 Indrajit Saha
Indrajit Saha Nimisha Ghosh
Nimisha Ghosh Debasree Maity3
Debasree Maity3 Dariusz Plewczynski
Dariusz Plewczynski
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
METHODS article
Front. Genet., 11 February 2021
Sec. Computational Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.569120
This article is part of the Research TopicSARS-CoV-2: From Genetic Variability to Vaccine DesignView all 13 articles
The COVID-19 disease for Novel coronavirus (SARS-CoV-2) has turned out to be a global pandemic. The high transmission rate of this pathogenic virus demands an early prediction and proper identification for the subsequent treatment. However, polymorphic nature of this virus allows it to adapt and sustain in different kinds of environment which makes it difficult to predict. On the other hand, there are other pathogens like SARS-CoV-1, MERS-CoV, Ebola, Dengue, and Influenza as well, so that a predictor is highly required to distinguish them with the use of their genomic information. To mitigate this problem, in this work COVID-DeepPredictor is proposed on the framework of deep learning to identify an unknown sequence of these pathogens. COVID-DeepPredictor uses Long Short Term Memory as Recurrent Neural Network for the underlying prediction with an alignment-free technique. In this regard, k-mer technique is applied to create Bag-of-Descriptors (BoDs) in order to generate Bag-of-Unique-Descriptors (BoUDs) as vocabulary and subsequently embedded representation is prepared for the given virus sequences. This predictor is not only validated for the dataset using -fold cross-validation but also for unseen test datasets of SARS-CoV-2 sequences and sequences from other viruses as well. To verify the efficacy of COVID-DeepPredictor, it has been compared with other state-of-the-art prediction techniques based on Linear Discriminant Analysis, Random Forests, and Gradient Boosting Method. COVID-DeepPredictor achieves 100% prediction accuracy on validation dataset while on test datasets, the accuracy ranges from 99.51 to 99.94%. It shows superior results over other prediction techniques as well. In addition to this, accuracy and runtime of COVID-DeepPredictor are considered simultaneously to determine the value of k in k-mer, a comparative study among k values in k-mer, Bag-of-Descriptors (BoDs), and Bag-of-Unique-Descriptors (BoUDs) and a comparison between COVID-DeepPredictor and Nucleotide BLAST have also been performed. The code, training, and test datasets used for COVID-DeepPredictor are available at http://www.nitttrkol.ac.in/indrajit/projects/COVID-DeepPredictor/.
The first case of COVID-19 surfaced in Wuhan, China in December 2019 (Huang et al., 2020; Meng et al., 2020; Yan L. et al., 2020). In no time it spread to 212 countries and territories (Worldometer, 2021) worldwide creating a pandemic in its wake. SARS-CoV-2 falls in the same family as SARS-CoV and MERS-CoV (all belong to the family of coronavirus) and mainly targets the respiratory system (Zhou et al., 2020). As of 8th January 2021, over 885 million cases of COVID-19 have been reported worldwide, with more than 1,906 thousand cases of death and 63.6 million cases of recovery (Worldometer, 2021).
SARS-CoV-2 is defined as an enveloped, positive-sense, single-stranded RNA virus with a genome of around 30 kilobases in length (Weiss and Navas-Martin, 2005; Su et al., 2016; Cui et al., 2019). RNA viruses generally have very high mutation rates (Jenkins et al., 2002; Woo et al., 2009). Genetic mutation can occur infrequently between viruses of the same species but of divergent lineages. The resulting mutated viruses may sometimes cause an outbreak of infection in humans e.g., the case of SARS-CoV-2. Coronavirus results from zoonotic transmission to human and shows symptoms of pneumonia, fever, and breathing difficulties (Guan et al., 2003; Alagaili et al., 2014). Human to human transmission has also been confirmed for SARS-CoV-2 (Chan et al., 2020; Huang et al., 2020). Next-generation sequencing using metagenomic analysis has recently been used to identify the genetic features of SARS-CoV-2 (Zhou et al., 2020).
There have been several analysis regarding SARS-CoV-2. This include whole genome analysis of a virus and viral protein-based comparisons which have resulted in the conclusion that SARS-CoV-2 is mostly related to two bat SARS-like coronaviruses (Chan et al., 2020; Lu et al., 2020). Phylogenetic analysis of full genome alignment and similarity plot show that SARS-CoV-2 has high similarity with bat coronavirus RaTG13 (Paraskevis et al., 2020). Furthermore, another study (Wan et al., 2020) has shown that spike protein receptor-binding domain (RBD) of SARS-CoV-2 binds with host receptor angiotensin-converting enzyme 2 (ACE2), just like other Sarbecovirus strains, thus making the claim that SARS-CoV-2 originated from bat very likely (Letko et al., 2020; Liu and Wang, 2020).
As the genomic structure of SARS-CoV-2 is similar to the other viruses of the same family, and it shows similar symptoms like them, the early prediction of SARS-CoV-2 is a very challenging task. Ozturk et al. (2020) have used deep neural networks with X-ray images for automated detection of SARS-CoV-2 cases. The results show that the method has a prediction accuracy of 98.08% for binary classes (COVID vs. No-Findings) and 87.02% for multiple classes (COVID vs. No-Findings vs. Pneumonia). Another work (Yan Q. et al., 2020) where deep learning has been used to predict age-related macular degeneration (AMD) which is a leading cause of blindness among the elderly population. The results show an average area under the curve (AUC) value of 0.85. On the other hand, the authors in Koohi-Moghadam et al. (2019) have used deep learning approach to predict disease-associated mutation of metal-binding sites in proteins. The prediction results depict AUC as 0.90 and an accuracy of 0.82. These encouraging results show that deep learning has the potential for highly accurate prediction. This led us to devise a predictor based on deep learning which uses genomic sequences of pathogenic viruses. In this work, a deep learning technique, viz. COVID-DeepPredictor based on Long-Short Term Memory (LSTM) (Hochreiter and Schmidhuber, 1997; Tang et al., 2019) is developed. Though, LSTM has been profusely used in many works for text classification (Jin et al., 2019; Liu et al., 2019; Zhang et al., 2019), to the best of the authors' knowledge, this is the first attempt to use LSTM for the prediction of SARS-CoV-2 using genomic sequences of virus considering alignment-free approach. For this purpose, k-mer technique is used to generate Bag-of-Descriptors (BoDs) and consequently Bag-of-Unique-Descriptors (BoUDs) as vocabulary. Subsequently embedded representation is prepared for the given virus sequences using BoDs and BoUDs. It is worth mentioning that, though SARS-CoV-2 is a single-stranded RNA virus, the genomic information of a virus is captured in the form of DNA sequence. These DNA sequences are used in this work to predict SARS-CoV-2 and other pathogenic viruses viz. SARS-CoV-1, MERS-CoV, Ebola, Dengue, and Influenza. COVID-DeepPredictor achieves 100% prediction accuracy on validation dataset while on test datasets, the accuracy ranges from 99.51 to 99.94%. COVID-DeepPredictor also shows superior results over the existing prediction techniques based on Linear Discriminant Analysis, Random Forests, and Gradient Boosting Method. Moreover, apart from prediction accuracy, critical analysis like the choice of k in k-mer by considering the accuracy and runtime of COVID-DeepPredictor simultaneously, a comparative study of Bag-of-Descriptors (BoDs) and Bag-of-Unique-Descriptors (BoUDs) for different values of k and a comparison between an alignment-based technique viz. Nucleotide Basic Local Alignment Search Tool (BLASTN) and COVID-DeepPredictor as alignment-free technique.
In this section, description of dataset preparation that has been used in this work are elucidated, a brief description of Long-Short Term Memory (LSTM) and the detailed discussion of proposed COVID-DeepPredictor are put forth.
The datasets of SARS-CoV-1, MERS-CoV, Ebola, Dengue, and Influenza have been downloaded from NCBI (National Center for Biotechnology Information)1. Dataset for SARS-CoV-2 has been downloaded from NCBI and GISAID (Global Initiative on Sharing All Influenza Data)2. The total number of complete or near-complete genomic sequences of all the pathogenic viruses amounted to 4,643, named as Initial dataset. Additionally, the recent complete or near-complete SARS-CoV-2 sequences of 3,030 during January 2020 to August 2020 are taken from NCBI whereas 2,410 (from February 2020 to July 2020) and 4,000 (from June 2020 to December 2020) sequences are considered from GISAID. For our training purpose, 1,500 samples from 4,643 sequences are taken randomly for training. To ensure that representatives from all the six pathogenic viruses are available and to avoid imbalance class problem, 250 samples from each pathogenic viruses are taken in the training dataset. In order to perform testing, five different test datasets are created and named as Testdata-1, Testdata-2, Testdata-3, Testdata-4, and Testdata-5. It is important to mention that Testdata-1 consists of the remaining 3,143 sequences out of 4,643 sequences, while Testdata-2 contains 200 sequences each for MERS-CoV, SARS-CoV-2, Ebola, Dengue, and Influenza and 90 sequences of SARS-CoV-1 from different sources. Moreover, Testdata-3, Testdata-4, and Tetsdata-5 comprise of recent SARS-CoV-2 sequences from NCBI and GISAID respectively along with other pathogenic viruses. The statistics of Initial dataset as well as training and testing datasets are given in Table 1. It is worth mentioning that in this work more than 10,000 SARS-CoV-2 genomic sequences have been used from January 2020 to December 2020 considering different sources in order to develop COVID-DeepPredictor.
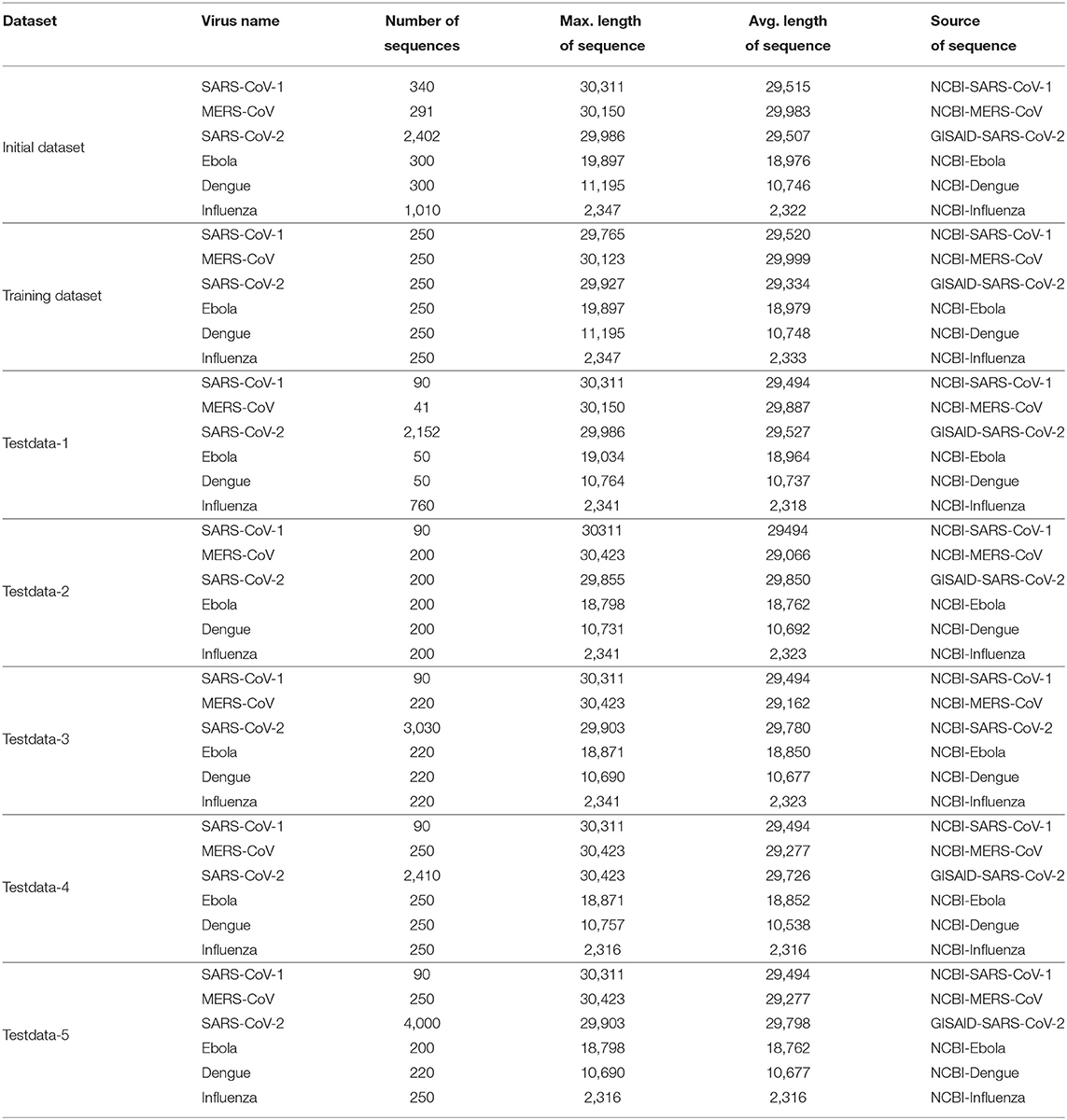
Table 1. Description of initial, training, and test datasets.
All the experiments are performed with the training and testing datasets as mentioned in Table 1. For the visualization of all the virus sequences (SARS-CoV-1, MERS-CoV, SARS-CoV-2, Ebola, Dengue, and Influenza), t-distributed Stochastic Neighbor Embedding (tSNE) (Hinton and Roweis, 2003) is applied on 4,643 sequences after generating the count vector (Khattak et al., 2019) using k-mer technique (Manekar and Sathe, 2018; Solis-Reyes et al., 2018). In this regard, the number of clusters known apriori is six and such embedded representation of virus sequences is shown in Figure 1A along with the distribution of initial SARS-CoV-2 sequences in 56 countries in Figure 1B. It is to be noted that COVID-DeepPredictor is developed in MATLAB R2020a.
