 Katarína Fabišíková1
*
Katarína Fabišíková1
* Olívia Hamidová2
Regína Lohajová Behulová2
Katarína Závodná2
Olívia Hamidová2
Regína Lohajová Behulová2
Katarína Závodná2
 Petra Priščáková1
Vanda Repiská1
Petra Priščáková1
Vanda Repiská1
- 1Faculty of Medicine, Institute of Medical Biology, Genetics and Clinical Genetics, Comenius University, Bratislava, Slovakia
- 2Department of Clinical Genetics, St. Elizabeth Cancer Institute, Bratislava, Slovakia
MUTYH-associated polyposis (MAP) is a rare hereditary condition caused by the biallelic mutation in the MUTYH gene encoding MUTYH glycosylase. This enzyme is a key member of the base excision repair (BER) pathway responsible for the repair of DNA lesions formed by reactive oxygen species (ROS). We report two cases of MAP. In case 1, a 67-year-old woman who presented with a personal history of colorectal and endometrial cancer and a family history of cancer syndromes underwent multigene panel testing that revealed a germline homozygous (biallelic) pathogenic variant c.1187G > A (p.Gly396Asp) in the MUTYH gene. Subsequent sequencing analysis performed in the offspring of the proband identified all three asymptomatic offspring as carriers of this pathogenic variant. In case 2, a 40-year-old woman with a strong family history of colorectal cancer [the proband’s sister was a carrier of the pathogenic variant c.536A > G (p.Tyr179Cys) of the MUTYH gene] and renal cancer underwent sequencing analysis of the MUTYH gene. The pathogenic heterozygous (monoallelic) variant c.536A > G (p.Tyr179Cys) of the MUTYH gene was identified in the proband. We found another pathogenic variant of the MUTYH gene—heterozygous (monoallelic) mutation c.1187G > A (p.Gly396Asp) in the genome of the proband’s husband. Molecular analysis of their offspring revealed that they are compound heterozygotes for MUTYH pathogenic variants c.536A > G (p.Tyr179Cys)/c.1187G > A (p.Gly396Asp). This paper shows the importance of genetic testing of asymptomatic relatives of the proband to ensure an early surveillance and management of individuals positive for pathogenic variant (s) in the MUTYH gene.
Introduction
Hereditary colorectal cancer (CRC) is mostly caused by mutations in the APC (adenomatous polyposis coli) gene or mismatch repair genes (MLH1, MSH2, MSH3, MSH6, PMS2, and EPCAM2).
In 2002, Al-Tassan et al. first described inherited variants in the MUTYH gene that led to an increased number of somatic inactivating mutations in the APC gene. Most of these somatic mutations were G:C to T:A transversions as a result of the impaired (defective) base excision repair (BER; Al-Tassan et al., 2002). The BER pathway, involving the DNA repair glycosylase MUTYH, repairs DNA lesions formed by reactive oxygen species (ROS).
ROS are natural by-products of normal aerobic metabolism. Elevated production of ROS arises from cellular exposure to toxins, ultraviolet light, and ionizing radiation. 8-Oxo-7,8-dihydroguanine (8-oxoG) is the most common, stable, and mutagenic oxidatively damaged guanine lesion (Banda et al., 2017). This nucleotide mispairs with adenine and MUTYH glycosylase removes these mispaired adenines (Terdiman, 2009).
MUTYH-associated polyposis (MAP) is an autosomal recessive disorder that can be diagnosed in patients with an attenuated colonic polyposis phenotype (Kidambi et al., 2018). Detection of germline MUTYH mutations is recommended in individuals affected by multiple colorectal adenomas in whom no mutation in the APC gene had been identified (Isidro et al., 2004). MAP is caused by biallelic pathogenic variants in the MUTYH gene and is characterized by the presence of 15–100 colorectal polyps and an increased carrier’s risk of colorectal adenomas and carcinomas. Patients are diagnosed with MAP at a mean age of 45 years. The chance of developing CRC reaches 20–80% in patients aged from 50 to 80 years (Lubbe et al., 2009; Castillejo et al., 2014). MAP is estimated to account for 0.7% of all CRC and between 0.5 and 6% of cohorts of familial or early-onset CRC in which affected individuals have a low number of adenomas (<15–20) (Sieber et al., 2003; Cleary et al., 2009; Lubbe et al., 2009; Landon et al., 2015; Pearlman et al., 2017). Seventy percent of MUTYH mutations involve c.1187G > A (p.Gly396Asp) and c.536A > G (p.Tyr179Cys) mutations (Sampson and Jones, 2009).
MUTYH mutations can contribute to the development of sporadic gastric cancer. The presence of MUTYH pathogenic variants is an independent predictor of poor prognosis in sporadic gastric cancer and in salivary gland secretory carcinoma, while its inhibition has been shown to reduce the survival of pancreatic ductal adenocarcinoma cells (Curia et al., 2020).
Biallelic (compound heterozygous or homozygous) MUTYH mutations inherited from both parents occur in 0.01–0.04% of the Caucasian population and are associated with an 18- to 100-fold increased risk of CRC compared with the general population. Carriers of biallelic MUTYH mutation have an increased risk of extracolonic cancers such as ovarian cancer, urinary bladder cancer, cancer of the upper gastrointestinal tracts, breast cancer, endometrial cancer, and skin cancer compared with the general population (Win et al., 2016; Table 1).
Monoallelic (heterozygous) MUTYH mutations, inherited from only one parent, occur in 1–2% of the Caucasian population and are associated with a moderately increased risk of CRC (Win et al., 2016). Carriers of monoallelic mutation have on average an approximately 2.5-fold increased risk of CRC compared with the general population (Win et al., 2014). It is estimated that there are an elevated risk of liver and gastric cancers and a slightly increased risk of breast cancer for carriers with monoallelic MUTYH mutation (Zhu et al., 2011; Rennert et al., 2012).
It is appropriate to perform molecular genetic analysis of asymptomatic relatives for the MUTYH pathogenic variants identified in the proband to ensure appropriate surveillance (beginning at the age of 10–15 years) and early identification of polyps (Nielsen et al., 2012, updated 2019).
Case Description
Case 1 Description
We performed multigene panel testing of the patient with a family history of cancer syndromes. The proband was a 67-year-old woman presenting with a personal history of CRC diagnosed at the age of 54 and endometrial cancer diagnosed at the age of 65. Immunohistochemistry (IHC) demonstrated intact expression of MLH1, MSH2, MSH6, and PMS2. Colorectal tumor that developed was microsatellite stable.
The grandmother of the patient developed breast cancer at the age of 48. The brother of the proband died aged 50 from an unknown gastrointestinal disease (probably gastric cancer; Figure 1).
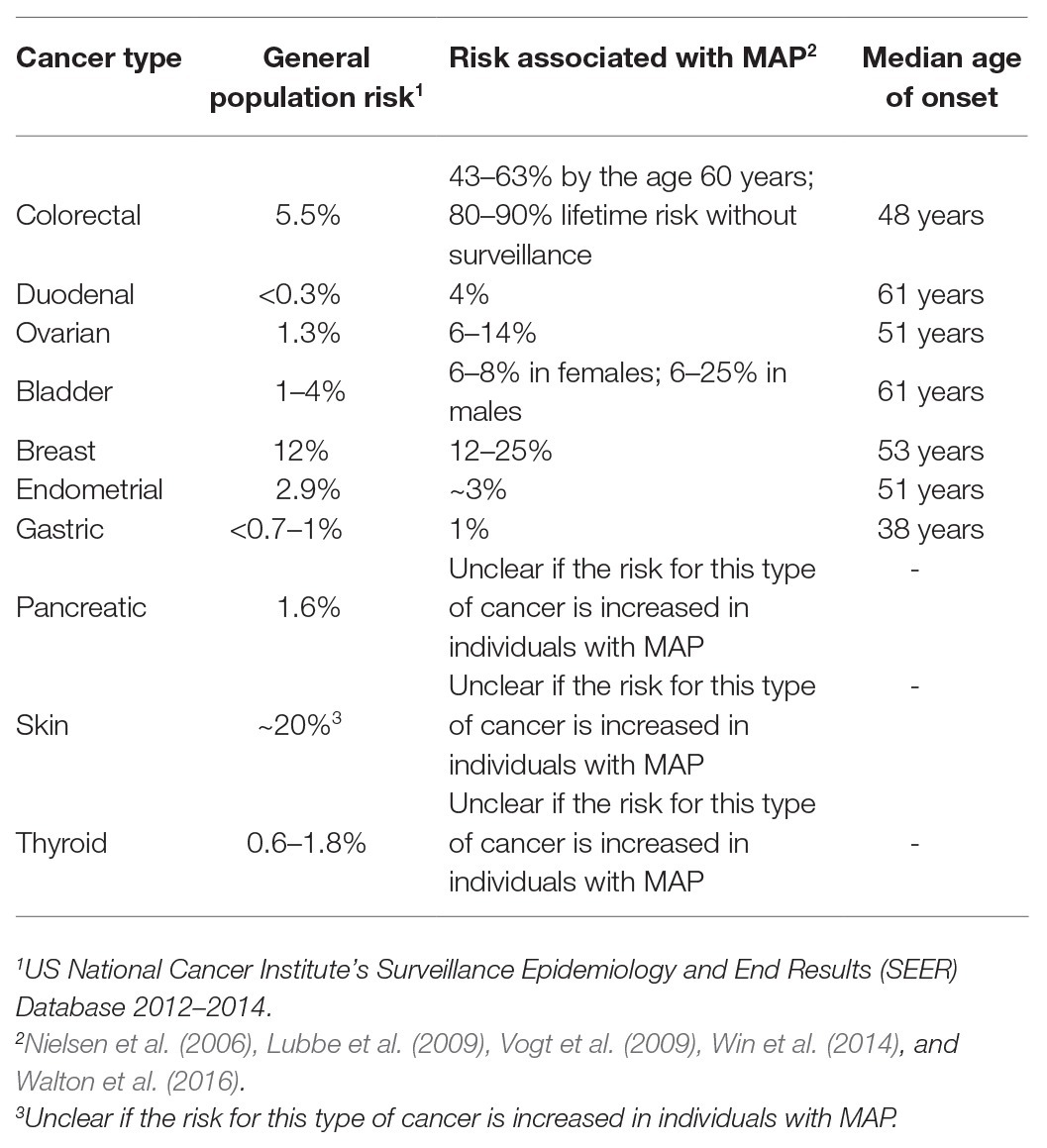
Table 1. Cancer risks in individuals with MUTYH polyposis compared with the general population (Nielsen et al., 2012, updated 2019).
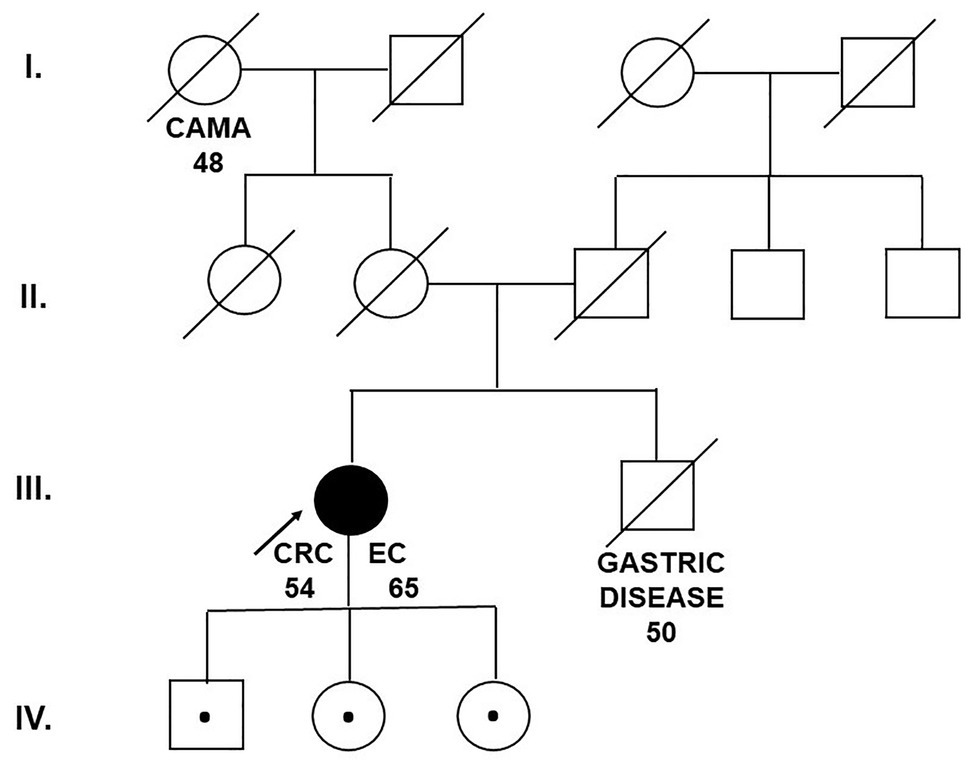
Figure 1. Pedigree of the family carrying the MUTYH mutation c.1187G>A (p.Gly396Asp). CAMA, breast cancer; CRC, colorectal cancer; EC, endometrial cancer (Department of Clinical Genetics, St. Elizabeth Cancer Institute, Bratislava, Slovakia).
A panel of 26 cancer predisposition genes identified a germline homozygous (biallelic) pathogenic variant c.1187G > A (p.Gly396Asp) in exon 13 of the MUTYH gene in the proband.
The c.1187G > A (p.Gly396Asp) variant is a common cause of MAP, and this missense variant disrupts MUTYH protein function. Furthermore, panel testing revealed also a heterozygous variant of the PALB2 gene [c.229 T > C (p.Cys77Arg)] and a heterozygous variant of the TP53 gene (c.1100 + 30A > T). However, according to the ACMG guidelines for the interpretation of sequence variants, both variants are of uncertain significance (VUS); thus, we suggest that they have no significant impact on the proband’s cancer etiology.
MUTYH molecular genetic testing was offered to the husband of the proband to determine the risk of MAP in the offspring, but he did not agree to being tested.
Sequencing analysis of the whole coding sequence of the MUTYH gene was subsequently conducted in the offspring of the proband: one son (aged 38) and two daughters (aged 47 and 39). The germline heterozygous (monoallelic) MUTYH mutation c.1187G > A (p.Gly396Asp) was found in all three asymptomatic offspring of the proband.
Case 2 Description
A 40-year-old woman was presented to the Department of Clinical Genetics for genetic counseling due to a strong family history of CRC and renal cell carcinoma (RCC). The proband’s sister was a carrier of the germline pathogenic heterozygous (monoallelic) mutation c.536A > G (p.Tyr179Cys) in exon 7 of the MUTYH gene (the result of the 26-gene panel examination) and she died from CRC at the age of 37. The proband’s father developed CRC at the age of 59. At the age of 61, he died after the recurrence of CRC. The brother of the proband’s father died from RCC at the age of 60 (Figure 2).
