 Jin Ok Yang1,2†
Jin Ok Yang1,2† Min-Hyuk Choi3,4†
Min-Hyuk Choi3,4† Ji-Yong Yoon3Jeong-Ju Lee3Sang Ook Nam5
Ji-Yong Yoon3Jeong-Ju Lee3Sang Ook Nam5 Soo Young Jun3Hyeok Hee Kwon6Sohyun Yun3Su-Jin Jeon3,4Iksu Byeon1Debasish Halder3Juhyun Kong5
Soo Young Jun3Hyeok Hee Kwon6Sohyun Yun3Su-Jin Jeon3,4Iksu Byeon1Debasish Halder3Juhyun Kong5 Byungwook Lee1
Byungwook Lee1 Jeehun Lee7
Jeehun Lee7 Joon-Won Kang8*
Joon-Won Kang8* Nam-Soon Kim3,4*
Nam-Soon Kim3,4*- 1Korea BioInformation Center, Korea Research Institute of Bioscience and Biotechnology, Daejeon, South Korea
- 2Department of Bio and Brain Engineering, Korea Advanced Institute of Science and Technology, Daejeon, South Korea
- 3Rare-Disease Research Center, Korea Research Institute of Bioscience and Biotechnology, Daejeon, South Korea
- 4Department of Functional Genomics, Korea University of Science and Technology, Daejeon, South Korea
- 5Department of Pediatrics, Pusan National University Children’s Hospital, Pusan National University School of Medicine, Yangsan, South Korea
- 6Department of Medical Science and Anatomy, Chungnam National University, Daejeon, South Korea
- 7Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, South Korea
- 8Department of Pediatrics and Medical Science, Chungnam National University Hospital, College of Medicine, Chungnam National University, Daejeon, South Korea
Lennox-Gastaut syndrome (LGS) is a severe type of childhood-onset epilepsy characterized by multiple types of seizures, specific discharges on electroencephalography, and intellectual disability. Most patients with LGS do not respond well to drug treatment and show poor long-term prognosis. Approximately 30% of patients without brain abnormalities have unidentifiable causes. Therefore, accurate diagnosis and treatment of LGS remain challenging. To identify causative mutations of LGS, we analyzed the whole-exome sequencing data of 17 unrelated Korean families, including patients with LGS and LGS-like epilepsy without brain abnormalities, using the Genome Analysis Toolkit. We identified 14 mutations in 14 genes as causes of LGS or LGS-like epilepsy. 64 percent of the identified genes were reported as LGS or epilepsy-related genes. Many of these variations were novel and considered as pathogenic or likely pathogenic. Network analysis was performed to classify the identified genes into two network clusters: neuronal signal transmission or neuronal development. Additionally, knockdown of two candidate genes with insufficient evidence of neuronal functions, SLC25A39 and TBC1D8, decreased neurite outgrowth and the expression level of MAP2, a neuronal marker. These results expand the spectrum of genetic variations and may aid the diagnosis and management of individuals with LGS.
Introduction
Lennox-Gastaut syndrome (LGS) is a severe form of childhood-onset epilepsy with a heterogeneous etiology, and epileptiform abnormalities may contribute to progressive dysfunction (Lund et al., 2014; Asadi-Pooya, 2018). The primary features of LGS are multiple types of seizures (generalized tonic, atonic, and atypical absence), generalized slow spike-and-wave or generalized paroxysmal fast activity discharges on electroencephalography, and intellectual disabilities (Camfield, 2011; Asadi-Pooya, 2018). In approximately 70% of patients with LGS, this disease is caused by brain damage, infection, and brain malformation. Thirty percent of LGS patients do not present abnormalities in brain imaging, and thus the cause of their condition is unclear (Asadi-Pooya, 2018). Functional magnetic resonance imaging studies showed that abnormal network connectivity in subcortical structures causes LGS (Pedersen et al., 2015). Hence, several researchers have focused on screening genetic risk factors from patients with LGS without abnormalities by next-generation sequencing.
Recently, several causative genetic variations related to LGS and epilepsy, which play important roles in the development of these syndromes, have been detected by whole-exome sequencing (WES; Allen et al., 2013, 2015; Lund et al., 2013; Terrone et al., 2014; Zerem et al., 2016; Wang et al., 2017; Asadi-Pooya, 2018; Dunn et al., 2018; Tumiene et al., 2018). Nevertheless, effective treatments and an understanding of the genetic basis of LGS are lacking because the biological mechanisms of LGS are not well-understood. The seizure frequency is either controlled by administering anti-epileptic medicine to patients or stimulating the vagus nerve. Therefore, comprehensive information from LGS-related genetic variations and networks is required to identify markers influencing LGS.
In this study, we examined novel candidate genetic variations and networks associated with LGS without brain abnormalities as genetic markers. First, we collected 58 WES datasets from 17 Korean families with a clinical history of LGS or LGS-like epilepsy without brain abnormalities. We investigated the causative variations in each family and relationships among genes and these variations, and then estimated the genetic risk factors for LGS. We found that these variations contained 14 mutations, including de novo, autosomal recessive (AR), and X-linked mutations. Several genes showed novel variations and were found to be associated with LGS or epilepsy. Depletion of two candidate genes with insufficient evidence of neuronal functions decreased neurite outgrowth in the SH-SY5Y cell line. This finding provides a more informative resource for LGS-related genetic variations and may contribute to the diagnosis of and therapeutic platform development for LGS and LGS-like epilepsy.
Materials and Methods
Clinical Specimens of Korean Patients With LGS
We collected 58 samples from 17 affected individuals and 41 unaffected individuals. These subjects were from 17 unrelated Korean families containing either 10 patients with LGS or 7 patients with LGS-like epilepsy (Figure 2). Patients with an incomplete phenotype of LGS were categorized as having LGS-like epilepsy. The magnetic resonance imaging results were normal (15 patients) or showed non-epileptogenic abnormalities (2 patients), including arachnoid cyst, posterior fossa, and brain atrophy. The mean age at seizure onset was 3.3 years (range 0.16–12 years). All patients diagnosed with LGS or LGS-like epilepsy were older than 3 years. Affected individuals presented with various seizure types: tonic (16/17), atonic (6/17), generalized tonic-clonic (4/17), myoclonic (4/17), and atypical absence (8/17; Table 1). All patients underwent neurologic and genetic evaluations based on the clinical criteria of LGS by an experienced neurologist (Camfield, 2011). LGS is characterized by: (1) a clinical triad of various types of generalized seizures, including generalized tonic, atonic, myoclonic, atypical absence seizures, and epileptic spasms; (2) generalized slow spikes and waves and/or generalized paroxysmal rapid activity for electroencephalography; and (3) progressive developmental regression after seizure onset. LGS-like epilepsy is broadly defined to result in at least two types of generalized seizures, including tonic seizures, or a combination of atonic and atypical absence seizures, learning disabilities, resistance to treatment, and bilateral synchronous epileptic discharges. Ethical approval for the study was obtained from the Institutional Review Board and Ethics Committee at the Chungnam National University Hospitals and Korea Research Institute of Bioscience and Biotechnology. Written informed consent was obtained from all participants or their legal representatives. Available clinical information on these patients is shown in Table 1.