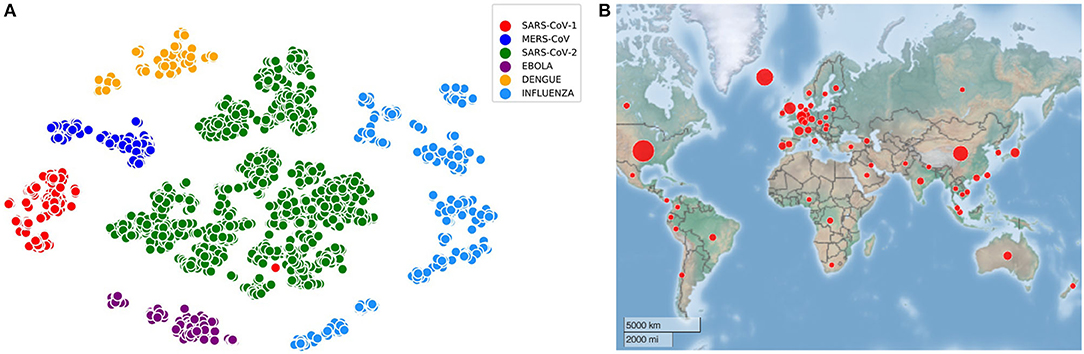
Figure 1. Visualization of virus sequences and their spread. (A) Embedded representation of SARS-CoV-1, MERS-CoV, SARS-CoV-2, Ebola, Dengue, and Influenza (k=3) (B) 56 countries considered for COVID-DeepPredictor are spread across the globe.
Long-Short Term Memory (LSTM) is a type of recurrent neural network (sub-branch of deep learning) which is capable of learning order dependence in sequence prediction problems. The main components of an LSTM network are sequence input layer and an LSTM layer. A sequence input layer provides text as an input into the LSTM network. An LSTM layer learns long-term association between steps of sequence data. Elaborately speaking, an LSTM network acquires a context vector from previous time step and an input vector from the given data. This is used to calculate the next context and gate vectors to control memory cell state vector (Kim et al., 2018). With an input data at time t and a context vector h, a raw cell vector and input vectors for each gate are created by one hidden layer. At the input gate, the cell vector is then multiplied by the input vector. The cell input is added to given previous cell vector weighted by the forget vector. Then the resultant vector is controlled by the output vector. The update of the cell is controlled by the control gate. LSTM is mainly trained using Back-propagation Through Time and mitigates the vanishing gradient problem that is quite rampant in RNN. In LSTM, the memory cells and the gates can store time and thus can eliminate old observations overcoming vanishing gradient problem.
To sum up, LSTM consists of four gates, input gate (it), forget gate (ft), control gate (Ct), and output gate (ot). Considering a sentence S = x1, x2, ..., xK, where K is the length of a sentence, the equations for LSTM can be depicted as:
Here, W are weight matrices, ht−1 is the hidden layer which is used updated by the output layer and is also responsible for updating the output and tanh and sigm, respectively represent the tanh-activation and sigmoid-activation functions.
The main objective of COVID-DeepPredictor is to correctly predict the virus classes based on the given genomic sequences of the different pathogenic viruses using an alignment-free technique. To achieve this, the entire genomic sequence is initially divided into descriptors of sequences called as Bag-of-Descriptors (BoDs) using the popular k-mer technique. Here, descriptors are patterns of the genomic sequences of length k. Thereafter, Bag-of-Unique-Descriptors (BoUDs) as vocabulary are created using such BoDs. With the use of BoDs and BoUDs, an embedded representation is created of size N × M where N is the number of genomic sequences and M is the indices of the descriptors in vocabulary. This embedded representation is then used to train COVID-DeepPredictor. Since we have divided the genomic sequences into descriptors and represented in the form of tokens, they behave like texts, thus boiling down to a text classification problem. The pipeline of the proposed COVID-DeepPredictor is shown in Figure 2.
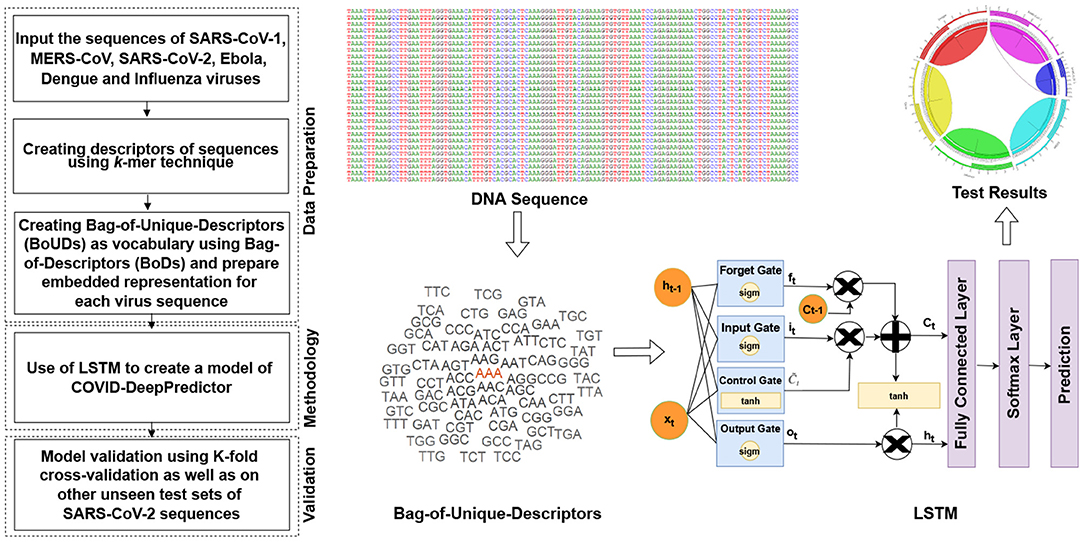
Figure 2. Pipeline of COVID-DeepPredictor. The pipeline is depicted in the form of a flowchart and then represented diagrammatically where the symbols are defined as above.
To validate COVID-DeepPredictor, experiments are conducted on genomic sequences of different pathogenic viruses. In this regard, MATLAB R2020a is used on an Intel Core i5-8250U CPU @ 1.80 GHz machine with 8 GB RAM and Windows 10 operating system. The parameters of the underlying predictor, LSTM of COVID-DeepPredictor have been set experimentally. In this regard, the number of hidden units for LSTM layer is set as 80. Next, to use the LSTM layer for a sequence-to-label prediction problem, the output mode is set to “last.” Finally, a fully connected layer with the same size as the number of classes, a softmax layer and a prediction layer are added as well. Mini-batch gradient descent is used to train LSTM. The mini-batch size is specified as 16 and the gradient threshold is set to 2. The COVID-DeepPredictor is compared with other predictors based on Linear Discriminant Analysis (LDA), Random Forests (RFs), and Gradient Boosting Method (GBM). For LDA, the discriminant type is considered to be pseudo-linear, for Random Forests, the number of trees taken are 50 and for GBM the maximum depth of the tree is 10 and maximum iterations are taken as 100. All these parameters are set experimentally.
Each predictor has been evaluated using -fold cross-validation ( = 10) technique followed by further validation on unseen test datasets. The cross-validation partition uses random non-stratified sampling method which is applied to prepare the training and validation datasets resulting in a total of 1,500 samples. The training and validation datasets consist of all the pathogenic virus classes; SARS-CoV-1, MERS-CoV, SARS-CoV-2, Ebola, Dengue, and Influenza. For each predictor, the descriptors of the sequences of the viruses are created using k-mer method. Thereafter to train the COVID-DeepPredictor and the other compared predictors, an embedded matrix of size N × M is created with the use of BoDs and BoUDs.