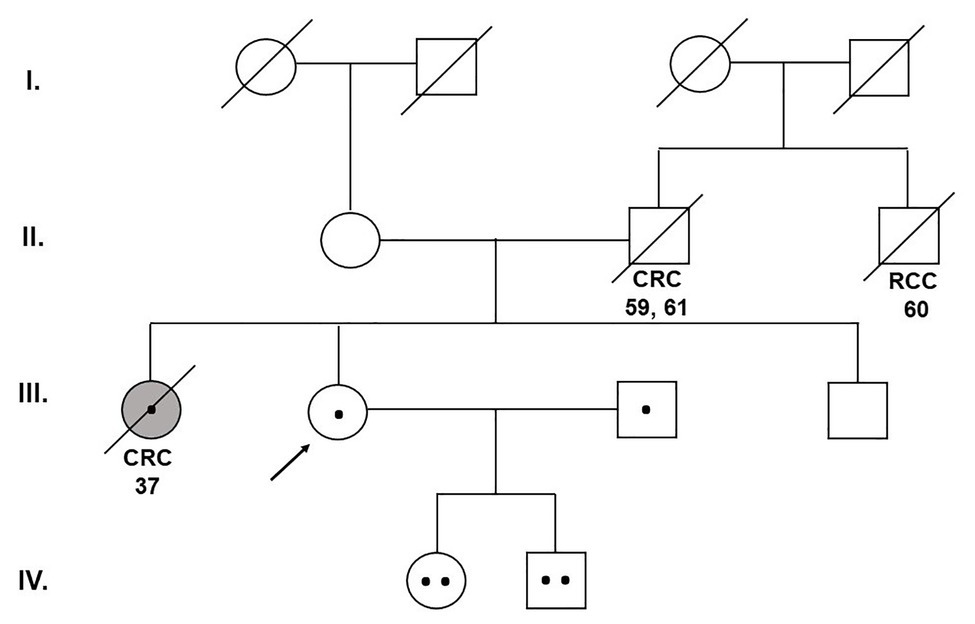
Figure 2. Pedigree of the family carrying the MUTYH mutations c.1187G>A (p.Gly396Asp) and c.536A>G (p.Tyr179Cys). CRC, colorectal cancer; RCC, renal cell cancer (Department of Clinical Genetics, St. Elizabeth Cancer Institute, Bratislava, Slovakia).
The proband underwent sequencing analysis of exon 7 for detection of the presence of MUTYH pathogenic variant c.536A > G (p.Tyr179Cys) found in her sister with a positive result—carrier of monoallelic MUTYH pathogenic variant. To determine the risk of MAP in the offspring, we performed sequencing analysis of the whole MUTYH gene also in the proband’s husband. We identified the pathogenic heterozygous (monoallelic) mutation c.1187G > A (p.Gly396Asp) in exon 13 of the MUTYH gene in the husband of the proband.
The proband’s asymptomatic offspring—one son aged 11 and one daughter aged 18—subsequently underwent genetic counseling and sequencing analysis. We identified that both son and daughter of the proband are carriers of germline pathogenic variants c.536A > G (p.Tyr179Cys; passed down to offspring from the mother carrying a mutation) and c.1187G > A (p.Gly396Asp; passed down to offspring from the father carrying a mutation) of the MUTYH gene. In this way, the offspring of the proband are combined heterozygotes and this genotype refers to the diagnosis of MAP requiring patient’s management.
Discussion
These two cases demonstrate the importance of genetic testing of asymptomatic partners and the proband or carrier offspring of the pathogenic variant. We identified in our probands and their relatives pathogenic variants in the MUTYH gene by next-generation sequencing (NGS) panel testing and Sanger sequencing.
Variants c.1187G > A (p.Gly396Asp) and c.536A > G (p.Tyr179Cys) are the two most common pathogenic variants in the MUTYH gene responsible for the MAP. MAP is associated with an increased lifetime risk of CRC development. Other features involve thyroid nodules, benign adrenal lesions, jawbone cysts, and congenital hypertrophy of the retinal pigment epithelium (Nielsen et al., 2012, updated 2019). The risk of CRC is strongly age dependent, with incomplete penetrance at the age of 60 (Lubbe et al., 2009). Several studies found the association between MUTYH variants and the risk of breast cancer. In a case-control study, 930 women with a high prevalence of MUTYH mutations were investigated for the two variants c.1187G > A (p.Gly396Asp) and c.536A > G (p.Tyr179Cys), and patients with breast cancer revealed a 6.7% prevalence of c.1187G > A (p.Gly396Asp) (Rennert et al., 2012).
