 Ping Siu Kee
Ping Siu Kee Paul Ken Leong Chin
Paul Ken Leong Chin Martin A. Kennedy
Martin A. Kennedy Simran D. S. Maggo
Simran D. S. Maggo- 1Gene Structure and Function Laboratory, Carney Centre for Pharmacogenomics, Department of Pathology and Biomedical Science, University of Otago, Christchurch, New Zealand
- 2Department of Medicine, University of Otago, Christchurch, New Zealand
Statins, a class of lipid-lowering medications, have been a keystone treatment in cardiovascular health. However, adverse effects associated with statin use impact patient adherence, leading to statin discontinuation. Statin-induced myotoxicity (SIM) is one of the most common adverse effects, prevalent across all ages, genders, and ethnicities. Although certain demographic cohorts carry a higher risk, the impaired quality of life attributed to SIM is significant. The pathogenesis of SIM remains to be fully elucidated, but it is clear that SIM is multifactorial. These factors include drug–drug interactions, renal or liver dysfunction, and genetics. Genetic-inferred risk for SIM was first reported by a landmark genome-wide association study, which reported a higher risk of SIM with a polymorphism in the SLCO1B1 gene. Since then, research associating genetic factors with SIM has expanded widely and has become one of the foci in the field of pharmacogenomics. This review provides an update on the genetic risk factors associated with SIM.
Introduction
Hydroxymethylglutaryl-coenzyme A reductase (HMG-CR) inhibitors, commonly known as statins, are indicated for hypercholesterolemia and atherosclerotic cardiovascular diseases (Grundy et al., 2019; Bartlomiejczyk et al., 2019). The pharmacological effects of statins include reducing blood plasma concentrations of total cholesterol, triglycerides, low-density lipoprotein (LDL) cholesterol, as well as increasing the level of high-density lipoprotein cholesterol (Sizar et al., 2019). Statins bind competitively to the HMG-CoA reductase enzyme and inhibit the endogenous production of cholesterol within hepatocytes (Figure 1). This reduces endogenous cholesterol levels, which in turn upregulates LDL receptors on the cell surface of hepatocytes, resulting in the removal of circulating cholesterol in the blood (Brown and Goldstein, 1981; Young and Fong, 2012). The first statin discovered was mevastatin, which was isolated from a fungus by Akira Endo in the 1970s (Endo, 2010). Since then, statins have been of interest to both academic and pharmaceutical fields, which has led to the discovery and synthesis of various statins available clinically today. While all statins share the same mechanism of action, they exhibit very different pharmacokinetic (PK) properties.
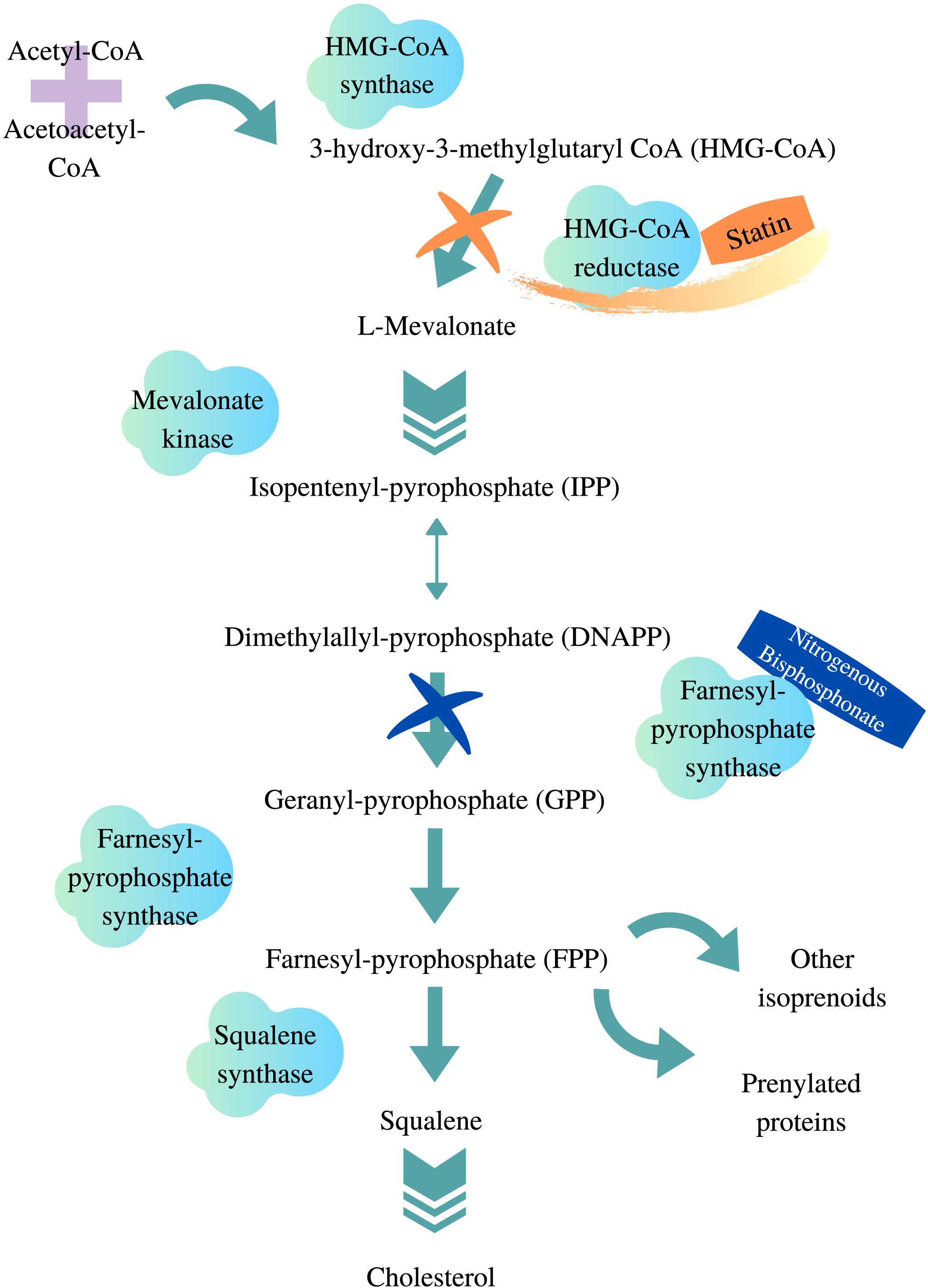
Figure 1. HMG-CoA reductase pathway. Adapted from van Beek et al. (2003), Gazzerro et al. (2012), designed with Canva.com.
To date, the only Food and Drug Administration (FDA)–approved route of administration for statins is oral (Korani et al., 2019). Generally, statins are rapidly absorbed following administration, reaching peak plasma concentration within 4 h in immediate-release formulations (Wiggins et al., 2016). All statins are administered in active hydroxy acid forms, except simvastatin and lovastatin, which require in vivo biotransformation from their lactone prodrug forms to exert pharmacological effects (Vickers et al., 1990; Vyas et al., 1990a; Taha et al., 2016). Based on their solubility, statins are transported systemically either through passive diffusion or actively assisted by endogenous transporters such as the adenosine triphosphate (ATP)–binding cassette (ABC) and solute carrier (SLC) transporters (Whirl-Carrillo et al., 2012). The liver is the site of action for statins, as well as for their metabolism. Some statins such as simvastatin (Kyrklund et al., 2000) and lovastatin (Zhu et al., 2011) also undergo intestinal degradation. Generally, statins are metabolized in hepatocytes prior to elimination via bile (Whirl-Carrillo et al., 2012). Cytochrome P450 (CYP450) enzymes are mainly responsible for oxidative biotransformation of statins (Brown et al., 2008), whereas conjugation through glucuronidation is commonly facilitated by the uridine 5′-diphospho-glucuronosyltransferase (UGT) family of enzymes (Schirris et al., 2015; Sanchez-Dominguez et al., 2018).
Structurally, all statins share a similar pharmacophore with the HMG-CR moiety as shown in Figure 2 (Arnaboldi and Corsini, 2010). Nonetheless, their interactions with the pocket binding site of HMG-CR enzyme vary, which contributes to their different potencies as HMG-CR inhibitors (Arnaboldi and Corsini, 2010). Among the statins available for prescription, pitavastatin is the most potent. However, rosuvastatin and atorvastatin achieved the greatest effect in lowering LDL cholesterol for the recommended dose range (Adams et al., 2020). In addition, the PK properties of statins also vary (Schachter, 2005). The different PK properties and transporter substrate specificity of statins are summarized in Table 1.
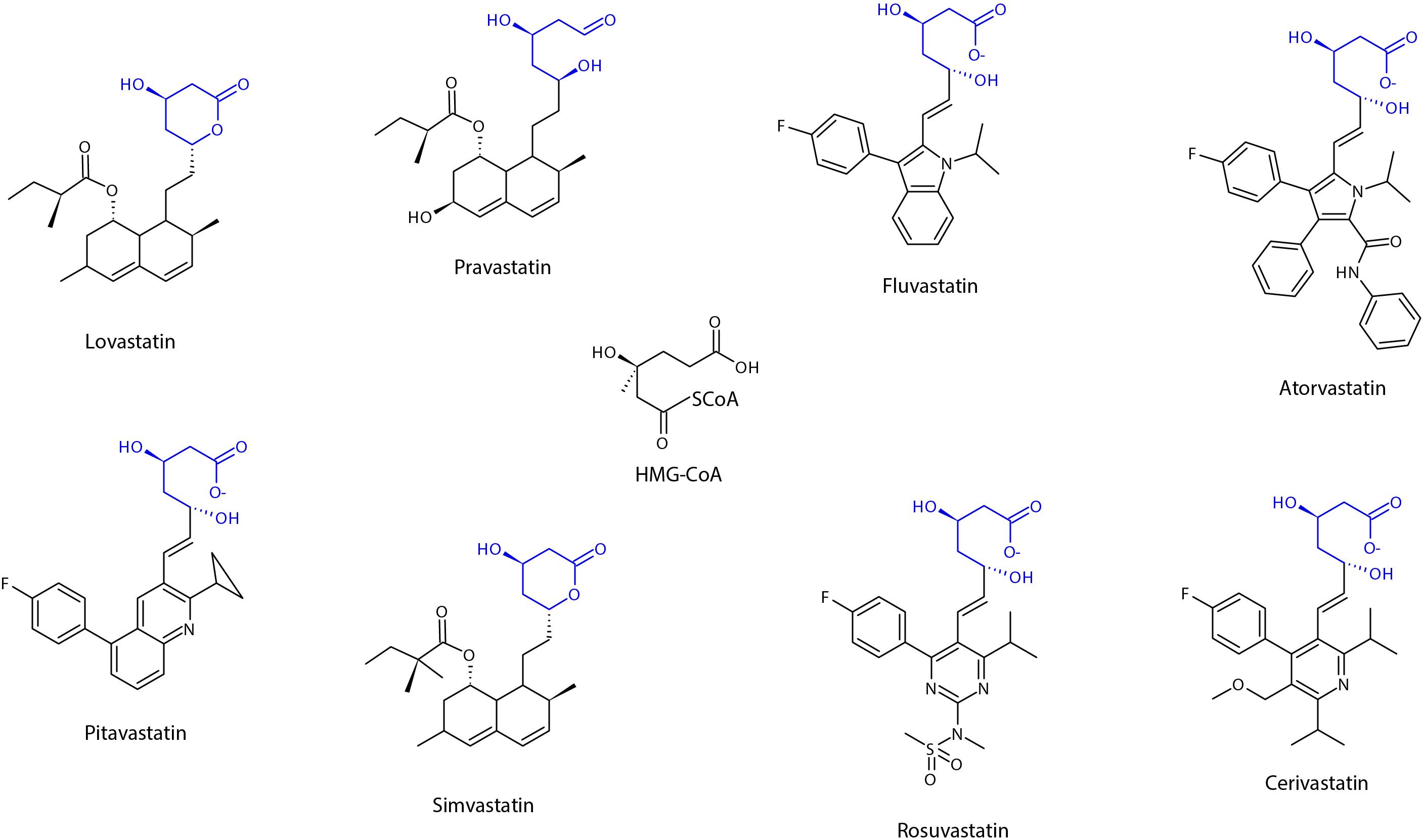
Figure 2. Chemical structure of statins, showing the shared pharmacophore of HMG-CoA. Adapted from Schachter (2005), designed with Chemsketch Version 2019.2.2 (C05E41) (ACD/Labs), Toronto, Ontario Canada.
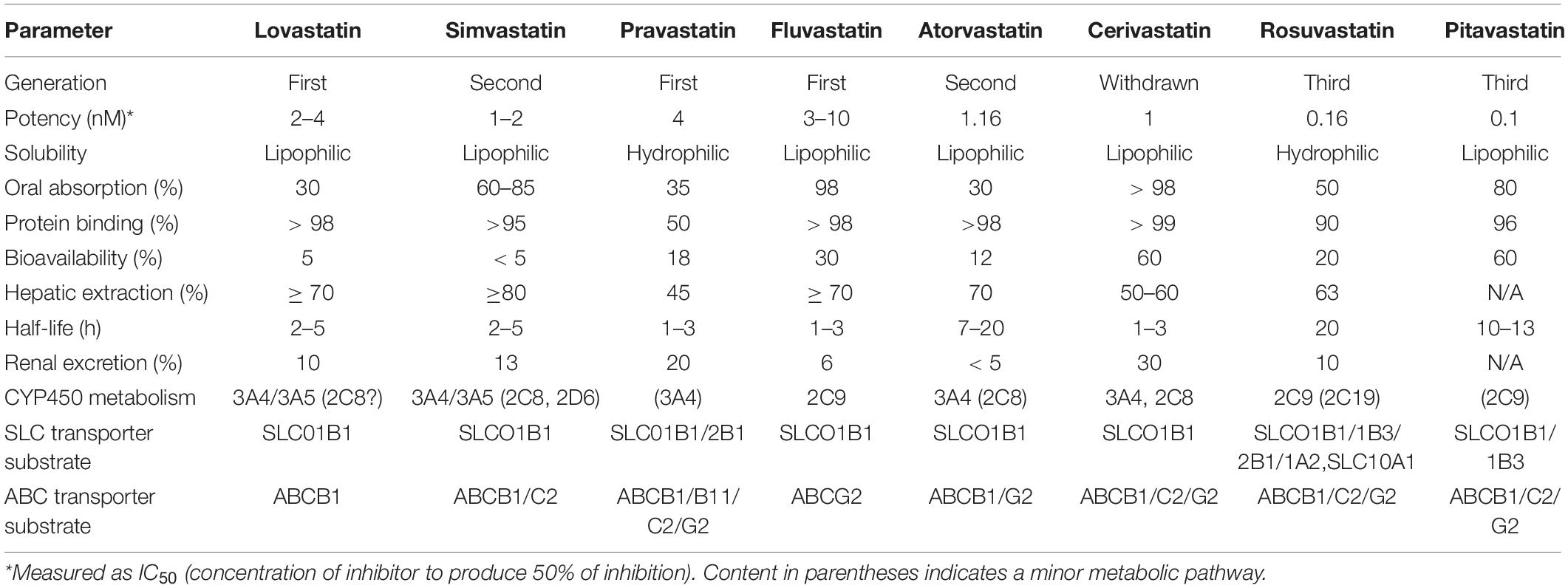
Table 1. Pharmacokinetic profile of statins [adapted and compiled from Bischoff et al. (1997), Hu et al. (2009), Sirtori et al. (2012), Maji et al. (2013)].
Statins, while generally considered to be a well-tolerated class of drugs, are associated with adverse effects. A systematic review and meta-analysis of randomized trials with more than 90,000 participants showed that statins significantly increased the relative and absolute risk of myopathy, renal dysfunction, and hepatic dysfunction (Yebyo et al., 2019). Statin-induced myotoxicity (SIM) and hepatotoxicity are common adverse effects with prevalence rates ranging from 7 to 30% and 2 to 5%, respectively (Bjornsson, 2017; du Souich et al., 2017). They were shown to be dose-dependent (McClure et al., 2007; Raju et al., 2013; Bjornsson, 2017). Unlike myotoxicity and hepatotoxicity, renal dysfunction associated with statin use is unclear and remains debatable (Verdoodt et al., 2018). Besides being highly incident, SIM is a major contributor to statin discontinuation (Ward et al., 2019). Therefore, our narrative review will focus on myotoxicity alone.
Statin-Induced Myotoxicity
Statin-induced myotoxicity presents as fatigue, muscle pain, muscle tenderness, muscle weakness, nocturnal cramping, or tendon pain (Sathasivam and Lecky, 2008). A retrospective investigation of 45 patients with statin-associated myopathy showed that the time to onset of muscle symptoms was 6 months on average, whereas time to symptom resolution was reported to be approximately 2 months after statin discontinuation (Hansen et al., 2005). SIM is concentration-dependent (Hu et al., 2012). Because of the complex interplay between PK and pharmacodynamics (PD), a drug’s exposure is often defined by the dose administered and its effects (Wright et al., 2011; Sandritter et al., 2017). McClure et al. (2007) identified myositis cases from the United Kingdom General Practice Research Database between the year 1999 and 2003. Using a 40 mg mean daily dose equivalent as cutoff threshold, subjects taking statins were classified into high-dose (> 40 mg) and low-dose (< 40 mg) groups. Their analysis showed that a high mean daily statin monotherapy dose conferred a sixfold greater myositis risk (McClure et al., 2007). Supporting that, an observational study [Prediction of Muscular Risk in Observational Conditions (PRIMO)] conducted in 2005 on nearly 8,000 hyperlipidemic patients revealed that SIM in high statin-dosage (80 mg fluvastatin, 40 or 80 mg atorvastatin, 40 mg pravastatin, 40 or 80 mg simvastatin) therapy is greatly under-reported (Bruckert et al., 2005).
It can be challenging to design and report studies on SIM, mainly due to the heterogeneity and non-standardized classification of SIM, especially myalgia. Clinically, SIM can be assessed subjectively and objectively. The most common example for subjective and objective assessment is a patient’s symptoms and plasma creatine kinase (CK) concentrations, respectively. While both subjective and objective parameters are often applied in diagnosing severe SIM such as myopathy and rhabdomyolysis, milder forms of myalgia are commonly diagnosed using symptoms alone. Therefore, the different approaches used may account for the variable incidence rate of SIM, as well as non-replicated findings. Hence, various efforts have been undertaken by the academic field to propose suitable definitions for SIM (Thompson et al., 2016). In 2014, a six category statin-related myotoxicity (SRM) classification was proposed by the European Phenotype Standardization Project (Table 2), which included a rare and recently discovered type of SIM: anti-HMGCR myopathy. It is a subtype of autoimmune-mediated necrotizing myositis, which is characterized by severe proximal weakness, myofiber necrosis, and occasional extramuscular involvement (Pinal-Fernandez et al., 2018). In addition to phenotype grouping, a statin myalgia index score algorithm was proposed by the National Lipid Association (Rosenson et al., 2014).
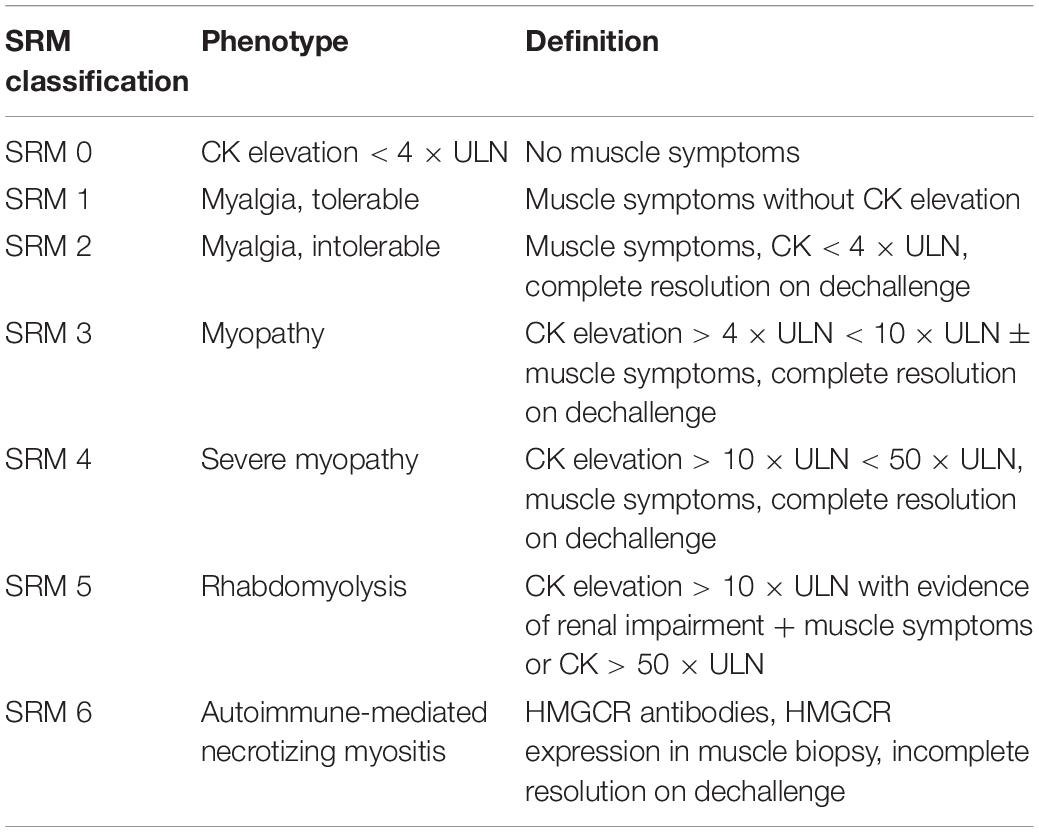
Table 2. Phenotype classification of statin-related myotoxicity [adapted from Alfirevic et al. (2014)].