To determine the performance of COVID-DeepPredictor and the other predictors, Confusion Matrix (Luque et al., 2019) is considered. In confusion matrix, True Positives (TP) refer to a data being correctly identified and they are represented by the diagonal elements. The remaining predictions lead to an error ϵ. Moreover, False Positives (FP) for a particular class refer to the sum of the values in the corresponding column, excluding the TP and False Negatives (FN) for a class is the sum of the values in the corresponding row, excluding the TP. Lastly, True Negatives (TN) for a class is the sum of all columns and row, barring the one for itself. To evaluate the results of COVID-DeepPredictor, metrics like Accuracy, Precision, Recall, and G-Mean have been considered which can be deduced from a confusion matrix. They can be calculated as:
Accuracy:
Precision:
Recall:
G-mean:
Different existing state-of-the-art predictors based on Linear Discriminant Analysis (LDA), Random Forests (RFs), and Gradient Boosting Method (GBM) are used in this work for comparison purposes. LDA is a very popular machine learning tool for prediction. In LDA, each dependent variable is expressed as a linear combination of other features. RFs are ensemble learning methods which build numerous decision trees during training and as an output produces the class that is the mode of the classes. GBM is also an ensemble learning model which produces a prediction model in the form of an ensemble weak prediction models, usually decision trees.
For conducting the experiments, first and foremost, we need to determine the value of k in k-mer. In order to do this, the experiments have been conducted on five test datasets as mentioned in section 2. The results are shown in Figures 3A–E, where k is varied from 3 to 15 with accuracy and running time of COVID-DeepPredictor. It can be seen from figures that the accuracy is higher at k = 3 for all the five test datasets. Although, the same accuracy can be found for other k values as well, e.g., in Figure 3A k = 9, 11, and 13 show the same accuracy, as we increase the k-mer value, the run time increases. This trend of increasing time with the increasing value of k-mer has also been shown in Solis-Reyes et al. (2018). Keeping this in context, we have taken the value of k in k-mer to be 3 as with this value, the run time is least. For the compared predictors based on LDA, RF, and GBM, the k values are similarly determined as 13, 4, and 4, respectively. In this work, -fold cross-validation with = 10 is used. The average results in terms of accuracy for the test datasets are shown in Figure 4A. Moreover, apart from accuracy, different metrics such as precision, recall and g-mean have also been computed for the test datasets and reported in Table 2. As can be seen from the results of Figure 4A, for COVID-DeepPredictor the accuracy ranges from 99.51 to 99.94%. Thus, the experiments establish the fact that COVID-DeepPredictor can detect SARS-CoV-2 with a very high accuracy. The confusion matrices as circos plots for Testdata-1 and Testdata-2 (k = 3) are shown in Figures 4B,C. It can be seen from Figures 4B,C that there is only one misprediction, where SARS-CoV-1 has been wrongly predicted as SARS-CoV-2. The confusion matrices for Testdata-3, Testdata-4, and Testdata-5 (k = 3) are shown in Supplementary Figure 2.
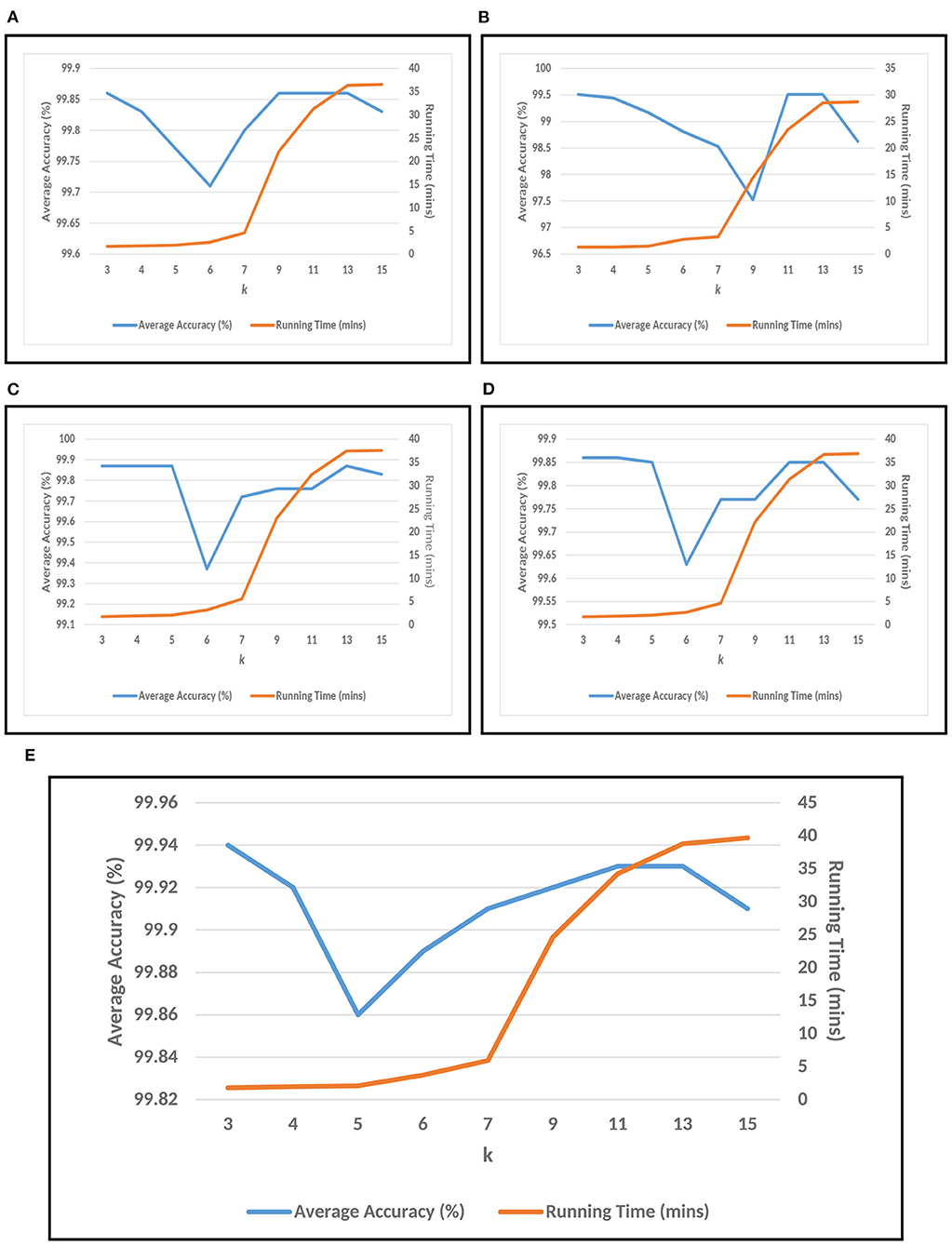
Figure 3. Choosing k value of k-mer for COVID-DeepPredictor based on accuracy and running time. (A) Testdata-1, (B) Testdata-2, (C) Testdata-3, (D) Testdata-4, (E) Testdata-5.