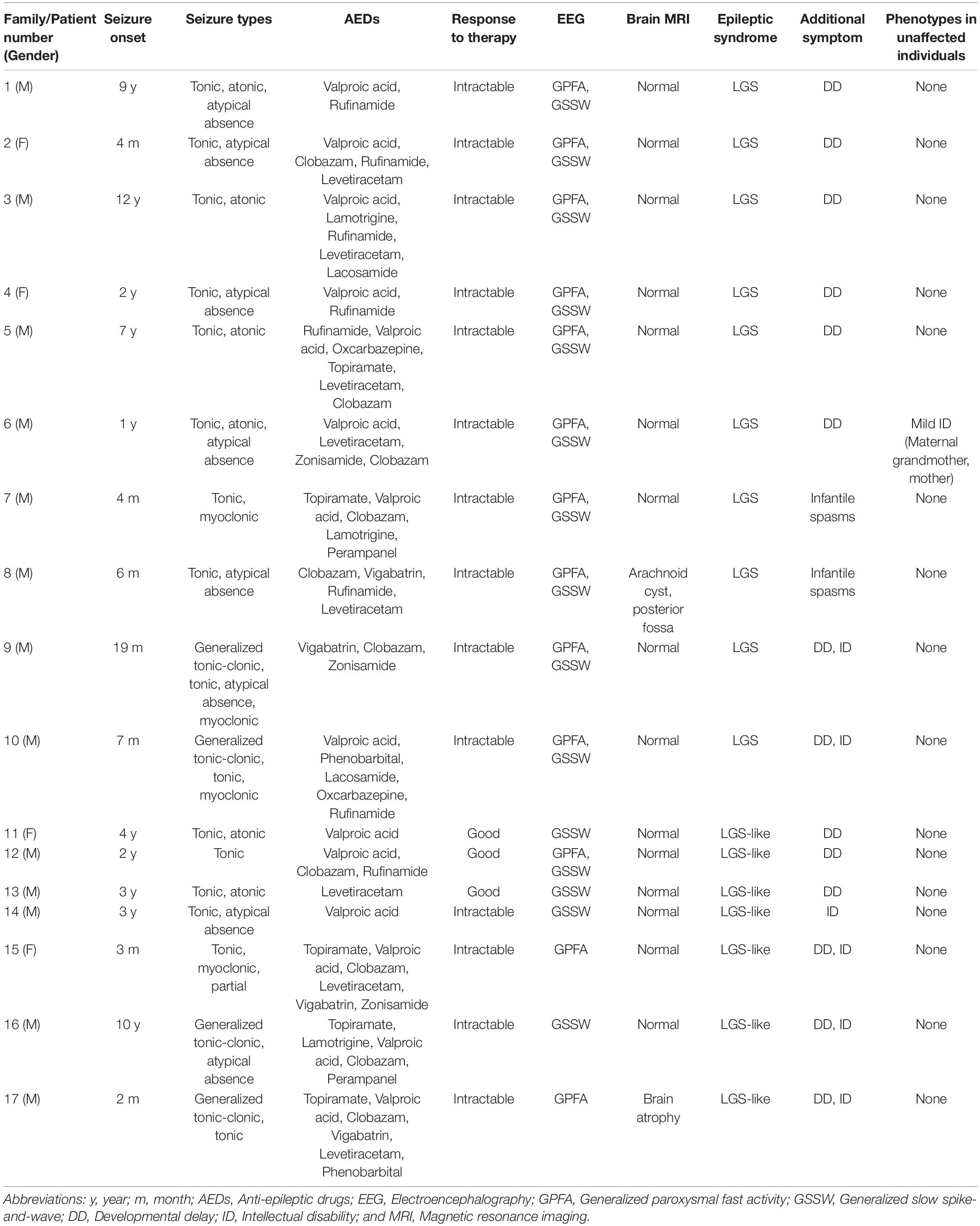
Table 1. Clinical features of patients with LGS and LGS-like epilepsy.
Analysis of Genetic Variations and Annotation
We obtained high-throughput WES data from all participants by HiSeq 2500 (Illumina, San Diego, CA, United States). All sequencing reads were mapped to the human reference genome GRCh38/hg38 by using Burrows-Wheeler Aligner software (v0.7.17). Variant calling and functional annotation were performed using the Genome Analysis Toolkit (GATK4, Broad Institute, MA, United States) and ANNOVAR (Version 2018Apr16), respectively, (McKenna et al., 2010; Wang et al., 2010). For the rare-disease study, mutations with less than 5% minor allele frequency in our data were selected using genetic variation data in the Single Nucleotide Polymorphism Database (dbSNP, https://www.ncbi.nlm.nih.gov/snp/), NHLBI Grand Opportunity Exome Sequencing Project database (ESP6500, https://evs.gs.washington.edu/EVS/), and 1000 Genomes Project1. We also examined the allele frequency of candidate genetic variations using the Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/) and Genome Aggregation Database (gnomAD, https://gnomad.broadinstitute.org/), and ethnic mutations were removed by referencing 1,055 healthy Korean WES data (KOVA v1, http://kobic.re.kr/kova/). To validate these genetic variants, we carried out Sanger sequencing using an ABI3730XL DNA sequencer (Applied Biosystems, Foster City, CA, United States). We also identified copy number variations (CNVs) by CODEX, which is based on a multi-sample normalization model (Jiang et al., 2015). It includes terms that specifically remove biases due to GC content, exon length, and targeting and amplification efficiency. We normalized the WES read count data of each patient with LGS and LGS-like epilepsy based on the read depth of unaffected individuals, after which we derived de novo CNVs. CNVs were validated by array-based SNP analysis (Illumina Infinium Omni2.5-8 BeadChip; Supplementary Methods). The family dataset without candidate markers was excluded. The summarized workflow is shown in Figure 1.
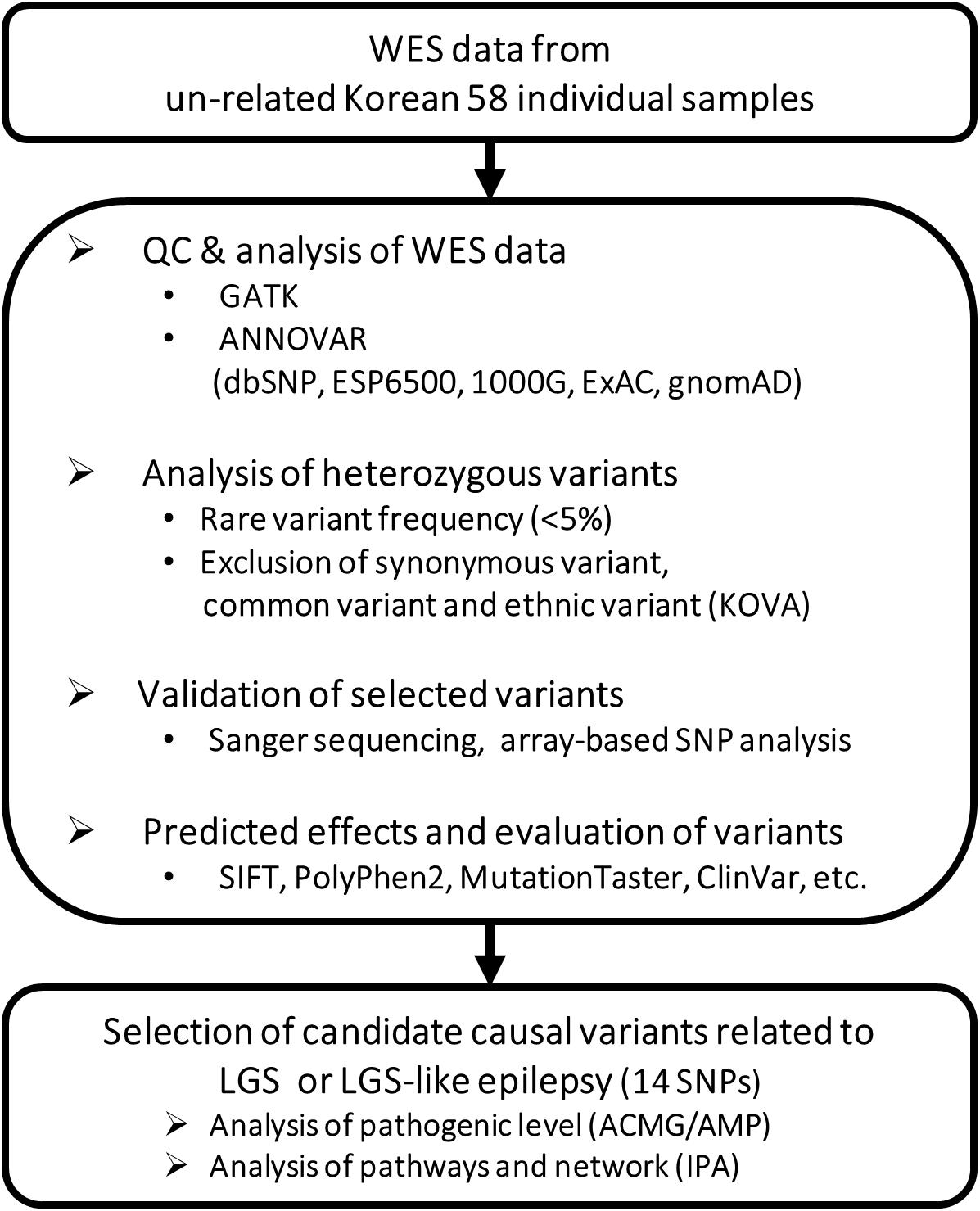
Figure 1. Workflow for identifying potential causative genetic variants related to LGS or LGS-like epilepsy.