Recently, it has been estimated that MUTYH mutations could cause some diseases that are not associated with polyposis, such as Parkinson’s disease, Alzheimer’s disease, Friedreich’s ataxia, Huntington’s disease, retinitis pigmentosa, and neurofibromatosis (Curia et al., 2020).
Several functional assays have shown that the MUTYH glycosylase activity is greatly reduced for the MUTYH pathogenic variant c.536A > G (p.Tyr179Cys) (a 98% reduction of glycosylase activity) compared with the MUTYH pathogenic variant c.1187G > A (p.Gly396Asp) (an 86% reduction of glycosylase activity) (Al-Tassan et al., 2002; Nielsen et al., 2009; Banda et al., 2017). MAP patients harboring homozygous MUTYH pathogenic variant c.536A > G (p.Tyr179Cys) showed more severe clinical features than those with homozygous MUTYH pathogenic variant c.1187G > A (p.Gly396Asp). Furthermore, patients with a homozygous c.1187G > A (p.Gly396Asp) mutation or compound heterozygous c.1187G > A (p.Gly396Asp)/c.536A > G (p.Tyr179Cys) mutations have a lower risk of CRC development as compared with patients with a homozygous c.536A > G (p.Tyr179Cys) mutation.
According to the functional studies mentioned above, the mean age of CRC diagnosis is 46 years for the homozygous c.536A > G (p.Tyr179Cys) mutation, 52 years for the heterozygous c.1187G > A (p.Gly396Asp)/c.536A > G (p.Tyr179Cys) mutation, and 58 years for the homozygous c.1187G > A (p.Gly396Asp) mutation.
It is suitable to begin surveillance earlier for p.Tyr179Cys homozygotes than for p.Gly396Asp homozygotes and p.Gly396Asp/p.Tyr179Cys compound heterozygotes (Nielsen et al., 2011).
According to the NCCN guidelines (2019), the management of monoallelic MUTYH pathogenic variant carriers involved undergoing colonoscopy every 5 years beginning at the age of 40 years. The identification of monoallelic carriers of MUTYH pathogenic variants is very important to select individuals who might benefit from preventive strategies.
It is appropriate to clarify the genetic status of apparently asymptomatic individuals to reduce morbidity and mortality in those who would benefit from appropriate surveillance (beginning at the age of 10–15 years) and early identification and treatment of polyps. That is why we recommended for both children to undergo colonoscopy and upper endoscopy (the frequency of colonoscopy is recommended every 1–2 years starting at the age of 25 in case of a negative result, the frequency of upper endoscopy every 3–4 years starting at the age of 30). We also recommended thyroid ultrasound and skin examination by a dermatologist for both children. For girls, we recommended regular gynecologic and mammalogical dispensarization starting at the 21st year.
Identification of mutations in cancer predisposition genes is a challenge for current cancer risk management, counseling, and treatment decision-making regarding patients and their families.
Conclusion
Polyposis syndromes are not only due to mutations in well-known and well-explored cancer predisposition genes. Here, we suggest the need for the extension of molecular analysis for genes that are mutated with a relatively low frequency but with a strong impact on CRC development. Mutations in the MUTYH gene are associated with an 18- to 100-fold increased risk of CRC and an elevated risk of extracolonic cancers in comparison with the general population (Win et al., 2016).
Genetic counseling and testing with a multigene panel could be considered for all patients with a personal or family history of cancer syndromes. Genetic screening can provide early diagnosis and improve prognosis.
