 Hugues Beauchemin1*
Hugues Beauchemin1* Tarik Möröy1,2,3*
Tarik Möröy1,2,3*- 1Institut de recherches cliniques de Montréal, Montreal, QC, Canada
- 2Division of Experimental Medicine, McGill University, Montreal, QC, Canada
- 3Département de Microbiologie, Infectiologie et Immunologie, Université de Montréal, Montreal, QC, Canada
Growth factor independence 1 (GFI1) and the closely related protein GFI1B are small nuclear proteins that act as DNA binding transcriptional repressors. Both recognize the same consensus DNA binding motif via their C-terminal zinc finger domains and regulate the expression of their target genes by recruiting chromatin modifiers such as histone deacetylases (HDACs) and demethylases (LSD1) by using an N-terminal SNAG domain that comprises only 20 amino acids. The only region that is different between both proteins is the region that separates the zinc finger domains and the SNAG domain. Both proteins are co-expressed in hematopoietic stem cells (HSCs) and, to some extent, in multipotent progenitors (MPPs), but expression is specified as soon as early progenitors and show signs of lineage bias. While expression of GFI1 is maintained in lymphoid primed multipotent progenitors (LMPPs) that have the potential to differentiate into both myeloid and lymphoid cells, GFI1B expression is no longer detectable in these cells. By contrast, GFI1 expression is lost in megakaryocyte precursors (MKPs) and in megakaryocyte-erythrocyte progenitors (MEPs), which maintain a high level of GFI1B expression. Consequently, GFI1 drives myeloid and lymphoid differentiation and GFI1B drives the development of megakaryocytes, platelets, and erythrocytes. How such complementary cell type- and lineage-specific functions of GFI1 and GFI1B are maintained is still an unresolved question in particular since they share an almost identical structure and very similar biochemical modes of actions. The cell type-specific accessibility of GFI1/1B binding sites may explain the fact that very similar transcription factors can be responsible for very different transcriptional programming. An additional explanation comes from recent data showing that both proteins may have additional non-transcriptional functions. GFI1 interacts with a number of proteins involved in DNA repair and lack of GFI1 renders HSCs highly susceptible to DNA damage-induced death and restricts their proliferation. In contrast, GFI1B binds to proteins of the beta-catenin/Wnt signaling pathway and lack of GFI1B leads to an expansion of HSCs and MKPs, illustrating the different impact that GFI1 or GFI1B has on HSCs. In addition, GFI1 and GFI1B are required for endothelial cells to become the first blood cells during early murine development and are among those transcription factors needed to convert adult endothelial cells or fibroblasts into HSCs. This role of GFI1 and GFI1B bears high significance for the ongoing effort to generate hematopoietic stem and progenitor cells de novo for the autologous treatment of blood disorders such as leukemia and lymphoma.
Introduction
GFI1 and GFI1B as Transcription Factors
The two members of the growth factor independence 1 transcription factor and proto-oncogene family, GFI1 and its paralogue GFI1B, are transcriptional repressors that play critical roles in hematopoiesis by regulating the cell fate of hematopoietic stem cells (HSCs), but also down the line in specific hematopoietic progenitor cells and precursors of more differentiated lineages. The Gfi1 gene was first identified almost three decades ago in a screen for Moloney murine leukemia virus (Mo-MuLV) insertions as a factor promoting IL-2-independent growth in a mouse T cell lymphoma cell line (Gilks et al., 1993), whereas GFI1B was identified a few years later in human through a homology screening using low-stringency hybridization with the chick and the mouse GFI1 zinc finger coding sequence (Rodel et al., 1998; Tong et al., 1998). Interestingly, the Gfi1b gene was also found later to be targeted by Mo-MuLV in c-Myc-dependent B-cell lymphomas in mouse (Mendrysa et al., 2010). Structurally, both GFI1 and GFI1B are constituted of three main domains that are very similar (Figure 1A). At the N-terminus of the proteins, there is a highly conserved SNAIL/GFI1 (SNAG) domain that is present in both GFI1 and GFI1B with ~90% homology, forming a sub-family on their own. This domain is shared with other transcriptional repressors such as SNAIL, SCRATCH, and SLUG forming a distinct but larger SNAG protein sub-family (Grimes et al., 1996; Manzanares et al., 2001; Katoh and Katoh, 2003, and reviewed in Chiang and Ayyanathan, 2013) and mediates transcriptional repression by recruiting chromatin modifier complexes to the regulatory regions of GFI1/GFI1B target genes (Tong et al., 1998; Saleque et al., 2007; Velinder et al., 2016; McClellan et al., 2019).
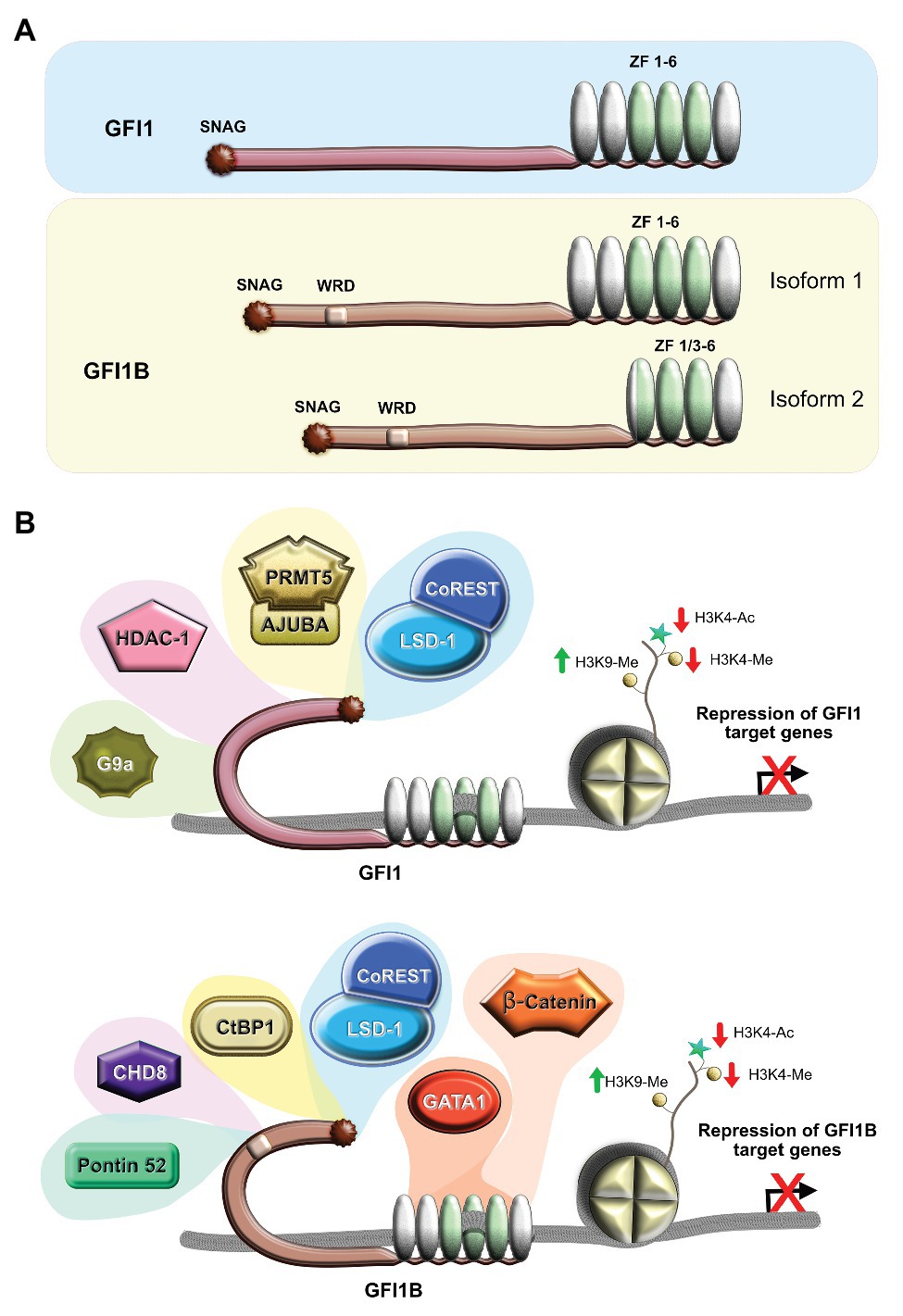
Figure 1. Structure and function of human GFI1 and GFI1B. (A) Schematic depiction of the structure of the proteins, showing the SNAG suppressor domain, the less characterized intermediate domain involved in protein/protein interactions, and the six zinc finger domains (ZF) localized at the C-terminal end with those three involved in DNA binding shown in green and the three other domains that play a role in interaction with other proteins shown in silver. The two isoforms of Gfi1b are shown with the longer megakaryocyte-specific isoform 1 that has the six zinc fingers and the short erythroid-specific isoform 2 that lacks two zinc fingers due to the fusion of ZF1 and ZF3. (B) Schematic representation of the GFI1 (top) and GFI1B (bottom) complexes with different partners that promote gene silencing by removal of open chromatin signatures and induction of marks that correlate with closed chromatin. WRD, Wnt regulatory domain.
Both GFI1 and GFI1B share six zinc finger domains at their C-terminal ends, of which three (zinc fingers 3, 4, and 5) form the DNA binding domain that recognizes a sequence containing the core motif AATC [aAATCac(ta)gc for GFI1 and aAATCacaGc for GFI1B] (Lee et al., 2010; Maiques-Diaz et al., 2018b; Shooshtarizadeh et al., 2019; Vinyard et al., 2019), whereas zinc fingers 1, 2, and 6 mediate protein/protein interactions with factors such as MIZ-1 and PU.1 that can recruit GFI1 to target genes independently of DNA binding (Dahl et al., 2007; Basu et al., 2009; Liu et al., 2010). The proteins that similarly bind to the zinc fingers of GFI1B are less defined than those binding to GFI1, although we have identified β-catenin as a factor whose interaction with GFI1B depends on GFI1B’s zinc finger domain (Shooshtarizadeh et al., 2019) whereas GATA 1 has also been demonstrated to interact with this domain (Figure 1B; Huang et al., 2005; Rodriguez et al., 2005). Interestingly, in human, GFI1B, but not GFI1, exists as two splicing isoforms (Figure 1A), of which the smaller one lacks zinc fingers 1 and 2 but is still able to repress transcription with an efficiency similar to, or better than, the long, major isoform that has all six zinc fingers, suggesting that the first two are not essential for the repressive function (Laurent et al., 2012; Beauchemin et al., 2019). However, it has been shown that the two isoforms play distinct roles in hematopoiesis, with the long isoform being important for megakaryocyte differentiation and maturation and the short isoform regulating erythropoiesis, indicating that zinc fingers 1 and 2 of GFI1B holds a megakaryocyte-specific function, probably by binding yet-to-be identified megakaryocyte factor(s) (Laurent et al., 2012; Polfus et al., 2016; Rabbolini et al., 2017; Schulze et al., 2017). If both the SNAG domain and the zinc-finger domains are highly conserved between GFI1 and GFI1B, the two domains are separated by non-conserved intermediate domains whose function is not yet fully understood but have been shown to mediate protein-protein interactions with other factors such as the EHMT2 (G9a) histone methyltransferase, ATXN1 (Ataxin 1), PIAS3 and U2AF26 in the case of GFI1 (Rodel et al., 2000; Duan et al., 2005; Tsuda et al., 2005; Heyd et al., 2006; Andrade et al., 2016), or CHD8, CTBP1, and RUVBL1 (Pontin 52) through a six-amino-acid motif called Wnt regulatory domain (WRD) in the case of GFI1B (Shooshtarizadeh et al., 2019; Figure 1B).