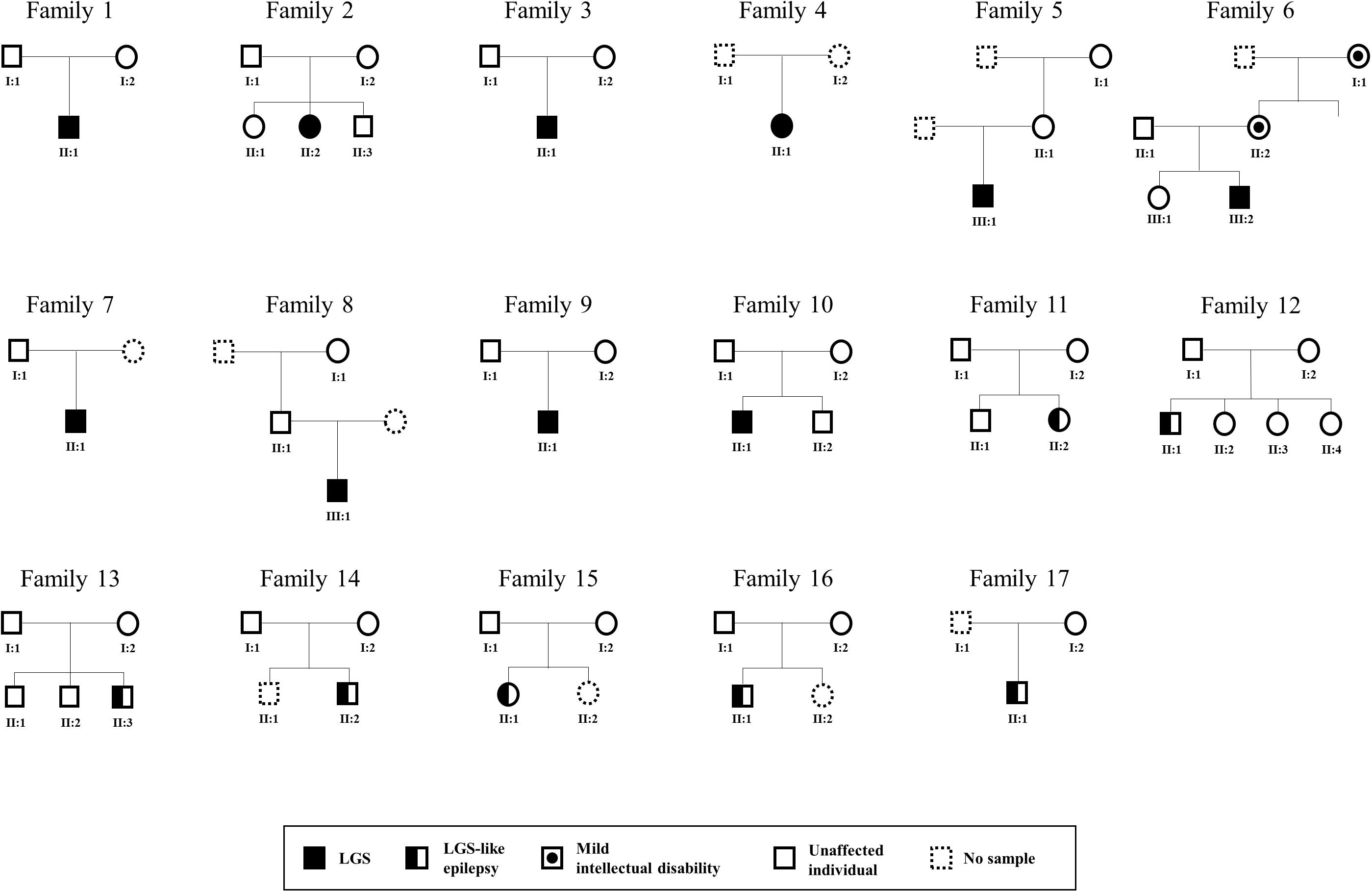
Figure 2. Pedigree of LGS or LGS-like epilepsy families. Males and females are represented as squares and circles, respectively. Patients with LGS are indicated as completely filled symbols, and those with LGS-like epilepsy are indicated as half-filled symbols. The circle with dots indicates females with mild intellectual disabilities.
Pathogenicity Prediction and Pathway Analysis
We investigated the pathogenicity of all mutations using the ClinVar database and prediction algorithms in ANNOVAR (PolyPhen2, SIFT, LRT, and MutationTaster; Chun and Fay, 2009; Adzhubei et al., 2010; Schwarz et al., 2010; Sim et al., 2012). Additionally, we evaluated pathogenicity using criteria described in the American College of Medical Genetics and Association for Molecular Pathology guidelines, after which we selected genes with damaging, possibly damaging, pathogenic, or likely pathogenic mutations as candidate genes of LGS and LGS-like epilepsy (Richards et al., 2015).
We performed Ingenuity Pathway Analysis (QIAGEN, Hilden, Germany) to explore the pathways and networks associated with selected candidate gene sets, and classified genes according to their functions by using Database for Annotation, Visualization and Integrated Discovery (DAVID, https://david.ncifcrf.gov).
Neurite Outgrowth Assay
Human SH-SY5Y neuroblastoma cells were transfected with target siRNAs for 24 h and then treated with retinoic acid for 48 h. Cells were then fixed in 4% paraformaldehyde for 1 h at room temperature (RT), permeabilized with 0.1% Triton X-100 in PBS for 15 min, and incubated in blocking reagent (5% normal fetal bovine serum in PBS) for 1 h. Cells were incubated with primary antibody against MAP2, a neuronal marker (Mouse Anti-MAP2, Abcam, Cambridge, United Kingdom), at 4°C overnight, followed by incubation with a secondary antibody (Alexa Fluor 488, Goat anti-mouse, Life Technologies, California, United States) at RT for 1 h. For nuclear counterstaining, the cells were incubated with DAPI solution (300 nM in PBS) for 5 min at RT and then observed with a fluorescence microscope (Eclipse Ti-S, Nikon, Tokyo, Japan). To measure neurite length in SH-SY5Y cells, neurite length was calculated as the longest neurite distance from the cell body (direct path to the soma) on each neuron showing MAP2 (green) using ImageJ software (10 calculated cells per group). The mRNA expression levels of target genes and MAP2 were analyzed by real-time PCR.
Results
Annotation of Genetic Variations as Candidates for LGS and LGS-Like Epilepsy
We identified 14 mutations as candidates for causing LGS and LGS-like epilepsy. Two mutations were in the splicing site and 12 mutations were in the coding region. Among the coding region mutations, there was one frameshift deletion, one non-frameshift insertion, and 10 single-nucleotide variations (Table 2 and Supplementary Figure 1).
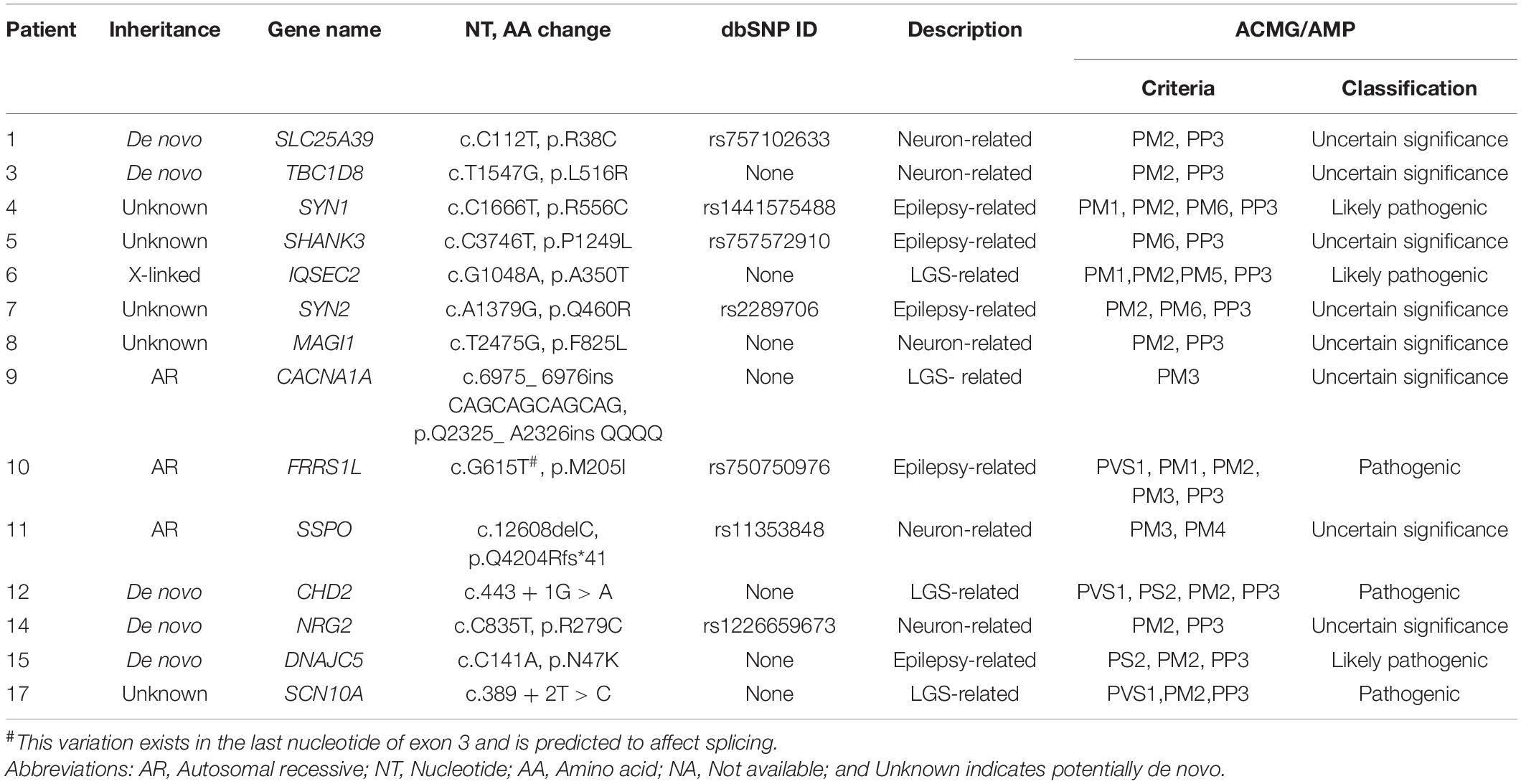
Table 2. Potential causative genetic variations in patients with LGS or LGS-like epilepsy.
