 Charikleia Karageorgiou
Charikleia Karageorgiou Rosa Tarrío*
Rosa Tarrío*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 09 October 2020
Sec. Evolutionary and Population Genetics
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.565836
This article is part of the Research Topic Coping with Climate Change: A Genomic Perspective on Thermal Adaptation View all 11 articles
Chromosome inversions are important contributors to standing genetic variation in Drosophila subobscura. Presently, the species is experiencing a rapid replacement of high-latitude by low-latitude inversions associated with global warming. Yet not all low-latitude inversions are correlated with the ongoing warming trend. This is particularly unexpected in the case of O7 because it shows a regular seasonal cycle that peaks in summer and rose with a heatwave. The inconsistent behavior of O7 across components of the ambient temperature suggests that is causally more complex than simply due to temperature alone. In order to understand the dynamics of O7, high-quality genomic data are needed to determine both the breakpoints and the genetic content. To fill this gap, here we generated a PacBio long read-based chromosome-scale genome assembly, from a highly homozygous line made isogenic for an O3+4+7 chromosome. Then we isolated the complete continuous sequence of O7 by conserved synteny analysis with the available reference genome. Main findings include the following: (i) the assembled O7 inversion stretches 9.936 Mb, containing > 1,000 annotated genes; (ii) O7 had a complex origin, involving multiple breaks associated with non-B DNA-forming motifs, formation of a microinversion, and ectopic repair in trans with the two homologous chromosomes; (iii) the O7 breakpoints carry a pre-inversion record of fragility, including a sequence insertion, and transposition with later inverted duplication of an Attacin immunity gene; and (iv) the O7 inversion relocated the major insulin signaling forkhead box subgroup O (foxo) gene in tight linkage with its antagonistic regulatory partner serine/threonine–protein kinase B (Akt1) and disrupted concerted evolution of the two inverted Attacin duplicates, reattaching them to dFOXO metabolic enhancers. Our findings suggest that O7 exerts antagonistic pleiotropic effects on reproduction and immunity, setting a framework to understand its relationship with climate change. Furthermore, they are relevant for fragility in genome rearrangement evolution and for current views on the contribution of breakage versus repair in shaping inversion-breakpoint junctions.
Chromosome inversions are arguably the genetic traits with the earliest and richest record of associations with climate (Hoffmann and Rieseberg, 2008). Research into evolutionary responses to contemporary global warming (Hughes, 2000; Parmesan, 2006) is therefore faced with the challenge of understanding how inversions originate and spread in populations (Kirkpatrick, 2010), while trying to determine their roles in climatic adaptation (Gienapp et al., 2008; Messer et al., 2016).
Chromosome inversions are ubiquitous chromosomal mutations consisting in the reversal of the orientation of a chromosome segment. They originate through either of two major mechanisms, each with its associated distinctive footprints. The first mechanism is intrachromatidal non-allelic homologous recombination (NAHR) between inversely oriented repeats. This mechanism generates inversions with duplications at their ends in both the inverted and uninverted states (Cáceres et al., 1999). The second mechanism is chromosomal breakage and ectopic repair via non-homologous end joining (NHEJ). This mechanism either does not generate duplications or generates them but at the ends of the inverted state only. These two types of NHEJ footprints have been explained in terms of differences in the mode of breakage. Two modes of breakage have been advanced: “cut-and-paste” via clean double-strand breaks (DSBs) that generate blunt ends and staggered. NHEJ inversions without duplications at their ends would originate via cut-and-paste (Wesley and Eanes, 1994), whereas those with inverted duplications at their ends would originate via staggered breaks in one or the two breakpoints. Two staggering models for the origin of the inverted duplications have been proposed (Kehrer-Sawatzki et al., 2005; Matzkin et al., 2005; Ranz et al., 2007): according to the isochromatid model, the duplications would be the filled-in single-stranded overhangs that would result from paired single strand breaks (SSBs) located staggered with each other on opposite strands of the same chromatid (Kehrer-Sawatzki et al., 2005), whereas according to the chromatid model, the duplications would result from unequal exchange between paired sister chromatids, each with one of two paired staggered DSBs at each breakpoint (Matzkin et al., 2005). Note that here the terms isochromatid and chromatid have switched meanings relative to how they are used in cytogenetics (Savage, 1976). The two staggering models are chromatid models because they assume that inversions originate from either single chromatids during premeiotic mitosis (isochromatid), or paired sister chromatids from the same chromosome during meiotic prophase (chromatid) (Ranz et al., 2007). The models cannot be distinguished based on the pattern of inverted duplications. Yet the chromatid model has been favored over the isochromatid model, because of the length of DNA that would need to be unwound by enzymatic activity in the latter model (Ranz et al., 2007). The chromatid model is also not without potential caveats because NHEJ was found to be suppressed during the meiotic prophase in Drosophila (Joyce et al., 2012; Hughes et al., 2018). The prevalence and distribution of the NAHR and NEHJ mechanisms of inversion formation within and across lineages are currently under debate (Ranz et al., 2007; Delprat et al., 2019). The NEHJ mechanism rests upon the occurrence of two or more DSBs. But the source of the DSBs (whether environmental, such as ionizing radiation, or spontaneous, such as non-B DNA-associated sequence instability, where non-B DNA denotes any DNA conformation that is not in the canonical right-handed B form; Lobachev et al., 2007; Zhao et al., 2010; Farré et al., 2015), the relative contributions of breakage versus repair to shaping breakpoint junctions (Ranz et al., 2007; Kramara et al., 2018; Scully et al., 2019), and the relative frequency with which the joined broken ends are from the same chromatid (isochromatid model) versus two distinct sisters (chromatid model) (Ranz et al., 2007) or even, as has been more recently suggested by Orengo et al. (2019), non-sister chromatids (chromosome model) are additional open questions.
Inversions can have direct or/and indirect functional effects (Kirkpatrick, 2010). Direct effects are those ascribable to the mutation per se, as it altered the structure or expression of functional sequences at the breakpoints, or the functional neighborhood of genes in the cell nucleus (McBroome et al., 2020). Indirect effects emanate from their associated recombination–suppression effects when in heterozygous condition, whereby they can bind together into close linkage association particular combinations of alleles at genetically distant loci. The evolutionary significance of polymorphic inversions is often thought to chiefly stem from their indirect effects (Dobzhansky, 1947; Wasserman, 1968; Kirkpatrick and Barton, 2006). Although data have been lacking on the relative importance of the two types of effects, there has been renewed interest in using genomics to determine mechanisms for the spread, establishment, and maintenance or fixation of inversions (Corbett-Detig and Hartl, 2012; Corbett-Detig, 2016; Fuller et al., 2016, 2017, 2019; Cheng et al., 2018; Said et al., 2018; Lowry et al., 2019). Because they usually involve many genes, chromosome inversions have enhanced potential for affecting multiple traits, which should expand the opportunities for their maintenance via balancing selection. The extent to which that is the case and the types and transience of the balancing selection mechanisms involved are only beginning to be elucidated (Kapun and Flatt, 2018; Wellenreuther and Bernatchez, 2018; Faria et al., 2019). Amid these unknowns, the inversion polymorphisms of Drosophila subobscura emerged among the first genetic traits identified as involved in a species’ adaptation to contemporary climate warming (Rodríguez-Trelles and Rodríguez, 1998, 2007; Balanyà et al., 2006; Rezende et al., 2010).
Drosophila subobscura is a native Palearctic species broadly distributed in Europe and the newly invaded areas of North and South America (reviewed in Krimbas, 1992), where it is found generally associated with woodland habitats. It belongs in the obscura group, within which it clusters with the recently derived small-island endemics Drosophila guanche and Drosophila madeirensis, forming the subobscura three-species subgroup (Bächli, 2020). D. subobscura has one of the smallest and least repetitive Drosophila reference genomes obtained thus far, which is distributed among five large telocentric chromosomes (A, J, U, E, and O) and one small dot (Karageorgiou et al., 2019). In stark contrast with its two insular relatives, the species has evolved highly rearranged chromosome sequences, which is due to having experienced accelerated fixation rates of paracentric inversions, especially the A sex chromosome. This situation has been interpreted as indicative of the inversions’ role in binding together adaptive alleles in the face of the species’ intense continent-wide gene flow (Karageorgiou et al., 2019). Presently, D. subobscura harbors a rich inversion polymorphism, with its five major chromosomes showing parallel adaptive variation patterns across latitude (Ayala et al., 1989), seasons (Rodríguez-Trelles et al., 1996, 2013), and through a heatwave (Rodríguez-Trelles et al., 2013), while rapidly shifting in close association with the ongoing rise in global temperatures (Rodríguez-Trelles and Rodríguez, 1998, 2010; Balanyà et al., 2006). Laboratory attempts to establish the causal nature of this association have, however, largely been inconclusive (Santos et al., 2005; Fragata et al., 2014). Ultimately, a complete understanding of the role of inversions in adaptation to contemporary climate warming in D. subobscura will necessarily include the identities and functional properties of the genome sequences affected by them. Advances along this line include the isolation and characterization of breakpoint sequences for 11 of the more than 65 large cytologically visible inversions known for the species, including A2 (Puerma et al., 2017), O3 (Papaceit et al., 2012), O4 and O8 (Puerma et al., 2016a), E1 and E2 (Puerma et al., 2014), E3 and E9 (Orengo et al., 2015), E12 (Puerma et al., 2016b), and U1 and U2 (Karageorgiou et al., 2019). An overall conclusion is that none of these inversion breakpoints disrupted any obvious candidate gene for direct adaptation to temperature, despite the fact that all but the E3 inversion are supposed to be involved in adaptation to climate (e.g., Menozzi and Krimbas, 1992; Rego et al., 2010; Arenas et al., 2018). Apart from the fact that thermal traits are genetically complex and that many of the genes that impinge on them are still unknown, the above conclusion supports that those inversions’ role in thermal adaptation would be through either position effects, indirect linkage generation effects, or both.
As part of a wider effort to develop a high-quality reference genome for D. subobscura encompassing the species’ rich chromosomal polymorphisms, here we focus on the O7 inversion. The breakpoints of this inversion were located cytologically at subsections 77B/C and 85E on the Kunze–Mühl and Müller standard map (Figure 1A; Kunze-Mühl and Müller, 1958; Götz, 1965). O7 is among the top 10% known largest D. subobscura inversions, stretching most of the centromere-proximal half of the O chromosome (Krimbas, 1992). In nature, it attains significant frequencies only in combination with the non-overlapping centromere-distal complex of two overlapping inversions O3+4, forming the chromosome arrangement O3+4+7 (Figure 1B). The tight association between O7 and O3+4 is likely maintained by an interaction between selection and the strongly reduced recombination between them (Pegueroles et al., 2010a).
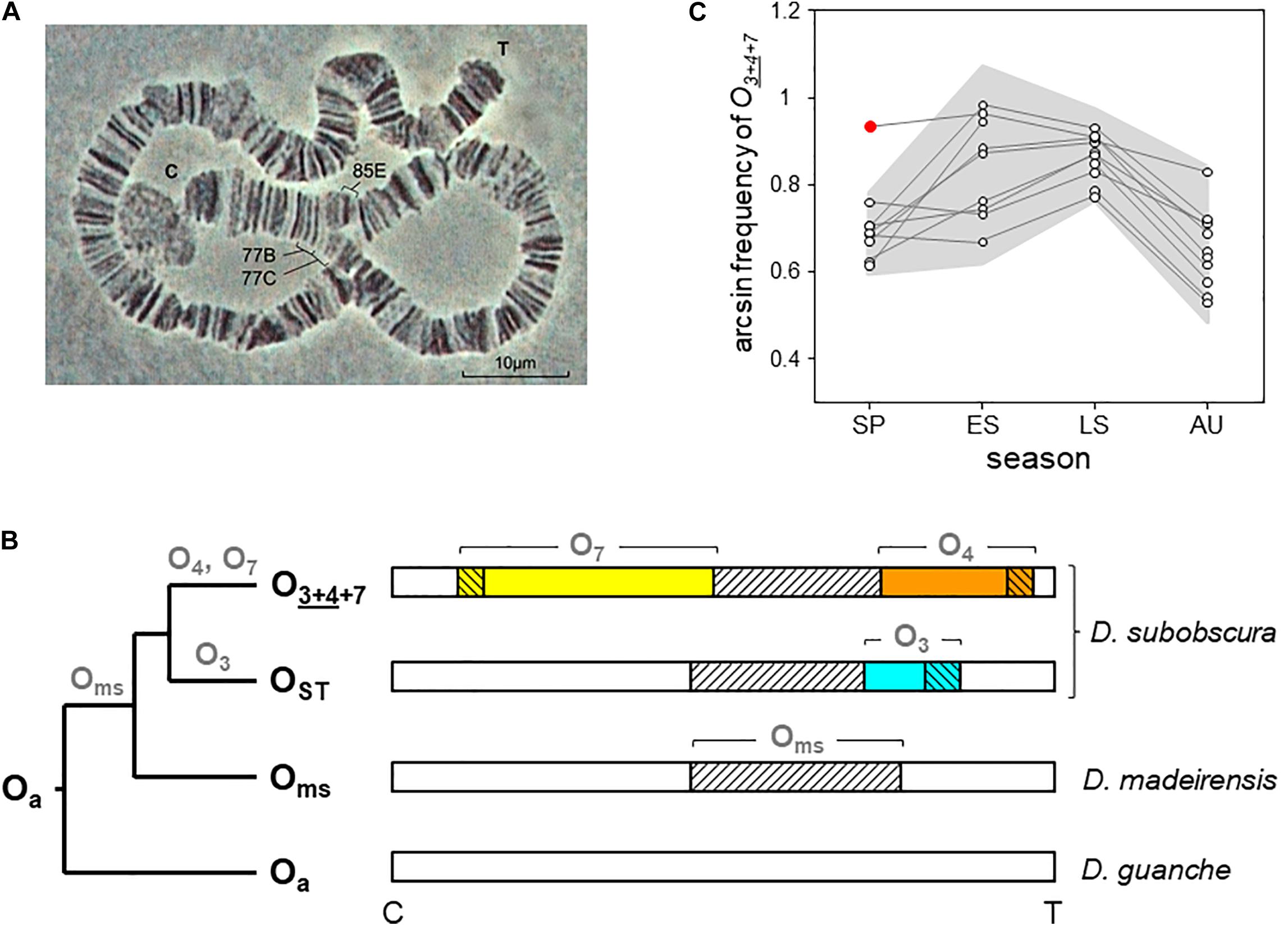
Figure 1. O7 inversion and O3+4+7 chromosome arrangement of D. subobscura. (A) Light micrograph (400 ×) of the O7 diagnostic loop from two paired polytene O chromosomes of a O3+4+7/O3+4 heterokaryotype, with indicated cytological map positions of the two inversion breakpoints (Kunze-Mühl and Müller, 1958; Götz, 1965). C and T denote centromere and telomere, respectively. (B) Phylogeny and chromosomal locations of the inversions forming the O3+4+7 arrangement in the subobscura subgroup. Names at the root and tips (bold black) and on branches (bold gray) denote chromosome arrangements and inversions, respectively. The ancestral O arrangement of the subgroup is Oa (Karageorgiou et al., 2019). The chromosome-central inversion Oms (diagonally hatched) is so-called because it became fixed in the last common ancestor of D. madeirensis and D. subobscura (Karageorgiou et al., 2019). In D. subobscura, O3 (blue) and O4 (orange) are two centromere-distal inversions with overlapping cytological map positions originated independently on separate Oms branches. The centromere-proximal inversion O7 (yellow) is assumed to have originated along the branch of O4. Oms became extinct as a single inversion in D. subobscura. Note that O3 is not in the path from Oa to O3+4+7, being the inversion that generated the OST arrangement. (C) Five decades of cyclic seasonal change of O3+4+7 at Mount Pedroso, Spain. Consecutive seasonal data (dots) from the same year are connected by lines. The gray background plots the ± 2σ confidence band around the seasonal averages, and the red dot the summer-like value recorded during the spring 2011 heatwave. Included are published data from 1976 to 1981 (Fontdevila et al., 1983), 1988 to 1991 (Rodríguez-Trelles et al., 1996), 2011 to 2012 (Rodríguez-Trelles et al., 2013), and our 2015 unpublished arcsin-transformed records for late summer (0.845) and autumn (0.574). SP, spring; ES and LS, early and late summer; AU, autumn.
O7 could be initially classified as a warm-climate inversion. In the Palearctic, it shows a southern distribution. In northwest Spain, where it has been longitudinally monitored starting in mid-1970s (Fontdevila et al., 1983; Rodríguez-Trelles et al., 1996, 2013), it shows a pronounced regular seasonal cycle (estimated to account for more than 60% of the inversion’s temporal variation; Rodríguez-Trelles et al., 1996) that peaks in summer and drops in winter (Figure 1C). In 2011, it rose to summer-like levels in spring during a heatwave, with the magnitude of the increase closely matching that of the thermal anomaly (Figure 1C; Rodríguez-Trelles et al., 2013). However, (i) the average annual frequency of O7 in northwest Spain remains unchanged after decades of sustained climate warming experienced by the region (Rodríguez-Trelles et al., 2013; our unpublished records). (ii) Following the 2011 heatwave, the inversion reached summer-like frequencies in April, but did not continue rising through the ensuing summer (Figure 1C), perhaps hampered by recessive deleterious alleles (Rodríguez-Trelles et al., 2013). (iii) The Palearctic distribution of O7 is disjointed between the peninsulas of Iberia and Turkey (Götz, 1967). These are similar latitude areas separated by ∼2,500 km within the continuous species’ range. Assuming that the inversion is molecularly the same in the two areas, this spatial pattern can hardly be explained on the sole basis of a postglacial expansion scenario (Menozzi and Krimbas, 1992), considering how rapidly it spread through the recently invaded areas of the New World (Prevosti et al., 1988). And (iv) in the more studied Iberian Peninsula, the distribution of the inversion has negative or no correlations with the geographical variation in temperature. For example, the average annual frequency of the inversion declines from ∼50% to near-zero values along the > 1,000-km stretching from the northwestern-most to the northeastern-most territories, despite the latter having a warmer climate than the former (de Frutos, 1972; Solé et al., 2002; Rodríguez-Trelles et al., 2013). The same is true for the West Atlantic fringe of the peninsula along which the inversion levels remain basically the same despite the fact that it stretches seven latitudinal degrees of steep thermal gradient (Brehm and Krimbas, 1988; Solé et al., 2002; Rodríguez-Trelles et al., 2013). The inconsistent patterns of O7 between components of the ambient temperature suggest that it is influenced by selective factors other than temperature alone.
The O chromosome offers the methodological advantage over the other D. subobscura chromosomes that there is an available balancer-strain called Varicose/Bare (Va/Ba) (Sperlich et al., 1977). In this study, we first used the Va/Ba strain to develop an isogenic line with two identical copies of a wild O chromosome carrying the O3+4+7 arrangement. Second, we used PacBio long-read technology to generate a high-quality annotated chromosome-scale genome sequence for the line. Third, we isolated the complete continuous nucleotide sequence of the inversion O7 by conserved synteny analysis of the obtained O3+4+7 chromosome with the available O chromosome from the species’ reference genome, which is structurally O3+4 (Karageorgiou et al., 2019). In addition, we also considered two other published sequences of the O chromosome, including a high-quality long-read–based sequence from D. subobscura (Bracewell et al., 2019), and an Illumina-based sequence from D. guanche (Puerma et al., 2018). We give an account of O7 main features, together with a detailed description of its mechanism of formation. Our findings provide clues to the mixed evidence for this inversion’s role in thermal adaptation.
Drosophila subobscura shows the ancestral karyotype configuration of the genus Drosophila, consisting of five large telocentric rods (Muller elements A-E) and one dot (Muller F) (Powell, 1997). The five rods include the sex chromosome (Muller A) and four autosomes of which the O chromosome (Muller E; homologous to chromosome arm 3R from D. melanogaster) is the largest (∼30 Mb), comprising around 25% of the species’ nuclear euchromatic genome (∼125 Mb; Karageorgiou et al., 2019).
An early landmark in the study of chromosomal inversion polymorphisms of D. subobscura was the development of structurally homozygous strains, as tools to identify new inversions by the location and shape of the loops formed in inversion heterozygotes (Zollinger, 1950; Maynard-Smith and Maynard-Smith, 1954; Zouros et al., 1974; Loukas et al., 1979). The “Küsnacht” strain, named after the Swiss locality of collection of the flies (Zollinger, 1950), became the first established (Koske and Maynard-Smith, 1954). The chromosomal arrangements of the strain, which happened to be those most common in Central Europe, were subscripted ST (for “standard”) and from them new inversions were designated with numeral subindices following their order of discovery (Kunze-Mühl and Sperlich, 1955). This naming system was not intended to convey polarity of evolutionary change. Accordingly, O3+4+7 is the arrangement that can be interconverted with OST by the two centromere-distal overlapping inversions O3 and O4 (denoted by the underline joining the subscripts; Zouros et al., 1974) and the centromere-proximal inversion O7. The ancestor-descendant relationships of these inversions are shown in Figure 1B.
O chromosome conserved synteny analysis was based on data from four whole-genome de novo assemblies, including three PacBio long-read–based assemblies from D. subobscura and one Illumina short-read–based assembly from D. guanche. Of the three D. subobscura assemblies, one was used as reference for inversion O7 and was newly generated in this study. The other two were used as references for the standard configuration [note that the distal breakpoint of O7 maps within inversion Oms (Karageorgiou et al., 2019), whereby is expected to exhibit opposite orientation in D. subobscura relative to D. guanche; Figure 1B] and were already available (Karageorgiou et al., 2019; Bracewell et al., 2019). Also available was the assembly from D. guanche (Puerma et al., 2018), which was used as an outgroup. Henceforth, we will refer to these four assemblies as Ds_7, Ds_ch-cu, Ds_B, and Dg, respectively.
To generate the Ds_7 assembly, we developed a line that is isogenic for an O3+4+7 arrangement from the wild and homokaryotypic and highly homozygous for the ST arrangements of the rest of the chromosomes (i.e., AST, JST, UST, EST, and O3+4+7). The O arrangement was first isolated by crossing wild males to virgin females from the cherry-curled (ch-cu) recessive marker stock; they were then submitted to nine generations of backcrossing with ch-cu females and finally isogenized using the Va/Ba balancer stock (Sperlich et al., 1977). The expression of the Ba gene is highly variable. Therefore, to prevent potential errors at sorting out phenotypically O3+4+7 homokaryotypes, the Va/Ba stock was previously selected for zero macrobristles on the scutum and scutellum. Crossing schemes and the methods for polytene chromosome staining and identification are described elsewhere (Rodríguez-Trelles et al., 1996). The assayed line was stored frozen at −80°C immediately upon obtention. The wild flies used to develop the line were derived from our survey of the natural population of Berbikiz (Spain; Lat.: 43,18949, Long.: –3,09025, Datum: WGS84, elevation: 219 m a.s.l) conducted in July 7, 2012 (Rodríguez-Trelles et al., 2013).
The remaining three assemblies were derived from strains homokaryotypic for all chromosomes. The Ds_ch-cu assembly was generated from the ch-cu strain of our laboratory (AST, JST, UST, EST, and O3+4; Karageorgiou et al., 2019) and the Ds_B assembly from an isofemale laboratory stock derived from a natural population from Eugene, Oregon, in 2006 (AST, JST, U1+2, EST, and O3+4; Bracewell et al., 2019). The Dg assembly was generated from an isofemale laboratory stock derived from a natural population from the Canary Islands, Spain, in winter 1999 (Puerma et al., 2018); it shows the chromosome configuration of the last common ancestor of the subobscura subgroup except for chromosome E, which carries the arrangement Eg1 (Aa, Ja, U1+2, Eg1, and Oa; Puerma et al., 2018; Karageorgiou et al., 2019; Bracewell et al., 2019).
High-quality high-molecular-weight gDNA was obtained from 60 mg of −80°C frozen adult females, using a modified version of the phenol/chloroform method of Chen et al. (2010) that yields ∼25 μg of high-quality DNA per assay, as assessed by NanoDrop ND1000 (NanoDrop Technologies Inc., Wilmington, DE, United States) spectrophotometer and standard agarose gel electrophoresis. The genome of the Ds_7 isogenic line was sequenced to nominal 66-fold genome coverage using PacBio (Pacific Biosciences, Menlo Park, CA, United States) Sequel single-molecule real-time (SMRT) technology from a 20-kb SMRTbell template library, using Polymerase 3.0 chemistry and two SMRT cells. Libraries construction and PacBio sequencing were outsourced to Macrogen (Macrogen Inc., Seoul, South Korea).
Raw PacBio reads were assembled using the Canu assembler (version 1.8; Koren et al., 2017) on recommended settings for read error correction, trimming and assembly, and genome size set at 150Mb based on previously published flow cytometry data (Karageorgiou et al., 2019). These analyses were performed on a 2.80-GHz 8-CPU Intel Xeon 64-bit 32GB-RAM computer running Ubuntu 18.04 LTS.
Chromosome-scale assembly and scaffolding followed the four steps outlined in Karageorgiou et al. (2019) as well as a fifth step, to improve genome completeness and contiguity, consisting of merging the Ds_7 assembly with a preselected set of segments from the reference Ds_ch-cu assembly using one round of quickmerge (Chakraborty et al., 2016), as follows: first, the CANU contigs that could be certainly anchored, ordered, and oriented on the nuclear chromosomes were aligned against the Ds_ch-cu reference using NUCmer (Kurtz et al., 2004). Second, the segments of Ds_ch-cu not overlapped by the CANU contigs, each extended 10 kb outward from each of its two ends, were extracted. Finally, separately for each chromosome, the extracted Ds_ch-cu segments, together with the CANU contigs set as the backbone, were fed into quickmerge. This approach was found to reduce the chances of misassembly and chimerism, while making it straightforward to trace the non-backbone sequence in the assembly. Dot plots of the merged assembly against the reference Ds_ch-cu assembly were used as a further step of misassembling correction. The obtained Ds_7 assembly was polished with 26 × mean coverage of 150–base-pair (bp) MP Illumina reads from the O3+4+7 isogenic line using two rounds of PILON (version 1.23; Walker et al., 2014).
Gene prediction and annotation of the assembled genome were conducted using the MAKER (version 3.01.02.-beta; Holt and Yandell, 2011; Campbell et al., 2014) annotation pipeline. Repetitive elements were identified using RepeatMasker (version 4.0.6; Smit et al., 2013/2015, at1) combined with three repeat libraries, including (i) the Drosophila genus–specific repeat library contained in the Repbase database (release 20170127; Bao et al., 2015); (ii) a library of subobscura subgroup specific satellites, sat290 and SGC-sat (Karageorgiou et al., 2019); and (iii) a library of de novo identified repeats generated using RepeatModeler (version1.0.11) on the assembly masked for the first two libraries. Novel long terminal repeats (LTRs), miniature inverted-repeat transposable elements (MITEs), tandem repeats, and rDNA and tDNA genes were identified using LTRharvest (GenomeTools version1.5.10; Ellinghaus et al., 2008), MITE Tracker (version 2.7.1; Crescente et al., 2018), Tandem Repeat Finder (TRF; version4.09; Benson, 1999), RNAmmer (version 1.2; Lagesen et al., 2007), and tRNAscan-SE (version 2.0; Lowe and Chan, 2016), respectively. All tools were run on default settings, except LTRharvest, for which we set -seed 100, -similar 90.0, and -mintsd 5, following Hill and Betancourt (2018). The quality of the annotation was controlled using the Annotation Edit Distance (AED) metric (Eilbeck et al., 2005). AED values are bounded between 0 and 1. An AED value of 0 indicates perfect agreement of the annotation to aligned evidence, and conversely, a value of 1 indicates no evidence support.
Functional annotation of MAKER-predicted proteins was made by BLASTP (version 2.6.0 +) searches against the Drosophila UniProt-SwissProt manually curated datasets (Apweiler et al., 2004). Prediction of protein functional domains was accomplished using InterProScan (version 5.29–68.0; Jones et al., 2014) on the Pfam (Finn et al., 2016), InterPro (Finn et al., 2017), and Gene Ontology (Ashburner et al., 2000; The Gene Ontology Consortium, 2017) domain databases. Genome assembly and annotation completeness were gauged using the Benchmarking Universal Single-Copy Orthologs (BUSCO) tool [BUSCO, version 4 (Seppey et al., 2019)], with the latest update of the dipteran gene set (diptera_odb10), which contains 3,285 highly conserved, single-copy genes expected to be present in any dipteran genome.
Suppose that +A|+B+C|+D and +A|−C−B|+D represent two chromosome arrangements whose gene orders differ only by the orientation of the segment between A and D (with symbols denoting A and D, the segments upstream from the centromere-proximal breakpoint and downstream from the centromere-distal breakpoint, respectively; vertical bars, breakpoint junctions; and plus/minus signs, orientation of the segment relative to the uninverted sequence). We proceeded in two steps. First, we isolated the regions containing the breakpoint junctions by chromosome conserved synteny analysis between the uninverted and inverted states using the Synteny Mapping and Analysis Program (SyMAP, version 4.2.; Soderlund et al., 2011) tool on default options, and NUCmer (see Karageorgiou et al., 2019). The O7 breakpoints were identified as the loci of interrupted synteny whose locations and distance from each other agree with the cytogenetic mapping data of the inversion (Karageorgiou et al., 2019). Second, we localized the breakpoint junctions at base-pair resolution and performed comparative analyses of their flanking sequences using the progressive guide tree-based MAFFT algorithm (version 72) with the accuracy-oriented method “L-INS-i” (Katoh et al., 2019). Each of the regions +A|+B and +C|+D from the uninverted state was aligned separately, first with +A|−C and then with −B|+D from the inverted state. From each of the four resulting alignments, we used the regions showing positional homology between the uninverted and inverted states to isolate segments A, B, C, and D, correspondingly. The remaining sequences of the uninverted state were submitted to a second round of comparative analysis among them, and with segments A to D to identify the homologies missed in the first round. As representatives of the uninverted state, we used Ds_ch-cu together with the previously published assemblies Ds_B and Dg, and this last one was set as the outgroup.
MAFFT-based tree reconstruction of the Attacin gene family in Drosophila was performed via maximum likelihood. Model selection and tree inference were conducted using IQ-Tree (Kalyaanamoorthy et al., 2017; Nguyen et al., 2015). Tree searches were conducted starting from sets of 100 initial maximum parsimony trees using nearest neighbor interchange with default perturbation strength and a stopping rule settings. Branch support was assessed using the ultrafast bootstrap approximation (UFboot; 1,000 replicates) (Hoang et al., 2018), and two single-branch tests including the Shimodaira–Hasegawa-like approximate likelihood ratio test (SH-aLRT; 1,000 replicates) (Guindon et al., 2010) and the approximate Bayes parametric test (Anisimova et al., 2011).
Scans for potential non-B DNA–forming sequences considered the following features: inverted repeats (IRs) (capable of forming hairpin and/or cruciform DNA), direct/tandem repeats (slipped/hairpin structures), mirror repeats (triplexes), alternate purine-pyrimidine tracts (left-handed Z-DNA), G4 motifs (tetraplex and G−quadruplex DNA), and A−phased repeats (static bending). Searches were conducted online using for IRs Palindrome Analyzer (Brázda et al., 20163; accessed January 24, 2020) with repeat length of 6-20 nt, spacer length ≤ 10 nt, and number of mismatches ≤ 1; for tandem repeats Tandem Repeat Finder (TRF version 4.09; Benson, 19994; accessed Jan 24, 2020) in basic mode; and for the remaining features nBMST (Cer et al., 20125; accessed January 24, 2020) with prefixed default settings. The propensity of IRs to adopt non-B conformation was assessed using the difference in free energy between the DNA sequence in the linear and cruciform structures, as implemented in Palindrome Analyser (Brázda et al., 2016).
Transcription start site (TSS) prediction was conducted using the NNPP method (Reese, 20016). Searches for putative binding sites for Relish (Rel), the heterodimer Dif/Rel, dFOXO, Dorsal (dl), and Serpent (srp) transcription factors in the 1-kb upstream region of the Attacin predicted TSSs were performed using the FIMO tool (Grant et al., 2011) from the MEME suite (Bailey et al., 2015). For Rel and Dif/Rel, and for dFOXO, we used the FootprintDB database (Sebastián and Contreras-Moreira, 20147) Drosophila melanogaster Major Position Matrix Motifs (DMMPMM) identified, respectively, by Senger et al. (2004) and Weirauch et al. (2014). For dl and srp, we used the REDfly database (version 5.5.3; Rivera et al., 20198) improved iDMMPMM motifs developed by Kulakovskiy and Makeev (2009). Searches were performed using a p value cutoff of 10–3.
The PacBio Sequel sequencing of the O3+4+7 isogenic line genome generated 2,457,493 reads, with mean and longest lengths of 11,257 bp and 117,750 bp, respectively. Canu correction and trimming retained a 42-fold genome coverage for the assembly. Of the 385 Canu-generated contigs, the 14 that could be confidently anchored, ordered, and oriented covered the complete reference genome, with an added length of 126.770 Mb and N50 of 10.587 Mb. Quickmerge of those 14 CANU contigs resulted in six chromosome-scale scaffolds, one per each of the major D. subobscura chromosomes (Table 1). Of note, chromosome O was built from two contigs only, with the centromere-proximal contig (tig00026085; 29.679 Mb) spanning almost all the chromosome length (96.9%) (Figure 2A). The Ds_7 assembly contained 13,459 MAKER-annotated genes, nearly all with well-supported predictions (AED50 = 99.3%). Only 2.6% (87) of the BUSCO genes were missing, indicating that the assembly is almost complete. The O chromosome contained 3,220 (23.9%) of the annotations of the assembly.
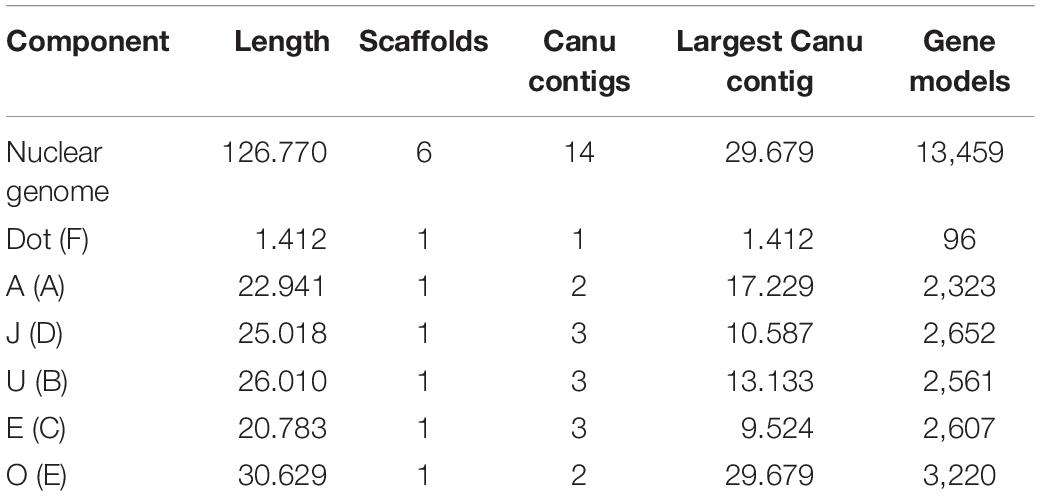
Table 1. Ds_7 assembly summary statistics (Muller elements are given in parenthesis, and lengths are given in megabases of sequence).
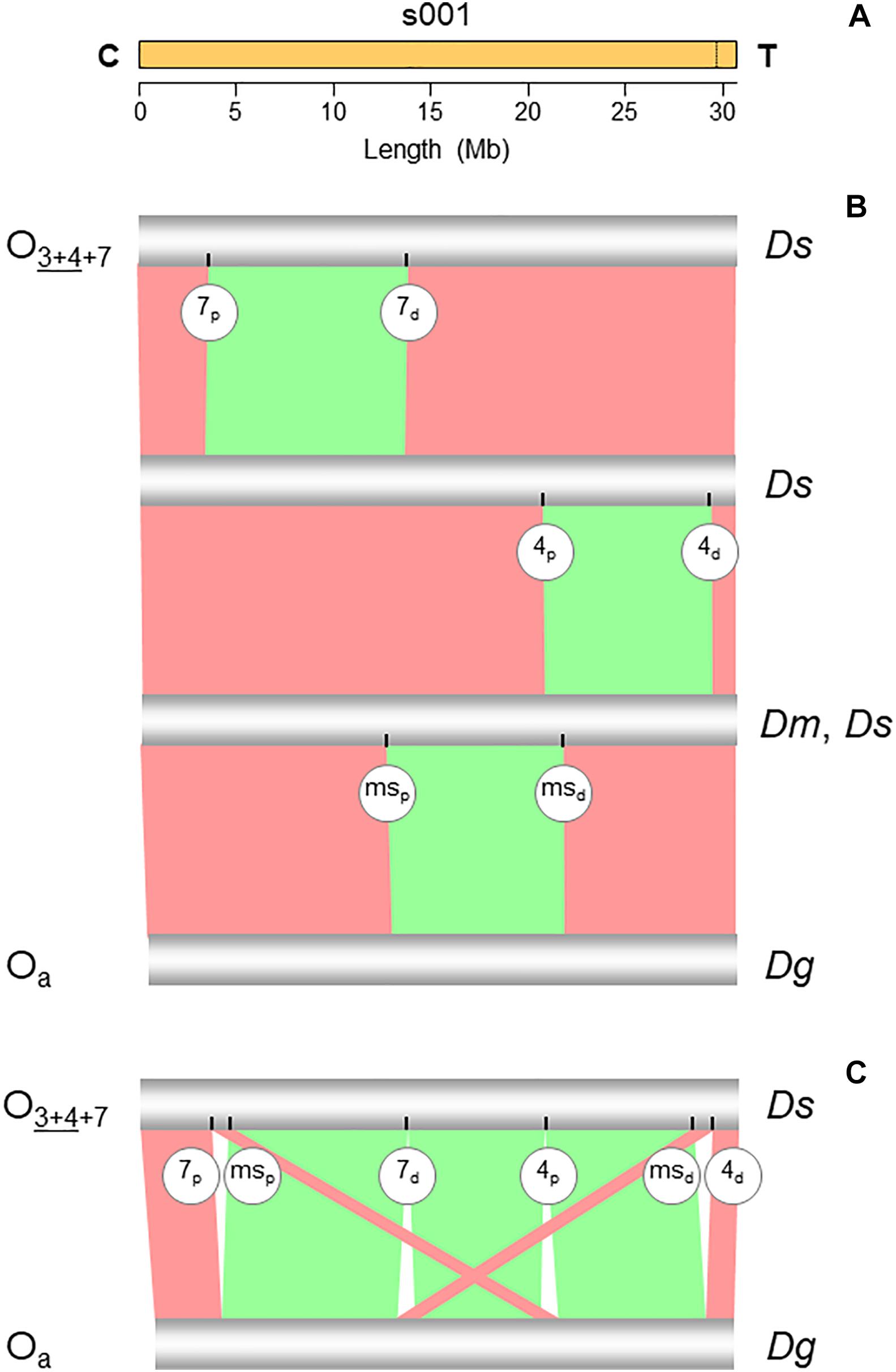
Figure 2. Chromosome conserved synteny analysis of the O7 breakpoints. (A) The long read-based O3+4+7 chromosome-scale scaffold (s001). The vertical dotted line near the telomere indicates the location of the stitch between the two Canu tigs. (B) SyMAP comparative synteny analysis showing the inversions found along the path from the ancestral arrangement of the subobscura subgroup (Oa) to O3+4+7 (see Figure 1B). In addition to O7, the two chromosomes also differ by inversions O4 and Oms. (C) SyMAP direct comparison of the Oa and O3+4+7 chromosome arrangements. Bands connecting the chromosomes denote uninverted (pink) and inverted (green) synteny blocks. Labeled ticks on chromosomes indicate proximal (p) and distal (d) inversion breakpoints. The remaining symbols are as in Figure 1.
The structural transition between the O chromosomes of the Ds_7 and Ds_ch-cu assemblies called for one large megabase-sized inversion (Figures 2B,C), whose breakpoints located cytologically precisely as it would be expected if they were from O7. Relative to the nearest of the available 140 cytologically mapped markers of the O chromosome (see Karageorgiou et al., 2019), the proximal breakpoint was located 44.5 kb downstream from Sb (Dmel\CG4316) and 117.4-kb upstream from microsatellite dsub02, and the distal breakpoint 111.8 kb downstream from rdx (Dmel\CG12537) and 29.3kb upstream from Abi (Dmel\CG9749). Sb and dsub02 have been respectively mapped to subsections 77B (Dolgova, 2013) and 77C (Santos et al., 2010), and rdx and Abi to subsection 85E (Dolgova, 2013; Pegueroles et al., 2013) of the Kunze-Mühl and Müller (1958) standard cytological map. Other than O7, no D. subobscura inversion maps to those positions.
Comparative analysis of the genes annotated in the regions immediately flanking the breakpoints in Ds_7, Ds_ch-cu and Ds_B with those in the outgroup Dg (Figure 3A) corroborated that Ds_ch-cu and Ds_B carried the uninverted state, whereas Ds_7 carried the inverted state. The assembled O7 has a size of 9,936,431 bp, totaling 32.4% of the chromosome (30,629,152 bp). It has a GC content (43.8%) below that of the O chromosome (44.9%) since it is located in the chromosome centromere-proximal half, which is relatively AT-rich (Karageorgiou et al., 2019). O7 was predicted to have 1,028 protein-coding genes, or 31.9% of the gene models of the O chromosome, in close agreement with its percent of chromosome length.
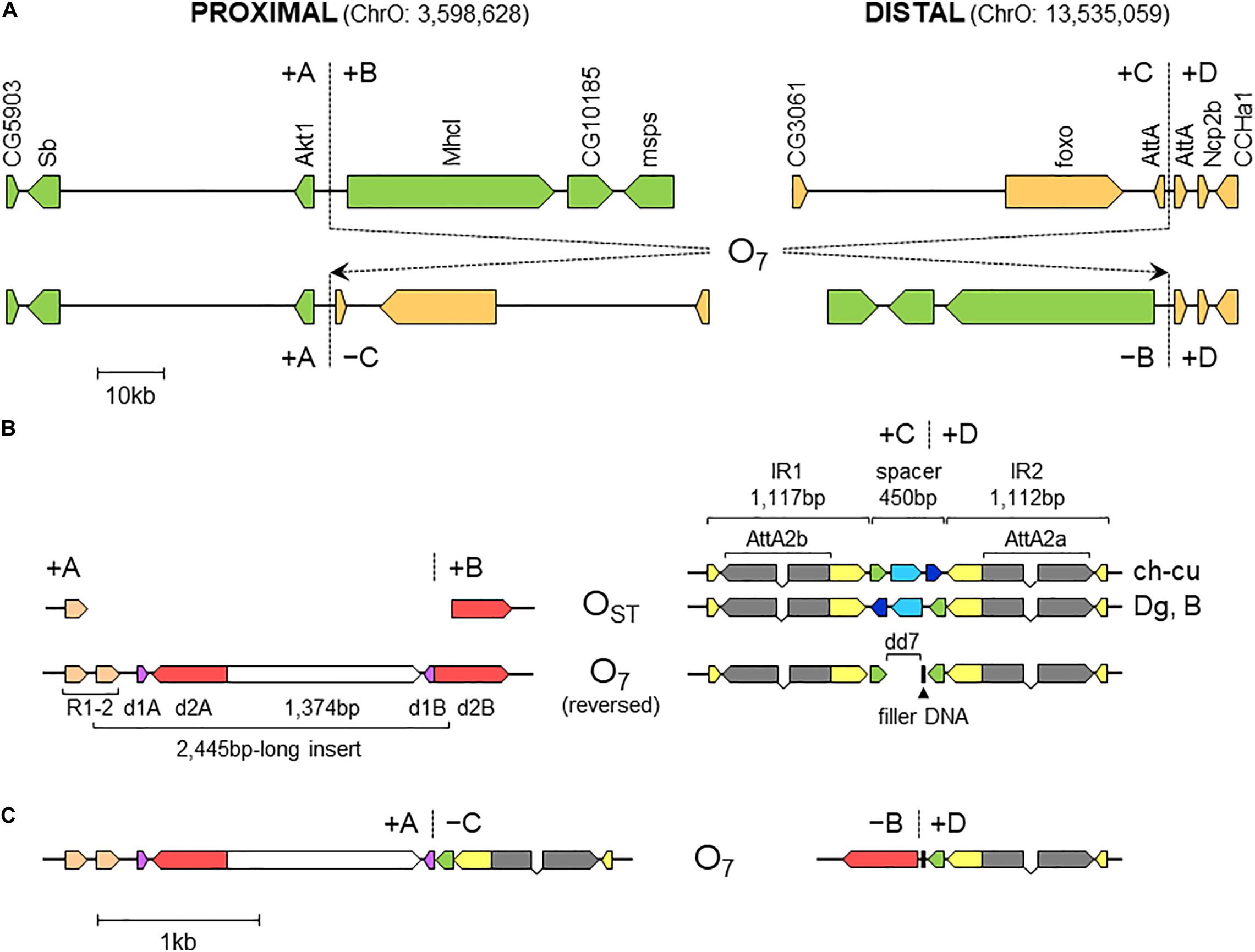
Figure 3. Sequence-annotated breakpoint regions in O7 and the standard uninverted state. (A) Gene scale (10-kb scale bar) depiction of the proximal and distal breakpoints in the uninverted state (+A|+B and +C|+D, respectively) versus O7 (+A|−C and −B|+D). In green are segments A and B, and in sepia C and D. Vertical broken lines indicate break junctions, and arrow boxes the size and direction of the genes labeled vertically using the names of the corresponding D. melanogaster orthologs. The coordinates of O7 in the assembled chromosome are given in parentheses. (B) Zoom-in (1-kb scale bar) on the regions immediately flanking the break junctions, with O7 oriented backward (i.e., +B+C, instead of −C−B) to better track the differences with the uninverted state. Arrow boxes indicate the size and direction of the sequence elements discussed in the text. Gray boxes (exons) linked by polygonal lines (introns) represent the two AttA2 paralogs oriented in the direction of transcription. In the distal breakpoint, the two alternative haplotypes of the uninverted states, namely, that from ch-cu and that from Dg and B, are shown. Note the reversal of the spacer in ch-cu versus Dg and B, and the mirror halves flanking dd7 in O7. (C) O7 represented as in (B), but in its actual orientation (i.e., -C-B).
The alignments used for isolation of the breakpoint junctions and their corresponding flanking regions A, B, C, and D are shown in Supplementary Figures 1, 2. Figure 3B provides a schematic representation of the +A|+B region based on the alignment of Supplementary Figure 3. In the case of O7, the region was reconstructed using the reverse complement of segment -B. The breakpoint junction is located within a 2,445-bp-long sequence stretch present only in the inverted state. The site of the insertion is flanked by multiple indels, which suggests that the insertion occurred in a region of prior sequence instability. Of the insertion length, 2,317 bp are on the + A segment and 128 bp on the + B segment. The insertion begins with a 153-bp-long direct repetition (R1-2) of the upstream flank. Proceeding downstream from this repeat, there are two inverted duplications named d1 and d2, each with copies A and B, with d1 shorter (59 bp long each of d1A and d1B) than d2 (534 and 540 bp for copies d2A and d2B, respectively). The two A copies (i.e., d1A and d2A) are separated from the two B copies (i.e., d1B and d2B) by an intervening sequence of 1,374 bp. The junction between + A and + B is precisely located between d1B and d2B. d2B extends 409 bp downward from the downstream end of the insertion into the region of resumed homology between O7 and the uninverted state, indicating the orientation of the parent copy.
The above pattern of sequence copy number, order, and orientation most parsimoniously indicates that the proximal breakpoint of O7 was formed on an insertion region that experienced two pairs of staggered SSBs, which resulted in two DSBs (Figure 4; but see section “DISCUSSION” for alternative models). The upstream-most DSB generated the proximal breakpoint of a 2,026-bp-long microinversion and the downstream-most DSB generated a junction flanked upstream by the distal end of the microinversion and downstream by the proximal end of O7. Accordingly, duplications d1 and d2 would, respectively, represent the filled-in staggered SSB-induced terminal single-stranded overhangs of the microinversion and inversion O7. Figure 3C shows O7’s segments A and B such as they are found in the inversion. That d2A and d2B show direct instead of reverse relative orientation as it would be expected if paired–staggered SSBs generate inversions with inverted repeated ends (Ranz et al., 2007) would be explained by the reversal in the orientation of d2A as a result of the microinversion.
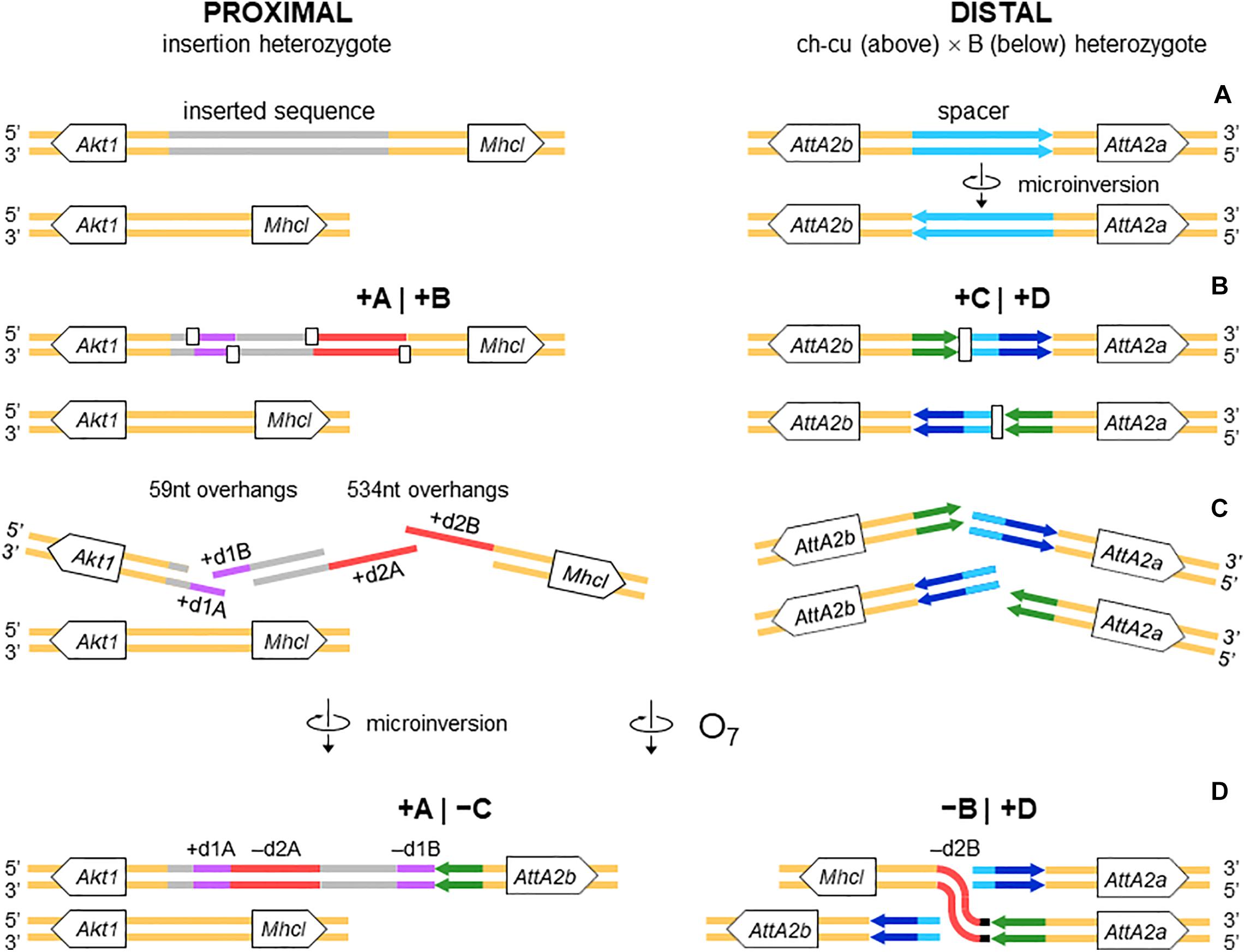
Figure 4. Isochromatid-chromosome mixed staggered model of O7 formation. (A) Start from an individual heterozygous (or homozygous) for the insertion at the proximal breakpoint, and heterozygous ch-cu–type (above)/B-type (below) for the orientation of the spacer at the distal breakpoint. (B) Occurrence of two pairs of isochromatidal staggered SSBs at the proximal breakpoint (open squares), and two staggered chromosomal blunt-ended DSBs at the distal breakpoint (open rectangles), with demarcation of breakpoints flanking segments +A|+B and +C|+D, respectively. (C) Emergence of broken ends with single stranded overhangs (+ d1A, + d1B and + d2A, + d2B) at the proximal breakpoint. (D) Microinversion formation with fill-in of the overhangs, resulting in the terminal inverted duplications + d1A and -d1B, and the reversed orientation of segment -d2A. And O7 formation by reversal of the +B+C segment with fill-in of the -d2B overhang, and distal breakpoint repair via rejoining in trans with the homologous chromosome.
Relative to the predicted nearest gene TSSs, the events took place in an intergenic region. Specifically, the upstream-most SSB occurred 1,364 bp downstream from Akt1 (CG4006; serine/threonine–protein kinase B) gene, and the downstream-most one 2,047 nt upstream from Mhcl (CG31045; myosin heavy chain-like) gene (Figure 3A). From our repeat annotation pipeline, the region around the breakages is a composite of repetitive sequences [16 in total, ranging in length from 21 bp of a (TTG)n simple-repeat to 532 bp of satellite rnd-4_family-179], interspersed with traces of transposable elements [84 bp from an LTR and 72 bp from a long interspersed nuclear element (LINE)]. Overall, no evidence of open reading frames and/or specific motifs could be found pointing to the observed breakages as directly caused by the insertion/excision of other sequences.
The role of non-B DNA as source of DSBs is well-established. Generally, DSBs are expected to colocalize with their causal non-B DNA motifs (e.g., Kolb et al., 2009; Lu et al., 2015). We used this prediction to investigate whether the local DNA conformational environment of the ancestral sequence could have acted as trigger or mediator of the complex rearrangement of the proximal breakpoint region. We proceeded in two steps: first, we reconstructed the region of the rearrangement before the breakages. It should be recalled that most of the rearranged sequence is embedded in an insertion that is absent in the ancestral non-rearranged state. Therefore, we reconstructed the prebreakages state by undoing the hypothetical rearrangement steps that generated the present sequence state. Specifically, we reversed the orientation of the microinversion (Supplementary Figure 4) and deleted one copy of each DSB-induced duplication (Supplementary Figure 5). The resulting sequence had the form: + d1, (+ 1,374 bp), |, + d2 (Figure 4B). Which copy of each of the two duplications to eliminate was inconsequential, because they are nearly identical to each other in the two cases (98.3% and 95.6%, for the identities between copies A and B of dup1 and dup2, respectively). Furthermore, the observed high level of identity (97.3%) between d2 and its homologous region in Ds_ch-cu and Ds_B suggested that the rearrangement is recent enough to allow assuming that the original conformational sequence features that could have mediated it are still observable. After establishing the prebreakage sequence, we next looked for sequences with the potential to form non-B DNA structures along a 10-kb window centered on it.
Figure 5 shows the distribution of the number of IRs capable of forming hairpin and cruciform structures along the target sequence. The highest density occurs immediately around the junction between the microinversion and inversion O7. In particular, the breakpoint is located within a ∼150-nt-long stretch of AT-rich sequence [simple repeat (ATTT)n, from our genome annotation pipeline] containing 15 IRs, of which one located 68 nt downstream the breakpoint junction ranked in the top 5% with highest likelihood of intrastrand annealing to form a hairpin (AATTTT AAAATT; ΔGS – ΔGL = 2.64). In addition, embedded in the IR cluster, there is one tandem repeat of 8.7 copies of the consensus heptanucleotide AATAAAT, and one mirror repeat of two 11 nt-long repeats separated by a 30-nt spacer, indicating that the proximal breakpoint of O7 occurred on an unstable sequence with potential for adopting multiple alternative non-B DNA conformations.
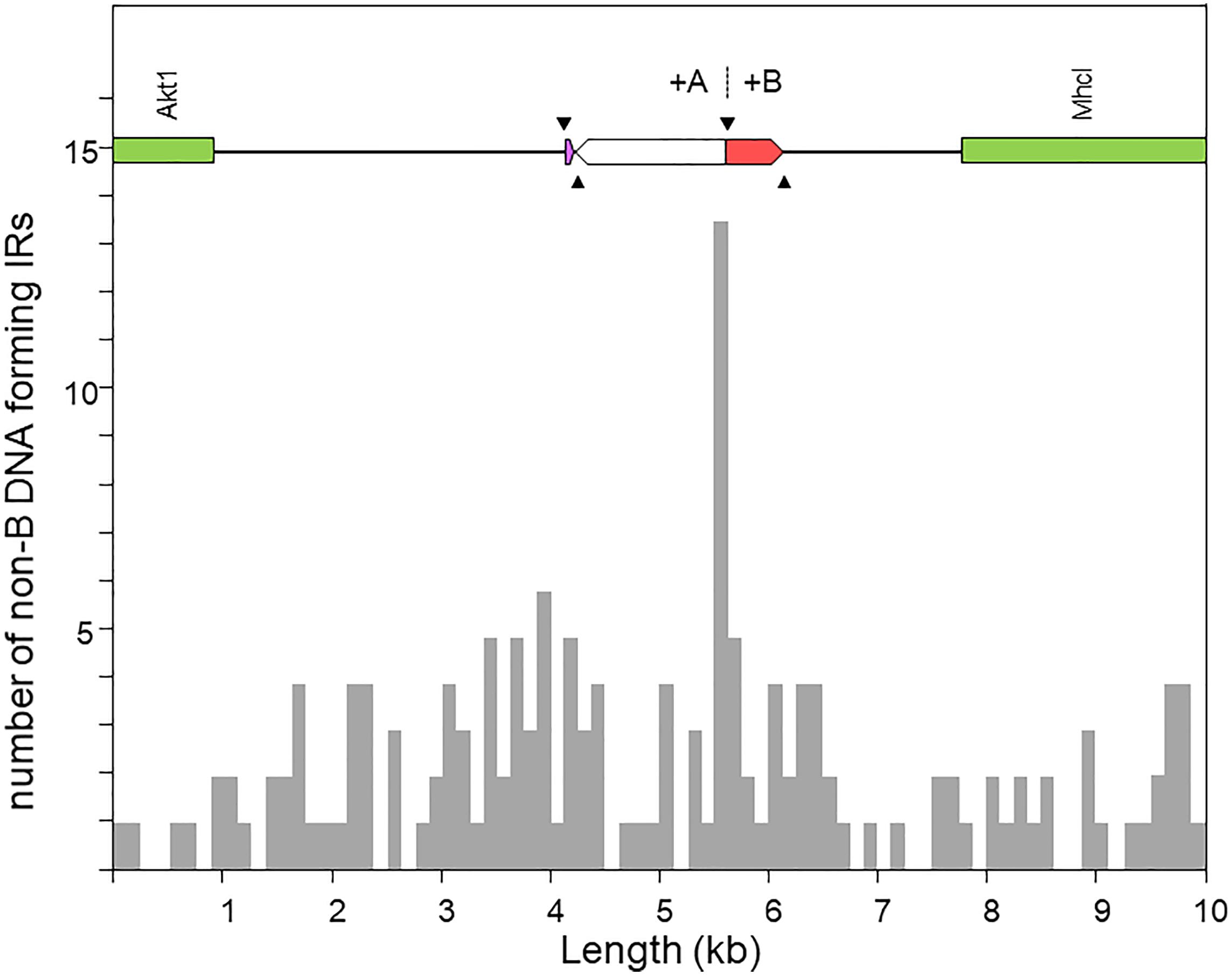
Figure 5. Distribution of inverted repeats (IRs) with potential to adopt non-B DNA hairpin/cruciform structures across a 10-kb window centered at the region of the O7 proximal breakpoint, as obtained using Palindrome Analyzer (Brázda et al., 2016). The region of interest is shown at scale above the plot. The purple and red boxes represent respectively the parental copies of d1 and d2 prior to their duplication as a result of the two pairs of staggered SSBs (black arrowheads). The highest concentration of IRs occurs around the junction of the O7 breakpoint (+A|+B).
Figure 3B provides a schematic representation of the +C|+D region based on the alignment of Supplementary Figure 6. In the case of O7, the region was reconstructed using the reverse complement of segment -C. From up to downstream, the breakpoint junction is located within a 450-aligned-sites-long gap-rich spacer region, spanning between two highly identical long IRs, IR1 and IR2, of 1,117 and 1,112 sites of alignment length, respectively. There is no evidence of duplicated sequence in Ds_7 relative to the other assemblies, indicating that the DSB either was a clean cut or did not involve significantly staggered SSBs. On the other hand, the spacer of Ds_7 was the shortest (250 nt) of all four lines (407, 317, and 343 nt for Ds_ch-cu, Ds_B, and Dg, respectively) because of a single deletion located precisely at the center of the region (hereon called dd7, for distal deletion of O7). A closer look at the pattern of pairwise sequence similarities along the spacer revealed two findings: (i) dd7 split the Ds_7 spacer in two mirror halves. For the upstream half, Ds_7 is almost identical (96.8%) to Ds_ch-cu while bearing no detectable homology to Ds_B, whereas for the downstream half, Ds_7 is almost identical (97,6%) to Ds_B while bearing no detectable homology to Ds_ch-cu; and (ii) the spacer of Ds_ch-cu is almost identical (95.4%; excluding indels) to that of Ds_B but in reversed orientation. The reversal occurred in Ds_ch-cu, because in Ds_B the spacer is oriented as in the outgroup Dg.
Altogether, the above observations can be understood as follows (Figure 4). Prior to the origination of the distal breakpoint of O7, a carrier of an uninverted chromosome of B-type experienced a reversal of the spacer region between the IRs, giving rise to the uninverted chromosome of ch-cu–type. Later on, a homokaryotype for the uninverted chromosome that was heterozygous for the microinversion of the spacer underwent at least two DSBs, one in each of two homologous non-sister chromatids, such that the DSB in the ch-cu–type chromatid occurred immediately before the first site of the dd7 and that in the B-type chromatid immediately after the last site of the dd7. Finally, the reversed + B end generated by the proximal staggered DSB in the ch-cu–type chromatid illegitimately joined with the + D end generated by the distal DSB in its homologous non-sister B-type chromatid, which resulted in a recombinant chromosome carrying the inversion O7 with the exact observed dd7 deletion.
Like the proximal breakpoint, the distal breakpoint occurred in an intergenic region yet at comparatively much shorter distance (∼390 bp) to the nearest genes. Specifically, the breakage separated two copies of an Attacin gene (CG10146; AttA) located opposite to each other on each of the two arms of the long IR. Our repeat annotation pipeline did not identify repetitive sequences in the vicinity of the distal breakpoint in Ds_ch-cu or Ds_B.
We searched the region of the spacer for potential non-B DNA–forming sequences in the vicinity of the breakpoint junctions in Ds_ch-cu and Ds_B. In both cases, we found that the IR with the highest propensity to form a hairpin was a perfect 14-bp-long palindromic sequence located next to the breakpoint junctions (ATGAACT AGTTCAT; ΔGS – ΔGL = = 2.05; located 13 and 2 bp upstream and downstream the junction in Ds_ch-cu and Ds_B, respectively). Apart from IRs, we did not detect additional potential non-B DNA sequences around the distal breakpoint.
All nucleotides in the +A|−C region of Ds_7 could be unambiguously ascribed to segment A or C. However, in the −B|+D region −B and +D are separated by 21 extra inserted nucleotides (i.e., GAGCACTCTCCACAGCAAAGT). We decided to ascribe this sequence to the distal breakpoint junction, because it contains an 8-bp substring (underlined) that resembles the beginning of the +D end (CATCAAAG), and hence it likely represents filler DNA generated by a microhomology-templated repair mechanism.
Previously, it was shown that the proximal breakage of O7 was preceded by an insertion. Likewise, the region of the distal breakage had a pre-inversion history of rearrangement, which run closely associated with a highly dynamic evolution of the Attacin immunity gene family in the obscura species group. This conclusion is based on phylogenetic analysis of the Attacin family in Drosophila (Supplementary Figure 7) using synteny to distinguish orthologous from paralogous copies (Supplementary Table 1). The results are summarized in Figures 6A–D. The most recent common ancestor of the Drosophila genus (Figure 6A) carried three copies of the gene with relationships [(A,C),D], of which the more distant D was located in Muller element E, and the closer to each other A and C in Muller element C. After it split from the melanogaster group (Figure 6B), the branch leading to the obscura group lost copy C and underwent an interchromosomal transposition of copy A from Muller element C to E. The daughter copy then underwent another, in this case intrachromosomal, transposition, which originated two new Attacin copies that we called AttA2 and AttA3, with AttA2 located between foxo and Npc2b, and AttA3 located ∼300 kb downstream from AttA2, between Cul5 and Sirt7. The two transpositions were genome-based duplications rather than retroposition events, because the new copies conserved the intron position of their parental gene. Before the split of the subobscura subgroup (Figure 6C), copy AttA2 underwent an inverted duplication that generated the two closely spaced copies AttA2b and AttA2a in head-to-head orientation, and transcribed in opposite directions. In D. subobscura (Figure 6D), the spacer between the IRs experienced a reversal of orientation generating the microinversion polymorphism of the distal breakpoint. Subsequently, a heterozygote for the microinversion underwent distal DSBs that allowed the formation of the recombinant O7 inversion via ectopic repair of non-sister chromatids.
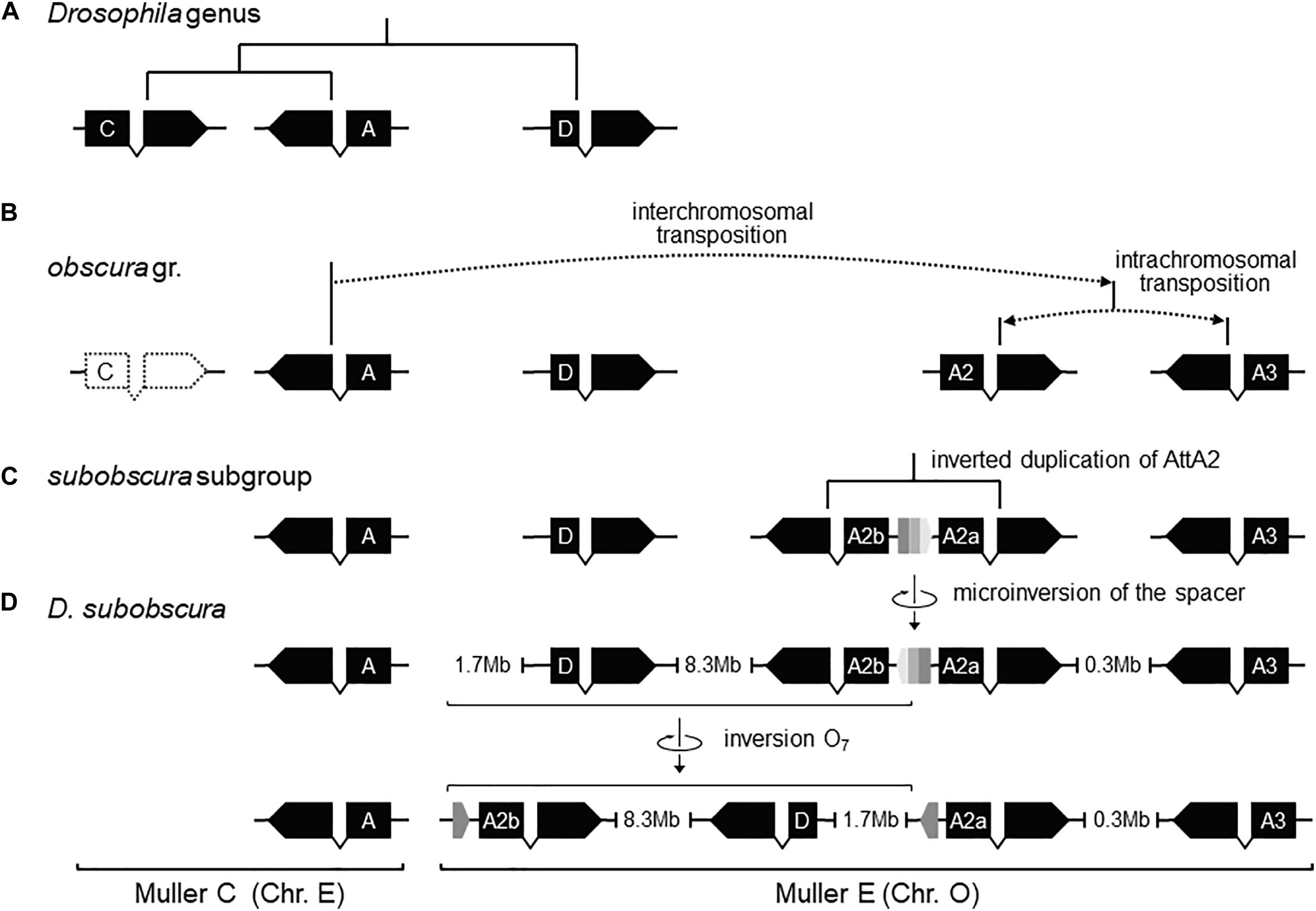
Figure 6. Pre-O7 history of instability of the distal breakpoint. (A) In the most recent common ancestor of the Drosophila genus, AttD was the only Attacin gene present in Muller element E. (B) Later, the ancestor of the obscura group lost AttC, and underwent DNA-based interchromosomal transposition of AttA (or a close paralog; see Supplementary Figure S7) from Muller C to Muller E, followed by DNA-based intrachromosomal transposition within Muller E, giving rise to AttA2 and AttA3 (whether simultaneously or sequentially and, if the latter, which was first is unknown). (C) Before the split of the subobscura subgroup, AttA2 was duplicated, giving rise to the inverted duplicates AttA2a (parent copy) and AttA2b (daughter) separated by a short central spacer. (D) In D. subobscura, the central spacer underwent a reversal, generating a microinversion polymorphism with segregating states B-type (ancestral) and ch-cu–type (derived). Genes are represented as solid black boxes (exons) linked by polygonal lines (introns), and oriented in the direction of transcription. The central spacer is represented as a box colored in three shades of gray pointing in the direction of its orientation.
The distal break of O7 disrupted concerted evolution between two subobscura subgroup-specific AttA2 duplicates. This conclusion is based on the previous section’s results, together with the phylonetwork of coding sequences shown in Figure 7. Accordingly, right after the duplication of AttA2, the two paralogs began to evolve in concert, converting each other to generate their present characteristic phylogenetic pattern of greater resemblance between paralogs from the same species (i.e., D. guanche and D. subobscura) than between orthologs from different species (e.g., Puig-Giribets et al., 2019). At one end of the resemblance, it is the ch-cu strain, whose two AttA2 copies are identical to each other, and at the other end O7, where the copy relocated by the inversion evolved significantly faster than the one that remained in place, owing exclusively to an acceleration of the synonymous substitution rate [P < 0.05; Tajima’s relative rate test (Tajima, 1993) using either of the remaining six sequences as outgroup], as the two copies are identical at the amino acid level. The acceleration took place in the direction of a slight decrease in codon bias in the relocated copy (Nc = 51.2 vs. 50.7, for the comparison AttA2b vs. AttA2a, respectively; where Nc is the improved effective number of codons index; Sun et al., 2013). The increased synonymous rate can be understood, in part because the inversion released the two Attacin copies from evolving in concert; and in part assuming that the expression of the paralogs shifted as a result of changes in regulatory environment associated with their relocation.
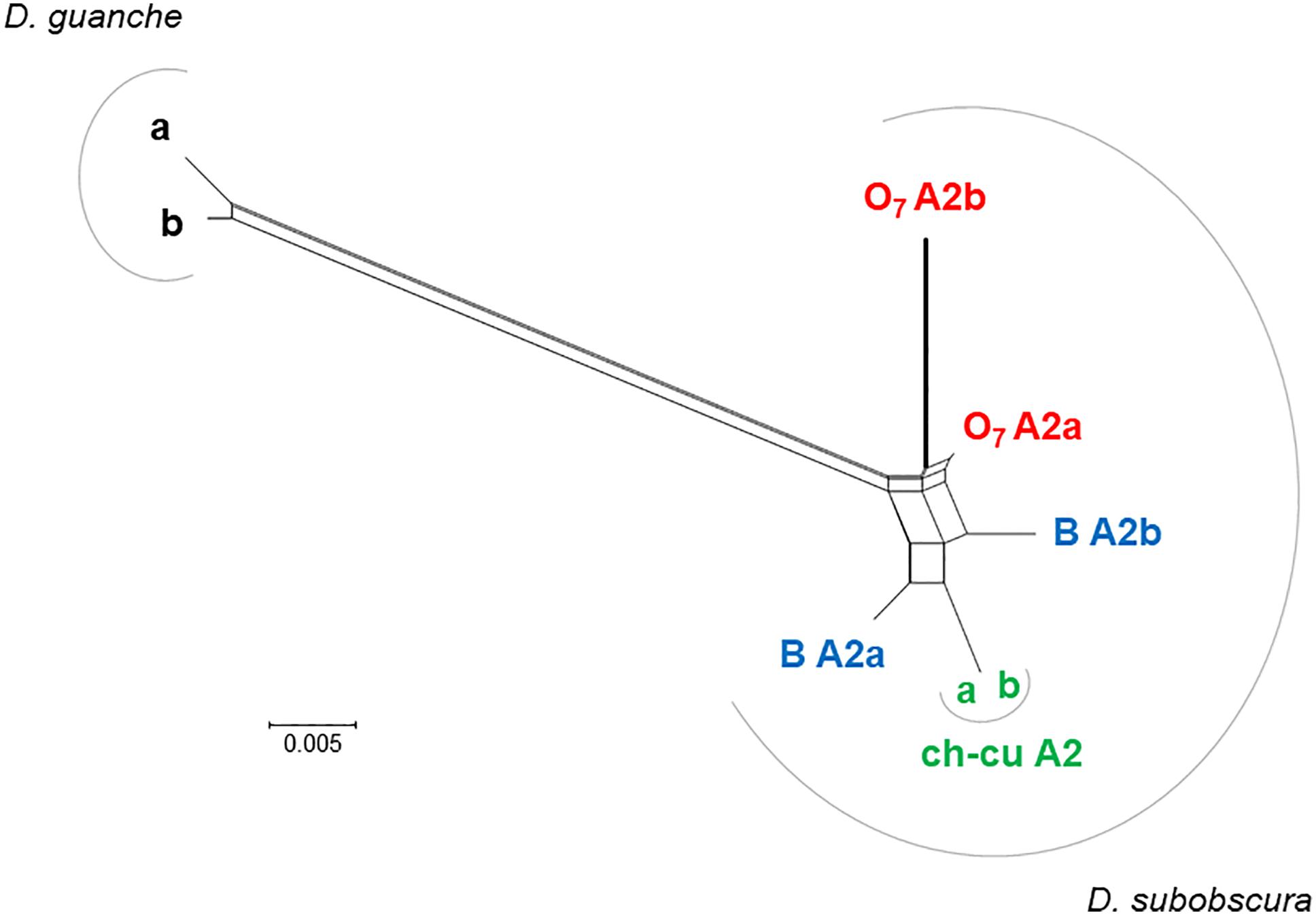
Figure 7. Phylogenetic network of AttA2 orthologous and paralogous sequences from the inverted (O7) and uninverted (B and ch-cu) states in D. guanche and D. subobscura. The duplication is older than the species, but the paralogs cluster within species owing to concerted evolution. Depicted thicker is the branch leading to the copy relocated by O7 (i.e., AttA2b), which is longer than that leading to the copy that remained in place (AttA2a) due to an acceleration of the synonymous substitution rate, likely as a result of having escaped concerted evolution. The split network was constructed using the NeighborNet method as implemented in SPLITSTREE version 4.14.5 (Huson and Bryant, 2006), on the JC69 + I (% of invariable sites 81.6) best-fit model distances obtained using the DIVEIN web server (https://indra.mullins.microbiol.washington.edu/DIVEIN/) (Deng et al., 2010). Sets of parallel edges represent conflicting topological signals.
Considering the short spacing between the two AttA2 paralogs in the uninverted chromosome (∼390 bp), it appeared likely that the inversion would have detached them from part of their promoters, binding them to new potentially cis-acting elements. To assess this possibility, we searched 1 kb upstream of the predicted TSS of each gene for putative transcription factor binding sites (TFBSs) for five transcription factors (TFs), including the nuclear factor κB factors dorsal (dl), dorsal-immunity related factor Dif and Relish (Rel), the GATA factor Serpent (srp), and the forkhead factor dFOXO. The first four TFs are under control of the Toll and immune deficiency (IMD) immunity pathways and regulate Attacin inducible expression in response to bacterial infection (Senger et al., 2004). dFOXO TF is controlled by the insulin/insulin-like growth factor signaling (IIS) metabolic pathway and regulates constitutive Attacin expression in non-infected flies suffering from energy shortage or stress (Becker et al., 2010). The results are shown in Figure 8. The AttA2 genes had predicted TFBSs for the immunity related factors in both uninverted and inverted chromosome states, but only the AttA2 genes of the inverted chromosome had TFBSs for the metabolic factor dFOXO. Furthermore, the dFOXO TFBSs were all contributed by the newly attached sequence. The fact that the AttA2 genes were conserved at the amino acid level in D. subobscura, together with the observed qualitative difference in predicted cis-acting sequence between uninverted and inverted chromosomes, suggests that the inversion O7 brought the AttA2 genes under the influence of the IIS metabolic pathway.
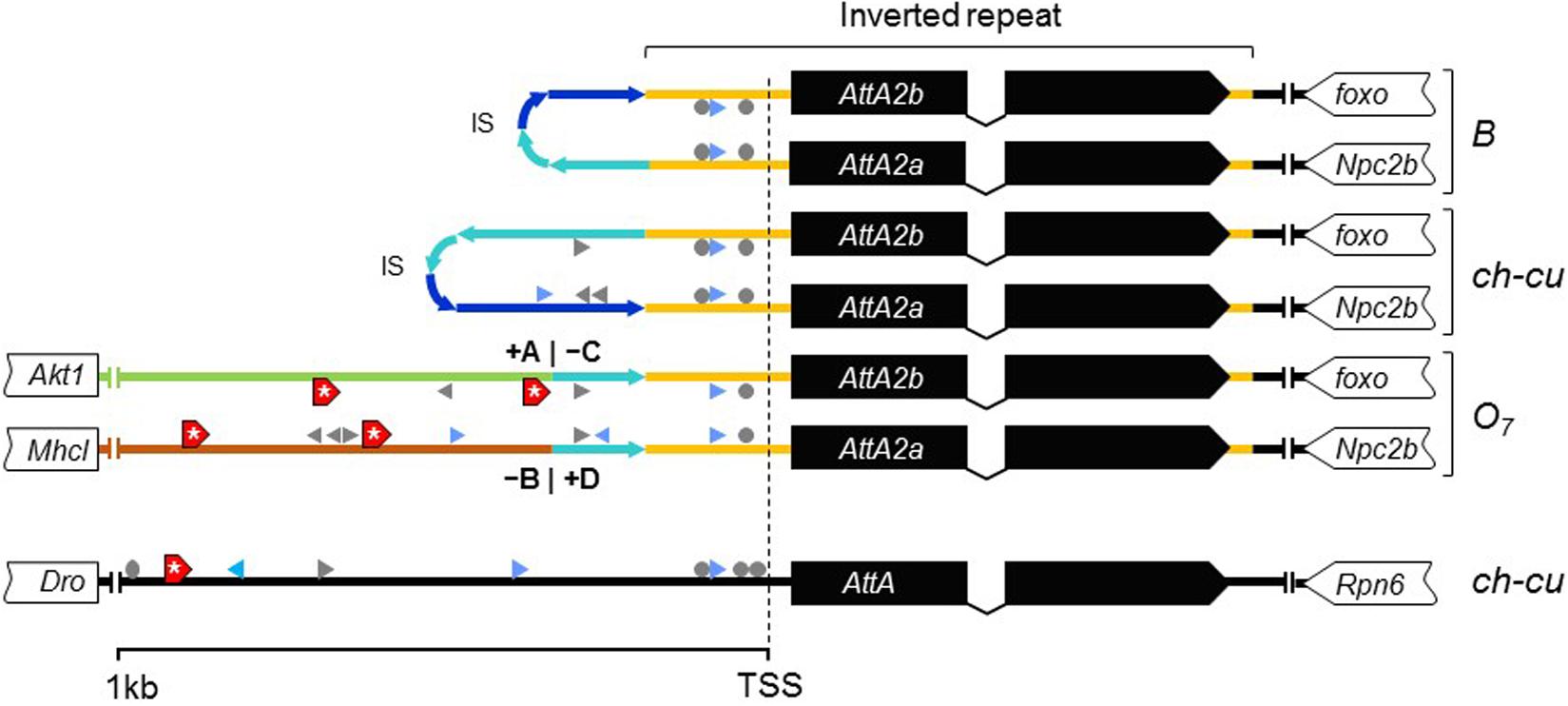
Figure 8. New dFOXO binding sites. Schematic representation of up to 1-kb sequence up and downstream predicted TSSs of AttA and the inverted duplicates of AttA2 (represented as in Figure 6) in O7 and the uninverted (B and ch-cu) states, including also the nearest flanking genes. Colored lines connecting genes designate the following: orange, region of the inverted repeats; dark and light blue, first and second halves of the spacer of the inverted repeats, respectively, oriented as the arrowheads; green and brown, the novel sequences to which the AttA2 copies became reattached by O7, with corresponding breakpoints (+A|−C and −B|+D) indicated. The inverted repeats of B and ch-cu are folded over each other. Putative TFBs are symbolized: gray arrowheads and circles (palindromic sites), for Dorsal, Relish, and Diff/Relish; blue arrowheads for Serpent, and red boxes with an asterisk for dFOXO, respectively. Only AttA and the two Att2A copies of O7 have TFBs for dFOXO.
In addition to the Attacin immunity genes, the breakpoint regions include Akt1 and foxo, two interacting core components of the IIS metabolic pathway identified by other studies as candidate for climate adaptation (Fabian et al., 2012; Paaby et al., 2014; Kapun et al., 2016; Durmaz et al., 2019). The roles of these genes and the potential impact of O7 on them are dealt with in the Discussion.
Sequence data on inversion formation in Drosophila have been interpreted in terms of two major mechanisms with associated distinctive footprints. The first mechanism is intrachromatidal NAHR between inversely oriented repeats. This mechanism generates inversions with duplications at their ends in both the inverted and uninverted states (Cáceres et al., 1999), which is not the case of O7.
The second mechanism is chromosomal breakage and ectopic repair via NHEJ. This mechanism either does not generate duplications or generates them but at the ends of the inverted state only. These two types of NHEJ footprints have been explained in terms of two alternative modes of breakage: cut-and-paste via clean DSBs that generate blunt ends and staggered on the same (isochromatidal) or different (chromatidal) sister chromatids (see Introduction). In the case of O7, it is not a cut-and-paste inversion, but neither is it a typical staggered breaks inversion. Thus, while the inversion proximal breakpoint could be either isochromatidal (Figure 4) or chromatidal (Figure 9), the distal breakpoint has to involve the two homologous chromosomes (Figures 4, 9). This latter pattern could be deduced because of the chanceful circumstance that our two representatives of the uninverted state (i.e., Ds_ch-cu and Ds_B) segregated for the microinversion of the spacer between the IRs flanking the distal breakpoint. Alternatively, the distal breakage could have occurred in a recombinant between chromosome types ch-cu and B. This, however, appears unlikely because crossover within microinversions should be extremely rare (Greig, 2007). Our conclusion agrees with a study of the genealogical relationships between inversions of the E chromosome in D. subobscura, which proposed that E9 arose in a heterokaryotype EST/E1+2 to accommodate a conflict between molecular and cytological data (Orengo et al., 2019). This and our results indicate that NHEJ inversions form through mechanisms that can incorporate information from the two homologous chromosomes (chromosome model), in addition to the previously proposed intrasister and intersister chromatidal exchanges.
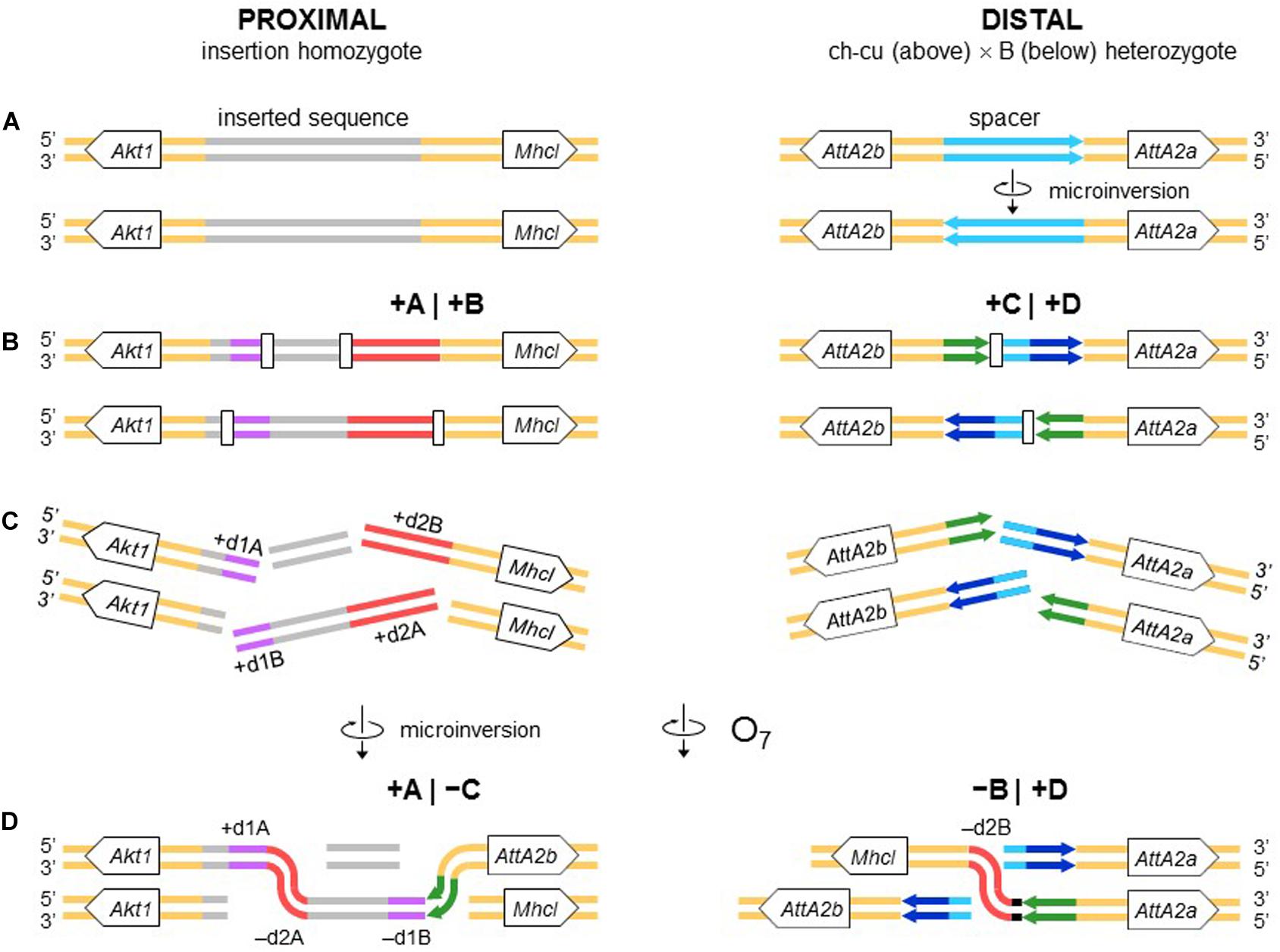
Figure 9. Chromosome model of O7 formation. (A) Start from an individual homozygous for the insertion at the proximal breakpoint, and as in Figure 4 at the distal breakpoint. (B) Occurrence of two pairs of staggered chromosomal blunt-ended DSBs (open rectangles) at the proximal breakpoint, and as in Figure 3 at the distal breakpoint. (C,D) Microinversion formation, and formation of O7 via rejoining in trans with the homologous chromosome as indicated. The model results in an order of the duplications (i.e., + d1A, -d2A, -d1B, -d2B) identical to that resulting from the model of Figure 4.
Inversion O7 provides, to our knowledge, the first compelling evidence for a role of non-B DNA in inversion formation in Drosophila. Previous studies had reported the presence of AT-rich sequences around the breakpoints of some fixed (Cirera et al., 1995; Richards et al., 2005) and polymorphic (Prazeres da Costa et al., 2009) inversions. In no instance, however, were particular sequences susceptible to adopt secondary structures identified. In the case of O7, the proximal break junction occurred just within a palindromic AT-rich repeat capable of adopting hairpin/cruciform, slipped and triplex DNA conformations. Likewise, the distal junctions are located next to perfect 14-bp-long hairpin/cruciform-forming palindromes.
The role of non-B DNA-forming sequences in causing genome instability is well-established (Wang and Vasquez, 2006; Lobachev et al., 2007; Aguilera and Gómez-González, 2008; Zhao et al., 2010). The shift from B to non-B DNA conformation occurs while DNA is in single-stranded form, e.g., behind replication forks, between Okazaki fragments, or in actively transcribed genes (Voineagu et al., 2008). Non B-DNA structures induce DSBs through, e.g., stalling replication and transcription (Mani and Chinnaiyan, 2010; Kaushal and Freudenreich, 2019). There are no specific predictions as to the type, number, and location of the DSBs generated by any given structure in any particular situation. Still, a single structure can induce multiple DSBs across hundreds of base pairs around it (Wang et al., 2006; McKinney et al., 2020), and stalled replication forks can accumulate up to 3 kb of single-stranded DNA (Sogo et al., 2002; Lopes et al., 2006). In the case of O7, this length is well over the size of the overhangs that would be generated by an isochromatid model of the proximal breakpoint (58 and 534 nt; see Figure 3 and Supplementary Figure 3).
All the aforementioned inverted duplication-generating NHEJ models are predicated upon the role of DNA breakage (Ranz et al., 2007). However, the inverted duplications at the ends of O7 could also be explained as a result exclusively of repair, with no need for invoking staggering of the breaks. DNA repair has emerged as a key factor capable of generating extremely complex breakpoint sequence rearrangements (reviewed in Scully et al., 2019). The spectrum of known error-prone repair mechanisms can be grossly classified as recombination-based, such as microhomology-mediated end-joining (MMEJ), and replication-based, such as break-induced replication (BIR) and microhomology-mediated BIR (MMBIR) (Lee et al., 2007; Zhang et al., 2009; Hastings et al., 2009). Here, the term microhomology is used to mean a short tract (∼1 – 25 bp) of chance similarity, rather than common descent. In the case of O7, three features suggest that what appear to be footprints of breakage by the staggering models could in fact be footprints of a replication-based mode of repair (reviewed in Kramara et al., 2018; Scully et al., 2019), including (i) presence of non-B DNA-forming sequences just in, or adjacent to breakpoint junctions (see below); (ii) spatial proximity of the breakpoint regions in the nucleus, as evinced by the fact that the genes flanking the junctions are closely related functionally (Farré et al., 2015; but see Sunder and Wilson, 2019); and (iii) multiple breaks concentrated in a short sequence segment. A fourth feature, namely, presence of microhomology at the distal breakpoint junction, would be also consistent with a recombination-based mechanism such as MMEJ. Overall, these features suggest that O7 arose as result of a non-B DNA-induced replication impairment, affecting at least its proximal breakpoint. It is known that this type of events can trigger BIR and MMBIR repair (Sakofsky et al., 2015). Of the two pathways, the second pathway has yet to be identified in Drosophila (Alexander et al., 2016; Bhandari et al., 2019). A possible scenario is detailed in Figure 10: first, non-B DNA-induced stalling of a replication fork at the proximal breakpoint of a ch-cu–type chromosome led to two DSBs generating three fragments. Second, the centromere-proximal fragment engaged in a BIR event using the homologous region of a B-type chromosome. Third, a second fork stalling triggered a switch from BIR to MMBIR with template switching to a downstream microhomology. Copying backward from the new template resulted in the rearrangement of the proximal breakpoint, including the inverted duplication of the O7 end (e.g., Lee et al., 2007; Smith et al., 2007; Carvalho et al., 2015; Tremblay-Belzile et al., 2015). Finally, the event was terminated by an MMEJ to the distal break-end of O7 from the original ch-cu-type chromosome (e.g., Scully et al., 2019).
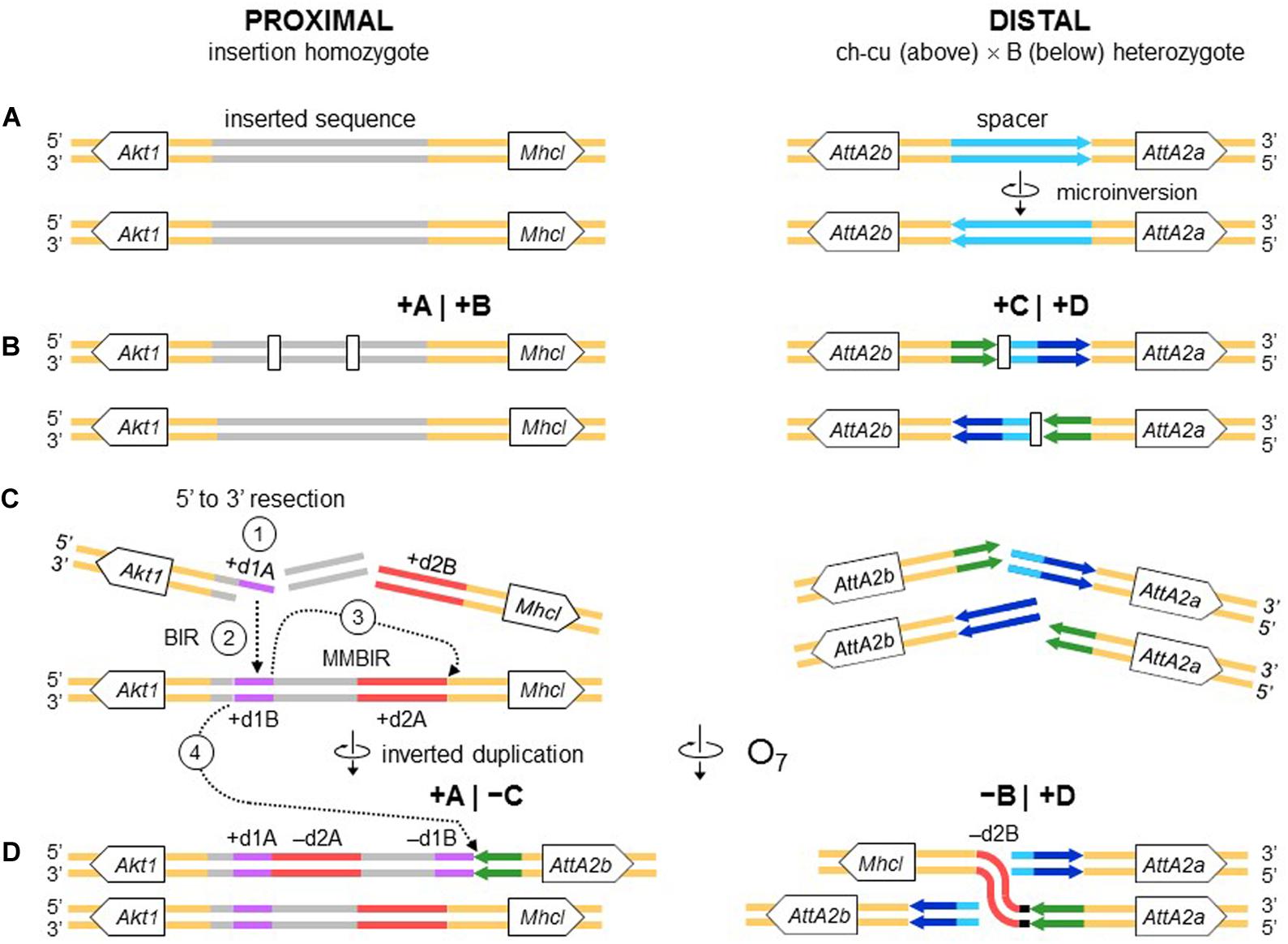
Figure 10. Chromosome and BIR/MMBIR repair model of O7 formation. (A) Start as in Figure 9. (B) Occurrence of one pair of blunt-ended DSBs (open rectangles) at the proximal breakpoint of the ch-cu type chromosome, and as in Figure 4 at the distal breakpoint. (C,D) Step 1: 5′ to 3′ resection generating a 3′ single stranded + d1A end. Step 2: beginning of a BIR event via strand invasion into the homologous region of the B-type chromosome. Step 3: switch from BIR to MMBIR, with forward template switching to the distal end of + d2A and backward copying. Step 4: MMEJ to the distal break-end of O7 from the original ch-cu–type chromosome. The distal breakpoint repaired as in Figure 9. The model results in an order of the duplications (i.e., + d1A, -d2A, -d1B, -d2B) identical to that resulting from the model of Figures 4, 9.
The breakpoint sequences of O7 had a record of instability prior to the origin of the inversion, as evinced by the fact that they are located within sequences inserted from elsewhere in the genome. This suggests that the regions that gained those insertions were relatively exposed in the nucleus (reviewed in Farré et al., 2015). In the case of the proximal breakpoint, that could be associated with high levels of transcriptional activity at the broadly expressed Akt1 gene (Andjelković et al., 1995; Slade and Staveley, 2016).
That the O7 junctions arose in fragile regions, beyond the proximate effects of their associated non-B DNA (see above), may be most apparent from the pre-inversion record of recurrent rearrangement of the IR at the distal breakpoint (Figure 6). This record is particularly amenable to reconstruction because the IR largely consists of two copies of the Attacin A gene that are highly conserved. It includes at least three rearrangements that occurred in the lineage of D. subobscura after its separation from that of the melanogaster group (see section “RESULTS”; Figure 6), namely, (i) insertion of AttA2 between the foxo and Npc2b genes; (ii) emergence of the IR by inverted duplication of the parental AttA2 (Figure 6B), which could have occurred through an event of forward template switching and backward copying by the DNA polymerase (Smith et al., 2007; Lee et al., 2007), as discussed above; and (iii) emergence of the ch-cu–type chromosome via inversion of the spacer between the IRs in a B-type chromosome (Figure 6D), which could be explained as an outcome of a stem-loop formation by the IR, followed by resolution of the strand-exchange junctions between the IR arms (see Figure 4 in Leigh Brown and Ish-Horowicz, 1981; Figure 3 in Kolb et al., 2009 and Zhao et al., 2010).
The pre-O7 insertion in the proximal breakpoint is specific to D. subobscura and is therefore much more recent than that of AttA2 in the distal breakpoint. Preliminary analyses indicate that it is internally rearranged relative to other paralogous copies, supporting that it carries recombinogenic potential. The origin and evolution of this inserted sequence, as well as its possible implication in the formation of other D. subobscura inversions, warrant further investigation (CK, RT, and FR-T; manuscript in preparation).
O7 changed foxo from being megabases (∼10 Mb) away from Akt1 to being tightly linked to it, with only the short AttA2b gene sandwiched between them. Akt1 and foxo are functionally conserved genes, which, in Drosophila, encode the serine/threonine–protein kinase B AKT/PKB, and the forkhead-box DNA-binding domain-containing TF dFOXO, respectively. The two genes are key antagonistic regulators of the IIS pathway (Teleman, 2010; Slade and Staveley, 2016), a major trigger of shifts in anabolic versus catabolic cellular activity in response to nutritional status (de Jong and Bochdanovits, 2003) and multiple other cues (Regan et al., 2020). In abundant nutrient conditions, AKT/PKB inactivates dFOXO, thus shifting food energy allocation toward reproduction and growth (the IIS pathway). Conversely, scarce nutrient conditions prevent AKT/PKB from inactivating dFOXO, which redirects metabolism toward mobilization of energy stores for somatic maintenance (FOXO pathway). Laboratory research using large effect mutants has shown that the IIS/FOXO pathway is extensively pleiotropic, with major evolutionary conserved effects on fitness-related life-history traits, including growth, size, reproduction, lifespan, and stress resistance (reviewed in Flatt and Partridge, 2018). Research from the field found IIS loci to harbor substantial genetic variation, which frequently exhibits spatiotemporal patterns that look as if they were shaped by selection on the associated IIS traits (Fabian et al., 2012; Paaby et al., 2014; Kapun et al., 2016). In a recent laboratory assay, two foxo alleles showing opposite latitudinal clines in D. melanogaster were compared on an otherwise homogeneous genetic background. The alleles showed contrasting effects on viability, size-related traits, starvation resistance, and fat content, whose directions were overall consistent with predictions from the clinal variation of the characters (Durmaz et al., 2019).
The O7 mutation could have altered Akt1 and/or foxo function via multiple non-mutually exclusive mechanisms, such as mutual regulatory interference, considering that they are antagonistic effectors; relocation to the sides of an immunity gene (i.e., AttA2b) expected to be under intense purifying selection on expression (see below); and alteration of the genes’ functional neighborhood at higher-order levels of chromatin organization (Farré et al., 2015; McBroome et al., 2020). It could be argued that the nuclear environment of the genes remained basically unaltered, if the reason why they became involved in the rearrangement was that they already were in close spatial proximity to each other in the nucleus. This, however, did not necessarily have to be the case, considering recent findings in yeast that rejoining of DNA break ends is not determined by the predamage spatial proximity of the DSBs (Sunder and Wilson, 2019). Be that as it may, bearing in mind that the seasonal increase of O7 occurs from early spring to midsummer, coinciding with the growth season, it seems more likely that whatever the effect of the inversion mutation on Akt1 and/or foxo, it occurred in the direction of an enhanced basal IIS versus dFOXO activity relative to the OST ancestral state. This would raise the question of why the O7 frequencies decrease (and those of OST increase) every year from late summer to winter.
The immune function is highly energy demanding in terms of both maintenance and, especially, rapid deployment upon infection (reviewed in Dolezal et al., 2019). Therefore, within a limited energy budget, a trade-off is expected between reproduction and immunity (Schwenke et al., 2016). The Drosophila innate immune response consists of a cellular and a humoral component. The humoral component involves the production of antimicrobial peptides, among which Attacins are active against gram-negative bacteria (Hanson and Lemaitre, 2020). The two main modes of Attacin production, including the induced (by a factor of even > 100) upon infection mode, and the basal in absence-of-infection mode link immunity with the Akt1/foxo IIS metabolic signaling pathway (Becker et al., 2010; Dolezal et al., 2019). The inducible mode is regulated primarily by the immunodeficiency Imd signaling pathway and to a lesser extent by the Toll signaling pathway. The two signaling pathways have the same effect of activating dFOXO, thus mobilizing resources toward the production of Attacins (Dionne et al., 2006; Dolezal et al., 2019). The basal mode is regulated directly by dFOXO activity when induced by starvation (Becker et al., 2010; Buchon et al., 2014). Immunity genes, including Attacins, are among the known most rapidly evolving genes and have frequently shown evidence of local adaptation in Drosophila (Lazzaro and Clark, 2001, 2003).
There would be a number of mechanisms by which the O7 mutation could have reduced Attacin genes’ expression. For example, the breakage of the invertedly transcribed AttA2 tandem duplicates could have impaired the inducibility of one or the two paralogs, or their separation could have made them lose gene expression coregulation, as might be surmised from the observations that they halted or slowed down evolving in concert, and that AttA2b shows decreased codon bias. These mechanisms could have acted synergistically with each other and with those already discussed in connection with Akt1 and foxo. Although this scenario could be partially offset by the increase in basal AttA2 transcript levels that may be expected from the duplicates having been reattached to dFOXO enhancers (Becker et al., 2010), all in all, the evidence suggests that (i) at its inception, O7 caused a rearrangement with partial disruption of a set of functionally related loci with overlapping pleiotropic effects on immunometabolic traits. If, in addition to these direct effects, there concurred indirect effects of linkage between locally, and given the functional relationship, likely epistatically interacting alleles warrant further investigation; and (ii) the resulting haplotype imparted a shifted pattern of resource allocation toward reproduction at a cost to immunity, compared to the OST ancestor. Such an opposing antagonistic pleiotropy would result in a seasonal frequency cycle qualitatively similar to that shown by the inversions, if reproduction is favored from early spring to midsummer, when O7 rises (and OST wanes), and immunity from late summer to winter, when it wanes (and OST rises). There is ample evidence that the qualitative and quantitative composition of temperate bacterial communities cycles seasonally (Lazzaro et al., 2015; Shigyo et al., 2019). Recently, a study using D. melanogaster from the eastern United States (Behrman et al., 2018) found a seasonal shift in immunocompetence, with the trait value declining every spring to autumn. The shift was interpreted as resulting from relaxed selection for immune response during the warm season, much like what we propose here for the O7/OST inversion polymorphism.
Prior data on temporal genetic variation within and between O inversions point to additional loci that would be consistent with the seasonal cycle of O7 being mediated by immunometabolic selection (Rodríguez-Trelles, 2003). The case of the Mpi gene encoding the key glycolytic enzyme mannose-6-phosphate isomerase (MPI) is noteworthy. From our assembly, Mpi is located 2.15 Mb outward from the distal breakpoint of O7, which is within the estimated region of the inversion-associated strong recombination–suppression effect (3.5 Mb; Pegueroles et al., 2010b). The MPI fast/slow electrophoretic polymorphism was found to be only moderately associated with the O7/OST polymorphism. Yet (i) the magnitude of the locus-by-inversion disequilibrium cycled seasonally, and (ii) the cycling occurred because the Fast allele increased in frequency every winter only within the O7 chromosomal class, but not within the OST class (Rodríguez-Trelles, 2003). The behavior of Mpi could be in part an outcome of hitch-hiking with other linked loci involved in seasonal adaptation. One such candidate could be the Na pumpα subunit (Atpα) gene, located only 0.13 Mb farther away from O7 than Mpi, and recently found to be under positive selection for defense against plant secondary compounds in D. subobscura (Pegueroles et al., 2016). Still, immune elicitation in Drosophila relies upon massive upregulation of glycolysis (Dolezal et al., 2019), which should place a strong demand on MPI activity (Shtraizent et al., 2017). In addition to the evidence from D. subobscura just discussed, Supplementary Table 2 provides additional loci found to exhibit seasonal variation in a genomic survey from other Drosophila, which may be candidates for being involved in the seasonal cycling of O7.
Previous work on the spatiotemporal distribution patterns of the inversion polymorphisms of D. subobscura indicated that O7 is driven by selective factors other than temperature alone. Here, we addressed this issue using a genome-based approach to isolate and characterize the O7 breakpoints. Our findings have general implications for current theories on the molecular mechanisms of formation of this common type of structural genomic change. Furthermore, they suggest that O7 may have altered fly’s immunometabolism through at least direct effects on core immunity and metabolism genes. This result could help to explain the inversion’s conflicting correlations with the seasonal and decadal climate changes, taking into account recent findings from microbial ecology, which indicate that microbial community responses to short- and long-term climate changes can be largely uncorrelated (Romero-Olivares et al., 2017). Considering its large size, it seems likely that O7’s evolution is also shaped by additional direct or/and indirect effects on genes other than those near its breakpoints. Further progress along this line will include development of functional tests of the identified genes on inverted versus uninverted chromosome backgrounds and use of the obtained assembly for building a SNP panel for O chromosome-wide scans of selection. We have incorporated the chromosome-scale sequence of O3+4+7 obtained here into our reference genome browser9 to facilitate the further use of this resource.
Datasets presented in this article are available at the European Nucleotide Archive (ENA) under the project ID: PRJEB38585.
CK, RT, and FR-T contributed to the design and implementation of the research, to the analysis of the results, and to the writing of the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by the Spanish Ministerio de Ciencia e Innovación grant CGL2017-89160P; and Generalitat de Catalunya grant 2017SGR 1379 to the Grup de Genòmica, Bioinformàtica i Biologia Evolutiva, Universitat Autònoma de Barcelona (Spain). CK was supported by a PIF Ph.D. fellowship from the Universitat Autònoma de Barcelona (Spain). Note that the funding agencies were not involved in the design of the study or in any aspect of the collection, analysis and interpretation of the data or paper writing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.565836/full#supplementary-material
Aguilera, A., and Gómez-González, B. (2008). Genome instability: a mechanistic view of its causes and consequences. Nat. Rev. Genet. 9, 204–217. doi: 10.1038/nrg2268
Alexander, J. L., Beagan, K., Orr-Weaver, T. L., and McVey, M. (2016). Multiple mechanisms contribute to double-strand break repair at rereplication forks in Drosophila follicle cells. Proc. Natl. Acad. Sci. U.S.A. 113, 13809–13814. doi: 10.1073/pnas.1617110113
Andjelković, M., Jones, P. F., Grossniklaus, U., Cron, P., Schier, A. F., Dick, M., et al. (1995). Developmental regulation of expression and activity of multiple forms of the Drosophila RAC protein kinase. J. Biol. Chem. 270, 4066–4075. doi: 10.1074/jbc.270.8.4066
Anisimova, M., Gil, M., Dufayard, J. F., Dessimoz, C., and Gascuel, O. (2011). Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst. Biol. 60, 685–699. doi: 10.1093/sysbio/syr041
Apweiler, R., Bairoch, A., Wu, C. H., Barker, W. C., Boeckmann, B., Ferro, S., et al. (2004). UniProt: the universal protein knowledgebase. Nucleic Acids Res. 32, D115–D119. doi: 10.1093/nar/gkw1099
Arenas, C., Zivanovic, G., and Mestres, F. (2018). Chromosomal thermal index: a comprehensive way to integrate the thermal adaptation of Drosophila subobscura whole karyotype. Genome 61, 73–78.
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., and Cherry, J. M. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Ayala, F. J., Serra, L., and Prevosti, A. (1989). A grand experiment in evolution: the Drosophila subobscura colonization of the Americas. Genome 31, 246–255. doi: 10.1139/g89-042
Bächli, G. (2020). TaxoDros: The Database on Taxonomy of Drosophilidae. Available online at: https://www.taxodros.uzh.ch (accessed February 21, 2020).
Bailey, T. L., Johnson, J., Grant, C. E., and Noble, W. S. (2015). The MEME Suite. Nucleic Acids Res. 43, W39–W49. doi: 10.1093/nar/gkv416
Balanyà, J., Oller, J. M., Huey, R. B., Gilchrist, G. W., and Serra, L. (2006). Global genetic change tracks global climate warming in Drosophila subobscura. Science 313, 1773–1775. doi: 10.1126/science.1131002
Bao, W., Kojima, K. K., and Kohany, O. (2015). Repbase update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6:11.
Becker, T., Loch, G., Beyer, M., Zinke, I., Aschenbrenner, A. C., Carrera, P., et al. (2010). FOXO-dependent regulation of innate immune homeostasis. Nature 463, 369–373. doi: 10.1038/nature08698
Behrman, E. L., Howick, V. M., Kapun, M., Staubach, F., Bergland, A. O., Petrov, D. A., et al. (2018). Rapid seasonal evolution in innate immunity of wild Drosophila melanogaster. Proc. Biol. Sci. 285:20172599. doi: 10.1098/rspb.2017.2599
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bhandari, J., Karg, T., and Golic, K. G. (2019). Homolog-dependent repair following dicentric chromosome breakage in Drosophila melanogaster. Genetics 212, 615–630. doi: 10.1534/genetics.119.302247
Bracewell, R., Chatla, K., Nalley, M. J., and Bachtrog, D. (2019). Dynamic turnover of centromeres drives karyotype evolution in Drosophila. eLife 8:e49002. doi: 10.7554/eLife.49002
Brázda, V., Kolomazník, J., Lýsek, J., Hároníková, L., Coufal, J., and Št’astný, J. (2016). Palindrome analyser - A new web-based server for predicting and evaluating inverted repeats in nucleotide sequences. Biochem. Biophys. Res. Commun. 478, 1739–1745. doi: 10.1016/j.bbrc.2016.09.015
Brehm, A., and Krimbas, C. B. (1988). The inversion polymorphism of Drosophila subobscura natural populations from Portugal. Genét. Ibér. 39, 235–248.
Buchon, N., Silverman, N., and Cherry, S. (2014). Immunity in Drosophila melanogaster –from microbial recognition to whole-organism physiology. Nat. Rev. Immunol. 14, 796–810. doi: 10.1038/nri3763
Cáceres, M., Ranz, J. M., Barbadilla, A., Long, M., and Ruiz, A. (1999). Generation of a widespread Drosophila inversion by a transposable element. Science 285, 415–418. doi: 10.1126/science.285.5426.415
Campbell, M. S., Holt, C., Moore, B., and Yandell, M. (2014). Genome annotation and curation using MAKER and MAKER-P. Curr. Protoc. Bioinformatics 48, 4.11.1–4.11.39. doi: 10.1002/0471250953.bi0411s48
Carvalho, C. M., Pfundt, R., King, D. A., Lindsay, S. J., Zuccherato, L. W., and Macville, M. V. (2015). Absence of heterozygosity due to template switching during replicative rearrangements. Am. J. Hum. Genet. 96, 555–564. doi: 10.1016/j.ajhg.2015.01.021
Cer, R. Z., Bruce, K. H., Donohue, D. E., Temiz, N. A., Mudunuri, U. S., Yi, M., et al. (2012). Searching for non-B DNA-forming motifs using nBMST (non-B DNA motif search tool). Curr. Protoc. Hum. Genet. 18, 11–22. doi: 10.1002/0471142905.hg1807s73
Chakraborty, M., Baldwin-Brown, J. G., Long, A. D., and Emerson, J. J. (2016). Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids. Res. 44:e147. doi: 10.1093/nar/gkw654
Chen, H., Rangasamy, M., Tan, S. Y., Wang, H., and Siegfried, B. D. (2010). Evaluation of five methods for total DNA extraction from Western corn rootworm beetles. PLoS One 5:e11963. doi: 10.1371/journal.pone.0011963
Cheng, C., Tan, J. C., Hahn, M. W., and Besansky, N. J. (2018). Systems genetic analysis of inversion polymorphisms in the malaria mosquito Anopheles gambiae. Proc. Natl. Acad. Sci. U.S.A. 115, E7005–E7014. doi: 10.1073/pnas.1806760115
Cirera, S., Martín-Campos, J. M., Segarra, C., and Aguadé, M. (1995). Molecular characterization of the breakpoints of an inversion fixed between Drosophila melanogaster and D. subobscura. Genetics 139, 321–326.
Corbett-Detig, R. B. (2016). Selection on inversion breakpoints favors proximity to pairing sensitive sites in Drosophila melanogaster. Genetics 204, 259–265. doi: 10.1534/genetics.116.190389
Corbett-Detig, R. B., and Hartl, D. L. (2012). Population genomics of inversion polymorphisms in Drosophila melanogaster. PLoS Genet. 8:e1003056. doi: 10.1371/journal.pgen.1003056
Crescente, J. M., Zavallo, D., Helguera, M., and Vanzetti, L. S. (2018). MITE Tracker: an accurate approach to identify miniature inverted-repeat transposable elements in large genomes. BMC Bioinformatics 19:348. doi: 10.1186/s12859-018-2376-y
de Frutos, R. (1972). Contribution to the study of chromosomal polymorphism in the Spanish populations of Drosophila subobscura. Genét. Ibér. 24, 123–140.
de Jong, G., and Bochdanovits, Z. (2003). Latitudinal clines in Drosophila melanogaster: body size, allozyme frequencies, inversion frequencies, and the insulin-signalling pathway. J. