
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 19 February 2021
Sec. Behavioral and Psychiatric Genetics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.553974
This article is part of the Research Topic Genetic and Epigenetic Mechanisms Underpinning Vulnerability to Developing Psychiatric Disorders View all 17 articles
 Hua Tao1,2†
Hua Tao1,2† Xu Zhou2†Jun Chen1
Xu Zhou2†Jun Chen1 Haihong Zhou1
Haihong Zhou1 Lidan Huang2Yujie Cai2Jiawu Fu1Zhou Liu2Yanyan Chen2Chaowen Sun1Bin Zhao2*Wangtao Zhong1*
Lidan Huang2Yujie Cai2Jiawu Fu1Zhou Liu2Yanyan Chen2Chaowen Sun1Bin Zhao2*Wangtao Zhong1* Keshen Li3,4*
Keshen Li3,4*Recent studies have reported patients who concurrently exhibit conditions of epilepsy and schizophrenia, indicating certain shared pathologies between them. This study aimed to investigate the genetic effects of the schizophrenia-related gene DTNBP1 in temporal lobe epilepsy (TLE). A total of 496 TLE patients and 528 healthy individuals were successfully genotyped for six DTNBP1 polymorphisms (rs760665, rs1011313, rs2619528, rs2619522, rs909706, and rs2619538), including 335 TLE patients and 325 healthy controls in cohort 1, and 161 TLE patients and 203 healthy controls in cohort 2. The frequency of the TT genotype at rs909706 T > C was lower in TLE patients than in normal controls in the initial cohort (cohort 1), which was confirmed in an independent cohort (cohort 2). However, the intronic T allele failed to be in linkage disequilibrium (LD) with any functional variations nearby; thus, together with the CCAC and TCAT haplotypes (rs1011313-rs2619528-rs2619522-rs909706) observed in the study, this allele acts only as a protective factor against susceptibility to TLE. Meanwhile, a novo mutant allele rs2619538 T > A was exclusively observed in TLE patients, and a dual-luciferase assay revealed that the mutant allele was increased by approximately 22% in the DTNBP2 promoter compared with the wild-type allele. Together with the trend of increasing DTNBP1 expression in epilepsy patients and animal models in this study, these are the first findings to demonstrate the genetic association of DTNBP1 with TLE. Homozygous mutation of rs2619538 T > A likely promotes DTNBP1 expression and facilitates subsequent processes in epilepsy pathologies. Thus, the role of DTNBP1 in TLE deserves further exploration in the future.
Epilepsy is a brain disorder characterized by recurrent seizures. Many gene mutations have been identified via exon sequencing as being responsible for epileptic seizures; these genes involve many ion and non-ion channels, such as KCNA2, KCNT1, GRIN2A, and CHD2 (He et al., 2019). Nevertheless, epilepsy pathologies are difficult to fully identify, making the development of efficient therapies challenging. Thus, almost one-third of patients, especially those with temporal lobe epilepsy (TLE), a main drug-resistant epilepsy, still suffer from recurrent seizures because of resistance to available treatments. Hence, more efforts should be made to explore epilepsy pathologies, and disclosing epileptic seizures in atypical conditions might be a novel strategy.
In clinical practice, typical symptoms of a disease are often seen as complications due to other diseases, underscoring certain shared mechanisms between them. Considering this phenomenon, some previous studies have explored the manifestation of epileptic seizures in Alzheimer’s disease and found that insufficient expression of ADAM10 is a driving force in epilepsy pathologies (Clement et al., 2008; Prox et al., 2013). The prevalence of epileptic seizures in psychosis has also attracted increased attention in recent years. According to a retrospective cohort study based on the Oxford Record Linkage Study and English national linked Hospital Episode Statistics, patients admitted to the hospital with schizophrenia have an elevated risk of epileptic seizures, and likewise, patients with epilepsy have an elevated risk of schizophrenia (Wotton and Goldacre, 2012). In addition, male adolescents with severe, refractory epilepsy have a high risk of schizophrenia (Andersen et al., 2019). Obviously, this evidence supports a bidirectional relationship and some shared mechanisms between epilepsy and schizophrenia.
Expanding experimental evidence supports the notion of shared mechanisms between epilepsy and schizophrenia. For example, Michelson et al. reported that familial partial trisomy 15q11-13 presented either as intractable epilepsy or as psychiatric illness (Michelson et al., 2011), and Stewart et al. observed that the frequency of copy number abnormalities and maternal duplication 15q11-q13 in patients with combined schizophrenia and epilepsy is significantly higher than in patients with schizophrenia (Stewart et al., 2011). Furthermore, schizophrenia- and epilepsy-related alterations were simultaneously observed in a mouse model with a microdeletion within 15q13.3 (Fejgin et al., 2014). Other studies revealed that abnormal expression of CYFIP1, VRK2, and metabotropic glutamate 2/3 (mGlu2/3) receptor is involved in both schizophrenia and epilepsy (Nebel et al., 2016; Azimi et al., 2018).
The DTNBP1 gene encodes dystrobrevin binding protein 1, which participates in organelle biogenesis, and recent evidence indicates that it plays key roles in brain development, neuronal excitability, and schizophrenia-related pathologies (Chen et al., 2008; Saggu et al., 2013; Wang et al., 2017; Konopaske et al., 2018; Mohamed et al., 2018), i.e., decreased expression of DTNBP1 was observed to reduce exocytosis of brain-derived neurotrophic factor (BDNF) from cortical excitatory neurons. The reduction in BDNF exocytosis resulted in a decreasing number of inhibitory synapses located on excitatory neurons, while the application of exogenous BDNF rescued these inhibitory synaptic deficits (Yuan et al., 2016). Interestingly, the hippocampus is an enriched region for DTNBP1 under normal conditions, and postmortem studies have shown a decreased level of DTNBP1 expression in the hippocampus of patients with schizophrenia (Wang et al., 2017), indicating that decreased DTNBP1 is a pathological condition in schizophrenia. Since the hippocampus is also crucial in TLE pathologies characterized by deregulation of neuronal excitability, it is likely that DTNBP1 plays a key role in both TLE and schizophrenia.
Considering that single-nucleotide polymorphisms are among the most important genetic variations that regulate gene expression and subsequent biological activity, this study selected candidate variations within the DTNBP1 gene region on the basis of biological plausibility in previous research, including rs2619522 involved in hippocampal gray matter volume (Trost et al., 2013) and rs2619538 in volumes of both gray and white matter (Tognin et al., 2011) in healthy subjects, rs760665 and rs909706 involved in the hippocampal glutamate concentration of healthy individuals (Wirth et al., 2012), and rs1011313 involved in volume reduction of multiple brain regions (Thirunavukkarasu et al., 2014) and rs2619528 in prefrontal function (Fallgatter et al., 2010) of schizophrenia patients. We aimed to clarify the genetic effects of DTNBP1 in TLE based on these six SNPs.
All experimental protocols involving human subjects were approved by the Ethics Committees of the Affiliated Hospital of Guangdong Medical University, The First Affiliated Hospital of Harbin Medical University, the Central People’s Hospital of Zhanjiang, the First People’s Hospital of Pingdingshan, and Beijing Tongren Hospital affiliated with Capital Medical University and were executed in accordance with the Declaration of Helsinki. Informed consent documents were signed by participants at the time of human subject enrollment.
A total of 335 TLE patients and 325 healthy controls from the First Affiliated Hospital of Harbin Medical University, the Affiliated Hospital of Guangdong Medical University and Beijing Tongren Hospital affiliated with Capital Medical University were consecutively enrolled in cohort 1. To confirm the initial statistics observed in cohort 1, a validating cohort (cohort 2), including 161 TLE patients and 203 healthy controls from the First People’s Hospital of Pingdingshan and the Central People’s Hospital of Zhanjiang, was consecutively established. The human subjects in both cohorts 1 and 2 were Han Chinese, and the combined cohort (cohorts 1 + 2) was composed of 496 TLE patients and 528 healthy controls.
Gender, age, age at onset, and severity of disease (seizure frequency and drug response) of all participants were recorded via field investigation or telephone interviews. The inclusion criteria for TLE were based on typical temporal auras and temporal discharges at seizure onset observed by video-electroencephalograph, and subjects who failed to be genotyped were excluded from the study. According to the consensus about drug-resistant epilepsy proposed in 2010 by the Commission of the International League Against Epilepsy (Kwan et al., 2010), the responses to drug treatments were grouped as follows: drug-resistant patients are subjects with the absence of a significant change, an insufficient reduction in seizure frequency (<60%), or even an augmentation after 1 year treatment with a schedule of not less than two tolerated and properly collected anti-epileptic medicines; the remaining were considered drug-sensitive patients.