CACNA1A, CHD2, IQSEC2, and SCN10A were LGS-related genes. DNAJC5, FRRS1L, SHANK3, SYN1, and SYN2 were epilepsy-related genes. MAGI1, NRG2, SLC25A39, SSPO, and TBC1D8 were functionally related to neurons or brain-expressed ion channels (Figure 3A and Table 2). Except for mutations in FRRS1L, NRG2, SHANK3, SLC25A39, SSPO, SYN1, and SYN2, the remaining mutations were novel.
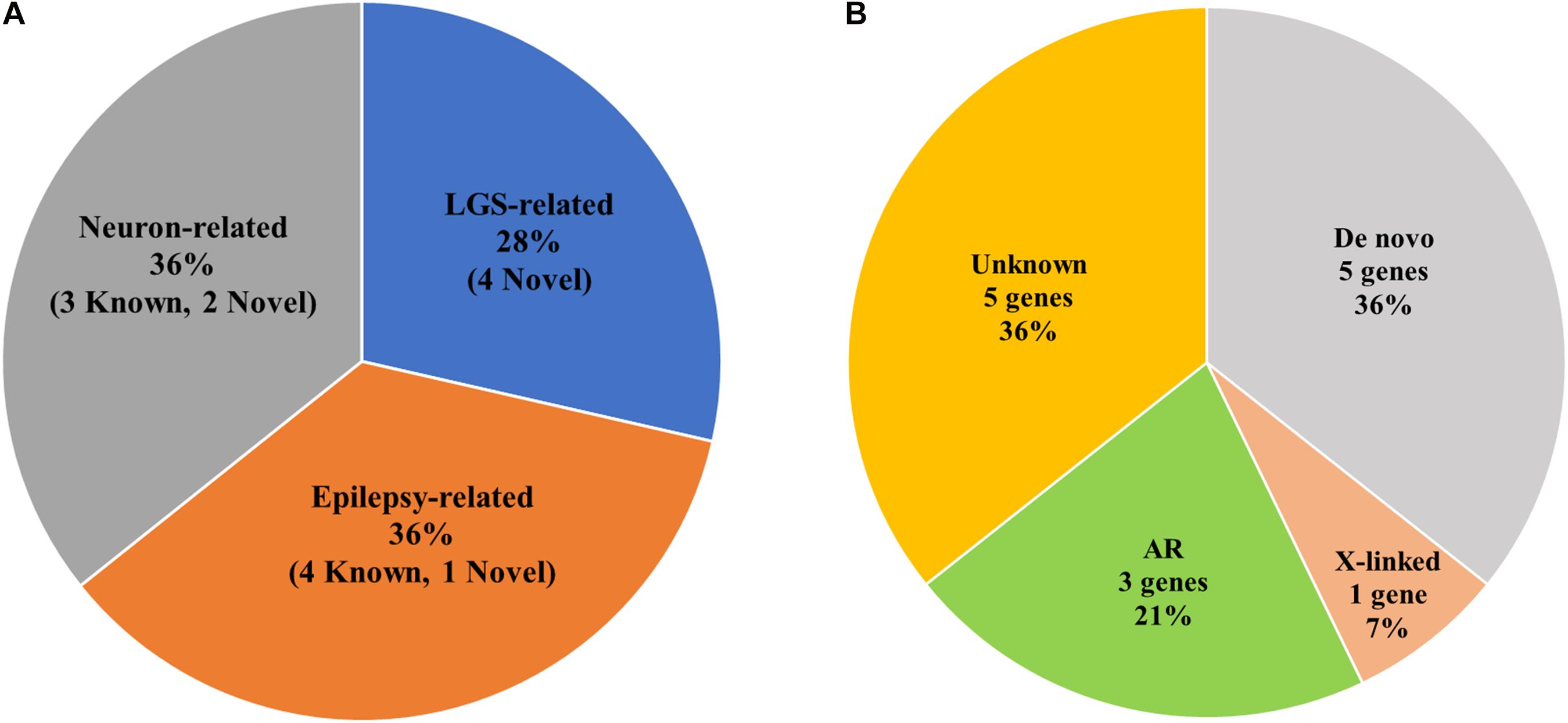
Figure 3. Statistics of genetic variations in candidate genes of LGS or LGS-like epilepsy. (A) Pie chart of the frequency of LGS, epilepsy, or neuron-related genes. () indicates the number of genetic variations in candidate genes. (B) Ratio of Mendelian inheritance patterns of candidate genes.
Mendelian Inheritance Pattern
Among the 17 families, one family contained members with mild intellectual disability, whereas disorders were not present in members of the other 16 families (Figure 2 and Table 1). We identified three AR (21%), one X-linked inherited (7%), five de novo (36%), and five potentially de novo mutations (36%; Figure 3B). Remarkably, as shown in Table 2, mutations in CACNA1A, FRRS1L, and SSPO were detected as AR in families 9, 10, and 11, in which unaffected parents possessed heterozygous mutations and the patients had homozygous mutations. The IQSEC2 mutation was inherited as X-linked in family 6, in which the maternal grandmother and mother of the patient had mild intellectual disability with heterozygous mutations, whereas the male patient had hemizygous mutations. Genes such as CHD2, DNAJC5, NRG2, SLC25A39, and TBC1D8 showed de novo mutations. MAGI1, SCN10A, SHANK3, SYN1, and SYN2 were considered as unknown, potentially de novo mutations, as they were found in patients from single-parent or adoptive families (Figure 3B).
Pathogenicity Validation for Genetic Variations
Mutations in FRRS1L, NRG2, SHANK3, SLC25A39, SSPO, SYN1, and SYN2 have been reported previously but their functional significance was not determined by ClinVar. In addition, other mutations were not reported in databases such as dbSNP, ClinVar, ExAC, ESP6500, gnomAD, and the 1000 Genomes Project. According to PolyPhen2, SIFT, LRT, and MutationTaster, damaging mutations were found in DNAJC5, FRRS1L, and IQSEC2 in all pathogenicity prediction results, with the remaining mutations classified as possibly damaging or of unknown pathogenicity. According to the American College of Medical Genetics/Association for Molecular Pathology guidelines, the CHD2, DNAJC5, FRRS1L, IQSEC2, SCN10A, and SYN1 mutations were sorted into pathogenic or likely pathogenic groups. Particularly, the FRRS1L mutation (c.615G > T, p.205M > I) at the last nucleotide of exon 3 may affect splicing. FRRS1L is related to epileptic encephalopathy, and its pathogenic variants have been reported previously (Madeo et al., 2016; Han et al., 2017). Based on these findings, the FRRS1L mutation was assigned to PVS1. The detailed criteria and classification results are presented in Table 2 and Supplementary Table 1.
