 Suleva Povea-Cabello1
Suleva Povea-Cabello1 Marina Villanueva-Paz2
Marina Villanueva-Paz2 Juan M. Suárez-Rivero1Mónica Álvarez-Córdoba1Irene Villalón-García1Marta Talaverón-Rey1Alejandra Suárez-Carrillo1Manuel Munuera-Cabeza1
Juan M. Suárez-Rivero1Mónica Álvarez-Córdoba1Irene Villalón-García1Marta Talaverón-Rey1Alejandra Suárez-Carrillo1Manuel Munuera-Cabeza1 José A. Sánchez-Alcázar1*
José A. Sánchez-Alcázar1*- 1Centro Andaluz de Biología del Desarrollo, Universidad Pablo de Olavide and Centro de Investigación Biomédica en Red: Enfermedades Raras, Instituto de Salud Carlos III, Seville, Spain
- 2Instituto de Investigación Biomédica de Málaga, Departamento de Farmacología y Pediatría, Facultad de Medicina, Universidad de Málaga, Málaga, Spain
Mitochondrial diseases are a heterogeneous group of rare genetic disorders that can be caused by mutations in nuclear (nDNA) or mitochondrial DNA (mtDNA). Mutations in mtDNA are associated with several maternally inherited genetic diseases, with mitochondrial dysfunction as a main pathological feature. These diseases, although frequently multisystemic, mainly affect organs that require large amounts of energy such as the brain and the skeletal muscle. In contrast to the difficulty of obtaining neuronal and muscle cell models, the development of induced pluripotent stem cells (iPSCs) has shed light on the study of mitochondrial diseases. However, it is still a challenge to obtain an appropriate cellular model in order to find new therapeutic options for people suffering from these diseases. In this review, we deepen the knowledge in the current models for the most studied mt-tRNA mutation-caused mitochondrial diseases, MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes) and MERRF (myoclonic epilepsy with ragged red fibers) syndromes, and their therapeutic management. In particular, we will discuss the development of a novel model for mitochondrial disease research that consists of induced neurons (iNs) generated by direct reprogramming of fibroblasts derived from patients suffering from MERRF syndrome. We hypothesize that iNs will be helpful for mitochondrial disease modeling, since they could mimic patient’s neuron pathophysiology and give us the opportunity to correct the alterations in one of the most affected cellular types in these disorders.
Introduction: Mitochondria, the Powerhouses of the Cell
Mitochondria are small, mobile, and plastic organelles located in the cytoplasm of most eukaryotic cells. These organelles are responsible for important cellular processes, such as regulation of apoptosis, calcium homeostasis, and reactive oxygen species (ROS) production. However, the main function of mitochondria is energy production through oxidative phosphorylation (OXPHOS), which takes place in the mitochondrial respiratory chain (MRC) (Russell and Turnbull, 2014). Mitochondria contain their own DNA, the mitochondrial DNA (mtDNA), which is circular and double stranded. The mitochondrial genome consists of 16,569 nucleotide pairs that encode 13 proteins, two ribosomal RNA components, and 22 transfer RNAs (tRNAs) (Alberts et al., 2002). Regarding mitochondrial structure, these organelles are composed by two membranes, the inner and the outer mitochondrial membranes (IMMs and OMMs, respectively) that delimit two main compartments: the internal matrix and the intermembrane space. The IMM contains many folds named cristae that protrude into the matrix and enlarge the IMM surface. This membrane can be subdivided into two compartments, the inner boundary membrane (IBM) and the cristae membrane (CM), that are connected via cristae junctions. Albeit IMM is considered a continuous membrane, lateral diffusion of membrane proteins is restricted and IBM and CM exhibit an asymmetric protein distribution. This heterogeneity is important for efficient OXPHOS, mitochondrial biogenesis, and remodeling (Wollweber et al., 2017; Busch, 2020). Although mainly separated, the inner and outer mitochondrial membranes are partially connected via contact sites that are involved in cristae organization (Bulthuis et al., 2019). Since the OMM is more permeable than the IMM, containing many copies of the transport protein porin, the intermembrane space composition is equivalent to the cytosolic one. However, due to the presence of cardiolipin, the IMM is specially impermeable to ions and it is selectively permeable to small molecules required by matrix enzymes, thanks to the presence of several transport proteins (Alberts et al., 2002).
For producing energy, mitochondria rely on the MRC components that are five multiprotein complexes whose polypeptides are encoded by mtDNA and nuclear DNA (nDNA), the latter contributing the most. For that reason, mitochondrial function relies on both genomes (Alberts et al., 2002). Four of these components (Complexes I–IV) are involved in electron transfer and proton pumping across the IMM, which generate an electrochemical proton gradient responsible for the mitochondrial membrane potential (ΔΨm), a marker of mitochondrial health. The flux of protons into the matrix through the ATP synthase (Complex V) drives ATP synthesis (Alberts et al., 2002). There is a current consensus about a higher level of organization of these components that are arranged in supercomplexes (SCs) (Brzezinski, 2020) that exist in a wide variety of stoichiometries (Cogliati et al., 2016). SC assembly and stability are determined by mitochondrial cristae shape since knockdown of OPA1, a protein that plays a key role in mitochondrial fusion, impairs SC formation (Cogliati et al., 2013; Jang and Javadov, 2020). In addition, cellular pathways such as the unfolding protein response (UPR) are involved in cristae density and SC assembly, improving mitochondrial respiratory function under nutrient stress conditions (Balsa et al., 2019). Thus, this higher level of organization is dynamic and adapts to the energetic status of the cell. Structural and modeling studies indicate that SC assembly facilitates electron transfer, prevents protein aggregation, and preserves the structural organization of MRC components (Jang and Javadov, 2020). However, despite extensive studies, SC functional relevance remains unknown (Brzezinski, 2020).
Defects in mitochondrial function have been linked not only to genetic mitochondrial diseases but also to cardiovascular diseases (Murphy et al., 2016) and neurodegenerative disorders such as Huntington’s and Parkinson’s diseases (Zeviani and Carelli, 2003; Martin-Jimenez et al., 2020).
Mitochondrial Diseases
Mitochondrial diseases are a heterogeneous group of rare genetic disorders caused by a partial or total dysfunction of mitochondria. These illnesses can be caused by mutations in nDNA or mtDNA. These mutations affect not only genes encoding for MRC components but also those that are involved in protein translation and assembly, mtDNA stability, as well as mutations in those nDNA-encoded proteins involved in the maintenance of mitochondrial nucleotide pools, nucleotide transport, mtDNA replication, RNA transcription, and mitochondrial dynamics (Schon et al., 2012; Chapman et al., 2020). Mitochondrial diseases are clinically heterogeneous; they may occur at any age, and patients manifest a wide variety of symptoms (Gorman et al., 2016). However, all of them share morphological and biochemical features. As a consequence of the MRC deficiency, cells manifest a reduced enzymatic function of MRC components, a reduction in oxygen consumption and ATP synthesis, and a ROS overproduction. In the case of patients, they suffer from lactic acidosis and elevated pyruvate levels in serum at rest and, specially, after moderate exercise. Additionally, patients’ muscle biopsies usually show ragged red fibers that reflect the proliferation of OXPHOS-defective mitochondria (Schon et al., 2012).
Mitochondrial Diseases Caused by Mutations in Mitochondrial Transfer RNAs
Mutations in mtDNA are associated with a wide variety of maternally inherited genetic disorders that provoke mitochondrial dysfunction (Kirino and Suzuki, 2005). Since 1988, when the first mtDNA mutations were identified (Holt et al., 1988; Wallace et al., 1988; Zeviani et al., 1988), more than 400 pathogenic mutations related to specific and non-specific diseases have been characterized (Lott et al., 2013). Among them, some frequent mitochondrial disorders caused by a point mutation in mtDNA are MELAS and MERRF syndromes. In most of the cases, MELAS syndrome is caused by a transition from adenine to guanine in the position 3243 in the mt-tRNALeu(UUR) (MT-TL1) gene (Schon et al., 2012). In the case of MERRF syndrome, the m.8344A > G mutation in the mt-tRNALys (MT-TK) gene is the most frequently associated with the disease (Shoffner et al., 1990; Yoneda et al., 1990; Kirino and Suzuki, 2005).
Pathogenic mutations in mt-tRNA affect mtDNA translation, causing a defect in protein synthesis and decreasing MRC complex function (Brisca et al., 2015). In the case of MELAS syndrome, the m.3243A > G mutation affects mt-tRNA structure stabilization, methylation, aminoacylation, and triplet recognition (Finsterer, 2007). This mutation causes a specific defect of UUG-rich gene translation such as ND6 gene that encodes a subunit of the NADH-coenzyme Q reductase complex, also known as Complex I. This translation defect results in specific Complex I deficiency due to a reduction in the synthesis of ND6 subunit that is characteristic of the MELAS syndrome (Kirino and Suzuki, 2005). On the other hand, m.8344A > G mutation, which accounts for the MERRF syndrome, affects both AAA and AAG codon translation, causing a defect of whole mitochondrial protein synthesis. This fact could explain some of the different symptoms that are associated with these two different diseases (Kirino and Suzuki, 2005).
Due to the multicopy nature of mtDNA, these mutations can be homoplasmic or heteroplasmic. Thus, MELAS and MERRF syndromes are heteroplasmic, which means that mutant and wild-type mtDNA copies coexist within the same cell. This feature is associated with the severity of the symptoms and hinders disease prognosis. In fact, it is thought that there is a threshold from which biochemical alterations are apparent (Zeviani and Carelli, 2007). This genetic heterogeneity gives rise to a wide variety of symptoms of diverse severity among patients. Both MELAS and MERRF syndromes are associated with neurological symptoms. MELAS syndrome affects several organs, and some of its manifestations include stroke-like episodes, dementia, epilepsy, lactic acidemia, myopathy, recurrent headaches, hearing impairment, diabetes, and short stature (El-Hattab et al., 2015). Stroke-like episodes are one of the main features of this syndrome, and patients’ brain MRIs usually show multiple stroke-like lesions in both occipital and temporoparietal areas (Angelini, 2014; El-Hattab et al., 2015). In the case of MERRF syndrome, the first symptom is usually myoclonus that is followed by generalized epilepsy, ataxia, weakness, and dementia. Other findings are hearing loss, short stature, optic atrophy, and cardiomyopathy (DiMauro, 2015).