The presently accepted model that describes the mode of action for both GFI1 and GFI1B is based on very robust biochemical evidence that both factors are able to recruit the histone demethylase LSD1, the histone methyltransferase G9a (in the case of Gfi1b also the methyltransferase SUV39H1), and the histone deacetylases (HDACs) or other enzymes with deacetylase activity such as Ajuba to target gene promoters or enhancers (Duan et al., 2005; Vassen et al., 2006; Saleque et al., 2007; Montoya-Durango et al., 2008). In the case of LSD1, the 20-amino-acid N-terminal SNAG domains of GFI1 and GFI1B are critical. This domain is in its sequence similar to the N-terminal end of the Histone H3 tail (Saleque et al., 2007; Chowdhury et al., 2013), and studies with the SNAG domain protein SNAIL1 have suggested that the affinity of the SNAG domain for LSD1 is higher than the histone H3 tail and therefore a binding of LSD1 to a SNAG transcription factor localized on chromatin is preferred over binding to histone H3 (Lin et al., 2010) and it is likely that the same is true for GFI1 and GFI1B. Transcriptional repression is achieved by the action of LSD1, which removes methyl groups from Histone H3 Lysine 4, thus contributing to a contraction of chromatin. HDACs that can bind to the domains of GFI1 and GFI1B located between the SNAG and zinc finger domains close accessibility to genomic sites by removing acetyl groups from histone H3 Lysine 9, thereby silencing transcription (Duan et al., 2005). The other histone-modifying enzymes, i.e., the methyl transferases G9a and SUV39H1 that also bind to regions of GFI1 or GFI1B located between the SNAG and the zinc finger domains, are part of this model as well. Both enzymes help to mediate the transcriptional repressor function of both GFI1 and GFI1B by catalyzing methylation of histone H3 Lysine 9 residues, which lead to contraction of chromatin or possibly even to the formation of transcriptionally inactive heterochromatin (Duan et al., 2005; Vassen et al., 2006).
However, parts of this model have been challenged by new findings using acute myeloid leukemia cells lines that indicated that the enzymatic activity of LSD1 as a demethylase is not per se required to mediate the function of GFI1, and by inference also of GFI1B, as transcriptional repressors. The study suggested that LSD1 functions rather as a scaffold protein enabling other enzymes such as HDACs to bind to GFI1 and GFIB and to act on histones in the vicinity of their target genes to silence transcription (Maiques-Diaz et al., 2018a,b). Evidence for this was provided among other data that an enzymatic inactive LSD1 can fulfill the same role as the wild-type enzyme. Future studies have to show how LSD1 exerts this scaffolding role, since it would suggest that HDACs associate with GFI1 and GFI1B indirectly via LSD1.
It is of interest to note that LSD1, which takes a central role in this model to mediate the transcriptional repressor function of GFI1 and GFI1B, has also other substrates than histones, one being the tumor suppressor and guardian of the genome p53. The p53 protein is methylated at lysine residues in its C-terminal regulatory region p53, which stabilizes it, restricts it to the nucleus, and enhances its ability to transcriptionally transactivate target genes (reviewed in Liu et al., 2019). LSD1 can remove these methyl groups and thus dampen p53 activity. In this context, a new function of GFI1 that is independent of its ability to bind DNA and act as a transcriptional regulator came to light. Proteome analyses revealed that GFI1 can bind to p53, and since both also bind to LSD1, a tripartite GFI1/p53/LSD1 complex is formed, which at least in lymphoid cells ensures the demethylation of the p53 C-terminal regulatory domain. In cells that lack GFI1, p53 remains methylated and is thus overactive and can induce apoptosis (Khandanpour et al., 2013; Vadnais et al., 2019). Similarly, GFI1 can also recruit other enzymes such as PRMT1 that methylate key DNA repair factors such as MRE11 or 53BP1 and regulate the response to DNA damage in this way (Vadnais et al., 2018). Similar abilities have not been reported for GFI1B, and it is thus likely that there may be major differences in molecular mechanisms that are employed by GFI1 versus GFI1B. Another difference between both factors may be the binding of the PIAS3 protein to GFI1 to affect the STAT3 signaling pathway, which has not been reported for GFI1B (Rodel et al., 2000). Moreover, the region in GFI1 that mediates the binding to PIAS3 contains a conserved type I SUMOylation consensus element, and recent reports suggest that SUMOylation of GFI1 favors the recruitment of LSD1 to target genes in hematopoietic cells (Andrade et al., 2016). By contrast, a mechanism that is employed by GFI1B, but for which no evidence exists that it may be also valid for GFI1, is the formation of a tripartite complex between GFI1B/β-catenin and LSD1. This complex occupies specific sites on chromatin and regulates β-catenin target genes in megakaryocytes, its precursors, and HSCs (Shooshtarizadeh et al., 2019). The fact that GFI1 and GFI1B are very similar proteins and in fact almost identical in sequence within their SNAG and zinc finger domains has raised the question how these factors can exert biological roles that are very different. The findings described in this last chapter and their very specific tissue and cell type-specific expression patterns provide first explanations for this conundrum, although more experimental data are needed to fully understand their differential regulatory potential.
Although both GFI1 and GFI1B seem to be strictly transcriptional repressors as there is no report published proposing a mechanism for an activating function, both of them are engaged in much more complex repression/activation loops due to the tight regulation of their expression either by themselves, as both GFI1 and GFI1B can repress their own expression, or from regulation through other hematopoietic master transcription factors such as RUNX1, GATA1/2, PU.1, and HMGB2 (Huang et al., 2005; Laurent et al., 2010; Wilson et al., 2010; Lancrin et al., 2012; Marneth et al., 2018). This tight regulation, characterized by waves of exclusive expression windows for GFI1 and GFI1B during the process of hematopoiesis from the emergence of hemogenic endothelial (HE) cells that occurs in the embryo to fully differentiated hematopoietic cells produced in adults, highlights a complex and multilayered function for these two key factors, which is discussed in depth in the next section.
GFI1/GFI1B in Hematopoiesis and the Programming of HSCs
From Endothelial-to-Hematopoietic Transition to Definitive Erythropoiesis
During the development, blood cells are produced in the embryo through three distinct waves called primitive, transient, and definitive hematopoiesis. During the first two waves of hematopoiesis that precede definitive hematopoiesis, blood cells are not derived from pluripotent HSCs, but rather from the conversion of special endothelial cells with hemogenic potential called HE cells or hemangioblasts that give rise to bipotent or multipotent progenitors (MPPs) that can colonize hematopoietic sites (reviewed in McGrath et al., 2015b; Gritz and Hirschi, 2016). The first wave that takes place as early as day e7.5 in the mouse is called primitive hematopoiesis and is often referred as to primitive erythropoiesis. The reason for this is the observation that this wave, which takes place in the blood islands of the yolk sac, produces essentially primitive nucleated erythrocytes and primitive megakaryocytes through conversion of hemangioblasts to primitive megakaryocyte-erythrocyte progenitors (MEPs) that are solely restricted to the erythroid and megakaryocyte lineages (Wong et al., 1986; Palis et al., 1999; Xu et al., 2001; Tober et al., 2007; Isern et al., 2011; Psaila et al., 2016).
The second wave that occurs shortly after the first, however, produces transient hematopoietic cells with an adult cell phenotype, or pro-definitive hematopoietic cells, including lymphoid cells. However, unlike cells produced during definitive hematopoiesis, those cells do not arise from single HSCs but rather from multipotent erythro-myeloid progenitors (EMPs) that seem to arise through hemogenic conversion of endothelial cells at different sites in the embryo such as vitelline, yolk sac, placenta, and the caudal intraembryonic splanchnopleura before colonizing the fetal liver (Godin et al., 1995; Cumano et al., 1996; Palis et al., 1999; McGrath et al., 2015a).
Definitive hematopoiesis is the process by which all blood cells are produced from ultimately a pluripotent HSC with long-term repopulating capacity (LT-HSC). This process occurs through successive differentiation along a continuum of progenitor cells that acquire various molecular and cellular identity leading to diverse levels of commitment and biases toward specific lineages (reviewed in Brown et al., 2018; Laurenti and Gottgens, 2018). Those LT-HSCs first arise during a narrow window in development by a process called endothelial-to-hematopoietic transition (EHT). In this process, a specific subset of endothelial cells, the so-called HE cells located in the ventral wall of the aorta-gonad-mesonephros region (AGM), undergo a morphological change. During EHT, HE cells lose their endothelial traits and acquire a round, non-adherent shape that allows them to leave the AGM and colonize at first the fetal liver and later the bone marrow where they establish permanently (Figure 2; Christensen et al., 2004; Zeng et al., 2019, and reviewed in Dejana et al., 2017; Howell and Speck, 2020).
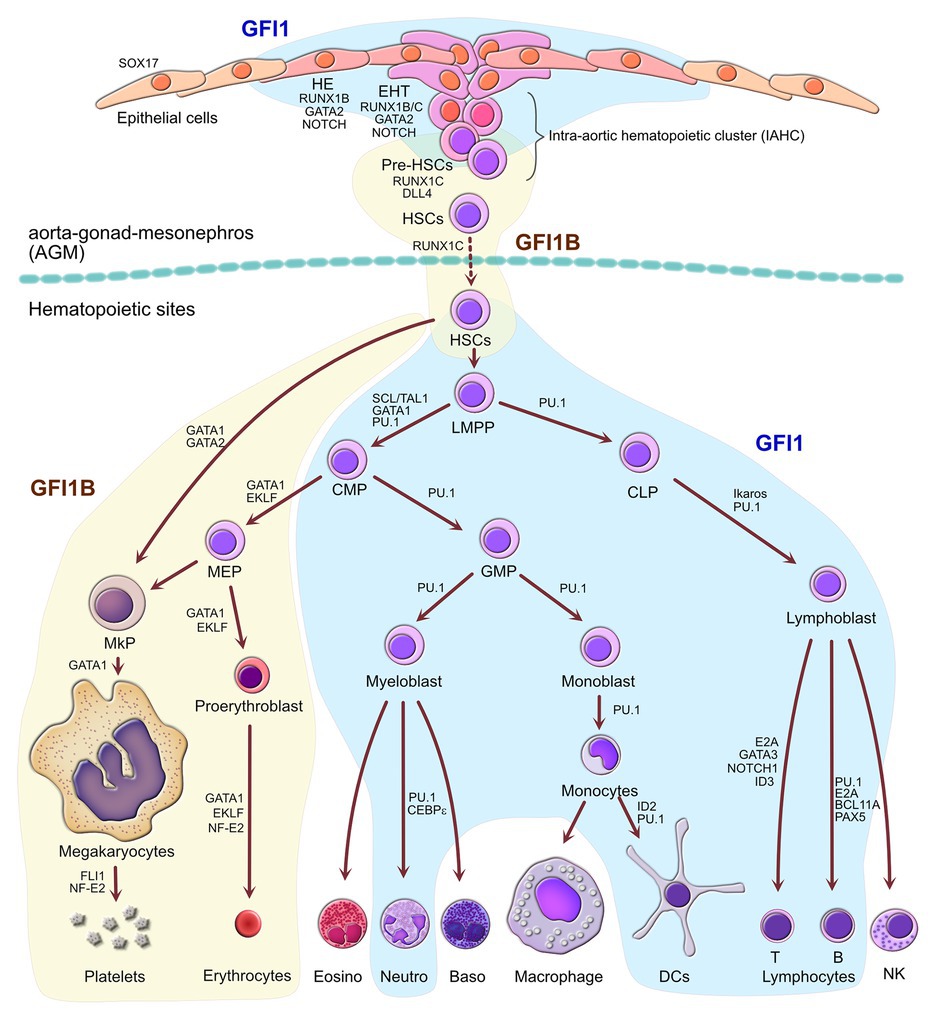
Figure 2. Separate roles of GFI1 and GFI1B during definitive hematopoiesis. Schematic representation of hematopoiesis from the initial production of primordial definitive hematopoietic stem cells (HSCs) from the hemogenic epithelial cells (HEs) found in the aorta-gonad-mesonephros (AGM) through endothelial-to-hematopoietic transition (EHT) during embryonic development until the production of all blood cells in the definitive hematopoietic sites such as fetal liver and adult bone marrow. Cellular compartments in which GFI1 plays a role are indicated as a blue area, whereas the compartment where GFI1B is important are indicated as the yellow areas, highlighting both overlapping and exclusive functions of GFI1 and GFI1B at all stages of hematopoiesis. Factors that contribute to cell fate decisions along with GFI1 or GFI1B are indicated. LMPP, lymphoid-primed multipotent progenitors; CMP, common myeloid progenitors; CLP, common lymphoid progenitors; GMP, granulocyte-monocyte progenitors; MEP, megakaryocyte-erythrocyte progenitors; MkP, megakaryocyte progenitors; Eosino, eosinophils; Neutro, neutrophils; Baso, basophils; DCs, dendritic cells; NK, natural killer cells.
Early experiments had shown that both GFI1 and GFI1B play often reciprocal yet complementary roles during hematopoiesis (Figure 2) and this is mainly due to the regulatory feedback loop that exists between the two transcription factors that can actively repress the expression of each other (Doan et al., 2004; Vassen et al., 2005, 2007; Anguita et al., 2010). GFI1, which is normally expressed at readily detectable levels in HSCs and is critical for the maintenance and proliferative control of this population (Hock et al., 2004; Zeng et al., 2004), sees its expression subsequently increased in MPP cells where it favors cells to engage into the lymphoid lineage, although its expression is sustained in both the lymphoid and more mature myeloid precursor cells such as the granulocyte-monocyte progenitors (GMPs). In GMPs, GFI1 expression drives the development of neutrophils by antagonizing eosinophilic differentiation, while expression of GFI1 also ceases in cells differentiating into the monocyte-macrophage lineage (Figure 2; Hock et al., 2003; Dahl et al., 2007; Spooner et al., 2009; Li et al., 2010; Liu and Dong, 2012; Phelan et al., 2013; Fraszczak et al., 2016; Zhang et al., 2019). GFI1B on the other hand is highly expressed already in the HSCs but sees its expression decrease in the MPP compartment where it is replaced by GFI1 whose expression starts to increase (Osawa et al., 2002; Vassen et al., 2007). Interestingly, GFI1B remains highly expressed in MEPs and in megakaryocyte precursors (MKPs) as well as during terminal differentiation of megakaryocytes and erythrocytes (Figure 2; Laurent et al., 2009). Studies with knockout mice have demonstrated that GFI1B plays a critical role in the biology of these two lineages (Saleque et al., 2002; Randrianarison-Huetz et al., 2010) and is pretty much absent from other cells of the myeloid and lymphoid compartment with the exception of a subset of early B-cells and rare plasmacytoid dendritic cells (pDC) where it seems to play a part in the regulation of the VD(J) recombination by repressing the Rag1 gene through repression of the transcription activator FOXO1 (Osawa et al., 2002; Vassen et al., 2007; Schulz et al., 2012; Chow et al., 2013). It is not clear yet if GFI1B could play some role in rare myeloid cells, but high expression of functional GFI1B not only in erythroid/megakaryocytic lineage but also in non-erythroid/megakaryocytic myeloid cells has been associated with both chronic and acute myeloid leukemias, suggesting that it can act as a proto-oncogene possibly by supporting cell survival in cells where it is not normally expressed (Elmaagacli et al., 2007; Vassen et al., 2009). Intriguingly, a somatic dominant-negative mutation D262N in the fourth zinc finger of GFI1B that decreases its function has been associated with malignant transformation in an acute myeloid leukemia patient. The study suggests that the D262N mutant GFI1B feeds back on the function of the wild-type, unmutated GFI1B with the consequence that myeloid differentiation of precursors is favored over erythroid differentiation resulting in higher cell survival in MDS patient samples, which would attribute a direct oncogenic role to this GF11B mutant. This oncogenic activity could be mediated by the Spi1 (PU.1), which is a direct GFI1B target gene and normally repressed by GFI1B. The GFI1B D262N mutant however leads to an increased expression of PU.1, leading the authors of this study to propose that a GFI1B-SPI1 interaction represents a critical element in the emergence of AML from MDS (Anguita et al., 2016). Such an association was not observed with any of the germ-line dominant-negative mutations identified in GFI1B, which represents a discrepancy that remains to be investigated (Stevenson et al., 2013; Monteferrario et al., 2014; Kitamura et al., 2016; van Oorschot et al., 2019). Similarly, loss of heterozygosity leading to low expression of GFI1B was also associated with myeloid leukemic transformation, suggesting that at normal level in early myeloid progenitors, GFI1B may act as a tumor suppressor, possibly by driving differentiation toward the erythroid lineage (Thivakaran et al., 2018).
The function of GFI1B in the terminal differentiation of both the erythroid lineage and megakaryocytes is critical but seems contradictory to some extent. The loss of GFI1B in MEPs leads on one side to an arrest in erythroid development at the erythroblast stage and ultimately to erythropoiesis failure, but also on the other side to an expansion of megakaryocytes that are however incapable to produce platelets due to a defect in proplatelet formation (Foudi et al., 2014; Vassen et al., 2014; Beauchemin et al., 2017; Shooshtarizadeh et al., 2019). Interestingly, overexpression of GFI1B in CD34+ human stem cells also leads to defects in erythroid differentiation with failure to produce cells beyond the proerythroblast stage, suggesting that a tight regulation of expression is required during the final maturation stages of red blood cells (Osawa et al., 2002). This dual, yet contradictory function raises questions on the biological role of GFI1B in cell fate decisions in MEPs that should either go toward the erythroid or the megakaryocyte lineages, in particular since high expression of GFI1B is required for both lineages. This apparent contradiction may have found its explanation in the recent discovery that megakaryocytes could derive directly through differentiation of HSCs therefore bypassing a multipotent EMP stage (Sanjuan-Pla et al., 2013; Shin et al., 2014, and reviewed in Yang et al., 2019).
Despite their critical role in definitive hematopoiesis, a role of GFI1 or GFI1B in primitive hematopoiesis or even during the transient hematopoiesis is still uncertain, and although such a role has been described for both GFI1 and GFI1B homologues Gfi1aa and Gfi1b in zebrafish, a requirement for either GFI1 or GFI1B in primitive erythropoiesis in mammals has not yet been identified (Cooney et al., 2013; Andrade et al., 2016; Velinder et al., 2017; Moore et al., 2018). Although mice lacking GFI1B die in utero at around day e14.5 because of a defect in erythropoiesis, the embryos survive until then because nucleated primitive erythrocytes are still generated although exhibiting some cellular abnormalities, indicating that GFI1B is dispensable to some extent at this stage for proper erythropoiesis to occur (Saleque et al., 2002). However, the absence of enucleated definitive erythrocytes in Gfi1b-knockout mice suggests that despite a definitive-like erythropoiesis occurring without HSCs during the transient hematopoiesis, GFI1B is required at this developmental stage (Saleque et al., 2002). Importantly, the simultaneous inactivation of GFI1 and GFI1B leads to a more severe reduction in pre-definitive erythrocytes and even defects in primitive erythrocytes, demonstrating that, indeed, GFI1 and GFI1B do play a role in the early hematopoietic development stages, albeit not as critical as during definitive hematopoiesis (Lancrin et al., 2012).
The critical role of both GFI1 and GFI1B in hematopoiesis is not restricted to the differentiation of HSCs into the different blood cell lineages but is already present in the process that gives rise to the first HSCs in the AGM of the embryo (Figure 2; Lancrin et al., 2012). Indeed, both proteins play an essential role in the repression of the endothelial genes within HE cells that undergo EHT to become HSCs, and it has been shown that this repression occurs through recruitment of LSD1 (Thambyrajah et al., 2016). Interestingly, during this process, GFI1 and GFI1B again exhibit mutually exclusive yet complementary expression patterns and functions. Although the role of master regulator of this conversion of HE cells pertains to RUNX1, its first action is to directly drive the expression of the GFI1/GFI1B factors, and it has been shown that forced expression of both can compensate for the loss of RUNX1 in this process (Lancrin et al., 2012). Indeed, the expression of RUNX1 is followed almost immediately by an increase in GFI1 in a subset of arterial endothelial cells of the AGM that defines them as hemogenic, despite the fact that they retain a fully endothelial phenotype (Thambyrajah et al., 2016; Porcheri et al., 2020). A subset of these GFI1-positive HE cells are then recruited in a Notch/DL4-dependent manner to the nascent intra-aortic hematopoietic cluster (IAHC) where they then start to express KIT (c-Kit). This marks the beginning of the EHT process during which cells silence their endothelial program and acquire a hematopoietic gene signature (Porcheri et al., 2020).
As the hematopoietic program gains in strength, GFI1 expression is gradually replaced by GFI1B, which along with the expression of CD45, marks the appearance of pre-HSCs. These cells begin to form clusters prior to their release into the bloodstream to colonize the fetal liver (Thambyrajah et al., 2016; Zeng et al., 2019). Intriguingly, the loss of either GFI1 or GFI1B does not prevent formation of IAHCs or the release of HSCs as does the loss of RUNX1 (Okuda et al., 1996; Saleque et al., 2002; Hock et al., 2004; Zeng et al., 2004; Zhang et al., 2019), but the loss of both GFI1 and GFI1B simultaneously completely abrogates the EHT process and leads to failure to produce any HSCs despite the formation of HE cells and the capacity of these cells to generate hematopoietic cells in vitro (Lancrin et al., 2012; Thambyrajah et al., 2016). A potential explanation for this observation could be the capacity, though limited, of GFI1 and GFI1B to compensate for the loss of each other (Fiolka et al., 2006). Indeed, the presence of either GFI1 or GFI1B, albeit in sub-optimal level, might just be sufficient to trigger EHT and to promote the recruitment of those transforming HE cells to the IAHC.
In vitro Hematopoietic Transdifferentiation From iPS/ES Cells or Non-stem Cells
Generating transplantable HSCs from either embryonic stem (ES) cells, induced pluripotent stem (iPS) cells, or even directly from somatic non-stem cells such as differentiated hematopoietic cells and fibroblasts still represents today one of the most important challenges in medicine. Because of the critical role of both GFI1 and GFI1B at all stages of hematopoiesis, including the earliest developmental stages, it is not surprising that both factors were used in attempts to drive hematopoietic differentiation in vitro from both multipotent and differentiated non-hematopoietic cells.
It is possible today to generate all hematopoietic cell types from iPSCs, including HSC-like cells that express CD34. Cells have even been generated through processes that recapitulate in vitro the EHT that takes place in the AGM as a way to ensure acquisition of stemness to produce pluripotent HSCs with the capacity to colonize hematopoietic sites. However, these cells still lack a robust capacity to engraft in a host (Wang et al., 2005; Lengerke et al., 2009; Doulatov et al., 2013; Riddell et al., 2014; Ruiz et al., 2019; Zhou et al., 2019). The first experiment that succeeds to produce engraftable HSCs from iPS cells achieved this by driving differentiation in vivo through teratoma formation, but even in this case, the efficiency was very low with less than 1–2% of engraftment and relied on complex procedures and cocktails of cytokines to achieve this (Amabile et al., 2013; Suzuki et al., 2013; Philipp et al., 2018). Other approaches using in vitro 3D culture systems produced HSCs with similar engraftment potential without the need to go through a teratoma stage in mouse (Shan et al., 2020). It was only recently that by overexpressing either the single fusion protein MLL-AF4 or a large panels of seven transcription factors (RUNX1, ERG, HOXA5, HOXA9, HOXA10, LCOR, and SP1) but not including GFI1/GFI1B that the differentiation of iPS cells into engraftable HSCs showed more promising results, despite an elevated risk of leukemic transformation (Sugimura et al., 2017; Tan et al., 2018).