First, 2 ml peripheral blood samples were collected from each human participant. DNA samples were extracted from blood samples using a Genomic DNA Extraction Kit (Tiangen Biotech, Beijing, China) and then used for genotyping the six DTNBP1 SNPs (rs760665, rs1011313, rs2619528, rs2619522, rs909706, and rs2619538) via an ABI PRISM SNaPshot system (Applied Biosystems, Carlsbad, CA, United States). The primers used in a multiplex polymerase chain reaction (PCR) for the amplification of the six target fragments (338, 354, 274, 164, 278, and 183 nucleotides, respectively) were as follows: rs760665, 5′- cagccttggcctccaaacat-3′ (forward primer) and 5′-ttagccccagaaatgttattgatttga-3′ (reverse primer); rs1011313, 5′-ccagcatggagacgccaagt-3′ (forward primer) and 5′-tctgaagccttgaacccctcaa-3′ (reverse primer); rs2619528, 5′-tggttgcctgggatggagag-3′ (forward primer) and 5′-cgcactgaaacag tcttttcccatt-3′ (reverse primer); rs2619522, 5′-tgtttgggctctta tgtctacctttcc-3′ (forward primer) and 5′-gcccatttatctgttttccagtgc-3′ (reverse primer); rs909706, 5′-ggtgacaggctaatggcacaca-3′ (forward primer) and 5′-ttctggagtgggctctggactg-3′ (reverse primer); and rs2619538, 5′-ttggatgaggccagtgaggtaa-3′ (forward primer) and 5′-gaggtggtcccccagctacaag-3′ (reverse primer). The primers used for one-nucleotide extension in SNaPshot PCR were 5′-ttttttttttttttttttttttttttacaatagccttgctactatttaggggataaaca-3′ (rs760665), 5′-tttttttttttttcacaggctacagaatggatgttgc-3′ (rs10113- 13), 5′-tttggattggatgcacaaccatgtgaa-3′ (rs2619528), 5′-tttttttttttttt ttttttttttgcagaagcagtgagtgagagctgaca-3′ (rs2619522), 5′-tttttttttttt ttttgtcaagtcagtttccaaggggttctaac-3′ (rs909706), and 5′-tttttttttttttt ttttttttttgcaaagcttaataagacagagcagtttacatc-3′ (rs2619538).
The multiplex PCR mix was composed of 1x HotStarTaq buffer, 0.3 mM dNTP, 3.0 mM Mg2+, 1 μl of primer mix, 1 U of HotStarTaq polymerase, and 1 μl of the DNA template. The reaction mix was then run as follows: 1 cycle for 95°C/120 s; 11 cycles for 94°C/20 s, 65°C/40 s, and 72°C/90 s; 24 cycles for 94°C/20 s, 59°C/30 s, and 72°C/90 s; and 1 cycle for 72°C/120 s. Subsequently, the reaction products were purified using shrimp alkaline phosphatase and exonuclease I. The SNaPshot PCR mix contained 5 μl of SNaPshot Multiplex Kit, 1 μl of primer mix, 2 μl of ultrapure H2O, and 2 μl of the purified reaction products. The experimental program for a one-nucleotide extension was as follows: 1 cycle of 96°C/60 s; 28 cycles of 96°C/10 s, 55°C/5 s, and 60°C/30 s; and 1 cycle of 4°C/120 s. After further purification by shrimp alkaline phosphatase, the final products were analyzed using an ABI 3730xl DNA Analyzer and GeneMapper 4.1 (Applied Biosystems, Carlsbad, CA, United States).
In light of the Homo sapiens chromosome 6, GRCh37.p13 primary assembly, a 2 kb sequence upstream of the DTNBP1 transcription start site harboring the T or A alleles at rs2619538 was cloned and individually ligated into pGL3 basic to create pDTNBP1-Promoter-Wildtype and pDTNBP1-Promoter-Mutant plasmids. In addition to the plasmids of the negative control (NC) and the positive control (PC), both of these plasmids were amplified in DH5α cells. Their positive clones were confirmed by further sequencing. Subsequently, HEK-293T cells were plated at 2 × 104 cells per well in 24-well dishes, and 24 h later, the cells were individually cotransfected with four types of plasmids with the assistance of X-tremeGENE HP reagent (Roche, Basel, Switzerland). These four plasmids included 1 μg of pGL3 basic as the NC, 1 μg of pGL3 promoter as the PC, 1 μg of pSMG6-Promoter-Wildtype (Wild-type), and 1 μg of pSMG6-Promoter-Mutant (Mutant). Each cotransfection reaction was replicated four times, and the pRL plasmid served as an internal reference. After 24 h, the cotransfected cells were identified using a Dual-Luciferase® Reporter Assay System (Promega, Madison, WI, United States), and both the firefly and Renilla luciferase activities were measured using a microplate luminometer (BioTek, VT, United States). The mutant impact of rs2619538 T > A was evaluated by dividing the averaged firefly/Renilla ratio of the mutant construct by the averaged firefly/Renilla ratio of the wild-type construct.
Referring to our previous research (Tao et al., 2018), a total of 15 male Sprague Dawley (SD) rats from the Animal Center of Guangdong Medical University, Zhanjiang, China, were bred for adapting to the experimental environment at a temperature between 22 and 26°C and a humidity between 55 and 65%. The light-dark cycle was synchronous with a natural day-and-night cycle. After a 1 week adaptation period with free access to food and water, the SD rats (310 ± 32 g) were used in the following experiments: 10 rats were randomly selected to be administered pentetrazol (PTZ, 60 mg/kg body weight, i.p.; Sigma-Aldrich, St. Louis, MO, United States). The severity of epileptic seizures was classified in five levels according to the Racine scale: (1) twitching of facial muscle; (2) nodding of the head; (3) unilateral forelimb with lifting or clonus; (4) bilateral forelimb with clonus when standing; (5) falling when standing or twisting (Glen et al., 2014; Orozco et al., 2014). The administration of PTZ was repeated every 20 min during a 2 h observation period (10 mg/kg body weight, i.p.) until seizures increased to level 4 or the total dose of PTZ reached 90 mg/kg body weight. Finally, with the exception of one rat reaching level 4 for a duration of only 3 min, nine rats exhibited seizures that reached level 4 with a duration of 60 min and were immediately enrolled in the experimental group, followed by the administration of diazepam (10 mg/kg body weight, i.p.; Sigma-Aldrich, St. Louis, MO, United States) every 5 min until seizure cessation to reduce unexpected deaths before being sacrificed. The remaining five rats were classified as the control group. All animal experiments in this study were performed in accordance with the “Guide for the Care and Use of Laboratory Animals” (He et al., 2006), which was approved by the Animal Ethics Committee of Guangdong Medical University, Zhanjiang, China.
After a 2 h observation period, all rats in the experimental and control groups were sacrificed through decapitation under deep anesthesia (3% chloral hydrate, 10 ml/kg body weight, i.p.; Sigma-Aldrich, St. Louis, MO, United States). Then, the left and right hippocampi of nine experimental rats and four control rats were rapidly collected for use in real-time quantitative PCR (qPCR) and enzyme-linked immunosorbent assay (ELISA), respectively: (a) real-time qPCR: total RNA was extracted via an RNA extraction kit (QIAGEN Sciences, Germantown, United States), and reverse transcription was performed by using a First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, United States) according to the manufacturer’s instructions. Subsequently, the cDNA products were amplified using a Light-Cycler 480 sequence detector system (Roche Applied Science, Penzberg, Germany), and the specific primers used for real-time qPCR were as follows: DTNBP1 forward primer, 5′-ttagcaggtatgaggatgcgt-3′, and reverse primer, 5′-ggtgcagcaaatggttctctac-3′. Relative expression levels were calculated by the 2–ΔΔCT method. And (b) ELISA: DTNBP1 concentrations in the hippocampi were measured using an ELISA kit (R&D Systems, Minneapolis, MN, United States) according to the manufacturer’s instructions. Absorbance was detected using an ELISA reader (Bio-Rad Laboratories, Hercules, CA, United States).
In addition to qPCR and ELISA, immunohistochemistry (IHC) was performed as follows: one experimental rat and one control rat under deep anesthesia were cardially perfused with physiological saline, followed by 4% paraformaldehyde, and their brains were then isolated and fixed in 4% paraformaldehyde at 4°C for 20 h. After dehydration and paraffin embedding, the specimens were cut into 4-μm-thick slices for subsequent histological staining. Paraffin-embedded slices were dewaxed with xylene and ethanol. Deparaffinized slices were rinsed using water, placed in a tissue slide, and heated in a microwave oven for antigen retrieval. Endogenous peroxidase was then blocked using 3% hydrogen peroxide. After three 5 min washes with phosphate buffer saline (PBS), the slices were blocked using 3% bovine serum albumin. Subsequently, the primary antibody (DTNBP1 polyclonal antibody, ABclonal, Boston, MA, United States) was diluted in blocking solution and incubated overnight at 4°C. The next day, the slices were washed three times for 5 min each in PBS and then incubated with the secondary antibody, washed, developed with 3-3′ diaminobenzidine tetrachloride, and washed to stop the reaction until brown color appeared. The slices were counterstained with hematoxylin, dehydrated and mounted, and finally photographed for biological analysis.
