 Alexandre Umpierrez Amaral1,2*
Alexandre Umpierrez Amaral1,2* Moacir Wajner1,3,4
Moacir Wajner1,3,4- 1Programa de Pós-Graduação em Ciências Biológicas: Bioquímica, Instituto de Ciências Básicas da Saúde, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
- 2Departamento de Ciências Biológicas, Universidade Regional Integrada do Alto Uruguai e das Missões, Erechim, Brazil
- 3Departamento de Bioquímica, Instituto de Ciências Básicas da Saúde, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
- 4Serviço de Genética Médica, Hospital de Clínicas de Porto Alegre, Porto Alegre, Brazil
Deficiencies of medium-chain acyl-CoA dehydrogenase, mitochondrial trifunctional protein, isolated long-chain 3-hydroxyacyl-CoA dehydrogenase, and very long-chain acyl-CoA dehydrogenase activities are considered the most frequent fatty acid oxidation defects (FAOD). They are biochemically characterized by the accumulation of medium-chain, long-chain hydroxyl, and long-chain fatty acids and derivatives, respectively, in tissues and biological fluids of the affected patients. Clinical manifestations commonly include hypoglycemia, cardiomyopathy, and recurrent rhabdomyolysis. Although the pathogenesis of these diseases is still poorly understood, energy deprivation secondary to blockage of fatty acid degradation seems to play an important role. However, recent evidence indicates that the predominant fatty acids accumulating in these disorders disrupt mitochondrial functions and are involved in their pathophysiology, possibly explaining the lactic acidosis, mitochondrial morphological alterations, and altered mitochondrial biochemical parameters found in tissues and cultured fibroblasts from some affected patients and also in animal models of these diseases. In this review, we will update the present knowledge on disturbances of mitochondrial bioenergetics, calcium homeostasis, uncoupling of oxidative phosphorylation, and mitochondrial permeability transition induction provoked by the major fatty acids accumulating in prevalent FAOD. It is emphasized that further in vivo studies carried out in tissues from affected patients and from animal genetic models of these disorders are necessary to confirm the present evidence mostly achieved from in vitro experiments.
Fatty Acid Oxidation Defects
Mitochondrial fatty acid oxidation is critical to provide ATP to mitochondria-enriched tissues with high energy demand, including the heart, skeletal muscle, and liver. Mutations in genes expressing enzymes or transport proteins involved in this catabolic pathway cause the fatty acid oxidation defects (FAOD) that are biochemically characterized by accumulation of specific patterns of fatty acids and acylcarnitine derivatives. Patients typically manifest hypoglycemia, cardiomyopathy, hepatopathy, recurrent rhabdomyolysis, and encephalopathy, whose pathogenesis is still poorly known, although energy deprivation secondary to blockage of fatty acid degradation seems to play an important role.
Deficiencies of medium-chain acyl-CoA dehydrogenase (MCAD), mitochondrial trifunctional protein (MTP), isolated long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD), and very long-chain acyl-CoA dehydrogenase (VLCAD) are the most common of these diseases. During acute episodes of metabolic decompensation usually associated with infections or fasting, the affected patients present hypoglycemia, encephalopathy, cardiomyopathy, rhabdomyolysis, and hepatopathy. Interestingly, during these catabolic crises, there is a high release of fatty acids from the adipose tissue and a significant increase of the accumulating fatty acids and derivatives due to the enzymatic defects, therefore suggesting the toxicity of these compounds (Rinaldo et al., 2002; Kompare and Rizzo, 2008; Olpin, 2013; Knottnerus et al., 2018; Anderson et al., 2020).
Medium-chain acyl-CoA dehydrogenase-deficient patients are generally asymptomatic, but 20–40% of them develop severe symptomatology along life during an acute episode of metabolic decompensation commonly triggered by prolonged fasting or infections (Merritt and Chang, 2019). During crises, they present hypoketotic hypoglycemia, vomiting that may progress to seizures and coma as well as elevation of liver enzymes and hepatomegaly, whereas a considerable percentage of them may have a fatal outcome (Grosse et al., 2006; Merritt and Chang, 2019; Anderson et al., 2020). Recurrent rhabdomyolysis is remarkably rare in this disease (Ruitenbeek et al., 1995; Schatz and Ensenauer, 2010), although long-term neurological complications are observed in up to 10–30% of patients (Maier, 2015). Tissue (blood) accumulation of medium-chain fatty acids (MCFA), especially octanoic acid (OA), decanoic acid (DA), and cis-4-decenoic acid (cDA), as well as their corresponding acylcarnitine derivatives hexanoylcarnitine, octanoylcarnitine (OC), decanoylcarnitine (DC), and cis-4-decenoylcarnitine and acylglycines (hexanoylglycine, suberylglycine, and phenylpropionylglycine) in urine is detected especially during episodes of acute metabolic decompensation (Dobrowolski et al., 2017; Anderson et al., 2020).
Mitochondrial trifunctional protein comprises three enzyme activities that catalyze the second, third, and fourth step of mitochondrial fatty acid oxidation cycle, namely, long-chain enoyl-CoA hydratase, LCHAD, and long-chain ketoacyl-CoA thiolase. Two inherited diseases are caused by defects of this complex protein, the isolated LCHAD deficiency and MTP deficiency. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency is caused by a common mutation (c.1528 G > C) in the HADHA gene, whereas all other HADHA and HADHB mutations in this complex enzyme lead to MTP deficiency (De Biase et al., 2017).
Untreated MTP and LCHAD deficiencies have high mortality and morbidity. The clinical features are usually manifested during fasting or metabolic stress triggered by common illness and mainly affect the heart and skeletal muscles that are highly dependent on fatty acids for their energy needs (Sykut-Cegielska et al., 2011; De Biase et al., 2017; Lotz-Havla et al., 2018). Individuals with MTP deficiency usually present a severe neonatal early onset form with elevated mortality caused by cardiomyopathy, as well as peripheral neuropathy, whereas patients with LCHAD deficiency commonly have a moderate late-onset phenotype with cardiomyopathy, retinal disease, and peripheral neuropathy during adulthood. Patients affected by these diseases may also present with hepatopathy and episodes of rhabdomyolysis during situations of intense lipolysis (Rocchiccioli et al., 1990; Tyni et al., 1997; Den Boer et al., 2002, 2003; Moczulski et al., 2009; De Biase et al., 2017). Long-term complications include rhabdomyolysis, cardiomyopathy, peripheral neuropathy, and retinopathy (Karall et al., 2015; Lotz-Havla et al., 2018). High levels of the long-chain 3-hydroxy fatty acids (LCHFA), 3-hydroxytetradecanoic acid (3HTA), 3-hydroxypalmitic acid (3HPA), and 3-hydroxytetradecanedioic acid (3HTDA) and their respective carnitine by-products accumulate in the patients, with 3-hydroxypalmitoylcarnitine (C16–OH) and 3-hydroxyoleoylcarnitine (C18:1–OH) being the primary biomarkers characteristically found at high concentrations in their blood (Hagenfeldt et al., 1990; Costa et al., 1998; Jones et al., 2001; Hintz et al., 2002; Olpin, 2005; Sander et al., 2005). High amounts of triglycerides containing long-chain fatty acids (LCFA) have been also shown in LCHAD-deficient patients (McCoin et al., 2016).
Very long-chain acyl-CoA dehydrogenase is the first and rate-limiting intramitochondrial step in the mitochondrial oxidation of LCFA. The deficiency of this enzyme activity causes the most prevalent defect of LCFA β-oxidation. The affected patients commonly manifest hypoglycemia, cardiomyopathy, and recurrent rhabdomyolysis. Hypoglycemia usually occurs during 2–4 years of life, whereas rhabdomyolysis is manifested later, and cardiomyopathy can occur at any age. Patients may present three different clinical phenotypes: (a) a severe early onset manifestation with life-threatening cardiomyopathy associated with arrhythmias, hypotonia, hepatomegaly, and intermittent hypoglycemia, (b) an early childhood presentation with hypoketotic hypoglycemia associated with liver alterations and hepatomegaly, or (c) a late-onset myopathic form with recurrent episodes of rhabdomyolysis and muscle pain usually induced by exercise (Spiekerkoetter, 2010; Diekman et al., 2014; Katz et al., 2017; Leslie et al., 2019; Rovelli et al., 2019). Recurrent rhabdomyolysis is a common and acute complication of this disease and should be treated with hydration and alkalization of the urine to prevent acute renal failure secondary to myoglobinuria. Elevated levels of myristic (Myr) and cis-5-tetradecenoic (Cis-5) acids, as well as their acylcarnitine derivatives tetradecenoyl-L-carnitine (C14:1), tetradecadienyl-L-carnitine (C14:2), tetradecanoyl-L-carnitine (C14), and dodecanoyl-L-carnitine (C12), are commonly found in blood (McHugh et al., 2011).
The diagnosis of MCAD, LCHAD/MTP, and VLCAD deficiencies is mainly performed by the detection of high concentrations of characteristic acylcarnitines in blood. The determination of enzyme activities in lymphocytes and/or fibroblasts and molecular analyses of mutations may be necessary for diagnosis confirmation. Since early diagnosis and prompt treatment are available in MCAD, LCHAD/MTP, and VLCAD deficiencies, these diseases have been included in the expanded newborn screening programs, allowing a much better outcome for the affected patients by significantly reducing morbidity and mortality (Wilcken et al., 2007; Spiekerkoetter et al., 2010; Maguolo et al., 2020).
The current treatments for these diseases include frequent meals and avoidance of catabolic stress situations caused by prolonged fasting or infectious illness. Fatty acid restriction, allied to medium-chain triglycerides (MCT) formulas and essential fatty acids to LCHAD/MTP and VLCAD deficiencies, as well as high oral or intravenous glucose administration to sustain anabolism is also critical in these disorders. The objective is therefore preventing hypoketotic hypoglycemia and metabolite accumulation. L-Carnitine supplementation should be used mainly to correct L-carnitine deficiency, but its beneficial effect to significantly increase the urinary excretion of potentially toxic fatty acids has still to be demonstrated (Spiekerkoetter et al., 2009, 2010). More recently, clinical trials have shown that bezafibrate, an agonist of peroxisome-proliferating activator receptor that increases mitochondrial biogenesis and the gene expression of mitochondrial fatty acid oxidation enzymes, may be useful in VLCAD (Yamada et al., 2018; Shiraishi et al., 2019) and MTP (Suyama et al., 2020) deficiencies. However, the clinical efficacy of bezafibrate is still disputed and needs to be further confirmed (Ørngreen et al., 2014, 2015). Furthermore, replacement of long-chain triglycerides by MCT and replenishment of citric acid cycle (CAC) intermediates by the seven-carbon fatty acid triglyceride (C7) triheptanoin to support ATP production have been recently demonstrated to improve the clinical outcome of these patients (Gillingham et al., 2017; Vockley et al., 2017, 2019).