Therapeutic Management of Mitochondrial Diseases
The development of useful therapies for mitochondrial diseases is challenging due to the difficulty of correcting the lack or dysfunction of essential mitochondrial proteins, the phenotypical heterogeneity of the diseases, and multisystem alteration. Furthermore, the brain, one of the most affected organs, is difficult to reach by potential therapies because it is protected by the blood–brain barrier. For those reasons, there are no effective treatments available for mitochondrial diseases and management of these diseases is mainly symptomatic. Still, several strategies that are summarized in this review (Viscomi and Zeviani, 2020) have been developed.
Pharmacological treatment options are generally focused on targeting cellular pathways, such as mitochondrial biogenesis or autophagy, or preventing oxidative damage. For these reasons, AMP-activated protein kinase (AMPK) and mammalian target of rapamycin complex 1 (mTORC1) signaling have been the main targets of these strategies. Some of the most known drugs for treating mitochondrial diseases are rapamycin and its analogs (rapalogs) that inhibit mTORC1. Rapamycin and/or rapalogs have been demonstrated to improve the symptoms of some patients (Sage-Schwaede et al., 2019), but individual cases should be evaluated and long-term side effects remain unknown (Viscomi and Zeviani, 2020). Another common compound that is usually used for mitochondrial disease treatment is coenzyme Q10 (CoQ10), an essential component in the mitochondrial electron transport and antioxidant in cell membranes (Santa, 2010). However, improvement of symptoms under CoQ10 treatment has been variable, and no sustained clinical benefits have been reported (Santa, 2010; Gorman et al., 2016). The activation of mitochondrial biogenesis through AMPK is another common therapeutic option for these diseases. 5-Aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR), an AMP analog that activates AMPK, has been demonstrated to ameliorate the clinical phenotype in mouse models of mitochondrial diseases (Viscomi and Zeviani, 2020), but it is still not clarified if all tissues will benefit from this treatment in the long term (Suomalainen and Battersby, 2018).
Particularly in mtDNA mutations, several supplements as antioxidants and cofactors are being used. In MELAS syndrome, these treatments are L-arginine, citrulline, CoQ10, creatine monohydrate, and L-carnitine (El-Hattab et al., 2015). In addition, oral taurine supplementation has been demonstrated to reduce the recurrence of stroke-like episodes and increase taurine modification in mt-tRNALeu(UUR) (Ohsawa et al., 2019). Moreover, idebenone, a CoQ10 analog, has shown promising results alone (Ikejiri et al., 1996) or in combination with riboflavin (Napolitano et al., 2000) when used in concrete patients. In Leber’s hereditary optic neuropathy (LHON), a mitochondrial disease commonly caused by a primary homoplasmic mutation in mtDNA (Meyerson et al., 2015), orphan designation (EU/3/07/434) was granted by the European Medicines Agency (EMA) for idebenone on 2007, and recently, commercialization of idebenone (Raxone) has been authorized by EMA for the treatment of visual impairment in adolescent and adult patients with this disease (EMA/480039/2015). In the case of MERRF syndrome, CoQ10, idebenone, or L-carnitine is frequently prescribed. Conventional anticonvulsant drugs, such as levetiracetam, are also used to treat seizures (DiMauro and Hirano, 1993; Finsterer, 2019). However, none of these treatments has demonstrated enough effectiveness and only partially ameliorate some symptoms.
Given the diversity of mutations and the different therapeutic options, a personalized therapeutic approach is required in mitochondrial diseases. For this reason, the development of cellular models derived from patients (discussed below) can be useful for both the evaluation of new drugs and the repositioning of existing ones.
Gene therapy is a promising alternative for treating mitochondrial diseases. Since pathogenic mtDNA mutations are usually heteroplasmic, reducing mutational load can be used as a therapeutic approach. There are several tools that could target mtDNA, but only two of them have been demonstrated to be successful: zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs). Both tools are delivered into mitochondria using a mitochondria localization signal, and they selectively target mtDNA sequences to create double-strand breaks (Reddy et al., 2020).
Zinc-finger nucleases have been demonstrated to reduce mutant mtDNA and consequently restore mitochondrial respiratory function in cytoplasmic hybrid (cybrid) cell models (Minczuk et al., 2008; Gammage et al., 2014). In addition, this tool has been able to specifically eliminate mutant mtDNA in the cardiac tissue of a mitochondrial disease mouse model (Gammage et al., 2018). ZFNs are small and, due to their similarity to mammalian transcription factors, they are thought to have low immunogenic properties. However, they are complex, expensive, and exhibit lower specificity and efficiency than TALENs (Reddy et al., 2020). TALENs have been widely used for genetic manipulation in different organisms. This tool has been demonstrated to reduce mutant mtDNA load and improve the pathophysiology in a cellular model of MERRF syndrome (Hashimoto et al., 2015) as well as to eliminate the m.3243A > G mutation in MELAS iPSCs and porcine oocytes (Yang et al., 2018). In addition, TALENs have been able to reduce mutant mtDNA load in a mouse model harboring a mutation in a mt-tRNA, reverting disease-related phenotypes (Bacman et al., 2018). These gene therapy tools could be used together with other therapies aiming the elimination or prevention of pathogenic mtDNA transfer from mother to child (Nissanka and Moraes, 2020).
Regarding prevention of these diseases, the only option available is transferring embryos below the threshold of clinical expression in order to avoid or at least reduce the risk of transmission of mtDNA mutations. The selection of these embryos is based on preimplantation genetic diagnosis (PGD) (Gorman et al., 2016). In addition, there is a new strategy, the mitochondrial donation, that consists of the substitution of mutant maternal mitochondria using enucleated donor oocytes (Gorman et al., 2016). However, this technique has raised ethical issues and remains controversial (Saxena et al., 2018).
Common Models for Mitochondrial Disease Study
Clinical research in mitochondrial diseases has been traditionally carried out in many patients worldwide. There are many articles regarding these disorders that are in fact case reports. These studies have provided valuable information about these diseases such as the clinical hallmarks, the mutations that cause the disease, the onset, and the effects of the available treatments and supplements. In addition, they help to infer the pathophysiological mechanisms underlying these diseases.
However, experimental research has been hindered by the lack of proper biological models. These models are necessary for understanding the pathophysiology of the diseases as well as for finding new therapeutic targets. Despite the difficulties to mimic the repercussions of mtDNA mutations, there are several models to study mitochondrial diseases, such as microorganism, animal, and cellular models (Figure 1).
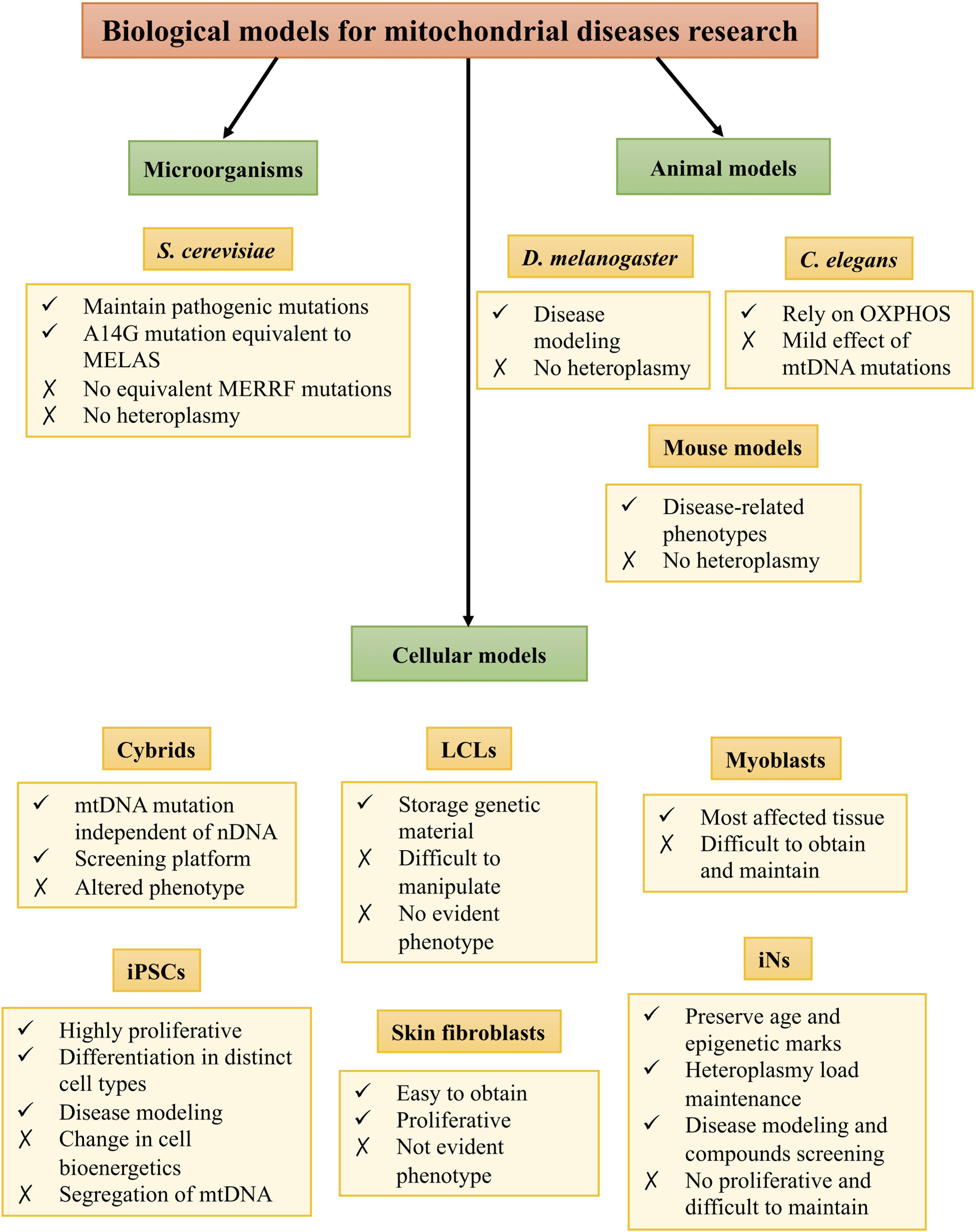
Figure 1. Biological models for mitochondrial disease research.