Despite the plethora of definitions of SIM, all agree that SIM can occur with or without CK elevation. Ranging from myalgia, myositis, myonecrosis, to rhabdomyolysis, it is understood that all of these symptoms represent a gradation along the same pathological pathway or pathways (Thompson et al., 2016). Unfortunately, the pathogenesis of SIM has not been clearly established, and this further complicates SIM categorization. To date, there are several mechanisms proposed, including the following:
1) A supply disruption of farnesyl and geranyl pyrophosphate, which results from statins blocking the downstream products of the mevalonate pathway (Jaskiewicz et al., 2018). These two end products are involved in maintaining cell growth and preventing apoptosis. This idea was proposed when squalene synthase inhibitor, which blocks cholesterol synthesis without affecting other end products of the mevalonate pathway, did not induce myotoxicity in vitro (Flint et al., 1997).
2) A localized impact on cholesterol biosynthesis. Statins have been shown to reduce cholesterol content in skeletal muscle cells (Nishimoto et al., 2003), which will disturb the stability of the cell membrane (Westwood et al., 2005). Because ions channels and transporters are embedded within the membrane, it was suggested that a change in the structure of skeletal cell membrane might disturb ion conductance, thus impairing muscle membrane excitability (Pierno et al., 1995).
3) Mitochondrial dysfunction resulting from coenzyme Q10 depletion (Marcoff and Thompson, 2007; Hargreaves, 2014) and calcium ion (Ca2+) leakage (Lotteau et al., 2019). The latter is a recent proposed pathway. It was shown that statin treatment dissociated FK506 binding protein from the ryanodine receptor 1 (RYR1), which is important for calcium regulation within the sarcoplasmic reticulum (SR) (Brillantes et al., 1994). This led to an excessive release of Ca2+ referred to as Ca2+ sparks (Lotteau et al., 2019). Impaired Ca2+ signaling pathway has been associated with several muscular disorders including myopathy and dystrophy (Agrawal et al., 2018). Because RYR1 is associated with proapoptotic signaling through reactive nitrogen or oxygen species, it was proposed that statin-mediated RNS/ROS-dependent destabilization of SR Ca2+ handling may initiate skeletal muscle toxicity (Lotteau et al., 2019).
While the lactone moiety of statins is suggested to induce SIM (Skottheim et al., 2008), it is important to note that some of the mechanisms were hypothesized based on the notion that statin lactones readily penetrate into muscle cells, facilitated by their lipophilicity (Taha et al., 2016). However, this does not explain SIM cases associated with the use of the hydrophilic statins (rosuvastatin and pravastatin) and simvastatin acid (Link et al., 2008). As discussed, SIM is a dose-dependent class effect of statins (Raju et al., 2013). Besides dose, there are other risk factors that have been identified to predispose a patient to develop SIM. These can be divided into genetic and non-genetic risk factors. Non-genetic factors include age 65 years or older (Schech et al., 2007), small body frame (Pasternak et al., 2002), Asian ethnicity (Birmingham et al., 2015), female gender (Puccetti et al., 2010), renal disease (Hedenmalm et al., 2010), and drug–drug interactions with concurrent administration of CYP3A4 inhibitors (Neuvonen, 2010). The focus of this review is genetic factors.
Improved understanding of relevant genetic factors may offer a strategy to predict the risk of SIM and allow improved targeting of statin therapy to the right patients. There is a wide range of genetic variation that is likely to be associated with the risk of developing SIM. This review aims to summarize the genetic evidence for SIM.
Pharmacogenetics of Statin-Induced Myotoxicity
The study of how genes can impact drug disposition is commonly referred to as pharmacogenetics. This can refer to an individual gene, a therapeutic area, or an individual drug. Variation in a drug’s PK and PD is the focus of pharmacogenetics, which in turn can impact on the safety and efficacy profile of a drug (Daly, 2017). The majority of pharmacogenetic associations were discovered using candidate gene association studies (Feng et al., 2012) or genome-wide association studies (GWAS). For SIM, the first pharmacogenetic association was reported with a hepatocyte uptake transporter: solute carrier organic anion transporter family member 1B1 (SLCO1B1) gene in a 2008 GWAS (Link et al., 2008). Over the subsequent years, studies exploring other genes and their association with SIM have expanded significantly. A list of GWAS studies discussed in this review is as summarized in Table 7. This review will detail the association of genetic variation in enzymes and transporters involved in statin disposition.
Pharmacokinetics
An important PK parameter in drug studies is the concentration of the drug’s moiety known for delivering either therapeutic response or resulting in adverse drug reactions (ADRs). Genetic variation in enzymes and transporters to be discussed here have been reported to influence the concentration of statins in the body, which in turn may increase the risk of developing SIM.
Metabolic Enzymes
Statin-induced myotoxicity is known to be dose-related (Barry et al., 2018). Drug-metabolizing enzymes that affect statin PK by impacting upon oral bioavailability and clearance therefore can alter the risk of SIM. Since CYP450 enzymes were discovered 50 years ago, these enzymes, and their genes, have been the focus of a great deal of research (Estabrook, 2003). The associations between CYP450 genetic variation and variable PK parameters of SIM are discussed below.
Cytochrome P450 2D6
The CYP2D6 gene is the most polymorphic among the CYP450 genes, with more than 100 different alleles that have been identified to date (Nofziger et al., 2020). Many of these variants influence CYP2D6 enzyme function and have been associated with toxicity and altered efficacy for a variety of drugs. The CYP2D6 enzyme metabolizes up to 25% of commonly prescribed medications. However, the role of CYP2D6 in statin metabolism is minor (Figure 3), and research that has investigated the association between genetic variation in CYP2D6 and SIM has reported mixed conclusions (Table 3).
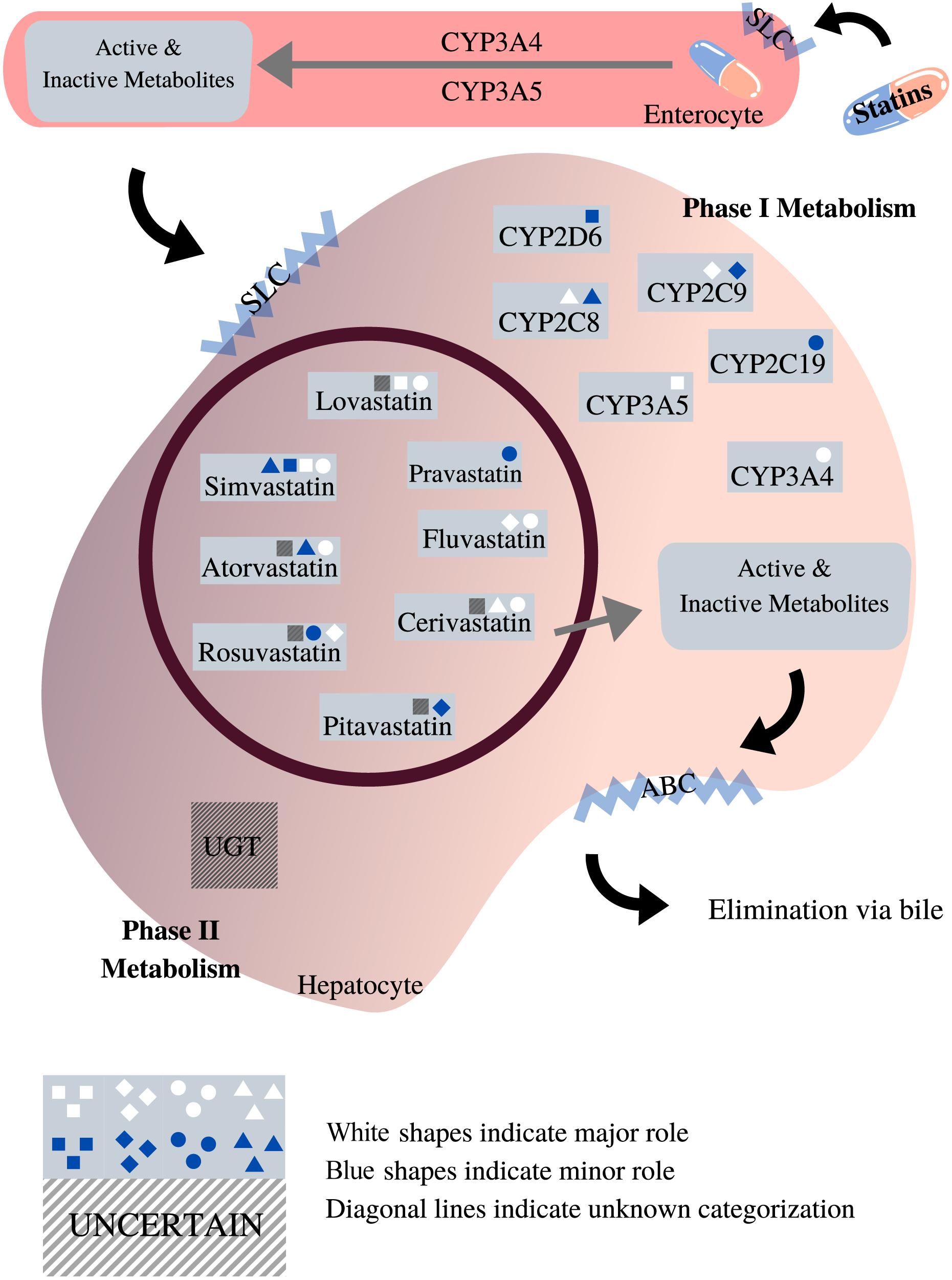
Figure 3. Roles of phases I and II enzymes in statin metabolism [adapted from Goldberg and Roth (1996), Bischoff et al. (1997), Hu et al. (2009), Sirtori et al. (2012), Whirl-Carrillo et al. (2012), Maji et al. (2013), Schirris et al. (2015)]. This diagram depicts the pharmacokinetic pathway of statins, facilitated by endogenous transporters, e.g., SLC and ABC transporters. All statins undergo both phases I and II metabolism prior to elimination. As shown, the impact exerted by different phase I metabolic enzymes on statins varies according to the type of statin. Unlike phase I, the role of phase II metabolic enzymes in statin metabolism is unclear, designed with Canva.com.
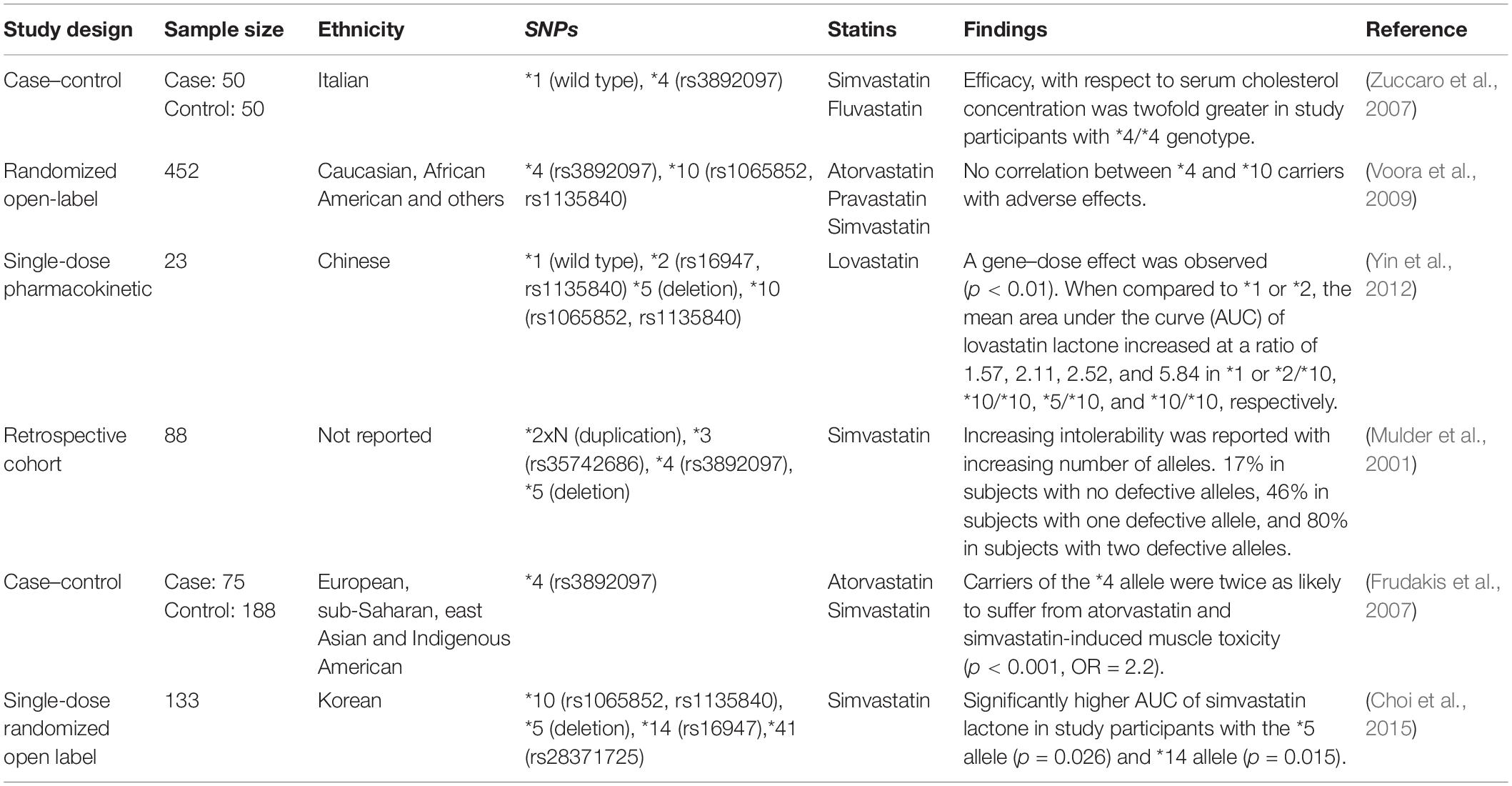
Table 3. Summary of CYP2D6 studies.
Evidence for the role of CYP2D6 in metabolizing statins was initially published by Nordin et al. (1997) where they reported a negative correlation between simvastatin-treated serum cholesterol and the debrisoquine metabolic ratio (ratio of urinary debrisoquine to its hydroxy metabolite). However, this research assessed only 10 samples of simvastatin. With respect to SIM, Frudakis et al. (2007) reported a significant (p = 0.001) association between atorvastatin treatment and SIM in samples with CYP2D6∗4 polymorphism.
As shown in Table 3, results reported by literature exploring the association of CYP2D6 single-nucleotide polymorphisms (SNPs) with SIM are mixed. It is more likely that CYP2D6 in combination with other metabolic pathways discussed below may account for the variation in response and muscle toxicity reported by statin users.
Cytochrome P450 2C9
The CYP2C family consists of CYP2C8, CYP2C9, CYP2C18, and CYP2C19 (Daly et al., 2017). CYP2C9 and CYP2C19 are responsible for metabolizing up to 20% of clinical drugs (Hirota et al., 2013). CYP2C9 is the main enzyme involved in the metabolism of fluvastatin (Fischer et al., 1999) and rosuvastatin (Martin et al., 2003; Luvai et al., 2012) and has a minor role in the metabolism of pitavastatin (Catapano, 2010). The CYP2C9 gene has more than 60 reported genetic variants (Gaedigk et al., 2018), of which two commonly studied CYP2C9 genotypes are the missense variants CYP2C9∗2 (rs1799853) and CYP2C9∗3 (rs1057910). CYP2C9 genetic variations have been reported to influence the PK profile of fluvastatin and pitavastatin. In an analysis of 12 healthy Chinese participants, ∗3 heterozygotes were shown to have a significantly higher area under the concentration–time curve (AUC) of pitavastatin acid (p = 0.000) and pitavastatin lactone (p = 0.001) (Zhou et al., 2013a). With respect to fluvastatin, Kirchheiner et al. (2003) recruited 24 healthy volunteers and the cohort received a 40 mg racemic dose of fluvastatin daily for 2 weeks. The measured AUC of 3S,5R-fluvastatin enantiomer increased in a gene-dose–dependent manner from non-carriers (227μg/L), ∗3 heterozygotes (360μg/L) to ∗3 homozygotes (1126μg/L) (Kirchheiner et al., 2003). In contrast, a PK study that assessed the impact of CYP2C9 genetic variation in 162 study participants reported no association (DeGorter et al., 2013). However, ∗3 allele carriers were shown to have a significantly (p < 0.01) lower plasma level for one of rosuvastatin’s metabolites, N-desmethyl rosuvastatin (Bai et al., 2019).
There are limited studies investigating the association between SIM (Frudakis et al., 2007; Zuccaro et al., 2007) and genetic variability in CYP2C9. With respect to ADRs, a study of 104 Croatian patients reported a 2.5-fold higher risk for homozygotes and heterozygotes of ∗2 and ∗3 alleles in developing ADRs (defined by the study as myotoxicity and hepatotoxicity) (p = 0.037) (Mirosevic Skvrce et al., 2013).
Therefore, CYP2C9 genetic variation may be a risk factor impacting the metabolism of fluvastatin, rosuvastatin and pitavastatin.
Cytochrome P450 2C19
CYP2C19 is the next most polymorphic CYP2C family member gene after CYP2C9. It has more than 30 star alleles in the Pharmvar database (Gaedigk et al., 2019). The two most commonly studied genetic variants that lead to reduced CYP2C19 function are CYP2C19∗2 (rs4244285) and CYP2C19∗3 (rs4986893). Another extensively researched variant is CYP2C19∗17 (rs12248560), which is known to increase the enzymatic activity of CYP2C19 (Scott et al., 2012).
CYP2C19 is reported to play a minor role in the metabolism of rosuvastatin (Bellosta et al., 2004). A PK study in 49 healthy Taiwanese participants investigated the impact of CYP2C19∗2 and ∗3 alleles on rosuvastatin and its metabolites. There were no reported differences in the AUC of rosuvastatin, rosuvastatin lactone, and N-desmethyl rosuvastatin between normal metabolizers of statins and participants with null function CYP2C19 (Finkelman et al., 2015). This result was supported by Lee et al. (2013), who also reported no effect of CYP2C19 variation on rosuvastatin metabolites in a Chinese cohort (Lee et al., 2013). While other statins are not known to be specific substrates of CYP2C19, studies have explored the association of CYP2C19 genotypes with PK and the safety profile of simvastatin and atorvastatin. Choi et al. (2015) showed that the AUC of simvastatin lactone increased significantly with the number of CYP2C19∗2 alleles (p = 0.022). However, no effects were observed with simvastatin acid. In addition, a further study reported that both the CYP2C19∗2 and ∗3 alleles were not associated with SIM induced by simvastatin or atorvastatin (Frudakis et al., 2007).
Cytochrome P450 2C8
The CYP2C8 enzyme is responsible for metabolizing an estimated 5% of prescribed medications such as montelukast, paclitaxel, and rosiglitazone (Naraharisetti et al., 2010; Hanioka et al., 2010; Karonen et al., 2012; Bazargan et al., 2017). To date, CYP2C8 is a known metabolic enzyme for one statin only—cerivastatin, which was withdrawn from the market in 2001 and is no longer in clinical use (Furberg and Pitt, 2001). Cerivastatin when coprescribed with a strong CYP2C8 inhibitor such as gemfibrozil significantly increased the risk of rhabdomyolysis (Kaspera et al., 2010). For this reason, cerivastatin was withdrawn from the market (Furberg and Pitt, 2001). Like other CYPs discussed, CYP2C8 is likely to play a minor role in the metabolism of other statins. With respect to CYP2C8 variation resulting in SIM, two studies identified showed no significant correlation between CYP2C8 SNPs (∗2, ∗3, and ∗4) and SIM (Voora et al., 2009; Marciante et al., 2011).
Cytochrome P450 3A4/3A5
The CYP3A subfamily is the largest human liver cytochrome enzyme system with a 29-fold variability in expression between individuals (Michaels and Wang, 2014). The CYP3A enzyme family comprises CYP3A4 and CYP3A5, which are the main metabolic pathways for most statins (Wang et al., 1991; Prueksaritanont et al., 1997, 2003; Igel et al., 2001; Neuvonen et al., 2008; Rowan et al., 2012). Simvastatin is reported to be preferentially metabolized by CYP3A4 (Prueksaritanont et al., 1997).
Simvastatin and lovastatin are administered as lactone prodrugs and require bioactivation by esterases through hydrolysis (Igel et al., 2001; Egom and Hafeez, 2016) before being oxidized by CYP3A4 into active metabolites (Huttunen et al., 2011). While other statins do not require bioactivation, CYP3A4 is the main enzyme involved in their metabolism (Liu et al., 2019). Administration of CYP3A4 inhibitors such as itraconazole concurrently with statins results in a 2.5-fold increase in the plasma concentration of atorvastatin (Mazzu et al., 2000). Similarly, concurrent administration of cimetidine and lovastatin reduced the formation of 6-hydroxy-lovastatin by 20% (Vyas et al., 1990b). Ciclosporin, a potent CYP3A4 inhibitor, is known to interact with most statins. Studies have reported that statins coadministered with ciclosporin resulted in an approximately 2-, 3-, 6-, and 20-fold increase in AUC for fluvastatin (Goldberg and Roth, 1996), simvastatin (Arnadottir et al., 1993), atorvastatin (Asberg et al., 2001), and lovastatin (Olbricht et al., 1997) respectively. In addition, ciclosporin is reported to impact other pathways important in statin disposition such as inhibiting SLC transporters (Kalliokoski and Niemi, 2009).
For CYP3A5, the most commonly studied polymorphism is CYP3A5∗3 (rs776746), which produces non-functional CYP3A5 enzyme (Kuehl et al., 2001; Lamba et al., 2012). Therefore, for individuals who express CYP3A5, the metabolism of substrate medication may be enhanced (Passey et al., 2011; Lamba et al., 2012). Shin et al. (2011) found that the AUC of atorvastatin lactone was 36% higher in non-expressors (CYP3A5∗3) than expressors (CYP3A5∗1) (p = 0.038). However, this was not replicated by Hermann et al. (2006), who reported a significant (p < 0.01) 2.4-fold higher AUC level of atorvastatin lactone in SIM cases but did not find any association with CYP3A5.
Unlike other CYPs, genes from the CYP3A family are not highly polymorphic (Wang and Sadee, 2012). This may also account for the inconsistent findings for association studies of genetic variation for both CYP3A4 and CYP3A5 with SIM. These studies are summarized in Table 4.