Treatment is effective to decrease mortality and morbidity for most FAOD, although it does not completely prevent long-term systemic and neurological complications. It is therefore expected that elucidation of the exact underlying mechanisms of pathogenesis of these disorders will potentially help in the development of novel treatments to improve the quality of life of the affected patients. In particular, observations of mitochondrial biochemical and morphological abnormalities in highly mitochondria-enriched tissues of MCAD-, LCHAD/MTP-, and VLCAD-deficient patients, such as the heart, liver, and skeletal muscle, indicate that disturbances of mitochondrial functions are probably involved in their pathophysiology. The present review will mainly focus on recent evidence indicating secondary alterations of important mitochondrial properties caused by major fatty acids accumulating in these disorders.
Mitochondrial Functions: Bioenergetics and Calcium Homeostasis
The mitochondria are dynamic organelles with essential roles in cellular physiology, particularly in tissues with high energy demand and oxidative metabolism, such as the heart, skeletal muscle, liver, and brain. The mitochondria regulate ATP production, redox status, cytosolic calcium concentrations, and apoptosis-induced cell death. These mitochondrial functions are closely associated with efficient dynamic processes, such as mitochondrial fission and fusion, biogenesis, and mitophagy.
Mitochondrial fatty acid oxidation is the main source of ATP generation in the heart, skeletal muscle, and liver. Acetyl-CoA originated from fatty acid degradation is oxidized in the CAC, providing the electrons to form NADH and FADH2, which are transferred through the respiratory chain complexes in the electron transfer system. Protons are pumped into the mitochondrial intermembrane space, generating an electrochemical gradient (mitochondrial membrane potential, ΔΨm). The energy of the electrochemical gradient is used by ATP synthase to support ATP production, and the whole process is called oxidative phosphorylation (OXPHOS; Nicholls and Ferguson, 2013).
Calcium regulates a significant number of critical intracellular events necessary to cell survival, particularly in the heart, skeletal muscle, and liver (Rizzuto et al., 2012). The mitochondria continuously promote calcium influx down the electrochemical gradient through the mitochondrial calcium uniporter (MCU) and efflux by the Na+/Ca2+ and H+/Ca2+ exchangers, contributing together with the endoplasmic reticulum to maintain the cytosolic concentrations of this ion that would be adequate for cell functioning (De Stefani et al., 2016). At physiological or pathological situations of calcium overload, the capacity of the mitochondria to retain calcium is particularly important (Williams et al., 2013; Granatiero et al., 2017; Nicholls, 2017).
Mitochondrial permeability transition (mPT) pore opening in the inner mitochondrial membrane occurs when the threshold of mitochondrial calcium retention capacity is exceeded. Mitochondrial permeability transition pore opening is deleterious to cell functioning and survival mainly due to non-selective mitochondrial permeabilization that leads to significant calcium, glutathione, and NADH and NAD(P)H release, inducing mitochondrial swelling, ΔΨm dissipation, disruption of ATP synthesis, and finally apoptotic cell death (Rasola and Bernardi, 2011; Bernardi and von Stockum, 2012; Giorgio et al., 2018).
Mitochondrial calcium homeostasis represents an important regulatory mechanism in muscular tissue physiology, being extremely important for normal cardiomyocyte (Drago et al., 2012; Eisner, 2014; Kohlhaas et al., 2017) and myocyte (Yi et al., 2011; Eisner, 2014) functioning. Dysregulation of intracellular calcium concentration due to mitochondrial dysfunction associated with mPT induction has been related to cardiac diseases (Gordan et al., 2016; Bravo-Sagua et al., 2017) and suggested as a pathophysiologic event leading to rhabdomyolysis (Hamel et al., 2015). Disturbance of mitochondrial calcium homeostasis and mPT pore opening has also been demonstrated to be involved in liver diseases (Ferreira et al., 2003; Brenner et al., 2013; Go et al., 2015).
On the other hand, balanced mitochondrial fusion (elongation) and fission (fragmentation) is essential for normal mitochondrial morphology, distribution, and function, being also necessary in response to the variable physiological demands of the cell (El-Hattab et al., 2018). These processes can restore or remove defective mitochondria and are mainly coordinated by the pro-fusion mitochondrial proteins optic atrophy 1 (OPA1), mitofusin 1 (MFN1), and mitofusin 2 (MFN2), as well as by the cytosolic pro-fission dynamin-related protein 1 (DRP1; Burté et al., 2015). The quantity and the quality of the mitochondria are regulated by the equilibrium between their formation (biogenesis) and removal (mitophagy). Upregulation of biogenesis improves mitochondrial function and has been related to various proteins, such as the peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) and sirtuin 1 (SIRT1), whereas mitophagy is dependent on the PTEN-induced putative kinase 1 (PINK1; Ploumi et al., 2017). PTEN-induced putative kinase 1 is highly expressed in injured mitochondria with collapsed ΔΨm, recruiting and activating mitophagy-related proteins, particularly ubiquitin ligase parkin. It is noteworthy that the defects in mitochondrial dynamics result in the impairment of mitochondrial bioenergetics and calcium homeostasis, potentially leading to cell death (Pernas and Scorrano, 2016; Kowaltowski et al., 2019). Furthermore, failure in mitochondrial dynamics has been related to various pathological processes, including myopathies, cardiomyopathy, hepatopathy, neurodegeneration, diabetes, and cancer (Archer, 2013; Vásquez-Trincado et al., 2016; Bartsakoulia et al., 2018; Mansouri et al., 2018; Ji and Yeo, 2019).
Fatty Acid Oxidation Defects and Mitochondrial Abnormalities
Table 1 summarizes the biochemical and morphological evidence of mitochondrial abnormalities in patients, genetic knockout mice, and cultured cell models of MCAD, LCHAD/MTP, and VLCAD deficiencies.
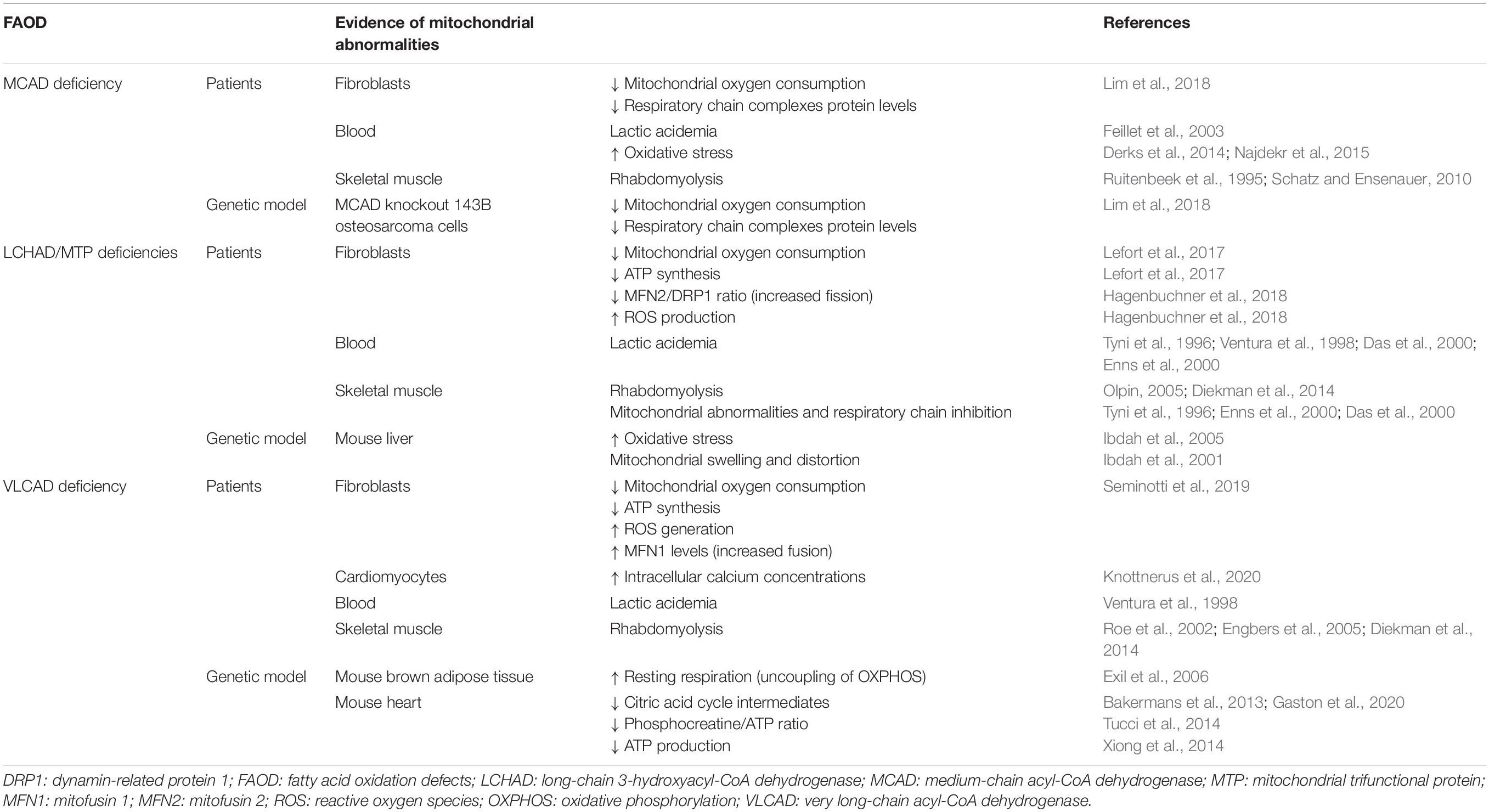
Table 1. Mitochondrial biochemical and morphological abnormalities observed in patients and genetic models of MCAD, LCHAD/MTP and VLCAD deficiencies.
Increased lactate formation commonly results from the impairment of mitochondrial bioenergetics, with lactic acidosis being therefore considered a biochemical hallmark of mitochondrial disorders (Kerr, 1991; Zeviani and Di Donato, 2004). However, it should be noted that, apart from being indicative of altered mitochondrial functions, lactic acidemia may be also a consequence of decreased lactate utilization due to liver dysfunction as found in some FAOD, particularly during episodes of metabolic decompensation. Patients with these diseases also present episodes of rhabdomyolysis that signalize severe disturbance of mitochondrial functions (Nance and Mammen, 2015). Oxidative stress has been also associated with mitochondrial dysfunction (Wang et al., 2014; Kudryavtseva et al., 2016; Rose et al., 2018) due to impairment of the mitochondrial electron flow through the respiratory chain, resulting in increased electron loss and subsequent reactive oxygen species (ROS) generation (Lambert and Brand, 2009; Nickel et al., 2014).
Although uncommon, lactic acidosis (Feillet et al., 2003), oxidative stress (Derks et al., 2014; Najdekr et al., 2015), and rhabdomyolysis (Ruitenbeek et al., 1995; Schatz and Ensenauer, 2010) have been observed in some patients with MCAD deficiency, particularly during episodes of metabolic decompensation. Reduced mitochondrial oxygen consumption, decreased respiratory chain complex protein levels, and increased ROS production were also found in fibroblasts from MCAD-deficient patients and in MCAD knockout 143B osteosarcoma cells (Lim et al., 2018), supporting a role for the disruption of bioenergetics and of redox homeostasis in the pathogenesis and progression of this disorder.
On the other hand, abnormal mitochondrial morphology and altered biochemical markers of mitochondrial functions, reflecting an impairment of bioenergetics, were commonly reported in isolated LCHAD and MTP deficiencies. In this scenario, lactic acidemia (Tyni et al., 1996; Ventura et al., 1998; Das et al., 2000; Enns et al., 2000), as well as rhabdomyolysis (Olpin, 2005; Diekman et al., 2014), mitochondrial morphological abnormalities, and inhibition of the respiratory chain electron flow (Tyni et al., 1996; Das et al., 2000; Enns et al., 2000; Hintz et al., 2002), was observed in skeletal muscle and cultured fibroblasts from patients with these diseases. Decreased mitochondrial oxygen consumption and reduced ATP synthesis were also detected in the fibroblasts of MTP-deficient patients (Lefort et al., 2017). Furthermore, an interesting study utilizing LCHAD-deficient fibroblasts demonstrated a dysregulation of the mitochondrial fusion/fission machinery as revealed by a decreased MFN2/DRP1 ratio, leading to the accumulation of fragmented mitochondria due to increased fission (Hagenbuchner et al., 2018). Mitochondrial swelling and distortion (Ibdah et al., 2001), as well as induction of oxidative stress, have been also reported in the liver of LCHAD-deficient mice (Ibdah et al., 2005).
As regards to VLCAD deficiency, lactic acidosis (Ventura et al., 1998) and rhabdomyolysis (Roe et al., 2002; Engbers et al., 2005; Diekman et al., 2014) were found in a considerable number of patients with this disorder. Decreased mitochondrial respiration associated with low ATP levels, as well as increased ROS generation, was verified in the fibroblasts of VLCAD-deficient patients (Seminotti et al., 2019). These fibroblasts revealed elevated mitochondrial mass and fusion, as well as an increased expression of MFN1, indicating alterations of mitochondrial dynamics besides the disruption of the endoplasmic reticulum–mitochondria crosstalk that is involved in cytosolic calcium homeostasis (Seminotti et al., 2019). This is in line with the observations of increased intracellular calcium concentrations that were correlated with fatty acid intermediate accumulation in fibroblasts from VLCAD-deficient patients differentiated into cardiomyocytes (Knottnerus et al., 2020). Furthermore, results obtained in the genetic mice model of VLCAD deficiency showed bioenergetics disruption. Thus, increased resting respiration suggesting uncoupling of OXPHOS was found in brown adipose tissue (Exil et al., 2006), and decreased phosphocreatine/ATP ratio (Tucci et al., 2014), ATP production (Xiong et al., 2014), and CAC intermediate pools (Bakermans et al., 2013; Gaston et al., 2020) were demonstrated in the heart of these animals.
Disturbance of Mitochondrial Bioenergetics and Calcium Homeostasis Caused by the Major Fatty Acids Accumulating in Fatty Acid Oxidation Defects
The pathophysiology of tissue damage in patients with MCAD, LCHAD/MTP, and VLCAD deficiencies has not yet been well established, although energy deprivation caused by fatty acid oxidation blockage, leading to hypoketotic hypoglycemia and sequestration of CoA and L-carnitine, was presumed to be mainly involved in the pathogenesis of these disorders (Spiekerkoetter and Wood, 2010; Olpin, 2013). More recently, alternative pathogenetic mechanisms have been hypothesized to contribute to their symptomatology, more particularly the toxicity of the accumulating metabolites. This is in accordance with the observations that clinical worsening occurs during stress catabolic situations characterized by intense lipolysis leading to the massive production of these compounds (Gregersen and Olsen, 2010; Olpin, 2013; Tein, 2013; Knottnerus et al., 2018).
In particular, there is mounting evidence of bioenergetics impairment in tissues that primarily consume fatty acids for their energy needs in patients and in the genetic models of MCAD, LCHAD/MTP, and VLCAD deficiencies (Table 1). Tables 2–4 summarize the updated data showing that the major metabolites, particularly MCFA, LCHFA, and LCFA, accumulating in these disorders provoke alterations of mitochondrial functions in the heart, liver and skeletal muscle of rats and also in cultured cell lines, mainly impairing mitochondrial bioenergetics and calcium homeostasis by distinct mechanisms. It is emphasized that, overall, the doses of the accumulating metabolites used in most of these studies were similar to the levels described in the blood of affected patients during metabolic decompensation. However, a significant impairment of mitochondrial functions was also achieved with lower concentrations of these compounds in LCHAD/MTP and VLCAD deficiencies that better approach the levels found in blood during periods of stable disease, therefore suggesting the chronic toxicity of these compounds in these inherited deficiencies.
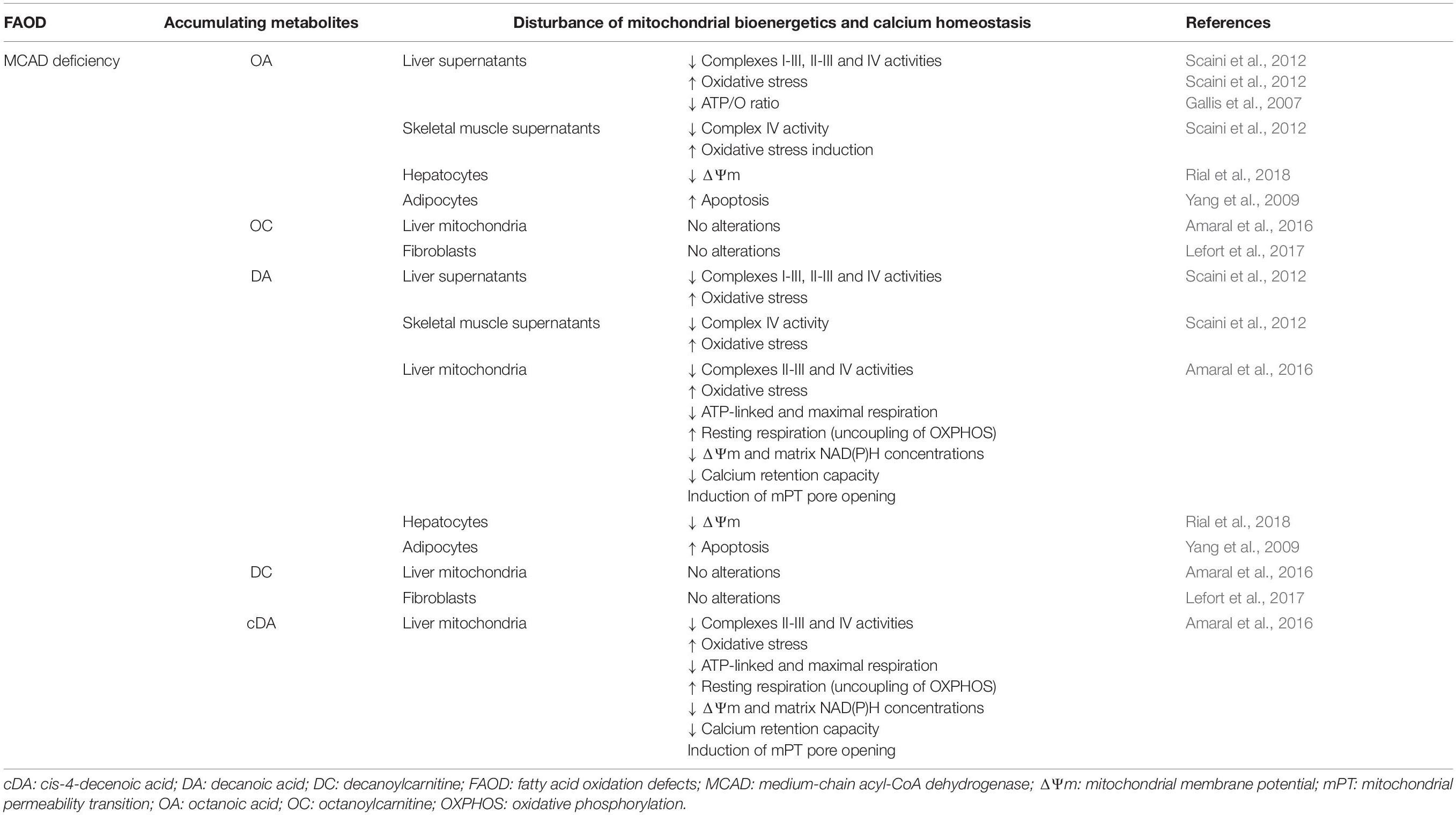
Table 2. Major metabolites accumulating in MCAD deficiency disturb mitochondrial bioenergetics and calcium homeostasis in liver and skeletal muscle of rats, as well as in cultured cell lines.
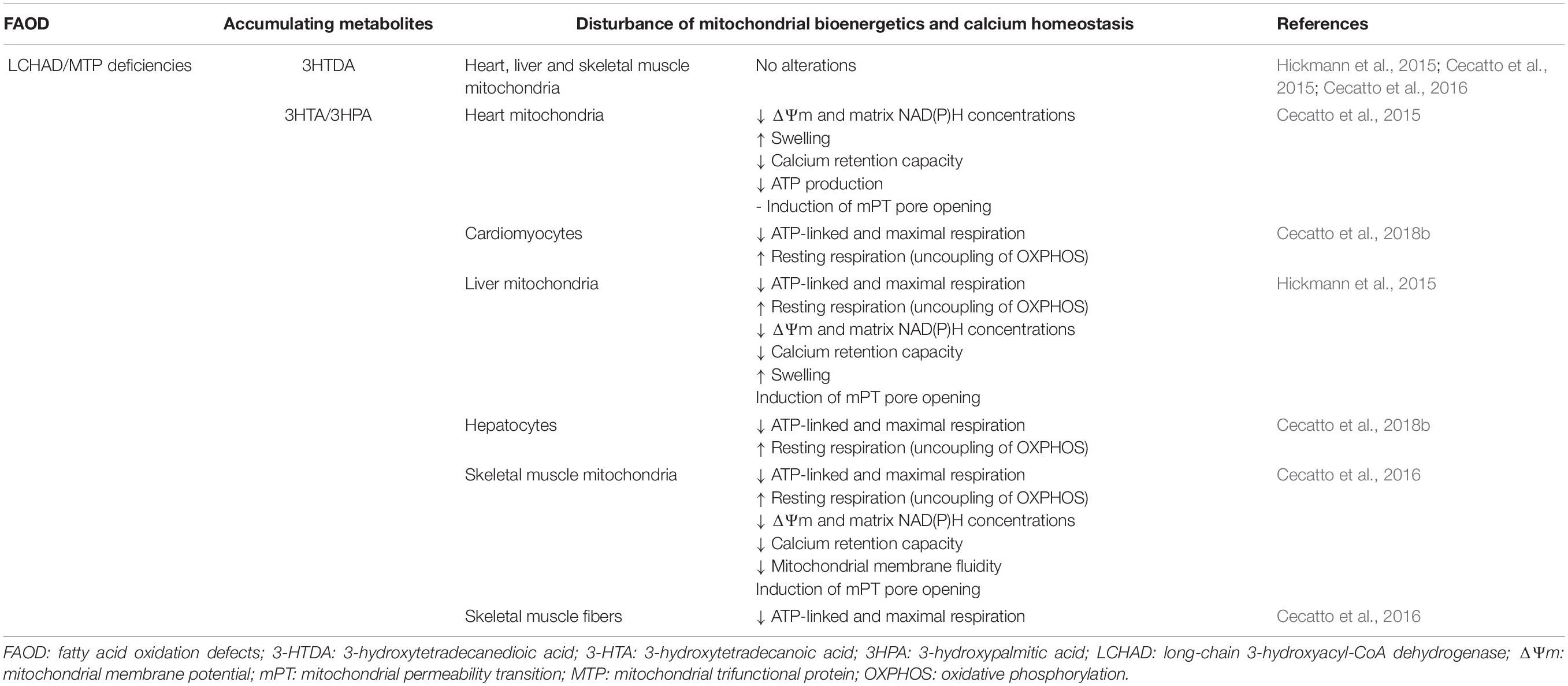
Table 3. Major metabolites accumulating in LCHAD/MTP deficiencies disturb mitochondrial bioenergetics and calcium homeostasis in heart, liver and skeletal muscle of rats, as well as in cultured cell lines.

Table 4. Major metabolites accumulating in VLCAD deficiency disturb mitochondrial bioenergetics and calcium homeostasis in heart, liver and skeletal muscle of rats, as well as in cultured cell lines.
Thus, OA, DA, and cDA, the MCFA that most commonly accumulate in MCAD deficiency, were shown to provoke mitochondrial dysfunction in the liver and skeletal muscle of rats. OA and DA were shown to inhibit the activities of the respiratory chain complexes I–III and II–III in rat liver, as well as of complex IV in the liver and skeletal muscle, besides inducing oxidative stress in these tissues (Scaini et al., 2012). In addition, DA and cDA severely inhibited ATP-linked (ADP-stimulated) and maximal (CCCP-stimulated) mitochondrial oxygen consumption, increased resting respiration (induced by the ATP synthase inhibitor oligomycin), and inhibited complexes II–III and IV activities, indicating metabolic inhibition and uncoupling of OXPHOS that lead to energy deficiency besides provoking a disruption of redox homeostasis in isolated liver mitochondria (Amaral et al., 2016). It is noteworthy that, in the same study, it was shown that DA and cDA decreased ΔΨm and matrix NAD(P)H content and stimulated the opening of cyclosporin A-sensitive mPT pore. Calcium retention capacity was also decreased by DA and cDA, probably as a result of mPT induction in the liver mitochondria (Amaral et al., 2016). Other studies reported that OA and DA caused ΔΨm dissipation in hepatocytes (Rial et al., 2018) and induced apoptosis in adipocytes (Yang et al., 2009), whereas OA was demonstrated to decrease the ATP/O ratio in perfused liver, indicating OXPHOS impairment (Gallis et al., 2007). In contrast, the carnitine derivatives of the corresponding MCFA tested, OC and DC, caused no changes in the mitochondrial parameters evaluated, implying that medium-chain acylcarnitine derivatives do not disturb these mitochondrial functions. This is consistent with a recent study showing no alterations of OC and DC on mitochondrial respiration in fibroblasts (Lefort et al., 2017).
As regards to LCHFA, it was demonstrated that 3HTA and 3HPA, which mostly accumulate in tissues and biological fluids of patients affected by LCHAD or MTP deficiencies, decrease ΔΨm, matrix NAD(P)H content, calcium retention capacity, and ATP synthesis besides inducing mitochondrial swelling in calcium-loaded liver and heart mitochondria (Cecatto et al., 2015; Hickmann et al., 2015). It was proposed that these effects were due to the induction of cyclosporin A-sensitive mPT pore opening as well as due to uncoupling and to the metabolic inhibition caused by these fatty acids (Cecatto et al., 2015; Hickmann et al., 2015). Similar results were achieved in cultured cell lines from the heart and liver (Cecatto et al., 2018b), as well as in skeletal muscle mitochondria and permeabilized muscle fibers (Cecatto et al., 2016). Altered mitochondrial membrane fluidity caused by the LCHFA was also shown in the skeletal muscle (Cecatto et al., 2016). However, the corresponding dicarboxylic 3HTDA caused no effect on these parameters, implying a selective toxicity of monocarboxylic LCHFA (Cecatto et al., 2015, 2016; Hickmann et al., 2015).
Disruption of mitochondrial functions has also been suggested as a relevant pathomechanism involved in the cardiomyopathy, hepatopathy, and myopathy in VLCAD deficiency since high concentrations of Myr and Cis-5, which mostly accumulate in this disorder, were shown to be toxic to mitochondrial functions by impairing bioenergetics and calcium homeostasis. In particular, previous works performed in isolated liver mitochondria showed that Myr uncouples OXPHOS through the involvement of the dicarboxylate carrier (Wieckowski and Wojtczak, 1998) and of the ANT (Bodrova et al., 2000). Induction of mPT pore opening was also demonstrated to be caused by Myr (Bodrova et al., 2003). Recent studies demonstrated that, apart from Myr, pathological concentrations of Cis-5 disturb mitochondrial bioenergetics and calcium homeostasis in the heart, liver, and skeletal muscle (Cecatto et al., 2018a, 2019, 2020). These results were obtained in different tissue preparations, such as isolated mitochondria, permeabilized cardiomyocytes and hepatocytes, and heart fibers. Myr and Cis-5 also inhibited complex I–III and α-ketoglutarate dehydrogenase activities and altered mitochondrial respiration, behaving as metabolic inhibitors and uncouplers of OXPHOS, leading to ATP depletion besides reducing ΔΨm and matrix NAD(P)H content. These LCFA also induced cyclosporin A-sensitive mPT in the presence of calcium, causing mitochondrial ΔΨm dissipation, reduction of calcium retention capacity, swelling, and cytochrome c release. These observations are in accordance with a previous study showing that monounsaturated LCFA (C14:1 and C16:1) accumulating in VLCAD deficiency decrease ΔΨm and induce apoptosis and necrosis in cardiomyocytes (Hoffmann et al., 2014). On the other hand, the toxicity of long-chain acylcarnitines (LCAC) was reported to be involved in long-chain FAOD pathogenesis (McCoin et al., 2015a, 2019). In particular, it was demonstrated that LCAC disturb cell calcium homeostasis in myocytes (McCoin et al., 2015b), cardiomyocytes (Berezhnov et al., 2008), and heart sarcoplasmic reticulum (Yamada et al., 2000) besides decreasing resting respiration (Lefort et al., 2017) and provoking ΔΨm dissipation (Nguyen et al., 2017) in fibroblasts. LCAC were also shown to reduce calcium retention capacity (De Villiers and Lochner, 1986; Baydoun et al., 1988), inhibit ATP-linked respiration, and generate ROS in the heart mitochondria (Liepinsh et al., 2016).
Figure 1 illustrates the main mechanisms of mitochondrial dysfunction caused by major fatty acids that accumulate in MCAD, LCHAD/MTP, and VLCAD deficiencies.
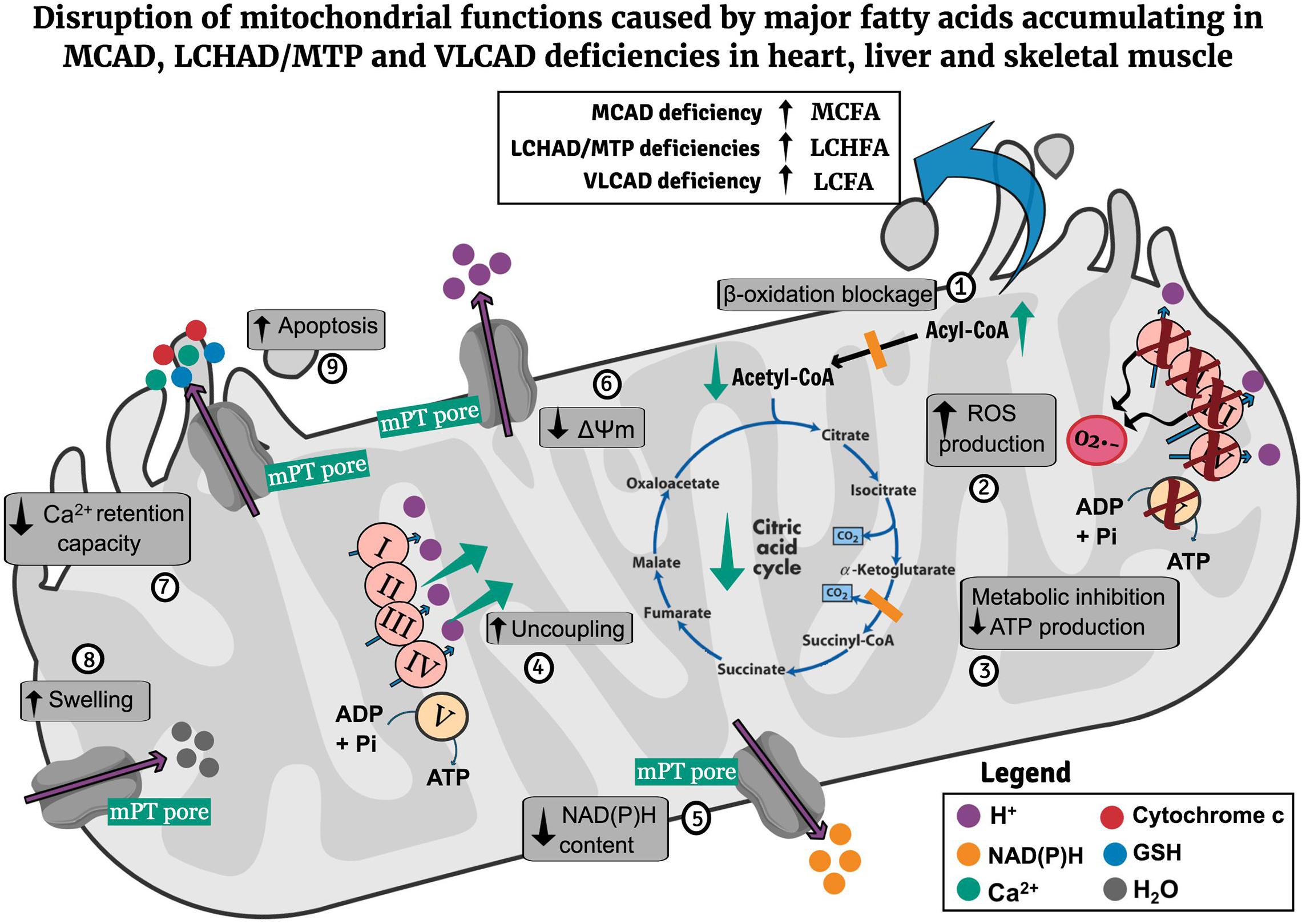
Figure 1. Potential pathomechanisms of mitochondrial dysfunction in the heart, liver, and skeletal muscle in MCAD, LCHAD/MTP, and VLCAD deficiencies. Mitochondrial β-oxidation blockage leads to the matrix accumulation of MCFA (MCAD deficiency), LCHFA (LCHAD/MTP deficiencies), and LCFA (VLCAD deficiency) as well as CoA depletion (1). The accumulating fatty acids inhibit respiratory chain complexes activities, leading to ROS generation (2), cause metabolic inhibition due to decrease of respiratory chain and citric acid cycle activities compromising ATP synthesis (3), uncouple oxidative phosphorylation (4), activate the mPT pore opening, provoking a decrease of mitochondrial NAD(P)H content (5), ΔΨm (6), and mitochondrial Ca2+ retention capacity (7), and induce mitochondrial swelling (8). Finally, mPT induction also promotes cytochrome c release, possibly contributing to apoptosis induction (9). CoA, coenzyme A; LCFA, long-chain fatty acids; LCHAD, long-chain hydroxyacyl-CoA dehydrogenase; LCHFA, long-chain 3-hydroxy fatty acids; MCAD, medium-chain acyl-CoA dehydrogenase; MCFA, medium-chain fatty acids; ΔΨm, mitochondrial membrane potential; mPT, mitochondrial permeability transition; MTP, mitochondrial trifunctional protein; ROS, reactive oxygen species; VLCAD, very long-chain acyl-CoA dehydrogenase.
Concluding Remarks
Mounting evidence of altered mitochondrial morphology, functions, and dynamics has been recently described in various tissues of patients with MCAD, LCHAD/MTP, and VLCAD deficiencies and in the genetic models of these diseases, suggesting that impairment of mitochondrial homeostasis may play a relevant role in the pathogenesis of these disorders. In particular, the major accumulating MCFA, LCHFA, and LCFA were demonstrated to severely disturb mitochondrial bioenergetics and calcium homeostasis in vitro in highly oxidative mitochondria-enriched tissues, such as the heart, liver, and skeletal muscle. These fatty acids behaved as metabolic inhibitors, uncouplers of OXPHOS, and inductors of mPT pore opening. It is therefore presumed that these pathomechanisms probably contribute to the mitochondrial alterations observed in patients and animal models with these pathological conditions. Interestingly, severe cardiomyopathy, hepatopathy, and skeletal muscle alterations are mainly manifested during catabolic stress situations in which the concentrations of the characteristic fatty acids dramatically increase in tissues and biological fluids, therefore supporting an acute toxicity for these endogenous compounds. However, the present evidence of bioenergetics disruption caused by the accumulating metabolites must be interpreted with caution since most available data were achieved by in vitro assays. Further studies performed preferentially in vivo in animal models and in patients affected by FAOD are therefore necessary to further clarify the underlying mechanisms of tissue damage in these disorders. Finally, it is expected that, besides restricting fat dietary intake and avoiding fasting, drugs that stimulate mitochondrial function such as bezafibrate and the anaplerotic compound triheptanoin may hopefully improve the clinical outcome of the affected patients (Gillingham et al., 2017; Vockley et al., 2017, 2019; Yamada et al., 2018; Shiraishi et al., 2019; Suyama et al., 2020).
Author Contributions
Both authors planned and wrote the review article.
Funding
The research activity in the authors’ laboratories was supported in part by research grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq)—425914/2016-0 and Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (FAPERGS)—17/2551-0000800-6.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the invaluable technical assistance of A.C. Roginski.
Abbreviations
CAC, citric acid cycle; cDA, cis-4-decenoic acid; Cis-5, cis-5-tetradecenoic acid; DA, decanoic acid; DRP1, dynamin-related protein 1; FAOD, fatty acid oxidation disorders; 3HTA, 3-hydroxytetradecanoic acid; 3HTDA, 3-hydroxytetradecanedioic acid; 3HPA, 3-hydroxypalmitic acid; MCAD, medium-chain acyl-CoA dehydrogenase; MCFA, medium-chain fatty acids; MCT, medium-chain triglycerides; LCAC, long-chain acylcarnitines; LCFA, long-chain fatty acids; LCHAD, long-chain 3-hydroxyacyl-CoA dehydrogenase; LCHFA, long-chain 3-hydroxy fatty acids; ΔΨ m, mitochondrial membrane potential; MCU, mitochondrial calcium uniporter; MFN1, mitofusin 1; MFN2, mitofusin 2; MTP, mitochondrial trifunctional protein; mPT, mitochondrial permeability transition; Myr, myristic acid; OA, octanoic acid; OPA1, optic atrophy 1; OXPHOS, oxidative phosphorylation; PGC-1 α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; PINK1, PTEN-induced putative kinase 1; ROS, reactive oxygen species; SIRT1, sirtuin 1; VLCAD, very long-chain acyl-CoA dehydrogenase.
References
Amaral, A. U., Cecatto, C., Da Silva, J. C., Wajner, A., Godoy, K. D. S., Ribeiro, R. T., et al. (2016). Cis-4-Decenoic and decanoic acids impair mitochondrial energy, redox and Ca2 + homeostasis and induce mitochondrial permeability transition pore opening in rat brain and liver: possible implications for the pathogenesis of MCAD deficiency. Biochim. Biophys. Acta 1857, 1363–1372. doi: 10.1016/j.bbabio.2016.05.007
Anderson, D. R., Viau, K., Botto, L. D., Pasquali, M., and Longo, N. (2020). Clinical and biochemical outcomes of patients with medium-chain acyl-CoA dehydrogenase deficiency. Mol. Genet. Metab. 129, 13–19. doi: 10.1016/j.ymgme.2019.11.006
Archer, S. L. (2013). Mitochondrial dynamics - mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 369, 2236–2251. doi: 10.1056/NEJMra1215233
Bakermans, A. J., Dodd, M. S., Nicolay, K., Prompers, J. J., Tyler, D. J., and Houten, S. M. (2013). Myocardial energy shortage and unmet anaplerotic needs in the fasted long-chain acyl-Co a dehydrogenase knockout mouse. Cardiovasc. Res. 100, 441–449. doi: 10.1093/cvr/cvt212
Bartsakoulia, M., Pyle, A., Troncoso-Chandía, D., Vial-Brizzi, J., Paz-Fiblas, M. V., Duff, J., et al. (2018). A novel mechanism causing imbalance of mitochondrial fusion and fission in human myopathies. Hum. Mol. Genet. 27, 1186–1195. doi: 10.1093/hmg/ddy033
Baydoun, A. R., Markham, A., Morgan, R. M., and Sweetman, A. J. (1988). Palmitoyl carnitine: an endogenous promotor of calcium efflux from rat heart mitochondria. Biochem. Pharmacol. 37, 3103–3107. doi: 10.1016/0006-2952(88)90307-3
Berezhnov, A. V., Fedotova, E. I., Nenov, M. N., Kokoz, I. M., Zinchenko, V. P., and Dynnik, V. V. (2008). Destabilization of the cytosolic calcium level and cardiomyocyte death in the presence of long-chain fatty acid derivatives. Biofizika 53, 1025–1032. doi: 10.1134/S0006350908060183
Bernardi, P., and von Stockum, S. (2012). The permeability transition pore as a Ca2+ release channel: new answers to an old question. Cell Calcium 52, 22–27. doi: 10.1016/j.ceca.2012.03.004
Bodrova, M. E., Brailovskaya, I. V., Efron, G. I., Starkov, A. A., and Mokhova, E. N. (2003). Cyclosporin A-sensitive decrease in the transmembrane potential across the inner membrane of liver mitochondria induced by low concentrations of fatty acids and Ca2+. Biochemistry 68, 391–398. doi: 10.1023/A:1023691628110
Bodrova, M. E., Dedukhova, V. I., Samartsev, V. N., and Mokhova, E. N. (2000). Role of the ADP/ATP-antiporter in fatty acid-induced uncoupling of Ca2+-loaded rat liver mitochondria. IUBMB Life 50, 189–194. doi: 10.1080/152165400300001507
Bravo-Sagua, R., Parra, V., López-Crisosto, C., Díaz, P., Quest, A. F. G., and Lavandero, S. (2017). Calcium transport and signaling in mitochondria. Compr. Physiol. 7, 623–634. doi: 10.1002/cphy.c160013
Brenner, C., Galluzzi, L., Kepp, O., and Kroemer, G. (2013). Decoding cell death signals in liver inflammation. J. Hepatol. 59, 583–594. doi: 10.1016/j.jhep.2013.03.033
Burté, F., Carelli, V., Chinnery, P. F., and Yu-Wai-Man, P. (2015). Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 11, 11–24. doi: 10.1038/nrneurol.2014.228
Cecatto, C., Amaral, A. U., da Silva, J. C., Wajner, A., Schimit, M. O. V, da Silva, L. H. R., et al. (2018a). Metabolite accumulation in VLCAD deficiency markedly disrupts mitochondrial bioenergetics and Ca(2+) homeostasis in the heart. FEBS J. 285, 1437–1455. doi: 10.1111/febs.14419
Cecatto, C., Amaral, A. U., Roginski, A. C., Castilho, R. F., and Wajner, M. (2019). Impairment of mitochondrial bioenergetics and permeability transition induction caused by major long-chain fatty acids accumulating in VLCAD deficiency in skeletal muscle as potential pathomechanisms of myopathy. Toxicol. Vitr. 62:104665. doi: 10.1016/j.tiv.2019.104665
Cecatto, C., Amaral, A. U., Wajner, A., Wajner, S. M., Castilho, R. F., and Wajner, M. (2020). Disturbance of mitochondrial functions associated with permeability transition pore opening induced by cis-5-tetradecenoic and myristic acids in liver of adolescent rats. Mitochondrion 50, 1–13. doi: 10.1016/j.mito.2019.09.008
Cecatto, C., Godoy, K. D. S., da Silva, J. C., Amaral, A. U., and Wajner, M. (2016). Disturbance of mitochondrial functions provoked by the major long-chain 3-hydroxylated fatty acids accumulating in MTP and LCHAD deficiencies in skeletal muscle. Toxicol. Vitr. 36, 1–9. doi: 10.1016/j.tiv.2016.06.007
Cecatto, C., Hickmann, F. H., Rodrigues, M. D., Amaral, A. U., and Wajner, M. (2015). Deregulation of mitochondrial functions provoked by long-chain fatty acid accumulating in long-chain 3-hydroxyacyl-CoA dehydrogenase and mitochondrial permeability transition deficiencies in rat heart–mitochondrial permeability transition pore opening as a potential contributing pathomechanism of cardiac alterations in these disorders. FEBS J. 282, 4714–4726. doi: 10.1111/febs.13526
Cecatto, C., Wajner, A., Vargas, C. R., Wajner, S. M., Amaral, A. U., and Wajner, M. (2018b). High vulnerability of the heart and liver to 3-hydroxypalmitic acid-induced disruption of mitochondrial functions in intact cell systems. J. Cell. Biochem. 119, 7678–7686. doi: 10.1002/jcb.27115
Costa, C. G., Dorland, L., Tavares De Almeida, I., Jakobs, C., Duran, M., and Poll-The, B. T. (1998). The effect of fasting, long-chain triglyceride load and carnitine load on plasma long-chain acylcarnitine levels in mitochondrial very long-chain acyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 21, 391–399. doi: 10.1023/A:1005354624735
