 Xiaojun Chen
Xiaojun Chen Fatao Liu2
Fatao Liu2- 1Department of Plastic and Reconstructive Surgery, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2Bio-X Institute, Shanghai Jiao Tong University, Shanghai, China
Hemifacial microsomia (HFM) is a rare congenital disease characterized by a spectrum of craniomaxillofacial malformations, including unilateral hypoplasia of the mandible and surrounding structures. Genetic predisposition for HFM is evident but the causative genes have not been fully understood. Thus, in the present study, we used whole-exome sequencing to screen 52 patients with HFM for rare germline mutations. We revealed 3,341 rare germline mutations in this patient cohort, including those in 13 genes previously shown to be associated with HFM. Among these HFM-related genes, NID2 was most frequently mutated (in 3/52 patients). PED4DIP, which has not been previously associated with HFM, exhibited rare variants most frequently (in 7/52 patients). Pathway enrichment analysis of genes that were mutated in >2 patients predicted the “laminin interactions” pathway to be most significantly disrupted, predominantly by mutations in ITGB4, NID2, or LAMA5. In summary, this study is the first to identify rare germline mutations in HFM. The likely disruptions in the signaling pathways due to the mutations reported here may be considered potential causes of HFM.
Introduction
Hemifacial microsomia (HFM), also known as craniofacial microsomia and oculo-auriculo-vertebral spectrum, is a rare congenital craniofacial malformation condition estimated to affect 1/3,000–1/5,000 live births (Birgfeld and Heike, 2019). Patients exhibit a broad spectrum of symptoms of varying severity, ranging from microtia to complex developmental defects of the face (e.g., maxillary and mandibular hypoplasia) and surrounding soft tissue. These can be further complicated by ipsilateral orbital anomalies, facial paralysis, transverse facial cleft, and other rare phenotypes (Tuin et al., 2015). HFM treatment is not only very painful and expensive but also challenging and predominantly symptomatic due to the complexity of induced defects.
The pathogenic mechanisms that underlie HFM remain unknown; however, HFM etiology is widely recognized as being multi-factorial, including both environmental and genetic factors (Beleza-Meireles et al., 2014). Despite the use of advanced gene sequencing technologies to identify chromosomal anomalies and candidate gene mutations in patients with HFM, the genetic causes of HFM remain elusive (Terhal et al., 2006; Kosaki et al., 2007; Zhu et al., 2007; Ala-Mello et al., 2008; Ou et al., 2008; Alasti and Van Camp, 2009; Rooryck et al., 2009; Huang et al., 2010; Northup et al., 2010; Rooryck et al., 2010; Su et al., 2012; Ballesta-Martinez et al., 2013; Quintero-Rivera and Martinez-Agosto, 2013; Torti et al., 2013; Zielinski et al., 2014; Beleza-Meireles et al., 2015; Colovati et al., 2015; Guida et al., 2015; Lopez et al., 2016; Zhang et al., 2016; Berenguer et al., 2017; Bragagnolo et al., 2018; Spineli-Silva et al., 2018). Thus, in the present study, we analyzed rare germline mutations by using the whole-exome sequencing (WES) technology to detect potential genetic causes and novel therapeutic targets for HFM.
Materials and Methods
Patients
Although no common HFM diagnostic criteria exist, most patients with HFM exhibit underdevelopment of the mandible, maxilla, ear, orbit, soft tissue, and/or facial nerve. In this study, our minimal diagnostic criterion was the presence of unilateral mandibular hypoplasia diagnosed by using computed tomography scans and excluded patients with extracranial symptoms to minimize differences caused by the presentation of multiple phenotypes. The study thus enrolled a cohort of 52 patients with a mean age of 4.99 years (range, 0.5–20 years) (Figure 1), male:female ratio of 30:22, and a right:left-side-affected ratio of 26:26. All patients were further classified by using the OMENS+ classification system for HFM (Tuin et al., 2015; Table 1). All patients (or patient guardians) provided written informed consent prior to their participation in the study, which was approved by the Ethics Committee of Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine.
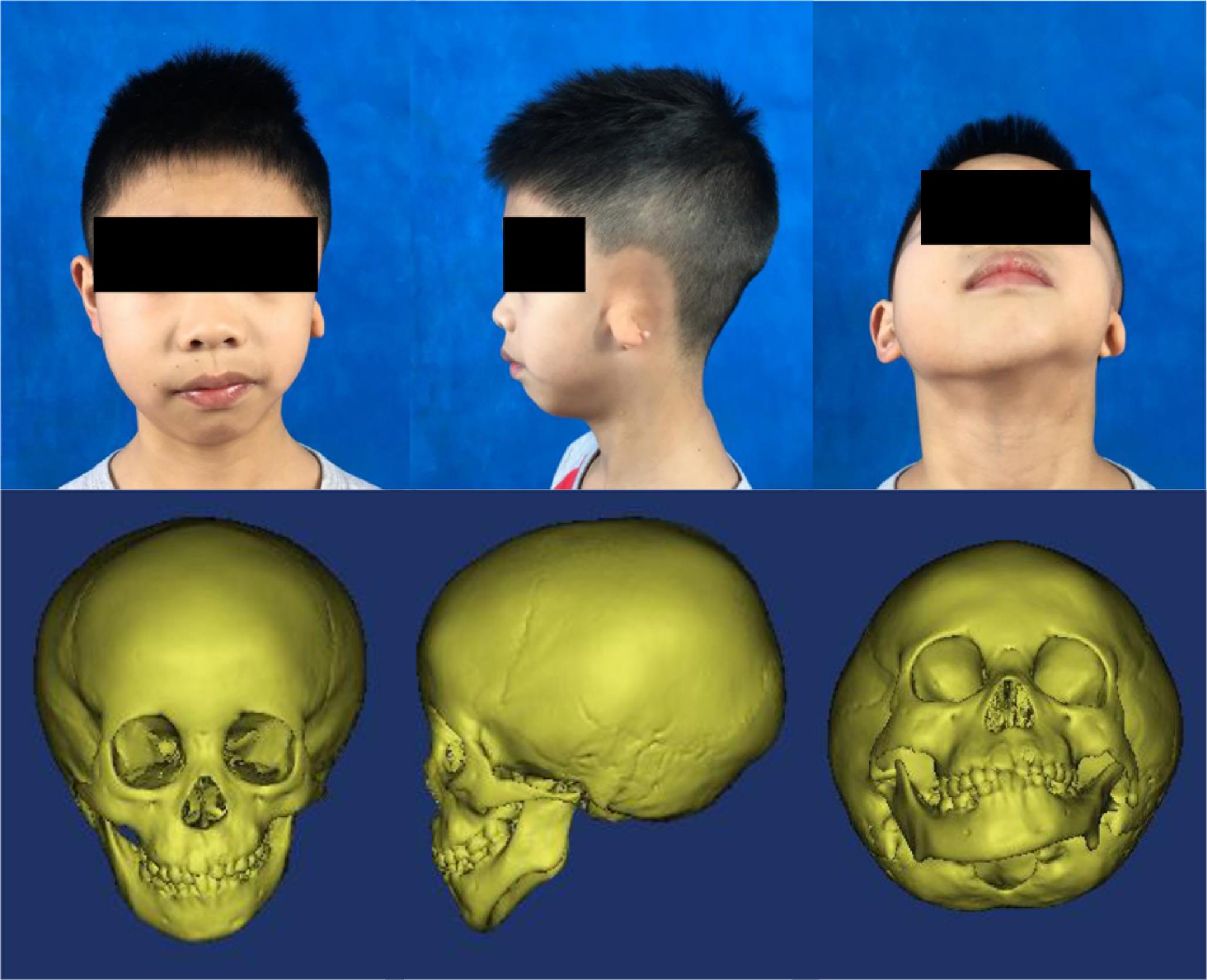
Figure 1. Typical photos and computed tomography scan images of a patient with hemifacial microsomia. From left to right: front, left-lateral, and upward view.