Microorganism Models
Saccharomyces cerevisiae is the most used microorganism model to study mitochondrial diseases. Mitochondrial functions are highly conserved between humans and S. cerevisiae, and this microorganism maintains pathogenic mutations that lead to mitochondrial dysfunction in humans. Therefore, yeasts are a good model to study mitochondrial diseases, providing insight into both physiological and pathophysiological processes (Baile and Claypool, 2013). The S. cerevisiae strain harboring the A14G mutation is equivalent to the human m.3243A > G mutation that causes MELAS syndrome. This model has been demonstrated to be suitable for compound screening (Garrido-Maraver et al., 2012). In the case of MERRF syndrome, there are no yeast strains with an equivalent mutation. However, the SLM3 gene in yeast is homologous to human TRMU, an enzyme involved in mt-tRNA wobble position thiolation. Mutations in SLM3 gene cause phenotypic features similar to those of m.8344A > G mutation, and consequently, yeasts harboring a defect in that gene have been used to study MERRF syndrome (Umeda et al., 2005). However, they do not maintain these mutations in a heteroplasmic state, so this aspect cannot be assessed using this model (Rinaldi et al., 2010).
Animal Models
To deepen the knowledge in the consequences of mtDNA mutations as well as develop new therapeutic strategies for mitochondrial diseases, animal models are necessary. However, there are only a few animal models for studying mitochondrial diseases, and none of them is specific for MELAS and MERRF syndromes.
The fruit fly Drosophila melanogaster is an excellent model organism for studying molecular mechanisms of human diseases and has been used for modeling neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases as well as for drug discovery (Pandey and Nichols, 2011). In the case of mitochondrial diseases, there are Drosophila mutants of MRC components that show neurodegeneration, motor dysfunction, increased ROS production, and abnormal mitochondrial morphology (Liu et al., 2007; Mast et al., 2008), as well as mutants of mtDNA, such as a model of Leigh syndrome (Celotto et al., 2006). However, currently, it is not possible to generate a Drosophila mutant with intermediate heteroplasmy for studying MELAS and MERRF syndromes (Palladino, 2010).
Mitochondrial composition, function, and mtDNA are well conserved between Caenorhabditis elegans and humans (Falk et al., 2009). C. elegans growth and development are energy-dependent, and the main ATP source for this organism is the MRC, which makes it interesting for studying mitochondrial diseases. In fact, there are worm strains that harbor mutations in MRC subunits encoded by nDNA (Ishii et al., 1998; Kayser et al., 2001; Tsang et al., 2001). However, mtDNA mutations have a mild effect in C. elegans (Tsang and Lemire, 2003), and they do not mimic the pathophysiological features of mtDNA mutation-caused mitochondrial diseases.
Mammalian models are necessary for studying pathogenesis or tissue-specific alterations that cannot be addressed using other models. Several murine models for OXPHOS deficiencies caused by mutations in nuclear genes encoding for MRC complexes, regulatory factors, and other components required for mitochondrial function have been created by transgenesis (Iommarini et al., 2015; Torraco et al., 2015). However, this is not the case for diseases caused by mutations in mtDNA, whose modeling is technically difficult (Tyynismaa and Suomalainen, 2009). Different mutagenesis methods to generate mutations in mouse mtDNA have been described, and these mutations can be transferred from cultured cells into mice, thereby creating transmitochondrial mice (mito-mice) (Inoue et al., 2000; Nakada et al., 2001; Fan et al., 2008; Kauppila et al., 2016). For example, a mouse model of LHON has been generated by introducing the equivalent of the human ND6 G14600A P25L mutation in homoplasmy into the mouse. Mutant mice showed a reduction in retinal function and neuronal accumulation of abnormal mitochondria among other events probably due to partial Complex I defects and increased ROS production (Lin et al., 2012). However, the generation of a mouse model with pathological heteroplasmic mtDNA mutations has proved challenging due to the multicopy nature of the mitochondrial genome. Furthermore, the transfection of plasmids or modified mtDNA into mouse mitochondria has not been successful (McGregor et al., 2001).
Cellular Models
Due to the lack of proper animal models for studying these diseases, cellular models have been extensively used in order to study MELAS and MERRF syndromes. Among them, we can find transmitochondrial cybrids, human B lymphoblastoid cell lines (LCLs), patient-derived cells such as myoblasts and dermal fibroblasts, and induced pluripotent stem cells (iPSCs) (Hu et al., 2019).
Transmitochondrial cybrids are generated by fusing enucleated cells that harbor wild-type or mutated mtDNA with ρ(0) cells, in which endogenous mtDNA has been depleted. For that reason, they are very useful for studying mtDNA mutations excluding the influence of nDNA variability (Vithayathil et al., 2012; Wilkins et al., 2014), and they have been used for studying MELAS and MERRF syndromes (Cotan et al., 2011; De la Mata et al., 2012; Garrido-Maraver et al., 2012; Villanueva-Paz et al., 2020). However, cybrid models show important limitations such as the need for a high mutational load to observe some pathophysiological features and the alteration in the cell behavior due to the loss of nDNA and mtDNA interactions (Dunbar et al., 1995).
Lymphoblastoid cell lines are generated by transformation of peripheral B lymphocytes and constitute a valuable source of mitochondria to study their function in mitochondrial diseases patients (Bourgeron et al., 1992). This model offers advantages such as the facility to obtain large quantities of lymphocytes and that they can be immortalized efficiently (Hu et al., 2019). For that reason, they have been used to study MERRF syndrome (Chang et al., 2013) and other mitochondrial diseases (Chin et al., 2018). Although this model is still the choice of storage for patients’ genetic material due to its low somatic mutation rate and ease of maintenance, LCLs have some limitations such as the presence of two different cellular stages and the different response in comparison with other cell types (Sie et al., 2009). Moreover, they are difficult to manipulate, and the phenotype is often not evident.
Myoblasts derived from patients’ biopsies are a very attractive model because they belong to one of the most affected tissues in these diseases. For that reason, they have been used to study the role of antioxidant enzymes and ATP levels in MELAS syndrome (Rusanen et al., 2000) and the distribution and expression of mutant mtDNAs in MERRF syndrome (Boulet et al., 1992). However, using myoblasts as a cellular model for mitochondrial diseases has several drawbacks. First, large quantities of proliferative myoblasts are difficult to isolate from a muscle tissue biopsy at later stages, so it is probable that more than one muscle biopsy is necessary to obtain enough cells for the analysis required. Moreover, primary myoblasts demand special conditions for optimal growth, and myoblast enrichment protocols are needed in order to obtain a pure cell culture.
Cultured fibroblasts are other patient-derived cells that have been a useful tool to study mitochondrial diseases. This model offers numerous advantages, since it is easy to obtain from a little invasive process such as a skin biopsy. In addition, fibroblast cultures are highly proliferative and provide a renewable source of cells (Hu et al., 2019). This model has been and still is widely used for studying cellular pathophysiology and as a screening tool for MELAS and MERRF syndromes (Wu et al., 2010; Cotan et al., 2011; De la Mata et al., 2012; Garrido-Maraver et al., 2015; Hayashi and Cortopassi, 2015; Villanueva-Paz et al., 2020) and other mitochondrial diseases. Nevertheless, it has some drawbacks as a cellular model for these diseases. First, they rely on glycolytic metabolism for energy production; therefore, they are not much vulnerable to energy-dependent defects resulting from mitochondrial dysfunction. In addition, they are difficult to maintain in culture and sometimes it is challenging to observe pathophysiological alterations, especially when the heteroplasmy load is low.
The most affected cell types in these diseases are the brain and the skeletal muscle cells, since they have a huge mitochondrial density due to high energy requirements and, consequently, they are more vulnerable to defects caused by mitochondrial dysfunction (DiMauro, 2007). In fact, neuron alteration is such that MELAS and MERRF syndromes are considered neurodegenerative mitochondriopathies in which there is neuronal cell death. MELAS neurodegeneration usually involves cortical territories in occipital and temporoparietal areas, as well as neurons in the Purkinje layer; meanwhile, in MERRF syndrome, the most affected areas are the Purkinje layer and dentate nucleus (Swerdlow, 2009).
The appearance of iPSCs in 2006 (Takahashi and Yamanaka, 2006) has led to numerous possibilities in the field of disease modeling. In fact, it has been a great step forward in the study of neurodegenerative diseases (Ebert et al., 2009; Lee et al., 2009; Kondo et al., 2013), as well as mitochondrial diseases (Cherry et al., 2013; Kodaira et al., 2015; Chou et al., 2016; Zhang et al., 2016; Inak et al., 2017; Lorenz et al., 2017; Yang et al., 2018). These iPSCs can be differentiated into somatic cell types such as neurons, which allow the study of these diseases in one of the most affected cellular types.
In the case of MERRF syndrome, neural progenitor cells (NPCs) derived from iPSCs have been demonstrated to reproduce pathophysiological features previously observed in other models of the disease, such as an impaired mitochondrial respiration, increased ROS production, altered antioxidant enzyme expression, as well as a fragmented mitochondrial network (Chou et al., 2016). Regarding MELAS syndrome, iPSC-derived neurons have made possible the study of not only common pathophysiological alterations but also neuron-specific alterations in the disease. For instance, in this work (Klein Gunnewiek et al., 2020), MELAS iPSC-derived neurons harboring a high heteroplasmy load showed lower dendrite complexity compared to control and low heteroplasmy neurons. These neurons also exhibited a reduced synaptic density, axonal mitochondrial abundance, and frequency of spontaneous excitatory activity, as well as an impaired neuronal network activity and synchronicity.
In addition, iPSC-derived neurons from mitochondrial disease patients can show different pathophysiological characteristics than parental patient-derived cells. For example, Hämäläinen et al. (2013) observed that parental MELAS fibroblast lines with intermediate heteroplasmy levels showed a combined deficiency of mtDNA-encoded Complexes I, III, and IV subunits; meanwhile, MELAS iPSC-derived neurons manifested a remarkable CI deficiency, which is typical for MELAS patient tissues and commonly reported upon mitochondria-associated neurodegeneration.
Reprogramming into iPSCs offers several advantages, since iPSCs can be cultured and a large quantity of starting material can be obtained. However, the protocol is complex, expensive, and time-consuming (Dolmetsch and Geschwind, 2011; Figure 2). Furthermore, this technique results in some disadvantages related to their use in mitochondrial disease research.