Das, A. M., Fingerhut, R., Wanders, R. J. A., and Ullrich, K. (2000). Secondary respiratory chain defect in a boy with long-chain 3- hydroxyacyl-CoA dehydrogenase deficiency: possible diagnostic pitfalls. Eur. J. Pediatr. 159, 243–246. doi: 10.1007/s004310050063
De Biase, I., Viau, K. S., Liu, A., Yuzyuk, T., Botto, L. D., Pasquali, M., et al. (2017). Diagnosis, treatment, and clinical outcome of patients with mitochondrial trifunctional protein/long-chain 3-hydroxy acyl-CoA dehydrogenase deficiency. JIMD Reports 31, 63–71. doi: 10.1007/8904_2016_558
De Stefani, D., Rizzuto, R., and Pozzan, T. (2016). Enjoy the trip: calcium in mitochondria back and forth. Annu. Rev. Biochem. 85, 161–192. doi: 10.1146/annurev-biochem-060614-034216
De Villiers, M., and Lochner, A. (1986). Mitochondrial Ca2+ fluxes: role of free fatty acids, acyl-CoA and acylcarnitine. Biochim. Biophys. Acta 876, 309–317. doi: 10.1016/0005-2760(86)90289-4
Den Boer, M. E. J., Dionisi-Vici, C., Chakrapani, A., Van Thuijl, A. O. J., Wanders, R. J. A., and Wijburg, F. A. (2003). Mitochondrial trifunctional protein deficiency: a severe fatty acid oxidation disorder with cardiac and neurologic involvement. J. Pediatr. 142, 684–689. doi: 10.1067/mpd.2003.231
Den Boer, M. E. J., Wanders, R. J. A., Morris, A. A. M., Ijlst, L., Heymans, H. S. A., and Wijburg, F. A. (2002). Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: clinical presentation and follow-up of 50 patients. Pediatrics 109, 99–104. doi: 10.1542/peds.109.1.99
Derks, T. G. J., Touw, C. M. L., Ribas, G. S., Biancini, G. B., Vanzin, C. S., Negretto, G., et al. (2014). Experimental evidence for protein oxidative damage and altered antioxidant defense in patients with medium-chain acyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 37, 783–789. doi: 10.1007/s10545-014-9700-0
Diekman, E. F., Van Der Pol, W. L., Nievelstein, R. A. J., Houten, S. M., Wijburg, F. A., and Visser, G. (2014). Muscle MRI in patients with long-chain fatty acid oxidation disorders. J. Inherit. Metab. Dis. 37, 405–413. doi: 10.1007/s10545-013-9666-3
Dobrowolski, S. F., Ghaloul-Gonzalez, L., and Vockley, J. (2017). Medium chain acyl-CoA dehydrogenase deficiency in a premature infant. Pediatr. Rep. 9, 7045. doi: 10.4081/pr.2017.7045
Drago, I., De Stefani, D., Rizzuto, R., and Pozzan, T. (2012). Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc. Natl. Acad. Sci. U. S. A. 109, 12986–12991. doi: 10.1073/pnas.1210718109
Eisner, D. (2014). Calcium in the heart: from physiology to disease. Exp. Physiol. 99, 1273–1282. doi: 10.1113/expphysiol.2013.077305
El-Hattab, A. W., Suleiman, J., Almannai, M., and Scaglia, F. (2018). Mitochondrial dynamics: biological roles, molecular machinery, and related diseases. Mol. Genet. Metab. 125, 315–321. doi: 10.1016/j.ymgme.2018.10.003
Engbers, H. M., Dorland, L., De Sain, M. G. M., Eskes, P. F., and Visser, G. (2005). Rhabdomyolysis in early-onset very long-chain acyl-CoA dehydrogenase deficiency despite normal glucose after fasting. J. Inherit. Metab. Dis. 28, 1151–1152. doi: 10.1007/s10545-005-0190-y
Enns, G. M., Bennett, M. J., Hoppel, C. L., Goodman, S. I., Weisiger, K., Ohnstad, C., et al. (2000). Mitochondrial respiratory chain complex I deficiency with clinical and biochemical features of long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. J. Pediatr. 136, 251–254. doi: 10.1016/S0022-3476(00)70111-9
Exil, V. J., Gardner, C. D., Rottman, J. N., Sims, H., Bartelds, B., Khuchua, Z., et al. (2006). Abnormal mitochondrial bioenergetics and heart rate dysfunction in mice lacking very-long-chain acyl-CoA dehydrogenase. Am. J. Physiol. Hear. Circ. Physiol. 290, H1289-H1297. doi: 10.1152/ajpheart.00811.2005
Feillet, F., Steinmann, G., Vianey-Saban, C., De Chillou, C., Sadoul, N., Lefebvre, E., et al. (2003). Adult presentation of MCAD deficiency revealed by coma and severe arrythmias. Intensive Care Med. 29, 1594–1597. doi: 10.1007/s00134-003-1871-3
Ferreira, F. M., Seiça, R., Oliveira, P. J., Coxito, P. M., Moreno, A. J., Palmeira, C. M., et al. (2003). Diabetes induces metabolic adaptations in rat liver mitochondria: role of coenzyme Q and cardiolipin contents. Biochim. Biophys. Acta 1639, 113–120. doi: 10.1016/j.bbadis.2003.08.001
Gallis, J. L., Tissier, P., Gin, H., and Beauvieux, M. C. (2007). Decrease in oxidative phosphorylation yield in presence of butyrate in perfused liver isolated from fed rats. BMC Physiol. 7, 8. doi: 10.1186/1472-6793-7-8
Gaston, G., Gangoiti, J. A., Winn, S., Chan, B., Barshop, B. A., Harding, C. O., et al. (2020). Cardiac tissue citric acid cycle intermediates in exercised very long-chain acyl-CoA dehydrogenase-deficient mice fed triheptanoin or medium-chain triglyceride. J. Inherit. Metab. Dis. 2020:12284. doi: 10.1002/jimd.12284
Gillingham, M. B., Heitner, S. B., Martin, J., Rose, S., Goldstein, A., El-Gharbawy, A. H., et al. (2017). Triheptanoin versus trioctanoin for long-chain fatty acid oxidation disorders: a double blinded, randomized controlled trial. J. Inherit. Metab. Dis. 40, 831–843. doi: 10.1007/s10545-017-0085-8
Giorgio, V., Guo, L., Bassot, C., Petronilli, V., and Bernardi, P. (2018). Calcium and regulation of the mitochondrial permeability transition. Cell Calcium 70, 56–63. doi: 10.1016/j.ceca.2017.05.004
Go, K. L., Lee, S., Zendejas, I., Behrns, K. E., and Kim, J. S. (2015). Mitochondrial Dysfunction and Autophagy in Hepatic Ischemia/Reperfusion Injury. Biomed Res. Int. 2015, 1–14. doi: 10.1155/2015/183469
Gordan, R., Fefelova, N., Gwathmey, J. K., and Xie, L. H. (2016). Involvement of mitochondrial permeability transition pore (mPTP) in cardiac arrhythmias: evidence from cyclophilin D knockout mice. Cell Calcium 60, 363–372. doi: 10.1016/j.ceca.2016.09.001
Granatiero, V., De Stefani, D., and Rizzuto, R. (2017). “Mitochondrial calcium handling in physiology and disease.” Adv Exp Med Biol. 982, 25–47. doi: 10.1007/978-3-319-55330-6_2
Gregersen, N., and Olsen, R. K. J. (2010). Disease mechanisms and protein structures in fatty acid oxidation defects. J. Inherit. Metab. Dis. 33, 547–553. doi: 10.1007/s10545-010-9046-1
Grosse, S. D., Khoury, M. J., Greene, C. L., Crider, K. S., and Pollitt, R. J. (2006). The epidemiology of medium chain acyl-CoA dehydrogenase deficiency: an update. Genet. Med. 8, 205–212. doi: 10.1097/01.gim.0000204472.25153.8d
Hagenbuchner, J., Scholl-Buergi, S., Karall, D., and Ausserlechner, M. J. (2018). very long-/and long chain-3-Hydroxy Acyl CoA Dehydrogenase eficiency correlates with deregulation of the mitochondrial fusion/fission machinery. Sci. Rep. 8:3254. doi: 10.1038/s41598-018-21519-2
Hagenfeldt, L., von Döbeln, U., Holme, E., Alm, J., Brandberg, G., Enocksson, E., et al. (1990). 3-hydroxydicarboxylic aciduria-a fatty acid oxidation defect with severe prognosis. J. Pediatr. 116, 387–392. doi: 10.1016/S0022-3476(05)82826-4
Hamel, Y., Mamoune, A., Mauvais, F. X., Habarou, F., Lallement, L., Romero, N. B., et al. (2015). Acute rhabdomyolysis and inflammation. J. Inherit. Metab. Dis. 38, 621–628. doi: 10.1007/s10545-015-9827-7
Hickmann, F. H., Cecatto, C., Kleemann, D., Monteiro, W. O., Castilho, R. F., Amaral, A. U., et al. (2015). Uncoupling, metabolic inhibition and induction of mitochondrial permeability transition in rat liver mitochondria caused by the major long-chain hydroxyl monocarboxylic fatty acids accumulating in LCHAD deficiency. Biochim. Biophys. Acta 1847, 620–628. doi: 10.1016/j.bbabio.2015.04.003
Hintz, S. R., Matern, D., Strauss, A., Bennett, M. J., Hoyme, H. E., Schelley, S., et al. (2002). Early neonatal diagnosis of long-chain 3-hydroxyacyl coenzyme a dehydrogenase and mitochondrial trifunctional protein deficiencies. Mol. Genet. Metab. 75, 120–127. doi: 10.1006/mgme.2001.3282
Hoffmann, L., Seibt, A., Herebian, D., and Spiekerkoetter, U. (2014). Monounsaturated 14:1n-9 and 16:1n-9 fatty acids but not 18:1n-9 induce apoptosis and necrosis in murine HL-1 cardiomyocytes. Lipids 49, 25–37. doi: 10.1007/s11745-013-3865-4
Ibdah, J. A., Paul, H., Zhao, Y., Binford, S., Salleng, K., Cline, M., et al. (2001). Lack of mitochondrial trifunctional protein in mice causes neonatal hypoglycemia and sudden death. J. Clin. Invest. 107, 1403–1409. doi: 10.1172/JCI12590
Ibdah, J. A., Perlegas, P., Zhao, Y., Angdisen, J., Borgerink, H., Shadoan, M. K., et al. (2005). Mice heterozygous for a defect in mitochondrial trifunctional protein develop hepatic steatosis and insulin resistance. Gastroenterology 128, 1381–1390. doi: 10.1053/j.gastro.2005.02.001
Ji, L. L., and Yeo, D. (2019). Mitochondrial dysregulation and muscle disuse atrophy. F1000 Res. 8:1621. doi: 10.12688/f1000research.19139.1
Jones, P. M., Moffitt, M., Joseph, D., Harthcock, P. A., Boriack, R. L., Ibdah, J. A., et al. (2001). Accumulation of free 3-hydroxy fatty acids in the culture media of fibroblasts from patients deficient in long-chain L-3-hydroxyacyl-CoA dehydrogenase: a useful diagnostic aid. Clin. Chem. 47, 1190–1194. doi: 10.1093/clinchem/47.7.1190
Karall, D., Brunner-Krainz, M., Kogelnig, K., Konstantopoulou, V., Maier, E. M., Möslinger, D., et al. (2015). Clinical outcome, biochemical and therapeutic follow-up in 14 austrian patients with long-chain 3-hydroxy acyl CoA dehydrogenase deficiency (LCHADD). Orphanet J. Rare Dis. 10:21. doi: 10.1186/s13023-015-0236-7