To identify whether DTNBP1 is abnormally expressed in human epilepsy patients, we used the online GSE datasets because of the lack of pathological brain samples of epilepsy patients in our laboratory. Unfortunately, after a series of searches in the online GSE datasets, we could not identify a single study that could be directly used. However, we found that GSE63808 was based on surgically acquired hippocampi from 129 TLE patients, and GSE29378 contained 32 autopsy samples of normal hippocampi. Moreover, both of these GSE datasets were acquired via the same technique, Illumina HumanHT-12 V3.0 expression BeadChip arrays (Johnson et al., 2015), and therefore, combining these two human transcriptome arrays makes it possible to study the expression of DTNBP1 between epilepsy patients and normal controls. Hence, GSE63808 and GSE29378 were downloaded from the online GSE datasets. In total, 129 TLE patients from the GSE63808 dataset and 32 normal controls form the GSE29378 dataset were individually enrolled in the TLE and control groups, respectively, in the present study. After normalization and testing of multiple hypotheses to reduce the false-positive rate, the average signals of DTNBP1 mRNA expression in the hippocampi of the TLE and control groups were compared for further analysis.
Variable data, reported as the mean ± SD, and attribute data were analyzed with a Student’s t-test and a chi-square test, respectively. Logistic regression was employed to correct for bias related to confounding factors, including age and gender, and q-values were used to adjust the false-positive results from multiple statistics by the Bonferroni correction. Statistical tests were performed using SPSS 19.0 (IBM, New York, NY, United States), and p ≤ 0.05 was considered statistically significant. In addition, haplotype construction and power analyses were carried out via Haploview 4.2 (Daly Lab, Cambridge, MA, United States) and Quanto 1.2 (University of Southern California, Los Angeles, CA, United States), respectively.
This study enrolled a total of 496 TLE patients and 528 healthy individuals. No significant differences in gender or age were observed between the TLE patients and the healthy controls in cohorts 1, 2, and 1 + 2 (all p > 0.05). Gender, age, duration of disease and severity of disease in cohorts 1 + 2 are listed in Table 1.
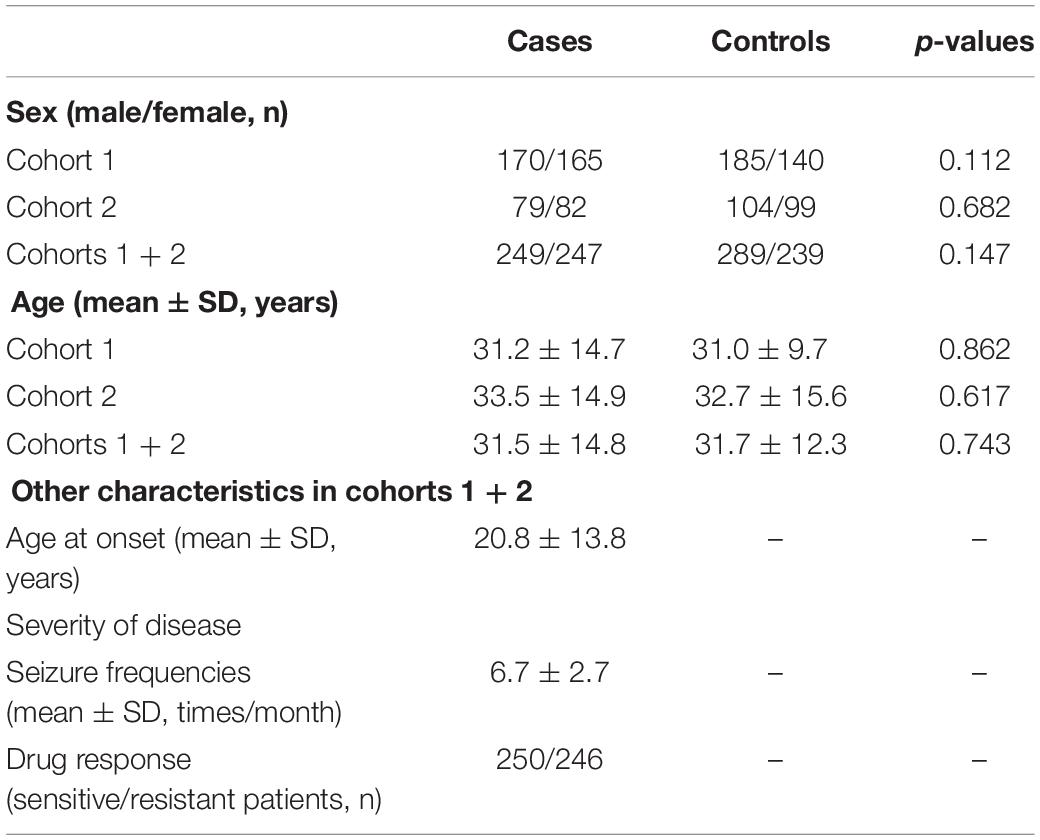
Table 1. General characteristics of enrolled cohorts.
In the present study, all enrolled patients and controls were successfully genotyped for the six DTNBP1 SNPs (rs760665, rs1011313, rs2619528, rs2619522, rs909706, and rs2619538). The frequency distributions of these SNPs complied with the Hardy-Weinberg equilibrium. Power analyses indicated that this study had 12.9% power for rs760665, 99.7% power for rs1011313, 97.5% power for rs2619528, 97.5% power for rs2619522, 99.6% power for rs909706 and 87.2% power for rs2619538 for detecting a recessive inheritance model with a genetic effect of 1.8 at a significance level of 0.05 for two-sided type 1 error.
In cohorts 1 and 2, the frequencies of the TT genotype at rs909706 T > C were consistently lower in the patients than in the controls (Table 2: p = 0.000 and 0.018, respectively). The same trend was observed after the Bonferroni correction for multiple comparisons in cohorts 1 + 2 (Table 3; OR = 1.770, p = 0.000 and q = 0.050), which confirmed the findings in cohorts 1 and 2. These results indicate that the TT genotype is associated with protection against TLE. In addition, no consistent differences were found for the remaining five SNPs (rs760665, rs1011313, rs2619528, rs2619522, and rs2619538) in cohorts 1, 2, and 1 + 2 (Tables 2, 3).
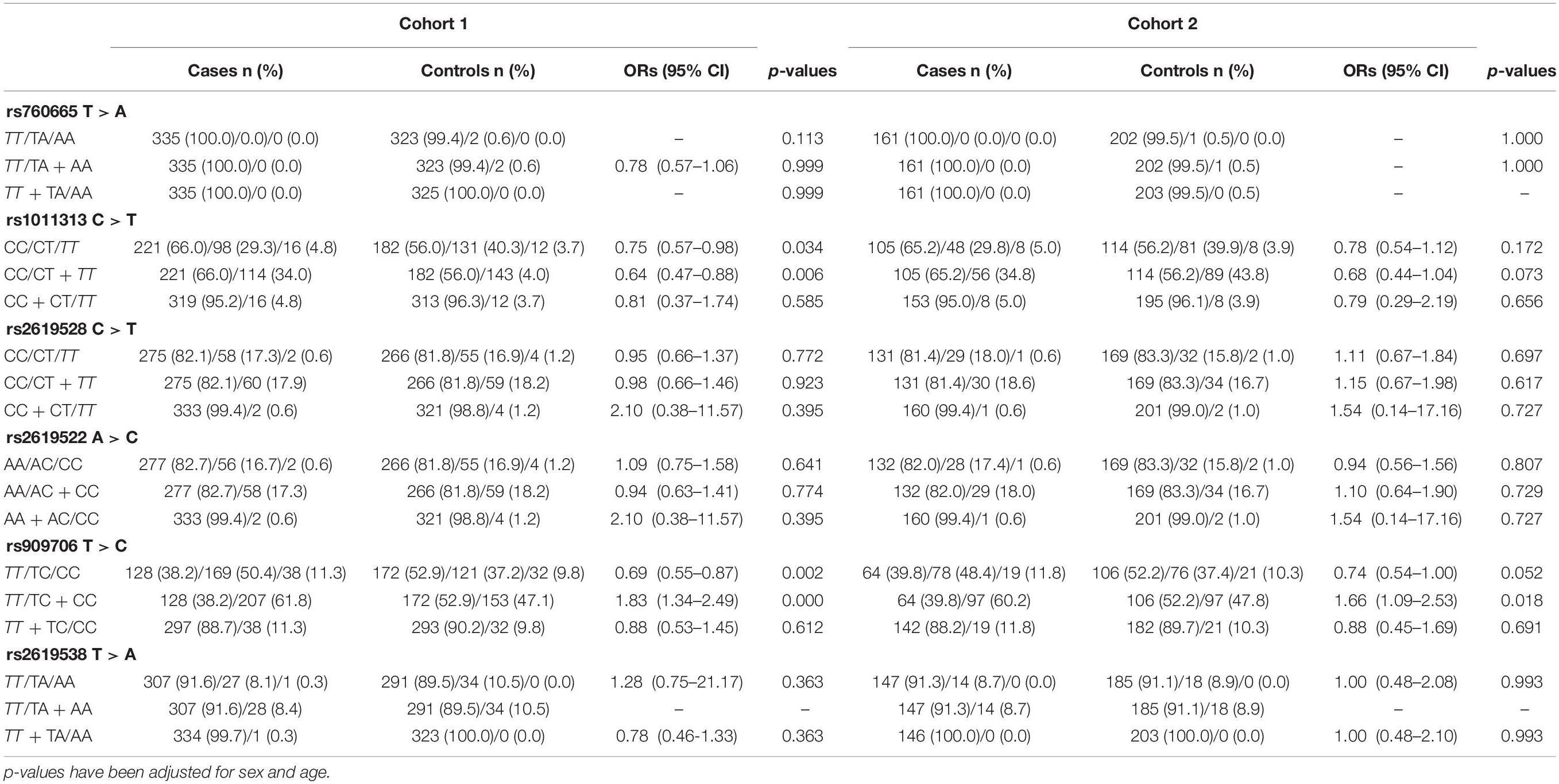
Table 2. Inheritance models of the DTNBP1 SNPs in cohorts 1 and 2.
