Selicorni et al., 2007). Additionally, individuals with a somatic heterozygous pathogenic mutation in NIPBL appear to be fully penetrant (Deardorff et al., 1993). Nevertheless, for 20–30% of affected individuals, molecular alterations cannot be found in the aforementioned genes (Pie et al., 2016; Wagner et al., 2019).
Whole-exome sequencing (WES), especially for the fetus–parental trio, has proven to be a feasible and effective approach for the identification of some rare disorders that are suggestive of genetic etiology but cannot be diagnosed using conventional testing methods. In two large prospective cohort studies, Petrovski and Lord and their respective teams identified an additional 8.5 and 10% diagnostic genetic variants, respectively, through WES for cases that tested negative in karyotype and chromosomal microarray analyses (CMA), which further confirmed the feasibility and potential value of prenatal WES in detecting genetic diseases (Lord et al., 2019; Petrovski et al., 2019). Currently, WES is a first-line detection method for hereditary diseases. In our study, we detected a de novo heterozygous variant of NIPBL in a fetus with bilateral upper-extremity malformations and cardiac defects via the application of WES, which provides a theoretical basis and guidance for this family in reproductive genetic counseling and prenatal diagnosis.
This research was approved by the Ethics Committee of the Nanjing Maternity and Child Health Care Hospital and adhered to the Declaration of Helsinki. Samples and information were collected from the parents after written informed consent was obtained. In addition, they agreed to the publication of the manuscript.
A 26-year-old pregnant Chinese woman gravida 2 para 0, who was referred to the Center of Prenatal Diagnosis at Nanjing Maternity and Child Health Care Hospital opted for genetic tests because of an ultrasonic abnormality. The woman and her husband were healthy and non-consanguineous, with no history of infection and/or exposure to teratogens. However, their family history was significant for a previous pregnancy that was terminated because of skeletal and cardiac dysplasia.
At 24 weeks of gestation, a prenatal ultrasound examination in our hospital indicated skeletal dysplasia of the bilateral upper limbs and congenital heart malformation. The bilateral humerus was short, ~1.93 cm, and only the ulna was angled with the humerus. Additionally, the fetus had congenital cardiac developmental abnormalities, and the structures of the ventricular outflow tract were unclear because of the small gestational age. After genetic counseling by the clinic's geneticist, the couple ultimately decided to terminate the pregnancy at 27+3 weeks because of the fetal malformations and uncertainty of prognosis. However, they did not consent to a pathological autopsy. To assess the risk of recurrence, they requested genetic testing to clarify the potential cause of the disease. Because of the negative results of CMA test, we applied WES for the proband and his healthy parents (Figure 1A) to search for potential variants. The detailed examinations of pregnant woman are listed in Supplementary Table 1.