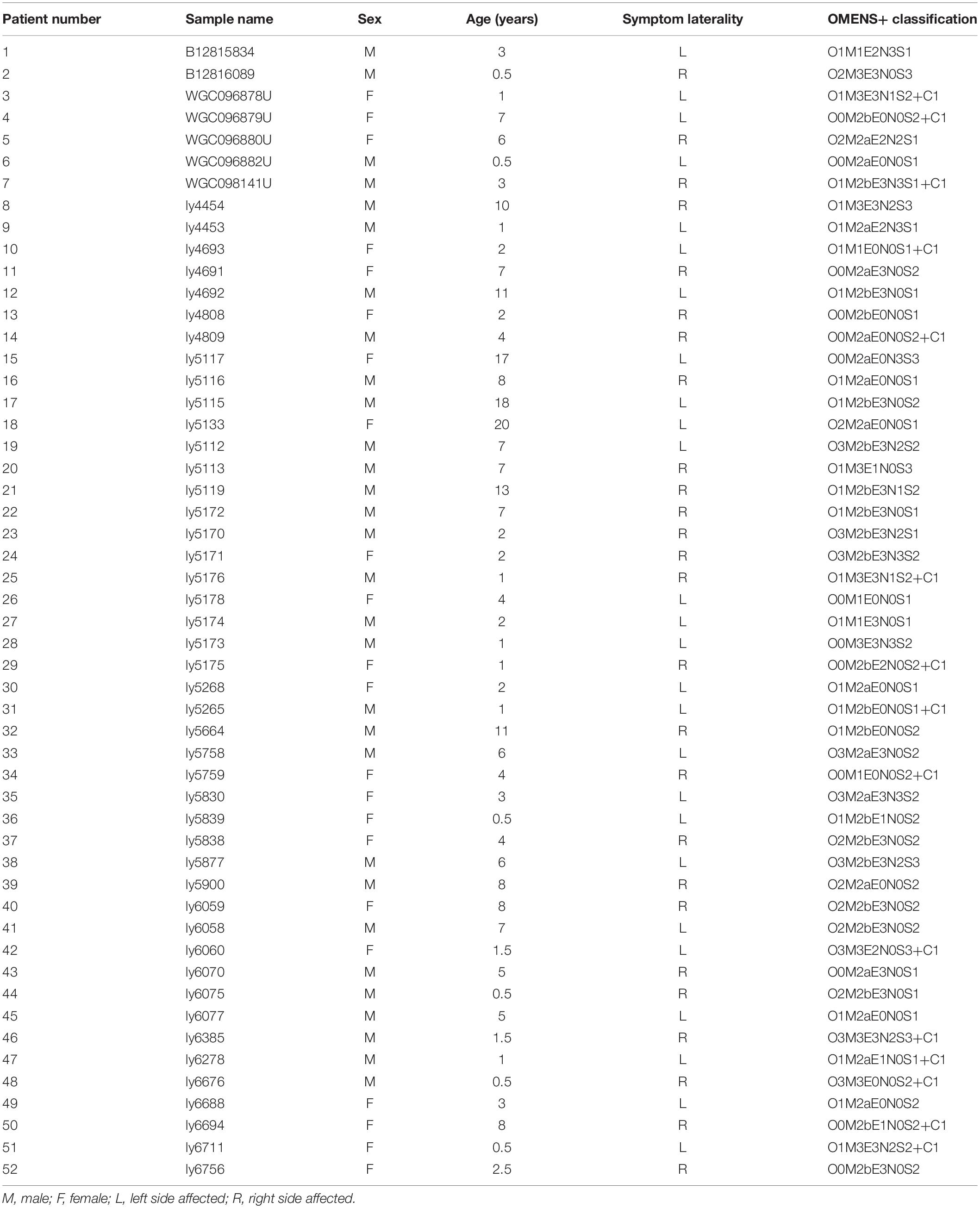
Table 1. Clinical information for the 52 analyzed patients with hemifacial microsomia.
WES
Whole-exome sequencing was conducted as previously described (Chen et al., 2018). Briefly, DNA was extracted from patient blood samples by using a Qiagen DNeasy Blood & Tissue Kit (QIAGEN, GmbH, Germany). RNaseA (QIAGEN, GmBH, Germany) was used to prevent RNA contamination. The purity and quality of the extracted DNA were determined by electrophoresis in a 1% agarose gel and by using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States) and a Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA, United States). The purified genomic DNA was then sheared, and the samples (500 ng) were subjected to further purification, end repair, 3’-end adenylation, indexed pair-end-adaptor ligation, ligation-product purification, and polymerase chain reaction (PCR) amplification. A WES library was constructed by using a SeqCap EZ capture kit (Roche) for exome capture and subjected to further PCR amplification, purification, validation, normalization, and pooling. An Illumina HiSeq Series Analyzer was used for library sequencing.