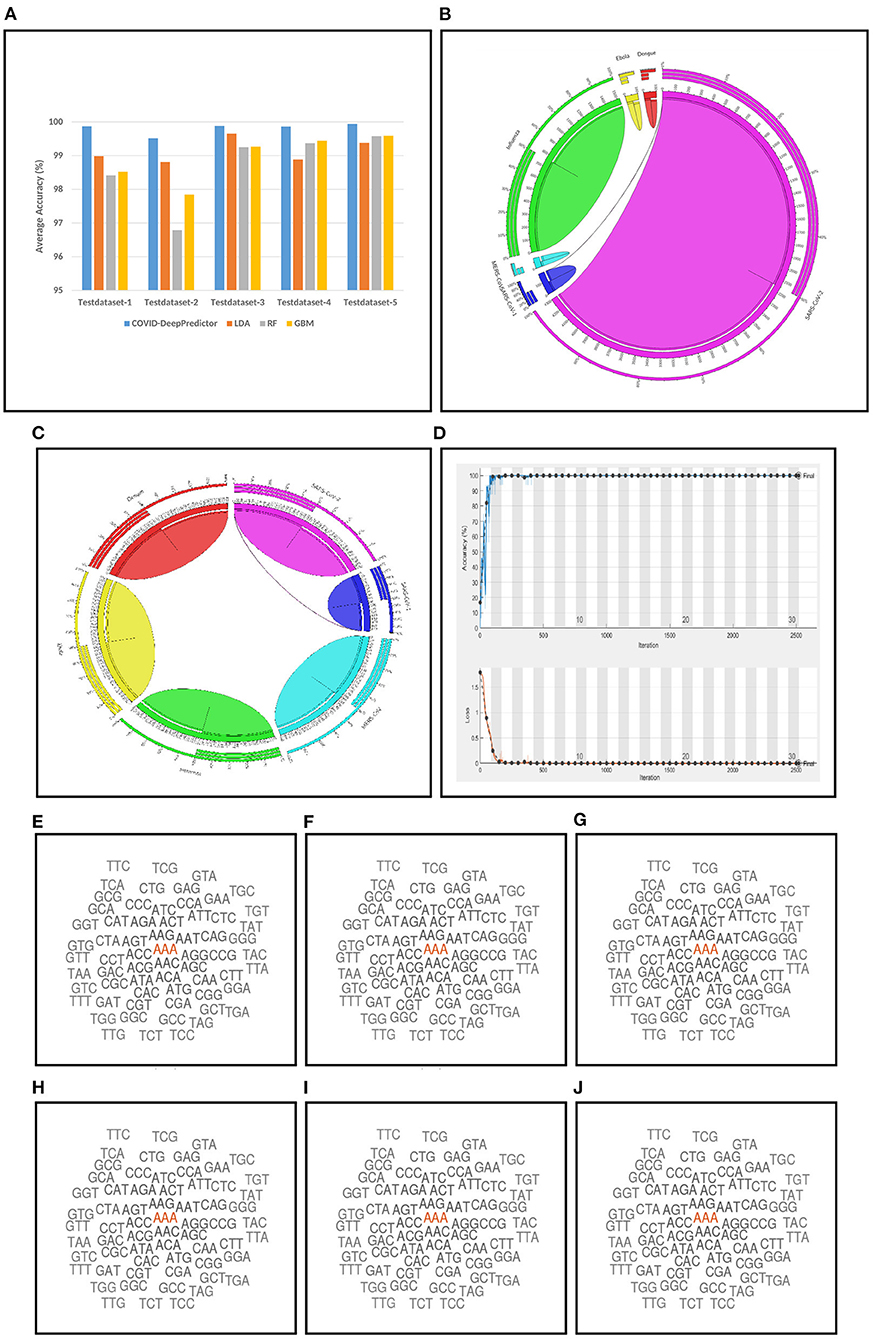
Figure 4. Results related to COVID-DeepPredictor. (A) Prediction performance of COVID-DeepPredictor and other compared methods in terms of average accuracy for the five test datasets. Circos plots of confusion matrix for COVID-DeepPredictor (k=3) for (B) Testdata-1 (C) Testdata-2. (D) Convergence plot of COVID-DeepPredictor. Word cloud of k-mer descriptors (k=3) of genome sequences for (E) SARS-CoV-1 (F) MERS-CoV (G) SARS-CoV-2 (H) Ebola (I) Dengue (J) Influenza.
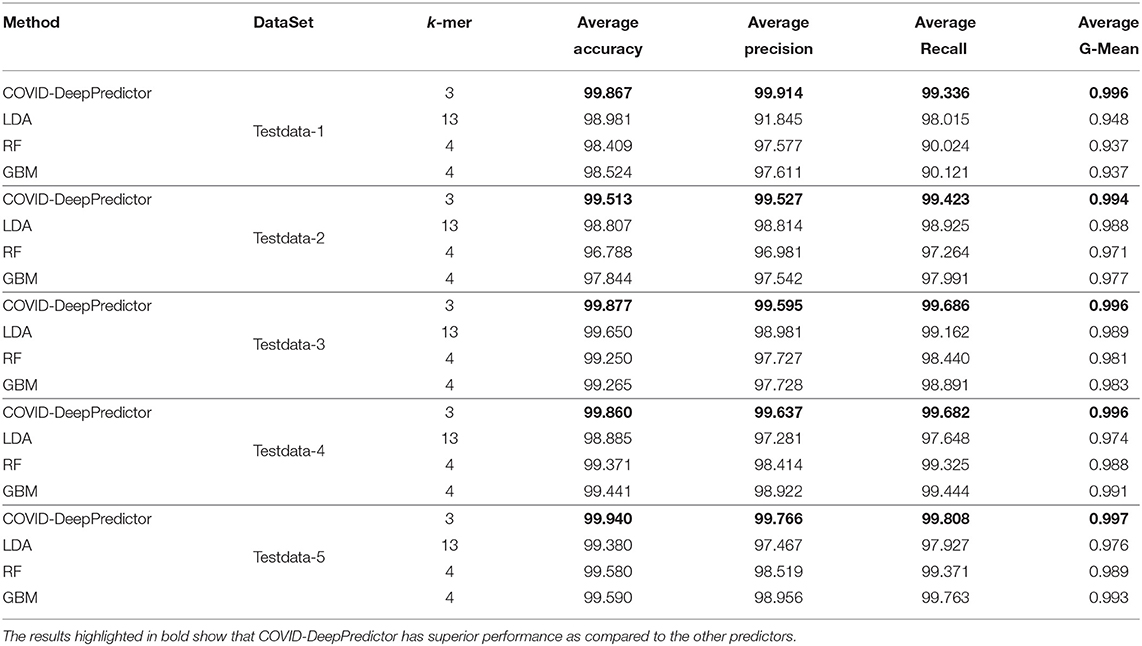
Table 2. Prediction performance of COVID-DeepPredictor and other compared methods on test datasets.