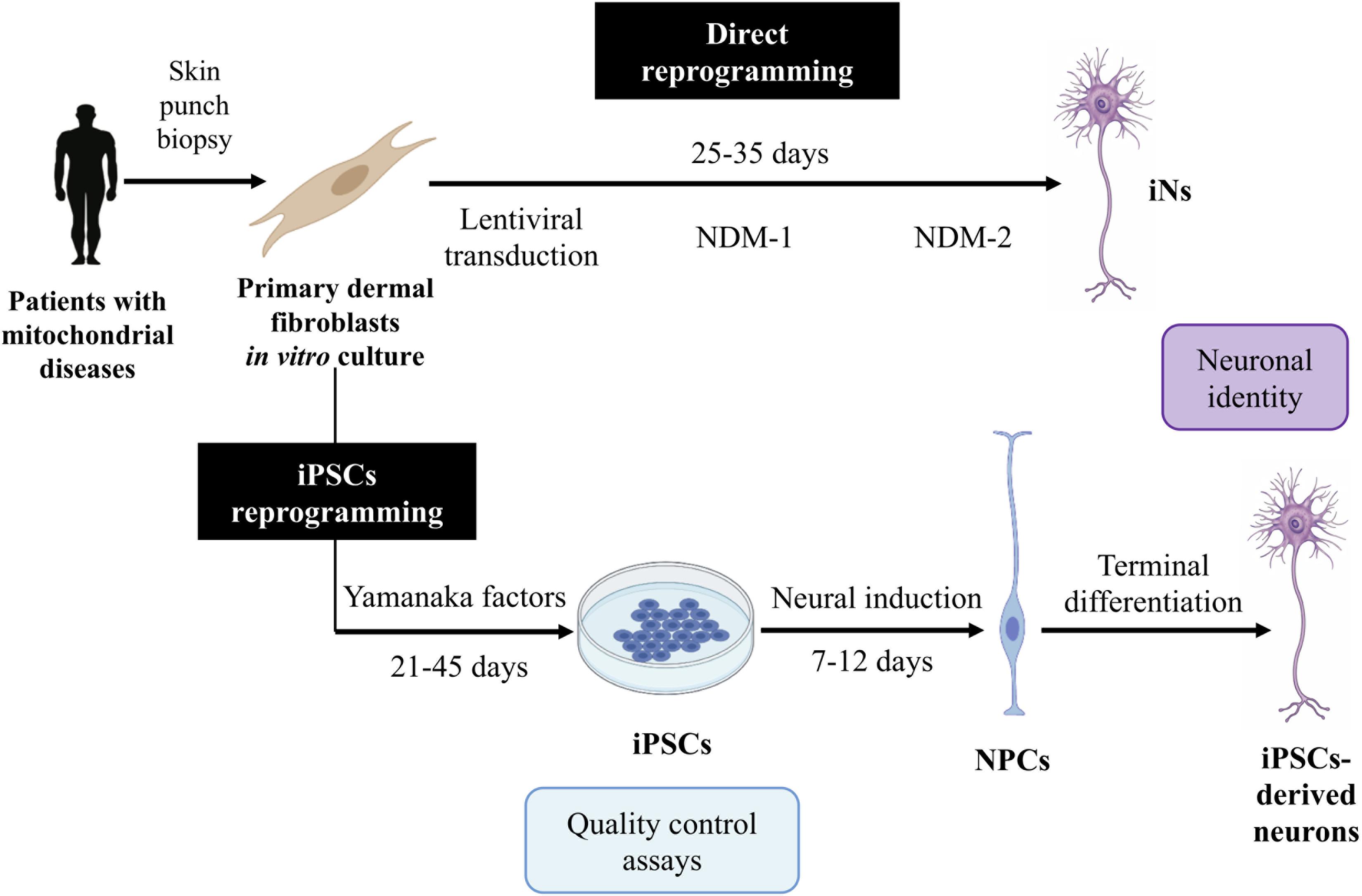
Figure 2. Induced neurons (iNs) and induced pluripotent stem cell (iPSC)-derived neuron generation. This figure summarizes the protocols for the generation of iNs and iPSC-derived neurons. For iN generation, following lentiviral transduction of neural transcription factors, cells are exposed to a neural differentiation medium (NDM) I containing specific small molecules and growth factors and then to an NDM II containing only the growth factors. iNs are obtained at 25–35 days post-infection (DPI). For iPSC-derived neuron generation, fibroblasts are transduced with Yamanaka factors to generate iPSCs. These iPSCs are converted into neural progenitor cells (NPCs) by defined factors, and these are then differentiated into the desired neuron subtype.
Several studies have demonstrated that reprogramming into iPSCs resets cellular age (Hsu et al., 2016), obtaining young neurons that could not show cell pathophysiology and, as a result, might not be useful to study neurodegenerative diseases. Furthermore, another study shows that this approach causes mitochondrial rejuvenation and an improvement in the cellular energy production capacity upon differentiation (Suhr et al., 2010). Another drawback would be that the use of these cells in cell replacement therapies is limited due to uncompleted differentiation and their propension to form tumors (Parmar and Jakobsson, 2011). In addition, analysis of mtDNA variants in iPSCs showed that low levels of potentially pathogenic mutations in the fibroblasts are revealed during reprogramming into iPSCs. Thus, mutant iPSCs are generated, causing a deleterious effect on their differentiated progeny and contributing to intra-person variability. This accumulation of mtDNA mutations can impact metabolic functions in iPSCs, hampering mitochondrial respiration (Kang et al., 2016; Perales-Clemente et al., 2016). These results highlight the need for monitoring mtDNA mutations and examining the metabolic status and quality of iPSCs intended for disease modeling or drug screening. Finally, reprogramming into iPSCs provokes an mtDNA segregation toward homoplasmy. In this way, after reprogramming, mutant and wild-type cells are obtained (Pickrell and Youle, 2013). These wild-type cells might be useful for cell therapy or as a syngeneic control, since they would be identical but will not express pathophysiological features. Therefore, the iPSC approach could be considered an autologous source of material suitable for cell therapy, without the risk of immune rejection (Smith et al., 2015). However, homoplasmic mutant iPSCs would not reflect patients’ cell pathophysiology, since they harbor the mutation in a heteroplasmic state. Thus, there are major limitations that we still have to overcome, and currently, iPSCs have no therapeutic use (Crow, 2019). All of the above reasons make necessary the generation of additional models for these diseases.
New Model for Mitochondrial Disease Research: Induced Neurons Generated by Direct Reprogramming
Direct reprogramming of fibroblasts into induced neurons (iNs) was achieved for the first time in 2010 (Vierbuchen et al., 2010). Using a combination of proneural transcription factors such as Ascl1, Brn2, and Myt1l, Vierbuchen et al. (2010) achieved the conversion of embryonic and postnatal murine fibroblasts into functional neurons in vitro. One year later, several advances allowed the improvement of the technique and increased the conversion efficiency. One of them was the utilization of microRNAs (miRNAs), in concrete miR-9/9∗ and miR-124, that leads to the conversion of fibroblasts into neurons even without the overexpression of proneural factors. However, since the reprogramming efficiency was low, these miRNAs were combined with the expression of NeuroD2 factor, Ascl1 and Myt1l (Yoo et al., 2011). Alternatively, using NeuroD1 factor together with the overexpression of Ascl1, Brn2, and Myt1l, direct reprogramming of fibroblasts into iNs was achieved with high efficiency (Pang et al., 2011). Additionally, compound screening has allowed the usage of a combination of small molecules and neural growth factors that increase reprogramming efficiency (Ladewig et al., 2012; Pfisterer et al., 2016).
Later, Drouin-Ouellet et al. (2017a) described a barrier for adult fibroblast reprogramming, the REST (RE-1 silencing transcription factor) complex, and developed one single lentiviral vector for the conversion of human adult fibroblasts into iNs in a very efficient manner. These iNs expressed neuron-specific proteins and exhibited electrophysiological properties. A recent paper from this group describes that the addition of the miRNAs indicated above, together with the overexpression of Ascl1 and Brn2, and the REST complex silencing, support neuronal maturation (Birtele et al., 2019). This technique allows the generation of a pan-neuronal population, but there are other strategies for reprogramming into neuronal subtypes, such as dopaminergic (Caiazzo et al., 2011), motor (Son et al., 2011; Qin et al., 2018), and serotonergic neurons (Xu et al., 2016). These approaches are valuable for some diseases in which there is one specific neuronal subtype affected or even different neuron subtypes may express distinct disease-related phenotypes.
Direct reprogramming brings numerous advantages. First, the procedure is relatively simple and fast (Ladewig et al., 2013; Figure 2). In addition, iNs maintain the age (Mertens et al., 2015) and the epigenetic marks of the donor (Horvath, 2013; Huh et al., 2016), which make them excellent models to study neurodegenerative diseases such as mitochondrial disorders. Furthermore, iNs have demonstrated to not cause tumorigenic processes after in vivo reprogramming (Torper et al., 2013), so they might be a promising tool for cell therapy. Thus, iN generation from patients’ fibroblasts is thought to be a useful approach for studying the pathogenesis of these diseases.
One of the main challenges of direct reprogramming is reaching a high conversion efficiency. It is defined as the percentage of iNs obtained over the number of cells plated for conversion and can be very variable depending on the starting cells and the protocol used. The first approaches using direct reprogramming obtained very poor conversion efficiencies (Yoo et al., 2011; Ladewig et al., 2013), but the single vector-based approach developed by Drouin-Ouellet et al. (2017a; 2017b) and Shrigley et al. (2018) can generate very high yields of iNs. However, it is also necessary to reach a high percentage of purity, which is the number of iNs in the final population over the cells remaining in the plate. These two parameters are crucial since neurons are post-mitotic cells not able to further expand.
In addition, maintaining iNs in long-term cultures is difficult and expensive, since cell death can be observed from 30 days post-infection (DPI). This fact may hinder electrophysiological characterization of iNs, since the earliest time point when spontaneous action potentials have been detected is at 80–100 DPI (Drouin-Ouellet et al., 2017a). Moreover, iNs tend to form clusters during the reprogramming process, hampering isolation of individual cells for further analysis.