NGS (Next-Generation Sequencing) Data Processing
Next-generation sequencing data processing was conducted as previously described (Chen et al., 2018), with minor changes. Briefly, BWA software (Li and Durbin, 2009) was used to map the generated sequencing reads to the human genome (hg19). VARSCAN2 software (Koboldt et al., 2012) was used to detect germline mutations in the BAM files (minimum coverage, 20; minimum variant frequency, 0.08; P-value, 0.05). The fpfilter module of VARSCAN was used to identify false-positive variations. The resulting high-quality mutations were annotated by using SNPEFF (Cingolani et al., 2012) and VEP (McLaren et al., 2016) software.
Mutation Classification and Pathway Analysis
Annotated mutations were selected for further analysis if they exhibited an alternative allele depth ≥ 10 and a population alternative-allele frequency < 0.0005 and were predicted to incur a ‘‘High’’ or ‘‘Moderate’’ severity impact. Small insertions and deletions (INDELs) and single nucleotide variants (SNVs) not predicted to be ‘‘benign’’ and ‘‘tolerated’’ by POLYPHEN21 (Adzhubei et al., 2010), SIFT2 (Kumar et al., 2009), and CADD (cadd.gs.washington.edu) (Rentzsch et al., 2019) software tools, respectively, were used for following analysis as rare, potentially causative mutations.
Pathway analysis was performed by using ConsensusPathDB software3 (Kamburov et al., 2013) by entering the official names of the detected SNVs and INDELs and by using all known human genes included in the software database as controls. The Reactome database4 was used as a reference database to evaluate pathway enrichment.
Results
Novel Mutations in the Known HFM Genes
PubMed database mining identified a number of gene and chromosome regions that have been previously associated with HFM (Table 2). Next, rare, potentially causative mutations that were detected in the 52 patients in the present study were screened to identify novel mutations in the known HFM genes. This analysis revealed 13 novel mutations in HFM-associated NID2, PARD3B, CACNA1C, ERC1, IQSEC3, LZTR1, MED15, PLCD3, SALL4, SCR1B, SHROOM3, SLC6A13, and TCOF1, which collectively affected 14 (26.92%) of the analyzed patients (Figure 2A). Among these, mutations in NID2 affected the largest number of patients (3/52). The identified NID2 mutations, Asn202His, Asn658Lys, and Leu1136Arg, were each located in different protein domains (Figure 2B). The PARD3B mutations Pro630fs and Arg773Gln were also detected in more than one patient (Figure 2C).

Table 2. Summary of previous genetic studies of hemifacial microsomia.
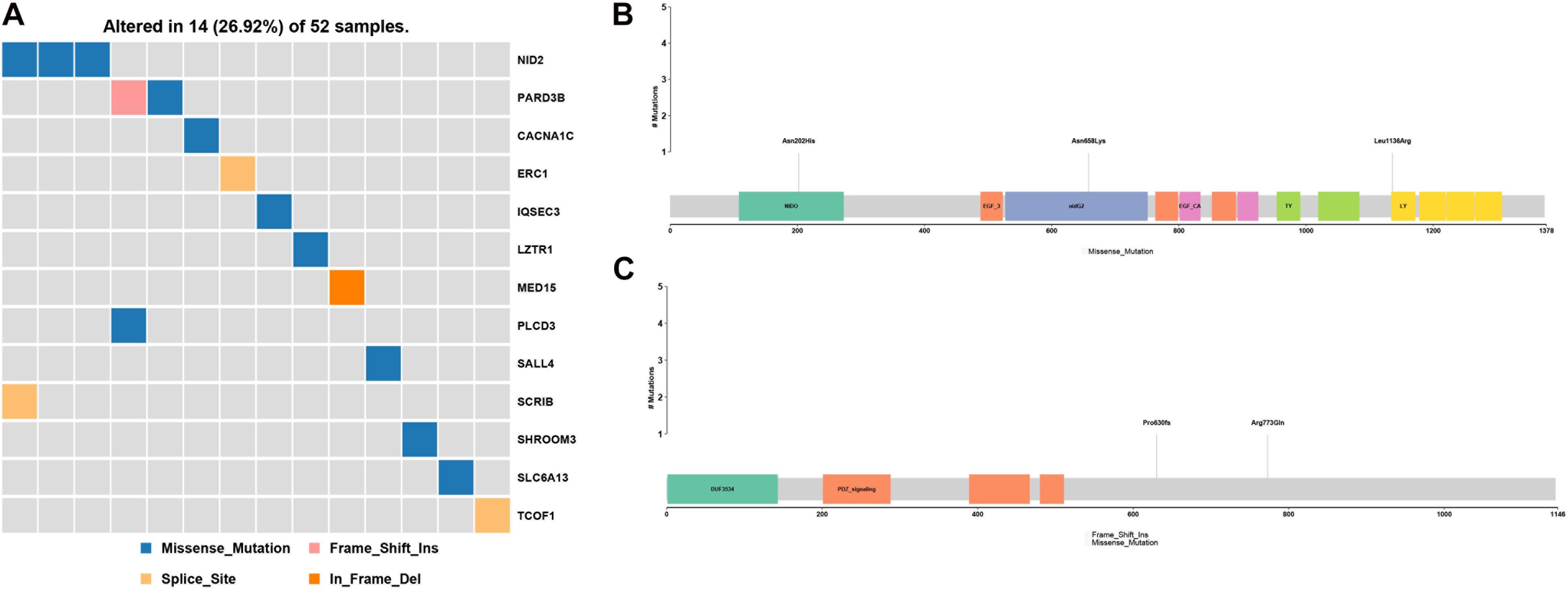
Figure 2. Gene mutations identified among the analyzed hemifacial microsomia (HFM) patient cohort. (A) A total of 13 known HFM genes were found to be mutated in the studied patient cohort, affecting 14 (16.92%) of the 52 analyzed patients. These included (B) three NID2 mutations, Asn202His, Asn658Lys, and Leu1136Arg, and (C) two PARD3B mutations, Pro630fs and Arg773Gln.
Genes With High-Frequency Rare, Potentially Causative Mutations in the HFM Patient Cohort
Genes with rare, potentially causative mutations that were detected in more than two patients were ranked by mutation frequency (Figure 3A). Of these, the most frequently mutated gene was PDE4DIP. Its mutations p.Ala141Thr, p.Lys154Arg, p.Cys19Gly, p.Asn1011Ser, p.Val120Ile, p.Gln535His, p.Pro2223Leu, and p.Gly2217Val affected the largest number of patients (7/52) (Figure 3B).