Using GFI1 or GFI1B to drive differentiation of iPS cells has been done only in one study by Tsukada et al. with moderate results (Tsukada et al., 2017). In this study, the authors drove expression of GFI1B together with FOS and GATA2 in murine iPS cells in a doxycycline-inducible way and were able to induce long-term HSC formation, but had to rely on teratoma formation to do so (Figure 3A). In their hands, forced expression of these sole three factors significantly improved the yield of engraftable HSCs that were produced compared to experiments that used unmodified iPS cells. Moreover, HSCs were obtained even without the need to co-inject supporting stromal cells along with the iPS cells or to provide hematopoietic cytokines, which are required to allow unmodified iPSC to transdifferentiate into putative HSCs (Amabile et al., 2013; Suzuki et al., 2013; Tsukada et al., 2017).
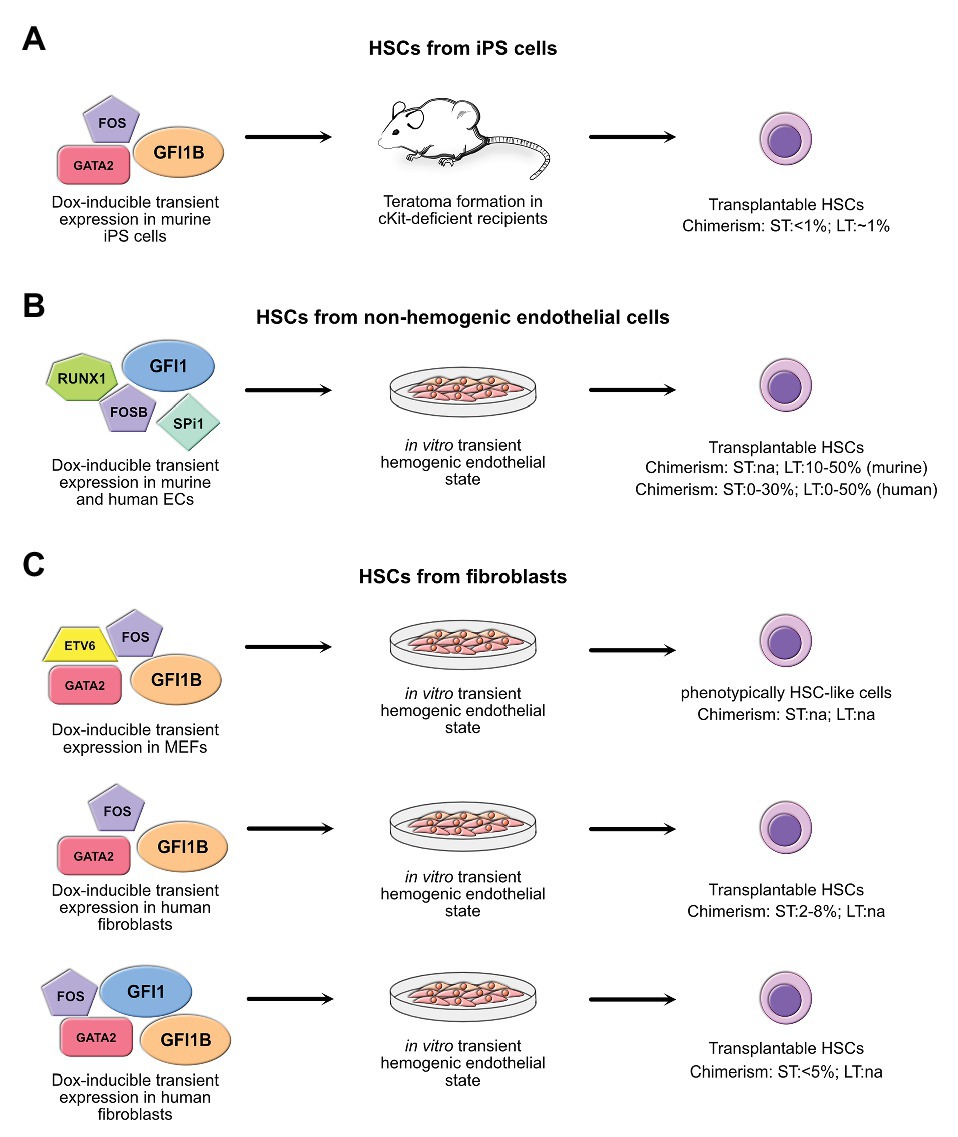
Figure 3. Summary of the strategies using overexpression of GFI1 and/or GFI1B. Shown are the combination of GFI1 and GFI1B with other factors to generate transplantable hematopoietic stem cells (HSCs) through trans-differentiation of non-hematopoietic cells, including induced pluripotent stem (iPS) cells (A), non-hemogenic endothelial cells (ECs) (B), and fibroblasts (C). All these strategies involve an intermediary hemogenic endothelial state either in vivo by means of teratoma formation or in vitro. When known, the efficiency of HSC generation as assessed by measuring chimerism in graft assay in recipient mice is indicated for both short-term (ST) and long-term (LT) engraftment. na, not assessed. References: iPS cells (Tsukada et al., 2017); non-hemogenic endothelial cells (Sandler et al., 2014; Lis et al., 2017; Barcia Duran et al., 2018); fibroblasts (Pereira et al., 2013; Gomes et al., 2018; Daniel et al., 2019).
Clearly, the differentiation of iPS cells into transplantable HSCs is of high interest, but it does also carry its share of imponderable risks. In particular, teratoma formation remains a significant threat and takes place if cells with iPS-like properties remain after their differentiation into HSCs. For this reason, researchers have considered the possibility to produce HSCs through a direct conversion of differentiated cells, such as differentiated hematopoietic cells, endothelial cells, or even fibroblasts without passing through a pluripotent state. De-differentiation of committed hematopoietic progenitor cells into HSCs may seem to be the easiest way to achieve this, as a complete reprogramming, which is needed when more differentiated cells are used, is not necessary. Indeed, such a de-differentiation of hematopoietic progenitor cells has been achieved through combined overexpression of defined sets of transcription factors in both myeloid and lymphoid committed progenitors (Riddell et al., 2014). Conversion of endothelial cells into HSCs is also a natural approach as HSCs arise from HE cells and several protocols are already established for this, most of which use doxycycline-inducible transient overexpression of either Gfi1 or Gfi1b simultaneously with the overexpression of other factors such as GATA2, FOS, RUNX1, and SPI1 using lentiviral expression vectors (Figure 3B; Sandler et al., 2014; Lis et al., 2017; Barcia Duran et al., 2018; Daniel et al., 2019).
Conversion of fibroblasts into HSCs capable to produce mature definitive hematopoietic cells in vitro has also been achieved through the transient expression of the transcription factors Oct4 altogether with a cytokine treatment, or with transcription factor cocktails that include GATA2, RUNX1, LMO2, and ERG. However, the efficiency to produce cells with engraftable potential was low, and these methods resulted in cells that lacked the ability to produce lymphoid cells (Szabo et al., 2010) or lead to the production of HSCs with only short-term reconstitution ability (Batta et al., 2014). Other approaches have shown that transient expression of the transcription factors SCL, LMO2, RUNX1, and BMI1 can also differentiate fibroblasts into both lymphoid and myeloid progenitors capable of short-term engraftment in mice (Cheng et al., 2016). Interestingly, in this experiment, a key step into hemogenic conversion of fibroblasts was the upregulation of Gfi1 controlled by LMO2 (Cheng et al., 2016). Importantly, generation of engraftable HSCs from fibroblasts was achieved by forcing transient expression of GFI1B in combination with GATA2 and FOS, either with or without ETV6, using pools of doxycycline-inducible lentiviruses (Pereira et al., 2013; Gomes et al., 2018). The efficiency of this approach was even further improved by adding GFI1 into the cocktail, allowing the generation of a similar number of hematopoietic stem-progenitor cells with short-term engraftable capacity in much shorter time, although long-term engraftment capacity was not assessed in this experiment (Figure 3C; Daniel et al., 2019). How GFI1/GFI1B forced transient expression contributes to the improvement of hemogenic conversion remains to be fully uncovered, but seems to be due to the capacity of at least GFI1B to directly cooperate with GATA2 by co-occupying promoters of critical non-hematopoietic genes leading to their silencing (Gomes et al., 2018). However, the lack of long-term hematopoietic reconstitution potential of cells from all these approaches demonstrates that despite a better understanding of hemogenic conversion, true reproduction of the initial generation of putative HSCs that occurs within the AGM during embryogenesis remains elusive.
GFI1/GFI1B in Non-hematopoietic Cells
GFI1 in Ear Hair Cells
Both GFI1 and GFI1B are prominently expressed in the hematopoietic system, but they are not entirely restricted to this compartment. Indeed, it has been shown that GFI1 for instance plays a critical role in the biogenesis of hair cells of the inner ear and that a loss of GFI1 leads to deafness and ataxia in mice (Wallis et al., 2003; Hertzano et al., 2004, and reviewed in Costa et al., 2017; Zhong et al., 2019). It has been reported that inner ear hair cells can be generated using forced transient expression of GFI1 in murine ES cells simultaneously with the transcription factor POU4F3 and its upstream hair cell master regulator ATHO1 using doxycycline-inducible polycistronic transgenes knocked into the HPRT locus to ensure balanced expression levels of all three factors (Hu et al., 2010; Masuda et al., 2011; Costa and Henrique, 2015; Costa et al., 2015; Figure 4). Likewise, direct conversion of otic epithelial cells from the inner ear into MYO7A-positive inner ear hair cell-like cells has also been achieved through simultaneous forced transient expression of GFI1, POU4F3, and ATOH1 in chicken embryos by in ovo electroporation of a similar doxycycline-inducible polycistronic transgene as the one used in mouse ES cells (Costa et al., 2015). Furthermore, recent experiments used transient overexpression of GFI1, ATHO1, and POU4F3, and included SIX1, a homeobox protein similar to D. melanogaster proteins called “sine oculis”, which is involved in autosomal dominant deafness type 23 (DFNA23) and has been shown to physically interact with the former three to target many hair cell regulators (Li et al., 2020). Either with or without SIX1, primary mouse embryonic and even adult human fibroblasts could be successfully converted into cells that exhibited most of the physiological traits of inner ear hair cells opening new avenues in potential treatment of deafness caused by the loss of these sensory cells (Figure 4; Duran Alonso et al., 2018; Menendez et al., 2020).
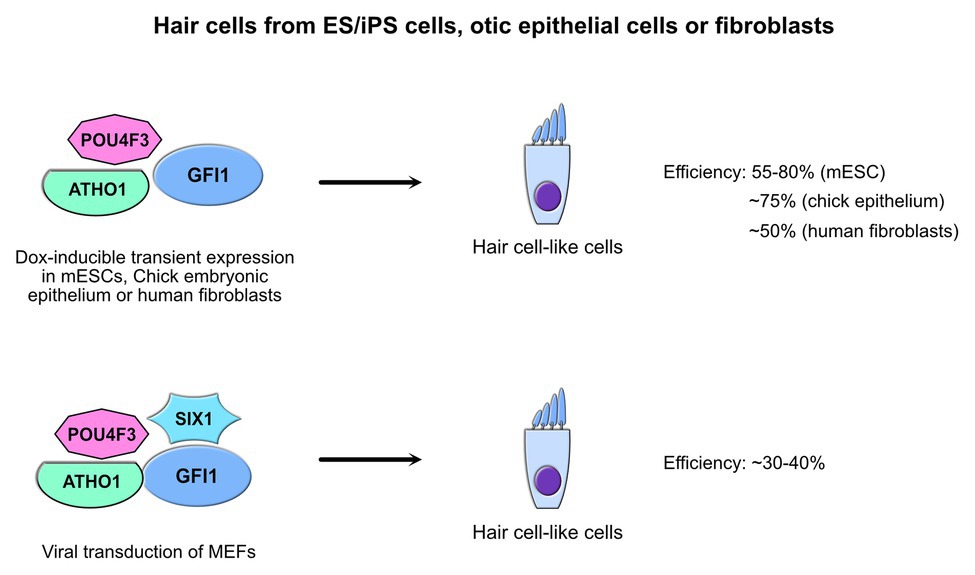
Figure 4. GFI1 and inner ear hair cells. Strategies to generate induced hair cell-like cells through trans-differentiation of non-hair cells in vitro by forced expression of GFI1 in combination with other factors. The efficiency to produce cells that harbor hair cell characteristics in vitro is indicated along the starting cell type used to achieve trans-differentiation (Costa and Henrique, 2015; Costa et al., 2015; Duran Alonso et al., 2018; Menendez et al., 2020).