Systematic Analysis of Candidate Genes
We examined gene interaction networks and functional annotations to comprehensively understand the relationships among the candidate genes of LGS and LGS-like epilepsy. Based on gene ontology annotations by DAVID, these gene sets were enriched in biological functions including synaptic transmission, ion transport, MAPK cascade, and transcription (Table 3). Ingenuity Pathway Analysis revealed that the candidate genes were divided into two clusters. The first cluster comprised 11 genes (CACNA1A, DNAJC5, FRRS1L, IQSEC2, MAGI1, NRG2, SHANK3, SLC25A39, SSPO, SYN1, and SYN2), which functionally belong to synaptogenesis signaling, calcium signaling, and AMPA signaling. This gene set was associated with neurotransmission (Figure 4A). Another cluster, consisting of three genes (CHD2, SCN10A, and TBC1D8), was associated with cellular assembly and organization, nervous system development and function, and ion channels, as well as neurological disease, skeletal and muscular disorders, and behavioral disorders (Figure 4B). These data indicate that the candidate genes play important roles in LGS development.
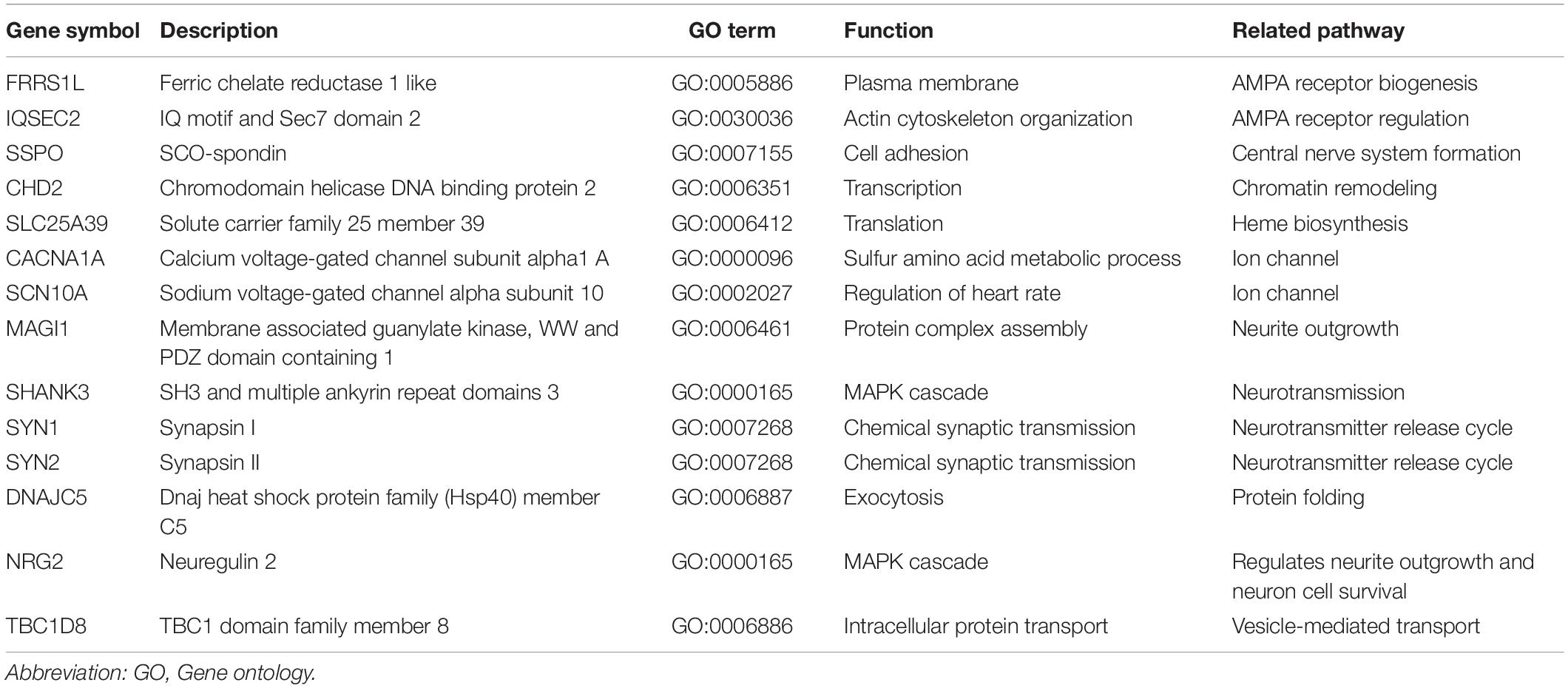
Table 3. Gene ontology results of candidate genes of LGS or LGS-like epilepsy.
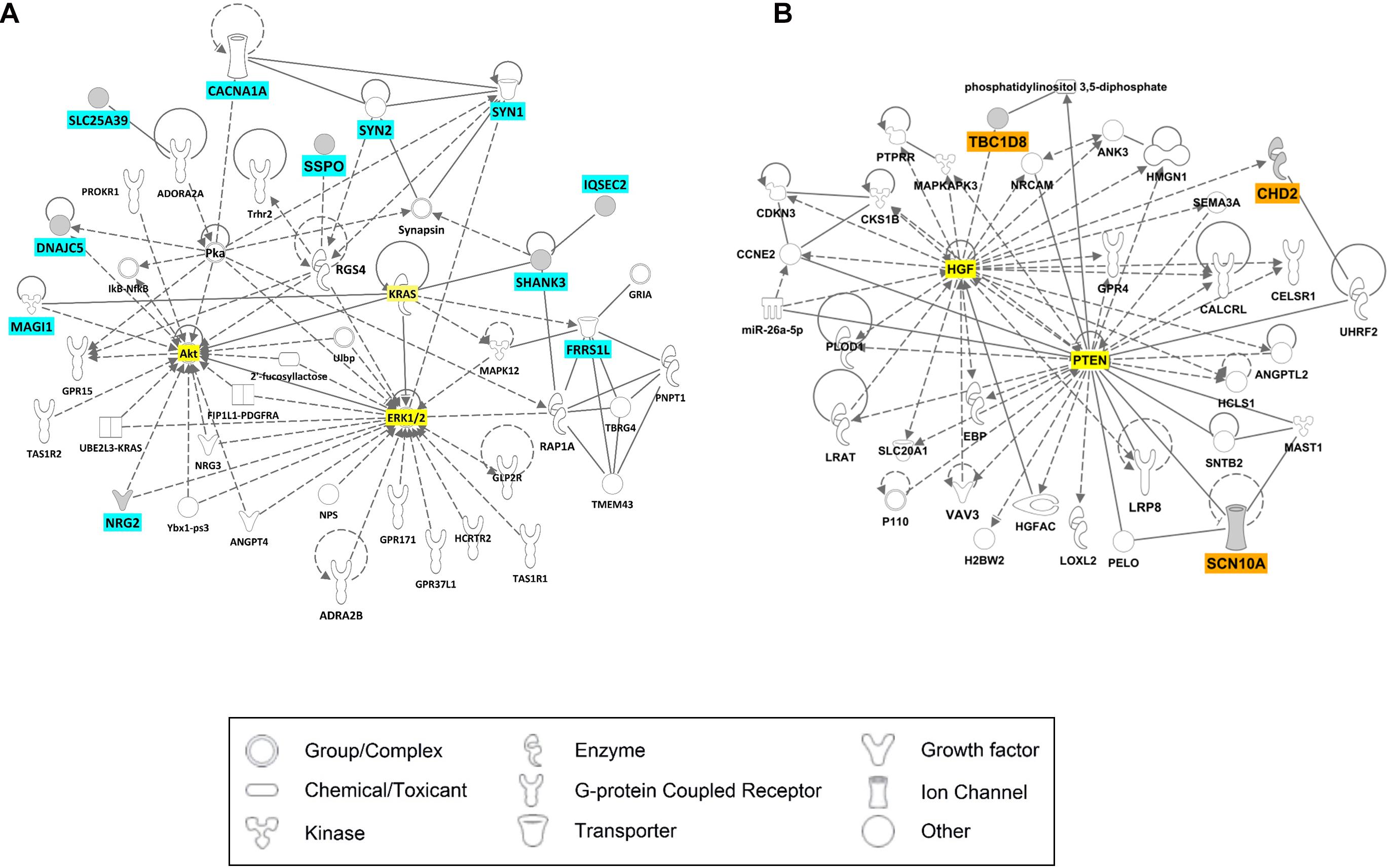
Figure 4. Ingenuity Pathway Analysis (IPA) results for candidate genes of LGS or LGS-like epilepsy. The genes with cyan or orange colors indicate candidate genes selected in this study. The genes with yellow color represent those most relevant to the candidate genes. (A) Cell signaling network. (B) Neurological disease network.