To our knowledge, the first work that uses iNs for mitochondrial disease study comes from our group. Following the protocol established for generating iNs using a single lentiviral vector (Shrigley et al., 2018), we successfully generated iNs from two MERRF patient-derived fibroblasts harboring the m.8344A > G mutation (Villanueva-Paz et al., 2019). The proportion of this mutation in the fibroblast cultures was 66% in MERRF 1 and 33% in MERRF 2 fibroblasts, and we demonstrated that iNs maintained these proportions after reprogramming. The maintenance of the heteroplasmy load is crucial if iNs are going to be used for disease modeling or as a screening platform. In addition, this fact makes direct reprogramming a more suitable approach for studying mitochondrial diseases than iPSC generation because, in the latter case, mtDNA segregation and a tendency to homoplasmy have been observed (Pickrell and Youle, 2013).
In this work (Villanueva-Paz et al., 2019), the presence of bouton-like structures and spine-like protrusions was observed in both control and MERRF iNs, suggesting neuronal maturation. Furthermore, in another paper from our group, we performed electrophysiological recordings, and iNs showed electrophysiological properties at 60–80 DPI (Villanueva-Paz et al., 2020). The presence of neuronal maturation markers and functional properties suggest that iNs are behaving like neurons and make them a good candidate for mimicking the alterations happening in the patients’ brain. We also characterized the iN areas, perimeters, neurite features, as well as mitochondrial morphology, and we observed differences between control and MERRF iNs, indicating that the maintenance of the mutational load was affecting these features (Villanueva-Paz et al., 2019).
In this regard, MERRF iNs also showed pathophysiological features that have been described in other models of the disease such as fibroblasts, cybrids (De la Mata et al., 2012; Villanueva-Paz et al., 2020), and NPCs derived from MERRF iPSCs (Chou et al., 2016). For instance, they showed a reduced ΔΨm, a ROS overproduction, a disrupted autophagy flux, and an increased mitophagy (Villanueva-Paz et al., 2019). This fact makes them a good model for studying the cellular alterations happening in one of the most affected cell types in the disease. MERRF iNs were also suitable for performing other experiments such as the assessment of cellular bioenergetics using an extracellular flux analyzer. In these experiments, MERRF iNs showed alterations in the bioenergetic status such as reduced basal and maximal respirations, spare respiratory capacity, and ATP production (Villanueva-Paz et al., 2019). Some of these alterations were able to be rescued by Guttaquinon CoQ10 (a water-soluble derivative of CoQ10) treatment, demonstrating that iNs are not only suitable for disease modeling but also for compound screening (Villanueva-Paz et al., 2020).
Conclusion and Future Perspectives
The generation of mitochondrial disease patient-derived iNs is a very promising starting point for the advance in the study of these illnesses and the search for new treatments. This model has been demonstrated to have neuronal identity as well as to reproduce pathophysiological features of the diseases (Villanueva-Paz et al., 2019). Among these characteristics, they have been able to show mitochondrial dysfunction that is a hallmark in mitochondrial disorders and other neurodegenerative illnesses such as Parkinson’s or Huntington’s diseases (Zeviani and Carelli, 2003). The presence of these features in iNs is even more important due to the fact that they are the most affected cell types in neurodegenerative disorders. This model also brings other advantages for studying this kind of diseases, such as the maintenance of the age and the epigenetic marks of the donor (Horvath, 2013; Mertens et al., 2015; Huh et al., 2016). For those reasons, we think that iNs are very valuable for modeling diseases that are accompanied by mitochondrial dysfunction. An interesting further step in heteroplasmic mitochondrial disease modeling would be the establishment of 3-dimensional (3D) cultures, such as cerebral organoids (Amin and Pasca, 2018), in which iNs as the main cellular component would presumably be maintained in culture for prolonged periods while remaining viable and retaining their specific activities. This 3D model has already been generated using iPSCs derived from mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) patients’ cells (Pacitti and Bax, 2018). In addition, iNs have been demonstrated to be suitable for compound screening, since bioenergetic status of MERRF iNs was able to be rescued by Guttaquinon CoQ10 treatment (Villanueva-Paz et al., 2020). However, we still have to go deeper into the characterization of these new models and their suitability for finding new therapeutic targets for mitochondrial diseases.
Author Contributions
SP-C wrote the manuscript. JS-R, MÁ-C, IV-G, MT-R, AS-C, and MM-C contributed to the literature search. MV-P and JS-A revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by PI19/00377 grant, Instituto de Salud Carlos III, Spain, and Fondo Europeo de Desarrollo Regional (FEDER-Unión Europea), Spanish Ministry of Education, Culture and Sport, “Ayudas para la Formación de Profesorado Universitario” (FPU), and AEPMI (Asociación de Enfermos de Patología Mitocondrial).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ΔΨ m, mitochondrial membrane potential; 3D, 3-dimensional; CM, cristae membrane; CoQ10, coenzyme Q10; DPI, days post-infection; EMA, European Medicines Agency; IBM, inner boundary membrane; IMM, inner mitochondrial membrane; iNs, induced neurons; iPSCs, induced pluripotent stem cells; LCLs, lymphoblastoid cell lines; LHON, Leber’s hereditary optic neuropathy; MELAS, mitochondrial encephalomyopathy lactic acidosis and stroke-like episodes; MERRF, myoclonic epilepsy with ragged red fibers; miRNAs, micro RNAs; MNGIE, mitochondrial neurogastrointestinal encephalomyopathy; MRC, mitochondrial respiratory chain; mtDNA, mitochondrial DNA; NDM, neural differentiation medium; nDNA, nuclear DNA; NPCs, neural progenitor cells; OMM, outer mitochondrial membrane; OXPHOS, oxidative phosphorylation; PGD, preimplantation genetic diagnosis; REST, RE-1 silencing transcription factor; ROS, reactive oxygen species; SCs, supercomplexes; TALENs, transcription activator-like effector nucleases; tRNA, transfer RNA; UPR, unfolding protein response; ZFNs, zinc-finger nucleases.
References
Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., and Walter, P. (2002). “The mitochondrion,” in Molecular Biology of the Cell, 4th Edn, (New York, NY: Garland Science).
Amin, N. D., and Pasca, S. P. (2018). Building models of brain disorders with three-dimensional organoids. Neuron 100, 389–405. doi: 10.1016/j.neuron.2018.10.007
Angelini, C. (2014). Genetic Neuromuscular Disorders: A Case-Based Approach. Switzerland: Springer International Publishing.
Bacman, S. R., Kauppila, J. H. K., Pereira, C. V., Nissanka, N., Miranda, M., Pinto, M., et al. (2018). MitoTALEN reduces mutant mtDNA load and restores tRNA(Ala) levels in a mouse model of heteroplasmic mtDNA mutation. Nat. Med. 24, 1696–1700. doi: 10.1038/s41591-018-0166-8
Baile, M. G., and Claypool, S. M. (2013). The power of yeast to model diseases of the powerhouse of the cell. Front. Biosci. 18:241–278. doi: 10.2741/4098
Balsa, E., Soustek, M. S., Thomas, A., Cogliati, S., Garcia-Poyatos, C., Martin-Garcia, E., et al. (2019). ER and nutrient stress promote assembly of respiratory chain Supercomplexes through the PERK-eIF2alpha axis. Mol Cell 74:877-890.e6. doi: 10.1016/j.molcel.2019.03.031
Birtele, M., Sharma, Y., Kidnapillai, S., Lau, S., Stoker, T. B., Barker, R. A., et al. (2019). Dual modulation of neuron-specific microRNAs and the REST complex promotes functional maturation of human adult induced neurons. FEBS Lett. 593, 3370–3380. doi: 10.1002/1873-3468.13612
Boulet, L., Karpati, G., and Shoubridge, E. A. (1992). Distribution and threshold expression of the tRNA(Lys) mutation in skeletal muscle of patients with myoclonic epilepsy and ragged-red fibers (MERRF). Am. J. Hum. Genet. 51, 1187–1200.