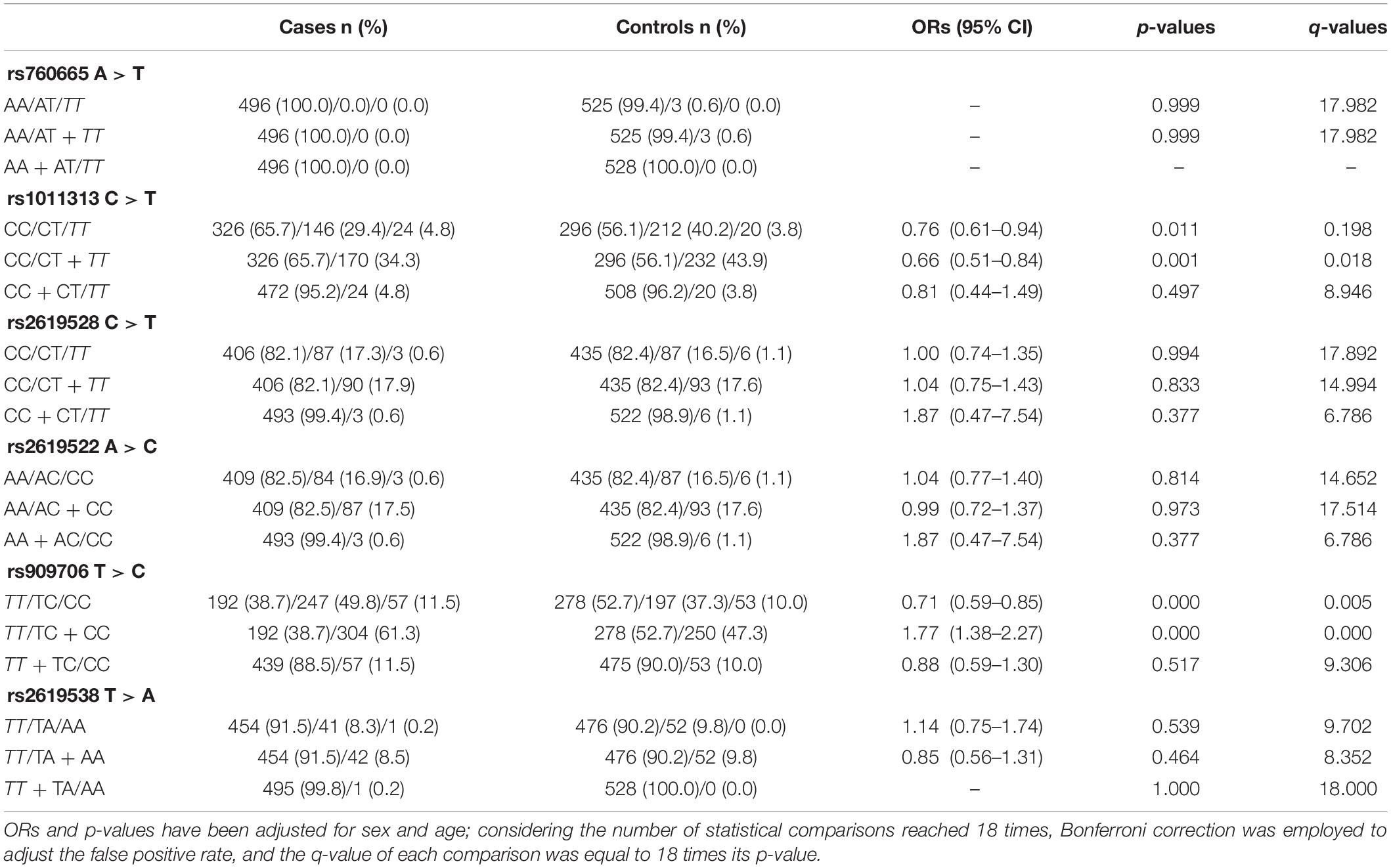
Table 3. Inheritance models of the SRR SNPs in cohorts 1 + 2.
Through the use of Haploview 4.2, a haplotype block (rs1011313-rs2619528-rs2619522-rs909706) approximately 27 kb in length was constructed based on all six SNPs including four haplotypes (CCAT, CCAC, TCAT, and CTCC). Their frequencies were then compared between the patients and the controls in cohorts 1 + 2. As displayed in Table 4, the patient ratios of the CCAC and TCAT haplotypes were lower than the control ratio (16.3 vs. 19.2%, p = 0.000, and 13.7 vs. 23.8%, p = 0.006), which was further confirmed by the Bonferroni correction for multiple comparisons (q = 0.000 and 0.025). Thus, the CCAC and TCAT haplotypes could be genetically protective markers against susceptibility to TLE.
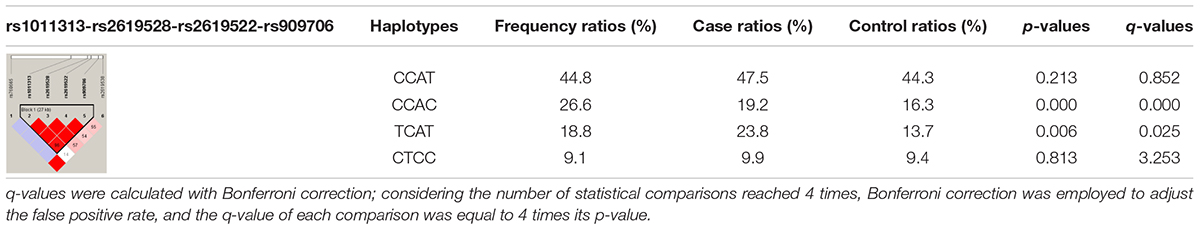
Table 4. Haplotypes of the DTNBP1 SNPs in cohorts 1 + 2.
Given the findings of case-control analysis, we further investigated the genetic impact of the wild-type homozygote at rs909706 T > C on age at onset, seizure frequency, and drug response in cohorts 1 + 2. As shown in Figure 1, no significant differences in age at onset, seizure frequency or drug response in the TLE patients were found between carriers with the TT genotype and those with the TT + TG genotypes (p = 0.221, 0.349, and 0.132), and similar results occurred after adjusting for gender and age (p = 0.202, 0.254, and 0.130).