COVID-DeepPredictor is performed on a validation dataset as well. Accuracy, precision, recall, and G-mean values of the prediction for the validation dataset are 100, 100, 100, and 1%, respectively (k=3). As we have used -fold cross-validation with = 10, ten convergence plots of COVID-DeepPredictor are generated. One of the corresponding convergence plots for COVID-DeepPredictor is given in Figure 4D. The blue line indicates the training accuracy and the black line is the validation accuracy. All the convergence plots are shown in Supplementary Figure 1. The Bag-of-Unique-Descriptors of the six virus classes, SARS-CoV-1, MERS-CoV, SARS-CoV-2, Ebola, Dengue, and Influenza are shown in Figures 4E–J for k=3.
SARS-CoV-2 is a global pandemic and since human to human transmission (Chan et al., 2020; Huang et al., 2020) is confirmed for SARS-CoV-2, the need for its early prediction has become imperative. Viral outbreaks of this kind call for timely and prompt analysis of the genomic sequences to help the prediction of the virus in its early stages. COVID-DeepPredictor can be used by pathogen laboratories for the prediction of SARS-CoV-2 very quickly and as concluded from the results, most accurately. It is worth mentioning over here that for COVID-DeepPredictor to be effective, there must be at least two virus classes present in the training input sequences.
COVID-DeepPredictor has two functions for: (a) training, testing, and accordingly saving an LSTM model [COVIDdeepPredictor()] and (b) loading a pre-trained LSTM model for testing on unseen test dataset [COVIDdeepPredictorLoad()]. There is a main code COVIDmain.m which loads both COVIDdeepPredictor() and COVIDdeepPredictorLoad(). If users want to have their own training model and also get the results for a test dataset, they need to use only COVIDdeepPredictor() and disable COVIDdeepPredictorLoad(). On the other hand, if they want to use a pre-trained model, they can disable COVIDdeepPredictor() and run only COVIDdeepPredictorLoad() to get the results for test datasets.
For ease of users, training and testing files are provided to make them acquainted with the functionalities of COVIDdeepPredictor(). Trainingdata.csv is the input file for training and any one of the test files among Testdata-1.csv, Testdata-2.csv, Testdata-3.csv, Testdata-4.csv, and Testdata-5.csv can be used for testing. The results of the prediction will have the sequence ID, predicted virus name, along with its sequence which will be stored in Results.csv.
On the other hand, in case of COVIDdeepPredictorLoad(), only any one of the test files needs to be provided to get the results in Results.csv. Similarly, new training and test datasets can be prepared by the users after following the same structures of the training and testing files as provided. This is important so that new training models of COVID-DeepPredictor can be prepared for different set of viruses or similar kind of tasks. It is to be noted that the pre-trained model is provided in Supplementary Material, where the value of k for k-mer is 3. The choice of k has been done experimentally as it takes computationally less amount of time and provides higher accuracy. Sample files for training, testing, pre-trained models for COVID-DeepPredictor and the code of the software are available in Supplementary Material for re-usability3.
Setting the appropriate value of k in k-mer is very important to achieve the desired results in a text classification problem. As this work is based on the underlying concept of text classification, k-mer has a very important role to play. Thus, to determine the value of k in k-mer, extensive experiments have been performed. It can be observed from Figures 3A–E that with the increasing value of k, the run time of COVID-DeepPredictor is also on the rise. Therefore, to choose the appropriate value of k, apart from the accuracy, the run time of COVID-DeepPredictor also needs to be taken into account. For Testdata-1, at k = 9, 11, and 13, the accuracy is same as at k = 3. Similarly, for Testdata-2, Testdata-3, Testdata-4, and Testdata-5, similar accuracies can be observed at k = 3, 11, 13, k = 3, 4, 5, 13, k = 3, 4, and k = 3, 13, respectively. Although, the accuracies are same at these k-mer values, run time is increasing as can been seen from Figures 3A–E. Thus, the smallest k-mer value has been chosen without compromising on the accuracy. From Table 2 and Figure 4A, it is quite evident that with k = 3, COVID-DeepPredictor shows the best results among all the compared predictors.
To understand the relation among k-mer, size of BoDs and BoUDs, Table 3 is reported. From this table, we can see that the sizes of both BoDs and BoUDs increase with the increase in k-mer for each virus class. In the table, “All” represents all the six virus classes taken together. For example, at k = 15 for training dataset of all virus classes, the sizes of BoDs and BoUDs are 30193594 and 518372, respectively for 1,500 sequences while for the same k, for Testdata-1, the sizes of BoDs and BoUDs are 70595908 and 581774 respectively for 3,143 sequences. On the other hand, for k = 3, less number of BoDs and BoUDs are generated. Here, as expected, the BoD values for “All” are the summation of the BoDs of the individual virus classes. On the contrary, BoUD is less than the summation of the BoUDs of the six virus classes. This can be attributed to the relatedness between different virus classes. For example, SARS-CoV-1, MERS-CoV, and SARS-CoV-2 are more related and thus they may share unique descriptors (BoUDs) resulting in the intersection of the BoUDs when all the virus classes are considered together. Apart from this, BoDs and BoUDs for the varying k have also an impact on the accuracy and run time of COVID-DeepPredictor as well which can be observed by combining Figure 3 and Table 3.
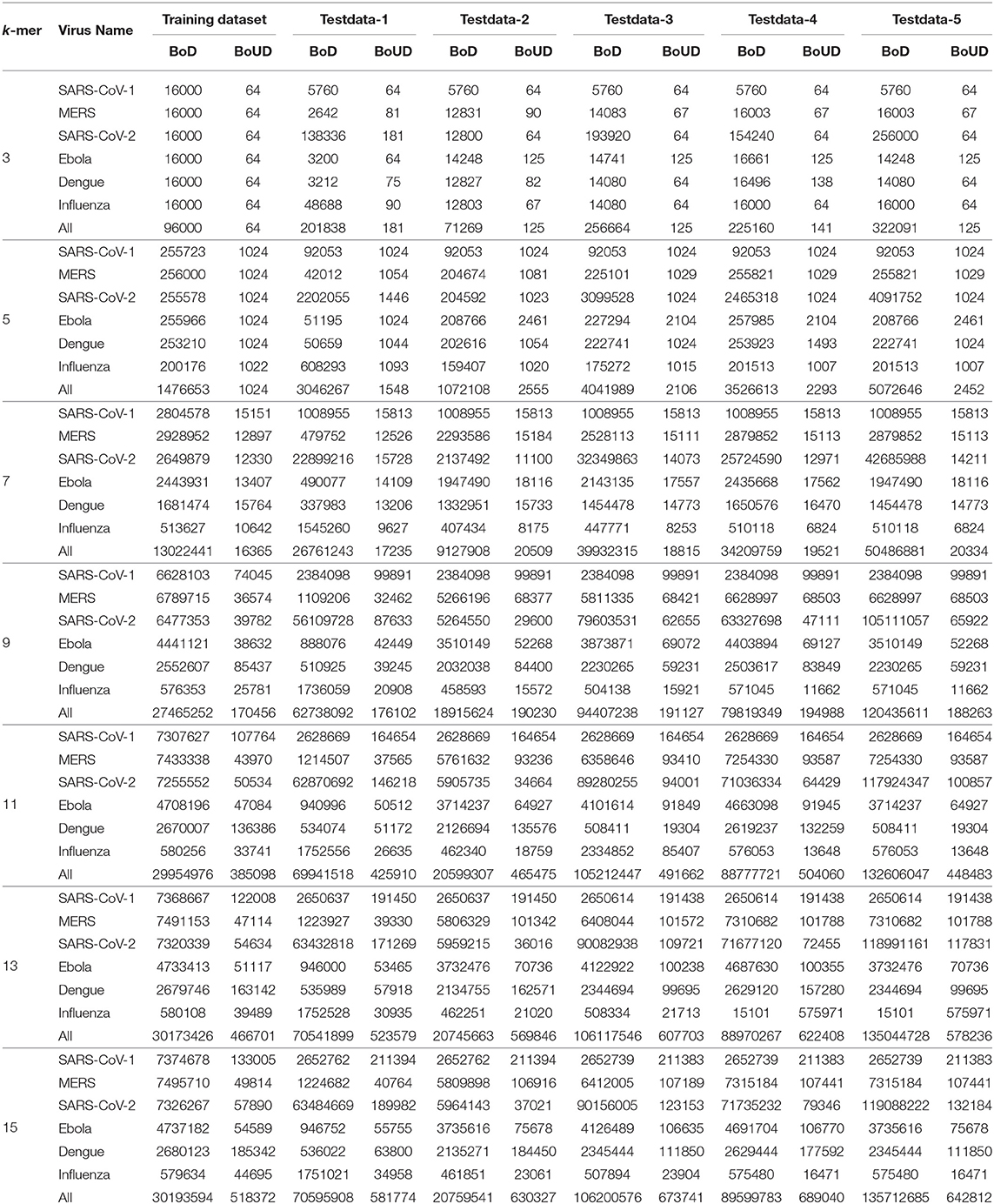
Table 3. Bag-of-Descriptors and Bag-of-Unique-Descriptors for each virus class.