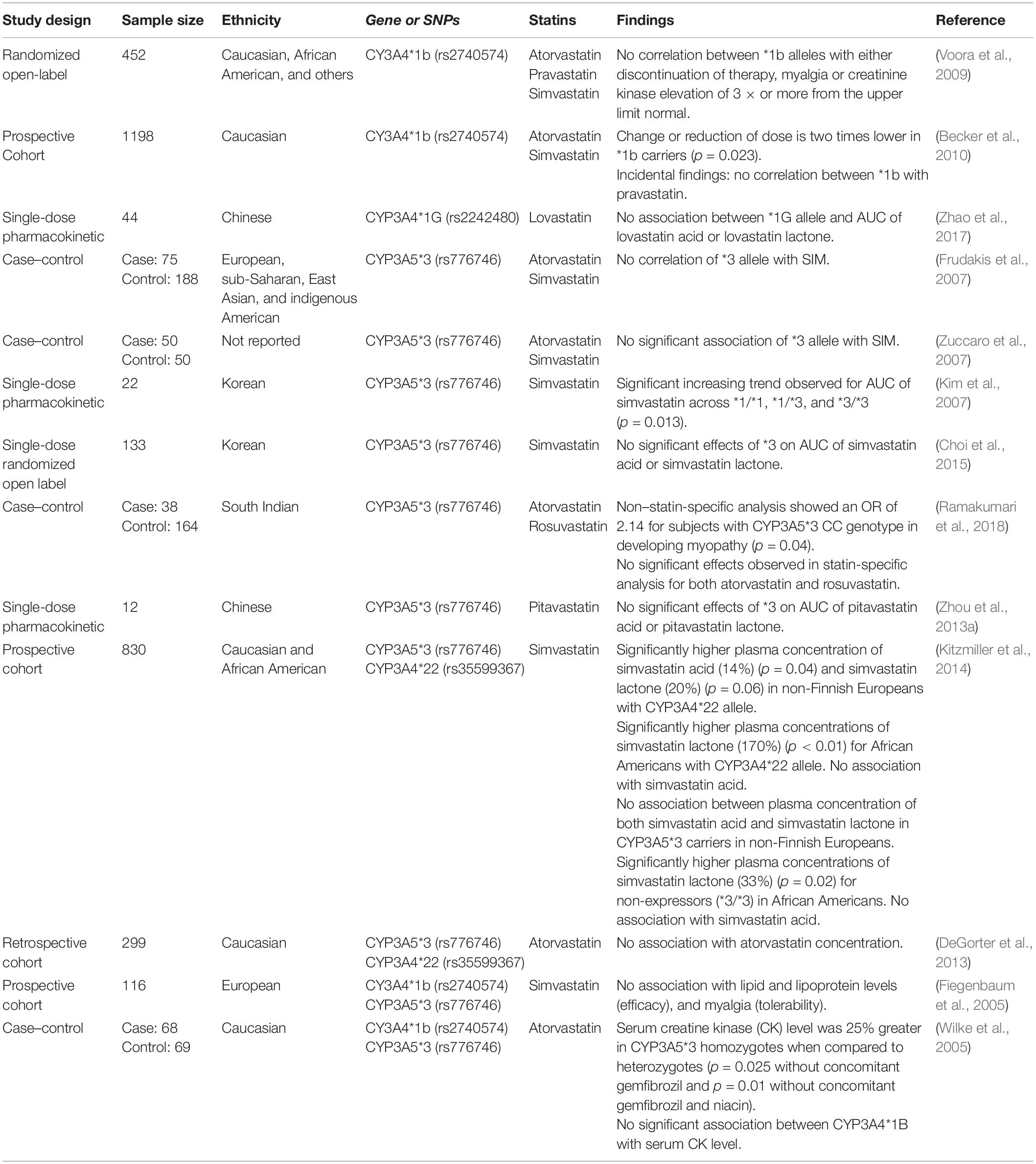
Table 4. Summary of CYP3A studies.
Uridine 5′-Diphosphoglucuronosyltransferase
The UGT is an important enzyme in phase II metabolism. It conjugates xenobiotics or endogenous compounds with glucuronic acid to facilitate their excretion from the body including the lactonization of statins (Schirris et al., 2015; Sanchez-Dominguez et al., 2018). An estimated 10% of administered simvastatin hydroxy acid undergoes lactonization through glucuronidation (Prueksaritanont et al., 2002). This superfamily consists of three subfamilies, UGT1A, UGT2A, and UGT2B (Mackenzie et al., 2005). The UGT1A gene undergoes alternative splicing resulting in the transcription of nine functional isoforms and four pseudogenes (Gong et al., 2001). Among the isoforms, UGT1A1 is the most commonly studied gene associated with drug toxicity (Liu et al., 2014) and disease (Servedio et al., 2005). While there are more than 100 UGT1A1 variants recorded (Barbarino et al., 2014), to date, only UGT1A1∗28 (rs3064744) and UGT1A∗80 (rs887829) have been associated with PK variability and toxicity of statins. UGT1A1∗28 is characterized by the presence of seven TA dinucleotide repeats, instead of the normal six repeats, in the promoter region, which results in decreased UGT1A1 expression (Sanchez-Dominguez et al., 2018). UGT1A∗80 is a SNP located within 300 base pairs of the UGT1A1∗28 TA repeat and in strong linkage disequilibrium with it (Gammal et al., 2016).
As mentioned earlier, the lactone forms of statins are reported to induce SIM (Skottheim et al., 2008). Several studies have investigated the association of UGT1A1 variants and their effect on PK parameters of statin lactones. Stormo et al. (2013) reported a significant reduction in atorvastatin lactone plasma concentrations in UGT1A1∗28 carriers when compared with non-carriers (p < 0.05). This observation was in contrast with Riedmaier et al. (2010), who showed a higher atorvastatin lactone plasma concentration in individuals with the UGT1A1∗28 polymorphism. Riedmaier et al. (2010) did both in vitro and in vivo tests on an estimated 150 human liver microsome samples and 56 Caucasian volunteers, respectively. The genetic analysis conducted in vitro reported a significant gene–dose effect between UGT1A1∗28 allele and the plasma concentration of atorvastatin lactone (p < 0.01). A recent GWAS investigated the levels of atorvastatin and its metabolites, 2-hydroxyatorvastatin, 2-hydroxyatorvastatin lactone, and atorvastatin lactone, in patients taking either 40 or 80 mg of atorvastatin. UGT1A1∗80 was significantly associated (p = 7.25 × 10–16) with a higher metabolic ratio of 2-hydroxyatorvastatin/atorvastatin and 2-hydroxyatorvastatin lactone/atorvastatin lactone (p = 3.95 × 10–15) (Turner et al., 2020).
UGT1A3, another UGT isoform, has also been correlated with the lactonization of atorvastatin. In a study with 24 Korean participants, an increasing trend was observed for the mean atorvastatin lactone AUC across carriers of the UGT1A3∗2 allele (Cho et al., 2012). However, this observation was not supported by Riedmaier et al. (2010). The in vivo test conducted by Riedmaier et al. (2010) showed that only the AUC of atorvastatin metabolite, 2-hydroxyatorvastatin lactone, was significantly (p < 0.05) associated with UGT1A3∗2 allele. No significance was observed with either atorvastatin or atorvastatin lactone (Riedmaier et al., 2010).
Transporters
Endogenous transporters play an important role in the PK of drugs and xenobiotics by controlling the influx of nutrients and efflux of waste material (Mao et al., 2018). ABC and SLC are two major families of transporters (Giacomini et al., 2010), where variations in the genes of these transporters have been shown to influence the disposition of statins and risk of SIM (Niemi, 2010).
Solute Carrier Organic Anion Transporter Family Member 1B1/Organic Anion Transporter Polypeptide-1B1
Previously known as organic anion transporter polypeptide 1B1 (OATP1B1), the SLCO1B1 gene encodes for a transporter located along the basolateral membrane of hepatocytes (Lee and Ho, 2017). This transporter mediates the hepatocellular intake of endogenous molecules and drugs such as statins (Oshiro et al., 2010). Statins exert their lipid-lowering mechanism and undergo metabolism, within hepatocytes (Stancu and Sima, 2001; Whirl-Carrillo et al., 2012). Therefore, SLCO1B1 polymorphisms are expected to impact upon statin disposition and contribute to SIM.
An association between the genetic variation of SLCO1B1 gene and SIM was initially discovered by a GWAS comprising of 85 participants with suspected simvastatin-myopathy and 90 matched controls. A significant (p = 4 × 10–9) association was found with the rs4363657 intronic SNP with an odds ratio (OR) of 4.3 per variant allele and 17.4 for both alleles. The rs4363657 (SLCO1B1) is in almost complete linkage disequilibrium with a missense variant, rs4149056. This SNP (rs4149056) was significantly associated (p < 2 × 109) with SIM, with an OR of 4.5 and 16.9 in heterozygotes and homozygotes, respectively. In a subsequent replication study conducted on 21 participants from the Heart Protection Study, rs4149056 association with SIM was found to be significant (p = 0.004). In addition, further SNPs discovered via the GWAS (rs2306283, rs11045819, and rs34671512) were reported to have marginal to no increased risk of SIM (Link et al., 2008).
Since then, the two most commonly studied SLCO1B1 SNPs associated with SIM have been rs4149056 and rs2306283. The SLCO1B1 alleles defined by these SNPs are ∗5 (rs4149056) and ∗1b (rs2306283), whereas individuals who carry both the alleles are assigned the ∗15 haplotype (Nozawa et al., 2002). The reference allele (wild type) for SLCO1B1 is defined as ∗1a with normal transporter function (Ramsey et al., 2014). SLCO1B1 ∗1b is reported to have functional activity parallel to ∗1a (Tirona et al., 2001; Nozawa et al., 2002), although substrate-dependent activity has been reported for pravastatin (Mwinyi et al., 2004; Maeda et al., 2006). Meanwhile, SLCO1B1 ∗5 is associated with reduced transporter function (Tirona et al., 2001). The ∗15 haplotype, initially discovered in a Japanese population, is reported to be a decreased function variant (Nozawa et al., 2002; Ramsey et al., 2014). A summary of commonly studied SLCO1B1 SNPs is listed in Table 5.
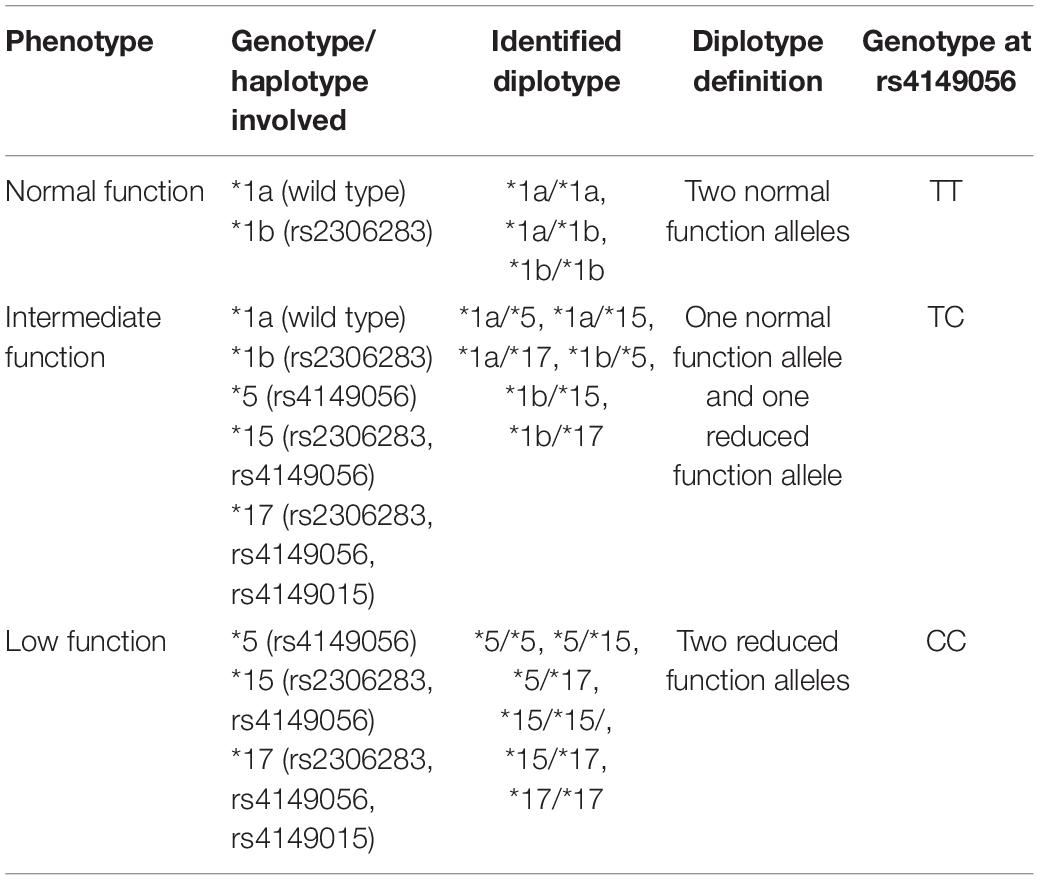
Table 5. Common SNPs of the SLCO1B1 gene associated with SIM [adapted from Ramsey et al. (2014)].
To elucidate the pharmacogenetic association of SLCO1B1 transporters and SIM, it is crucial to understand the PK relationship between statins and SLCO1B1 genotype. Mwinyi et al. (2004) conducted a PK study of pravastatin in 30 healthy male subjects. The AUC in participants homozygous for ∗1a and ∗1b did not show any significant differences. However, the AUC in individuals carrying the ∗5 allele was twofold higher than ∗1b (p = 0.002) and ∗1a (p = 0.049) (Mwinyi et al., 2004). Similarly, several studies have reported no significant difference between wild type and ∗1b homozygotes in the AUC measured for rosuvastatin (Choi et al., 2008; Lee et al., 2013; Liu et al., 2016) and simvastatin acid (Choi et al., 2015). However, when ∗1b was analyzed in 299 participants taking atorvastatin or rosuvastatin, a lower atorvastatin plasma concentration was reported (p < 0.01) in carriers of ∗1b (DeGorter et al., 2013). Pasanen et al. (2006) genotyped 32 young healthy Caucasians after a single dose of 40 mg simvastatin and showed that the peak plasma concentration of active simvastatin acid in ∗5 homozygotes was 162 and 200% higher when compared with ∗5 heterozygotes and non-∗5 carriers. In addition, further studies have supported the association of significantly elevated simvastatin acid AUC with the SLCO1B1∗5 allele (Choi et al., 2008; Voora et al., 2009; Jiang et al., 2017).
Furthermore, de Keyser et al. (2014) analyzed the SLCO1B1 genotype from an estimated 1,900 subjects and found that those currently prescribed simvastatin and also carriers of two ∗5 alleles had a 1.7-fold higher risk of statin dose reduction or change of cholesterol lowering drug due to statin-associated ADRs (p = 0.0033). When compared with wild type, ∗5 homozygotes were observed to achieve 144, 100, and 65% greater mean AUC for atorvastatin, 2 hydroxy-atorvastatin, and rosuvastatin respectively (Pasanen et al., 2007).
As discussed earlier, the GWAS conducted by Turner et al. (2020) also reported a significant association between the ∗5 allele and the level of atorvastatin (p = 2.21 × 10–6) and its metabolite, 2-hydroxy atorvastatin (p = 1.09 × 10–6). The observed significant association between ∗5 alleles and the plasma concentration of statin was supported by other studies on pravastatin (p = 0.01) (Ho et al., 2007), rosuvastatin (p = 0.019) (Lee et al., 2013), atorvastatin (p < 0.05) (DeGorter et al., 2013), and the AUC of both pitavastatin acid (p = 0.00) and pitavastatin lactone (p = 0.007) (Zhou et al., 2013a). Birmingham et al. (2015) reported that the average AUC of atorvastatin, rosuvastatin, and simvastatin acid was higher in SLCO1B1 ∗5 allele carriers. This trend was similar across all three ethnic groups (Chinese, Japanese, and European). Meanwhile, no effect was reported with simvastatin lactone.
Because both ∗5 and ∗15 result in impaired SLCO1B1 transporter function, the PK impact of ∗15 on statins is expected to be similar to that of ∗5. Choi et al. (2008) found that Korean carriers of ∗15 had a twofold higher AUC of rosuvastatin compared with ∗1a and ∗1b homozygotes. This observation was supported by a recent study in a Chinese population (p = 0.015) (Liu et al., 2016). For pravastatin and pitavastatin, carriers of the ∗15 haplotype had a twofold and threefold higher AUC when compared with ∗1a, respectively (Deng et al., 2008). Other PK studies on either pitavastatin (Oh et al., 2013) or pravastatin (Niemi et al., 2004; Ho et al., 2007) also reported significant associations with the ∗15 allele. With respect to atorvastatin and lovastatin, significantly higher AUCs of atorvastatin acid (p = 0.0018), 2-hydroxyatorvastatin acid (p = 0.0123), and lovastatin acid (p < 0.005, p = 0.072) were associated with the ∗5 or ∗15 alleles (Tornio et al., 2015). No significant association was found for the lactone forms of both atorvastatin and lovastatin (Lee et al., 2010; Tornio et al., 2015; Zhao et al., 2017).
A recent study conducted with Korean patients investigated the impact of SLCO1B1 genetic variation on rosuvastatin according to phenotypes (Table 5), demonstrating that the mean plasma concentration of rosuvastatin showed an increasing trend across groups of normal function, intermediate function to low function (Kim et al., 2019).
Solute carrier organic anion transporter family member 1B1 polymorphisms and statin-induced myotoxicity
A recent GWAS that aimed to validate genetic risk factors for statin-induced myopathy in 128 myopathy cases successfully replicated the prior association of the SLCO1B1∗5 allele with SIM (Carr et al., 2019). This SNP was significantly associated with statin-induced myopathy both in the initial case–control discovery cohort (p = 2.5 × 10–9), the replication study (p = 0.001), simvastatin cohort validation (p = 1.3 × 10–11), and final meta-analysis (p = 2.63 × 10–18). The replication cohort consisted of 19 myopathy cases and 585 statin-tolerant controls. The OR of SIM reported from this replication study was 3.98 for SLCO1B1∗5 allele. Meanwhile, the meta-analysis combined the discovery and replication cohorts, resulting in a comparison of 271 myopathy cases with 7,493 controls. The OR of statin-associated SIM with the ∗5 allele was 2.99 in the meta-analysis (Carr et al., 2019). This finding has been supported by other studies, which reported an increased risk of SIM in ∗5 allele carriers. One study compared 12 cases and 39 controls and reported a threefold increased risk of myopathy in carriers of the ∗5 allele (p = 0.032) (Brunham et al., 2012). Meanwhile, Carr et al. (2013) showed that ∗5 heterozygote carriers have an OR of 2.13 for the risk of myopathy (p = 0.014). In addition, a recent GWAS on the circulating levels of atorvastatin and its metabolites reported SLCO1B1 ∗5 allele was significantly associated with increased muscular symptoms (p = 0.016) and atorvastatin intolerance (p = 0.014) at an OR of 4 and 1.5, respectively (Turner et al., 2020).
A study that investigated ∗1b, ∗5, and ∗15 found that the presence of the ∗5 allele was associated with statin intolerance. In this study, intolerance was defined as any biochemical abnormality (CK or alanine aminotransferase levels) or prescribing change (statin switch or dose change) recorded. This study showed that the ∗5 allele was significantly (p = 0.02) associated with statin intolerance with an OR of 1.14 (Donnelly et al., 2011). In addition, a recent case–control study in a cohort of 606 Europeans reported that of 12 SNPs in nine candidate genes, only the ∗5 allele was significantly associated with SIM (OR = 1.73, p = 0.01) (Bakar et al., 2018).
There are several meta-analyses assessing the association of SLCO1B1∗5 with SIM. The majority of them reported a significant association between the ∗5 allele and increased risk of SIM with an OR ranging from 1.57 to 2.09 (Hou et al., 2015; Lee and Chun, 2018; Xiang et al., 2018). Interestingly, another meta-analysis that investigated 13 studies showed that SLCO1B1 ∗5 allele was associated with overall statin-induced adverse reactions, including SIM (Jiang et al., 2016). Furthermore, a meta-analysis on simvastatin specifically with 1,360 cases and 3,082 controls showed that ∗5 allele carriers had a threefold higher risk of SIM compared with non-carriers (Hou et al., 2015). Similarly, Xiang et al. (2018) with a larger number of cases (3265) and controls (7743) reported an OR of 2.35 for SIM in individuals receiving simvastatin.
Simvastatin is the most investigated statin with respect to SIM. However, other statins have been reported to significantly induce SIM. An observational case–control study in 76 European cases of muscular intolerance reported a threefold (OR = 2.7) risk for developing SIM with atorvastatin (p < 0.001) in individuals carriers of the ∗5 allele (Puccetti et al., 2010). For rosuvastatin, a significant association (p = 0.007) with SIM was observed in a Chinese population study consisting of 148 myotoxicity cases and 255 controls (Liu et al., 2017). In addition, Bai et al. (2019) reported a significant (p = 0.0052) association between ∗5 alleles and rosuvastatin-induced myopathy in an observational study of 758 Chinese patients with coronary artery disease. There is limited evidence with respect to SLCO1B1∗5 and SIM in patients prescribed fluvastatin or pravastatin (Liu et al., 2017).
However, there have also been inconsistent findings reported for both SIM and PK variability with SLCO1B1 polymorphisms. A PK study conducted with atorvastatin-related myopathy cases found that elevated AUCs of atorvastatin lactone and p-hydroxyatorvastatin lactone among patients suffering from atorvastatin-related myopathy were not associated with SLCO1B1 polymorphisms or CK levels (Hermann et al., 2006). In addition, further studies have reported no significant association between SLCO1B1∗5 and SIM for simvastatin (Khine et al., 2016; Liu et al., 2017), atorvastatin (Brunham et al., 2012; Carr et al., 2013; Hou et al., 2015; Liu et al., 2017; Ramakumari et al., 2018), and rosuvastatin (Puccetti et al., 2010; Danik et al., 2013; Ramakumari et al., 2018). Hubáček et al. (2015a) investigated the SNP rs4363657, which is in linkage disequilibrium with ∗5, and they too, did not find an association with SIM. Two possible factors for the discordant results may be ethnic-dependent variation (Lee and Ho, 2017) and SLCO1B1 transporter substrate specificity (Niemi et al., 2011).