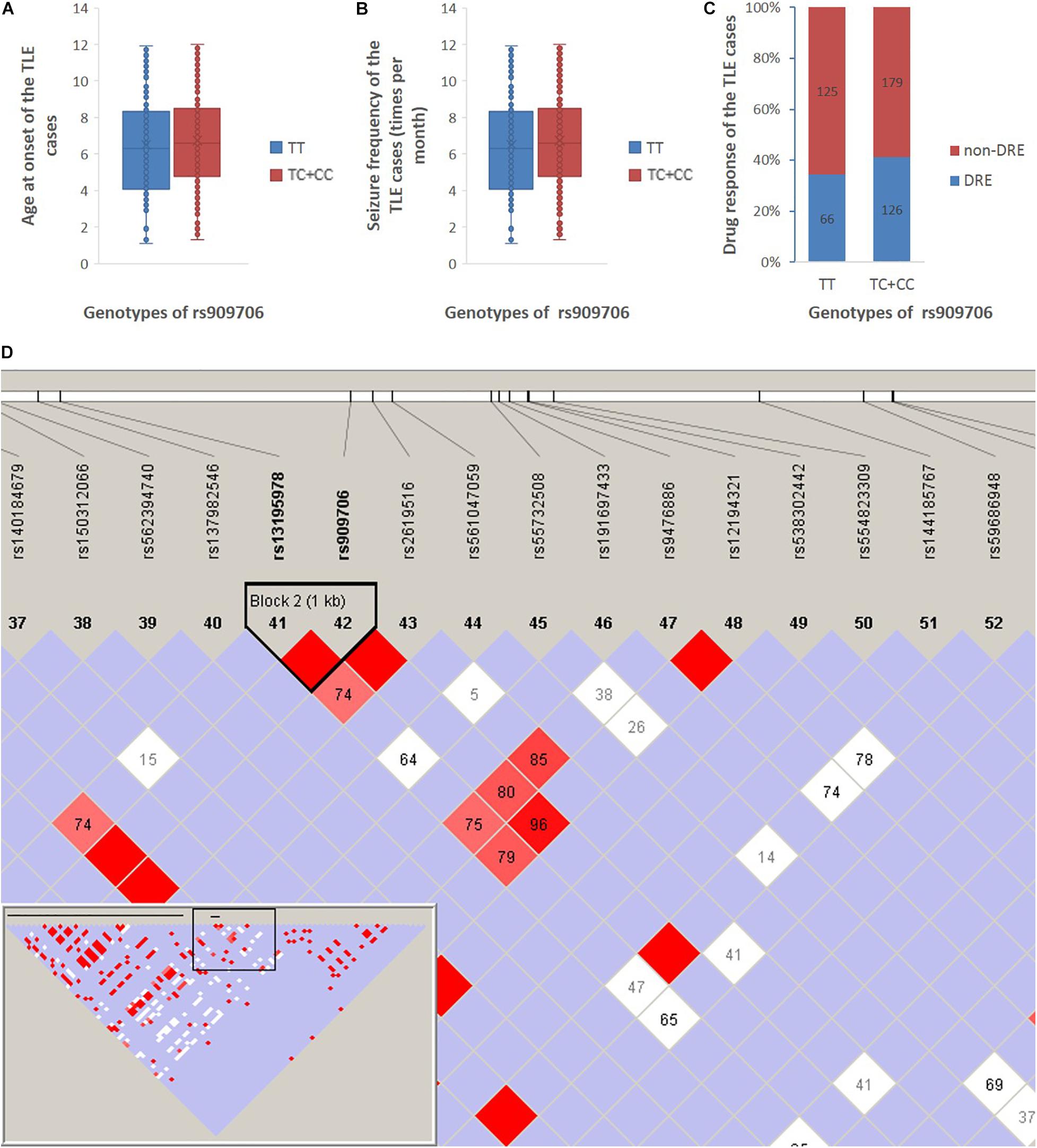
Figure 1. Age at onset, seizure frequency, and drug response between the TT genotype and the TC + CC genotypes in all TLE patients in cohort 1 + 2. (A) The age at onset of the TLE patients was 22 ± 16 and 20 ± 13 years among those with the TT genotype and the TC + CC genotypes, respectively. (B) Seizure frequency of patients with the TT genotype and the TC + CC genotypes was 6.5 ± 2.7 and 6.7 ± 2.6 per month, respectively. (C) Drug-resistant incidence of patients with the TT genotype and the TC + CC genotypes was 34.4 and 41.1%, respectively. (D) According to the 1,000 Genomes Project, SNPs are downloaded between 10,000 nucleotides downstream and 10,000 nucleotides upstream of the rs909706 locus (Chromosome 6, NC_000006.11 (15650871..15670871), and according to the analysis using Haploview 4.2, only rs13195978 was observed to be in LD with rs909706 (D’ = 1.0).
According to the assembly of the human genome GRCh38.p7, rs909706 is located in the first intron of the DTNBP1 gene. However, introns are non-coding sequences, and variations within them rarely modulate gene function. Thus, we decided to investigate whether other functional SNPs are in linkage disequilibrium (LD) with variations at rs909706. As shown in Figure 1, rs13195978 was found to be in LD with rs909706, but rs13195978 is still non-functional due to its locus in the second intron of the DTNBP1 gene. Therefore, there are no clues to explain the protective role of the TT genotype at rs909706 against susceptibility to TLE.
As shown in Figure 2, among all the SNPs genotyped in the study, rs2619538 T > A is the only SNP located in the predicted promoter region of the DTNBP1 gene; thus, its variation might affect DTNBP1 transcription. Notably, homozygous mutation of rs2619538 T > A was rarely observed in the patient group (1/325) but not in the control group (0/335), suggesting that rs2619538 T > A might be a functional site involved in modulation of DTNBP1 transcription. Given these clues, we further constructed a genealogical chart of the mutant patient and genotyped the same site of his parents and brothers/sisters, and we identified a novo mutant allele in his father. Hence, we precluded its potential impact on the activity of the DTNBP1 gene promoter.
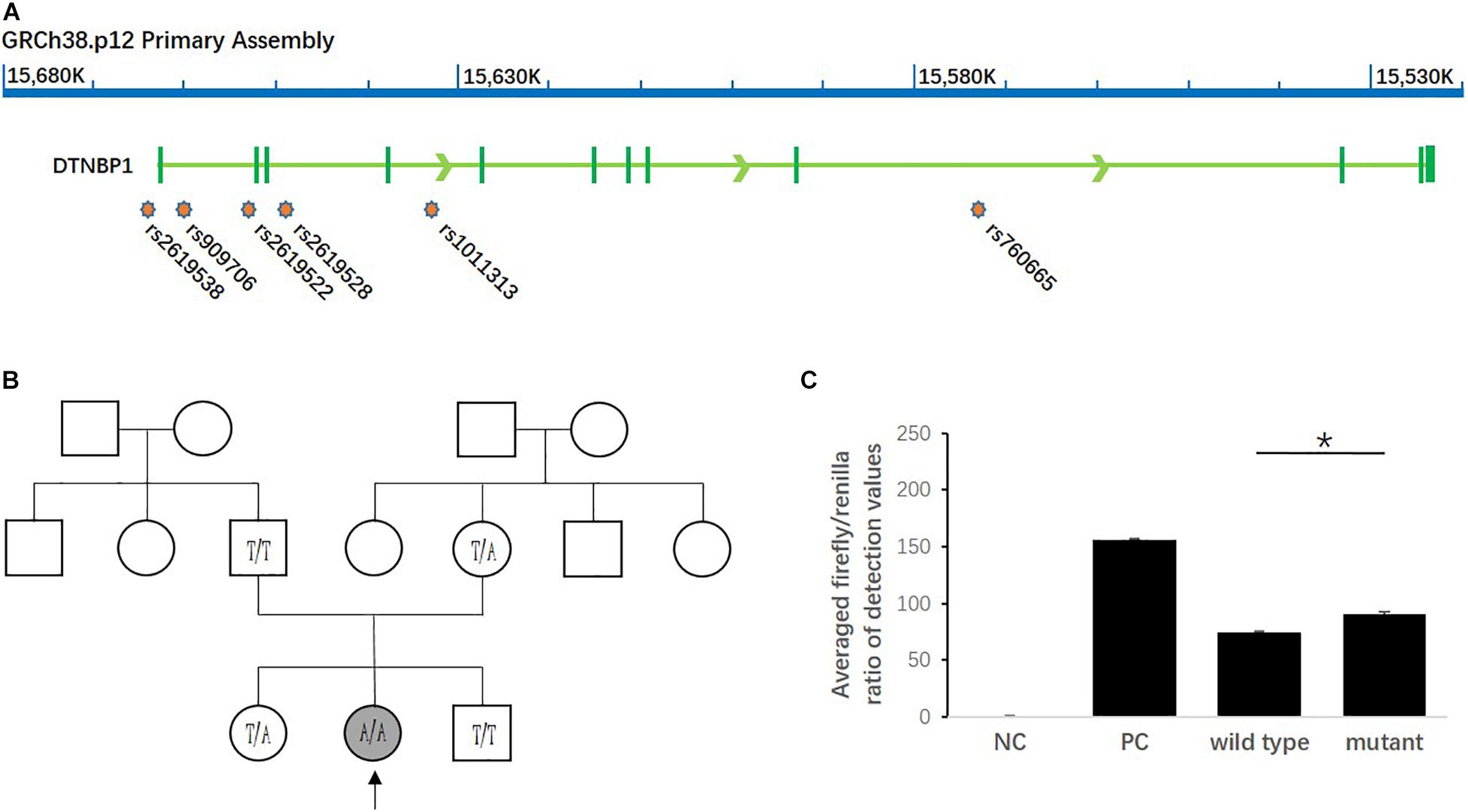
Figure 2. Impact of the homozygous mutation of rs2619538 T > A on DTNBP1 transcription. (A) Relative loci of rs2619538 T > A and other SNPs genotyped in this study are shown in the DNTBP1 gene region. Only rs2619538 T > A is potentially functional because of its location in the predicted promoter of the DNTBP1 gene, and yet the remaining SNPs are located within several introns of the DTNBP1 gene. (B) The TLE patient carrying the homozygous mutation of rs2619538 T > A was a 33-years-old male (age at onset, 32 years). After observation of the treatment for 1 year, seizure frequencies decreased from three times per month to one time per half year. A genealogical chart of his family was also constructed, and his parents consented to genotyping of the same site at rs2619538 T > A; a novo mutant allele probably coming from his father was observed. (C) The effects of rs2619538 T > A mutant on the activity of the DTNBP1 gene promoter. Compared with the averaged firefly/renilla ratios of the pGL3 basic negative control (NC), the ratios of the pGL3 promoter positive control (PC), pDTNBP1-Promoter-Wildtype (Wildtype) and pDTNBP1-Promoter-Mutant (Mutant) for rs2619538 T > A were 155.74 ± 1.43, 74.07 ± 0.99, and 90.48 ± 2.09, respectively. *p < 0.05.