Neurite Alteration in SLC25A39 and TBC1D8 Knockdown Cell Lines
Among neuron-related genes, the direct neuronal functions of SLC25A39 and TBC1D8 have not been reported: Shawn, the Drosophila homolog of SLC25A39 and SLC25A40, reportedly plays a role in neurotransmitter release (Slabbaert et al., 2016) and TBC1D24, a member of the protein family of TBC1D8, regulates neuronal migration (Riazuddin et al., 2017). To examine the effects of SLC25A39 and TBC1D8 on neuronal function, we depleted SLC25A39 and TBC1D8 from the SH-SY5Y neuroblastoma cell line and examined neurite outgrowth. siRNA treatment of target genes decreased SLC25A39 and TBC1D8 expression by approximately 40%. Under retinoic acid-induced neuronal differentiation conditions, the mRNA level of MAP2 was significantly decreased in SLC25A39 or TBC1D8 knockdown cells (Figure 5A). Additionally, the neurite length was reduced by 30% in the TBC1D8 knockdown group and by 40% in the SLC25A39 knockdown group compared to in the control, indicating impaired neuronal development (Figures 5B,C). These results suggest that SLC25A39 and TBC1D8 are involved in the neurite extension of neuronal cells.
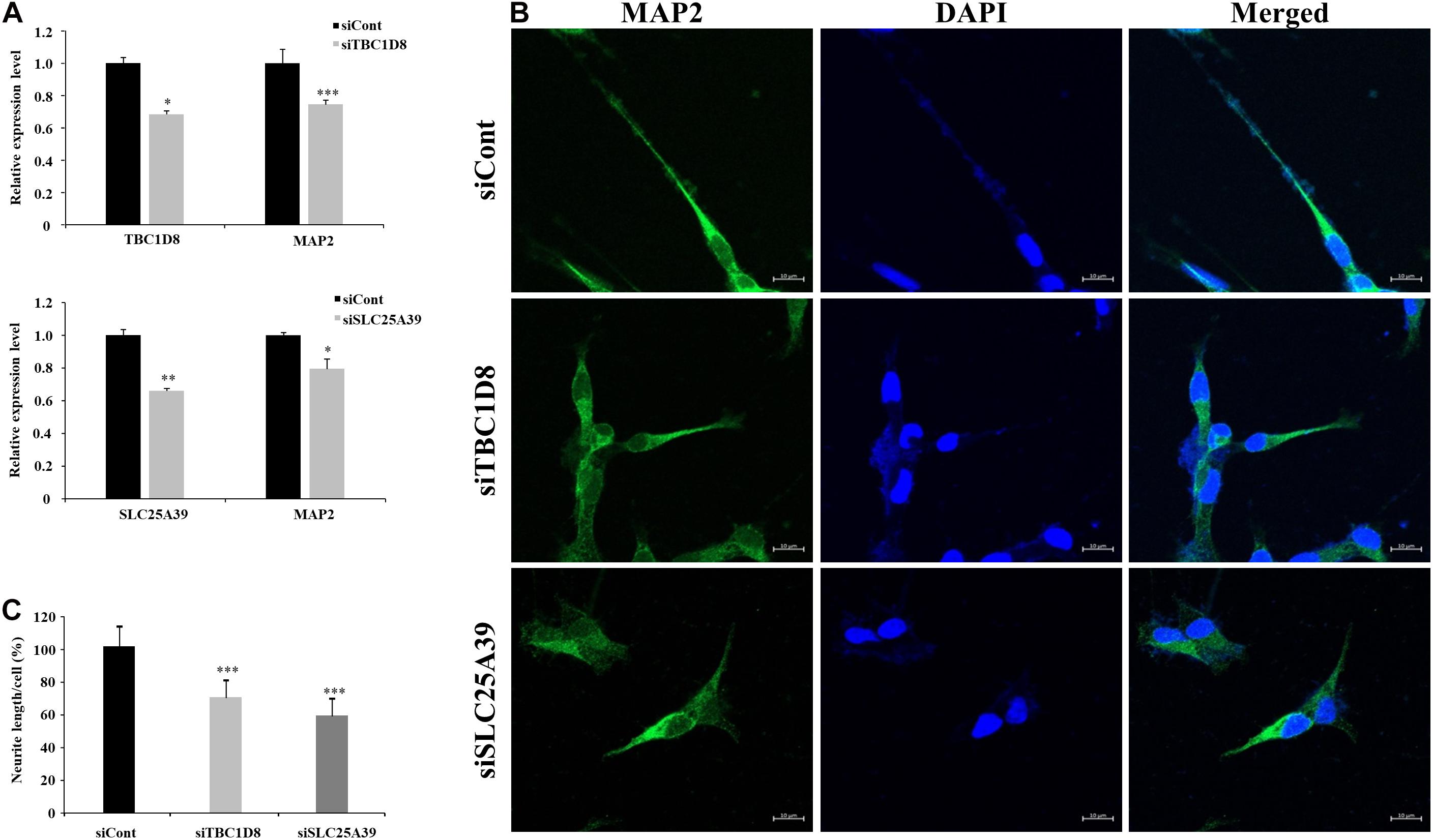
Figure 5. Neurite length analysis by knockdown of candidate genes in neuronal cells. Human SH-SY5Y neuroblastoma cells were transfected with siRNA for TBC1D8 and SLC25A39 on day 3 in vitro (DIV 3). MAP2 was used as a neuronal marker. (A) Expression levels of TBC1D8, SLC25A39, and MAP2 were analyzed by RT-PCR (n = 3). (B) After retinoic acid stimulation for 48 h, the cells were stained for MAP2 (green) and nuclei (blue). Scale bars: 30 μm. (C) Neurite length was measured in more than 10 cells in three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and Student’s t-test compared with the control group.
Discussion
Genetic variations play an important role in the development of epileptic syndromes, including LGS without brain abnormalities, by altering neurotransmission or neuronal development (Falace et al., 2014; Lund et al., 2014; Fedele et al., 2018). In this study, we identified 14 candidate genes and genetic variations related to neuronal development or neurotransmission as biomarkers of LGS and LGS-like epilepsy with unknown causes. Of these, seven mutations were novel. In addition, two genes showed insufficient evidence for epilepsy or neuronal functions; however, we demonstrated that these genes affect neurite outgrowth in a human neuroblastoma cell line.
Pathogenicity evaluation of genetic variations in our candidate genes, with probability used as a risk factor for LGS and LGS-like epilepsy, revealed that several variations were pathogenic or likely pathogenic. In addition, variations reported in four genes, IQSEC2, SYN1, SYN2, and FRRSIL, were in a domain position similar to those in the candidate genes and classified as pathogenic or of uncertain significance according to ClinVar classification (Supplementary Table 2). Furthermore, the protein stability of the 10 candidate genes was altered by isoelectric point changes via side chain modifications of amino acids by genetic variations, implicating the effect of protein function (Supplementary Table 3). Collectively, these results indicate that our variations are associated with LGS and LGS-like epilepsy. However, genotype-phenotype data and gene-specific mutation rates for our candidate genes were not provided because these variants were rare.
Among the genes with 14 genetic variations, including seven novel mutations, MAGI1, NRG2, SSPO, SLC25A39, and TBC1D8 were not associated with LGS or epilepsy. However, they were selected as candidate genes because of their roles in neuronal development. MAGI1, NRG2, and SSPO exert neuron-related functions. MAGI1 plays a role in regulating neurite outgrowth (Ito et al., 2012). SSPO promotes neuronal survival and differentiation and is required during early brain development (Goncalves-Mendes et al., 2004; Vera et al., 2013). NRG2 promotes neuronal survival and neurite extension (Nakano et al., 2016). SLC25A39 resides in susceptibility loci for epilepsy with heterogeneous phenotypes but not as an epilepsy-related gene because its variant has not been identified in families with epilepsy (Siren et al., 2010). However, a variant of SLC25A39 was identified in our LGS data, and depletion of SLC25A39 decreased neurite outgrowth in human neuroblastoma cells. These data suggest a relationship between LGS and epilepsy with SLC25A39. These findings are also supported by previous studies in which mutations in Shawn, the Drosophila homolog of SLC25A39 and SLC25A40, increase neurotransmitter release (Slabbaert et al., 2016). TBC1D24, a member of the same protein family as TBC1D8, regulates neuronal migration and its epilepsy-related mutation is in the Rab-GAP TBC domain, which is the same domain containing the mutation evaluated in our study (Falace et al., 2014; Banuelos et al., 2017). Alternatively, mutations in TBC1D8 have been detected in patients with intellectual disability without an epilepsy phenotype (Riazuddin et al., 2017). However, our data revealed mutations of TBC1D8 in Korean patients with LGS as well as decreased neurite outgrowth in human neuroblastoma cells by depletion of TBC1D8, suggesting that TBC1D8 is a candidate gene for LGS or epilepsy. Epileptic seizure is defined as the transient occurrence of signs and/or symptoms because of abnormal excessive or synchronous neutral activity in the brain. Previous studies showed that epilepsy or LGS phenotypes may occur when neurotransmission and neuronal development in the brain are disrupted (Hermann et al., 2002; Staley, 2015; Fukata and Fukata, 2017; Falco-Walter et al., 2018; Jahngir et al., 2018). Collectively, these findings indicate that mutations in these five genes can trigger the LGS phenotype by disrupting neuronal development.