Gfi1 in the Pancreas
Besides the inner hair cells, GFI1 has also been suggested to play a role in pancreatic acinar cells, as one study has shown that its ablation results in defects in exocrine cells of the pancreas, characterized by a reduction of normal centroacinar cell markers and reduced exocrine function (Qu et al., 2015).
GFI1 in the Intestine
In addition, an ATOH1-dependent role of GFI1 in Paneth and mucus-secreting goblet cells of the intestine has also been suggested by the observation that the fate of these cells is shifted toward enteroendocrine cells in either Gfi1-knockout mice or Atoh1-knockout mice in which Gfi1 expression is also turned off by the loss of ATOH1 (Shroyer et al., 2005; Bjerknes and Cheng, 2010; Gracz et al., 2018).
Gfi1b in the Intestine
GFI1 has been shown to be dispensable in intestinal tuft cells that do not depend on ATOH1 but mainly on POU2F3 for their survival and differentiation (Gerbe et al., 2011, 2016). These rare cells might be however more dependent on GFI1B instead, as they specifically express GFI1B at much higher levels than the GFI1-expressing cells that do not express GFI1B (Gracz et al., 2018; Wang et al., 2020). Although it is not known at this point whether expression of GFI1B is required for tuft cells or not, these cells are the only intestinal cells that express GFI1B in a POU2F3-dependent manner (Bjerknes et al., 2012; Huang et al., 2018; Wang et al., 2020). This was further supported by the observation that these two factors (GFI1B and POU2F3) are always co-expressed in tuft cells (Gerbe et al., 2016). This exclusive yet complementary expression pattern of GFI1 and GFI1B in intestinal endothelium cells is interesting as it replicates the specificity observed in the hematopoietic cells and demonstrates that this mutually exclusive expression regulation may be a typical feature of the GFI1 family of transcription factors.
Conclusion
Producing engraftable HSCs from non-hematopoietic cells has represented the holy grail for many years and would mark a major breakthrough for clinical medicine. While the in vitro generation of almost all hematopoietic cell types is readily achievable to date, obtaining true engraftable HSCs remains elusive. It is of interest that GFI1 and GFI1B, two factors that are known to play important roles in all stages of hematopoiesis, including EHT during the initial production of definitive HSCs, might provide a way to generate those elusive HSCs. Although many obstacles have still to be overcome, considering the progress achieved during the past few years, it is conceivable that with the help of these two factors, a success in producing HSCs may be in reach in the foreseeable future. If such a breakthrough would be achieved, a number of new avenues would open up to treat a large spectrum of blood cell deficiencies as well as leukemia and lymphoma.
Author Contributions
HB wrote text, collected references, and made the first version of the Figures. TM drew up the concept of the review, wrote text, and revised the Figures. Both the authors contributed to the article and approved the submitted version.
Funding
Work in the laboratory of the authors is supported by a CIHR Foundation grant (FGN-148372) to TM, a Tier 1 Canadian Research Chair to TM, by a grant from AmorChem Inc. to TM.
Conflict of Interest
TM receives funding from the Canadian Institute of Health Research (CIHR, FDN-148372) and from a Canada Research Chair (CRC Tier 1) for work on GFI1 and GFI1B. Both are public Canadian funding organizations. TM acknowledges also funding from the Cancer Research Society (CRS), a Canadian Charity, for studies on Burkitt Lymphoma and from a VC fund (AmorChem) for work unrelated to the subject matter treated in this article.
References
Amabile, G., Welner, R. S., Nombela-Arrieta, C., D’alise, A. M., Di Ruscio, A., Ebralidze, A. K., et al. (2013). In vivo generation of transplantable human hematopoietic cells from induced pluripotent stem cells. Blood 121, 1255–1264. doi: 10.1182/blood-2012-06-434407
Andrade, D., Velinder, M., Singer, J., Maese, L., Bareyan, D., Nguyen, H., et al. (2016). SUMOylation regulates growth factor Independence 1 in transcriptional control and hematopoiesis. Mol. Cell. Biol. 36, 1438–1450. doi: 10.1128/MCB.01001-15
Anguita, E., Gupta, R., Olariu, V., Valk, P. J., Peterson, C., Delwel, R., et al. (2016). A somatic mutation of GFI1B identified in leukemia alters cell fate via a SPI1 (PU.1) centered genetic regulatory network. Dev. Biol. 411, 277–286. doi: 10.1016/j.ydbio.2016.02.002
Anguita, E., Villegas, A., Iborra, F., and Hernandez, A. (2010). GFI1B controls its own expression binding to multiple sites. Haematologica 95, 36–46. doi: 10.3324/haematol.2009.012351
Barcia Duran, J. G., Lis, R., Lu, T. M., and Rafii, S. (2018). In vitro conversion of adult murine endothelial cells to hematopoietic stem cells. Nat. Protoc. 13, 2758–2780. doi: 10.1038/s41596-018-0060-3
Basu, S., Liu, Q., Qiu, Y., and Dong, F. (2009). Gfi-1 represses CDKN2B encoding p15INK4B through interaction with Miz-1. Proc. Natl. Acad. Sci. U. S. A. 106, 1433–1438. doi: 10.1073/pnas.0804863106
Batta, K., Florkowska, M., Kouskoff, V., and Lacaud, G. (2014). Direct reprogramming of murine fibroblasts to hematopoietic progenitor cells. Cell Rep. 9, 1871–1884. doi: 10.1016/j.celrep.2014.11.002
Beauchemin, H., Shooshtarizadeh, P., Vadnais, C., Vassen, L., Pastore, Y. D., and Moroy, T. (2017). Gfi1b controls integrin signaling-dependent cytoskeleton dynamics and organization in megakaryocytes. Haematologica 102, 484–497. doi: 10.3324/haematol.2016.150375
Beauchemin, H., Shooshtharizadeh, P., Pinder, J., Dellaire, G., and Moroy, T. (2019). Dominant negative Gfi1b mutations cause moderate thrombocytopenia and an impaired stress thrombopoiesis associated with mild erythropoietic abnormalities in mice. Haematologica. doi: 10.3324/haematol.2019.222596 [Epub ahead of print]
Bjerknes, M., and Cheng, H. (2010). Cell lineage metastability in Gfi1-deficient mouse intestinal epithelium. Dev. Biol. 345, 49–63. doi: 10.1016/j.ydbio.2010.06.021
Bjerknes, M., Khandanpour, C., Moroy, T., Fujiyama, T., Hoshino, M., Klisch, T. J., et al. (2012). Origin of the brush cell lineage in the mouse intestinal epithelium. Dev. Biol. 362, 194–218. doi: 10.1016/j.ydbio.2011.12.009
Brown, G., Tsapogas, P., and Ceredig, R. (2018). The changing face of hematopoiesis: a spectrum of options is available to stem cells. Immunol. Cell Biol. 96, 898–911. doi: 10.1111/imcb.12055
Cheng, H., Ang, H. Y., El Farran, C. A., Li, P., Fang, H. T., Liu, T. M., et al. (2016). Reprogramming mouse fibroblasts into engraftable myeloerythroid and lymphoid progenitors. Nat. Commun. 7:13396. doi: 10.1038/ncomms13396
Chiang, C., and Ayyanathan, K. (2013). Snail/Gfi-1 (SNAG) family zinc finger proteins in transcription regulation, chromatin dynamics, cell signaling, development, and disease. Cytokine Growth Factor Rev. 24, 123–131. doi: 10.1016/j.cytogfr.2012.09.002
Chow, K. T., Schulz, D., McWhirter, S. M., and Schlissel, M. S. (2013). Gfi1 and gfi1b repress rag transcription in plasmacytoid dendritic cells in vitro. PLoS One 8:e75891. doi: 10.1371/journal.pone.0075891
Chowdhury, A. H., Ramroop, J. R., Upadhyay, G., Sengupta, A., Andrzejczyk, A., and Saleque, S. (2013). Differential transcriptional regulation of meis1 by Gfi1b and its co-factors LSD1 and CoREST. PLoS One 8:e53666. doi: 10.1371/journal.pone.0053666
Christensen, J. L., Wright, D. E., Wagers, A. J., and Weissman, I. L. (2004). Circulation and chemotaxis of fetal hematopoietic stem cells. PLoS Biol. 2:E75. doi: 10.1371/journal.pbio.0020075
Cooney, J. D., Hildick-Smith, G. J., Shafizadeh, E., McBride, P. F., Carroll, K. J., Anderson, H., et al. (2013). Teleost growth factor independence (gfi) genes differentially regulate successive waves of hematopoiesis. Dev. Biol. 373, 431–441. doi: 10.1016/j.ydbio.2012.08.015
Costa, A., and Henrique, D. (2015). Transcriptome profiling of induced hair cells (iHCs) generated by combined expression of Gfi1, Pou4f3 and Atoh1 during embryonic stem cell differentiation. Genom Data 6, 77–80. doi: 10.1016/j.gdata.2015.08.017
Costa, A., Powell, L. M., Lowell, S., and Jarman, A. P. (2017). Atoh1 in sensory hair cell development: constraints and cofactors. Semin. Cell Dev. Biol. 65, 60–68. doi: 10.1016/j.semcdb.2016.10.003
Costa, A., Sanchez-Guardado, L., Juniat, S., Gale, J. E., Daudet, N., and Henrique, D. (2015). Generation of sensory hair cells by genetic programming with a combination of transcription factors. Development 142, 1948–1959. doi: 10.1242/dev.119149
Cumano, A., Dieterlen-Lievre, F., and Godin, I. (1996). Lymphoid potential, probed before circulation in mouse, is restricted to caudal intraembryonic splanchnopleura. Cell 86, 907–916. doi: 10.1016/s0092-8674(00)80166-x
Dahl, R., Iyer, S. R., Owens, K. S., Cuylear, D. D., and Simon, M. C. (2007). The transcriptional repressor GFI-1 antagonizes PU.1 activity through protein-protein interaction. J. Biol. Chem. 282, 6473–6483. doi: 10.1074/jbc.M607613200
Daniel, M. G., Sachs, D., Bernitz, J. M., Fstkchyan, Y., Rapp, K., Satija, N., et al. (2019). Induction of human hemogenesis in adult fibroblasts by defined factors and hematopoietic coculture. FEBS Lett. 593, 3266–3287. doi: 10.1002/1873-3468.13621
Dejana, E., Hirschi, K. K., and Simons, M. (2017). The molecular basis of endothelial cell plasticity. Nat. Commun. 8:14361. doi: 10.1038/ncomms14361
Doan, L. L., Porter, S. D., Duan, Z., Flubacher, M. M., Montoya, D., Tsichlis, P. N., et al. (2004). Targeted transcriptional repression of Gfi1 by GFI1 and GFI1B in lymphoid cells. Nucleic Acids Res. 32, 2508–2519. doi: 10.1093/nar/gkh570