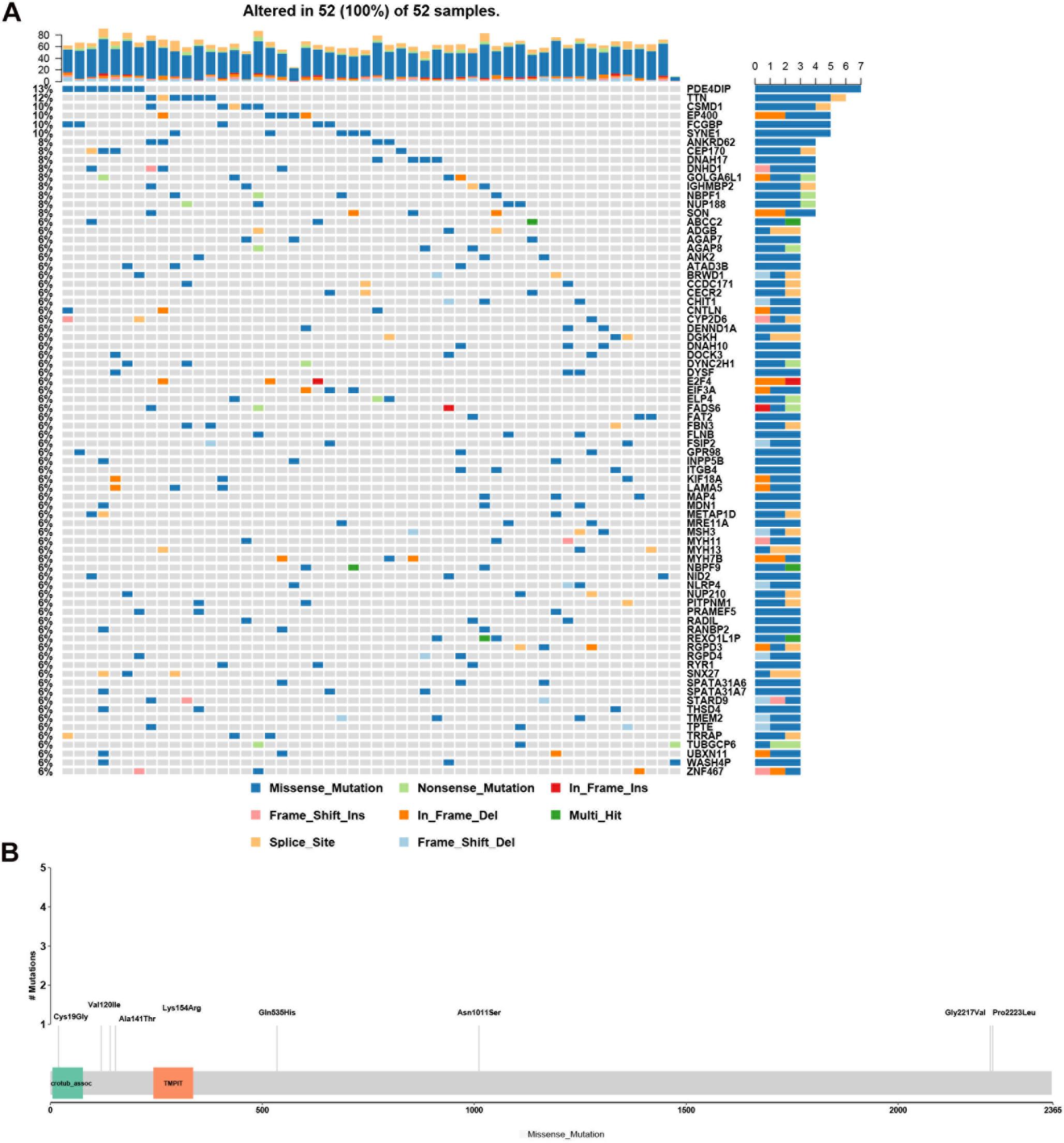
Figure 3. Analysis of rare, potentially causative mutations identified in the analyzed hemifacial microsomia (HFM) patient cohort. (A) List of genes found to harbor variants in >2 patients, ranked by mutation frequency. (B) Details of mutations (p.Ala141Thr, p.Lys154Arg, p.Cys19Gly, p.Asn1011Ser, p.Val120Ile, p.Gln535His, p.Pro2223Leu, and p.Gly2217Val.) in PDE4DIP that exhibited the greatest number of mutations and was the most frequently mutated gene among the analyzed patients.
Pathway Analysis of Frequently Mutated Genes in the HFM Patient Cohort
Next, a pathway analysis was conducted for all genes detected to harbor mutations among the 52 analyzed patients. This analysis showed that the most enriched pathways in the list of mutated genes were “extracellular-matrix organization,” “collagen-chain trimerization,” and “collagen formation” (Supplementary Figure 1). Genes mutated in >2 patients were also subjected to the ConsensusPathDB enrichment analysis (Figure 4), which found that the “laminin-interaction” pathway most frequently affected (17/52, 32.69%), predominantly by mutations in ITGB4, LAMA5, or NID2 (Figure 5).
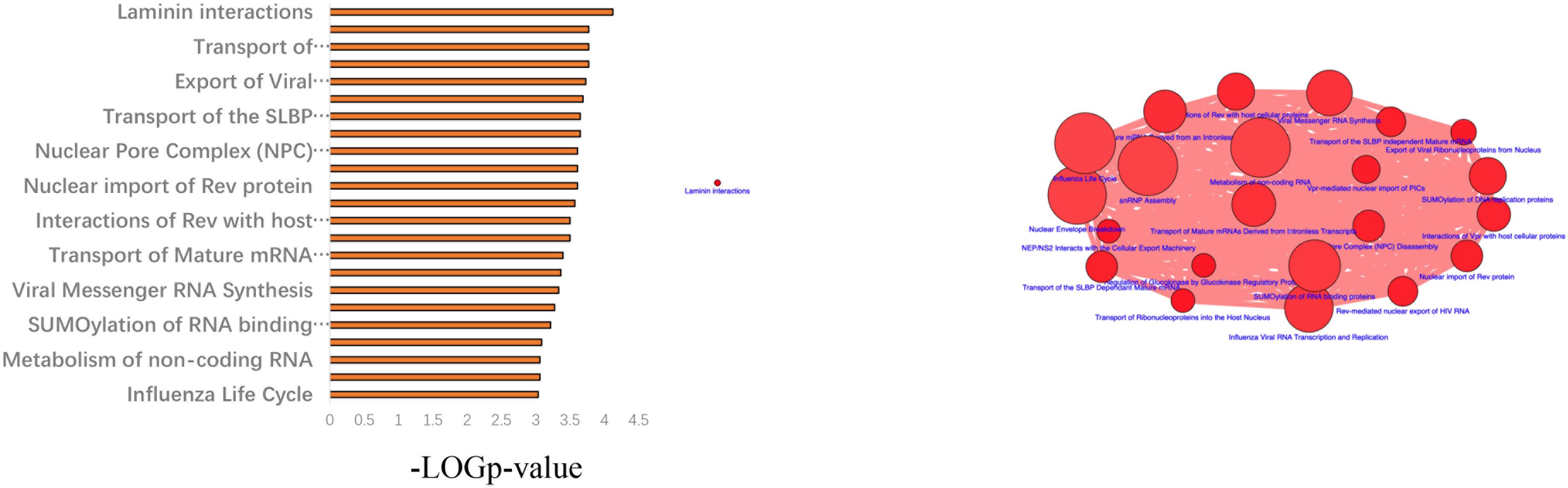
Figure 4. Results of the ConsensusPathDB enrichment analysis of genes found to be mutated in >2 patients with hemifacial microsomia (HFM).