Katz, S., Landau, Y., Pode-Shakked, B., Pessach, I. M., Rubinshtein, M., Anikster, Y., et al. (2017). Cardiac failure in very long chain acyl-CoA dehydrogenase deficiency requiring extracorporeal membrane oxygenation (ECMO) treatment: a case report and review of the literature. Mol. Genet. Metab. Reports 10, 5–7. doi: 10.1016/j.ymgmr.2016.11.008
Kerr, D. S. (1991). Lactic acidosis and mitochondrial disorders. Clin. Biochem. 24, 331–336. doi: 10.1016/0009-9120(91)80007-P
Knottnerus, S. J. G., Bleeker, J. C., Wüst, R. C. I., Ferdinandusse, S., IJlst, L., Wijburg, F. A., et al. (2018). Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle. Rev. Endocr. Metab. Disord. 19, 93–106. doi: 10.1007/s11154-018-9448-1
Knottnerus, S. J. G., Mengarelli, I., Wüst, R. C. I., Baartscheer, A., Bleeker, J. C., Coronel, R., et al. (2020). Electrophysiological abnormalities in VLCAD deficient hiPSC-cardiomyocytes can be improved by lowering accumulation of fatty acid oxidation intermediates. Int. J. Mol. Sci. 21:2589. doi: 10.3390/ijms21072589
Kohlhaas, M., Nickel, A. G., and Maack, C. (2017). Mitochondrial energetics and calcium coupling in the heart. J. Physiol. 595, 3753–3763. doi: 10.1113/JP273609
Kompare, M., and Rizzo, W. B. (2008). Mitochondrial fatty-acid oxidation disorders. Semin. Pediatr. Neurol. 15, 140–149. doi: 10.1016/j.spen.2008.05.008
Kowaltowski, A. J., Menezes-Filho, S. L., Assali, E. A., Gonçalves, I. G., Cabral-Costa, J. V., Abreu, P., et al. (2019). Mitochondrial morphology regulates organellar Ca2+ uptake and changes cellular Ca2+ homeostasis. FASEB J. 33, 13176–13188. doi: 10.1096/fj.201901136R
Kudryavtseva, A. V., Krasnov, G. S., Dmitriev, A. A., Alekseev, B. Y., Kardymon, O. L., Sadritdinova, A. F., et al. (2016). Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 7, 44879–44905. doi: 10.18632/oncotarget.9821
Lambert, A. J., and Brand, M. D. (2009). Reactive oxygen species production by mitochondria. Methods Mol. Biol. 554, 165–181. doi: 10.1007/978-1-59745-521-3_11
Lefort, B., Gouache, E., Acquaviva, C., Tardieu, M., Benoist, J. F., Dumas, J. F., et al. (2017). Pharmacological inhibition of carnitine palmitoyltransferase 1 restores mitochondrial oxidative phosphorylation in human trifunctional protein deficient fibroblasts. Biochim. Biophys. Acta 1863, 1292–1299. doi: 10.1016/j.bbadis.2017.04.005
Leslie, N. D., Valencia, C. A., Strauss, A. W., Connor, J., and Zhang, K. (2019). “Very Long-Chain Acyl-Coenzyme A Dehydrogenase Deficiency,” in GeneReviews® eds M. P. Ardinger, Adam H. H. R. A. Pagon, S. E. Wallace, L. J. Bean, K. Stephens, et al. (Seattle (WA): University of Washington), 1993–2020.
Liepinsh, E., Makrecka-Kuka, M., Volska, K., Kuka, J., Makarova, E., Antone, U., et al. (2016). Long-chain acylcarnitines determine ischaemia/reperfusion-induced damage in heart mitochondria. Biochem. J. 473, 1191–1202. doi: 10.1042/BCJ20160164
Lim, S. C., Tajika, M., Shimura, M., Carey, K. T., Stroud, D. A., Murayama, K., et al. (2018). Loss of the mitochondrial fatty acid β-oxidation protein medium-chain Acyl-Coenzyme a dehydrogenase disrupts oxidative phosphorylation protein complex stability and function. Sci. Rep. 8:153. doi: 10.1038/s41598-017-18530-4
Lotz-Havla, A. S., Röschinger, W., Schiergens, K., Singer, K., Karall, D., Konstantopoulou, V., et al. (2018). Fatal pitfalls in newborn screening for mitochondrial trifunctional protein (MTP)/long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency. Orphanet J. Rare Dis. 13:122. doi: 10.1186/s13023-018-0875-6
Maguolo, A., Rodella, G., Dianin, A., Nurti, R., Monge, I., Rigotti, E., et al. (2020). Diagnosis, genetic characterization and clinical follow up of mitochondrial fatty acid oxidation disorders in the new era of expanded newborn screening: a single centre experience. Mol. Genet. Metab. Reports 24;100632. doi: 10.1016/j.ymgmr.2020.100632
Maier, E. M. (2015). Neonatal screening for medium-chain acyl-CoA deficiency—insights and unexpected challenges. Int. J. Neonatal Screen. 1, 79–88. doi: 10.3390/ijns1030079
Mansouri, A., Gattolliat, C. H., and Asselah, T. (2018). Mitochondrial dysfunction and signaling in chronic liver diseases. Gastroenterology 155, 629–647. doi: 10.1053/j.gastro.2018.06.083
McCoin, C. S., Gillingham, M. B., Knotts, T. A., Vockley, J., Ono-Moore, K. D., Blackburn, M. L., et al. (2019). Blood cytokine patterns suggest a modest inflammation phenotype in subjects with long-chain fatty acid oxidation disorders. Physiol. Rep. 7:e14037. doi: 10.14814/phy2.14037
McCoin, C. S., Knotts, T. A., and Adams, S. H. (2015a). Acylcarnitines-old actors auditioning for new roles in metabolic physiology. Nat. Rev. Endocrinol. 11, 617–625. doi: 10.1038/nrendo.2015.129
McCoin, C. S., Knotts, T. A., Ono-Moore, K. D., Oort, P. J., and Adams, S. H. (2015b). Long-chain acylcarnitines activate cell stress and myokine release in C2C12 myotubes: calcium-dependent and -independent effects. Am. J. Physiol. Endocrinol. Metab. 308, E990-E1000. doi: 10.1152/ajpendo.00602.2014
McCoin, C. S., Piccolo, B. D., Knotts, T. A., Matern, D., Vockley, J., Gillingham, M. B., et al. (2016). Unique plasma metabolomic signatures of individuals with inherited disorders of long-chain fatty acid oxidation. J. Inherit. Metab. Dis. 39, 399–408. doi: 10.1007/s10545-016-9915-3
McHugh, D. M. S., Cameron, C. A., Abdenur, J. E., Abdulrahman, M., Adair, O., Al Nuaimi, S. A., et al. (2011). Clinical validation of cutoff target ranges in newborn screening of metabolic disorders by tandem mass spectrometry: a worldwide collaborative project. Genet. Med. 13, 230–254. doi: 10.1097/GIM.0b013e31820d5e67
Merritt, J. L. 2nd, and Chang, I. J. (2019). “Medium-Chain Acyl-Coenzyme A Dehydrogenase Deficiency,” in GeneReviews® eds M. P. Ardinger, Adam H. H. R. A. Pagon, S. E. Wallace, L. J. Bean, K. Stephens, et al. (Seattle: (WA): University of Washington), 1993–2020.
Moczulski, D., Majak, I., and Mamczur, D. (2009). An overview of beta-oxidation disorders. Postepy Hig. Med. Dosw. 63, 266–277.
Najdekr, L., Gardlo, A., Mádrová, L., Friedecký, D., Janečková, H., Correa, E. S., et al. (2015). Oxidized phosphatidylcholines suggest oxidative stress in patients with medium-chain acyl-CoA dehydrogenase deficiency. Talanta 139, 62–66. doi: 10.1016/j.talanta.2015.02.041
Nance, J. R., and Mammen, A. L. (2015). Diagnostic evaluation of rhabdomyolysis. Muscle and Nerve 51, 793–810. doi: 10.1002/mus.24606
Nguyen, T. B., Louie, S. M., Daniele, J. R., Tran, Q., Dillin, A., Zoncu, R., et al. (2017). DGAT1-dependent lipid droplet biogenesis protects mitochondrial function during starvation-induced autophagy. Dev. Cell 42, 9-21. doi: 10.1016/j.devcel.2017.06.003
Nicholls, D. G. (2017). Brain mitochondrial calcium transport: origins of the set-point concept and its application to physiology and pathology. Neurochem. Int. 109, 5–12. doi: 10.1016/j.neuint.2016.12.018
Nickel, A., Kohlhaas, M., and Maack, C. (2014). Mitochondrial reactive oxygen species production and elimination. J. Mol. Cell. Cardiol. 73, 26–33. doi: 10.1016/j.yjmcc.2014.03.011
Olpin, S. E. (2005). Fatty acid oxidation defects as a cause of neuromyopathic disease in infants and adults. Clin. Lab. 51, 289–306. doi:Google Scholar
Olpin, S. E. (2013). Pathophysiology of fatty acid oxidation disorders and resultant phenotypic variability. J. Inherit. Metab. Dis. 36, 645–658. doi: 10.1007/s10545-013-9611-5
Ørngreen, M. C., Madsen, K. L., Preisler, N., Andersen, G., Vissing, J., and Laforêt, P. (2014). Bezafibrate in skeletal muscle fatty acid oxidation disorders: a randomized clinical trial. Neurology 82, 607–613. doi: 10.1212/WNL.0000000000000118
Ørngreen, M. C., Vissing, J., and Laforét, P. (2015). No effect of bezafibrate in patients with CPTII and VLCAD deficiencies. J. Inherit. Metab. Dis. 38, 373–374. doi: 10.1007/s10545-014-9779-3
Pernas, L., and Scorrano, L. (2016). Mito-Morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu. Rev. Physiol. 78, 505–531. doi: 10.1146/annurev-physiol-021115-105011
Ploumi, C., Daskalaki, I., and Tavernarakis, N. (2017). Mitochondrial biogenesis and clearance: a balancing act. FEBS J. 284, 183–195. doi: 10.1111/febs.13820
Rasola, A., and Bernardi, P. (2011). Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium 50, 222–233. doi: 10.1016/j.ceca.2011.04.007
Rial, S., Ravaut, G., Malaret, T., Bergeron, K.-F., and Mounier, C. (2018). Hexanoic, octanoic and decanoic acids promote basal and insulin-induced phosphorylation of the Akt-mTOR axis and a balanced lipid metabolism in the HepG2 hepatoma cell line. Molecules 23:2315. doi: 10.3390/molecules23092315
Rinaldo, P., Matern, D., and Bennett, M. J. (2002). Fatty acid oxidation disorders. Annu. Rev. Physiol. 64, 477–502. doi: 10.1146/annurev.physiol.64.082201.154705
Rizzuto, R., De Stefani, D., Raffaello, A., and Mammucari, C. (2012). Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 13, 566–578. doi: 10.1038/nrm3412
Rocchiccioli, F., Wanders, R. J. A., Aubourg, P., Vianey-Liaud, C., Ijlst, L., Fabre, M., et al. (1990). Deficiency of long-chain 3-hydroxyacyl-CoA dehydrogenase: a cause of lethal myopathy and cardiomyopathy in early childhood. Pediatr. Res. 28, 657–662. doi: 10.1203/00006450-199012000-00023