Potential future implementation of the exome sequencing indicates the discovery and investigation of new candidate genes involved in the etiology of the CRC.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
Ethical approval was not provided for this study on human participants because this research was performed as a part of a routine molecular diagnostic of patients. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s) legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
All authors listed have made substantial, direct and intellectual contribution to the work and approved it for publication.
Funding
This article was created with the support of the OP Integrated Infrastructure for the project: Long-term strategic research and development focused on the occurrence of Lynch syndrome in the Slovak population and possibilities of prevention of tumors associated with this syndrome. ITMS: 313011V578, co-financed by the European Regional Development Fund. This work was created also with the support of the Grant UK 2020 (UK/208/2020).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Al-Tassan, N., Chmiel, N. H., Maynard, J., Fleming, N., Livingston, A. L., Williams, G. T., et al. (2002). Inherited variants of MYH associated with somatic G:C-->T:A mutations in colorectal tumors. Nat. Genet. 30, 227–232. doi: 10.1038/ng828
Banda, D. M., Nuñez, N. N., Burnside, M. A., Bradshaw, K. M., and David, S. S. (2017). Repair of 8-oxoG:a mismatches by the MUTYH glycosylase: mechanism, metals and medicine. Free Radic. Biol. Med. 107, 202–215. doi: 10.1016/j.freeradbiomed.2017.01.008
Castillejo, A., Vargas, G., Castillejo, M. I., Navarro, M., Barberá, V. M., González, S., et al. (2014). Prevalence of germline MUTYH mutations among lynch-like syndrome patients. Eur. J. Cancer 50, 2241–2250. doi: 10.1016/j.ejca.2014.05.022
Cleary, S. P., Cotterchio, M., Jenkins, M. A., Kim, H., Bristow, R., Green, R., et al. (2009). Germline MutY human homologue mutations and colorectal cancer: a multisite case-control study. Gastroenterology 136, 1251–1260. doi: 10.1053/j.gastro.2008.12.050
Curia, M. C., Catalano, T., and Aceto, G. M. (2020). MUTYH: Not just polyposis. World J. Clin. Oncol. 11, 428–449. doi: 10.5306/wjco.v11.i7.428
Isidro, G., Laranjeira, F., Pires, A., Leite, J., and Regateiro, F., Castro e Sousa, F. et al. (2004). Germline MUTYH (MYH) mutations in Portuguese individuals with multiple colorectal adenomas. Hum. Mutat. 24, 353–354. doi: 10.1002/humu.9282
Kidambi, T. D., Goldberg, D., Nussbaum, R., Blanco, A., Umetsu, S. E., Terdiman, J. P., et al. (2018). Novel variant of unknown significance in MUTYH in a patient with MUTYH-associated polyposis: a case to reclassify. Clin. J. Gastroenterol. 11, 457–460. doi: 10.1007/s12328-018-0870-4
Landon, M., Ceulemans, S., Saraiya, D. S., Strike, B., Arnell, C., Burbidge, L. A., et al. (2015). Analysis of current testing practices for biallelic MUTYH mutations in MUTYH-associated polyposis. Clin. Genet. 87, 368–372. doi: 10.1111/cge.12375
Lubbe, S. J., Di Bernardo, M. C., Chandler, I. P., and Houlston, R. S. (2009). Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J. Clin. Oncol. 27, 3975–3980. doi: 10.1200/JCO.2008.21.6853
Nielsen, M., Infante, E., and Brand, R. (2012). “MUTYH polyposis [Updated 2019 Oct 10]” in GeneReviews® [Internet]. eds. M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, and K. Stephens, et al. (Seattle, WA: University of Washington, Seattle), 1993–2020.