Doulatov, S., Vo, L. T., Chou, S. S., Kim, P. G., Arora, N., Li, H., et al. (2013). Induction of multipotential hematopoietic progenitors from human pluripotent stem cells via respecification of lineage-restricted precursors. Cell Stem Cell 13, 459–470. doi: 10.1016/j.stem.2013.09.002
Duan, Z., Zarebski, A., Montoya-Durango, D., Grimes, H. L., and Horwitz, M. (2005). Gfi1 coordinates epigenetic repression of p21Cip/WAF1 by recruitment of histone lysine methyltransferase G9a and histone deacetylase 1. Mol. Cell. Biol. 25, 10338–10351. doi: 10.1128/MCB.25.23.10338-10351.2005
Duran Alonso, M. B., Lopez Hernandez, I., de La Fuente, M. A., Garcia-Sancho, J., Giraldez, F., and Schimmang, T. (2018). Transcription factor induced conversion of human fibroblasts towards the hair cell lineage. PLoS One 13:e0200210. doi: 10.1371/journal.pone.0200210
Elmaagacli, A. H., Koldehoff, M., Zakrzewski, J. L., Steckel, N. K., Ottinger, H., and Beelen, D. W. (2007). Growth factor-independent 1B gene (GFI1B) is overexpressed in erythropoietic and megakaryocytic malignancies and increases their proliferation rate. Br. J. Haematol. 136, 212–219. doi: 10.1111/j.1365-2141.2006.06407.x
Fiolka, K., Hertzano, R., Vassen, L., Zeng, H., Hermesh, O., Avraham, K. B., et al. (2006). Gfi1 and Gfi1b act equivalently in haematopoiesis, but have distinct, non-overlapping functions in inner ear development. EMBO Rep. 7, 326–333. doi: 10.1038/sj.embor.7400618
Foudi, A., Kramer, D. J., Qin, J., Ye, D., Behlich, A. S., Mordecai, S., et al. (2014). Distinct, strict requirements for Gfi-1b in adult bone marrow red cell and platelet generation. J. Exp. Med. 211, 909–927. doi: 10.1084/jem.20131065
Fraszczak, J., Helness, A., Chen, R., Vadnais, C., Robert, F., Khandanpour, C., et al. (2016). Threshold levels of Gfi1 maintain E2A activity for B cell commitment via repression of Id1. PLoS One 11:e0160344. doi: 10.1371/journal.pone.0160344
Gerbe, F., Sidot, E., Smyth, D. J., Ohmoto, M., Matsumoto, I., Dardalhon, V., et al. (2016). Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 529, 226–230. doi: 10.1038/nature16527
Gerbe, F., van Es, J. H., Makrini, L., Brulin, B., Mellitzer, G., Robine, S., et al. (2011). Distinct ATOH1 and Neurog3 requirements define tuft cells as a new secretory cell type in the intestinal epithelium. J. Cell Biol. 192, 767–780. doi: 10.1083/jcb.201010127
Gilks, C. B., Bear, S. E., Grimes, H. L., and Tsichlis, P. N. (1993). Progression of interleukin-2 (IL-2)-dependent rat T cell lymphoma lines to IL-2-independent growth following activation of a gene (Gfi-1) encoding a novel zinc finger protein. Mol. Cell. Biol. 13, 1759–1768. doi: 10.1128/mcb.13.3.1759
Godin, I., Dieterlen-Lievre, F., and Cumano, A. (1995). Emergence of multipotent hemopoietic cells in the yolk sac and paraaortic splanchnopleura in mouse embryos, beginning at 8.5 days postcoitus. Proc. Natl. Acad. Sci. U. S. A. 92, 773–777. doi: 10.1073/pnas.92.3.773
Gomes, A. M., Kurochkin, I., Chang, B., Daniel, M., Law, K., Satija, N., et al. (2018). Cooperative transcription factor induction mediates Hemogenic reprogramming. Cell Rep. 25, 2821.e7–2835.e7. doi: 10.1016/j.celrep.2018.11.032
Gracz, A. D., Samsa, L. A., Fordham, M. J., Trotier, D. C., Zwarycz, B., Lo, Y. H., et al. (2018). Sox4 promotes Atoh1-independent intestinal secretory differentiation toward tuft and Enteroendocrine fates. Gastroenterology 155, 1508.e10–1523.e10. doi: 10.1053/j.gastro.2018.07.023
Grimes, H. L., Chan, T. O., Zweidler-McKay, P. A., Tong, B., and Tsichlis, P. N. (1996). The Gfi-1 proto-oncoprotein contains a novel transcriptional repressor domain, SNAG, and inhibits G1 arrest induced by interleukin-2 withdrawal. Mol. Cell. Biol. 16, 6263–6272. doi: 10.1128/mcb.16.11.6263
Gritz, E., and Hirschi, K. K. (2016). Specification and function of hemogenic endothelium during embryogenesis. Cell. Mol. Life Sci. 73, 1547–1567. doi: 10.1007/s00018-016-2134-0
Hertzano, R., Montcouquiol, M., Rashi-Elkeles, S., Elkon, R., Yucel, R., Frankel, W. N., et al. (2004). Transcription profiling of inner ears from Pou4f3(ddl/ddl) identifies Gfi1 as a target of the Pou4f3 deafness gene. Hum. Mol. Genet. 13, 2143–2153. doi: 10.1093/hmg/ddh218
Heyd, F., Ten Dam, G., and Moroy, T. (2006). Auxiliary splice factor U2AF26 and transcription factor Gfi1 cooperate directly in regulating CD45 alternative splicing. Nat. Immunol. 7, 859–867. doi: 10.1038/ni1361
Hock, H., Hamblen, M. J., Rooke, H. M., Schindler, J. W., Saleque, S., Fujiwara, Y., et al. (2004). Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature 431, 1002–1007. doi: 10.1038/nature02994
Hock, H., Hamblen, M. J., Rooke, H. M., Traver, D., Bronson, R. T., Cameron, S., et al. (2003). Intrinsic requirement for zinc finger transcription factor Gfi-1 in neutrophil differentiation. Immunity 18, 109–120. doi: 10.1016/s1074-7613(02)00501-0
Howell, E. D., and Speck, N. A. (2020). Forks in the road to the first hematopoietic stem cells. Cell Res. 30, 457–458. doi: 10.1038/s41422-020-0331-8
Hu, X., Huang, J., Feng, L., Fukudome, S., Hamajima, Y., and Lin, J. (2010). Sonic hedgehog (SHH) promotes the differentiation of mouse cochlear neural progenitors via the Math1-Brn3.1 signaling pathway in vitro. J. Neurosci. Res. 88, 927–935. doi: 10.1002/jnr.22286
Huang, Y. H., Klingbeil, O., He, X. Y., Wu, X. S., Arun, G., Lu, B., et al. (2018). POU2F3 is a master regulator of a tuft cell-like variant of small cell lung cancer. Genes Dev. 32, 915–928. doi: 10.1101/gad.314815.118
Huang, D. Y., Kuo, Y. Y., and Chang, Z. F. (2005). GATA-1 mediates auto-regulation of Gfi-1B transcription in K562 cells. Nucleic Acids Res. 33, 5331–5342. doi: 10.1093/nar/gki838
Isern, J., He, Z., Fraser, S. T., Nowotschin, S., Ferrer-Vaquer, A., Moore, R., et al. (2011). Single-lineage transcriptome analysis reveals key regulatory pathways in primitive erythroid progenitors in the mouse embryo. Blood 117, 4924–4934. doi: 10.1182/blood-2010-10-313676
Katoh, M., and Katoh, M. (2003). Identification and characterization of human SNAIL3 (SNAI3) gene in silico. Int. J. Mol. Med. 11, 383–388. doi: 10.3892/ijmm.11.3.383
Khandanpour, C., Phelan, J. D., Vassen, L., Schutte, J., Chen, R., Horman, S. R., et al. (2013). Growth factor independence 1 antagonizes a p53-induced DNA damage response pathway in lymphoblastic leukemia. Cancer Cell 23, 200–214. doi: 10.1016/j.ccr.2013.01.011
Kitamura, K., Okuno, Y., Yoshida, K., Sanada, M., Shiraishi, Y., Muramatsu, H., et al. (2016). Functional characterization of a novel GFI1B mutation causing congenital macrothrombocytopenia. J. Thromb. Haemost. 14, 1462–1469. doi: 10.1111/jth.13350
Lancrin, C., Mazan, M., Stefanska, M., Patel, R., Lichtinger, M., Costa, G., et al. (2012). GFI1 and GFI1B control the loss of endothelial identity of hemogenic endothelium during hematopoietic commitment. Blood 120, 314–322. doi: 10.1182/blood-2011-10-386094
Laurent, B., Randrianarison-Huetz, V., Frisan, E., Andrieu-Soler, C., Soler, E., Fontenay, M., et al. (2012). A short Gfi-1B isoform controls erythroid differentiation by recruiting the LSD1-CoREST complex through the dimethylation of its SNAG domain. J. Cell Sci. 125, 993–1002. doi: 10.1242/jcs.095877
Laurent, B., Randrianarison-Huetz, V., Kadri, Z., Romeo, P. H., Porteu, F., and Dumenil, D. (2009). Gfi-1B promoter remains associated with active chromatin marks throughout erythroid differentiation of human primary progenitor cells. Stem Cells 27, 2153–2162. doi: 10.1002/stem.151
Laurent, B., Randrianarison-Huetz, V., Marechal, V., Mayeux, P., Dusanter-Fourt, I., and Dumenil, D. (2010). High-mobility group protein HMGB2 regulates human erythroid differentiation through trans-activation of GFI1B transcription. Blood 115, 687–695. doi: 10.1182/blood-2009-06-230094
Laurenti, E., and Gottgens, B. (2018). From haematopoietic stem cells to complex differentiation landscapes. Nature 553, 418–426. doi: 10.1038/nature25022
Lee, S., Doddapaneni, K., Hogue, A., McGhee, L., Meyers, S., and Wu, Z. (2010). Solution structure of Gfi-1 zinc domain bound to consensus DNA. J. Mol. Biol. 397, 1055–1066. doi: 10.1016/j.jmb.2010.02.006
Lengerke, C., Grauer, M., Niebuhr, N. I., Riedt, T., Kanz, L., Park, I. H., et al. (2009). Hematopoietic development from human induced pluripotent stem cells. Ann. N. Y. Acad. Sci. 1176, 219–227. doi: 10.1111/j.1749-6632.2009.04606.x
Li, H., Ji, M., Klarmann, K. D., and Keller, J. R. (2010). Repression of Id2 expression by Gfi-1 is required for B-cell and myeloid development. Blood 116, 1060–1069. doi: 10.1182/blood-2009-11-255075
Li, J., Zhang, T., Ramakrishnan, A., Fritzsch, B., Xu, J., Wong, E. Y. M., et al. (2020). Dynamic changes in cis-regulatory occupancy by Six1 and its cooperative interactions with distinct cofactors drive lineage-specific gene expression programs during progressive differentiation of the auditory sensory epithelium. Nucleic Acids Res. 48, 2880–2896. doi: 10.1093/nar/gkaa012
Lin, Y., Wu, Y., Li, J., Dong, C., Ye, X., Chi, Y. I., et al. (2010). The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 29, 1803–1816. doi: 10.1038/emboj.2010.63
Lis, R., Karrasch, C. C., Poulos, M. G., Kunar, B., Redmond, D., Duran, J. G. B., et al. (2017). Conversion of adult endothelium to immunocompetent haematopoietic stem cells. Nature 545, 439–445. doi: 10.1038/nature22326
Liu, Q., Basu, S., Qiu, Y., Tang, F., and Dong, F. (2010). A role of Miz-1 in Gfi-1-mediated transcriptional repression of CDKN1A. Oncogene 29, 2843–2852. doi: 10.1038/nature22326
Liu, Q., and Dong, F. (2012). Gfi-1 inhibits the expression of eosinophil major basic protein (MBP) during G-CSF-induced neutrophilic differentiation. Int. J. Hematol. 95, 640–647. doi: 10.1007/s12185-012-1078-x