In support of the mutant impact of rs2619538 T > A on the DTNBP1 gene promoter, we synthesized reporter gene constructs containing T (wild-type) or A (mutant) alleles in the context of the full-length promoter of DTNBP1 (2 kb sequence upstream of the transcription start site). The dual-luciferase assay revealed that both wild-type and mutant constructs have biological activities in the full-length promoter of DTNBP1, and the latter increased by approximately 22% in comparison with the former (Figure 2). This finding indicates that the homozygous mutation at rs2619538 T > A could upregulate DTNBP1 transcription.
To determine whether DTNBP1 expression is abnormal in epileptic activity, we established a rat epilepsy model according to a PTZ-induced protocol. DTNBP1 mRNA and protein expression was detected in the hippocampi by qPCR and ELISA. Compared with control rats, the epilepsy model showed an approximately 87% increase (p = 0.000) in the expression of DTNBP1 mRNA, similar to the increased rate of DTNBP1 protein (approximately 35%, p = 0.044). In addition, IHC confirmed the abnormal expression of DTNBP1 in the epilepsy model, indicating that hyperfunction of DTNBP1 is involved in epileptic seizures (Figure 3).
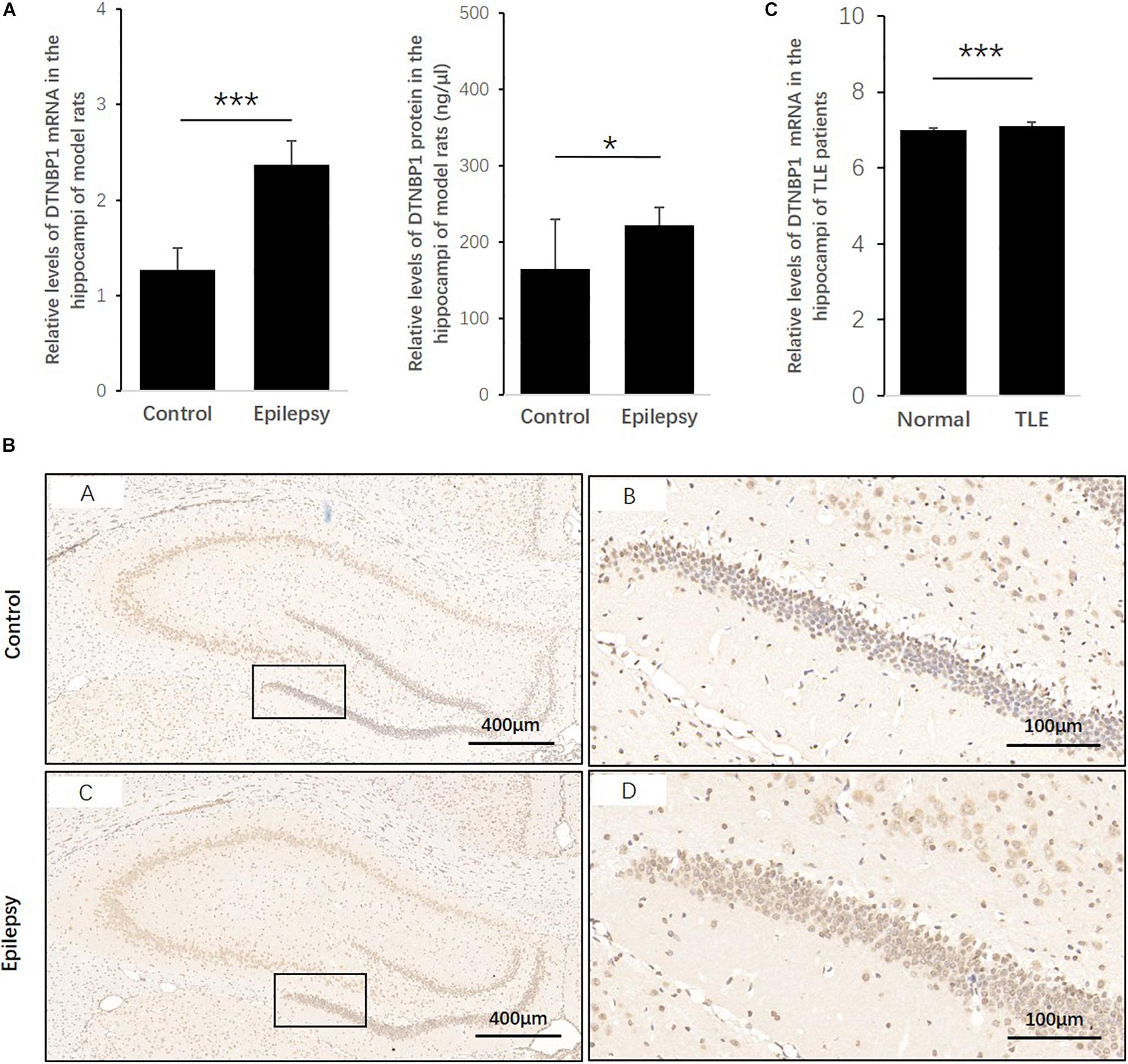
Figure 3. Expression patterns of DTNBP1 in an epilepsy rat model and TLE patients. (A) According to qPCR, the relative levels of DTNBP1 mRNA in the hippocampi of normal rats and epilepsy model rats were 1.27 ± 0.23 and 2.37 ± 0.25, respectively. (B) According to ELISA, the concentrations of DTNBP1 protein in the hippocampi of normal rats and epilepsy model rats were 164.60 ± 64.93 and 222.42 ± 23.20 ng/μl, respectively. (C) A total of 129 TLE patients from the GSE63808 dataset and 32 normal controls from the GSE29378 dataset were enrolled in the TLE and control groups. After normalization and multiple hypothesis testing to reduce the false-positive rate, the average DTNBP1 mRNA expression in the hippocampi of the TLE and control groups was 7.11 ± 0.09 and 6.99 ± 0.07, respectively. (D) IHC staining is shown individually for global and local regions of the hippocampi. Hematoxylin staining was used to mark the cell nuclei (blue), and immunostaining was used to show DTNBP1 expression (brown). *p < 0.05, ***p < 0.001.
To further identify whether DTNBP1 is abnormally expressed in human epilepsy patients, we used online GSE datasets of pathological brain samples from TLE patients and normal controls that were acquired by the same technique, Illumina HumanHT-12 V3.0 expression BeadChip arrays. Despite the small amplitude increase of 1.74%, the level of DTNBP1 expression in the hippocampi of the TLE patients was significantly higher than that of the normal controls (p = 9.316 × 10–9), suggesting that homozygous mutation at rs2619538 T > A might be involved in epileptic activity by upregulating DTNBP1 expression.
This study first observed that the frequencies of the TT genotype at rs909706 T > C and the CCAC and TCAT haplotypes (rs1011313-rs2619528-rs2619522-rs909706) were significantly lower in TLE patients than in controls. A previous study reported that the concentration of hippocampal glutamate was significantly affected by variations in rs909706 T > C (Wirth et al., 2012), whereas in our analyses, the intronic TT genotype was not related to age at onset or severity of disease and was not in LD with any functional variations nearby. Hence, these findings are considered to indicate protective markers against genetic susceptibility to TLE.
Meanwhile, the homozygous mutation of rs2619538 T > A was rarely observed in TLE patients, and the novo mutant allele came from the father. The dual-luciferase assay revealed that DTNBP2 promoter activity in patients with the mutant allele increased by approximately 22% compared with patients with the wild-type allele. Together with the trend of increased DTNBP1 expression in epilepsy patients and animal models in the present study, we hypothesized that the homozygous mutation of rs2619538 T > A functions by promoting DTNBP1 expression and subsequent processes in epilepsy pathologies.
According to previous research, the homozygous mutation at rs2619538 T > A is associated with reduced volumes of both gray and white matter in healthy children as young as 10–12 years old (Tognin et al., 2011). Structural volume deficits were observed in cortical regions, the subiculum and dentate gyrus, and the striatum of DTNBP1 mutant mice (Wirth et al., 2012). These findings indicate a key role of DTNBP1 in brain development, and abnormalities of brain development often lead to cortical dysplasia and subsequent early conset seizures (Palmini and Holthausen, 2013; Liu et al., 2015; Shaker et al., 2016; Maynard et al., 2017). On the other hand, DTNBP1 inhibits the release of glutamate (Chen et al., 2008; Saggu et al., 2013), which is crucial for AMPAR-mediated synaptic transmission and plasticity and NMDAR-dependent synaptic potentiation in the hippocampus (Glen et al., 2014; Orozco et al., 2014). In addition, DTNBP1 null gene mutation was observed to influence the developmental switch between GluN2B and GluN2A in the mouse cortex and hippocampus (Sinclair et al., 2016). These findings further indicate that DTNBP1 should be a critical modulator in excitatory signal transduction of glutamate, as well as brain development. Considering that an epilepsy model induced by PTZ is usually used to observe changes of excitatory/inhibitory regulatory molecules during the period of acute convulsions, this study successfully used the model to confirm increased DTNBP1 expression in the PTZ model, which indicates that the model can be used for disclosing potential mechanisms of DTNBP1 in future experiments.