Ethnicity plays a major role in genetic variation. With regard to SLCO1B1 polymorphisms, the prevalence of ∗5 is lower in African Americans or individuals of African descent (Ho et al., 2007; Santos et al., 2011; Khine et al., 2016), but higher in American Indians, where the genotype frequency observed is doubled (Santos et al., 2011). Interestingly, the association of SIM differs between ethnicities despite having similar allele frequencies. Although the genome aggregation database (gnomAD) reports the allele frequency of SLCO1B1∗5 (rs4149056) in both non-Finnish Europeans and East Asians to be similar (∼15%), the significant association between rosuvastatin and SIM reported by Liu et al. (2017) among Chinese ∗5 allele carriers was not observed in individuals of European ancestry (Puccetti et al., 2010; Danik et al., 2013). However, other factors (study design, adequate sample size, and also the unexplainable higher plasma exposure of rosuvastatin observed in Asians when compared to non-Finnish Europeans) should be considered during interpreting ethnicity-driven differences (Lee et al., 2005). Another potential factor is the physical properties of different statins. Hydrophilic statins require functional SLC transporters in order to move across membranes or into cells. In contrast, lipophilic statins should be able to diffuse through the lipid membrane into extrahepatic tissues such as muscle, possibly increasing the risk of SIM (Bruckert et al., 2005; Neuvonen et al., 2006). This hypothesis is supported by PK studies reporting the impact of SLCO1B1 polymorphisms on the acid form of simvastatin and rosuvastatin but not their lipophilic lactone structures (Pasanen et al., 2006; Choi et al., 2008).
A further factor that has been associated with SLCO1B1 polymorphisms and SIM is an individual’s vitamin D status. Studies have reported an association between serum levels of vitamin D and SIM (Ahmed et al., 2009; Morioka et al., 2015; Glueck et al., 2017). Linde et al. (2010) genotyped a cohort of 46 patients on statin treatment and measured 25-hydroxyvitamin D levels. This analysis showed that carriers of the ∗5 allele had a threefold higher risk (p = 0.07) of developing myalgia regardless of their vitamin D status (sufficient or deficient). In contrast, Alghalyini et al. (2018) showed that the ∗5 allele increased the risk of SIM in vitamin D deficient subjects only (p = 0.022). Despite the inconsistency, these studies show the detrimental effect of the ∗5 allele.
From this review, it is evident that there is a strong association between SLCO1B1 rs4149056 and SIM. The Clinical Pharmacogenetics Implementation Consortium (CPIC) has conducted a review on the available evidence and published a peer-reviewed guideline to assist in prescribing simvastatin in individuals who carry a higher risk of developing SIM (Ramsey et al., 2014). The clinical implementation of SLCO1B1 testing is discussed later in this review.
Other Solute Carrier Transporters
SLCO1B3 and SLCO2B1 are two other members of the SLC transporter family. Similar to SLCO1B1, both SLCO1B3 and SLCO2B1 can be transporters for statins (Grube et al., 2006; Alam et al., 2018). However, their roles in statin disposition are still unclear because of the poor classification of genetic variation in these transporter genes. A PK study on rosuvastatin plasma concentration found no association with SLCO1B3 (rs7311358) and SLCO2B1 (rs12422149) (DeGorter et al., 2013). Furthermore, a study in 758 Chinese participants reported that SLCO1B3 (rs7311358) was not significantly associated with variation in the plasma concentration of rosuvastatin and its two metabolites (Bai et al., 2019).
ATP-Binding Cassette Subfamily B Member 1
The ATP-binding cassette subfamily B member 1 (ABCB1), commonly known as P-glycoprotein or multidrug resistance protein 1 (Dean et al., 2001), is an efflux pump transporting compounds out of the cell (Chang, 2003). Initially known to be responsible for chemoresistance (Juliano and Ling, 1976; Sharom, 2008), the focus on its role in drug disposition has expanded from cytotoxic medications to include many other drugs (Leschziner et al., 2007).
There are more than 3,000 ABCB1 variants, with SNPs 1236T > C (rs1128503), 2677T > G/A (rs2032582), and 3435T > C (rs1045642) being the three most studied (Wang et al., 2005). These SNPs have not been sufficiently characterized to establish a confident genotype-to-phenotype prediction, largely due to discordant study results. For example, studies of the rs1045642 SNP have reported an increase (Hitzl et al., 2001) as well as decrease (Nakamura et al., 2002) in ABCB1 expression.
The three SNPs mentioned above are in strong linkage disequilibrium, which leads to common haplotypes of 1236C:2677G:3435C and 1236T:2677T:3435T (Wang et al., 2005). These haplotypes are defined by two common star allele designations: ABCB1∗1 and ABCB1∗2. However, the lack of standardization in referencing and assigning ABCB1 haplotypes has resulted in inconsistent nomenclature (Hodges et al., 2011; Kalman et al., 2016), and therefore, assessment of the impact of ABCB1 genetic variation on statin PK has been variable (Table 6).
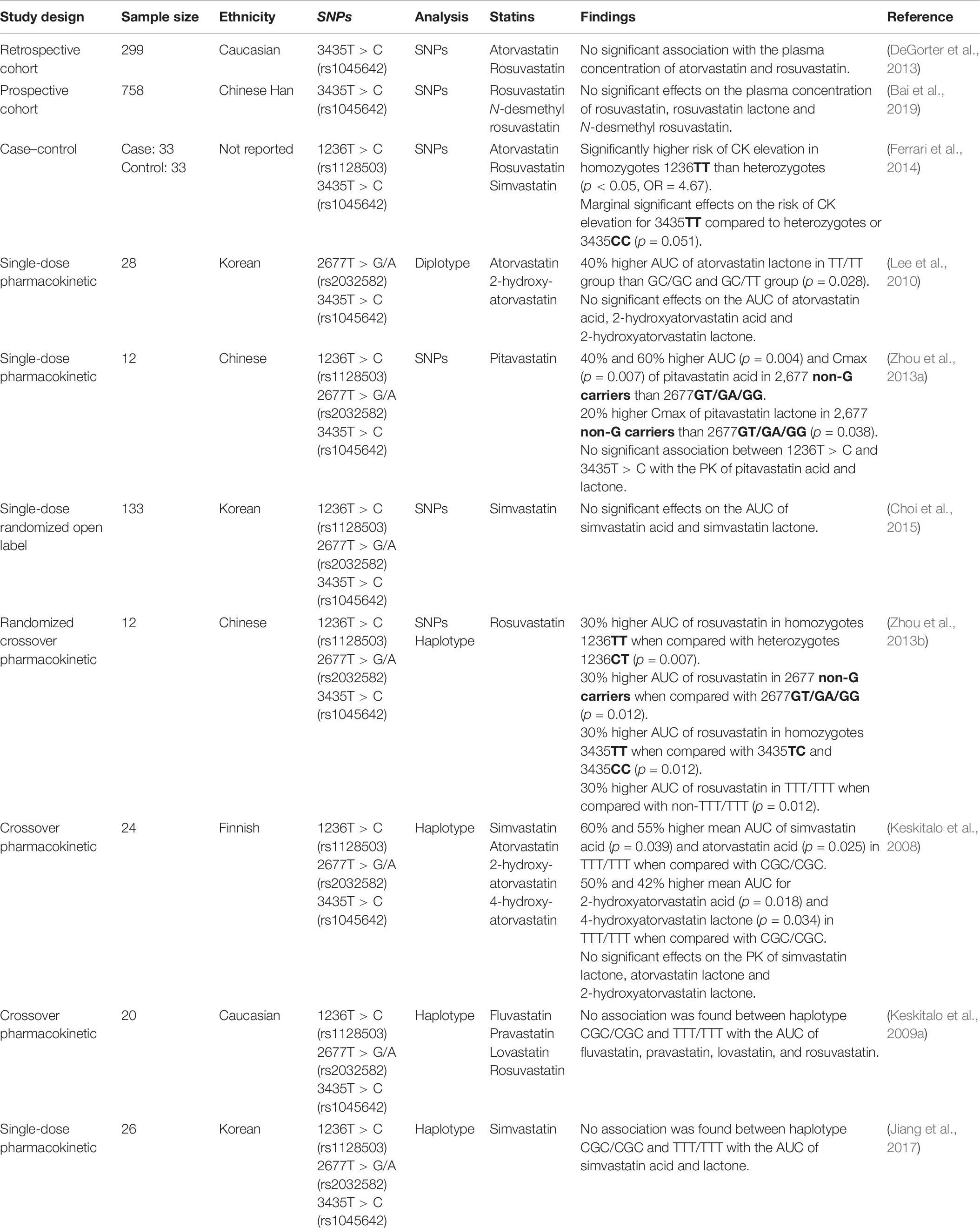
Table 6. Summary ABCB1 studies.

Table 7. List of GWAS studies reviewed.
Similarly, the association between ABCB1 genotypes and SIM is unclear. For the SNP 1236T > C (rs1128503), one study showed a significant (p = 0.049) association between the C allele and development of myalgia (Fiegenbaum et al., 2005). In contrast, a study conducted by Becker et al. (2010) showed no association of the 1236T > C SNP (rs1128503) and dose reduction or statin change during simvastatin or atorvastatin therapy. Similarly, no association was found in a study by Hermann et al. (2006), which compared 15 controls and 14 cases of atorvastatin-induced myopathy. These studies also analyzed SNPs 2677T > G/A (rs2032582) and 3435T > C (rs1045642). While no significant results were observed for these SNPs in two studies (Hermann et al., 2006; Becker et al., 2010), Fiegenbaum and colleagues (2005) (Fiegenbaum et al., 2005) reported that the SNPs 3435T > C (rs1045642) and 2677T > G/A (rs2032582) were significantly (p = 0.03) associated with myalgia development (Fiegenbaum et al., 2005). In addition, Morimoto et al. (2005) reported a significant association between the 2677T > G/A SNP (rs2032582) and simvastatin or atorvastatin-induced myopathy (p < 0.05). With respect to SNPs 3435T > C (rs1045642), the results reported by Fiegenbaum et al. (2005) were not replicated by two further studies. Hoenig et al. (2011) investigated the impact of SNPs 3435T > C (rs1045642) on the efficacy and safety of atorvastatin. Of 154 participants enrolled in the study, 10 reported myalgia. Analysis showed that the T allele of 3435T > C (rs1045642) was significantly (p = 0.043) associated with atorvastatin-induced muscle symptoms (Hoenig et al., 2011). Similarly, this observation was supported by Becker et al. (2010), who found that the T allele of 3435T > C (rs1045642) resulted in a higher risk of dose reduction or statin change during simvastatin or atorvastatin therapy. However, the association did not reach significance.
In addition to individual SNP analysis, Fiegenbaum et al. (2005) investigated the association of SIM with eight haplotypes. Only one haplotype (1236T:2677non-G:3435T) showed a significant association (p = 0.027).
The genetic variations of ABCB1 have been reported to affect statin disposition. However, functional characterization of ABCB1 SNPs is necessary to understand its impact on statin PK and subsequently the association with the risk of SIM.
ATP-Binding Cassette Subfamily G Member 2
ATP-binding cassette subfamily G member 2 (ABCG2) belongs to one of the seven subfamilies across the 48 ABC transporters in humans (Dean et al., 2001), with physiological roles in regulating the movement of substances across human tissues (Robey et al., 2009; Cuperus et al., 2014). To date, more than 200 substrates of ABCG2 have been identified including antibiotics, statins, and anticoagulants (Horsey et al., 2016; Heyes et al., 2018). The most extensively studied variant is a missense variant 421C > A (rs2231142). This SNP reduces the transporter activity of ABCG2 by enhancing proteasomal degradation, which leads to impaired protein expression (Yanase et al., 2006; Furukawa et al., 2009). Another common SNP is 34G > A (rs2231137). However, this SNP does not affect protein expression levels when compared with wild type (Imai et al., 2002; Kondo et al., 2004; Yanase et al., 2006; Tamura et al., 2007). Similarly, PharmGKB, an established pharmacogenomic online database, have reported no functional effects for this SNP (Fohner et al., 2017).
The statins, atorvastatin, fluvastatin, pitavastatin, pravastatin, and rosuvastatin are known substrates of ABCG2 (Fohner et al., 2017). Rosuvastatin is the most studied statin with respect to SNP 421C > A (rs2231142). In a PK study in 14 healthy Chinese participants, a single oral dose of 20 mg rosuvastatin showed a doubling in AUC of rosuvastatin in carriers of the A allele (p = 0.018) (Zhang et al., 2006). This was replicated in a bigger Chinese cohort consisting of 62 samples by Wan et al. (2015). Interestingly, the positive PK association between ABCG2 421C > A (rs2231142) with rosuvastatin in Chinese is well replicated in the European population. For example, a gene–dose effect was observed in a PK study on 32 healthy Finnish subjects. The AUC of rosuvastatin in homozygote A allele carriers was 50% (p = 0.004) and 60% (p < 0.001) higher than heterozygotes and non-carriers, respectively (Keskitalo et al., 2009b). This is supported by further studies, which reported a higher plasma concentration of rosuvastatin in carriers of the A allele (DeGorter et al., 2013; Birmingham et al., 2015). A recent study by Bai et al. (2019) investigated the impact of ABCG2 421C > A (rs2231142) on rosuvastatin and its metabolites: rosuvastatin lactone and N-desmethyl rosuvastatin. The AUC measured in A allele homozygotes was significantly higher when compared with heterozygotes and non-carriers for all three compounds (Bai et al., 2019). The association observed with rosuvastatin and N-desmethyl rosuvastatin was also supported by Lee et al. (2013).
The association between ABCG2 421C > A (rs2231142) and the AUC of statin is evident in other studies with respect to rosuvastatin (Zhou et al., 2013b; Liu et al., 2016; Kim et al., 2019), atorvastatin (Keskitalo et al., 2009b; Birmingham et al., 2015), and simvastatin acid (Birmingham et al., 2015; Choi et al., 2015). However, contrasting and negative findings have also been published. No association of rs2231142 with AUC was found for rosuvastatin (Kim et al., 2017), atorvastatin (DeGorter et al., 2013), pitavastatin (Ieiri et al., 2007; Oh et al., 2013; Zhou et al., 2013b), simvastatin lactone (Birmingham et al., 2015; Choi et al., 2015), lovastatin acid (Zhao et al., 2017), and lovastatin lactone (Zhao et al., 2017). Furthermore, no association between this SNP and CK elevation was reported in a study cohort taking either atorvastatin, rosuvastatin, or simvastatin (Ferrari et al., 2014). With respect to SIM, the findings were contrary. While there is a significant association between ABCG2 421C > A (rs2231142) and the PK of rosuvastatin, the results were not significantly associated with SIM. As an example, in a study of 758 participants, 51 patients developed myopathy after rosuvastatin treatment. No significant association was found between the 421C > A SNP (rs2231142) and SIM (Bai et al., 2019).
In contrast, Mirosevic and colleagues (2015) (Mirosevic Skvrce et al., 2015) genotyped and compared 60 patients who experienced dose-related ADRs with atorvastatin with 90 matching controls. The ADRs included myotoxicity and hepatotoxicity, where myotoxicity constituted a majority of the events. Carriers of the A allele were determined to have a 2.9-fold greater risk of developing dose-related atorvastatin ADRs (p = 0.016) (Mirosevic Skvrce et al., 2015). This effect was more prominent with fluvastatin, where the risk of ADRs associated with A allele carriers was 4.76-fold when compared with non–A-allele carriers (CC homozygotes) (p = 0.014) (Mirosevic Skvrce et al., 2013). However, this is a cohort targeted study where all of the samples studied were renal transplant patients taking fluvastatin. Therefore, interpreting and generalizing the results should be done with caution due to confounding factors such as the concurrent administration of immunosuppressants, which might be substrates or inhibitors of ABCG2 transporters.
As for 34G > A ABCG2 SNP (rs2231137), Wan et al. (2015) reported a significantly higher AUC of rosuvastatin in homozygotes of the A allele (p < 0.01) among 62 healthy Chinese volunteers. However, this association with rosuvastatin was not replicated by Zhou et al. (2013b). Furthermore, no significant effects on pitavastatin acid and its lactone form were reported (Zhou et al., 2013a).
ATP-Binding Cassette Subfamily C Member 2
Adenosine triphosphate-binding cassette subfamily C member 2 (ABCC2) or multidrug resistance protein 2 is also a member of the wide ABC transporter family. Highly expressed in the liver, kidney, and intestine (Nguyen et al., 2013), this efflux membrane transporter holds important roles in drug disposition. Although all statins (simvastatin, pravastatin, pitavastatin, fluvastatin, atorvastatin, rosuvastatin, lovastatin) are substrates of ABCC2 (Ellis et al., 2013), PK studies on statins and genetic variation in the ABCC2 transporter are lacking. The most commonly studied ABCC2 SNP with respect to statin disposition is 1249G > A (rs2273697). However, the functional characterization of this SNP is unclear, although it may impair the transporter function of glutathione and glucuronide-conjugated substrates (Kim et al., 2010; Megaraj et al., 2011).
Studies investigating the effect of the 1249G > A SNP (rs2273697) on the PK profile of statins reported no association between the variant allele and plasma concentration or AUC of rosuvastatin (DeGorter et al., 2013), atorvastatin (DeGorter et al., 2013), pravastatin (Niemi et al., 2004; Ho et al., 2007), and pitavastatin (Oh et al., 2013). A similar trend was observed with SIM. Becker et al. (2013) studied the impact of ABCC2 genotypes on any dose reduction or statin switching events during simvastatin and atorvastatin therapy. With a sample of 1,014 subjects (789 simvastatin users, 225 atorvastatin users) from the Rotterdam study, they reported that the number of dose reduction or statin switching events significantly (p = 0.045) increased with the number of A alleles. However, this finding was not replicated in a separate statin-specific analysis on simvastatin and atorvastatin (Becker et al., 2013).
Another promising variant is ABCC2 -24C > T (rs717620). Oh et al. (2013) reported a significant association between the AUC of pitavastatin with SNPs ABCC2 -24C > T (rs717620). Participants homozygous for the T allele had a lower AUC of pitavastatin when compared with non–T-allele carriers (p = 0.028).
Pharmacodynamics and Other Important Genetic Factors
To this point, genetic variation affecting the PK of statins has been discussed. However, there are other contributors reported to have a role in SIM. This includes proteins which are involved in the PD of statins, synthesis of muscle toxicity biomarkers, or immunologically associated ADRs. These are briefly discussed below.
Glycine Amidinotransferase
Glycine amidinotransferase (GATM) gene encodes for the L-arginine:glycine amidinotransferase enzyme. This enzyme converts L-arginine into guanidinoacetate, a rate-limiting step in creatine synthesis (Wyss and Kaddurah-Daouk, 2000). The potential role of GATM genetic variations contributing to SIM was highlighted by Mangravite et al. (2013), this study explored expression quantitative trait loci (eQTL) in SIM cases. In this study, lymphoblastoid cell lines were exposed to simvastatin and the expression of various genes assessed. The change in expression of the GATM gene was strongly associated with statin exposure, and a further analysis of SNPs across the GATM locus revealed rs9806699 as being the most significantly associated differential eQTL (deQTL) after in vitro exposure to simvastatin. To test the relationship of this deQTL with SIM, an association analysis of rs9806699 was carried out in two patient cohorts, the first comparing 72 myopathy cases with 220 matched controls, and the second including 100 myopathy cases. A significant association was observed in both studies independently, and a meta-analysis of these two studies showed that the A allele of rs9806699 was significantly associated with a reduced risk of SIM (OR = 0.6, p = 6 × 10–4) (Mangravite et al., 2013).
However, these results have not been replicated. Luzum et al. (2015) genotyped 609 cases and 106 controls in a multicenter, case–control study. The analysis was conducted on three different arms: mild SIM, severe SIM, and combined mild and severe SIM groups. No significant associations were found between GATM and SIM in all three groups. This study also did a meta-analysis on related studies and reported a non-significant association between GATM and SIM (OR = 0.82, p = 0.072) (Luzum et al., 2015). This was supported by further research on 150 SIM cases and 587 statin-exposed control patients (Carr et al., 2014). No significant differences were found between the frequency of the A allele with myopathy (OR = 0.94, p = 0.68) or severe myopathy (OR = 0.94, p = 0.83). Further analysis in patients receiving only simvastatin (99 cases and 344 controls) did not reveal a significant association between either myopathy (OR = 1.12, p = 0.49) or severe myopathy (OR = 1.42, p = 0.24) with GATM (Carr et al., 2014). In addition, a case–control study of 175 cerivastatin-induced rhabdomyolysis cases and 645 statin users as controls found no association between GATM rs9806699 and the risk of rhabdomyolysis (OR = 1.01, p = 0.96) (Floyd et al., 2014).
There are several factors that might lead to the differing results observed for GATM. One, which has been discussed before, is the different definitions of myopathy and case inclusion criteria. Ethnicity is another important factor to consider; Bai et al. (2019) assessed SIM incidents in 758 Chinese rosuvastatin users with coronary artery disease. The A allele of GATM rs9806699 was reported to have a marginally protective effect on SIM (OR = 0.617, p = 0.024). Therefore, ethnicity-dependent effects should be considered when assessing the association of GATM with SIM (Bai et al., 2019).
Coenzyme Q2
Coenzyme Q2 (COQ2) is one of the many genes required for the biosynthesis of coenzyme Q10 (COQ10). At least 13 genes are involved in this pathway and COQ2 encodes for 4-hydroxybenzoate prenyl-transferase, which mediates the second last step in the reaction synthesis of COQ10 (Forsgren et al., 2004; Doimo et al., 2014). COQ10 or ubiquinone plays an important role in producing ATP, which drives the mitochondrial respiratory chain, pyrimidine biosynthesis, and apoptosis regulation (Doimo et al., 2014). As briefly mentioned in the pathogenesis of SIM, statins reduce COQ10 levels by up to 40% in both healthy volunteers and hypercholesteremic patients (Ghirlanda et al., 1993; Thompson et al., 2003). While several reviews and a clinical trial did not support the role of COQ10 supplementation in preventing SIM (Marcoff and Thompson, 2007; Schaars and Stalenhoef, 2008; Banach et al., 2015; Taylor et al., 2015), a recent meta-analysis on 12 randomized controlled trials concluded that COQ10 supplementation provided symptomatic relief for patients with SIM (Qu et al., 2018).