The results of network analysis revealed that the 11 genes CACNA1A, DNAJC5, FRRS1L, IQSEC2, MAGI1, NRG2, SHANK3, SLC25A39, SSPO, SYN1, and SYN2 are directly or indirectly associated with ERK1/2, AKT, and KRAS, which play important roles in neuronal survival, neural stem cell proliferation, and neurotransmission by α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-d-aspartate receptors (Nateri et al., 2007; Subramaniam and Unsicker, 2010; Bender et al., 2015). Therefore, genetic variations in genes from this group can induce seizure phenotypes of LGS because of both disrupted neuronal development and neurotransmission via ion channels in the brain. Another group of genes, CHD2, SCN10A, and TBC1D8, was associated with HGF and PTEN; these two genes are related to synaptic plasticity, neuronal cell survival, and CNS development, as well as related to epilepsy (Bae et al., 2010; Tonges et al., 2011; Sperow et al., 2012; Knafo and Esteban, 2017; Godale and Danzer, 2018). These results indicate that variations within candidate genes can impair protein function, directly or indirectly affect neurotransmission or neuronal development pathways, and subsequently induce LGS or LGS-like epilepsy. Our results are in accordance with a previous study in which homozygous mutations in STXBP1 were identified in two siblings diagnosed with LGS. The mutations resulted in impairment of protein stability leading to reduced synaptic transmission (Lammertse et al., 2020).
Children with LGS have a high percentage of severe injuries as well as other comorbidities, with a potential increased risk of death (Autry et al., 2010). These children were 14-fold more likely than those of the general population to die, indicating that the genetic variation causing LGS is difficult to detect in the next generation. This is likely because there was no LGS family with AD in our study. In addition, the two patients with SYN2 and MAGI1 variations in our datasets experienced infantile spasms. The variations may have been responsible for these effects and subsequently led to LGS, as 30% of LGS patients can gradually progress from infantile spasms (Cross et al., 2017).
In summary, we identified mutations in 14 genes as potential causative markers of both LGS and LGS-like epilepsy. Many of these candidate genes are generally pathogenic and are associated with neurotransmission or neuronal function. These results expand the spectrum of variations in LGS and LGS-like epilepsy and are crucial for understanding their biological mechanisms for patient-specific therapeutic development. Further animal model studies are required to determine whether these genetic variations can disrupt neuronal function and cause the LGS phenotype.
Data Availability Statement
The dataset generated for this study was submitted to the Seqeuence Read Archive (SRA) and can be accessed by searching the BioProject ID PRJNA601231 on the NCBI website (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA601231).
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Review Board and Ethics Committe at the Chungnam National University Hospitals and the Korea Research Institute of Bioscience and Biotechnology (KRIBB). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
N-SK and J-WK conceived and designed the study. JY, M-HC, J-YY, and N-SK drafted the manuscript. JY performed breakpoints verification. JY, M-HC, J-YY, J-JL, and JL participated in the data analysis. SY, S-JJ, SJ, IB, DH, and BL reviewed and edited the manuscript and contributed to the discussions. SN, JK, HK, and JL participated in clinical data collection. N-SK supervised the study. All authors reviewed and approved this submission.
Funding
This study was supported by the Basic Science Research Program of the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (NRF-2014M3A9A5034157, 2014M3C9A2064619, and 2020R1A2C2006752) and the KRIBB Research Initiative Program.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the patients and their families for participating in our research.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.590924/full#supplementary-material
Footnotes
References
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. doi: 10.1038/nmeth0410-248
Allen, A. S., Berkovic, S. F., Coe, B. P., Cook, J., Cossette, P., Delanty, N., et al. (2015). Copy number variant analysis from exome data in 349 patients with epileptic encephalopathy. Ann. Neurol. 78, 323–328. doi: 10.1002/ana.24457
Allen, A. S., Berkovic, S. F., Cossette, P., Delanty, N., Dlugos, D., Eichler, E. E., et al. (2013). De novo mutations in epileptic encephalopathies. Nature 501, 217–221. doi: 10.1038/nature12439
Asadi-Pooya, A. A. (2018). Lennox-Gastaut syndrome: a comprehensive review. Neurol. Sci. 39, 403–414. doi: 10.1007/s10072-017-3188-y
Autry, A. R., Trevathan, E., Van Naarden Braun, K., and Yeargin-Allsopp, M. (2010). Increased risk of death among children with Lennox-Gastaut syndrome and infantile spasms. J. Child Neurol. 25, 441–447. doi: 10.1177/0883073809348355
Bae, M. H., Bissonette, G. B., Mars, W. M., Michalopoulos, G. K., Achim, C. L., Depireux, D. A., et al. (2010). Hepatocyte growth factor (HGF) modulates GABAergic inhibition and seizure susceptibility. Exp. Neurol. 221, 129–135. doi: 10.1016/j.expneurol.2009.10.011
Banuelos, E., Ramsey, K., Belnap, N., Krishnan, M., Balak, C., Szelinger, S., et al. (2017). Case Report: Novel mutations in TBC1D24 are associated with autosomal dominant tonic-clonic and myoclonic epilepsy and recessive Parkinsonism, psychosis, and intellectual disability. F1000Res 6:553. doi: 10.12688/f1000research.10588.1
Bender, R. H., Haigis, K. M., and Gutmann, D. H. (2015). Activated k-ras, but not h-ras or N-ras, regulates brain neural stem cell proliferation in a raf/rb-dependent manner. Stem Cells 33, 1998–2010. doi: 10.1002/stem.1990
Camfield, P. R. (2011). Definition and natural history of Lennox-Gastaut syndrome. Epilepsia 52(Suppl. 5), 3–9. doi: 10.1111/j.1528-1167.2011.03177.x
Chun, S., and Fay, J. C. (2009). Identification of deleterious mutations within three human genomes. Genome Res. 19, 1553–1561. doi: 10.1101/gr.092619.109
Cross, J. H., Auvin, S., Falip, M., Striano, P., and Arzimanoglou, A. (2017). Expert Opinion on the Management of Lennox-Gastaut Syndrome: Treatment Algorithms and Practical Considerations. Front. Neurol. 8:505. doi: 10.3389/fneur.2017.00505
Dunn, P., Albury, C. L., Maksemous, N., Benton, M. C., Sutherland, H. G., Smith, R. A., et al. (2018). Next Generation Sequencing Methods for Diagnosis of Epilepsy Syndromes. Front. Genet. 9:20. doi: 10.3389/fgene.2018.00020
Falace, A., Buhler, E., Fadda, M., Watrin, F., Lippiello, P., Pallesi-Pocachard, E., et al. (2014). TBC1D24 regulates neuronal migration and maturation through modulation of the ARF6-dependent pathway. Proc. Natl. Acad. Sci. U S A. 111, 2337–2342. doi: 10.1073/pnas.1316294111
Falco-Walter, J. J., Scheffer, I. E., and Fisher, R. S. (2018). The new definition and classification of seizures and epilepsy. Epilepsy Res. 139, 73–79. doi: 10.1016/j.eplepsyres.2017.11.015
Fedele, L., Newcombe, J., Topf, M., Gibb, A., Harvey, R. J., and Smart, T. G. (2018). Disease-associated missense mutations in GluN2B subunit alter NMDA receptor ligand binding and ion channel properties. Nat. Commun. 9:957. doi: 10.1038/s41467-018-02927-4
Fukata, Y., and Fukata, M. (2017). Epilepsy and synaptic proteins. Curr. Opin. Neurobiol. 45, 1–8. doi: 10.1016/j.conb.2017.02.001
Godale, C. M., and Danzer, S. C. (2018). Signaling Pathways and Cellular Mechanisms Regulating Mossy Fiber Sprouting in the Development of Epilepsy. Front. Neurol. 9:298. doi: 10.3389/fneur.2018.00298
Goncalves-Mendes, N., Blanchon, L., Meiniel, A., Dastugue, B., and Sapin, V. (2004). Placental expression of SCO-spondin during mouse and human development. Gene Expr. Patterns 4, 309–314. doi: 10.1016/j.modgep.2003.10.004
Han, W., Wang, H., Li, J., Zhang, S., and Lu, W. (2017). Ferric Chelate Reductase 1 Like Protein (FRRS1L) Associates with Dynein Vesicles and Regulates Glutamatergic Synaptic Transmission. Front. Mol. Neurosci. 10:402. doi: 10.3389/fnmol.2017.00402
Hermann, B., Seidenberg, M., Bell, B., Rutecki, P., Sheth, R., Ruggles, K., et al. (2002). The neurodevelopmental impact of childhood-onset temporal lobe epilepsy on brain structure and function. Epilepsia 43, 1062–1071.