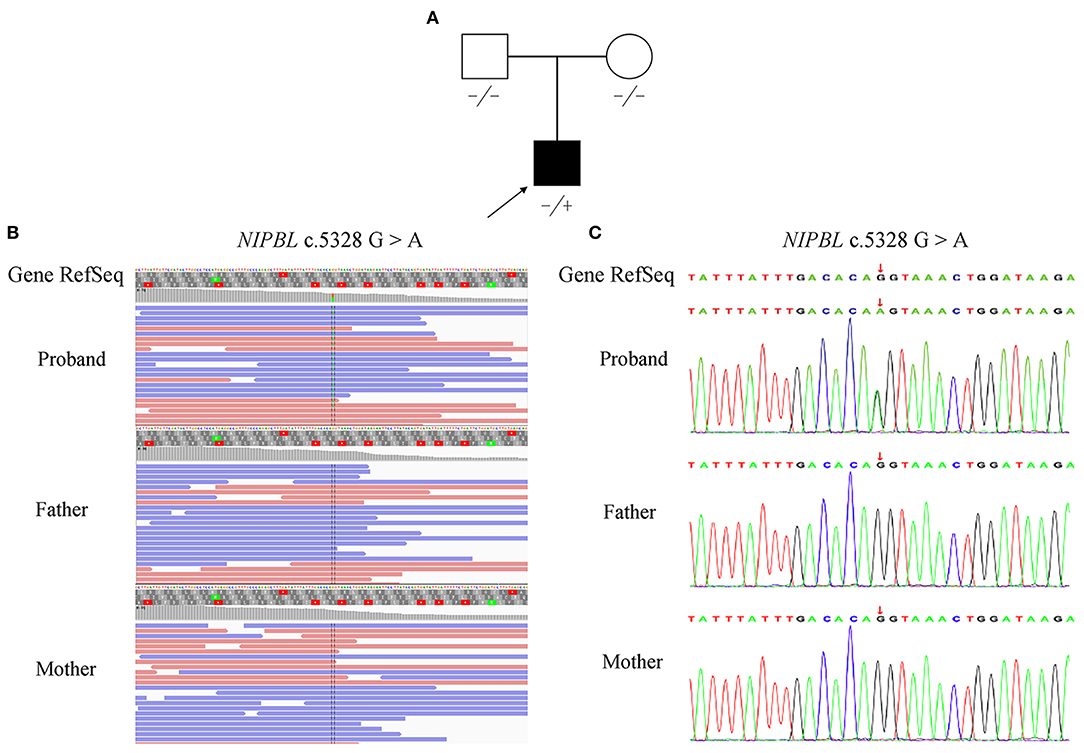
Figure 1. Pedigree and mutation analysis of the family. (A) Pedigree of the family with segregation of the identified NIPBL mutation. The square represents the male, and circle represents the female. A filled symbol indicates the proband. (B) Visualization of the mutation in NIPBL is shown with an integrative genomics viewer. The variant (c.5328G>A) was heterozygous in the proband. (C) Confirmation of the mutation (c.5328G>A), which was not found in his parents, was conducted by Sanger sequencing.
Genomic DNA was extracted from the proband's tissue sample after the pregnancy was terminated and from the parental peripheral blood using an Automated Nucleic Acid Extractor (RBCBioscience) following standard procedures for WES analysis. First, genomic DNA was sheared and amplified to generate a fragment library with a read length of 150 bp that was captured by the IDT The xGen Exome Research Panel v1.0 according to the manufacturer's protocols. Sequencing was subsequently carried out on the Illumina NovaSeq 6000 (Illumina, Inc.) platform, and the resultant reads were mapped against GRCh37/hg19 assembly. Single-nucleotide variants (SNVs) and small indels (<50 bp) were identified using the Genome Analysis Toolkit 3.4 (GATK). Novel mutations were filtered against the 1000 Genomes database (1000 genomes release phase 3, http://www.1000genomes.org/), dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi), and the genome aggregation database (gnomad.broadinstitute.org). Finally, polymerase chain reaction (PCR) was performed to amplify the affected fragment of NIPBL using specific primers (forward primer: 5′-TACACCCGGCCGGAGAACCTAAAA-3′ and reverse primer: 5′-ATGCATCGAGCTGAAAACCGAAAA-3′), and the purified PCR products were applied to Sanger sequencing to affirm the mutation.
MaxEntScan (Wappenschmidt et al., 2012; Afzal et al., 2019) and dbscSNV (Jian et al., 2014) were used to predict whether the variant affects splice site. To detect aberrant transcripts, total RNA was isolated from fetal skin and control cells using TRIzol™ Reagent (Invitrogen; Thermo Fisher Scientific) according to the manufacturer's protocols. Complementary DNA (cDNA) was synthesized from the total RNA using a PrimeScript™ RT reagent Kit with gDNA Eraser (TAKARA), which was further amplified to obtain PCR products using specific primers (forward primer: 5′-AGGAACACATCATGCAAAGGA-3′ and reverse primer: 5′-ACAGCAACAACCTCAGACAA-3′). PCR products were ultimately separated on a 2% agarose gel and validated by Sanger sequencing.