The two common SNPs investigated in the COQ2 gene are COQ2 A > G (rs6535454) and COQ2 G > A/C/T (rs4693075) (Oh et al., 2007). Oh et al. (2007) compared 133 European subjects on statin monotherapy who suffered from myotoxicity with 158 matched controls. They reported a significant association between statin intolerance and the SNPs rs6535454 (p = 0.047) and rs4693075 (p = 0.019). This was supported by Puccetti et al. (2010), who showed that the rs4693075 SNP was associated with atorvastatin or rosuvastatin-induced SIM. Subjects taking rosuvastatin and atorvastatin had a significantly (p < 0.001) higher risk (2.6- and 3.1-fold) of being statin intolerant with muscular symptoms and CK elevation, respectively (Puccetti et al., 2010). However, there are several studies that did not find a significant association between COQ2 SNPs and SIM (Carr et al., 2013; Hubacek et al., 2017; Ramakumari et al., 2018). The populations studied varied across these three reports from Caucasian (Carr et al., 2013), South Indian (Ramakumari et al., 2018) to Czech (Hubacek et al., 2017).
Human Leukocyte Antigen and Leukocyte Immunoglobulin-Like Receptor Subfamily B5
Human leukocyte antigen (HLA) has been associated with human disease for decades with its role in body physiology, protective immunity, and disease-causing autoimmune reactivity (Dendrou et al., 2018). As mentioned previously, statin-induced anti–HMGCR-associated myopathy is a rare condition associated with statins. A significant association between HMGCR autoantibodies and HLA locus (HLA-DRB1∗11) has been reported by Limaye et al. (2015) with an OR of 50 (p < 0.0001). This is supported by Mammen et al. (2012), who studied 28 European and African American patients with HMGCR autoantibodies. When compared with 654 controls, HLA-DRB1∗11 was identified in 70% and 18% of the European cases and controls, respectively (p = 1.2 × 10–6). Similarly, 88% and 21% out of the African cases and controls had HLA-DRB1∗11 (p = 0.0002) (Mammen et al., 2012).
Leukocyte immunoglobulin-like receptor subfamily B5 (LILRB5) gene is another immunogenic risk factor associated with SIM (Siddiqui et al., 2017). To date, only one variant (rs12975366 T>C), which is also referred to as 247Asp > Gly within the LILRB5 locus, has been reported as being associated with statin intolerance and myalgia. A study in nearly 12,000 people from the Scottish GoDARTS cohort reported a twofold risk in developing general statin intolerance in participants homozygous for the T allele. A replication study conducted in the JUPITER trial showed an OR of 1.35 for developing myalgia (p = 0.04). A meta-analysis of the initial and replication studies showed a significant association of the homozygous T allele with a 1.5-fold higher risk of developing statin intolerance (p = 7 × 10–5) (Siddiqui et al., 2017).
Other Genes
There are other genes not discussed here that have been reported to have an association with the risk of developing SIM. They include apolipoprotein E (APOE) (Hubáček et al., 2015b; Liu et al., 2017), HMGCR (Frudakis et al., 2007), vitamin D receptor (VDR) (Ovesjo et al., 2016), serotonin receptors HTR3B and HTR7 (Ruano et al., 2007), chloride channel protein 1 (CLCN1) (Neroldova et al., 2016), eyes shut homolog (EYS) (Isackson et al., 2011; Stranecky et al., 2016), glycosyl transferase-like protein (LARGE) (Stranecky et al., 2016), ryanodine receptor 1 and 2 (RYR1 and RYR2) (Marciante et al., 2011; Isackson et al., 2018), and calcium voltage-gated channel subunit alpha 1S (CACNA1S) (Isackson et al., 2018).
The genetic variations in these genes are not well studied, and their association with SIM is not widely replicated. Therefore, their roles as candidate genes in exploring SIM remain unclear.
Discussion and Conclusion
For decades, statins have remained as an important drug in cardiovascular health. Despite emerging alternatives such as proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors (Wang and Liu, 2019) and bempedoic acid (Laufs et al., 2019), statins as affordable and effective cholesterol-lowering agents are likely to remain as first-line in the treatment of hypercholesterolemia. In the era of personalized medicine, genomics offers promising approaches for research and development, especially in optimizing efficacy and reducing ADRs.
As reviewed, there are several genetic targets that may increase the risk of developing SIM. While all of the CYP enzymes discussed are involved in statin metabolism, it is likely that genetic variations in enzymes with major metabolism roles have a significant effect on statin disposition. They are CYP2C9, CYP3A4, and CYP3A5. As for phase II metabolism, the role of UGT in statin metabolism and SIM should be further explored. With respect to transporters, SLCO1B1, SLCO2B1, and ABCG2 are good candidates, whereas proper functional characterization of ABCB1 is necessary prior to applying any strong conclusions of their association with SIM.
While the discovery of rs4149056 in the SLCO1B1 transporter gene is an example of a significant and initially striking finding, several further studies have failed to replicate this finding in different cohorts (Puccetti et al., 2010; Brunham et al., 2012; Carr et al., 2013; Danik et al., 2013; Hou et al., 2015; Hubáček et al., 2015a; Khine et al., 2016; Liu et al., 2017; Ramakumari et al., 2018). Similarly, the impact of genetic variation in pharmacogenes (CYP-450) and other genes (GATM, CoQ2, etc.) have been largely inconclusive. The ambiguity in findings shows that more research in elucidating the pharmacogenomic impact of these genes on SIM is warranted. Several factors may contribute to these discordant results, and the lack of clear and robust findings, which are listed below:
1) SIM has a complex and inexplicit pathogenesis.
2) SIM is heterogeneous and lacks standardized phenotype definition.
3) There are limited genetic association studies on statin-induced myalgia, which is the most commonly occurring form of SIM (e.g., Ruano et al., 2011; Siddiqui et al., 2017; Elam et al., 2017).
4) Metabolic enzymes exhibit substrate specificity (e.g., fluvastatin is a substrate of CYP2C9, but not CYP3A4).
5) Unclear functional characterization of SNPs in genes of interest (e.g., ABCB1 gene).
6) The role of non-genetic factors in SIM (e.g., environmental influences and comorbidity).
Furthermore, an important covariate in genetic studies with SIM is ethnicity. Two examples discussed in this review are SLCO1B1 rs4149056 with SIM (Liu et al., 2017) and the protective effect of GATM rs9806699 (Bai et al., 2019), both these studies investigated rosuvastatin-induced myotoxicity in a Chinese population. As discussed, Asian populations prescribed rosuvastatin have generally been reported having a higher plasma exposure of rosuvastatin compared to non-Finnish Europeans. This led to the FDA recommendation of reducing the starting dose of rosuvastatin by 50% in Asian patients (Lee et al., 2005; Liao, 2007). The higher plasma exposure in Asians is still unclear and not associated with genetic variation in SLCO1B1 (Lee et al., 2005; Tomita et al., 2013; Birmingham et al., 2015). Because SIM is dose-related, plasma concentration exposure is one of the risk factors in developing SIM. Therefore, study results derived from a specific ethnic population should be replicated and validated before generalizing the findings to other populations.
Clearly, further research is required to understand the genetic association with SIM. One promising approach is the generation of a polygenic risk score (PRS). PRS is defined as an individualized risk burden, quantitatively measured from a range of common, intermediate and rare risk alleles, identified by large-scale genome sequencing in predisposing phenotype susceptibility (Chatterjee et al., 2016; Torkamani et al., 2018). In addition to that, PRS may be combined with clinical and lifestyle factors. Since its introduction in 2007 (Wray et al., 2007), PRS have been trialed by researchers for risk stratification to inform disease screening and therapeutic intervention. SIM may be a good candidate for PRS application because of its multifactorial causal mechanisms associated with genetic variation.
Ultimately, a clearly elucidated association will assist in the clinical implementation of statin pharmacogenetics. CPIC has published a clinical guideline on the management of patients who have specific SLCO1B1 genetic variants, specifically with respect to statin prescription and dosing. A recent systematic review reported that genotype testing may influence the prescribing of statins among high-risk patients, but the impact of SIM or cardiovascular events was not reported (Vassy et al., 2019). Peyser et al. (2018) conducted a prospective randomized SLCO1B1 genotype–guided statin therapy trial in 159 patients who stopped taking statins because of a history of statin myalgia. The study reported that when compared with the cohort receiving standard care, SLCO1B1 pharmacogenetic testing improved statin reinitiation and lowering of LDL among identified SLCO1B1∗5 carriers. However, there was no improvement on self-reported statin adherence (Peyser et al., 2018). Because SIM is one of the main factors leading to poor statin adherence and subsequently higher risk of cardiovascular events (De Vera et al., 2014), educating and providing patients with clear information along with SLCO1B1 pharmacogenetic testing may improve statin adherence (Lansberg et al., 2018).
While it may be feasible to incorporate SLCO1B1 pharmacogenetic testing clinically, further well-designed randomized controlled trials are necessary to investigate its clinical utility. A recent pragmatic trial conducted by Brunette et al. (2020) describes a promising approach. By referring to the PRagmatic-Explanatory Continuum Indicator Summary 2 (PRECIS-2) framework, statin-naive patients were randomized into two arms: with or without SLCO1B1 rs4149056 genotype result available to healthcare providers through electronic health records. The clinical impact of SLCO1B1 genotyping in reducing SIM was one of the outcomes measured. However, it was not reported in this publication (Brunette et al., 2020).
Although it is clear that the rs4149056 SNP is strongly associated with simvastatin-induced myopathy, the broader association of other genetic variation in pharmacogenes remains a challenge with respect to understanding their role in SIM. Despite a great deal of existing research, we may still be some way from the ability to apply genomic knowledge clinically to predict and ideally prevent the occurrence of SIM.
Author Contributions
PK wrote the first draft of this article. SM and MK aided in the editing of this manuscript. PC provided clinical insight into this review and helped with the editing of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
SM’s salary is funded by the Jim and Mary Carney Charitable Trust, and support was provided by Health Research Council of New Zealand project grant (16/226). PK was supported by a doctoral scholarship from the University of Otago.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ABC, ATP-binding cassette; ABCB1, ATP-binding cassette subfamily B member 1; ABCC2, ATP-binding cassette subfamily C member 2; ADRs, adverse drug reactions; APOE, apolipoprotein E; ATP, adenosine triphosphate; AUC, area under the concentration–time curve; CACNA1S, calcium voltage-gated channel subunit alpha 1S; CK, creatine kinase; CLCN1, chloride channel protein 1; COQ10, coenzyme Q10; COQ2, coenzyme Q2; CPIC, Clinical Pharmacogenetics Implementation Consortium; CYP450, cytochrome P450; deQTL, differential expression quantitative trait loci; eQTL, expression quantitative trait loci; EYS, eyes shut homolog; FDA, Food and Drug Administration; GATM, glycine amidinotransferase; gnomAD, genome aggregation database; GoDARTS, Genetics of Diabetic Audit and Research; GWAS, genome-wide association studies; HLA, human leukocyte antigen; HLA-DRB1, human leukocyte antigen DR-isotope beta chain; HMG-CR, hydroxymethylglutaryl-coenzyme A reductase; HTR3B, 5-hydroxytryptamine receptor 3B; HTR7, 5-hydroxytryptamine receptor 7; JUPITER, Justification for the Use of Statins in Primary Prevention, An Intervention Trial Evaluating Rosuvastatin; LARGE, glycosyl transferase-like protein; LDL, low-density lipoprotein; LILRB5, leukocyte immunoglobulin-like receptor subfamily B5; OR, Odds ratio; PCSK9, proprotein convertase subtilisin/kexin type 9; PD, pharmacodynamics; PK, pharmacokinetics; PRIMO, Prediction of Muscular Risk in Observational Conditions; PRS, polygenic risk score; RYR1, ryanodine receptor 1; RYR2, ryanodine receptor 2; SIM, statin-induced myotoxicity; SLC, solute carrier; SLCO1B1, solute carrier organic anion transporter family member 1B1; SNPs, single nucleotide polymorphisms; SR, sarcoplasmic reticulum; SRM, statin-related myotoxicity; UGT, uridine 5 ′-diphospho (UDP)-glucuronosyltransferase; ULN, upper limit normal; VDR, vitamin D receptor.
References
Adams, S. P., Alaeiilkhchi, N., and Wright, J. M. (2020). Pitavastatin for lowering lipids. Cochrane Database Syst. Rev. 6:CD012735.
Agrawal, A., Suryakumar, G., and Rathor, R. (2018). Role of defective Ca(2+) signaling in skeletal muscle weakness: pharmacological implications. J. Cell Commun. Signal. 12, 645–659. doi: 10.1007/s12079-018-0477-z
Ahmed, W., Khan, N., Glueck, C. J., Pandey, S., Wang, P., Goldenberg, N., et al. (2009). Low serum 25 (OH) vitamin D levels (<32 ng/mL) are associated with reversible myositis-myalgia in statin-treated patients. Transl. Res. 153, 11–16. doi: 10.1016/j.trsl.2008.11.002
Alam, K., Crowe, A., Wang, X., Zhang, P., Ding, K., Li, L., et al. (2018). Regulation of organic anion transporting polypeptides (OATP) 1B1- and OATP1B3-mediated transport: an updated review in the context of OATP-mediated drug-drug interactions. Int. J. Mol. Sci. 19:855. doi: 10.3390/ijms19030855
Alfirevic, A., Neely, D., Armitage, J., Chinoy, H., Cooper, R. G., Laaksonen, R., et al. (2014). Phenotype standardization for statin-induced myotoxicity. Clin. Pharmacol. Ther. 96, 470–476. doi: 10.1038/clpt.2014.121
Alghalyini, B., El Shamieh, S., Salami, A., Visvikis Siest, S., Fakhoury, H. M., and Fakhoury, R. (2018). Effect of SLCO1B1 gene polymorphisms and vitamin D on statin-induced myopathy. Drug Metab. Pers. Ther. 33, 41–47. doi: 10.1515/dmpt-2017-0030
Arnaboldi, L., and Corsini, A. (2010). Do structural differences in statins correlate with clinical efficacy? Curr. Opin. Lipidol. 21, 298–304. doi: 10.1097/mol.0b013e32833b776c
Arnadottir, M., Eriksson, L. O., Thysell, H., and Karkas, J. D. (1993). Plasma concentration profiles of simvastatin 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase inhibitory activity in kidney transplant recipients with and without ciclosporin. Nephron 65, 410–413. doi: 10.1159/000187521
Asberg, A., Hartmann, A., Fjeldsa, E., Bergan, S., and Holdaas, H. (2001). Bilateral pharmacokinetic interaction between cyclosporine A and atorvastatin in renal transplant recipients. Am. J. Transplant. 1, 382–386. doi: 10.1034/j.1600-6143.2001.10415.x
Bai, X., Zhang, B., Wang, P., Wang, G. L., Li, J. L., Wen, D. S., et al. (2019). Effects of SLCO1B1 and GATM gene variants on rosuvastatin-induced myopathy are unrelated to high plasma exposure of rosuvastatin and its metabolites. Acta Pharmacol. Sin. 40, 492–499. doi: 10.1038/s41401-018-0013-y
Bakar, N. S., Neely, D., Avery, P., Brown, C., Daly, A. K., and Kamali, F. (2018). Genetic and clinical factors are associated with statin-related myotoxicity of moderate severity: a case-control study. Clin. Pharmacol. Ther. 104, 178–187. doi: 10.1002/cpt.887
Banach, M., Serban, C., Sahebkar, A., Ursoniu, S., Rysz, J., Muntner, P., et al. (2015). Effects of coenzyme Q10 on statin-induced myopathy: a meta-analysis of randomized controlled trials. Mayo Clin. Proc. 90, 24–34.
Barbarino, J. M., Haidar, C. E., Klein, T. E., and Altman, R. B. (2014). PharmGKB summary: very important pharmacogene information for UGT1A1. Pharmacogenet. Genomics 24, 177–183. doi: 10.1097/fpc.0000000000000024
Barry, A. R., Beach, J. E., and Pearson, G. J. (2018). Prevention and management of statin adverse effects: a practical approach for pharmacists. Can. Pharm. J. 151, 179–188. doi: 10.1177/1715163518768534
Bartlomiejczyk, M. A., Penson, P., and Banach, M. (2019). Worldwide dyslipidemia guidelines. Curr. Cardiovasc. Risk Rep. 13:2.
Bazargan, M., Foster, D. J., Davey, A. K., and Muhlhausler, B. S. (2017). Rosiglitazone metabolism in human liver microsomes using a substrate depletion method. Drugs R D 17, 189–198.
Becker, M. L., Elens, L. L., Visser, L. E., Hofman, A., Uitterlinden, A. G., van Schaik, R. H., et al. (2013). Genetic variation in the ABCC2 gene is associated with dose decreases or switches to other cholesterol-lowering drugs during simvastatin and atorvastatin therapy. Pharmacogenomics J. 13, 251–256. doi: 10.1038/tpj.2011.59
Becker, M. L., Visser, L. E., van Schaik, R. H., Hofman, A., Uitterlinden, A. G., and Stricker, B. H. (2010). Influence of genetic variation in CYP3A4 and ABCB1 on dose decrease or switching during simvastatin and atorvastatin therapy. Pharmacoepidemiol. Drug Saf. 19, 75–81. doi: 10.1002/pds.1866
Bellosta, S., Paoletti, R., and Corsini, A. (2004). Safety of statins: focus on clinical pharmacokinetics and drug interactions. Circulation 109(23 Suppl. 1), Iii50–Iii57.
Birmingham, B. K., Bujac, S. R., Elsby, R., Azumaya, C. T., Wei, C., Chen, Y., et al. (2015). Impact of ABCG2 and SLCO1B1 polymorphisms on pharmacokinetics of rosuvastatin, atorvastatin and simvastatin acid in Caucasian and Asian subjects: A class effect? Eur. J. Clin. Pharmacol. 71, 341–355.
Bischoff, H., Angerbauer, R., Bender, J., Bischoff, E., Faggiotto, A., Petzinna, D., et al. (1997). Cerivastatin: pharmacology of a novel synthetic and highly active HMG-CoA reductase inhibitor. Atherosclerosis 135, 119–130. doi: 10.1016/s0021-9150(97)00188-3
Bjornsson, E. S. (2017). Hepatotoxicity of statins and other lipid-lowering agents. Liver Int. 37, 173–178. doi: 10.1111/liv.13308
Brillantes, A. B., Ondrias, K., Scott, A., Kobrinsky, E., Ondriasová, E., Moschella, M. C., et al. (1994). Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell 77, 513–523. doi: 10.1016/0092-8674(94)90214-3
Brown, C. M., Reisfeld, B., and Mayeno, A. N. (2008). Cytochromes P450: a structure-based summary of biotransformations using representative substrates. Drug Metab. Rev. 40, 1–100.
Brown, M. S., and Goldstein, J. L. (1981). Lowering plasma cholesterol by raising LDL receptors. N. Engl. J. Med. 305, 515–517. doi: 10.1056/nejm198108273050909
Bruckert, E., Hayem, G., Dejager, S., Yau, C., and Begaud, B. (2005). Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients–the PRIMO study. Cardiovasc. Drugs Ther. 19, 403–414. doi: 10.1007/s10557-005-5686-z
Brunette, C. A., Miller, S. J., Majahalme, N., Hau, C., MacMullen, L., Advani, S., et al. (2020). Pragmatic trials in genomic medicine: the integrating pharmacogenetics in clinical care (I-PICC) Study. Clin. Transl. Sci. 13, 381–390. doi: 10.1111/cts.12723
Brunham, L. R., Lansberg, P. J., Zhang, L., Miao, F., Carter, C., Hovingh, G. K., et al. (2012). Differential effect of the rs4149056 variant in SLCO1B1 on myopathy associated with simvastatin and atorvastatin. Pharmacogenomics J. 12, 233–237. doi: 10.1038/tpj.2010.92
Carr, D. F., Alfirevic, A., Johnson, R., Chinoy, H., van Staa, T., and Pirmohamed, M. (2014). GATM gene variants and statin myopathy risk. Nature 513:E1.
Carr, D. F., Francis, B., Jorgensen, A. L., Zhang, E., Chinoy, H., Heckbert, S. R., et al. (2019). Genomewide association study of statin-induced myopathy in patients recruited using the UK clinical practice research datalink. Clin. Pharmacol. Ther. 106, 1353–1361. doi: 10.1002/cpt.1557
Carr, D. F., O’Meara, H., Jorgensen, A. L., Campbell, J., Hobbs, M., McCann, G., et al. (2013). SLCO1B1 genetic variant associated with statin-induced myopathy: a proof-of-concept study using the clinical practice research datalink. Clin. Pharmacol. Ther. 94, 695–701. doi: 10.1038/clpt.2013.161
Catapano, A. L. (2010). Pitavastatin - pharmacological profile from early phase studies. Atheroscler. Suppl. 11, 3–7. doi: 10.1016/s1567-5688(10)71063-1
Chatterjee, N., Shi, J., and Garcia-Closas, M. (2016). Developing and evaluating polygenic risk prediction models for stratified disease prevention. Nat. Rev. Genet. 17, 392–406. doi: 10.1038/nrg.2016.27
Cho, S. K., Oh, E. S., Park, K., Park, M. S., and Chung, J. Y. (2012). The UGT1A3∗2 polymorphism affects atorvastatin lactonization and lipid-lowering effect in healthy volunteers. Pharmacogenet. Genomics 22, 598–605. doi: 10.1097/fpc.0b013e3283544085
Choi, H. Y., Bae, K. S., Cho, S. H., Ghim, J. L., Choe, S., Jung, J. A., et al. (2015). Impact of CYP2D6, CYP3A5, CYP2C19, CYP2A6, SLCO1B1, ABCB1, and ABCG2 gene polymorphisms on the pharmacokinetics of simvastatin and simvastatin acid. Pharmacogenet. Genomics 25, 595–608. doi: 10.1097/fpc.0000000000000176
Choi, J. H., Lee, M. G., Cho, J. Y., Lee, J. E., Kim, K. H., and Park, K. (2008). Influence of OATP1B1 genotype on the pharmacokinetics of rosuvastatin in Koreans. Clin. Pharmacol. Ther. 83, 251–257. doi: 10.1038/sj.clpt.6100267
Cuperus, F. J., Claudel, T., Gautherot, J., Halilbasic, E., and Trauner, M. (2014). The role of canalicular ABC transporters in cholestasis. Drug Metab. Dispos. 42, 546–560. doi: 10.1124/dmd.113.056358
Daly, A. K. (2017). Pharmacogenetics: a general review on progress to date. Br. Med. Bull. 124, 65–79.