Bourgeron, T., Chretien, D., Rotig, A., Munnich, A., and Rustin, P. (1992). Isolation and characterization of mitochondria from human B lymphoblastoid cell lines. Biochem. Biophys. Res. Commun. 186, 16–23. doi: 10.1016/s0006-291x(05)80769-7
Brisca, G., Fiorillo, C., Nesti, C., Trucco, F., Derchi, M., Andaloro, A., et al. (2015). Early onset cardiomyopathy associated with the mitochondrial tRNALeu((UUR)) 3271T>C MELAS mutation. Biochem. Biophys. Res. Commun. 458, 601–604. doi: 10.1016/j.bbrc.2015.01.157
Brzezinski, P. (2020). New structures reveal interaction dynamics in respiratory Supercomplexes. Trends Biochem. Sci. 45, 3–5. doi: 10.1016/j.tibs.2019.10.011
Bulthuis, E. P., Adjobo-Hermans, M. J. W., Willems, P., and Koopman, W. J. H. (2019). Mitochondrial Morphofunction in mammalian cells. Antioxid. Redox. Signal. 30, 2066–2109. doi: 10.1089/ars.2018.7534
Busch, K. B. (2020). Inner mitochondrial membrane compartmentalization: dynamics across scales. Int. J. Biochem. Cell Biol. 120:105694. doi: 10.1016/j.biocel.2020.105694
Caiazzo, M., Dell’Anno, M. T., Dvoretskova, E., Lazarevic, D., Taverna, S., Leo, D., et al. (2011). Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 476, 224–227. doi: 10.1038/nature10284
Celotto, A. M., Frank, A. C., McGrath, S. W., Fergestad, T., Van Voorhies, W. A., Buttle, K. F., et al. (2006). Mitochondrial encephalomyopathy in Drosophila. J. Neurosci. 26, 810–820. doi: 10.1523/JNEUROSCI.4162-05.2006
Chang, J. C., Liu, K. H., Chuang, C. S., Su, H. L., Wei, Y. H., Kuo, S. J., et al. (2013). Treatment of human cells derived from MERRF syndrome by peptide-mediated mitochondrial delivery. Cytotherapy 15, 1580–1596. doi: 10.1016/j.jcyt.2013.06.008
Chapman, J., Ng, Y. S., and Nicholls, T. J. (2020). The maintenance of Mitochondrial DNA integrity and dynamics by Mitochondrial membranes. Life 10:164. doi: 10.3390/life10090164
Cherry, A. B., Gagne, K. E., McLoughlin, E. M., Baccei, A., Gorman, B., Hartung, O., et al. (2013). Induced pluripotent stem cells with a mitochondrial DNA deletion. Stem Cells 31, 1287–1297. doi: 10.1002/stem.1354
Chin, R. M., Panavas, T., Brown, J. M., and Johnson, K. K. (2018). Patient-derived lymphoblastoid cell lines harboring mitochondrial DNA mutations as tool for small molecule drug discovery. BMC Res. Notes 11:205. doi: 10.1186/s13104-018-3297-6
Chou, S. J., Tseng, W. L., Chen, C. T., Lai, Y. F., Chien, C. S., Chang, Y. L., et al. (2016). Impaired ROS scavenging system in human induced Pluripotent stem cells generated from patients with MERRF Syndrome. Sci. Rep. 6:23661. doi: 10.1038/srep23661
Cogliati, S., Calvo, E., Loureiro, M., Guaras, A. M., Nieto-Arellano, R., Garcia-Poyatos, C., et al. (2016). Mechanism of super-assembly of respiratory complexes III and IV. Nature 539, 579–582. doi: 10.1038/nature20157
Cogliati, S., Frezza, C., Soriano, M. E., Varanita, T., Quintana-Cabrera, R., Corrado, M., et al. (2013). Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160–171. doi: 10.1016/j.cell.2013.08.032
Cotan, D., Cordero, M. D., Garrido-Maraver, J., Oropesa-Avila, M., Rodriguez-Hernandez, A., Gomez Izquierdo, L., et al. (2011). Secondary coenzyme Q10 deficiency triggers mitochondria degradation by mitophagy in MELAS fibroblasts. FASEB J. 25, 2669–2687. doi: 10.1096/fj.10-165340
Crow, D. (2019). Could iPSCs Enable “Off-the-Shelf”. Cell Therapy? Cell 177, 1667–1669. doi: 10.1016/j.cell.2019.05.043
De la Mata, M., Garrido-Maraver, J., Cotan, D., Cordero, M. D., Oropesa-Avila, M., Izquierdo, L. G., et al. (2012). Recovery of MERRF fibroblasts and cybrids pathophysiology by coenzyme Q10. Neurotherapeutics 9, 446–463. doi: 10.1007/s13311-012-0103-3
DiMauro, S. (2007). Mitochondrial DNA medicine. Biosci. Rep. 27, 5–9. doi: 10.1007/s10540-007-9032-5
DiMauro, S., and Hirano, M. (1993). “MERRF,” in GeneReviews(®), eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, et al. (Seattle, WA: University of Washington).
DiMauro, S. H. M. (2015). “MERRF,” in GeneReviews, eds A. H. Adam, H. H Ardinger, R. A. Pagon, and S. E Wallace (Seattle, WA: University of Washington).
Dolmetsch, R., and Geschwind, D. H. (2011). The human brain in a dish: the promise of iPSC-derived neurons. Cell 145, 831–834. doi: 10.1016/j.cell.2011.05.034
Drouin-Ouellet, J., Lau, S., Brattas, P. L., Rylander Ottosson, D., Pircs, K., Grassi, D. A., et al. (2017a). REST suppression mediates neural conversion of adult human fibroblasts via microRNA-dependent and -independent pathways. EMBO Mol. Med. 9, 1117–1131. doi: 10.15252/emmm.201607471
Drouin-Ouellet, J., Pircs, K., Barker, R. A., Jakobsson, J., and Parmar, M. (2017b). Direct neuronal reprogramming for disease modeling studies using patient-derived neurons: what have we learned? Front. Neurosci. 11:530. doi: 10.3389/fnins.2017.00530
Dunbar, D. R., Moonie, P. A., Jacobs, H. T., and Holt, I. J. (1995). Different cellular backgrounds confer a marked advantage to either mutant or wild-type mitochondrial genomes. Proc. Natl. Acad. Sci. U.S.A. 92, 6562–6566. doi: 10.1073/pnas.92.14.6562
Ebert, A. D., Yu, J., Rose, F. F. Jr., Mattis, V. B., Lorson, C. L., et al. (2009). Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 457, 277–280. doi: 10.1038/nature07677
El-Hattab, A. W., Adesina, A. M., Jones, J., and Scaglia, F. (2015). MELAS syndrome: clinical manifestations, pathogenesis, and treatment options. Mol. Genet. Metab. 116, 4–12. doi: 10.1016/j.ymgme.2015.06.004
Falk, M. J., Rosenjack, J. R., Polyak, E., Suthammarak, W., Chen, Z., Morgan, P. G., et al. (2009). Subcomplex Ilambda specifically controls integrated mitochondrial functions in Caenorhabditis elegans. PLoS One 4:e6607. doi: 10.1371/journal.pone.0006607
Fan, W., Waymire, K. G., Narula, N., Li, P., Rocher, C., Coskun, P. E., et al. (2008). A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science 319, 958–962. doi: 10.1126/science.1147786
Finsterer, J. (2007). Genetic, pathogenetic, and phenotypic implications of the mitochondrial A3243G tRNALeu(UUR) mutation. Acta Neurol. Scand. 116, 1–14. doi: 10.1111/j.1600-0404.2007.00836.x
Finsterer, J. (2019). Pharmacotherapeutic management of epilepsy in MERRF syndrome. Exp. Opin. Pharmacother. 20, 1289–1297. doi: 10.1080/14656566.2019.1609941
Gammage, P. A., Rorbach, J., Vincent, A. I., Rebar, E. J., and Minczuk, M. (2014). Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol. Med. 6, 458–466. doi: 10.1002/emmm.201303672
Gammage, P. A., Viscomi, C., Simard, M. L., Costa, A. S. H., Gaude, E., Powell, C. A., et al. (2018). Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med. 24, 1691–1695. doi: 10.1038/s41591-018-0165-9
Garrido-Maraver, J., Cordero, M. D., Monino, I. D., Pereira-Arenas, S., Lechuga-Vieco, A. V., Cotan, D., et al. (2012). Screening of effective pharmacological treatments for MELAS syndrome using yeasts, fibroblasts and cybrid models of the disease. Br. J. Pharmacol. 167, 1311–1328. doi: 10.1111/j.1476-5381.2012.02086.x
Garrido-Maraver, J., Paz, M. V., Cordero, M. D., Bautista-Lorite, J., Oropesa-Avila, M., de la Mata, M., et al. (2015). Critical role of AMP-activated protein kinase in the balance between mitophagy and mitochondrial biogenesis in MELAS disease. Biochim. Biophys. Acta 1852, 2535–2553. doi: 10.1016/j.bbadis.2015.08.027
Gorman, G. S., Chinnery, P. F., DiMauro, S., Hirano, M., Koga, Y., McFarland, R., et al. (2016). Mitochondrial diseases. Nat. Rev. Dis. Primers 2:16080. doi: 10.1038/nrdp.2016.80
Hämäläinen, R. H., Manninen, T., Koivumaki, H., Kislin, M., Otonkoski, T., and Suomalainen, A. (2013). Tissue- and cell-type-specific manifestations of heteroplasmic mtDNA 3243A>G mutation in human induced pluripotent stem cell-derived disease model. Proc. Natl. Acad. Sci. U.S.A. 110, E3622–E3630. doi: 10.1073/pnas.1311660110
Hashimoto, M., Bacman, S. R., Peralta, S., Falk, M. J., Chomyn, A., Chan, D. C., et al. (2015). MitoTALEN: a general approach to reduce Mutant mtDNA loads and restore oxidative Phosphorylation Function in Mitochondrial diseases. Mol. Ther. 23, 1592–1599. doi: 10.1038/mt.2015.126
Hayashi, G., and Cortopassi, G. (2015). Oxidative stress in inherited mitochondrial diseases. Free Radic. Biol. Med. 88(Pt A), 10–17. doi: 10.1016/j.freeradbiomed.2015.05.039
Holt, I. J., Harding, A. E., and Morgan-Hughes, J. A. (1988). Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 331, 717–719. doi: 10.1038/331717a0
Horvath, S. (2013). DNA methylation age of human tissues and cell types. Genome Biol. 14:R115. doi: 10.1186/gb-2013-14-10-r115
Hsu, Y. C., Chen, C. T., and Wei, Y. H. (2016). Mitochondrial resetting and metabolic reprogramming in induced pluripotent stem cells and mitochondrial disease modeling. Biochim. Biophys. Acta 1860, 686–693. doi: 10.1016/j.bbagen.2016.01.009
Hu, S. Y., Zhuang, Q. Q., Qiu, Y., Zhu, X. F., and Yan, Q. F. (2019). Cell models and drug discovery for mitochondrial diseases. J. Zhejiang Univ. Sci. B 20, 449–456. doi: 10.1631/jzus.B1900196