We first examined the CMA for the proband, but no chromosomal abnormalities or submicroscopic chromosomal imbalances were detected at the whole genome level. Then, we performed sequencing and subsequent alignment of the whole exome and obtained 77.82 million reads with a read length of 150 bp and average sequencing depth of 99.59 × and 98.93% of the whole exome target region covered at ≥20 ×. There were 32,561 single-nucleotide polymorphisms (SNPs), including 3,432 non-synonymous SNPs and 1,725 synonymous SNPs in the coding sequence, as well as 35 SNPs in the splice sites. In addition, there were 244 indels in the coding sequence.
Following the prioritized filtration strategy (Roy et al., 2018), we selected nine candidate variants, including seven missense mutations and two frameshift mutations (Supplementary Table 2). Notably, a plausible point mutation in NIPBL [chr5:37020979; NM_133433.4: c.5328G>A, p. (Gln1776=)] may explain the features described above, considering the inheritance model of the disease and phenotypes of the gene. By bioinformatics, the variant is predicted to affect splicing according to MaxEntScan and dbscSNV (Supplementary Table 3). Next, we performed RNA transcript analysis using specific RT-PCR to explore the implication of this aberrant variant on the splicing of NIPBL. The resultant PCR products of the control RNA showed that a single amplified cDNA fragment of 300 bp was expected of a normal transcript, whereas the proband showed two bands comprising an aberrant band (197 bp) that was smaller than that of the control cells (Figure 2A). Sanger sequencing cDNA data analysis revealed that the aberrant transcript was affected by the variant, thus leading to complete skipping of exon 27 (103 bp) (Figure 2B). Although the variant was located at the last nucleotide of exon 27 and not within the canonical splice sites, the variant affects splicing generating an aberrant transcript with complete absence of exon 27 characterized by altered protein reading frame and premature stop codons (ACMG variant evidence PVS1). Using Sanger sequencing, the variant was further verified as a de novo heterozygous synonymous mutation in the proband, which means that it was not identified in either of the non-consanguineous parents (ACMG variant evidence PS2) (Figures 1B,C). Furthermore, this variant was absent from the 1000 Genomes, ExAC, or dbSNP databases (ACMG variant evidence PM2). Moreover, the probability of loss-of-function intolerance (pLI) is 1 in the ExAC Browser, which suggests that NIPBL is highly intolerant to heterozygous loss-of-function variants. Considered together, the variant (c.5328G>A) was classified as a pathogenic mutation according to the ACMG guidelines (Richards et al., 2015).
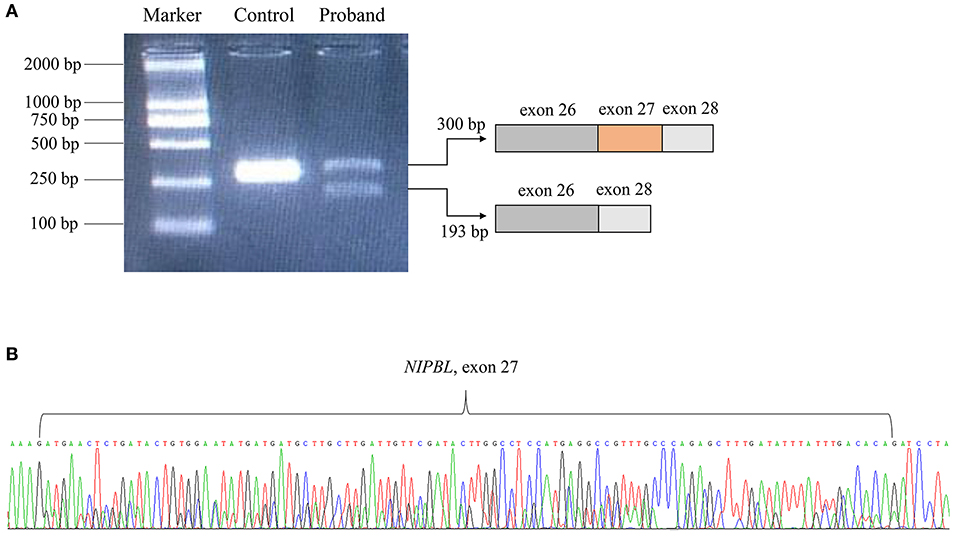
Figure 2. RNA transcript analysis using specific RT-PCR. (A) Gel image showing the PCR products of NIPBL cDNA fragments. (B) The deletion of exon 27 in NIPBL mRNA was confirmed by Sanger sequencing.