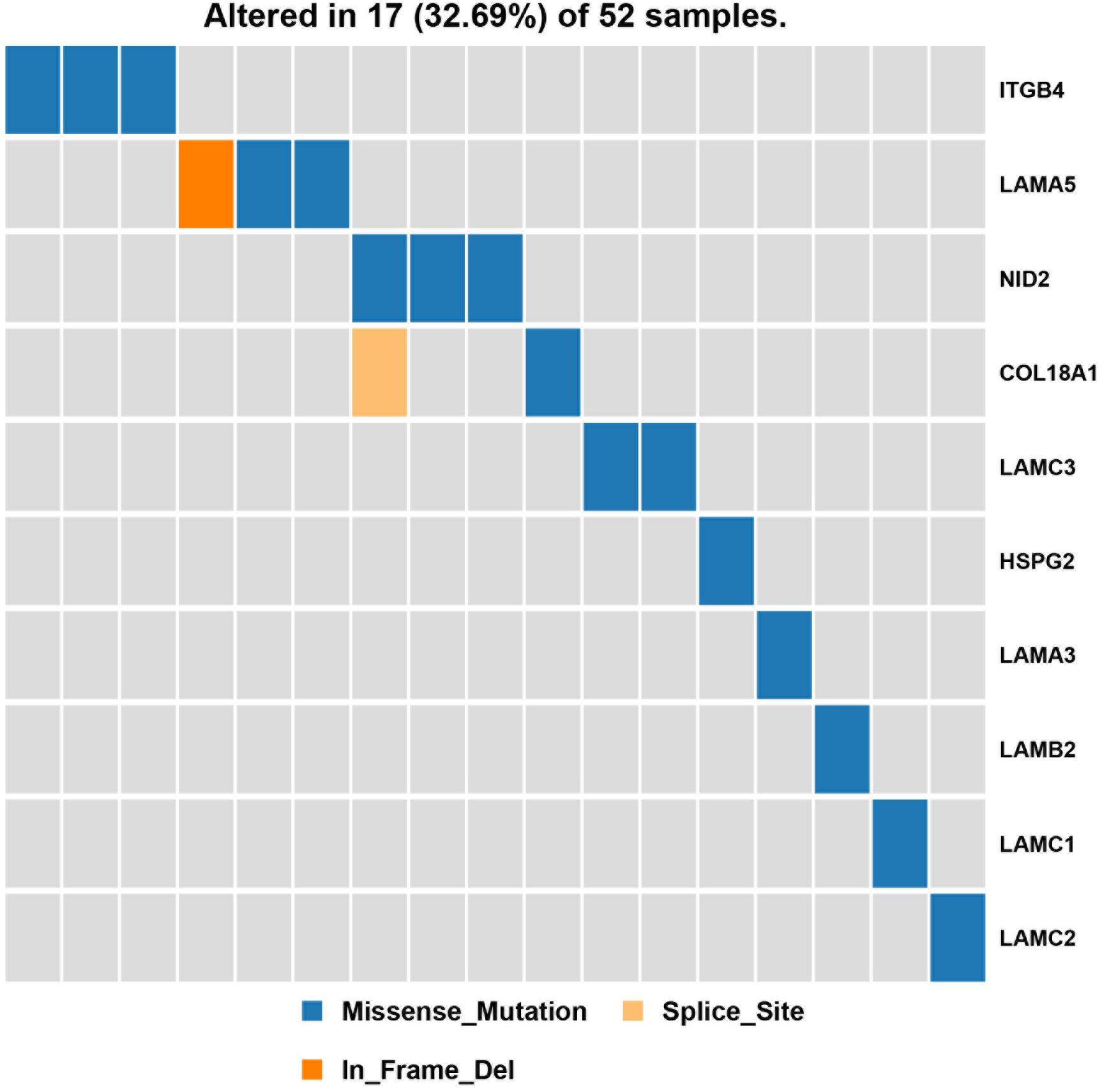
Figure 5. Results of the pathway enrichment analysis of genes found to be mutated in the analyzed hemifacial microsomia (HFM) patient cohort. The “laminin-interaction” pathway was predicted to be most significantly affected by the identified mutations (most often in ITGB4, LAMA5, or NID2) and was likely disrupted in 17 (32.69%) of the analyzed patients.
Discussion
To date, the molecular mechanisms that underlie HFM pathogenesis remain unclear; however, strong evidence suggests that these include genetic factors, as evidenced by the fact that several chromosomal abnormalities and gene mutations have been previously reported in patients with HFM (Table 2). The largest study conducted to date was a genome-wide association study (GWAS) that was performed in 2016 (Zhang et al., 2016). However, that study encompassed all immature derivatives of the first and second pharyngeal arches. Thus, its results may not be closely related to the mandibular malformation. Furthermore, it is worth noting that very few risk variants identified by GWASs to date have been located in functionally significant protein regions (e.g., in the exons or the 5’-untranslated region) (McClellan and King, 2010). Moreover, the GWAS method is generally considered insufficiently sensitive to detect rare variants for rare diseases (McClellan and King, 2010). Given these shortcomings, NGS technologies such as WES have become increasingly important tools for studies aiming to elucidate the pathogenesis of rare diseases (Abul-Husn et al., 2016; Bilguvar et al., 2010; Bowden et al., 2010; Dewey et al., 2016; Lupo et al., 2019). Here, we have performed the first ever WES study of a large cohort of patients with HFM.