Liu, Y., Tavana, O., and Gu, W. (2019). p53 modifications: exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 11, 564–577. doi: 10.1093/jmcb/mjz060
Maiques-Diaz, A., Lynch, J. T., Spencer, G. J., and Somervaille, T. C. P. (2018a). LSD1 inhibitors disrupt the GFI1 transcription repressor complex. Mol. Cell. Oncol. 5:e1481813. doi: 10.1080/23723556.2018.1481813
Maiques-Diaz, A., Spencer, G. J., Lynch, J. T., Ciceri, F., Williams, E. L., Amaral, F. M. R., et al. (2018b). Enhancer activation by pharmacologic displacement of LSD1 from GFI1 induces differentiation in acute myeloid leukemia. Cell Rep. 22, 3641–3659. doi: 10.1016/j.celrep.2018.03.012
Manzanares, M., Locascio, A., and Nieto, M. A. (2001). The increasing complexity of the Snail gene superfamily in metazoan evolution. Trends Genet. 17, 178–181. doi: 10.1016/s0168-9525(01)02232-6
Marneth, A. E., Botezatu, L., Hones, J. M., Israel, J. C. L., Schutte, J., Vassen, L., et al. (2018). GFI1 is required for RUNX1/ETO positive acute myeloid leukemia. Haematologica 103, e395–e399. doi: 10.3324/haematol.2017.180844
Masuda, M., Dulon, D., Pak, K., Mullen, L. M., Li, Y., Erkman, L., et al. (2011). Regulation of POU4F3 gene expression in hair cells by 5' DNA in mice. Neuroscience 197, 48–64. doi: 10.1016/j.neuroscience.2011.09.033
McClellan, D., Casey, M. J., Bareyan, D., Lucente, H., Ours, C., Velinder, M., et al. (2019). Growth factor Independence 1B-mediated transcriptional repression and lineage allocation require lysine-specific demethylase 1-dependent recruitment of the BHC complex. Mol. Cell. Biol. 39, e00020–e00019. doi: 10.1128/MCB.00020-19
McGrath, K. E., Frame, J. M., Fegan, K. H., Bowen, J. R., Conway, S. J., Catherman, S. C., et al. (2015a). Distinct sources of hematopoietic progenitors emerge before HSCs and provide functional blood cells in the mammalian embryo. Cell Rep. 11, 1892–1904. doi: 10.1016/j.celrep.2015.05.036
McGrath, K. E., Frame, J. M., and Palis, J. (2015b). Early hematopoiesis and macrophage development. Semin. Immunol. 27, 379–387. doi: 10.1016/j.smim.2016.03.013
Mendrysa, S. M., Akagi, K., Roayaei, J., Lien, W. H., Copeland, N. G., Jenkins, N. A., et al. (2010). An integrated genetic-genomic approach for the identification of novel Cancer loci in mice sensitized to c-Myc-induced apoptosis. Genes Cancer 1, 465–479. doi: 10.1177/1947601910374875
Menendez, L., Trecek, T., Gopalakrishnan, S., Tao, L., Markowitz, A. L., Yu, H. V., et al. (2020). Generation of inner ear hair cells by direct lineage conversion of primary somatic cells. elife 9:e55249. doi: 10.7554/eLife.55249
Monteferrario, D., Bolar, N. A., Marneth, A. E., Hebeda, K. M., Bergevoet, S. M., Veenstra, H., et al. (2014). A dominant-negative GFI1B mutation in the gray platelet syndrome. N. Engl. J. Med. 370, 245–253. doi: 10.1056/NEJMoa1308130
Montoya-Durango, D. E., Velu, C. S., Kazanjian, A., Rojas, M. E., Jay, C. M., Longmore, G. D., et al. (2008). Ajuba functions as a histone deacetylase-dependent co-repressor for autoregulation of the growth factor-independent-1 transcription factor. J. Biol. Chem. 283, 32056–32065. doi: 10.1074/jbc.M802320200
Moore, C., Richens, J. L., Hough, Y., Ucanok, D., Malla, S., Sang, F., et al. (2018). Gfi1aa and Gfi1b set the pace for primitive erythroblast differentiation from hemangioblasts in the zebrafish embryo. Blood Adv. 2, 2589–2606. doi: 10.1182/bloodadvances.2018020156
Okuda, T., van Deursen, J., Hiebert, S. W., Grosveld, G., and Downing, J. R. (1996). AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 84, 321–330. doi: 10.1016/s0092-8674(00)80986-1
Osawa, M., Yamaguchi, T., Nakamura, Y., Kaneko, S., Onodera, M., Sawada, K., et al. (2002). Erythroid expansion mediated by the Gfi-1B zinc finger protein: role in normal hematopoiesis. Blood 100, 2769–2777. doi: 10.1182/blood-2002-01-0182
Palis, J., Robertson, S., Kennedy, M., Wall, C., and Keller, G. (1999). Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. Development 126, 5073–5084.
Pereira, C. F., Chang, B., Qiu, J., Niu, X., Papatsenko, D., Hendry, C. E., et al. (2013). Induction of a hemogenic program in mouse fibroblasts. Cell Stem Cell 13, 205–218. doi: 10.1016/j.stem.2013.05.024
Phelan, J. D., Saba, I., Zeng, H., Kosan, C., Messer, M. S., Olsson, H. A., et al. (2013). Growth factor independent-1 maintains Notch1-dependent transcriptional programming of lymphoid precursors. PLoS Genet. 9:e1003713. doi: 10.1371/journal.pgen.1003713
Philipp, F., Selich, A., Rothe, M., Hoffmann, D., Rittinghausen, S., Morgan, M. A., et al. (2018). Human teratoma-derived hematopoiesis is a highly polyclonal process supported by human umbilical vein endothelial cells. Stem Cell Rep. 11, 1051–1060. doi: 10.1016/j.stemcr.2018.09.010
Polfus, L. M., Khajuria, R. K., Schick, U. M., Pankratz, N., Pazoki, R., Brody, J. A., et al. (2016). Whole-exome sequencing identifies loci associated with blood cell traits and reveals a role for alternative GFI1B splice variants in human hematopoiesis. Am. J. Hum. Genet. 99, 481–488. doi: 10.1016/j.ajhg.2016.06.016
Porcheri, C., Golan, O., Calero-Nieto, F. J., Thambyrajah, R., Ruiz-Herguido, C., Wang, X., et al. (2020). Notch ligand Dll4 impairs cell recruitment to aortic clusters and limits blood stem cell generation. EMBO J. 39:e104270. doi: 10.15252/embj.2019104270
Psaila, B., Barkas, N., Iskander, D., Roy, A., Anderson, S., Ashley, N., et al. (2016). Single-cell profiling of human megakaryocyte-erythroid progenitors identifies distinct megakaryocyte and erythroid differentiation pathways. Genome Biol. 17:83. doi: 10.1186/s13059-016-0939-7
Qu, X., Nyeng, P., Xiao, F., Dorantes, J., and Jensen, J. (2015). Growth factor independence-1 (Gfi1) is required for pancreatic acinar unit formation and centroacinar cell differentiation. Cell. Mol. Gastroenterol. Hepatol. 1, 233.e1–247.e1. doi: 10.1016/j.jcmgh.2014.12.004
Rabbolini, D. J., Morel-Kopp, M. C., Chen, Q., Gabrielli, S., Dunlop, L. C., Chew, L. P., et al. (2017). Thrombocytopenia and CD34 expression is decoupled from alpha-granule deficiency with mutation of the first growth factor-independent 1B zinc finger. J. Thromb. Haemost. 15, 2245–2258. doi: 10.1111/jth.13843
Randrianarison-Huetz, V., Laurent, B., Bardet, V., Blobe, G. C., Huetz, F., and Dumenil, D. (2010). Gfi-1B controls human erythroid and megakaryocytic differentiation by regulating TGF-beta signaling at the bipotent erythro-megakaryocytic progenitor stage. Blood 115, 2784–2795. doi: 10.1182/blood-2009-09-241752
Riddell, J., Gazit, R., Garrison, B. S., Guo, G., Saadatpour, A., Mandal, P. K., et al. (2014). Reprogramming committed murine blood cells to induced hematopoietic stem cells with defined factors. Cell 157, 549–564. doi: 10.1016/j.cell.2014.04.006
Rodel, B., Tavassoli, K., Karsunky, H., Schmidt, T., Bachmann, M., Schaper, F., et al. (2000). The zinc finger protein Gfi-1 can enhance STAT3 signaling by interacting with the STAT3 inhibitor PIAS3. EMBO J. 19, 5845–5855. doi: 10.1093/emboj/19.21.5845
Rodel, B., Wagner, T., Zornig, M., Niessing, J., and Moroy, T. (1998). The human homologue (GFI1B) of the chicken GFI gene maps to chromosome 9q34.13-A locus frequently altered in hematopoietic diseases. Genomics 54, 580–582. doi: 10.1006/geno.1998.5601
Rodriguez, P., Bonte, E., Krijgsveld, J., Kolodziej, K. E., Guyot, B., Heck, A. J., et al. (2005). GATA-1 forms distinct activating and repressive complexes in erythroid cells. EMBO J. 24, 2354–2366. doi: 10.1038/sj.emboj.7600702
Ruiz, J. P., Chen, G., Haro Mora, J. J., Keyvanfar, K., Liu, C., Zou, J., et al. (2019). Robust generation of erythroid and multilineage hematopoietic progenitors from human iPSCs using a scalable monolayer culture system. Stem Cell Res. 41:101600. doi: 10.1016/j.scr.2019.101600
Saleque, S., Cameron, S., and Orkin, S. H. (2002). The zinc-finger proto-oncogene Gfi-1b is essential for development of the erythroid and megakaryocytic lineages. Genes Dev. 16, 301–306. doi: 10.1101/gad.959102
Saleque, S., Kim, J., Rooke, H. M., and Orkin, S. H. (2007). Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol. Cell 27, 562–572. doi: 10.1016/j.molcel.2007.06.039
Sandler, V. M., Lis, R., Liu, Y., Kedem, A., James, D., Elemento, O., et al. (2014). Reprogramming human endothelial cells to haematopoietic cells requires vascular induction. Nature 511, 312–318. doi: 10.1038/nature13547
Sanjuan-Pla, A., Macaulay, I. C., Jensen, C. T., Woll, P. S., Luis, T. C., Mead, A., et al. (2013). Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature 502, 232–236. doi: 10.1038/nature12495
Schulz, D., Vassen, L., Chow, K. T., McWhirter, S. M., Amin, R. H., Moroy, T., et al. (2012). Gfi1b negatively regulates rag expression directly and via the repression of FoxO1. J. Exp. Med. 209, 187–199. doi: 10.1038/nature12495
Schulze, H., Schlagenhauf, A., Manukjan, G., Beham-Schmid, C., Andres, O., Klopocki, E., et al. (2017). Recessive grey platelet-like syndrome with unaffected erythropoiesis in the absence of the splice isoform GFI1B-p37. Haematologica 102, e375–e378. doi: 10.3324/haematol.2017.167957
Shan, W., Wang, B., Xu, Y., Li, X., Li, X., Wang, H., et al. (2020). Generation of hematopoietic cells from mouse pluripotent stem cells in a 3D culture system of self-assembling peptide hydrogel. J. Cell. Physiol. 235, 2080–2090. doi: 10.1002/jcp.29110
Shin, J. Y., Hu, W., Naramura, M., and Park, C. Y. (2014). High c-Kit expression identifies hematopoietic stem cells with impaired self-renewal and megakaryocytic bias. J. Exp. Med. 211, 217–231. doi: 10.1084/jem.20131128
Shooshtarizadeh, P., Helness, A., Vadnais, C., Brouwer, N., Beauchemin, H., Chen, R., et al. (2019). Gfi1b regulates the level of Wnt/beta-catenin signaling in hematopoietic stem cells and megakaryocytes. Nat. Commun. 10:1270. doi: 10.1038/s41467-019-09273-z