Roe, C. R., Sweetman, L., Roe, D. S., David, F., and Brunengraber, H. (2002). Treatment of cardiomyopathy and rhabdomyolysis in long-chain fat oxidation disorders using an anaplerotic odd-chain triglyceride. J. Clin. Invest. 110, 259–269. doi: 10.1172/JCI0215311
Rose, S., Niyazov, D. M., Rossignol, D. A., Goldenthal, M., Kahler, S. G., and Frye, R. E. (2018). Clinical and molecular characteristics of mitochondrial dysfunction in autism spectrum disorder. Mol. Diagnosis Ther. 22, 571–593. doi: 10.1007/s40291-018-0352-x
Rovelli, V., Manzoni, F., Viau, K., Pasquali, M., and Longo, N. (2019). Clinical and biochemical outcome of patients with very long-chain acyl-CoA dehydrogenase deficiency. Mol. Genet. Metab. 127, 64–73. doi: 10.1016/j.ymgme.2019.04.001
Ruitenbeek, W., Poels, P. J. E., Turnbull, D. M., Garavaglia, B., Chalmers, R. A., Taylor, R. W., et al. (1995). Rhabdomyolysis and acute encephalopathy in late onset medium chain acyl-CoA dehydrogenase deficiency. J. Neurol. Neurosurg. Psychiatry 58, 209–214. doi: 10.1136/jnnp.58.2.209
Sander, J., Sander, S., Steuerwald, U., Janzen, N., Peter, M., Wanders, R. J. A., et al. (2005). Neonatal screening for defects of the mitochondrial trifunctional protein. Mol. Genet. Metab. 85, 108–114. doi: 10.1016/j.ymgme.2005.02.002
Scaini, G., Simon, K. R., Tonin, A. M., Busanello, E. N. B., Moura, A. P., Ferreira, G. C., et al. (2012). Toxicity of octanoate and decanoate in rat peripheral tissues: evidence of bioenergetic dysfunction and oxidative damage induction in liver and skeletal muscle. Mol. Cell. Biochem. 361, 329–335. doi: 10.1007/s11010-011-1119-4
Schatz, U. A., and Ensenauer, R. (2010). The clinical manifestation of MCAD deficiency: challenges towards adulthood in the screened population. J. Inherit. Metab. Dis. 33, 513–520. doi: 10.1007/s10545-010-9115-5
Seminotti, B., Leipnitz, G., Karunanidhi, A., Kochersperger, C., Roginskaya, V. Y., Basu, S., et al. (2019). Mitochondrial energetics is impaired in very long-chain acyl-CoA dehydrogenase deficiency and can be rescued by treatment with mitochondria-targeted electron scavengers. Hum. Mol. Genet. 28, 928–941. doi: 10.1093/hmg/ddy403
Shiraishi, H., Yamada, K., Oki, E., Ishige, M., Fukao, T., Hamada, Y., et al. (2019). Open-label clinical trial of bezafibrate treatment in patients with fatty acid oxidation disorders in Japan; 2nd report QOL survey. Mol. Genet. Metab. Reports 20:100496. doi: 10.1016/j.ymgmr.2019.100496
Spiekerkoetter, U. (2010). Mitochondrial fatty acid oxidation disorders: clinical presentation of long-chain fatty acid oxidation defects before and after newborn screening. J. Inherit. Metab. Dis. 33, 527–532. doi: 10.1007/s10545-010-9090-x
Spiekerkoetter, U., Bastin, J., Gillingham, M., Morris, A., Wijburg, F., and Wilcken, B. (2010). Current issues regarding treatment of mitochondrial fatty acid oxidation disorders. J. Inherit. Metab. Dis. 33, 555–561. doi: 10.1007/s10545-010-9188-1
Spiekerkoetter, U., Lindner, M., Santer, R., Grotzke, M., Baumgartner, M. R., Boehles, H., et al. (2009). Treatment recommendations in long-chain fatty acid oxidation defects: consensus from a workshop. J. Inherit. Metab. Dis. 32, 498–505. doi: 10.1007/s10545-009-1126-8
Spiekerkoetter, U., and Wood, P. A. (2010). Mitochondrial fatty acid oxidation disorders: pathophysiological studies in mouse models. J. Inherit. Metab. Dis. 33, 539–546. doi: 10.1007/s10545-010-9121-7
Suyama, T., Shimura, M., Fushimi, T., Kuranobu, N., Ichimoto, K., Matsunaga, A., et al. (2020). Efficacy of bezafibrate in two patients with mitochondrial trifunctional protein deficiency. Mol. Genet. Metab. Reports 24:100610. doi: 10.1016/j.ymgmr.2020.100610
Sykut-Cegielska, J., Gradowska, W., Piekutowska-Abramczuk, D., Andresen, B. S., Olsen, R. K. J., Ołtarzewski, M., et al. (2011). Urgent metabolic service improves survival in long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency detected by symptomatic identification and pilot newborn screening. J. Inherit. Metab. Dis. 34, 185–195. doi: 10.1007/s10545-010-9244-x
Tein, I. (2013). Disorders of fatty acid oxidation. Handb. Clin. Neurol. 113, 1675–1688. doi: 10.1016/B978-0-444-59565-2.00035-6
Tucci, S., Flögel, U., Hermann, S., Sturm, M., Schäfers, M., and Spiekerkoetter, U. (2014). Development and pathomechanisms of cardiomyopathy in very long-chain acyl-CoA dehydrogenase deficient (VLCAD-/-) mice. Biochim. Biophys. Acta 1842, 677–685. doi: 10.1016/j.bbadis.2014.02.001
Tyni, T., Majander, A., Kalimo, H., Rapola, J., and Pihko, H. (1996). Pathology of skeletal muscle and impaired respiratory chain function in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency with the G1528C mutation. Neuromuscul. Disord. 6, 327–337. doi: 10.1016/0960-8966(96)00352-5
Tyni, T., Rapola, J., Paetau, A., Palotie, A., and Pihko, H. (1997). Pathology of long-chain 3-hydroxyacyl-coa dehydrogenase deficiency caused by the G1528C mutation. Pediatr. Pathol. Lab. Med. 17, 427–447. doi: 10.1080/15513819709168585
Vásquez-Trincado, C., García-Carvajal, I., Pennanen, C., Parra, V., Hill, J. A., Rothermel, B. A., et al. (2016). Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 594, 509–525. doi: 10.1113/JP271301
Ventura, F. V., Ruiter, J. P. N., IJlst, L., Tavares De Almeida, I., and Wanders, R. J. A. (1998). Lactic acidosis in long-chain fatty acid β-oxidation disorders. J. Inherit. Metab. Dis. 21, 645–654. doi: 10.1023/A:1005480516801
Vockley, J., Burton, B., Berry, G. T., Longo, N., Phillips, J., Sanchez-Valle, A., et al. (2017). UX007 for the treatment of long chain-fatty acid oxidation disorders: safety and efficacy in children and adults following 24 weeks of treatment. Mol. Genet. Metab. 120, 370–377. doi: 10.1016/j.ymgme.2017.02.005
Vockley, J., Burton, B., Berry, G. T., Longo, N., Phillips, J., Sanchez-Valle, A., et al. (2019). Results from a 78-week, single-arm, open-label phase 2 study to evaluate UX007 in pediatric and adult patients with severe long-chain fatty acid oxidation disorders (LC-FAOD). J. Inherit. Metab. Dis. 42, 169–177. doi: 10.1002/jimd.12038
Wang, X., Wang, W., Li, L., Perry, G., Lee, H-g., and Zhu, X. (2014). Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 1842, 1240–1247. doi: 10.1016/j.bbadis.2013.10.015
Wieckowski, M. R., and Wojtczak, L. (1998). Fatty acid-induced uncoupling of oxidative phosphorylation is partly due to opening of the mitochondrial permeability transition pore. FEBS Lett. 423, 339–342. doi: 10.1016/S0014-5793(98)00118-5
Wilcken, B., Haas, M., Joy, P., Wiley, V., Chaplin, M., Black, C., et al. (2007). Outcome of neonatal screening for medium-chain acyl-CoA dehydrogenase deficiency in Australia: a cohort study. Lancet 369, 37–42. doi: 10.1016/S0140-6736(07)60029-4
Williams, G. S. B., Boyman, L., Chikando, A. C., Khairallah, R. J., and Lederer, W. J. (2013). Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. U. S. A. 110, 10479–10486. doi: 10.1073/pnas.1300410110
Xiong, D., He, H., James, J., Tokunaga, C., Powers, C., Huang, Y., et al. (2014). Cardiac-specific VLCAD deficiency induces dilated cardiomyopathy and cold intolerance. Am. J. Physiol. Hear. Circ. Physiol. 306, H326–H338. doi: 10.1152/ajpheart.00931.2012
Yamada, K. A., Kanter, E. M., and Newatia, A. (2000). Long-chain acylcarnitine induces Ca2+ efflux from the sarcoplasmic reticulum. J. Cardiovasc. Pharmacol. 36, 14–21. doi: 10.1097/00005344-200007000-00002
Yamada, K., Shiraishi, H., Oki, E., Ishige, M., Fukao, T., Hamada, Y., et al. (2018). Open-label clinical trial of bezafibrate treatment in patients with fatty acid oxidation disorders in Japan. Mol. Genet. Metab. Reports 15, 55–63. doi: 10.1016/j.ymgmr.2018.02.003
Yang, J.-Y., Rayalam, S., Della-Fera, M. A., Ambati, S., and Baile, C. A. (2009). Octanoate and Decanoate Induce Apoptosis in 3T3-L1 Adipocytes. J. Med. Food 12, 959–966. doi: 10.1089/jmf.2008.0262
Yi, J., Ma, C., Li, Y., Weisleder, N., Ríos, E., Ma, J., et al. (2011). Mitochondrial calcium uptake regulates rapid calcium transients in skeletal muscle during excitation-contraction (E-C) coupling. J. Biol. Chem. 286, 32436–32443. doi: 10.1074/jbc.M110.217711
Keywords: fatty acid oxidation defects, bioenergetics, calcium homeostasis, mitochondrial permeability transition, mitochondrial functions
Citation: Amaral AU and Wajner M (2020) Recent Advances in the Pathophysiology of Fatty Acid Oxidation Defects: Secondary Alterations of Bioenergetics and Mitochondrial Calcium Homeostasis Caused by the Accumulating Fatty Acids. Front. Genet. 11:598976. doi: 10.3389/fgene.2020.598976
Received: 26 August 2020; Accepted: 03 November 2020;
Published: 27 November 2020.
Edited by:
Grant M. Hatch, University of Manitoba, CanadaReviewed by:
Jerry Vockley, University of Pittsburgh, United StatesPeter Bross, Aarhus University, Denmark
Sander Michel Houten, Icahn School of Medicine at Mount Sinai, United States
Copyright © 2020 Amaral and Wajner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandre Umpierrez Amaral, YWxleGFuZHJlYW1hcmFsQHVyaWNlci5lZHUuYnI=