In addition, the remaining eight candidate variants were excluded because of insufficient evidence. COL11A2, DCSH1, POC1A, and WDR60 follow the model of autosomal recessive inheritance, but the proband with heterozygosity variants was not affected. Although the variant of RAB40AL was assessed as likely pathogenic, its syndrome phenotype was not consistent with the ultrasonic phenotype of the fetus. Furthermore, the mutations of IFIH1, WHSC1, and TBX4 were evaluated as variants of uncertain significance (Supplementary Table 2).
Here, we report a fetus with CdLS whose diagnosis was prompted by abnormal sonographic findings of upper extremity anomalies and congenital heart defects. The cause of the disease was a previously undescribed de novo heterozygous synonymous variant in NIPBL (c.5328G>A) that was identified by WES. The variant located in the exon–intron border disrupted the normal splicing process as predicted by bioinformatics. By further validation using RT-PCR and Sanger sequencing, we detected the deletion of exon 27 (103 bp, 1,776–1,810 aa) at the mRNA level in only the proband.
NIPBL encodes the Nipped-B-like protein (delangin), which plays a pivotal role in loading the cohesin complex onto DNA and is crucial for sister chromatid cohesion and regulation of other proteins (Gause et al., 2008; Bot et al., 2017). To our knowledge, NIPBL is expressed in developing limbs and subsequently in cartilage primordia of the ulna and various hand bones, as well as in the developing heart, especially in the atrial and ventricular myocardium during the embryo stage (Ben-Asher and Lancet, 2004; Tonkin et al., 2004; Kang et al., 2018). In the case presented here, a prenatal ultrasound suggested that the bilateral humerus of the fetus was shorter than expected at standard gestational age, and the metacarpal bone could not be clearly identified. Furthermore, the fetus showed congenital cardiac developmental abnormalities. Combining the bias of NIPBL expression regions and the phenotypic characteristics of the fetus, we inferred that the mutation of NIPBL is correlated with the aberrant fetal phenotypes.
It is universally acknowledged that synonymous mutations are often neglected as silent variants, which may be because of a lack of alteration in the composition of amino acids caused by the degeneracy of the genetic code. To narrow down candidate mutations, synonymous mutations are usually eliminated first as benign variants in clinical analysis. However, with the advancement of research, mounting evidence illustrates the pathogenic mechanism of synonymous mutation by affecting the translation rate, mRNA stability, protein folding, and splicing accuracy in human diseases (Cartegni et al., 2002; Nackley et al., 2006; Sauna and Kimchi-Sarfaty, 2011). Previous studies have demonstrated that synonymous mutations could affect the splicing accuracy. According to a study by Faa et al. (2010), a synonymous mutation (c.2811G>T) in CFTR of an Italian patient with cystic fibrosis created a new 5′ splice site within exon 15, which generated a transcript lacking 76 amino acid residues. The aberrant protein may affect the structure and regulation of the chloride (Cl−) channel (Faa et al., 2010). Furthermore, Kim et al. (2013) also reported a novel synonymous mutation (c.1803G>A) in SLC26A4 that resulted in complete skipping of exon 16 by affecting the splicing accuracy in two members of a Korean family suffering from hearing impairment (Kim et al., 2013). All these described cases reveal that synonymous mutations should be evaluated separately in clinical diagnoses as they usually affect splicing. As described in our case, the absence of exon 27 was caused by a synonymous mutation, which was suspected to affect the splice-donor site of NIPBL. To some degree, exon 27 skipping may impact the patient's phenotypes.