Ito, H., Morishita, R., Sudo, K., Nishimura, Y. V., Inaguma, Y., Iwamoto, I., et al. (2012). Biochemical and morphological characterization of MAGI-1 in neuronal tissue. J. Neurosci. Res. 90, 1776–1781. doi: 10.1002/jnr.23074
Jahngir, M. U., Ahmad, M. Q., and Jahangir, M. (2018). Lennox-Gastaut Syndrome: In a Nutshell. Cureus 10:e3134. doi: 10.7759/cureus.3134
Jiang, Y., Oldridge, D. A., Diskin, S. J., and Zhang, N. R. (2015). CODEX: a normalization and copy number variation detection method for whole exome sequencing. Nucleic Acids Res. 43:e39. doi: 10.1093/nar/gku1363
Knafo, S., and Esteban, J. A. (2017). PTEN: Local and Global Modulation of Neuronal Function in Health and Disease. Trends Neurosci. 40, 83–91. doi: 10.1016/j.tins.2016.11.008
Lammertse, H. C. A., van Berkel, A. A., Iacomino, M., Toonen, R. F., Striano, P., Gambardella, A., et al. (2020). Homozygous STXBP1 variant causes encephalopathy and gain-of-function in synaptic transmission. Brain 143, 441–451. doi: 10.1093/brain/awz391
Lund, C., Brodtkorb, E., Oye, A. M., Rosby, O., and Selmer, K. K. (2014). CHD2 mutations in Lennox-Gastaut syndrome. Epilepsy Behav. 33, 18–21. doi: 10.1016/j.yebeh.2014.02.005
Lund, C., Brodtkorb, E., Rosby, O., Rodningen, O. K., and Selmer, K. K. (2013). Copy number variants in adult patients with Lennox-Gastaut syndrome features. Epilepsy Res. 105, 110–117. doi: 10.1016/j.eplepsyres.2013.01.009
Madeo, M., Stewart, M., Sun, Y., Sahir, N., Wiethoff, S., Chandrasekar, I., et al. (2016). Loss-of-Function Mutations in FRRS1L Lead to an Epileptic-Dyskinetic Encephalopathy. Am. J. Hum. Genet. 98, 1249–1255. doi: 10.1016/j.ajhg.2016.04.008
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Nakano, N., Kanekiyo, K., Nakagawa, T., Asahi, M., and Ide, C. (2016). NTAK/neuregulin-2 secreted by astrocytes promotes survival and neurite outgrowth of neurons via ErbB3. Neurosci. Lett. 622, 88–94. doi: 10.1016/j.neulet.2016.04.050
Nateri, A. S., Raivich, G., Gebhardt, C., Da Costa, C., Naumann, H., Vreugdenhil, M., et al. (2007). ERK activation causes epilepsy by stimulating NMDA receptor activity. EMBO J. 26, 4891–4901. doi: 10.1038/sj.emboj.7601911
Pedersen, M., Curwood, E. K., Archer, J. S., Abbott, D. F., and Jackson, G. D. (2015). Brain regions with abnormal network properties in severe epilepsy of Lennox-Gastaut phenotype: Multivariate analysis of task-free fMRI. Epilepsia 56, 1767–1773. doi: 10.1111/epi.13135
Riazuddin, S., Hussain, M., Razzaq, A., Iqbal, Z., Shahzad, M., Polla, D. L., et al. (2017). Exome sequencing of Pakistani consanguineous families identifies 30 novel candidate genes for recessive intellectual disability. Mol. Psychiatry 22, 1604–1614. doi: 10.1038/mp.2016.109
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Schwarz, J. M., Rodelsperger, C., Schuelke, M., and Seelow, D. (2010). MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576. doi: 10.1038/nmeth0810-575
Sim, N. L., Kumar, P., Hu, J., Henikoff, S., Schneider, G., and Ng, P. C. (2012). SIFT web server: predicting effects of amino acid substitutions on proteins. Nucl. Acids Res. 40, W452–W457. doi: 10.1093/nar/gks539
Siren, A., Polvi, A., Chahine, L., Labuda, M., Bourgoin, S., Anttonen, A. K., et al. (2010). Suggestive evidence for a new locus for epilepsy with heterogeneous phenotypes on chromosome 17q. Epilepsy Res. 88, 65–75. doi: 10.1016/j.eplepsyres.2009.09.022
Slabbaert, J. R., Kuenen, S., Swerts, J., Maes, I., Uytterhoeven, V., Kasprowicz, J., et al. (2016). Shawn, the Drosophila Homolog of SLC25A39/40, Is a Mitochondrial Carrier That Promotes Neuronal Survival. J. Neurosci. 36, 1914–1929. doi: 10.1523/JNEUROSCI.3432-15.2016
Sperow, M., Berry, R. B., Bayazitov, I. T., Zhu, G., Baker, S. J., and Zakharenko, S. S. (2012). Phosphatase and tensin homologue (PTEN) regulates synaptic plasticity independently of its effect on neuronal morphology and migration. J. Physiol. 590, 777–792. doi: 10.1113/jphysiol.2011.220236
Staley, K. (2015). Molecular mechanisms of epilepsy. Nat. Neurosci. 18, 367–372. doi: 10.1038/nn.3947
Subramaniam, S., and Unsicker, K. (2010). ERK and cell death: ERK1/2 in neuronal death. FEBS J. 277, 22–29. doi: 10.1111/j.1742-4658.2009.07367.x
Terrone, G., Bienvenu, T., Germanaud, D., Barthez-Carpentier, M. A., Diebold, B., Delanoe, C., et al. (2014). A case of Lennox-Gastaut syndrome in a patient with FOXG1-related disorder. Epilepsia 55, 116–119e. doi: 10.1111/epi.12800
Tonges, L., Ostendorf, T., Lamballe, F., Genestine, M., Dono, R., Koch, J. C., et al. (2011). Hepatocyte growth factor protects retinal ganglion cells by increasing neuronal survival and axonal regeneration in vitro and in vivo. J. Neurochem. 117, 892–903. doi: 10.1111/j.1471-4159.2011.07257.x
Tumiene, B., Maver, A., Writzl, K., Hodzic, A., Cuturilo, G., Kuzmanic-Samija, R., et al. (2018). Diagnostic exome sequencing of syndromic epilepsy patients in clinical practice. Clin. Genet. 93, 1057–1062. doi: 10.1111/cge.13203
Vera, A., Stanic, K., Montecinos, H., Torrejon, M., Marcellini, S., and Caprile, T. (2013). SCO-spondin from embryonic cerebrospinal fluid is required for neurogenesis during early brain development. Front. Cell Neurosci. 7:80. doi: 10.3389/fncel.2013.00080
Wang, J., Lin, Z. J., Liu, L., Xu, H. Q., Shi, Y. W., Yi, Y. H., et al. (2017). Epilepsy-associated genes. Seizure 44, 11–20. doi: 10.1016/j.seizure.2016.11.030
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucl. Acids Res. 38:e164. doi: 10.1093/nar/gkq603
Keywords: Lennox-Gastaut syndrome, epilepsy, whole-exome sequencing, genetic variation, Rare-diseases
Citation: Yang JO, Choi M-H, Yoon J-Y, Lee J-J, Nam SO, Jun SY, Kwon HH, Yun S, Jeon S-J, Byeon I, Halder D, Kong J, Lee B, Lee J, Kang J-W and Kim N-S (2021) Characteristics of Genetic Variations Associated With Lennox-Gastaut Syndrome in Korean Families. Front. Genet. 11:590924. doi: 10.3389/fgene.2020.590924
Received: 03 August 2020; Accepted: 31 December 2020;
Published: 20 January 2021.
Edited by:
Oliver Semler, University Hospital of Cologne, GermanyReviewed by:
Pasquale Striano, University of Genoa, ItalyAglaia Vignoli, University of Milan, Italy
Copyright © 2021 Yang, Choi, Yoon, Lee, Nam, Jun, Kwon, Yun, Jeon, Byeon, Halder, Kong, Lee, Lee, Kang and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nam-Soon Kim, bnNraW0zN0BrcmliYi5yZS5rcg==; Joon-Won Kang, Y2hpbGRsb3ZlQGNudS5hYy5rcg==
†These authors have contributed equally to this work