Daly, A. K., Rettie, A. E., Fowler, D. M., and Miners, J. O. (2017). Pharmacogenomics of CYP2C9: functional and clinical considerations. J. Pers. Med. 8:1. doi: 10.3390/jpm8010001
Danik, J. S., Chasman, D. I., MacFadyen, J. G., Nyberg, F., Barratt, B. J., and Ridker, P. M. (2013). Lack of association between SLCO1B1 polymorphisms and clinical myalgia following rosuvastatin therapy. Am. Heart J. 165, 1008–1014. doi: 10.1016/j.ahj.2013.01.025
de Keyser, C. E., Peters, B. J., Becker, M. L., Visser, L. E., Uitterlinden, A. G., Klungel, O. H., et al. (2014). The SLCO1B1 c.521T>C polymorphism is associated with dose decrease or switching during statin therapy in the Rotterdam Study. Pharmacogenet. Genomics 24, 43–51. doi: 10.1097/fpc.0000000000000018
De Vera, M. A., Bhole, V., Burns, L. C., and Lacaille, D. (2014). Impact of statin adherence on cardiovascular disease and mortality outcomes: a systematic review. Br. J. Clin. Pharmacol. 78, 684–698. doi: 10.1111/bcp.12339
Dean, M., Rzhetsky, A., and Allikmets, R. (2001). The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 11, 1156–1166. doi: 10.1101/gr.gr-1649r
DeGorter, M. K., Tirona, R. G., Schwarz, U. I., Choi, Y. H., Dresser, G. K., Suskin, N., et al. (2013). Clinical and pharmacogenetic predictors of circulating atorvastatin and rosuvastatin concentrations in routine clinical care. Circ. Cardiovasc. Genet. 6, 400–408. doi: 10.1161/circgenetics.113.000099
Dendrou, C. A., Petersen, J., Rossjohn, J., and Fugger, L. (2018). HLA variation and disease. Nat. Rev. Immunol. 18, 325–339.
Deng, J. W., Song, I. S., Shin, H. J., Yeo, C. W., Cho, D. Y., Shon, J. H., et al. (2008). The effect of SLCO1B1∗15 on the disposition of pravastatin and pitavastatin is substrate dependent: the contribution of transporting activity changes by SLCO1B1∗15. Pharmacogenet. Genomics 18, 424–433. doi: 10.1097/fpc.0b013e3282fb02a3
Doimo, M., Desbats, M. A., Cerqua, C., Cassina, M., Trevisson, E., and Salviati, L. (2014). Genetics of coenzyme q10 deficiency. Mol. Syndromol. 5, 156–162.
Donnelly, L. A., Doney, A. S., Tavendale, R., Lang, C. C., Pearson, E. R., Colhoun, H. M., et al. (2011). Common nonsynonymous substitutions in SLCO1B1 predispose to statin intolerance in routinely treated individuals with type 2 diabetes: a go-DARTS study. Clin. Pharmacol. Ther. 89, 210–216. doi: 10.1038/clpt.2010.255
du Souich, P., Roederer, G., and Dufour, R. (2017). Myotoxicity of statins: mechanism of action. Pharmacol. Ther. 175, 1–16. doi: 10.1016/j.pharmthera.2017.02.029
Egom, E. E., and Hafeez, H. (2016). Biochemistry of Statins. Adv. Clin. Chem. 73, 127–168. doi: 10.1016/bs.acc.2015.10.005
Elam, M. B., Majumdar, G., Mozhui, K., Gerling, I. C., Vera, S. R., Fish-Trotter, H., et al. (2017). Patients experiencing statin-induced myalgia exhibit a unique program of skeletal muscle gene expression following statin re-challenge. PLoS One 12:e0181308. doi: 10.1371/journal.pone.0181308
Ellis, L. C., Hawksworth, G. M., and Weaver, R. J. (2013). ATP-dependent transport of statins by human and rat MRP2/Mrp2. Toxicol. Appl. Pharmacol. 269, 187–194. doi: 10.1016/j.taap.2013.03.019
Endo, A. (2010). A historical perspective on the discovery of statins. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 86, 484–493. doi: 10.2183/pjab.86.484
Estabrook, R. W. (2003). A passion for P450s (rememberances of the early history of research on cytochrome P450). Drug Metab. Dispos. 31, 1461–1473. doi: 10.1124/dmd.31.12.1461
Feng, Q., Wilke, R. A., and Baye, T. M. (2012). Individualized risk for statin-induced myopathy: current knowledge, emerging challenges and potential solutions. Pharmacogenomics 13, 579–594. doi: 10.2217/pgs.12.11
Ferrari, M., Guasti, L., Maresca, A., Mirabile, M., Contini, S., Grandi, A. M., et al. (2014). Association between statin-induced creatine kinase elevation and genetic polymorphisms in SLCO1B1, ABCB1 and ABCG2. Eur. J. Clin. Pharmacol. 70, 539–547. doi: 10.1007/s00228-014-1661-6
Fiegenbaum, M., da Silveira, F. R., Van der Sand, C. R., Van der Sand, L. C., Ferreira, M. E., Pires, R. C., et al. (2005). The role of common variants of ABCB1, CYP3A4, and CYP3A5 genes in lipid-lowering efficacy and safety of simvastatin treatment. Clin. Pharmacol. Ther. 78, 551–558. doi: 10.1016/j.clpt.2005.08.003
Finkelman, R. D., Wang, T. D., Wang, Y., Azumaya, C. T., Birmingham, B. K., Wissmar, J., et al. (2015). Effect of CYP2C19 polymorphism on the pharmacokinetics of rosuvastatin in healthy Taiwanese subjects. Clin. Pharmacol. Drug Dev. 4, 33–40. doi: 10.1002/cpdd.135
Fischer, V., Johanson, L., Heitz, F., Tullman, R., Graham, E., Baldeck, J. P., et al. (1999). The 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor fluvastatin: effect on human cytochrome P-450 and implications for metabolic drug interactions. Drug Metab. Dispos. 27, 410–416.
Flint, O. P., Masters, B. A., Gregg, R. E., and Durham, S. K. (1997). Inhibition of cholesterol synthesis by squalene synthase inhibitors does not induce myotoxicity in vitro. Toxicol. Appl. Pharmacol. 145, 91–98. doi: 10.1006/taap.1997.8131
Floyd, J. S., Bis, J. C., Brody, J. A., Heckbert, S. R., Rice, K., and Psaty, B. M. (2014). GATM locus does not replicate in rhabdomyolysis study. Nature 513, E1–E3.
Fohner, A. E., Brackman, D. J., Giacomini, K. M., Altman, R. B., and Klein, T. E. (2017). PharmGKB summary: very important pharmacogene information for ABCG2. Pharmacogenet. Genomics 27, 420–427. doi: 10.1097/fpc.0000000000000305
Forsgren, M., Attersand, A., Lake, S., Grunler, J., Swiezewska, E., Dallner, G., et al. (2004). Isolation and functional expression of human COQ2, a gene encoding a polyprenyl transferase involved in the synthesis of CoQ. Biochem. J. 382(Pt 2), 519–526. doi: 10.1042/bj20040261
Frudakis, T. N., Thomas, M. J., Ginjupalli, S. N., Handelin, B., Gabriel, R., and Gomez, H. J. (2007). CYP2D6∗4 polymorphism is associated with statin-induced muscle effects. Pharmacogenet. Genomics 17, 695–707. doi: 10.1097/fpc.0b013e328012d0a9
Furberg, C. D., and Pitt, B. (2001). Withdrawal of cerivastatin from the world market. Curr. Control. Trials Cardiovasc. Med. 2, 205–207.
Furukawa, T., Wakabayashi, K., Tamura, A., Nakagawa, H., Morishima, Y., Osawa, Y., et al. (2009). Major SNP (Q141K) variant of human ABC transporter ABCG2 undergoes lysosomal and proteasomal degradations. Pharm. Res. 26, 469–479. doi: 10.1007/s11095-008-9752-7
Gaedigk, A., Ingelman-Sundberg, M., Miller, N. A., Leeder, J. S., Whirl-Carrillo, M., and Klein, T. E. (2018). The Pharmacogene Variation (PharmVar) Consortium: incorporation of the Human Cytochrome P450 (CYP) Allele Nomenclature Database. Clin. Pharmacol. Ther. 103, 399–401. doi: 10.1002/cpt.910
Gaedigk, A., Sangkuhl, K., Whirl-Carrillo, M., Twist, G. P., Klein, T. E., and Miller, N. A. (2019). The evolution of PharmVar. Clin. Pharmacol. Ther. 105, 29–32. doi: 10.1002/cpt.1275
Gammal, R. S., Court, M. H., Haidar, C. E., Iwuchukwu, O. F., Gaur, A. H., Alvarellos, M., et al. (2016). Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for UGT1A1 and Atazanavir Prescribing. Clin. Pharmacol. Ther. 99, 363–369. doi: 10.1002/cpt.269
Gazzerro, P., Proto, M. C., Gangemi, G., Malfitano, A. M., Ciaglia, E., Pisanti, S., et al. (2012). Pharmacological actions of statins: a critical appraisal in the management of cancer. Pharmacol. Rev. 64, 102–146. doi: 10.1124/pr.111.004994
Ghirlanda, G., Oradei, A., Manto, A., Lippa, S., Uccioli, L., Caputo, S., et al. (1993). Evidence of plasma CoQ10-lowering effect by HMG-CoA reductase inhibitors: a double-blind, placebo-controlled study. J. Clin. Pharmacol. 33, 226–229. doi: 10.1002/j.1552-4604.1993.tb03948.x
Giacomini, K. M., Huang, S. M., Tweedie, D. J., Benet, L. Z., Brouwer, K. L., Chu, X., et al. (2010). Membrane transporters in drug development. Nat. Rev. Drug Discov. 9, 215–236. doi: 10.1038/nrd3028
Glueck, C. J., Lee, K., Prince, M., Milgrom, A., Makadia, F., and Wang, P. (2017). Low serum vitamin D, statin associated muscle symptoms, vitamin D supplementation. Atherosclerosis 256, 125–127. doi: 10.1016/j.atherosclerosis.2016.11.027
Goldberg, R., and Roth, D. (1996). Evaluation of fluvastatin in the treatment of hypercholesterolemia in renal transplant recipients taking cyclosporine. Transplantation 62, 1559–1564. doi: 10.1097/00007890-199612150-00005
Gong, Q. H., Cho, J. W., Huang, T., Potter, C., Gholami, N., Basu, N. K., et al. (2001). Thirteen UDPglucuronosyltransferase genes are encoded at the human UGT1 gene complex locus. Pharmacogenetics 11, 357–368. doi: 10.1097/00008571-200106000-00011
Grube, M., Kock, K., Oswald, S., Draber, K., Meissner, K., Eckel, L., et al. (2006). Organic anion transporting polypeptide 2B1 is a high-affinity transporter for atorvastatin and is expressed in the human heart. Clin. Pharmacol. Ther. 80, 607–620. doi: 10.1016/j.clpt.2006.09.010
Grundy, S. M., Stone, N. J., Bailey, A. L., Beam, C., Birtcher, K. K., Blumenthal, R. S., et al. (2019). 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the management of blood cholesterol: executive summary: a report of the american college of cardiology/American heart association task force on clinical practice guidelines. Circulation 139, e1046–e1081. doi: 10.1161/CIR.0000000000000624
Hanioka, N., Matsumoto, K., Saito, Y., and Narimatsu, S. (2010). Functional characterization of CYP2C8.13 and CYP2C8.14: catalytic activities toward paclitaxel. Basic Clin. Pharmacol. Toxicol. 107, 565–569. doi: 10.1111/j.1742-7843.2010.00543.x
Hansen, K. E., Hildebrand, J. P., Ferguson, E. E., and Stein, J. H. (2005). Outcomes in 45 patients with statin-associated myopathy. Arch. Intern. Med. 165, 2671–2676. doi: 10.1001/archinte.165.22.2671
Hargreaves, I. P. (2014). Coenzyme Q10 as a therapy for mitochondrial disease. Int. J. Biochem. Cell Biol. 49, 105–111. doi: 10.1016/j.biocel.2014.01.020
Hedenmalm, K., Alvan, G., Ohagen, P., and Dahl, M. L. (2010). Muscle toxicity with statins. Pharmacoepidemiol. Drug Saf. 19, 223–231. doi: 10.1002/pds.1895
Hermann, M., Bogsrud, M. P., Molden, E., Asberg, A., Mohebi, B. U., Ose, L., et al. (2006). Exposure of atorvastatin is unchanged but lactone and acid metabolites are increased several-fold in patients with atorvastatin-induced myopathy. Clin. Pharmacol. Ther. 79, 532–539. doi: 10.1016/j.clpt.2006.02.014
Heyes, N., Kapoor, P., and Kerr, I. D. (2018). Polymorphisms of the Multidrug Pump ABCG2: a systematic review of their effect on protein expression, function, and drug pharmacokinetics. Drug Metab. Dispos. 46, 1886–1899. doi: 10.1124/dmd.118.083030
Hirota, T., Eguchi, S., and Ieiri, I. (2013). Impact of genetic polymorphisms in CYP2C9 and CYP2C19 on the pharmacokinetics of clinically used drugs. Drug Metab. Pharmacokinet. 28, 28–37. doi: 10.2133/dmpk.dmpk-12-rv-085
Hitzl, M., Drescher, S., van der Kuip, H., Schaffeler, E., Fischer, J., Schwab, M., et al. (2001). The C3435T mutation in the human MDR1 gene is associated with altered efflux of the P-glycoprotein substrate rhodamine 123 from CD56+ natural killer cells. Pharmacogenetics 11, 293–298. doi: 10.1097/00008571-200106000-00003
Ho, R. H., Choi, L., Lee, W., Mayo, G., Schwarz, U. I., Tirona, R. G., et al. (2007). Effect of drug transporter genotypes on pravastatin disposition in European- and African-American participants. Pharmacogenet. Genomics 17, 647–656. doi: 10.1097/fpc.0b013e3280ef698f
Hodges, L. M., Markova, S. M., Chinn, L. W., Gow, J. M., Kroetz, D. L., Klein, T. E., et al. (2011). Very important pharmacogene summary: ABCB1 (MDR1, P-glycoprotein). Pharmacogenet. Genomics 21, 152–161. doi: 10.1097/fpc.0b013e3283385a1c
Hoenig, M. R., Walker, P. J., Gurnsey, C., Beadle, K., and Johnson, L. (2011). The C3435T polymorphism in ABCB1 influences atorvastatin efficacy and muscle symptoms in a high-risk vascular cohort. J. Clin. Lipidol. 5, 91–96. doi: 10.1016/j.jacl.2011.01.001
Horsey, A. J., Cox, M. H., Sarwat, S., and Kerr, I. D. (2016). The multidrug transporter ABCG2: still more questions than answers. Biochem. Soc. Trans. 44, 824–830. doi: 10.1042/bst20160014
Hou, Q., Li, S., Li, L., Li, Y., Sun, X., and Tian, H. (2015). Association between SLCO1B1 Gene T521C polymorphism and statin-related myopathy risk: a meta-analysis of case-control studies. Medicine 94:e1268. doi: 10.1097/md.0000000000001268
Hu, M., Cheung, B. M., and Tomlinson, B. (2012). Safety of statins: an update. Ther. Adv. Drug Saf. 3, 133–144. doi: 10.1177/2042098612439884
Hu, M. C., Mak, V. W. L., Chu, T. T. W., Waye, M. M. Y., and Tomlinson, B. (2009). Pharmacogenetics of HMG-CoA reductase inhibitors: optimizing the prevention of coronary heart disease. Curr. Pharmacogenomics Pers. Med. 7, 1–26. doi: 10.2174/187569209787582349
Hubacek, J. A., Adamkova, V., Zlatohlavek, L., Steiner-Mrazova, L., and Vrablik, M. (2017). COQ2 polymorphisms are not associated with increased risk of statin-induced myalgia/myopathy in the Czech population. Drug Metab. Pers. Ther. 32, 177–182.
Hubáček, J. A., Dlouhá, D., Adámková, V., Zlatohlavek, L., Viklický, O., Hrubá, P., et al. (2015a). SLCO1B1 polymorphism is not associated with risk of statin-induced myalgia/myopathy in a Czech population. Med. Sci. Monit. 21, 1454–1459. doi: 10.12659/msm.893007
Hubáček, J. A., Schwarzova, L., Zlatohlavek, L., Adámková, V., Češka, R., and Vrablík, M. (2015b). Apolipoprotein E polymorphism is not associated with statin induced myalgia/myopathy. Austin J. Cardiovasc. Dis. Atheroscler. 2:1009.
Huttunen, K. M., Raunio, H., and Rautio, J. (2011). Prodrugs–from serendipity to rational design. Pharmacol. Rev. 63, 750–771. doi: 10.1124/pr.110.003459
Ieiri, I., Suwannakul, S., Maeda, K., Uchimaru, H., Hashimoto, K., Kimura, M., et al. (2007). SLCO1B1 (OATP1B1, an uptake transporter) and ABCG2 (BCRP, an efflux transporter) variant alleles and pharmacokinetics of pitavastatin in healthy volunteers. Clin. Pharmacol. Ther. 82, 541–547. doi: 10.1038/sj.clpt.6100190
Igel, M., Sudhop, T., and von Bergmann, K. (2001). Metabolism and drug interactions of 3-hydroxy-3-methylglutaryl coenzyme A-reductase inhibitors (statins). Eur. J. Clin. Pharmacol. 57, 357–364. doi: 10.1007/s002280100329
Imai, Y., Nakane, M., Kage, K., Tsukahara, S., Ishikawa, E., Tsuruo, T., et al. (2002). C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low-level drug resistance. Mol. Cancer Ther. 1, 611–616.
Isackson, P. J., Ochs-Balcom, H. M., Ma, C., Harley, J. B., Peltier, W., Tarnopolsky, M., et al. (2011). Association of common variants in the human eyes shut ortholog (EYS) with statin-induced myopathy: evidence for additional functions of EYS. Muscle Nerve 44, 531–538. doi: 10.1002/mus.22115
Isackson, P. J., Wang, J., Zia, M., Spurgeon, P., Levesque, A., Bard, J., et al. (2018). RYR1 and CACNA1S genetic variants identified with statin-associated muscle symptoms. Pharmacogenomics 19, 1235–1249. doi: 10.2217/pgs-2018-0106
Jaskiewicz, A., Pajak, B., Litwiniuk, A., Urbanska, K., and Orzechowski, A. (2018). Geranylgeraniol Prevents Statin-Dependent Myotoxicity in C2C12 Muscle Cells through RAP1 GTPase Prenylation and Cytoprotective Autophagy. Oxid. Med. Cell Longev. 2018:6463807.
Jiang, F., Choi, J. Y., Lee, J. H., Ryu, S., Park, Z. W., Lee, J. G., et al. (2017). The influences of SLCO1B1 and ABCB1 genotypes on the pharmacokinetics of simvastatin, in relation to CYP3A4 inhibition. Pharmacogenomics 18, 459–469. doi: 10.2217/pgs-2016-0199
Jiang, J., Tang, Q., Feng, J., Dai, R., Wang, Y., Yang, Y., et al. (2016). Association between SLCO1B1 -521T>C and -388A>G polymorphisms and risk of statin-induced adverse drug reactions: a meta-analysis. Springerplus 5:1368.
Juliano, R. L., and Ling, V. (1976). A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta 455, 152–162. doi: 10.1016/0005-2736(76)90160-7
Kalliokoski, A., and Niemi, M. (2009). Impact of OATP transporters on pharmacokinetics. Br. J. Pharmacol. 158, 693–705. doi: 10.1111/j.1476-5381.2009.00430.x
Kalman, L. V., Agundez, J., Appell, M. L., Black, J. L., Bell, G. C., Boukouvala, S., et al. (2016). Pharmacogenetic allele nomenclature: international workgroup recommendations for test result reporting. Clin. Pharmacol. Ther. 99, 172–185. doi: 10.1002/cpt.280
Karonen, T., Neuvonen, P. J., and Backman, J. T. (2012). CYP2C8 but not CYP3A4 is important in the pharmacokinetics of montelukast. Br. J. Clin. Pharmacol. 73, 257–267. doi: 10.1111/j.1365-2125.2011.04086.x
Kaspera, R., Naraharisetti, S. B., Tamraz, B., Sahele, T., Cheesman, M. J., Kwok, P. Y., et al. (2010). Cerivastatin in vitro metabolism by CYP2C8 variants found in patients experiencing rhabdomyolysis. Pharmacogenet. Genomics 20, 619–629. doi: 10.1097/fpc.0b013e32833ecace
Keskitalo, J. E., Kurkinen, K. J., Neuvonen, M., Backman, J. T., Neuvonen, P. J., and Niemi, M. (2009a). No significant effect of ABCB1 haplotypes on the pharmacokinetics of fluvastatin, pravastatin, lovastatin, and rosuvastatin. Br. J. Clin. Pharmacol. 68, 207–213. doi: 10.1111/j.1365-2125.2009.03440.x
Keskitalo, J. E., Kurkinen, K. J., Neuvoneni, P. J., and Niemi, M. (2008). ABCB1 haplotypes differentially affect the pharmacokinetics of the acid and lactone forms of simvastatin and atorvastatin. Clin. Pharmacol. Ther. 84, 457–461. doi: 10.1038/clpt.2008.25
Keskitalo, J. E., Zolk, O., Fromm, M. F., Kurkinen, K. J., Neuvonen, P. J., and Niemi, M. (2009b). ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 86, 197–203. doi: 10.1038/clpt.2009.79
Khine, H., Yuet, W. C., Adams-Huet, B., and Ahmad, Z. (2016). Statin-associated muscle symptoms and SLCO1B1 rs4149056 genotype in patients with familial hypercholesterolemia. Am. Heart J. 179, 1–9. doi: 10.1016/j.ahj.2016.05.015
Kim, K. A., Park, P. W., Lee, O. J., Kang, D. K., and Park, J. Y. (2007). Effect of polymorphic CYP3A5 genotype on the single-dose simvastatin pharmacokinetics in healthy subjects. J. Clin. Pharmacol. 47, 87–93. doi: 10.1177/0091270006295063
Kim, T. E., Shin, D., Gu, N., Jung, B. H., Kim, J., Cho, Y. M., et al. (2017). The effect of genetic polymorphisms in SLCO2B1 on the lipid-lowering efficacy of rosuvastatin in healthy adults with elevated low-density lipoprotein. Basic Clin. Pharmacol. Toxicol. 121, 195–201. doi: 10.1111/bcpt.12826
Kim, W. J., Lee, J. H., Yi, J., Cho, Y. J., Heo, K., Lee, S. H., et al. (2010). A nonsynonymous variation in MRP2/ABCC2 is associated with neurological adverse drug reactions of carbamazepine in patients with epilepsy. Pharmacogenet. Genomics 20, 249–256.