Huh, C. J., Zhang, B., Victor, M. B., Dahiya, S., Batista, L. F., Horvath, S., et al. (2016). Maintenance of age in human neurons generated by microRNA-based neuronal conversion of fibroblasts. eLife 5:e18648. doi: 10.7554/eLife.18648
Ikejiri, Y., Mori, E., Ishii, K., Nishimoto, K., Yasuda, M., and Sasaki, M. (1996). Idebenone improves cerebral mitochondrial oxidative metabolism in a patient with MELAS. Neurology 47, 583–585. doi: 10.1212/wnl.47.2.583
Inak, G., Lorenz, C., Lisowski, P., Zink, A., Mlody, B., and Prigione, A. (2017). Concise review: induced Pluripotent stem cell-based drug discovery for Mitochondrial disease. Stem Cells 35, 1655–1662. doi: 10.1002/stem.2637
Inoue, K., Nakada, K., Ogura, A., Isobe, K., Goto, Y., Nonaka, I., et al. (2000). Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nat. Genet. 26, 176–181. doi: 10.1038/82826
Iommarini, L., Peralta, S., Torraco, A., and Diaz, F. (2015). Mitochondrial Diseases Part II: mouse models of OXPHOS deficiencies caused by defects in regulatory factors and other components required for mitochondrial function. Mitochondrion 22, 96–118. doi: 10.1016/j.mito.2015.01.008
Ishii, N., Fujii, M., Hartman, P. S., Tsuda, M., Yasuda, K., Senoo-Matsuda, N., et al. (1998). A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature 394, 694–697. doi: 10.1038/29331
Jang, S., and Javadov, S. (2020). OPA1 regulates respiratory supercomplexes assembly: the role of mitochondrial swelling. Mitochondrion 51, 30–39. doi: 10.1016/j.mito.2019.11.006
Kang, E., Wang, X., Tippner-Hedges, R., Ma, H., Folmes, C. D., Gutierrez, N. M., et al. (2016). Age-related accumulation of somatic Mitochondrial DNA mutations in adult-derived human iPSCs. Cell Stem Cell 18, 625–636. doi: 10.1016/j.stem.2016.02.005
Kauppila, J. H. K., Baines, H. L., Bratic, A., Simard, M. L., Freyer, C., Mourier, A., et al. (2016). A Phenotype-Driven approach to generate mouse models with Pathogenic mtDNA mutations causing mitochondrial disease. Cell Rep. 16, 2980–2990. doi: 10.1016/j.celrep.2016.08.037
Kayser, E. B., Morgan, P. G., Hoppel, C. L., and Sedensky, M. M. (2001). Mitochondrial expression and function of GAS-1 in Caenorhabditis elegans. J. Biol. Chem. 276, 20551–20558. doi: 10.1074/jbc.M011066200
Kirino, Y., and Suzuki, T. (2005). Human mitochondrial diseases associated with tRNA wobble modification deficiency. RNA Biol. 2, 41–44. doi: 10.4161/rna.2.2.1610
Klein Gunnewiek, T. M., Van Hugte, E. J. H., Frega, M., Guardia, G. S., Foreman, K., Panneman, D., et al. (2020). m.3243A > G-Induced Mitochondrial Dysfunction impairs human neuronal development and reduces neuronal network activity and synchronicity. Cell Rep. 31:107538. doi: 10.1016/j.celrep.2020.107538
Kodaira, M., Hatakeyama, H., Yuasa, S., Seki, T., Egashira, T., Tohyama, S., et al. (2015). Impaired respiratory function in MELAS-induced pluripotent stem cells with high heteroplasmy levels. FEBS Open Biol. 5, 219–225. doi: 10.1016/j.fob.2015.03.008
Kondo, T., Asai, M., Tsukita, K., Kutoku, Y., Ohsawa, Y., Sunada, Y., et al. (2013). Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell 12, 487–496. doi: 10.1016/j.stem.2013.01.009
Ladewig, J., Koch, P., and Brustle, O. (2013). Leveling Waddington: the emergence of direct programming and the loss of cell fate hierarchies. Nat. Rev. Mol. Cell Biol. 14, 225–236. doi: 10.1038/nrm3543
Ladewig, J., Mertens, J., Kesavan, J., Doerr, J., Poppe, D., Glaue, F., et al. (2012). Small molecules enable highly efficient neuronal conversion of human fibroblasts. Nat. Methods 9, 575–578. doi: 10.1038/nmeth.1972
Lee, G., Papapetrou, E. P., Kim, H., Chambers, S. M., Tomishima, M. J., Fasano, C. A., et al. (2009). Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature 461, 402–406. doi: 10.1038/nature08320
Lin, C. S., Sharpley, M. S., Fan, W., Waymire, K. G., Sadun, A. A., Carelli, V., et al. (2012). Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proc. Natl. Acad. Sci. U.S.A. 109, 20065–20070. doi: 10.1073/pnas.1217113109
Liu, W., Gnanasambandam, R., Benjamin, J., Kaur, G., Getman, P. B., Siegel, A. J., et al. (2007). Mutations in cytochrome c oxidase subunit VIa cause neurodegeneration and motor dysfunction in Drosophila. Genetics 176, 937–946. doi: 10.1534/genetics.107.071688
Lorenz, C., Lesimple, P., Bukowiecki, R., Zink, A., Inak, G., Mlody, B., et al. (2017). Human iPSC-derived neural progenitors are an effective drug discovery model for neurological mtDNA disorders. Cell Stem Cell 20, 659–674.e9. doi: 10.1016/j.stem.2016.12.013
Lott, M. T., Leipzig, J. N., Derbeneva, O., Xie, H. M., Chalkia, D., Sarmady, M., et al. (2013). mtDNA variation and analysis using Mitomap and Mitomaster. Curr. Protoc. Bioinform. 44, 21–26. doi: 10.1002/0471250953.bi0123s44
Martin-Jimenez, R., Lurette, O., and Hebert-Chatelain, E. (2020). Damage in Mitochondrial DNA associated with Parkinson’s disease. DNA Cell Biol. 39, 1421–1430. doi: 10.1089/dna.2020.5398
Mast, J. D., Tomalty, K. M., Vogel, H., and Clandinin, T. R. (2008). Reactive oxygen species act remotely to cause synapse loss in a Drosophila model of developmental mitochondrial encephalopathy. Development 135, 2669–2679. doi: 10.1242/dev.020644
McGregor, A., Temperley, R., Chrzanowska-Lightowlers, Z. M., and Lightowlers, R. N. (2001). Absence of expression from RNA internalised into electroporated mammalian mitochondria. Mol. Genet. Genomics 265, 721–729. doi: 10.1007/s004380100469
Mertens, J., Paquola, A. C. M., Ku, M., Hatch, E., Bohnke, L., Ladjevardi, S., et al. (2015). Directly reprogrammed human neurons retain aging-associated Transcriptomic signatures and reveal age-related Nucleocytoplasmic defects. Cell Stem Cell 17, 705–718. doi: 10.1016/j.stem.2015.09.001
Meyerson, C., Van Stavern, G., and McClelland, C. (2015). Leber hereditary optic neuropathy: current perspectives. Clin. Ophthalmol. 9, 1165–1176. doi: 10.2147/OPTH.S62021
Minczuk, M., Papworth, M. A., Miller, J. C., Murphy, M. P., and Klug, A. (2008). Development of a single-chain, quasi-dimeric zinc-finger nuclease for the selective degradation of mutated human mitochondrial DNA. Nucleic Acids Res. 36, 3926–3938. doi: 10.1093/nar/gkn313
Murphy, E., Ardehali, H., Balaban, R. S., DiLisa, F., Dorn, G. W. II, Kitsis, R. N., et al. (2016). Mitochondrial function, biology, and role in disease: a scientific statement from the american heart association. Circ. Res. 118, 1960–1991. doi: 10.1161/RES.0000000000000104
Nakada, K., Inoue, K., and Hayashi, J. I. (2001). Mito-mice: animal models for mitochondrial DNA-based diseases. Semin. Cell Dev. Biol. 12, 459–465. doi: 10.1006/scdb.2001.0283
Napolitano, A., Salvetti, S., Vista, M., Lombardi, V., Siciliano, G., and Giraldi, C. (2000). Long-term treatment with idebenone and riboflavin in a patient with MELAS. Neurol Sci. 21(5 Suppl.), S981–S982. doi: 10.1007/s100720070015
Nissanka, N., and Moraes, C. T. (2020). Mitochondrial DNA heteroplasmy in disease and targeted nuclease-based therapeutic approaches. EMBO Rep. 21:e49612. doi: 10.15252/embr.201949612
Ohsawa, Y., Hagiwara, H., Nishimatsu, S. I., Hirakawa, A., Kamimura, N., Ohtsubo, H., et al. (2019). Taurine supplementation for prevention of stroke-like episodes in MELAS: a multicentre, open-label, 52-week phase III trial. J. Neurol. Neurosurg. Psychiatry 90, 529–536. doi: 10.1136/jnnp-2018-317964
Pacitti, D., and Bax, B. E. (2018). The development of an in vitro cerebral organoid model for investigating the pathomolecular mechanisms associated with the central nervous system involvement in Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE). Nucleosides Nucleotides Nucleic Acids 37, 603–617. doi: 10.1080/15257770.2018.1492139
Palladino, M. J. (2010). Modeling mitochondrial encephalomyopathy in Drosophila. Neurobiol. Dis. 40, 40–45. doi: 10.1016/j.nbd.2010.05.009
Pandey, U. B., and Nichols, C. D. (2011). Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol. Rev. 63, 411–436. doi: 10.1124/pr.110.003293
Pang, Z. P., Yang, N., Vierbuchen, T., Ostermeier, A., Fuentes, D. R., Yang, T. Q., et al. (2011). Induction of human neuronal cells by defined transcription factors. Nature 476, 220–223. doi: 10.1038/nature10202
Parmar, M., and Jakobsson, J. (2011). Turning skin into dopamine neurons. Cell Res. 21, 1386–1387. doi: 10.1038/cr.2011.130
Perales-Clemente, E., Cook, A. N., Evans, J. M., Roellinger, S., Secreto, F., Emmanuele, V., et al. (2016). Natural underlying mtDNA heteroplasmy as a potential source of intra-person hiPSC variability. EMBO J. 35, 1979–1990. doi: 10.15252/embj.201694892
Pfisterer, U., Ek, F., Lang, S., Soneji, S., Olsson, R., and Parmar, M. (2016). Small molecules increase direct neural conversion of human fibroblasts. Sci. Rep. 6:38290. doi: 10.1038/srep38290
Pickrell, A. M., and Youle, R. J. (2013). Mitochondrial disease: mtDNA and protein segregation mysteries in iPSCs. Curr. Biol. 23, R1052–R1054. doi: 10.1016/j.cub.2013.10.048
Qin, H., Zhao, A., Ma, K., and Fu, X. (2018). Chemical conversion of human and mouse fibroblasts into motor neurons. Sci. China Life Sci. 61, 1151–1167. doi: 10.1007/s11427-018-9359-8