Notably, DTNBP1 facilitates neurite outgrowth by promoting the transcriptional activity of p53 (Ma et al., 2011), and nucleocytoplasmic shuttling of DTNBP1 regulates synapsin I expression (Fei et al., 2010). However, neither overexpression (Dys1A-Tg) nor underexpression (Sandy) of DTNBP1 cause epileptic seizures in mice (Shintani et al., 2014). Because these prior studies focused on schizophrenia, more attention was paid to the effects of low-level DTNBP1 expression. In contrast, the expression of DTNBP1 was demonstrated to be increased in TLE patients and an animal model in this study; thus, behavioral observations and electrophysiological results of epileptic seizures in Dys1A-Tg mice and the related mechanisms should be further evaluated in the future.
Several limitations of the present study should be noted. First, DTNBP1 encodes dysbindin-1, which is composed of three spliceosomes, but only dysbindin-1B, not dysbindin-1A or dysbindin-1C, displays a tendency for toxic aggregation. In postmortem brains, dysbindin-1B not only aggregates with itself but also aggregates with proteins that interact with it (Xu et al., 2015; Zhu et al., 2015; Yang et al., 2016). Another study observed that increased expression of dysbindin-1A resulted in a selective deficit in NMDA receptor signaling in the hippocampus (Jeans et al., 2011). Additionally, dysbindin-1C is required for the survival of hilar mossy cells and the maturation of adult newborn neurons in the dentate gyrus (Wang et al., 2014), and dysbindin-1C deficiency could result in impaired autophagy (Yuan et al., 2015). Therefore, the expression patterns of the three spliceosomes most likely interfere with the function of DTNBP1 and influence genetic susceptibility to TLE. Second, when we consider 1,000 genomes, the minor allele frequencies of rs2619538 T > A are 0.027 among those of east Asian ancestry, 0.645 among those of African ancestry, 0.549 among those of European ancestry and 0.42 among those of American ancestry, which indicates that the mutant effects of rs2619538 T > A in the Han Chinese population included in the present study probably represent a mechanism that is distinct from those of other ancestries. Thus, one should be cautious in generalizing our findings to other races. Third, in addition to SNPs at rs2619538 T > A, DNA methylation of the DTNBP1 promoter also plays a key role in the expression of DTNBP1 and related pathological activities (Wockner et al., 2014; Abdolmaleky et al., 2015); thus, DNA methylation is another potential factor that could interfere with the genetic effects of rs2619538 T > A.
This study first demonstrated the association of DTNBP1 with TLE from a genetic perspective. In particular, the homozygous mutation rs2619538 T > A was observed in TLE patients but not in healthy controls. The increased activities of the DTNBP1 promoter with the A allele in dual-luciferase assays and increased DTNBP1 expression in epilepsy patients and animal models suggest that the mutation likely functions by promoting transcription of the DTNBP1 gene and facilitating subsequent processes in epilepsy pathologies. Hence, the role of DTNBP1 in TLE deserves further exploration in the future.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
All experimental protocols involving human subjects were approved by the Ethics Committees of the Affiliated Hospital of Guangdong Medical University, The First Affiliated Hospital of Harbin Medical University, the Central People’s Hospital of Zhanjiang, the First People’s Hospital of Pingdingshan, and Beijing Tongren Hospital affiliated with Capital Medical University. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by the Ethics Committees of Guangdong Medical University. Written informed consent was obtained from the owners for the participation of their animals in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
HT and XZ undertook data analyses and wrote the manuscript. JC, HZ, LH, and YCa carried out biological experiments. JF, ZL, and YCh carried out specimen collection. CS carried out epilepsy model. BZ, WZ, and KL conceptualized the hypothesis and designed the study. All authors contributed to the article and approved the submitted version.
This study was funded by the Postdoctoral Science Foundation of Guangdong Medical University (Grant No. 701z20150002) and the Natural Science Foundation of Guangdong Province (Grant No. 2017A030310658).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abdolmaleky, H., Pajouhanfar, S., Faghankhani, M., Joghataei, M., Mostafavi, A., and Thiagalingam, S. (2015). Antipsychotic drugs attenuate aberrant DNA methylation of DTNBP1 (dysbindin) promoter in saliva and post-mortem brain of patients with schizophrenia and Psychotic bipolar disorder. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 168, 687–696. doi: 10.1002/ajmg.b.32361
Andersen, K., Petersen, L., Vestergaard, M., Pedersen, C., and Christensen, J. (2019). Premature mortality in persons with epilepsy and schizophrenia: a population-based nationwide cohort study. Epilepsia 60, 1200–1208. doi: 10.1111/epi.15158
Azimi, T., Ghafouri-Fard, S., Davood Omrani, M., Mazdeh, M., Arsang-Jang, S., Sayad, A., et al. (2018). Vaccinia related Kinase 2 (VRK2) expression in neurological disorders: schizophrenia, epilepsy and multiple sclerosis. Multi. Sclerosis Relat. Disord. 19, 15–19. doi: 10.1016/j.msard.2017.10.017
Chen, X., Feng, Y., Hao, C., Guo, X., He, X., Zhou, Z., et al. (2008). DTNBP1, a schizophrenia susceptibility gene, affects kinetics of transmitter release. J. Cell Biol. 181, 791–801. doi: 10.1083/jcb.200711021
Clement, A. B., Hanstein, R., Schröder, A., Nagel, H., Endres, K., Fahrenholz, F., et al. (2008). Effects of neuron-specific ADAM10 modulation in an in vivo model of acute excitotoxic stress. Neuroscience 152, 459–468.
Fallgatter, A. J., Ehlis, A., Herrmann, M., Hohoff, C., Reif, A., Freitag, C., et al. (2010). DTNBP1 (dysbindin) gene variants modulate prefrontal brain function in schizophrenic patients–support for the glutamate hypothesis of schizophrenias. Genes Brain Behav. 9, 489–497. doi: 10.1111/j.1601-183X.2010.00574.x
Fei, E., Ma, X., Zhu, C., Xue, T., Yan, J., Xu, Y., et al. (2010). Nucleocytoplasmic shuttling of dysbindin-1, a schizophrenia-related protein, regulates synapsin I expression. J. Biol. Chem. 285, 38630–38640. doi: 10.1074/jbc.M110.107912
Fejgin, K., Nielsen, J., Birknow, M., Bastlund, J., Nielsen, V., Lauridsen, J., et al. (2014). A mouse model that recapitulates cardinal features of the 15q13.3 microdeletion syndrome including schizophrenia- and epilepsy-related alterations. Biol. Psychiatry 76, 128–137. doi: 10.1016/j.biopsych.2013.08.014
Glen, W., Horowitz, B., Carlson, G., Cannon, T., Talbot, K., Jentsch, J., et al. (2014). Dysbindin-1 loss compromises NMDAR-dependent synaptic plasticity and contextual fear conditioning. Hippocampus 24, 204–213. doi: 10.1002/hipo.22215
He, N., Lin, Z., Wang, J., Wei, F., Meng, H., Liu, X., et al. (2019). Evaluating the pathogenic potential of genes with de novo variants in epileptic encephalopathies. Genet. Med. 21, 17–27. doi: 10.1038/s41436-018-0011-y
He, Z., Li, G., Zhu, D., and Lu, S. (2006). Guide for the Care and Use of Laboratory Animals, Science Press, 1–469.
Jeans, A., Malins, R., Padamsey, Z., Reinhart, M., and Emptage, N. (2011). Increased expression of dysbindin-1A leads to a selective deficit in NMDA receptor signaling in the hippocampus. Neuropharmacology 61, 1345–1353. doi: 10.1016/j.neuropharm.2011.08.007
Johnson, M. R., Behmoaras, J., Bottolo, L., Krishnan, M. L., Pernhorst, K., Santoscoy, P. L. M., et al. (2015). Systems genetics identifies Sestrin 3 as a regulator of a proconvulsant gene network in human epileptic hippocampus. Nat. Commun. 6:6031. doi: 10.1038/ncomms7031
Konopaske, G., Balu, D., Presti, K., Chan, G., Benes, F., and Coyle, J. (2018). Dysbindin-1 contributes to prefrontal cortical dendritic arbor pathology in schizophrenia. Schizophren. Res. 201, 270–277. doi: 10.1016/j.schres.2018.04.042
Kwan, P., Arzimanoglou, A., Berg, A. T., Brodie, M. J., Allen Hauser, W., Mathern, G., et al. (2010). Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE commission on therapeutic strategies. Epilepsia 51, 1069–1077. doi: 10.1111/j.1528-1167.2009.02397.x
Liu, W., An, D., Xiao, J., Li, J., Hao, N., and Zhou, D. (2015). Malformations of cortical development and epilepsy: a cohort of 150 patients in western China. Seizure 32, 92–99.