To our knowledge, the most common effect of splicing site changes is skipping of the next exon. In our case, this novel variant c.5328G>A occurred at the terminal nucleotide of exon 27 and generated an aberrant transcript with an exon 27 deletion by affecting the splicing accuracy. Previously, Teresa-Rodrigo et al. (2014) discovered that splice site mutations account for ~17% of NIPBL variants. The variant (c.5328+1G>A) in NIPBL that they reported was at a different in genomic location from that reported in our case (c.5328G>A), but coincidentally generated an identical splicing effect with the skipping of exon 27. In addition, the patient seemed to have the same severe structural anomalies of the limbs as in our case, such as bilateral aplasia of the ulna and distal humerus (Teresa-Rodrigo et al., 2014). Exon 27 is located in an evolutionarily conserved protein domain called HEAT repeats (H1: 1,767–1,805 aa, H2: 1,843–1,881 aa, H3: 1,945–1,984 aa, H4: 2,227–2,267 aa, H5: 2,313–2,351 aa), which is an essential component of the NIPBL gene product (delangin) and function in protein–protein interaction motifs (Neuwald and Hirano, 2000; Mannini et al., 2013; Li et al., 2020). On the one hand, absence of exon 27 might disrupt the integrity of the HEAT repeats. A previous study showed that the C-terminal region containing HEAT repeats and the heterochromatin protein 1 (HP1) interacting motif could recruit NIPBL to the sites of DNA damage, thus indicating the HEAT repeats are essential to repair DNA damage (Oka et al., 2011). In another research, Gause et al. (2008) proposed that some of the HEAT repeats are crucial for gene expression and sister chromatid cohesion. On the other hand, exon 27 skipping disrupted the reading frame and inserted premature stop codons that may have resulted in degradation of the corresponding transcript by nonsense-mediated mRNA decay. Consequently, the putative pathogenic mechanism in this case would be haploinsufficiency. Further exploration is needed to attain a detailed understanding of the effects of this variant at the transcript and protein level.
Although the mother of the proband experienced the adverse outcome of terminated pregnancy because of skeletal and cardiac dysplasia in her first pregnancy, we found a de novo explicit mutation in COL1A1 [chr17:48264402; NM_000088.4: c.3505G>A, p. (Gly1169Ser)] in the first fetus by trio WES. Furthermore, the variant was already reported as pathogenic in ClinVar: SCV000695351.1 (Pathogenic*-Osteogenesis imperfecta), SCV000883622.1 (Pathogenic*-not provided), SCV000955221.1 (Pathogenic*-Osteogenesis imperfecta type I), SCV000574580.1 (Pathogenic*-Osteogenesis imperfecta type I). Therefore, the different causes of two adverse outcomes contribute to us ruling out the possibility of gonadal mosaicism. For future pregnancy, prenatal diagnosis is necessary for the fetus, especially to detect the above-mentioned mutations in these two genes.
Overall, this study presented the case of a fetus with CdLS with a de novo heterozygous mutation (c.5328G>A) in NIPBL. The synonymous mutation affects the normal splicing process and produces a frameshift that results in premature stop codons. Thus, the variant identified in our case is valuable for comprehending possible pathogenic mechanisms of NIPBL, which not only broadens the mutation spectrum of NIPBL but also contributes to supportive genetic counseling and timely management for pregnancies with a structural abnormality. Finally, we cannot ignore novel synonymous variants as silent mutations in clinical analysis; sometimes these variants elucidate the potential pathogenic mechanism of a disease.
All datasets for this study are included in the article/Supplementary Tables.