In the analyzed patient cohort comprising 52 individuals, we detected mutations in 13 genes that were previously associated with HFM, of which NID2, which encodes a member of the nidogen family of basement membrane proteins, was most frequently mutated. Previous studies have inferred a relationship between NID2 and osteogenic processes, consistent with the fact that the HFM phenotypic spectrum includes mandibular hypoplasia. For example, a previous study showed that NID2 was markedly upregulated in demineralized osseous surfaces compared to its levels in mineralized osseous surfaces, suggesting that NID2 may act as a temporal migration guide (Wischmann et al., 2018). Furthermore, NID2 has been implicated in osteoclastogenesis. TRAP5α, which is involved in osteoclast signaling and RANK signaling in osteoclasts, has been shown to interact with NID2 in cultured 3T3-L1 mouse pre-adipocytes (Patlaka et al., 2014). The fact that patients harboring NID2 mutations develop mandibular hypoplasia rather than mandibular agenesis is likely due to the fact that NID1 and NID2 are found in all vertebrates and exhibit partial functional redundancy. Consistent with this, genetic deletion of either gene alone induces only mild defects in the mouse (Bose et al., 2006).
Among the genes with rare, potentially causative mutations detected in the present study, those in PDE4DIP were found to affect the greatest number of patients in the analyzed cohort. Mutations in PDE4DIP have been previously identified predominantly during sequencing of tumor samples, e.g., in prostate (Gupta et al., 2017), ovarian (Er et al., 2016), or lung cancer (Li et al., 2015), as well as in adult pineoblastoma (Snuderl et al., 2018). To date, however, to the best of our knowledge, there have been no published reports implicating PDE4DIP mutations in HFM or bone formation.
Most previous studies investigating HFM pathogenesis have focused on a single gene mutation or mutations identified in a single patient; however, this approach is problematic given that the etiology of HFM is thought to be highly heterogeneous and dependent on genetic, epigenetic, and environmental factors. Thus, instead of focusing on single genes, in the present study, we conducted a pathway-based association analysis of generated WES data to identify common biological pathways that are likely to be affected in multiple (unrelated) patients with HFM. We suggest that this type of pathway analysis, which combines genomic and functional data and assesses the effect of multiple gene mutations, may be a better method to investigate the pathogenesis of rare diseases. Thus, we explored the distribution of signaling pathways known to be associated with genes that were mutated in >2 sporadic HFM cases. The “laminin-interaction” signaling pathway was predicted to be most frequently disrupted in the present cohort of patients with HFM, predominantly by mutations in ITGB4, NID2, or LAMA5. This is consistent with the fact that previous reports have demonstrated a close relationship (either positive or negative) between laminin interaction and bone formation during both osteogenesis and osteoclastogenesis (Langen et al., 2017; Susek et al., 2018).
The limitations of the present study are that candidate genes and pathways were not confirmed; further functional experiments are needed to confirm the candidate genes and pathways. Furthermore, it should be noted that the diverse phenotypic spectrum of HFM is thought to be the result of gene-environment interactions and etiologic heterogeneity, which collectively cause incomplete penetrance and variable expression (Birgfeld and Heike, 2019). Heterozygous mutations in reference alleles causing organ specificity in HFM may be affected by environmental and other factors. Many additional studies, such as RNA sequencing or whole-genome methylation studies, are needed to provide further information regarding this issue. In addition, we unfortunately failed to collect biological samples from the parents of the individuals involved. This was because the majority of patients came to our department with only one parent, and some of them refused to provide biological samples.
Conclusion
In summary, this was the first study of rare germline mutations in a cohort of individuals with HFM. The likely disruptions in the signaling pathways due to the described mutations may be considered potential pathogenic causes of HFM and therefore, may serve as promising therapeutic targets.
Data Availability Statement
The data presented in this study are deposited in NCBI online repositories. The BioProject ID is PRJNA626382. The SRA accession numbers for these isolates are from SAMN14639160 to 14639211.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
XC, FL, YZ, and GC contributed to conception and design of the study. XC and ZM collected the clinical data and samples. FL performed the bioinformatics analysis. XC wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This study was funded by Shanghai Municipal Key Clinical Specialty-shslczdzk00901 (ZWJCB18), Science and Technology Commission of Shanghai Municipality (19441912300 and 18DZ2201900), and Clinical Research Program of the 9th People’s Hospital, Shanghai Jiao Tong University School of Medicine (JYLJ031).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Editage (www.editage.com) for English language editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.580761/full#supplementary-material
Supplementary Table 1 | Detailed information about all the mutations mentioned in the manuscript.
Footnotes
- ^ http://genetics.bwh.harvard.edu/pph2/
- ^ http://sift.jcvi.org
- ^ http://consensuspathdb.org
- ^ https://reactome.org/
References
Abul-Husn, N. S., Manickam, K., Jones, L. K., Wright, E. A., Hartzel, D. N., Gonzaga-Jauregui, C., et al. (2016). Genetic identification of familial hypercholesterolemia within a single U.S. health care system. Science 354:aaf7000.
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods. 7, 248–249. doi: 10.1038/nmeth0410-248
Ala-Mello, S., Siggberg, L., Knuutila, S., von Koskull, H., Taskinen, M., and Peippo, M. (2008). Further evidence for a relationship between the 5p15 chromosome region and the oculoauriculovertebral anomaly. Am. J. Med. Genet. A 146A, 2490–2494. doi: 10.1002/ajmg.a.32479
Alasti, F., and Van Camp, G. (2009). Genetics of microtia and associated syndromes. J. Med. Genet. 46, 361–369. doi: 10.1136/jmg.2008.062158
Ballesta-Martinez, M. J., Lopez-Gonzalez, V., Dulcet, L. A., Rodriguez-Santiago, B., Garcia-Minaur, S., and Guillen-Navarro, E. (2013). Autosomal dominant oculoauriculovertebral spectrum and 14q23.1 microduplication. Am. J. Med. Genet. A 161A, 2030–2035. doi: 10.1002/ajmg.a.36007
Beleza-Meireles, A., Clayton-Smith, J., Saraiva, J. M., and Tassabehji, M. (2014). Oculo-auriculo-vertebral spectrum: a review of the literature and genetic update. J. Med. Genet. 51, 635–645. doi: 10.1136/jmedgenet-2014-102476
Beleza-Meireles, A., Hart, R., Clayton-Smith, J., Oliveira, R., Reis, C. F., Venancio, M., et al. (2015). Oculo-auriculo-vertebral spectrum: clinical and molecular analysis of 51 patients. Eur. J. Med. Genet. 58, 455–465.