Kim, Y., Yoon, S., Choi, Y., Yoon, S. H., Cho, J. Y., Jang, I. J., et al. (2019). Influence of OATP1B1 and BCRP polymorphisms on the pharmacokinetics and pharmacodynamics of rosuvastatin in elderly and young Korean subjects. Sci. Rep. 9:19410.
Kirchheiner, J., Kudlicz, D., Meisel, C., Bauer, S., Meineke, I., Roots, I., et al. (2003). Influence of CYP2C9 polymorphisms on the pharmacokinetics and cholesterol-lowering activity of (-)-3S,5R-fluvastatin and (+)-3R,5S-fluvastatin in healthy volunteers. Clin. Pharmacol. Ther. 74, 186–194. doi: 10.1016/s0009-9236(03)00121-8
Kitzmiller, J. P., Luzum, J. A., Baldassarre, D., Krauss, R. M., and Medina, M. W. (2014). CYP3A4∗22 and CYP3A5∗3 are associated with increased levels of plasma simvastatin concentrations in the cholesterol and pharmacogenetics study cohort. Pharmacogenet. Genomics 24, 486–491. doi: 10.1097/fpc.0000000000000079
Kondo, C., Suzuki, H., Itoda, M., Ozawa, S., Sawada, J., Kobayashi, D., et al. (2004). Functional analysis of SNPs variants of BCRP/ABCG2. Pharm. Res. 21, 1895–1903. doi: 10.1023/b:pham.0000045245.21637.d4
Korani, S., Bahrami, S., Korani, M., Banach, M., Johnston, T. P., and Sahebkar, A. (2019). Parenteral systems for statin delivery: a review. Lipids Health Dis. 18:193.
Kuehl, P., Zhang, J., Lin, Y., Lamba, J., Assem, M., Schuetz, J., et al. (2001). Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat. Genet. 27, 383–391. doi: 10.1038/86882
Kyrklund, C., Backman, J. T., Kivistö, K. T., Neuvonen, M., Laitila, J., and Neuvonen, P. J. (2000). Rifampin greatly reduces plasma simvastatin and simvastatin acid concentrations. Clin. Pharmacol. Ther. 68, 592–597. doi: 10.1067/mcp.2000.111414
Lamba, J., Hebert, J. M., Schuetz, E. G., Klein, T. E., and Altman, R. B. (2012). PharmGKB summary: very important pharmacogene information for CYP3A5. Pharmacogenet. Genomics 22, 555–558. doi: 10.1097/fpc.0b013e328351d47f
Lansberg, P., Lee, A., Lee, Z. V., Subramaniam, K., and Setia, S. (2018). Nonadherence to statins: individualized intervention strategies outside the pill box. Vasc. Health Risk Manag. 14, 91–102. doi: 10.2147/vhrm.s158641
Laufs, U., Banach, M., Mancini, G. B. J., Gaudet, D., Bloedon, L. T., Sterling, L. R., et al. (2019). Efficacy and safety of bempedoic acid in patients with hypercholesterolemia and statin intolerance. J. Am. Heart Assoc. 8: e011662.
Lee, E., Ryan, S., Birmingham, B., Zalikowski, J., March, R., Ambrose, H., et al. (2005). Rosuvastatin pharmacokinetics and pharmacogenetics in white and Asian subjects residing in the same environment. Clin. Pharmacol. Ther. 78, 330–341. doi: 10.1016/j.clpt.2005.06.013
Lee, H. H., and Ho, R. H. (2017). Interindividual and interethnic variability in drug disposition: polymorphisms in organic anion transporting polypeptide 1B1 (OATP1B1 SLCO1B1). Br. J. Clin. Pharmacol. 83, 1176–1184. doi: 10.1111/bcp.13207
Lee, H. K., Hu, M., Lui, S., Ho, C. S., Wong, C. K., and Tomlinson, B. (2013). Effects of polymorphisms in ABCG2, SLCO1B1, SLC10A1 and CYP2C9/19 on plasma concentrations of rosuvastatin and lipid response in Chinese patients. Pharmacogenomics 14, 1283–1294. doi: 10.2217/pgs.13.115
Lee, Y. J., Lee, M. G., Lim, L. A., Jang, S. B., and Chung, J. Y. (2010). Effects of SLCO1B1 and ABCB1 genotypes on the pharmacokinetics of atorvastatin and 2-hydroxyatorvastatin in healthy Korean subjects. Int. J. Clin. Pharmacol. Ther. 48, 36–45. doi: 10.5414/cpp48036
Lee, Y. S., and Chun, P. S. (2018). Effect of SLCO1B1 T521C on statin-induced myotoxicity: a systematic review and meta-analysis. Korean J. Clin. Pharm. 28, 320–332. doi: 10.24304/kjcp.2018.28.4.320
Leschziner, G. D., Andrew, T., Pirmohamed, M., and Johnson, M. R. (2007). ABCB1 genotype and PGP expression, function and therapeutic drug response: a critical review and recommendations for future research. Pharmacogenomics J. 7, 154–179. doi: 10.1038/sj.tpj.6500413
Liao, J. K. (2007). Safety and efficacy of statins in Asians. Am. J. Cardiol. 99, 410–414. doi: 10.1016/j.amjcard.2006.08.051
Limaye, V., Bundell, C., Hollingsworth, P., Rojana-Udomsart, A., Mastaglia, F., Blumbergs, P., et al. (2015). Clinical and genetic associations of autoantibodies to 3-hydroxy-3-methyl-glutaryl-coenzyme a reductase in patients with immune-mediated myositis and necrotizing myopathy. Muscle Nerve 52, 196–203. doi: 10.1002/mus.24541
Linde, R., Peng, L., Desai, M., and Feldman, D. (2010). The role of vitamin D and SLCO1B1∗5 gene polymorphism in statin-associated myalgias. Dermatoendocrinology 2, 77–84. doi: 10.4161/derm.2.2.13509
Link, E., Parish, S., Armitage, J., Bowman, L., Heath, S., Matsuda, F., et al. (2008). SLCO1B1 variants and statin-induced myopathy–a genomewide study. N. Engl. J. Med. 359, 789–799. doi: 10.1056/nejmoa0801936
Liu, A., Wu, Q., Guo, J., Ares, I., Rodriguez, J. L., Martinez-Larranaga, M. R., et al. (2019). Statins: adverse reactions, oxidative stress and metabolic interactions. Pharmacol. Ther.195, 54–84. doi: 10.1016/j.pharmthera.2018.10.004
Liu, J. E., Liu, X. Y., Chen, S., Zhang, Y., Cai, L. Y., Yang, M., et al. (2017). SLCO1B1 521T > C polymorphism associated with rosuvastatin-induced myotoxicity in Chinese coronary artery disease patients: a nested case-control study. Eur. J. Clin. Pharmacol. 73, 1409–1416. doi: 10.1007/s00228-017-2318-z
Liu, M., Wu, X. J., Zhao, G. L., Zhang, T., Xu, S. S., Sun, Y. X., et al. (2016). Effects of Polymorphisms in NR1H4, NR1I2, SLCO1B1, and ABCG2 on the pharmacokinetics of rosuvastatin in healthy chinese volunteers. J. Cardiovasc. Pharmacol. 68, 383–390. doi: 10.1097/fjc.0000000000000426
Liu, X., Cheng, D., Kuang, Q., Liu, G., and Xu, W. (2014). Association of UGT1A1∗28 polymorphisms with irinotecan-induced toxicities in colorectal cancer: a meta-analysis in Caucasians. Pharmacogenomics J. 14, 120–129. doi: 10.1038/tpj.2013.10
Lotteau, S., Ivarsson, N., Yang, Z., Restagno, D., Colyer, J., Hopkins, P., et al. (2019). A mechanism for statin-induced susceptibility to myopathy. JACC Basic Transl. Sci. 4, 509–523.
Luvai, A., Mbagaya, W., Hall, A. S., and Barth, J. H. (2012). Rosuvastatin: a review of the pharmacology and clinical effectiveness in cardiovascular disease. Clin. Med. Insights Cardiol. 6, 17–33.
Luzum, J. A., Kitzmiller, J. P., Isackson, P. J., Ma, C., Medina, M. W., Dauki, A. M., et al. (2015). GATM polymorphism associated with the risk for statin-induced myopathy does not replicate in case-control analysis of 715 dyslipidemic individuals. Cell Metab. 21, 622–627. doi: 10.1016/j.cmet.2015.03.003
Mackenzie, P. I., Bock, K. W., Burchell, B., Guillemette, C., Ikushiro, S., Iyanagi, T., et al. (2005). Nomenclature update for the mammalian UDP glycosyltransferase (UGT) gene superfamily. Pharmacogenet. Genomics 15, 677–685. doi: 10.1097/01.fpc.0000173483.13689.56
Maeda, K., Ieiri, I., Yasuda, K., Fujino, A., Fujiwara, H., Otsubo, K., et al. (2006). Effects of organic anion transporting polypeptide 1B1 haplotype on pharmacokinetics of pravastatin, valsartan, and temocapril. Clin. Pharmacol. Ther. 79, 427–439. doi: 10.1016/j.clpt.2006.01.011
Maji, D., Shaikh, S., Solanki, D., and Gaurav, K. (2013). Safety of statins. Indian J. Endocrinol. Metab. 17, 636–646.
Mammen, A. L., Gaudet, D., Brisson, D., Christopher-Stine, L., Lloyd, T. E., Leffell, M. S., et al. (2012). Increased frequency of DRB1∗11:01 in anti-hydroxymethylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Care Res. 64, 1233–1237.
Mangravite, L. M., Engelhardt, B. E., Medina, M. W., Smith, J. D., Brown, C. D., Chasman, D. I., et al. (2013). A statin-dependent QTL for GATM expression is associated with statin-induced myopathy. Nature 502, 377–380. doi: 10.1038/nature12508
Mao, Q., Lai, Y., and Wang, J. (2018). Drug Transporters in Xenobiotic Disposition and Pharmacokinetic Prediction. Drug Metab. Dispos. 46, 561–566. doi: 10.1124/dmd.118.081356
Marciante, K. D., Durda, J. P., Heckbert, S. R., Lumley, T., Rice, K., McKnight, B., et al. (2011). Cerivastatin, genetic variants, and the risk of rhabdomyolysis. Pharmacogenet. Genomics 21, 280–288.
Marcoff, L., and Thompson, P. D. (2007). The role of coenzyme Q10 in statin-associated myopathy: a systematic review. J. Am. Coll. Cardiol. 49, 2231–2237. doi: 10.1016/j.jacc.2007.02.049
Martin, P. D., Warwick, M. J., Dane, A. L., Hill, S. J., Giles, P. B., Phillips, P. J., et al. (2003). Metabolism, excretion, and pharmacokinetics of rosuvastatin in healthy adult male volunteers. Clin. Ther. 25, 2822–2835. doi: 10.1016/s0149-2918(03)80336-3
Mazzu, A. L., Lasseter, K. C., Shamblen, E. C., Agarwal, V., Lettieri, J., and Sundaresen, P. (2000). Itraconazole alters the pharmacokinetics of atorvastatin to a greater extent than either cerivastatin or pravastatin. Clin. Pharmacol. Ther. 68, 391–400. doi: 10.1067/mcp.2000.110537
McClure, D. L., Valuck, R. J., Glanz, M., Murphy, J. R., and Hokanson, J. E. (2007). Statin and statin-fibrate use was significantly associated with increased myositis risk in a managed care population. J. Clin. Epidemiol. 60, 812–818. doi: 10.1016/j.jclinepi.2006.11.006
Megaraj, V., Zhao, T., Paumi, C. M., Gerk, P. M., Kim, R. B., and Vore, M. (2011). Functional analysis of nonsynonymous single nucleotide polymorphisms of multidrug resistance-associated protein 2 (ABCC2). Pharmacogenet. Genomics 21, 506–515. doi: 10.1097/fpc.0b013e328348c786
Michaels, S., and Wang, M. Z. (2014). The revised human liver cytochrome P450 “Pie”: absolute protein quantification of CYP4F and CYP3A enzymes using targeted quantitative proteomics. Drug Metab. Dispos. 42, 1241–1251. doi: 10.1124/dmd.114.058040
Mirosevic Skvrce, N., Bozina, N., Zibar, L., Barisic, I., Pejnovic, L., and Macolic Sarinic, V. (2013). CYP2C9 and ABCG2 polymorphisms as risk factors for developing adverse drug reactions in renal transplant patients taking fluvastatin: a case-control study. Pharmacogenomics 14, 1419–1431. doi: 10.2217/pgs.13.135
Mirosevic Skvrce, N., Macolic Sarinic, V., Simic, I., Ganoci, L., Muacevic Katanec, D., and Bozina, N. (2015). ABCG2 gene polymorphisms as risk factors for atorvastatin adverse reactions: a case-control study. Pharmacogenomics 16, 803–815. doi: 10.2217/pgs.15.47
Morimoto, K., Ueda, S., Seki, N., Igawa, Y., Kameyama, Y., Shimizu, A., et al. (2005). OATP-C (OATP01B1) ∗15 is associated with statin-induced myopathy in hypercholesterolemic patients. Clin. Pharmacol. Ther. 77:P21.
Morioka, T. Y., Lee, A. J., Bertisch, S., and Buettner, C. (2015). Vitamin D status modifies the association between statin use and musculoskeletal pain: a population based study. Atherosclerosis 238, 77–82. doi: 10.1016/j.atherosclerosis.2014.11.012
Mulder, A. B., van Lijf, H. J., Bon, M. A., van den Bergh, F. A., Touw, D. J., Neef, C., et al. (2001). Association of polymorphism in the cytochrome CYP2D6 and the efficacy and tolerability of simvastatin. Clin. Pharmacol. Ther. 70, 546–551. doi: 10.1067/mcp.2001.120251
Mwinyi, J., Johne, A., Bauer, S., Roots, I., and Gerloff, T. (2004). Evidence for inverse effects of OATP-C (SLC21A6) 5 and 1b haplotypes on pravastatin kinetics. Clin. Pharmacol. Ther. 75, 415–421. doi: 10.1016/j.clpt.2003.12.016
Nakamura, T., Sakaeda, T., Horinouchi, M., Tamura, T., Aoyama, N., Shirakawa, T., et al. (2002). Effect of the mutation (C3435T) at exon 26 of the MDR1 gene on expression level of MDR1 messenger ribonucleic acid in duodenal enterocytes of healthy Japanese subjects. Clin. Pharmacol. Ther. 71, 297–303. doi: 10.1067/mcp.2002.122055
Naraharisetti, S. B., Lin, Y. S., Rieder, M. J., Marciante, K. D., Psaty, B. M., Thummel, K. E., et al. (2010). Human liver expression of CYP2C8: gender, age, and genotype effects. Drug Metab. Dispos. 38, 889–893. doi: 10.1124/dmd.109.031542
Neroldova, M., Stranecky, V., Hodanova, K., Hartmannova, H., Piherova, L., Pristoupilova, A., et al. (2016). Rare variants in known and novel candidate genes predisposing to statin-associated myopathy. Pharmacogenomics 17, 1405–1414. doi: 10.2217/pgs-2016-0071
Neuvonen, P. J. (2010). Drug interactions with HMG-CoA reductase inhibitors (statins): the importance of CYP enzymes, transporters and pharmacogenetics. Curr. Opin. Investig. Drugs 11, 323–332.
Neuvonen, P. J., Backman, J. T., and Niemi, M. (2008). Pharmacokinetic comparison of the potential over-the-counter statins simvastatin, lovastatin, fluvastatin and pravastatin. Clin. Pharmacokinet. 47, 463–474. doi: 10.2165/00003088-200847070-00003
Neuvonen, P. J., Niemi, M., and Backman, J. T. (2006). Drug interactions with lipid-lowering drugs: mechanisms and clinical relevance. Clin. Pharmacol. Ther. 80, 565–581. doi: 10.1016/j.clpt.2006.09.003
Nguyen, T. D., Markova, S., Liu, W., Gow, J. M., Baldwin, R. M., Habashian, M., et al. (2013). Functional characterization of ABCC2 promoter polymorphisms and allele-specific expression. Pharmacogenomics J. 13, 396–402. doi: 10.1038/tpj.2012.20
Niemi, M. (2010). Transporter pharmacogenetics and statin toxicity. Clin. Pharmacol. Ther. 87, 130–133. doi: 10.1038/clpt.2009.197
Niemi, M., Pasanen, M. K., and Neuvonen, P. J. (2011). Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol. Rev. 63, 157–181.
Niemi, M., Schaeffeler, E., Lang, T., Fromm, M. F., Neuvonen, M., Kyrklund, C., et al. (2004). High plasma pravastatin concentrations are associated with single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide-C (OATP-C, SLCO1B1). Pharmacogenetics 14, 429–440.
Nishimoto, T., Tozawa, R., Amano, Y., Wada, T., Imura, Y., and Sugiyama, Y. (2003). Comparing myotoxic effects of squalene synthase inhibitor, T-91485, and 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors in human myocytes. Biochem. Pharmacol. 66, 2133–2139.
Nofziger, C., Turner, A. J., Sangkuhl, K., Whirl-Carrillo, M., Agundez, J. A. G., Black, J. L., et al. (2020). PharmVar GeneFocus: CYP2D6. Clin. Pharmacol. Ther. 107, 154–170.
Nordin, C., Dahl, M. L., Eriksson, M., and Sjoberg, S. (1997). Is the cholesterol-lowering effect of simvastatin influenced by CYP2D6 polymorphism? Lancet 350, 29–30. doi: 10.1016/s0140-6736(05)66238-1
Nozawa, T., Nakajima, M., Tamai, I., Noda, K., Nezu, J., Sai, Y., et al. (2002). Genetic polymorphisms of human organic anion transporters OATP-C (SLC21A6) and OATP-B (SLC21A9): allele frequencies in the Japanese population and functional analysis. J. Pharmacol. Exp. Ther. 302, 804–813. doi: 10.1124/jpet.302.2.804
Oh, E. S., Kim, C. O., Cho, S. K., Park, M. S., and Chung, J. Y. (2013). Impact of ABCC2, ABCG2 and SLCO1B1 polymorphisms on the pharmacokinetics of pitavastatin in humans. Drug Metab. Pharmacokinet. 28, 196–202. doi: 10.2133/dmpk.dmpk-12-rg-068
Oh, J., Ban, M. R., Miskie, B. A., Pollex, R. L., and Hegele, R. A. (2007). Genetic determinants of statin intolerance. Lipids Health Dis. 6:7. doi: 10.1186/1476-511x-6-7
Olbricht, C., Wanner, C., Eisenhauer, T., Kliem, V., Doll, R., Boddaert, M., et al. (1997). Accumulation of lovastatin, but not pravastatin, in the blood of cyclosporine-treated kidney graft patients after multiple doses. Clin. Pharmacol. Ther. 62, 311–321. doi: 10.1016/s0009-9236(97)90034-5
Oshiro, C., Mangravite, L., Klein, T., and Altman, R. (2010). PharmGKB very important pharmacogene: SLCO1B1. Pharmacogenet. Genomics 20, 211–216. doi: 10.1097/fpc.0b013e328333b99c
Ovesjo, M. L., Skilving, I., Bergman, P., Rane, A., Ekstrom, L., and Bjorkhem-Bergman, L. (2016). Low Vitamin D levels and genetic polymorphism in the Vitamin D receptor are associated with increased risk of statin-induced myopathy. Basic Clin. Pharmacol. Toxicol. 118, 214–218. doi: 10.1111/bcpt.12482
Pasanen, M. K., Neuvonen, M., Neuvonen, P. J., and Niemi, M. (2006). SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet. Genomics 16, 873–879. doi: 10.1097/01.fpc.0000230416.82349.90
Pasanen, M. K., Fredrikson, H., Neuvonen, P. J., and Niemi, M. (2007). Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 82, 726–733. doi: 10.1038/sj.clpt.6100220
Passey, C., Birnbaum, A. K., Brundage, R. C., Oetting, W. S., Israni, A. K., and Jacobson, P. A. (2011). Dosing equation for tacrolimus using genetic variants and clinical factors. Br. J. Clin. Pharmacol. 72, 948–957. doi: 10.1111/j.1365-2125.2011.04039.x
Pasternak, R. C., Smith, S. C. Jr., Bairey-Merz, C. N., Grundy, S. M., Cleeman, J. I., and Lenfant, C. (2002). ACC/AHA/NHLBI clinical advisory on the use and safety of statins. J. Am. Coll. Cardiol. 40, 567–572. doi: 10.1016/s0735-1097(02)02030-2
Peyser, B., Perry, E. P., Singh, K., Gill, R. D., Mehan, M. R., Haga, S. B., et al. (2018). Effects of delivering SLCO1B1 pharmacogenetic information in randomized trial and observational settings. Circ. Genom. Precis. Med. 11:e002228.