This research was approved by the Ethics Committee of the Nanjing Maternity and Child Health Care Hospital and adhered to the Declaration of Helsinki. Samples and information were collected from fetal parents after written informed consent was obtained and they also agreed to the publication of the manuscript.
FQ and CZ performed the experiments, wrote the manuscript, and analyzed the WES data. YW and BS collected the clinical information and provided the genetics counseling. GL conducted the bioinformatics analysis. PH and ZX designed the study and revised the manuscript. All authors read and approved the final manuscript.
This work was supported by the National Key R&D Program of China (No. 2018YFC1002402), National Natural Science Foundation of China (81602300, 81770236, 81801445, 81801373), Natural Science Foundation of Jiangsu Province (BK20181121), and Project on Maternal and Child Health Talents of Jiangsu Province (FRC201791).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We were thankful for the members of the group who participated and assisted us in the study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.628890/full#supplementary-material
Afzal, R., Firasat, S., Kaul, H., Ahmed, B., Siddiqui, S. N., Zafar, S. N., et al. (2019). Mutational analysis of the CYP1B1 gene in Pakistani primary congenital glaucoma patients: Identification of four known and a novel causative variant at the 3′ splice acceptor site of intron 2. Congenit. Anom. (Kyoto) 59, 152–161. doi: 10.1111/cga.12312
Avagliano, L., Bulfamante, G. P., and Massa, V. (2017). Cornelia de Lange syndrome: to diagnose or not to diagnose in utero? Birth Defects Res. 109, 771–777. doi: 10.1002/bdr2.1045
Baquero-Montoya, C., Gil-Rodriguez, M. C., Hernandez-Marcos, M., Teresa-Rodrigo, M. E., Vicente-Gabas, A., Bernal, M. L., et al. (2014). Severe ipsilateral musculoskeletal involvement in a Cornelia de Lange patient with a novel NIPBL mutation. Eur. J. Med. Genet. 57, 503–509. doi: 10.1016/j.ejmg.2014.05.006
Ben-Asher, E., and Lancet, D. (2004). NIPBL gene responsible for Cornelia de Lange syndrome, a severe developmental disorder. Isr. Med. Assoc. J. 6, 571–572.
Bot, C., Pfeiffer, A., Giordano, F., Manjeera, D. E., Dantuma, N. P., and Strom, L. (2017). Independent mechanisms recruit the cohesin loader protein NIPBL to sites of DNA damage. J. Cell Sci. 130, 1134–1146. doi: 10.1242/jcs.197236
Brachmann, W. (1916). Ein fall von symmetrischer monodaktylie durch Ulnadefekt, mit symmetrischer flughautbildung in den ellenbeugen, sowie anderen abnormitaten (zwerghaftogkeit, halsrippen, behaarung). Jarb. Kinder Phys. Erzie. 84, 225–235.
Cartegni, L., Chew, S. L., and Krainer, A. R. (2002). Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat. Rev. Genet. 3, 285–298. doi: 10.1038/nrg775
de Lange, C. (1933). Sur un type nouveau de degenerescence (typus Amstelodamensis). Arch. Med. Enfants 36, 713–719.
Deardorff, M. A., Noon, S. E., and Krantz, I. D. (1993). “Cornelia de Lange syndrome,” in GeneReviews((R)), eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, et al. (Seattle, WA: U.S. National Library of Medicine).
Faa, V., Coiana, A., Incani, F., Costantino, L., Cao, A., and Rosatelli, M. C. (2010). A synonymous mutation in the CFTR gene causes aberrant splicing in an italian patient affected by a mild form of cystic fibrosis. J. Mol. Diagn. 12, 380–383. doi: 10.2353/jmoldx.2010.090126
Gause, M., Webber, H. A., Misulovin, Z., Haller, G., Rollins, R. A., Eissenberg, J. C., et al. (2008). Functional links between Drosophila Nipped-B and cohesin in somatic and meiotic cells. Chromosoma 117, 51–66. doi: 10.1007/s00412-007-0125-5
Ireland, M., and Burn, J. (1993). Cornelia de Lange syndrome–photo essay. Clin. Dysmorphol. 2, 151–160.