Berenguer, M., Tingaud-Sequeira, A., Colovati, M., Melaragno, M. I., Bragagnolo, S., Perez, A. B. A., et al. (2017). A novel de novo mutation in MYT1, the unique OAVS gene identified so far. Eur. J. Hum. Genet. 25, 1083–1086. doi: 10.1038/ejhg.2017.101
Bilguvar, K., Ozturk, A. K., Louvi, A., Kwan, K. Y., Choi, M., Tatli, B., et al. (2010). Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature 467, 207–210. doi: 10.1038/nature09327
Bose, K., Nischt, R., Page, A., Bader, B. L., Paulsson, M., and Smyth, N. (2006). Loss of nidogen-1 and -2 results in syndactyly and changes in limb development. J. Biol. Chem. 281, 39620–39629. doi: 10.1074/jbc.m607886200
Bowden, D. W., An, S. S., Palmer, N. D., Brown, W. M., Norris, J. M., Haffner, S. M., et al. (2010). Molecular basis of a linkage peak: exome sequencing and family-based analysis identify a rare genetic variant in the ADIPOQ gene in the IRAS Family Study. Hum. Mol. Genet. 19, 4112–4120. doi: 10.1093/hmg/ddq327
Bragagnolo, S., Colovati, M. E. S., Souza, M. Z., Dantas, A. G., de Soares, M. F. F., Melaragno, M. I., et al. (2018). Clinical and cytogenomic findings in OAV spectrum. Am. J. Med. Genet. A 176, 638–648. doi: 10.1002/ajmg.a.38576
Chen, X., Xu, F., Liu, F., Aung, Z. M., Chen, W., Han, W., et al. (2018). Whole-exome sequencing for monozygotic twins discordant for hemifacial microsomia. J. Craniomaxillofac. Surg. 46, 802–807. doi: 10.1016/j.jcms.2018.02.005
Cingolani, P., Platts, A., Wang le, L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. doi: 10.4161/fly.19695
Colovati, M. E., Bragagnolo, S., Guilherme, R. S., Dantas, A. G., Soares, M. F., Kim, C. A., et al. (2015). Atypical 581-kb 22q11.21 deletion in a patient with oculo-auriculo-vertebral spectrum phenotype. Cytogenet. Genome Res. 147, 130–134. doi: 10.1159/000444228
Dewey, F. E., Murray, M. F., Overton, J. D., Habegger, L., Leader, J. B., Fetterolf, S. N., et al. (2016). Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 354:aaf6814.
Er, T. K., Su, Y. F., Wu, C. C., Chen, C. C., Wang, J., Hsieh, T. H., et al. (2016). Targeted next-generation sequencing for molecular diagnosis of endometriosis-associated ovarian cancer. J. Mol. Med. 94, 835–847. doi: 10.1007/s00109-016-1395-2
Guida, V., Sinibaldi, L., Pagnoni, M., Bernardini, L., Loddo, S., Margiotti, K., et al. (2015). A de novo proximal 3q29 chromosome microduplication in a patient with oculo auriculo vertebral spectrum. Am. J. Med. Genet. A 167A, 797–801. doi: 10.1002/ajmg.a.36951
Gupta, S., Li, J., Kemeny, G., Bitting, R. L., Beaver, J., Somarelli, J. A., et al. (2017). Whole genomic copy number alterations in circulating tumor cells from men with abiraterone or enzalutamide-resistant metastatic castration-resistant prostate cancer. Clin. Cancer Res. 23, 1346–1357. doi: 10.1158/1078-0432.ccr-16-1211
Huang, X.-S., Xiao, L., Li, X., Xie, Y., Jiang, H.-O., Tan, C., et al. (2010). Two neighboring microdeletions of 5q13.2 in a child with oculo-auriculo-vertebral spectrum. Eur. J. Med. Genet. 53, 153–158. doi: 10.1016/j.ejmg.2010.03.004
Kamburov, A., Stelzl, U., Lehrach, H., and Herwig, R. (2013). The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 41, D793–D800.
Koboldt, D. C., Zhang, Q., Larson, D. E., Shen, D., McLellan, M. D., Lin, L., et al. (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. doi: 10.1101/gr.129684.111
Kosaki, R., Fujimaru, R., Samejima, H., Yamada, H., Izumi, K., Iijima, K., et al. (2007). Wide phenotypic variations within a family with SALL1 mutations: Isolated external ear abnormalities to Goldenhar syndrome. Am. J. Med. Genet. A 143A, 1087–1090. doi: 10.1002/ajmg.a.31700
Kumar, P., Henikoff, S., and Ng, P. C. (2009). Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081. doi: 10.1038/nprot.2009.86
Langen, U. H., Pitulescu, M. E., Kim, J. M., Enriquez-Gasca, R., Sivaraj, K. K., Kusumbe, A. P., et al. (2017). Cell-matrix signals specify bone endothelial cells during developmental osteogenesis. Nat. Cell Biol. 19, 189–201. doi: 10.1038/ncb3476
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, S., Wang, L., Ma, Z., Ma, Y., Zhao, J., Peng, B. O., et al. (2015). Sequencing study on familial lung squamous cancer. Oncol. Lett. 10, 2634–2638. doi: 10.3892/ol.2015.3583
Lopez, E., Berenguer, M., Tingaud-Sequeira, A., Marlin, S., Toutain, A., Denoyelle, F., et al. (2016). Mutations in MYT1, encoding the myelin transcription factor 1, are a rare cause of OAVS. J. Med. Genet. 53, 752–760. doi: 10.1136/jmedgenet-2016-103774
Lupo, P. J., Mitchell, L. E., and Jenkins, M. M. (2019). Genome-wide association studies of structural birth defects: a review and commentary. Birth Defects Res. 111, 1329–1342. doi: 10.1002/bdr2.1606
McClellan, J., and King, M. C. (2010). Genetic heterogeneity in human disease. Cell 141, 210–217. doi: 10.1016/j.cell.2010.03.032
McLaren, W., Gil, L., Hunt, S. E., Riat, H. S., Ritchie, G. R., Thormann, A., et al. (2016). The ensembl variant effect predictor. Genome Biol. 17:122.