Pierno, S., De Luca, A., Tricarico, D., Roselli, A., Natuzzi, F., Ferrannini, E., et al. (1995). Potential risk of myopathy by HMG-CoA reductase inhibitors: a comparison of pravastatin and simvastatin effects on membrane electrical properties of rat skeletal muscle fibers. J. Pharmacol. Exp. Ther. 275, 1490–1496.
Pinal-Fernandez, I., Casal-Dominguez, M., and Mammen, A. L. (2018). Immune-mediated necrotizing myopathy. Curr. Rheumatol. Rep. 20:21.
Prueksaritanont, T., Gorham, L. M., Ma, B., Liu, L., Yu, X., Zhao, J. J., et al. (1997). In vitro metabolism of simvastatin in humans [SBT]identification of metabolizing enzymes and effect of the drug on hepatic P450s. Drug Metab. Dispos. 25, 1191–1199.
Prueksaritanont, T., Ma, B., and Yu, N. (2003). The human hepatic metabolism of simvastatin hydroxy acid is mediated primarily by CYP3A, and not CYP2D6. Br. J. Clin. Pharmacol. 56, 120–124. doi: 10.1046/j.1365-2125.2003.01833.x
Prueksaritanont, T., Subramanian, R., Fang, X., Ma, B., Qiu, Y., Lin, J. H., et al. (2002). Glucuronidation of statins in animals and humans: a novel mechanism of statin lactonization. Drug Metab. Dispos. 30, 505–512. doi: 10.1124/dmd.30.5.505
Puccetti, L., Ciani, F., and Auteri, A. (2010). Genetic involvement in statins induced myopathy. Preliminary data from an observational case-control study. Atherosclerosis 211, 28–29. doi: 10.1016/j.atherosclerosis.2010.02.026
Qu, H., Guo, M., Chai, H., Wang, W. T., Gao, Z. Y., and Shi, D. Z. (2018). Effects of coenzyme Q10 on Statin-Induced Myopathy: an updated meta-analysis of randomized controlled trials. J. Am. Heart Assoc. 7:e009835.
Raju, S. B., Varghese, K., and Madhu, K. (2013). Management of statin intolerance. Indian J. Endocrinol. Metab. 17, 977–982. doi: 10.4103/2230-8210.122602
Ramakumari, N., Indumathi, B., Katkam, S. K., and Kutala, V. K. (2018). Impact of pharmacogenetics on statin-induced myopathy in South-Indian subjects. Indian Heart J. 70(Suppl. 3), S120–S125.
Ramsey, L. B., Johnson, S. G., Caudle, K. E., Haidar, C. E., Voora, D., Wilke, R. A., et al. (2014). The clinical pharmacogenetics implementation consortium guideline for SLCO1B1 and simvastatin-induced myopathy: 2014 update. Clin. Pharmacol. Ther. 96, 423–428. doi: 10.1038/clpt.2014.125
Riedmaier, S., Klein, K., Hofmann, U., Keskitalo, J. E., Neuvonen, P. J., Schwab, M., et al. (2010). UDP-glucuronosyltransferase (UGT) polymorphisms affect atorvastatin lactonization in vitro and in vivo. Clin. Pharmacol. Ther. 87, 65–73. doi: 10.1038/clpt.2009.181
Robey, R. W., To, K. K., Polgar, O., Dohse, M., Fetsch, P., Dean, M., et al. (2009). ABCG2: a perspective. Adv. Drug Deliv. Rev. 61, 3–13.
Rosenson, R. S., Baker, S. K., Jacobson, T. A., Kopecky, S. L., Parker, B. A., and The National, et al. (2014). An assessment by the Statin Muscle Safety Task Force: update. J. Clin. Lipidol. 8(3 Suppl.), S58–S71.
Rowan, C. G., Brunelli, S. M., Munson, J., Flory, J., Reese, P. P., Hennessy, S., et al. (2012). Clinical importance of the drug interaction between statins and CYP3A4 inhibitors: a retrospective cohort study in The Health Improvement Network. Pharmacoepidemiol. Drug Saf. 21, 494–506. doi: 10.1002/pds.3199
Ruano, G., Thompson, P. D., Windemuth, A., Seip, R. L., Dande, A., Sorokin, A., et al. (2007). Physiogenomic association of statin-related myalgia to serotonin receptors. Muscle Nerve 36, 329–335. doi: 10.1002/mus.20871
Ruano, G., Windemuth, A., Wu, A. H., Kane, J. P., Malloy, M. J., Pullinger, C. R., et al. (2011). Mechanisms of statin-induced myalgia assessed by physiogenomic associations. Atherosclerosis 218, 451–456. doi: 10.1016/j.atherosclerosis.2011.07.007
Sanchez-Dominguez, C. N., Gallardo-Blanco, H. L., Salinas-Santander, M. A., and Ortiz-Lopez, R. (2018). Uridine 5’-diphospho-glucronosyltrasferase: its role in pharmacogenomics and human disease. Exp. Ther. Med. 16, 3–11.
Sandritter, T. L., McLaughlin, M., Artman, M., and Lowry, J. (2017). The Interplay between Pharmacokinetics and Pharmacodynamics. Pediatr. Rev. 38, 195–206. doi: 10.1542/pir.2016-0101
Santos, P. C., Soares, R. A., Nascimento, R. M., Machado-Coelho, G. L., Mill, J. G., Krieger, J. E., et al. (2011). SLCO1B1 rs4149056 polymorphism associated with statin-induced myopathy is differently distributed according to ethnicity in the Brazilian general population: amerindians as a high risk ethnic group. BMC Med. Genet. 12:136. doi: 10.1186/1471-2350-12-136
Schaars, C. F., and Stalenhoef, A. F. (2008). Effects of ubiquinone (coenzyme Q10) on myopathy in statin users. Curr. Opin. Lipidol. 19, 553–557. doi: 10.1097/mol.0b013e3283168ecd
Schachter, M. (2005). Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam. Clin. Pharmacol. 19, 117–125. doi: 10.1111/j.1472-8206.2004.00299.x
Schech, S., Graham, D., Staffa, J., Andrade, S. E., La Grenade, L., Burgess, M., et al. (2007). Risk factors for statin-associated rhabdomyolysis. Pharmacoepidemiol. Drug Saf. 16, 352–358. doi: 10.1002/pds.1287
Schirris, T. J., Ritschel, T., Bilos, A., Smeitink, J. A., and Russel, F. G. (2015). Statin Lactonization by Uridine 5’-Diphospho-glucuronosyltransferases (UGTs). Mol. Pharm. 12, 4048–4055. doi: 10.1021/acs.molpharmaceut.5b00474
Scott, S. A., Sangkuhl, K., Shuldiner, A. R., Hulot, J. S., Thorn, C. F., Altman, R. B., et al. (2012). PharmGKB summary: very important pharmacogene information for cytochrome P450, family 2, subfamily C, polypeptide 19. Pharmacogenet. Genomics 22, 159–165. doi: 10.1097/fpc.0b013e32834d4962
Servedio, V., d’Apolito, M., Maiorano, N., Minuti, B., Torricelli, F., Ronchi, F., et al. (2005). Spectrum of UGT1A1 mutations in Crigler-Najjar (CN) syndrome patients: identification of twelve novel alleles and genotype-phenotype correlation. Hum. Mutat. 25:325. doi: 10.1002/humu.9322
Sharom, F. J. (2008). ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics 9, 105–127. doi: 10.2217/14622416.9.1.105
Shin, J., Pauly, D. F., Pacanowski, M. A., Langaee, T., Frye, R. F., and Johnson, J. A. (2011). Effect of cytochrome P450 3A5 genotype on atorvastatin pharmacokinetics and its interaction with clarithromycin. Pharmacotherapy 31, 942–950. doi: 10.1592/phco.31.10.942
Siddiqui, M. K., Maroteau, C., Veluchamy, A., Tornio, A., Tavendale, R., Carr, F., et al. (2017). A common missense variant of LILRB5 is associated with statin intolerance and myalgia. Eur. Heart J. 38, 3569–3575. doi: 10.1093/eurheartj/ehx467
Sirtori, C. R., Mombelli, G., Triolo, M., and Laaksonen, R. (2012). Clinical response to statins: mechanism(s) of variable activity and adverse effects. Ann. Med. 44, 419–432. doi: 10.3109/07853890.2011.582135
Skottheim, I. B., Gedde-Dahl, A., Hejazifar, S., Hoel, K., and Asberg, A. (2008). Statin induced myotoxicity: the lactone forms are more potent than the acid forms in human skeletal muscle cells in vitro. Eur. J. Pharm. Sci. 33, 317–325. doi: 10.1016/j.ejps.2007.12.009
Stancu, C., and Sima, A. (2001). Statins: mechanism of action and effects. J. Cell. Mol. Med. 5, 378–387. doi: 10.1111/j.1582-4934.2001.tb00172.x
Stormo, C., Bogsrud, M. P., Hermann, M., Asberg, A., Piehler, A. P., Retterstol, K., et al. (2013). UGT1A1∗28 is associated with decreased systemic exposure of atorvastatin lactone. Mol. Diagn. Ther. 17, 233–237. doi: 10.1007/s40291-013-0031-x
Stranecky, V., Neroldova, M., Hodanova, K., Hartmannova, H., Piherova, L., Zemankova, P., et al. (2016). Large copy-number variations in patients with statin-associated myopathy affecting statin myopathy-related loci. Physiol. Res. 65, 1005–1011. doi: 10.33549/physiolres.933284
Taha, D. A., De Moor, C. H., Barrett, D. A., Lee, J. B., Gandhi, R. D., Hoo, C. W., et al. (2016). The role of acid-base imbalance in statin-induced myotoxicity. Transl. Res. 174, 140–160.e14.
Tamura, A., Wakabayashi, K., Onishi, Y., Takeda, M., Ikegami, Y., Sawada, S., et al. (2007). Re-evaluation and functional classification of non-synonymous single nucleotide polymorphisms of the human ATP-binding cassette transporter ABCG2. Cancer Sci. 98, 231–239. doi: 10.1111/j.1349-7006.2006.00371.x
Taylor, B. A., Lorson, L., White, C. M., and Thompson, P. D. (2015). A randomized trial of coenzyme Q10 in patients with confirmed statin myopathy. Atherosclerosis 238, 329–335. doi: 10.1016/j.atherosclerosis.2014.12.016
Thompson, P. D., Clarkson, P., and Karas, R. H. (2003). Statin-associated myopathy. JAMA 289, 1681–1690. doi: 10.1001/jama.289.13.1681
Thompson, P. D., Panza, G., Zaleski, A., and Taylor, B. (2016). Statin-associated side effects. J. Am. Coll. Cardiol. 67, 2395–2410. doi: 10.1016/j.jacc.2016.02.071
Tirona, R. G., Leake, B. F., Merino, G., and Kim, R. B. (2001). Polymorphisms in OATP-C: identification of multiple allelic variants associated with altered transport activity among European- and African-Americans. J. Biol. Chem. 276, 35669–35675. doi: 10.1074/jbc.m103792200
Tomita, Y., Maeda, K., and Sugiyama, Y. (2013). Ethnic variability in the plasma exposures of OATP1B1 substrates such as HMG-CoA reductase inhibitors: a kinetic consideration of its mechanism. Clin. Pharmacol. Ther. 94, 37–51. doi: 10.1038/clpt.2012.221
Torkamani, A., Wineinger, N. E., and Topol, E. J. (2018). The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 19, 581–590. doi: 10.1038/s41576-018-0018-x
Tornio, A., Vakkilainen, J., Neuvonen, M., Backman, J. T., Neuvonen, P. J., and Niemi, M. (2015). SLCO1B1 polymorphism markedly affects the pharmacokinetics of lovastatin acid. Pharmacogenet. Genomics 25, 382–387. doi: 10.1097/fpc.0000000000000148
Turner, R. M., Fontana, V., Zhang, J. E., Carr, D., Yin, P., FitzGerald, R., et al. (2020). A genome-wide association study of circulating levels of atorvastatin and its major metabolites. Clin. Pharmacol. Ther. 108, 287–297. doi: 10.1002/cpt.1820
van Beek, E. R., Cohen, L. H., Leroy, I. M., Ebetino, F. H., Löwik, C. W., and Papapoulos, S. E. (2003). Differentiating the mechanisms of antiresorptive action of nitrogen containing bisphosphonates. Bone 33, 805–811. doi: 10.1016/j.bone.2003.07.007
Vassy, J. L., Chun, S., Advani, S., Ludin, S. A., Smith, J. G., and Alligood, E. C. (2019). Impact of SLCO1B1 pharmacogenetic testing on patient and healthcare outcomes: a systematic review. Clin. Pharmacol. Ther. 106, 360–373. doi: 10.1002/cpt.1223
Verdoodt, A., Honore, P. M., Jacobs, R., De Waele, E., Van Gorp, V., De Regt, J., et al. (2018). Do statins induce or protect from acute kidney injury and chronic kidney disease: an update review in 2018. J. Transl. Int. Med. 6, 21–25. doi: 10.2478/jtim-2018-0005
Vickers, S., Duncan, C. A., Vyas, K. P., Kari, P. H., Arison, B., Prakash, S. R., et al. (1990). In vitro and in vivo biotransformation of simvastatin, an inhibitor of HMG CoA reductase. Drug Metab. Dispos. 18, 476–483.
Voora, D., Shah, S. H., Spasojevic, I., Ali, S., Reed, C. R., Salisbury, B. A., et al. (2009). The SLCO1B1∗5 genetic variant is associated with statin-induced side effects. J. Am. Coll. Cardiol. 54, 1609–1616. doi: 10.1016/j.jacc.2009.04.053
Vyas, K. P., Kari, P. H., Pitzenberger, S. M., Halpin, R. A., Ramjit, H. G., Arison, B., et al. (1990a). Biotransformation of lovastatin. I. Structure elucidation of in vitro and in vivo metabolites in the rat and mouse. Drug Metab Dispos. 18, 203–211.
Vyas, K. P., Kari, P. H., Wang, R. W., and Lu, A. Y. (1990b). Biotransformation of lovastatin–III. Effect of cimetidine and famotidine on in vitro metabolism of lovastatin by rat and human liver microsomes. Biochem. Pharmacol. 39, 67–73. doi: 10.1016/0006-2952(90)90649-6
Wan, Z., Wang, G., Li, T., Xu, B., Pei, Q., Peng, Y., et al. (2015). Marked Alteration of Rosuvastatin Pharmacokinetics in Healthy Chinese with ABCG2 34G>A and 421C>A Homozygote or Compound Heterozygote. J. Pharmacol. Exp. Ther. 354, 310–315. doi: 10.1124/jpet.115.225045
Wang, D., Johnson, A. D., Papp, A. C., Kroetz, D. L., and Sadee, W. (2005). Multidrug resistance polypeptide 1 (MDR1, ABCB1) variant 3435C>T affects mRNA stability. Pharmacogenet. Genomics 15, 693–704. doi: 10.1097/01.fpc.0000178311.02878.83
Wang, D., and Sadee, W. (2012). The Making of a CYP3A biomarker panel for guiding drug therapy. J. Pers. Med. 2, 175–191. doi: 10.3390/jpm2040175
Wang, R. W., Kari, P. H., Lu, A. Y., Thomas, P. E., Guengerich, F. P., and Vyas, K. P. (1991). Biotransformation of lovastatin. IV. Identification of cytochrome P450 3A proteins as the major enzymes responsible for the oxidative metabolism of lovastatin in rat and human liver microsomes. Arch. Biochem. Biophys. 290, 355–361.
Wang, Y., and Liu, Z. P. (2019). PCSK9 Inhibitors: novel therapeutic strategies for lowering LDLCholesterol. Mini Rev. Med. Chem. 19, 165–176. doi: 10.2174/1389557518666180423111442
Westwood, F. R., Bigley, A., Randall, K., Marsden, A. M., and Scott, R. C. (2005). Statin-induced muscle necrosis in the rat: distribution, development, and fibre selectivity. Toxicol. Pathol. 33, 246–257. doi: 10.1080/01926230590908213
Whirl-Carrillo, M., McDonagh, E. M., Hebert, J. M., Gong, L., Sangkuhl, K., Thorn, C. F., et al. (2012). Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 92, 414–417.
Wiggins, B. S., Saseen, J. J., Page, R. L. II, Reed, B. N., Sneed, K., Kostis, J. B., et al. (2016). Recommendations for management of clinically significant drug-drug interactions with statins and select agents used in patients with cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 134, e468–e495.
Wilke, R. A., Moore, J. H., and Burmester, J. K. (2005). Relative impact of CYP3A genotype and concomitant medication on the severity of atorvastatin-induced muscle damage. Pharmacogenet. Genomics 15, 415–421. doi: 10.1097/01213011-200506000-00007
Wray, N. R., Goddard, M. E., and Visscher, P. M. (2007). Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res. 17, 1520–1528. doi: 10.1101/gr.6665407
Wright, D. F., Winter, H. R., and Duffull, S. B. (2011). Understanding the time course of pharmacological effect: a PKPD approach. Br. J. Clin. Pharmacol. 71, 815–823. doi: 10.1111/j.1365-2125.2011.03925.x
Wyss, M., and Kaddurah-Daouk, R. (2000). Creatine and creatinine metabolism. Physiol. Rev. 80, 1107–1213.
Xiang, Q., Chen, S. Q., Ma, L. Y., Hu, K., Zhang, Z., Mu, G. Y., et al. (2018). Association between SLCO1B1 T521C polymorphism and risk of statin-induced myopathy: a meta-analysis. Pharmacogenomics J. 18, 721–729. doi: 10.1038/s41397-018-0054-0
Yanase, K., Tsukahara, S., Mitsuhashi, J., and Sugimoto, Y. (2006). Functional SNPs of the breast cancer resistance protein-therapeutic effects and inhibitor development. Cancer Lett. 234, 73–80. doi: 10.1016/j.canlet.2005.04.039
Yebyo, H. G., Aschmann, H. E., Kaufmann, M., and Puhan, M. A. (2019). Comparative effectiveness and safety of statins as a class and of specific statins for primary prevention of cardiovascular disease: a systematic review, meta-analysis, and network meta-analysis of randomized trials with 94,283 participants. Am. Heart J. 210, 18–28. doi: 10.1016/j.ahj.2018.12.007
Yin, O. Q., Mak, V. W., Hu, M., Fok, B. S., Chow, M. S., and Tomlinson, B. (2012). Impact of CYP2D6 polymorphisms on the pharmacokinetics of lovastatin in Chinese subjects. Eur. J. Clin. Pharmacol. 68, 943–949. doi: 10.1007/s00228-011-1202-5
Young, S. G., and Fong, L. G. (2012). Lowering plasma cholesterol by raising LDL receptors–revisited. N. Engl. J. Med. 366, 1154–1155. doi: 10.1056/nejme1202168
Zhang, W., Yu, B. N., He, Y. J., Fan, L., Li, Q., Liu, Z. Q., et al. (2006). Role of BCRP 421C>A polymorphism on rosuvastatin pharmacokinetics in healthy Chinese males. Clin. Chim. Acta 373, 99–103. doi: 10.1016/j.cca.2006.05.010
Zhao, G., Liu, M., Wu, X., Li, G., Qiu, F., Gu, J., et al. (2017). Effect of polymorphisms in CYP3A4, PPARA, NR1I2, NFKB1, ABCG2 and SLCO1B1 on the pharmacokinetics of lovastatin in healthy Chinese volunteers. Pharmacogenomics 18, 65–75. doi: 10.2217/pgs.16.31
Zhou, Q., Chen, Q. X., Ruan, Z. R., Yuan, H., Xu, H. M., and Zeng, S. (2013a). CYP2C9∗3(1075A > C), ABCB1 and SLCO1B1 genetic polymorphisms and gender are determinants of inter-subject variability in pitavastatin pharmacokinetics. Pharmazie 68, 187–194.
Zhou, Q., Ruan, Z. R., Yuan, H., Xu, D. H., and Zeng, S. (2013b). ABCB1 gene polymorphisms, ABCB1 haplotypes and ABCG2 c.421c > A are determinants of inter-subject variability in rosuvastatin pharmacokinetics. Pharmazie 68, 129–134.
Zhu, Y., D’Agostino, J., and Zhang, Q. Y. (2011). Role of intestinal cytochrome P450 (P450) in modulating the bioavailability of oral lovastatin: insights from studies on the intestinal epithelium-specific P450 reductase knockout mouse. Drug Metab. Dispos. 39, 939–943. doi: 10.1124/dmd.110.037861
Zuccaro, P., Mombelli, G., Calabresi, L., Baldassarre, D., Palmi, I., and Sirtori, C. R. (2007). Tolerability of statins is not linked to CYP450 polymorphisms, but reduced CYP2D6 metabolism improves cholesteraemic response to simvastatin and fluvastatin. Pharmacol. Res. 55, 310–317. doi: 10.1016/j.phrs.2006.12.009
Keywords: statins, pharmacogenetics, pharmacogenomics, muscle toxicity, myotoxicity, adverse effects, SLCO1B1
Citation: Kee PS, Chin PKL, Kennedy MA and Maggo SDS (2020) Pharmacogenetics of Statin-Induced Myotoxicity. Front. Genet. 11:575678. doi: 10.3389/fgene.2020.575678
Received: 24 June 2020; Accepted: 26 August 2020;
Published: 16 October 2020.
Edited by:
Ulrich M. Zanger, Dr. Margarete Fischer-Bosch Institut für Klinische Pharmakologie (IKP), GermanyReviewed by:
Brian Tomlinson, The Chinese University of Hong Kong, ChinaMing Ta Michael Lee, Geisinger Health System, United States
Copyright © 2020 Kee, Chin, Kennedy and Maggo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simran D. S. Maggo, c2ltcmFuLm1hZ2dvQG90YWdvLmFjLm56