Jang, M. A., Lee, C. W., Kim, J. K., and Ki, C. S. (2015). Novel pathogenic variant (c.3178G>A) in the SMC1A gene in a family with Cornelia de Lange syndrome identified by exome sequencing. Ann. Lab Med. 35, 639–642. doi: 10.3343/alm.2015.35.6.639
Jian, X., Boerwinkle, E., and Liu, X. (2014). In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 42, 13534–13544. doi: 10.1093/nar/gku1206
Kang, M. J., Ahn, S. M., and Hwang, I. T. (2018). A novel frameshift mutation (c.5387_5388insTT) in NIPBL in Cornelia de Lange syndrome with severe phenotype. Ann. Clin. Lab Sci. 48, 106–109.
Kim, Y., Kim, H. R., Kim, J., Shin, J. W., Park, H. J., Choi, J. Y., et al. (2013). A novel synonymous mutation causing complete skipping of exon 16 in the SLC26A4 gene in a Korean family with hearing loss. Biochem. Biophys. Res. Commun. 430, 1147–1150. doi: 10.1016/j.bbrc.2012.12.022
Kinjo, T., Mekaru, K., Nakada, M., Nitta, H., Masamoto, H., and Aoki, Y. (2019). A Case of Cornelia de Lange syndrome: difficulty in prenatal diagnosis. Case Rep. Obstet. Gynecol. 2019:4530491. doi: 10.1155/2019/4530491
Kline, A. D., Moss, J. F., Selicorni, A., Bisgaard, A. M., Deardorff, M. A., Gillett, P. M., et al. (2018). Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement. Nat. Rev. Genet. 19, 649–666. doi: 10.1038/s41576-018-0031-0
Lalatta, F., Russo, S., Gentilin, B., Spaccini, L., Boschetto, C., Cavalleri, F., et al. (2007). Prenatal/neonatal pathology in two cases of Cornelia de Lange syndrome harboring novel mutations of NIPBL. Genet. Med. 9, 188–194. doi: 10.1097/gim.0b013e31803183dd
Li, S., Miao, H., Yang, H., Wang, L., Gong, F., Chen, S., et al. (2020). A report of 2 cases of Cornelia de Lange syndrome (CdLS) and an analysis of clinical and genetic characteristics in a Chinese CdLS cohort. Mol. Genet. Genomic Med. 8:e1066. doi: 10.1002/mgg3.1066
Lord, J., McMullan, D. J., Eberhardt, R. Y., Rinck, G., Hamilton, S. J., Quinlan-Jones, E., et al. (2019). Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet 393, 747–757. doi: 10.1016/S0140-6736(18)31940-8
Mannini, L., Cucco, F., Quarantotti, V., Krantz, I. D., and Musio, A. (2013). Mutation spectrum and genotype-phenotype correlation in Cornelia de Lange syndrome. Hum. Mutat. 34, 1589–1596. doi: 10.1002/humu.22430
Nackley, A. G., Shabalina, S. A., Tchivileva, I. E., Satterfield, K., Korchynskyi, O., Makarov, S. S., et al. (2006). Human catechol-O-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science 314, 1930–1933. doi: 10.1126/science.1131262
Neuwald, A. F., and Hirano, T. (2000). HEAT repeats associated with condensins, cohesins, and other complexes involved in chromosome-related functions. Genome Res. 10, 1445–1452. doi: 10.1101/gr.147400
Oka, Y., Suzuki, K., Yamauchi, M., Mitsutake, N., and Yamashita, S. (2011). Recruitment of the cohesin loading factor NIPBL to DNA double-strand breaks depends on MDC1, RNF168 and HP1gamma in human cells. Biochem. Biophys. Res. Commun. 411, 762–767. doi: 10.1016/j.bbrc.2011.07.021
Petrovski, S., Aggarwal, V., Giordano, J. L., Stosic, M., Wou, K., Bier, L., et al. (2019). Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet. 393, 758–767. doi: 10.1016/S0140-6736(18)32042-7