Northup, J. K., Matalon, D., Hawkins, J. C., Matalon, R., and Velagaleti, G. V. (2010). Pericentric inversion, inv(14)(p11.2q22.3), in a 9-month old with features of Goldenhar syndrome. Clin. Dysmorphol. 19, 185–189. doi: 10.1097/mcd.0b013e3283359386
Ou, Z., Martin, D. M., Bedoyan, J. K., Cooper, M. L., Chinault, A. C., Stankiewicz, P., et al. (2008). Branchiootorenal syndrome and oculoauriculovertebral spectrum features associated with duplication of SIX1, SIX6, and OTX2 resulting from a complex chromosomal rearrangement. Am. J. Med. Genet. A 146A, 2480–2489. doi: 10.1002/ajmg.a.32398
Patlaka, C., Mai, H. A., Lang, P., and Andersson, G. (2014). The growth factor-like adipokine tartrate-resistant acid phosphatase 5a interacts with the rod G3 domain of adipocyte-produced nidogen-2. Biochem. Biophys. Res. Commun. 454, 446–452. doi: 10.1016/j.bbrc.2014.10.112
Quintero-Rivera, F., and Martinez-Agosto, J. A. (2013). Hemifacial microsomia in cat-eye syndrome: 22q11.1-q11.21 as candidate loci for facial symmetry. Am. J. Med. Genet. A 161A, 1985–1991. doi: 10.1002/ajmg.a.35895
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J., and Kircher, M. (2019). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894.
Rooryck, C., Souakri, N., Cailley, D., Bouron, J., Goizet, C., Delrue, M. A., et al. (2010). Array-CGH analysis of a cohort of 86 patients with oculoauriculovertebral spectrum. Am. J. Med. Genet. A 152A, 1984–1989. doi: 10.1002/ajmg.a.33491
Rooryck, C., Stef, M., Burgelin, I., Simon, D., Souakri, N., Thambo, J. B., et al. (2009). 2.3 Mb terminal deletion in 12p13.33 associated with oculoauriculovertebral spectrum and evaluation of WNT5B as a candidate gene. Eur. J. Med. Genet. 52, 446–449. doi: 10.1016/j.ejmg.2009.08.005
Snuderl, M., Kannan, K., Pfaff, E., Wang, S., Stafford, J. M., Serrano, J., et al. (2018). Recurrent homozygous deletion of DROSHA and microduplication of PDE4DIP in pineoblastoma. Nat. Commun. 9:2868.
Spineli-Silva, S., Bispo, L. M., Gil-da-Silva-Lopes, V. L., and Vieira, T. P. (2018). Distal deletion at 22q11.2 as differential diagnosis in Craniofacial microsomia: case report and literature review. Eur. J. Med. Genet. 61, 262–268. doi: 10.1016/j.ejmg.2017.12.013
Su, P. H., Liu, Y. F., Yu, J. S., Chen, J. Y., Chen, S. J., and Lai, Y. J. (2012). Facial asymmetry and clinical manifestations in patients with novel insertion of the TCOF1 gene. Clin. Genet. 82, 460–465. doi: 10.1111/j.1399-0004.2011.01765.x
Susek, K. H., Korpos, E., Huppert, J., Wu, C., Savelyeva, I., Rosenbauer, F., et al. (2018). Bone marrow laminins influence hematopoietic stem and progenitor cell cycling and homing to the bone marrow. Matrix Biol. 67, 47–62. doi: 10.1016/j.matbio.2018.01.007
Terhal, P., Rosler, B., and Kohlhase, J. (2006). A family with features overlapping Okihiro syndrome, hemifacial microsomia and isolated Duane anomaly caused by a novel SALL4 mutation. Am. J. Med. Genet. A 140, 222–226. doi: 10.1002/ajmg.a.31060
Torti, E. E., Braddock, S. R., Bernreuter, K., and Batanian, J. R. (2013). Oculo-auriculo-vertebral spectrum, cat eye, and distal 22q11 microdeletion syndromes: a unique double rearrangement. Am. J. Med. Genet. A 161A, 1992–1998. doi: 10.1002/ajmg.a.35918
Tuin, A. J., Tahiri, Y., Paine, K. M., Paliga, J. T., Taylor, J. A., and Bartlett, S. P. (2015). Clarifying the relationships among the different features of the OMENS+ classification in craniofacial microsomia. Plast. Reconstruct. Surg. 135, 149e–156e.
Wischmann, J., Lenze, F., Thiel, A., Bookbinder, S., Querido, W., Schmidt, O., et al. (2018). Matrix mineralization controls gene expression in osteoblastic cells. Exp. Cell Res. 372, 25–34. doi: 10.1016/j.yexcr.2018.09.005
Zhang, Y. B., Hu, J., Zhang, J., Zhou, X., Li, X., Gu, C., et al. (2016). Genome-wide association study identifies multiple susceptibility loci for craniofacial microsomia. Nat. Commun. 7:10605.
Zhu, L., Peng, J. L., Harutyunyan, K. G., Garcia, M. D., Justice, M. J., and Belmont, J. W. (2007). Craniofacial, skeletal, and cardiac defects associated with altered embryonic murine Zic3 expression following targeted insertion of a PGK-NEO cassette. Front. Biosci. 12:1680–1690. doi: 10.2741/2180
Keywords: hemifacial microsomia, whole-exome sequencing, rare germline mutations, pathway enrichment analysis, mandibular hypoplasia
Citation: Chen X, Liu F, Mar Aung Z, Zhang Y and Chai G (2021) Whole-Exome Sequencing Reveals Rare Germline Mutations in Patients With Hemifacial Microsomia. Front. Genet. 12:580761. doi: 10.3389/fgene.2021.580761
Received: 07 July 2020; Accepted: 06 April 2021;
Published: 17 May 2021.
Edited by:
Loredana Bury, University of Perugia, ItalyReviewed by:
Leslie Matalonga, National Center for Genomic Analysis, Center for Genomic Regulation (CRG), SpainNelson L. S. Tang, The Chinese University of Hong Kong, China
Copyright © 2021 Chen, Liu, Mar Aung, Zhang and Chai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gang Chai, Y2hhaWcxMDgxQHNoOWhvc3BpdGFsLm9yZy5jbg==; Yan Zhang, emhhbmd5MTMzMEBzaDlob3NwaXRhbC5vcmcuY24=