Shroyer, N. F., Wallis, D., Venken, K. J., Bellen, H. J., and Zoghbi, H. Y. (2005). Gfi1 functions downstream of Math1 to control intestinal secretory cell subtype allocation and differentiation. Genes Dev. 19, 2412–2417. doi: 10.1101/gad.1353905
Spooner, C. J., Cheng, J. X., Pujadas, E., Laslo, P., and Singh, H. (2009). A recurrent network involving the transcription factors PU.1 and Gfi1 orchestrates innate and adaptive immune cell fates. Immunity 31, 576–586. doi: 10.1016/j.immuni.2009.07.011
Stevenson, W. S., Morel-Kopp, M. C., Chen, Q., Liang, H. P., Bromhead, C. J., Wright, S., et al. (2013). GFI1B mutation causes a bleeding disorder with abnormal platelet function. J. Thromb. Haemost. 11, 2039–2047. doi: 10.1111/jth.12368
Sugimura, R., Jha, D. K., Han, A., Soria-Valles, C., da Rocha, E. L., Lu, Y. F., et al. (2017). Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature 545, 432–438. doi: 10.1038/nature22370
Suzuki, N., Yamazaki, S., Yamaguchi, T., Okabe, M., Masaki, H., Takaki, S., et al. (2013). Generation of engraftable hematopoietic stem cells from induced pluripotent stem cells by way of teratoma formation. Mol. Ther. 21, 1424–1431. doi: 10.1038/mt.2013.71
Szabo, E., Rampalli, S., Risueno, R. M., Schnerch, A., Mitchell, R., Fiebig-Comyn, A., et al. (2010). Direct conversion of human fibroblasts to multilineage blood progenitors. Nature 468, 521–526. doi: 10.1038/nature09591
Tan, Y. T., Ye, L., Xie, F., Beyer, A. I., Muench, M. O., Wang, J., et al. (2018). Respecifying human iPSC-derived blood cells into highly engraftable hematopoietic stem and progenitor cells with a single factor. Proc. Natl. Acad. Sci. U. S. A. 115, 2180–2185. doi: 10.1073/pnas.1718446115
Thambyrajah, R., Mazan, M., Patel, R., Moignard, V., Stefanska, M., Marinopoulou, E., et al. (2016). GFI1 proteins orchestrate the emergence of haematopoietic stem cells through recruitment of LSD1. Nat. Cell Biol. 18, 21–32. doi: 10.1038/ncb3276
Thivakaran, A., Botezatu, L., Hones, J. M., Schutte, J., Vassen, L., Al-Matary, Y. S., et al. (2018). Gfi1b: a key player in the genesis and maintenance of acute myeloid leukemia and myelodysplastic syndrome. Haematologica 103, 614–625. doi: 10.3324/haematol.2017.167288
Tober, J., Koniski, A., McGrath, K. E., Vemishetti, R., Emerson, R., de Mesy-Bentley, K. K., et al. (2007). The megakaryocyte lineage originates from hemangioblast precursors and is an integral component both of primitive and of definitive hematopoiesis. Blood 109, 1433–1441. doi: 10.1182/blood-2006-06-031898
Tong, B., Grimes, H. L., Yang, T. Y., Bear, S. E., Qin, Z., Du, K., et al. (1998). The Gfi-1B proto-oncoprotein represses p21WAF1 and inhibits myeloid cell differentiation. Mol. Cell. Biol. 18, 2462–2473. doi: 10.1128/mcb.18.5.2462
Tsuda, H., Jafar-Nejad, H., Patel, A. J., Sun, Y., Chen, H. K., Rose, M. F., et al. (2005). The AXH domain of Ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/senseless proteins. Cell 122, 633–644. doi: 10.1016/j.cell.2005.06.012
Tsukada, M., Ota, Y., Wilkinson, A. C., Becker, H. J., Osato, M., Nakauchi, H., et al. (2017). In vivo generation of engraftable murine hematopoietic stem cells by Gfi1b, c-Fos, and Gata2 overexpression within teratoma. Stem Cell Rep. 9, 1024–1033. doi: 10.1016/j.stemcr.2017.08.010
Vadnais, C., Chen, R., Fraszczak, J., Hamard, P. J., Manfredi, J. J., and Moroy, T. (2019). A novel regulatory circuit between p53 and GFI1 controls induction of apoptosis in T cells. Sci. Rep. 9:6304. doi: 10.1038/s41598-019-41684-2
Vadnais, C., Chen, R., Fraszczak, J., Yu, Z., Boulais, J., Pinder, J., et al. (2018). GFI1 facilitates efficient DNA repair by regulating PRMT1 dependent methylation of MRE11 and 53BP1. Nat. Commun. 9:1418. doi: 10.1038/s41467-018-03817-5
van Oorschot, R., Hansen, M., Koornneef, J. M., Marneth, A. E., Bergevoet, S. M., van Bergen, M., et al. (2019). Molecular mechanisms of bleeding disorder-associated GFI1BQ287* mutation and its affected pathways in megakaryocytes and platelets. Haematologica 104, 1460–1472. doi: 10.3324/haematol.2018.194555
Vassen, L., Beauchemin, H., Lemsaddek, W., Krongold, J., Trudel, M., and Moroy, T. (2014). Growth factor independence 1b (gfi1b) is important for the maturation of erythroid cells and the regulation of embryonic globin expression. PLoS One 9:e96636. doi: 10.1371/journal.pone.0096636
Vassen, L., Fiolka, K., Mahlmann, S., and Moroy, T. (2005). Direct transcriptional repression of the genes encoding the zinc-finger proteins Gfi1b and Gfi1 by Gfi1b. Nucleic Acids Res. 33, 987–998. doi: 10.1093/nar/gki243
Vassen, L., Fiolka, K., and Moroy, T. (2006). Gfi1b alters histone methylation at target gene promoters and sites of gamma-satellite containing heterochromatin. EMBO J. 25, 2409–2419. doi: 10.1038/sj.emboj.7601124
Vassen, L., Khandanpour, C., Ebeling, P., van Der Reijden, B. A., Jansen, J. H., Mahlmann, S., et al. (2009). Growth factor independent 1b (Gfi1b) and a new splice variant of Gfi1b are highly expressed in patients with acute and chronic leukemia. Int. J. Hematol. 89, 422–430. doi: 10.1007/s12185-009-0286-5
Vassen, L., Okayama, T., and Moroy, T. (2007). Gfi1b:green fluorescent protein knock-in mice reveal a dynamic expression pattern of Gfi1b during hematopoiesis that is largely complementary to Gfi1. Blood 109, 2356–2364. doi: 10.1182/blood-2006-06-030031
Velinder, M., Singer, J., Bareyan, D., Meznarich, J., Tracy, C. M., Fulcher, J. M., et al. (2016). GFI1 functions in transcriptional control and cell fate determination require SNAG domain methylation to recruit LSD1. Biochem. J. 473, 3355–3369. doi: 10.1042/BCJ20160558
Velinder, M., Singer, J., Bareyan, D., Meznarich, J., Tracy, C. M., Fulcher, J. M., et al. (2017). GFI1 functions in transcriptional control and cell fate determination require SNAG domain methylation to recruit LSD1. Biochem. J. 474:2951. doi: 10.1042/BCJ-2016-0558_COR
Vinyard, M. E., Su, C., Siegenfeld, A. P., Waterbury, A. L., Freedy, A. M., Gosavi, P. M., et al. (2019). CRISPR-suppressor scanning reveals a nonenzymatic role of LSD1 in AML. Nat. Chem. Biol. 15, 529–539. doi: 10.1038/s41589-019-0263-0
Wallis, D., Hamblen, M., Zhou, Y., Venken, K. J., Schumacher, A., Grimes, H. L., et al. (2003). The zinc finger transcription factor Gfi1, implicated in lymphomagenesis, is required for inner ear hair cell differentiation and survival. Development 130, 221–232. doi: 10.1242/dev.00190
Wang, L., Menendez, P., Shojaei, F., Li, L., Mazurier, F., Dick, J. E., et al. (2005). Generation of hematopoietic repopulating cells from human embryonic stem cells independent of ectopic HOXB4 expression. J. Exp. Med. 201, 1603–1614. doi: 10.1084/jem.20041888
Wang, Y., Song, W., Wang, J., Wang, T., Xiong, X., Qi, Z., et al. (2020). Single-cell transcriptome analysis reveals differential nutrient absorption functions in human intestine. J. Exp. Med. 217:e20191130. doi: 10.1084/jem.20191130
Wilson, N. K., Timms, R. T., Kinston, S. J., Cheng, Y. H., Oram, S. H., Landry, J. R., et al. (2010). Gfi1 expression is controlled by five distinct regulatory regions spread over 100 kilobases, with Scl/Tal1, Gata2, PU.1, erg, Meis1, and Runx1 acting as upstream regulators in early hematopoietic cells. Mol. Cell. Biol. 30, 3853–3863. doi: 10.1128/MCB.00032-10
Wong, P. M., Chung, S. W., Chui, D. H., and Eaves, C. J. (1986). Properties of the earliest clonogenic hemopoietic precursors to appear in the developing murine yolk sac. Proc. Natl. Acad. Sci. U. S. A. 83, 3851–3854. doi: 10.1073/pnas.83.11.3851
Xu, M. J., Matsuoka, S., Yang, F. C., Ebihara, Y., Manabe, A., Tanaka, R., et al. (2001). Evidence for the presence of murine primitive megakaryocytopoiesis in the early yolk sac. Blood 97, 2016–2022. doi: 10.1182/blood.v97.7.2016
Yang, J., Zhao, S., and Ma, D. (2019). Biological characteristics and regulation of early megakaryocytopoiesis. Stem Cell Rev. Rep. 15, 652–663. doi: 10.1007/s12015-019-09905-3
Zeng, Y., He, J., Bai, Z., Li, Z., Gong, Y., Liu, C., et al. (2019). Tracing the first hematopoietic stem cell generation in human embryo by single-cell RNA sequencing. Cell Res. 29, 881–894. doi: 10.1038/s41422-019-0228-6
Zeng, H., Yucel, R., Kosan, C., Klein-Hitpass, L., and Moroy, T. (2004). Transcription factor Gfi1 regulates self-renewal and engraftment of hematopoietic stem cells. EMBO J. 23, 4116–4125. doi: 10.1038/sj.emboj.7600419
Zhang, Y., Hu, N., and Dong, F. (2019). Gfi1-mediated repression of c-Fos, Egr-1 and Egr-2, and inhibition of ERK1/2 signaling contribute to the role of Gfi1 in granulopoiesis. Sci. Rep. 9:737. doi: 10.1038/s41598-018-37402-z
Zhong, C., Fu, Y., Pan, W., Yu, J., and Wang, J. (2019). Atoh1 and other related key regulators in the development of auditory sensory epithelium in the mammalian inner ear: function and interplay. Dev. Biol. 446, 133–141. doi: 10.1016/j.ydbio.2018.12.025
Keywords: GFI1, GFI1B, hemogenic epithelium, hematopoietic stem cells, transdifferentiation, hair cells
Citation: Beauchemin H and Möröy T (2020) Multifaceted Actions of GFI1 and GFI1B in Hematopoietic Stem Cell Self-Renewal and Lineage Commitment. Front. Genet. 11:591099. doi: 10.3389/fgene.2020.591099
Edited by:
Rachita Yadav, Massachusetts General Hospital and Harvard Medical School, United StatesReviewed by:
Nicola Wilson, University of Cambridge, United KingdomMaaike Van Bergen, Radboud University Nijmegen Medical Centre, Netherlands
Copyright © 2020 Beauchemin and Möröy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hugues Beauchemin, aHVndWVzLmJlYXVjaGVtaW5AaXJjbS5xYy5jYQ==; Tarik Möröy, dGFyaWsubW9yb3lAaXJjbS5xYy5jYQ==