Pie, J., Puisac, B., Hernandez-Marcos, M., Teresa-Rodrigo, M. E., Gil-Rodriguez, M., Baquero-Montoya, C., et al. (2016). Special cases in Cornelia de Lange syndrome: the Spanish experience. Am. J. Med. Genet. C Semin. Med. Genet. 172, 198–205. doi: 10.1002/ajmg.c.31501
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Roy, S., Coldren, C., Karunamurthy, A., Kip, N. S., Klee, E. W., Lincoln, S. E., et al. (2018). Standards and guidelines for validating next-generation sequencing bioinformatics pipelines: a joint recommendation of the Association for Molecular Pathology and the College of American Pathologists. J. Mol. Diagn. 20, 4–27. doi: 10.1016/j.jmoldx.2017.11.003
Sauna, Z. E., and Kimchi-Sarfaty, C. (2011). Understanding the contribution of synonymous mutations to human disease. Nat. Rev. Genet. 12, 683–691. doi: 10.1038/nrg3051
Selicorni, A., Russo, S., Gervasini, C., Castronovo, P., Milani, D., Cavalleri, F., et al. (2007). Clinical score of 62 Italian patients with Cornelia de Lange syndrome and correlations with the presence and type of NIPBL mutation. Clin. Genet. 72, 98–108. doi: 10.1111/j.1399-0004.2007.00832.x
Teresa-Rodrigo, M. E., Eckhold, J., Puisac, B., Dalski, A., Gil-Rodriguez, M. C., Braunholz, D., et al. (2014). Functional characterization of NIPBL physiological splice variants and eight splicing mutations in patients with Cornelia de Lange syndrome. Int. J. Mol. Sci. 15, 10350–10364. doi: 10.3390/ijms150610350
Tonkin, E. T., Wang, T. J., Lisgo, S., Bamshad, M. J., and Strachan, T. (2004). NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat. Genet. 36, 636–641. doi: 10.1038/ng1363
Wagner, V. F., Hillman, P. R., Britt, A. D., Ray, J. W., and Farach, L. S. (2019). A de novo HDAC2 variant in a patient with features consistent with Cornelia de Lange syndrome phenotype. Am. J. Med. Genet. A. 179, 852–856. doi: 10.1002/ajmg.a.61101
Wappenschmidt, B., Becker, A. A., Hauke, J., Weber, U., Engert, S., Kohler, J., et al. (2012). Analysis of 30 putative BRCA1 splicing mutations in hereditary breast and ovarian cancer families identifies exonic splice site mutations that escape in silico prediction. PLoS ONE. 7:e50800. doi: 10.1371/journal.pone.0050800
Keywords: Cornelia de Lange syndrome, whole-exome sequencing, NIPBL, splicing mutation, prenatal diagnosis
Citation: Qiao F, Zhang C, Wang Y, Liu G, Shao B, Hu P and Xu Z (2021) Case Report: Prenatal Whole-Exome Sequencing to Identify a Novel Heterozygous Synonymous Variant in NIPBL in a Fetus With Cornelia de Lange Syndrome. Front. Genet. 12:628890. doi: 10.3389/fgene.2021.628890
Received: 13 November 2020; Accepted: 19 January 2021;
Published: 09 February 2021.
Edited by:
Natália Duarte Linhares, Genuity Science, IrelandReviewed by:
Palma Finelli, University of Milan, ItalyCopyright © 2021 Qiao, Zhang, Wang, Liu, Shao, Hu and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhengfeng Xu, emhlbmdmZW5nX3h1X25qQDE2My5jb20=; Ping Hu, bmpmeWJqeWh1cGluZ0AxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.