Ma, X., Fei, E., Fu, C., Ren, H., and Wang, G. (2011). Dysbindin-1, a schizophrenia-related protein, facilitates neurite outgrowth by promoting the transcriptional activity of p53. Mol. Psychiatry 16, 1105–1116. doi: 10.1038/mp.2011.43
Maynard, L., Leach, J., Horn, P., Spaeth, C., Mangano, F., Holland, K., et al. (2017). Epilepsy prevalence and severity predictors in MRI-identified focal cortical dysplasia. Epilepsy Res. 132, 41–49.
Michelson, M., Eden, A., Vinkler, C., Leshinsky-Silver, E., Kremer, U., Lerman-Sagie, T., et al. (2011). Familial partial trisomy 15q11-13 presenting as intractable epilepsy in the child and schizophrenia in the mother. Eur. J. Paediatr. Neurol. 15, 230–233. doi: 10.1016/j.ejpn.2010.11.001
Mohamed, Z., Tee, S., and Tang, P. (2018). Association of functional polymorphisms in 3′-untranslated regions of COMT, DISC1, and DTNBP1 with schizophrenia: a meta-analysis. Psychiatr. Genet. 28, 110–119. doi: 10.1097/ypg.0000000000000210
Nebel, R., Zhao, D., Pedrosa, E., Kirschen, J., Lachman, H., Zheng, D., et al. (2016). Reduced CYFIP1 in human neural progenitors results in dysregulation of schizophrenia and epilepsy gene networks. PLoS One 11:e0148039. doi: 10.1371/journal.pone.0148039
Orozco, I. J., Koppensteiner, P., Ninan, I., and Arancio, O. (2014). The schizophrenia susceptibility gene DTNBP1 modulates AMPAR synaptic transmission and plasticity in the hippocampus of juvenile DBA/2J mice. Mol. Cell. Neurosci. 58, 76–84. doi: 10.1016/j.mcn.2013.12.003
Palmini, A., and Holthausen, H. (2013). Focal malformations of cortical development: a most relevant etiology of epilepsy in children. Handb. Clin. Neurol. 111, 549–565.
Prox, J., Bernreuther, C., Altmeppen, H., Grendel, J., Glatzel, M., D’Hooge, R., et al. (2013). Postnatal disruption of the disintegrin/metalloproteinase ADAM10 in brain causes epileptic seizures, learning deficits, altered spine morphology, and defective synaptic functions. J. Neurosci. 33, 12915–12928.
Saggu, S., Cannon, T., Jentsch, J., and Lavin, A. (2013). Potential molecular mechanisms for decreased synaptic glutamate release in dysbindin-1 mutant mice. Schizophren. Res. 146, 254–263. doi: 10.1016/j.schres.2013.01.037
Shaker, T., Bernier, A., and Carmant, L. (2016). Focal cortical dysplasia in childhood epilepsy. Semin. Pediatr. Neurol. 23, 108–119.
Shintani, N., Onaka, Y., Hashimoto, R., Takamura, H., Nagata, T., Umeda-Yano, S., et al. (2014). Behavioral characterization of mice overexpressing human dysbindin-1. Mol. Brain 7:74. doi: 10.1186/s13041-014-0074-x
Sinclair, D., Cesare, J., McMullen, M., Carlson, G., Hahn, C., and Borgmann-Winter, K. (2016). Effects of sex and DTNBP1 (dysbindin) null gene mutation on the developmental GluN2B-GluN2A switch in the mouse cortex and hippocampus. J. Neurodev. Disord. 8:14. doi: 10.1186/s11689-016-9148-7
Stewart, L., Hall, A., Kang, S., Shaw, C., and Beaudet, A. (2011). High frequency of known copy number abnormalities and maternal duplication 15q11-q13 in patients with combined schizophrenia and epilepsy. BMC Med. Genet. 12:154. doi: 10.1186/1471-2350-12-154
Tao, H., Zhou, X., Xie, Q., Ma, Z., Sun, F., Cui, L., et al. (2018). SRR intronic variation inhibits expression of its neighbouring SMG6 gene and protects against temporal lobe epilepsy. J. Cell. Mol. Med. 22, 1883–1893. doi: 10.1111/jcmm.13473
Thirunavukkarasu, P., Vijayakumari, A., John, J., Halahalli, H., Paul, P., Sen, S., et al. (2014). An exploratory association study of the influence of dysbindin and neuregulin polymorphisms on brain morphometry in patients with schizophrenia and healthy subjects from South India. Asian J. Psychiatry 10, 62–68. doi: 10.1016/j.ajp.2014.04.002
Tognin, S., Viding, E., McCrory, E., Taylor, L., O’Donovan, M., McGuire, P., et al. (2011). Effects of DTNBP1 genotype on brain development in children. J. Child Psychol. Psychiatry All. Discipl. 52, 1287–1294. doi: 10.1111/j.1469-7610.2011.02427.x
Trost, S., Platz, B., Usher, J., Scherk, H., Wobrock, T., Ekawardhani, S., et al. (2013). The DTNBP1 (dysbindin-1) gene variant rs2619522 is associated with variation of hippocampal and prefrontal grey matter volumes in humans. Eur. Archiv. Psychiatry Clin. Neurosci. 263, 53–63. doi: 10.1007/s00406-012-0320-0
Wang, H., Xu, J., Lazarovici, P., and Zheng, W. (2017). Dysbindin-1 involvement in the etiology of schizophrenia. Intern. J. Mol. Sci. 18:2044. doi: 10.3390/ijms18102044
Wang, H., Yuan, Y., Zhang, Z., Yan, H., Feng, Y., and Li, W. (2014). Dysbindin-1C is required for the survival of hilar mossy cells and the maturation of adult newborn neurons in dentate gyrus. J. Biol. Chem. 289, 29060–29072. doi: 10.1074/jbc.M114.590927
Wirth, C., Schubert, F., Lautenschlager, M., Brühl, R., Klär, A., Majic, T., et al. (2012). DTNBP1 (dysbindin) gene variants: in vivo evidence for effects on hippocampal glutamate status. Curr. Pharm. Biotechnol. 13, 1513–1521. doi: 10.2174/138920112800784952
Wockner, L., Noble, E., Lawford, B., Young, R., Morris, C., Whitehall, V., et al. (2014). Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Transl. Psychiatry 4:e339. doi: 10.1038/tp.2013.111
Wotton, C. J., and Goldacre, M. (2012). Coexistence of schizophrenia and epilepsy: record-linkage studies. Epilepsia 53, e71–e74. doi: 10.1111/j.1528-1167.2011.03390.x
Xu, Y., Sun, Y., Ye, H., Zhu, L., Liu, J., Wu, X., et al. (2015). Increased dysbindin-1B isoform expression in schizophrenia and its propensity in aggresome formation. Cell Discov. 1:15032. doi: 10.1038/celldisc.2015.32
Yang, W., Zhu, C., Shen, Y., and Xu, Q. (2016). The pathogenic mechanism of dysbindin-1B toxic aggregation: BLOC-1 and intercellular vesicle trafficking. Neuroscience 333, 78–91. doi: 10.1016/j.neuroscience.2016.07.008
Yuan, Q., Yang, F., Xiao, Y., Tan, S., Husain, N., Ren, M., et al. (2016). Regulation of brain-derived neurotrophic factor exocytosis and gamma-aminobutyric acidergic interneuron synapse by the schizophrenia susceptibility gene Dysbindin-1. Biol. Psychiatry 80, 312–322. doi: 10.1016/j.biopsych.2015.08.019
Yuan, Y., Wang, H., Wei, Z., and Li, W. (2015). Impaired autophagy in hilar mossy cells of the dentate gyrus and its implication in schizophrenia. J. Genet. Genom. 42, 1–8. doi: 10.1016/j.jgg.2014.12.001
Keywords: DTNBP1, schizophrenia, genetic mutation, temporal lobe epilepsy, polymorphism
Citation: Tao H, Zhou X, Chen J, Zhou H, Huang L, Cai Y, Fu J, Liu Z, Chen Y, Sun C, Zhao B, Zhong W and Li K (2021) Genetic Effects of the Schizophrenia-Related Gene DTNBP1 in Temporal Lobe Epilepsy. Front. Genet. 12:553974. doi: 10.3389/fgene.2021.553974
Received: 20 April 2020; Accepted: 15 January 2021;
Published: 19 February 2021.
Edited by:
Elena Martín-García, Pompeu Fabra University, SpainReviewed by:
Gilberto Vargas Alarcón, Instituto Nacional de Cardiologia Ignacio Chavez, MexicoCopyright © 2021 Tao, Zhou, Chen, Zhou, Huang, Cai, Fu, Liu, Chen, Sun, Zhao, Zhong and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bin Zhao, Ymluemhhb2VAc29odS5jb20=; Wangtao Zhong, emhvbmd3YW5ndGFvNTEyQGFsaXl1bi5jb20=; Keshen Li, bGlrZXNoZW4xOTcxQDEyNi5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.