Reddy, P., Vilella, F., Izpisua Belmonte, J. C., and Simón, C. (2020). Use of customizable nucleases for gene editing and other novel applications. Genes 11:976. doi: 10.3390/genes11090976
Rinaldi, T., Dallabona, C., Ferrero, I., Frontali, L., and Bolotin-Fukuhara, M. (2010). Mitochondrial diseases and the role of the yeast models. FEMS Yeast Res. 10, 1006–1022. doi: 10.1111/j.1567-1364.2010.00685.x
Rusanen, H., Majamaa, K., and Hassinen, I. E. (2000). Increased activities of antioxidant enzymes and decreased ATP concentration in cultured myoblasts with the 3243A–>G mutation in mitochondrial DNA. Biochim. Biophys. Acta 1500, 10–16. doi: 10.1016/s0925-4439(99)00081-2
Russell, O., and Turnbull, D. (2014). Mitochondrial DNA disease-molecular insights and potential routes to a cure. Exp. Cell Res. 325, 38–43. doi: 10.1016/j.yexcr.2014.03.012
Sage-Schwaede, A., Engelstad, K., Salazar, R., Curcio, A., Khandji, A., Garvin, J. H., et al. (2019). Exploring mTOR inhibition as treatment for mitochondrial disease. Ann. Clin. Transl. Neurol 6, 1877–1881. doi: 10.1002/acn3.50846
Santa, K. M. (2010). Treatment options for mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome. Pharmacotherapy 30, 1179–1196. doi: 10.1592/phco.30.11.1179
Saxena, N., Taneja, N., Shome, P., and Mani, S. (2018). Mitochondrial donation: a boon or curse for the treatment of incurable mitochondrial diseases. J. Hum. Reprod. Sci. 11, 3–9. doi: 10.4103/jhrs.JHRS_54_17
Schon, E. A., DiMauro, S., and Hirano, M. (2012). Human mitochondrial DNA: roles of inherited and somatic mutations. Nat. Rev. Genet. 13, 878–890. doi: 10.1038/nrg3275
Shoffner, J. M., Lott, M. T., Lezza, A. M., Seibel, P., Ballinger, S. W., and Wallace, D. C. (1990). Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell 61, 931–937. doi: 10.1016/0092-8674(90)90059-n
Shrigley, S., Pircs, K., Barker, R. A., Parmar, M., and Drouin-Ouellet, J. (2018). Simple generation of a high yield culture of induced neurons from human adult skin fibroblasts. J. Vis. Exp. 132:56904. doi: 10.3791/56904
Sie, L., Loong, S., and Tan, E. K. (2009). Utility of lymphoblastoid cell lines. J. Neurosci. Res. 87, 1953–1959. doi: 10.1002/jnr.22000
Smith, C., Abalde-Atristain, L., He, C., Brodsky, B. R., Braunstein, E. M., Chaudhari, P., et al. (2015). Efficient and allele-specific genome editing of disease loci in human iPSCs. Mol. Ther. 23, 570–577. doi: 10.1038/mt.2014.226
Son, E. Y., Ichida, J. K., Wainger, B. J., Toma, J. S., Rafuse, V. F., Woolf, C. J., et al. (2011). Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell 9, 205–218. doi: 10.1016/j.stem.2011.07.014
Suhr, S. T., Chang, E. A., Tjong, J., Alcasid, N., Perkins, G. A., Goissis, M. D., et al. (2010). Mitochondrial rejuvenation after induced pluripotency. PLoS One 5:e14095. doi: 10.1371/journal.pone.0014095
Suomalainen, A., and Battersby, B. J. (2018). Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 19, 77–92. doi: 10.1038/nrm.2017.66
Swerdlow, R. H. (2009). The neurodegenerative mitochondriopathies. J. Alzheimers Dis. 17, 737–751. doi: 10.3233/JAD-2009-1095
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi: 10.1016/j.cell.2006.07.024
Torper, O., Pfisterer, U., Wolf, D. A., Pereira, M., Lau, S., Jakobsson, J., et al. (2013). Generation of induced neurons via direct conversion in vivo. Proc. Natl. Acad. Sci. U.S.A. 110, 7038–7043. doi: 10.1073/pnas.1303829110
Torraco, A., Peralta, S., Iommarini, L., and Diaz, F. (2015). Mitochondrial Diseases Part I: mouse models of OXPHOS deficiencies caused by defects in respiratory complex subunits or assembly factors. Mitochondrion 21, 76–91. doi: 10.1016/j.mito.2015.01.009
Tsang, W. Y., and Lemire, B. D. (2003). The role of mitochondria in the life of the nematode, Caenorhabditis elegans. Biochim. Biophys. Acta 1638, 91–105. doi: 10.1016/s0925-4439(03)00079-6
Tsang, W. Y., Sayles, L. C., Grad, L. I., Pilgrim, D. B., and Lemire, B. D. (2001). Mitochondrial respiratory chain deficiency in Caenorhabditis elegans results in developmental arrest and increased life span. J. Biol. Chem. 276, 32240–32246. doi: 10.1074/jbc.M103999200
Tyynismaa, H., and Suomalainen, A. (2009). Mouse models of mitochondrial DNA defects and their relevance for human disease. EMBO Rep. 10, 137–143. doi: 10.1038/embor.2008.242
Umeda, N., Suzuki, T., Yukawa, M., Ohya, Y., Shindo, H., Watanabe, K., et al. (2005). Mitochondria-specific RNA-modifying enzymes responsible for the biosynthesis of the wobble base in mitochondrial tRNAs. Implications for the molecular pathogenesis of human mitochondrial diseases. J. Biol. Chem. 280, 1613–1624. doi: 10.1074/jbc.M409306200
Vierbuchen, T., Ostermeier, A., Pang, Z. P., Kokubu, Y., Sudhof, T. C., and Wernig, M. (2010). Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041. doi: 10.1038/nature08797
Villanueva-Paz, M., Povea-Cabello, S., Villalon-Garcia, I., Alvarez-Cordoba, M., Suarez-Rivero, J. M., Talaveron-Rey, M., et al. (2020). Parkin-mediated mitophagy and autophagy flux disruption in cellular models of MERRF syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 1866:165726. doi: 10.1016/j.bbadis.2020.165726
Villanueva-Paz, M., Povea-Cabello, S., Villalon-Garcia, I., Suarez-Rivero, J. M., Alvarez-Cordoba, M., de la Mata, M., et al. (2019). Pathophysiological characterization of MERRF patient-specific induced neurons generated by direct reprogramming. Biochim. Biophys. Acta Mol. Cell Res. 1866, 861–881. doi: 10.1016/j.bbamcr.2019.02.010
Viscomi, C., and Zeviani, M. (2020). Strategies for fighting mitochondrial diseases. J. Intern. Med. 287, 665–684. doi: 10.1111/joim.13046
Vithayathil, S. A., Ma, Y., and Kaipparettu, B. A. (2012). Transmitochondrial cybrids: tools for functional studies of mutant mitochondria. Methods Mol. Biol. 837, 219–230. doi: 10.1007/978-1-61779-504-6_15
Wallace, D. C., Singh, G., Lott, M. T., Hodge, J. A., Schurr, T. G., Lezza, A. M., et al. (1988). Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 242, 1427–1430. doi: 10.1126/science.3201231
Wilkins, H. M., Carl, S. M., and Swerdlow, R. H. (2014). Cytoplasmic hybrid (cybrid) cell lines as a practical model for mitochondriopathies. Redox Biol. 2, 619–631. doi: 10.1016/j.redox.2014.03.006
Wollweber, F., von der Malsburg, K., and van der Laan, M. (2017). Mitochondrial contact site and cristae organizing system: a central player in membrane shaping and crosstalk. Biochim. Biophys. Acta Mol. Cell Res. 1864, 1481–1489. doi: 10.1016/j.bbamcr.2017.05.004
Wu, S. B., Ma, Y. S., Wu, Y. T., Chen, Y. C., and Wei, Y. H. (2010). Mitochondrial DNA mutation-elicited oxidative stress, oxidative damage, and altered gene expression in cultured cells of patients with MERRF syndrome. Mol. Neurobiol. 41, 256–266. doi: 10.1007/s12035-010-8123-7
Xu, Z., Jiang, H., Zhong, P., Yan, Z., Chen, S., and Feng, J. (2016). Direct conversion of human fibroblasts to induced serotonergic neurons. Mol. Psychiatry 21, 62–70. doi: 10.1038/mp.2015.101
Yang, Y., Wu, H., Kang, X., Liang, Y., Lan, T., Li, T., et al. (2018). Targeted elimination of mutant mitochondrial DNA in MELAS-iPSCs by mitoTALENs. Protein Cell 9, 283–297. doi: 10.1007/s13238-017-0499-y
Yoneda, M., Tanno, Y., Horai, S., Ozawa, T., Miyatake, T., and Tsuji, S. (1990). A common mitochondrial DNA mutation in the t-RNA(Lys) of patients with myoclonus epilepsy associated with ragged-red fibers. Biochem. Int. 21, 789–796.
Yoo, A. S., Sun, A. X., Li, L., Shcheglovitov, A., Portmann, T., Li, Y., et al. (2011). MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 476, 228–231. doi: 10.1038/nature10323
Zeviani, M., and Carelli, V. (2003). Mitochondrial disorders. Curr. Opin. Neurol. 16, 585–594. doi: 10.1097/01.wco.0000093101.34793.c8
Zeviani, M., and Carelli, V. (2007). Mitochondrial disorders. Curr. Opin. Neurol. 20, 564–571. doi: 10.1097/WCO.0b013e3282ef58cd
Zeviani, M., Moraes, C. T., DiMauro, S., Nakase, H., Bonilla, E., Schon, E. A., et al. (1988). Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology 38, 1339–1346. doi: 10.1212/wnl.38.9.1339
Keywords: mitochondrial diseases, mtDNA, disease modeling, direct reprogramming, induced neurons
Citation: Povea-Cabello S, Villanueva-Paz M, Suárez-Rivero JM, Álvarez-Córdoba M, Villalón-García I, Talaverón-Rey M, Suárez-Carrillo A, Munuera-Cabeza M and Sánchez-Alcázar JA (2021) Advances in mt-tRNA Mutation-Caused Mitochondrial Disease Modeling: Patients’ Brain in a Dish. Front. Genet. 11:610764. doi: 10.3389/fgene.2020.610764
Received: 27 September 2020; Accepted: 26 November 2020;
Published: 12 January 2021.
Edited by:
Guilhian Leipnitz, Federal University of Rio Grande do Sul, BrazilReviewed by:
Josef Finsterer, K.A. Rasmussen, NorwayAyman W. El Hattab, Tawam Hospital, United Arab Emirates
Alessandra Maresca, IRCCS Institute of Neurological Sciences of Bologna (ISNB), Italy
Copyright © 2021 Povea-Cabello, Villanueva-Paz, Suárez-Rivero, Álvarez-Córdoba, Villalón-García, Talaverón-Rey, Suárez-Carrillo, Munuera-Cabeza and Sánchez-Alcázar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José A. Sánchez-Alcázar, amFzYW5hbGNAdXBvLmVz