Nielsen, M., Joerink-van de Beld, M. C., Jones, N., Vogt, S., Tops, C. M., Vasen, H. F., et al. (2009). Analysis of MUTYH genotypes and colorectal phenotypes in patients with MUTYH-associated polyposis. Gastroenterology 136, 471–476. doi: 10.1053/j.gastro.2008.10.056
Nielsen, M., Morreau, H., Vasen, H. F., and Hes, F. J. (2011). MUTYH-associated polyposis (MAP). Crit. Rev. Oncol. Hematol. 79, 1–16. doi: 10.1016/j.critrevonc.2010.05.011
Nielsen, M., Poley, J. W., Verhoef, S., van Puijenbroek, M., Weiss, M. M., Burger, G. T., et al. (2006). Duodenal carcinoma in MUTYH-associated polyposis. J. Clin. Pathol. 59, 212–215. doi: 10.1136/jcp.2005.031757
Pearlman, R., Frankel, W. L., Swanson, B., Zhao, W., Yilmaz, A., Miller, K., et al. (2017). Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol. 3, 464–471. doi: 10.1001/jamaoncol.2016.5194
Rennert, G., Lejbkowicz, F., Cohen, I., Pinchev, M., Rennert, H. S., and Barnett-Griness, O. (2012). MutYH mutation carriers have increased breast cancer risk. Cancer 118, 1989–1993. doi: 10.1002/cncr.26506
Sampson, J. R., and Jones, N. (2009). MUTYH-associated polyposis. Best Pract. Res. Clin. Gastroenterol. 23, 209–218. doi: 10.1016/j.bpg.2009.03.006
Sieber, O. M., Lipton, L., Crabtree, M., Heinimann, K., Fidalgo, P., Phillips, R. K., et al. (2003). Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N. Engl. J. Med. 348, 791–799. doi: 10.1056/NEJMoa025283
Terdiman, J. P. (2009). MYH-associated disease: attenuated adenomatous polyposis of the colon is only part of the story. Gastroenterology 137, 1883–1886. doi: 10.1053/j.gastro.2009.10.017
Vogt, S. J., Jones, N., Christian, D., Engel, C., Nielsen, M., Kaufmann, A., et al. (2009). Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology 137, 1976–1985.e1–10. doi: 10.1053/j.gastro.2009.08.052
Walton, S. J., Kallenberg, F. G., Clark, S. K., Dekker, E., and Latchford, A. (2016). Frequency and features of duodenal adenomas in patients with MUTYH-associated polyposis. Clin. Gastroenterol. Hepatol. 14, 986–992. doi: 10.1016/j.cgh.2016.02.020
Win, A. K., Dowty, J. G., Cleary, S. P., Kim, H., Buchanan, D. D., Young, J. P., et al. (2014). Risk of colorectal cancer for carriers of mutations in MUTYH, with and without a family history of cancer. Gastroenterology 146, 1208-11.e1–1208-11.e5. doi: 10.1053/j.gastro.2014.01.022
Win, A. K., Reece, J. C., Dowty, J. G., Buchanan, D. D., Clendenning, M., Rosty, C., et al. (2016). Risk of extracolonic cancers for people with biallelic and monoallelic mutations in MUTYH. Int. J. Cancer 139, 1557–1563. doi: 10.1002/ijc.30197 Erratum in: Int J Cancer. (2017). 141: 12, E7.
Keywords: case report, MUTYH glycosylase, MUTYH gene, compound heterozygote, germline mutation, pathogenic variant, base excision repair
Citation: Fabišíková K, Hamidová O, Behulová RL, Závodná K, Priščáková P and Repiská V (2020) Case Report: The Role of Molecular Analysis of the MUTYH Gene in Asymptomatic Individuals. Front. Genet. 11:590486. doi: 10.3389/fgene.2020.590486
Edited by:
Enrico Baruffini, University of Parma, ItalyReviewed by:
Margherita Bignami, Istituto Superiore di Sanità, ItalySue Clark, St. Mark’s Hospital, United Kingdom
Copyright © 2020 Fabišíková, Hamidová, Behulová, Závodná, Priščáková and Repiská. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katarína Fabišíková, a2F0YXJpbmEuZmFiaXNpa292YUBnbWFpbC5jb20=