The main advantage of COVID-DeepPredictor is that it uses k-mer technique which is an alignment-free technique. Most analysis based works attempted so far have used alignment based techniques. Although, they are highly successful in detecting similarities in sequences of viruses, they take a lot of computational time. Also, alignment based techniques have the underlying constraint of homologous sequences which may not be the case every time. To mitigate these problems of alignment based techniques, alignment-free techniques (Kari et al., 2015) can be used. Alignment-free techniques are meant to be fast and can work with a large number of sequences. To prove the advantage of COVID-DeepPredictor over BLASTN4, which is an alignment-based technique, Table 4 is reported where different input sequences of size 50, 100, 200, 300, and 400 of SARS-CoV-2 are taken. For 50 sequences, BLASTN takes 1 h 15 min to align the sequences and to produce the subsequent results. Thereafter, such results are further required to be analyzed by machine intelligence technique to predict the virus class which takes some additional time as well. On the contrary, COVID-DeepPredictor successfully completes the job of training and testing, which involves prediction, in just 1.26 min. Similar results are also seen for the other varying sequences as well. Thus, we can conclude that an alignment-free technique is significantly faster than an alignment based technique.
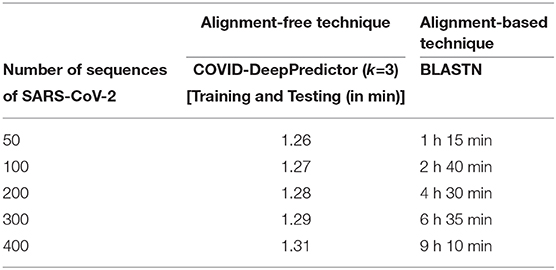
Table 4. Runtime comparison of COVID-DeepPredictor and BLASTN.
In the current scenario of global pandemic, it has become very important to predict SARS-CoV-2 as early as possible as both the affected and the number of death cases are increasing exponentially everyday. However, polymorphic nature of SARS-CoV-2 allows it to adapt and sustain in different kinds of environment which makes SARS-CoV-2 very hard to predict. In such scenarios, the proposed COVID-DeepPredictor can be very useful for predicting SARS-CoV-2 and other kinds of pathogenic viruses based on their genomic information very quickly as it uses an alignment-free technique. The results for COVID-DeepPredictor are highly encouraging as it shows prediction accuracy in the range of 99.51 to 99.94% for test datasets. Human health being the main concern of this work, the code for COVID-DeepPredictor along with the pre-trained model are also provided so that the scientific community can reap as much benefit as possible from it. Apart from SARS-CoV-2, COVID-DeepPredictor can also be used by pathogen laboratories to recognize the other five pathogenic viruses (SARS-CoV-1, MERS-CoV, Ebola, Dengue, and Influenza) very easily4 and accurately from a given genomic sequence. To achieve good performance, data preprocessing and the experiments are carried out on real-life datasets. Moreover, comparisons with popular existing prediction methods based on Linear Discriminant Analysis, Random Forests, and Gradient Boosting Method are also performed to show the superiority of COVID-DeepPredictor. Additionally, accuracy and runtime of COVID-DeepPredictor are taken together to determine the value of k in k-mer, comparison among k values in k-mer, Bag-of-Descriptors (BoDs) and Bag-of-Unique-Descriptors (BoUDs) is considered along with a comparative study between COVID-DeepPredictor and Nucleotide BLAST.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
IS designed the research. IS, NG, DM, AS, and DP analyzed data and wrote the manuscript. NG performed the experiments and collected results. All authors reviewed and approved the final version of the manuscript.
This work has been partially supported by CRG short term research grant on COVID-19 (CVD/2020/000991) from Science and Engineering Research Board (SERB), Department of Science and Technology, Govt. of India. In addition to this, this work has been supported by Polish National Science Centre (2019/35/O/ST6/02484), Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund (TEAM to DP). This research was co-funded by IDUB against COVID-19 project granted by Warsaw University of Technology under the program Excellence Initiative: Research University (IDUB).
AS was employed by company Cognizant Technology Solutions.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank all those who have contributed sequences to GISAID and NCBI databases. We are also thankful to the reviewers for providing valuable comments to improve the paper.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.569120/full#supplementary-material
1. ^https://www.ncbi.nlm.nih.gov/genome/viruses
2. ^https://gisaid.org/CoV2020
3. ^http://www.nitttrkol.ac.in/indrajit/projects/COVID-DeepPredictor/
4. ^https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&BLAST_SPEC=&LINK_LOC=blasttab&LAST_PAGE=blastn
Alagaili, A. N., Briese, T., Mishra, N., Kapoor, V., Sameroff, S. C., de Wit, E., et al. (2014). Middle east respiratory syndrome coronavirus infection in dromedary camels in Saudi Arabia. MBio 5:e00884-14. doi: 10.1128/mBio.01002-14
Chan, J. F.-W., Yuan, S., Kok, K.-H., Kai-Wang, K., Chu, H., Yang, J., et al. (2020). A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet 395, 514–523. doi: 10.1016/S0140-6736(20)30154-9
Cui, J., Li, F., and Shi, Z.-L. (2019). Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 17, 181–192. doi: 10.1038/s41579-018-0118-9
Guan, Y., Zheng, B., He, Y., Liu, X., Zhuang, Z., Cheung, C., et al. (2003). Isolation and characterization of viruses related to the sars coronavirus from animals in southern China. Science 302, 276–278. doi: 10.1126/science.1087139
Hinton, G. E., and Roweis, S. T. (2003). “Stochastic neighbor embedding,” in Advances in Neural Information Processing Systems (Vancouver, BC), 857–864.
Hochreiter, S., and Schmidhuber, J. (1997). Long short-term memory. Neural Comput. 9, 1735–1780. doi: 10.1162/neco.1997.9.8.1735
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., et al. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506. doi: 10.1016/S0140-6736(20)30183-5
Jenkins, G. M., Rambaut, A., Pybus, O. G., and Holmes, E. C. (2002). Rates of molecular evolution in RNA viruses: a quantitative phylogenetic analysis. J. Mol. Evol. 54, 156–165. doi: 10.1007/s00239-001-0064-3
Jin, Y., Luo, C., Guo, W., Xie, J., Wu, D., and Wang, R. (2019). Text classification based on conditional reflection. IEEE Access 7, 76712–76719. doi: 10.1109/ACCESS.2019.2921976
Kari, L., Hill, K. A., Sayem, A. S., Karamichalis, R., Bryans, N., Davis, K., et al. (2015). Mapping the space of genomic signatures. PLoS ONE 10:e119815. doi: 10.1371/journal.pone.0119815
Khattak, F. K., Jeblee, S., Pou-Prom, C., Abdalla, M., Meaney, C., and Rudzicz, F. (2019). A survey of word embeddings for clinical text. J. Biomed. Informatics 4:100057. doi: 10.1016/j.yjbinx.2019.100057
Kim, K., Kim, D., Noh, J., and Kim, M. (2018). Stable forecasting of environmental time series via long short term memory recurrent neural network. IEEE Access 6, 75216–75228. doi: 10.1109/ACCESS.2018.2884827
Koohi-Moghadam, M., Wang, H., Wang, Y., Yang, X., Li, H., Wang, J., et al. (2019). Predicting disease-associated mutation of metal-binding sites in proteins using a deep learning approach. Nat. Mach. Intell. 1, 561–567. doi: 10.1038/s42256-019-0119-z
Letko, M., Marzi, A., and Munster, V. (2020). Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage b betacoronaviruses. Nat. Microbiol. 5, 562–569. doi: 10.1038/s41564-020-0688-y
Liu, J., Xia, C., Yan, H., Xie, Z., and Sun, J. (2019). Hierarchical comprehensive context modeling for Chinese text classification. IEEE Access 7, 154546–154559. doi: 10.1109/ACCESS.2019.2949175
Liu, X., and Wang, X.-J. (2020). Potential inhibitors against 2019-nCoV coronavirus m protease from clinically approved medicines. J. Genet. Genomics. 47, 119–121. doi: 10.1016/j.jgg.2020.02.001
Lu, R., Zhao, X., Li, J., Niu, P., Yang, B., Wu, H., et al. (2020). Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395, 565–574. doi: 10.1016/S0140-6736(20)30251-8
Luque, A., Carrasco, A., Martín, A., and de las Heras, A. (2019). The impact of class imbalance in classification performance metrics based on the binary confusion matrix. Pattern Recogn. 91, 216–231. doi: 10.1016/j.patcog.2019.02.023
Manekar, S., and Sathe, S. (2018). A benchmark study of k-mer counting methods for high-throughput sequencing. Gigascience 7:giy125. doi: 10.1093/gigascience/giy125
Meng, Y., Wu, P., Lu, W., Liu, K., Ma, K., Huang, L., et al. (2020). Sex-specific clinical characteristics and prognosis of coronavirus disease-19 infection in Wuhan, China: a retrospective study of 168 severe patients. PLoS Pathol. 16:e1008520. doi: 10.1371/journal.ppat.1008520
Ozturk, T., Talo, M., Yildirim, E. A., Baloglu, U. B., Yildirim, O., and Rajendra Acharya, U. (2020). Automated detection of covid-19 cases using deep neural networks with x-ray images. Comput. Biol. Med. 121:103792. doi: 10.1016/j.compbiomed.2020.103792
Paraskevis, D., Kostaki, E., Magiorkinis, G., Panayiotakopoulos, G., Sourvinos, G., and Tsiodras, S. (2020). Full-genome evolutionary analysis of the novel corona virus (2019-nCoV) rejects the hypothesis of emergence as a result of a recent recombination event. Infect. Genet. Evol. 79:104212. doi: 10.1016/j.meegid.2020.104212
Solis-Reyes, S., Avino, M., Poon, A., and Kari, L. (2018). An open-source k-mer based machine learning tool for fast and accurate subtyping of HIV-1 genomes. PLoS ONE 13:e206409. doi: 10.1371/journal.pone.0206409
Su, S., Wong, G., Shi, W., Liu, J., Lai, A. C., Zhou, J., et al. (2016). Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 24, 490–502. doi: 10.1016/j.tim.2016.03.003
Tang, B., Pan, Z., Yin, K., and Khateeb, A. (2019). Recent advances of deep learning in bioinformatics and computational biology. Front. Genet. 10:214. doi: 10.3389/fgene.2019.00214
Wan, Y., Shang, J., Graham, R., Baric, R. S., and Li, F. (2020). Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 94:e00127-20. doi: 10.1128/JVI.00127-20
Weiss, S., and Navas-Martin, S. (2005). Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 4, 635–664. doi: 10.1128/MMBR.69.4.635-664.2005
Woo, P. C., Lau, S. K., Huang, Y., and Yuen, K.-Y. (2009). Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. 234, 1117–1127. doi: 10.3181/0903-MR-94
Worldometer (2021). Coronavirus Disease 2019 (COVID-19) Cases. Available online at: https://www.worldometers.info/coronavirus (accessed January 8, 2021).
Yan, L., Zhang, H.-T., Goncalves, J., Xiao, Y., Wang, M., Guo, Y., et al. (2020). An interpretable mortality prediction model for covid-19 patients. Nat. Mach. Intell. 2, 283–288. doi: 10.1038/s42256-020-0180-7
Yan, Q., Weeks, D. E., Xin, H., Swaroop, A., Y. E. Chew, E., Huang, H., et al. (2020). Deep-learning-based prediction of late age-related macular degeneration progression. Nat. Mach. Intell. 2, 141–150. doi: 10.1038/s42256-020-0154-9
Zhang, Y., Zheng, J., Jiang, Y., Huang, G., and Chen, R. (2019). A text sentiment classification modeling method based on coordinated CNN-LSTM-attention model. Chinese J. Electron. 28, 120–126. doi: 10.1049/cje.2018.11.004
Keywords: long-short term memory, SARS-CoV-2, sequence analysis, virus prediction, genomic information
Citation: Saha I, Ghosh N, Maity D, Seal A and Plewczynski D (2021) COVID-DeepPredictor: Recurrent Neural Network to Predict SARS-CoV-2 and Other Pathogenic Viruses. Front. Genet. 12:569120. doi: 10.3389/fgene.2021.569120
Received: 03 June 2020; Accepted: 13 January 2021;
Published: 11 February 2021.
Edited by:
Xian-Tao Zeng, Wuhan University, ChinaReviewed by:
Sarath Chandra Janga, Indiana University, Purdue University Indianapolis, United StatesCopyright © 2021 Saha, Ghosh, Maity, Seal and Plewczynski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Indrajit Saha, aW5kcmFqaXRAbml0dHRya29sLmFjLmlu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.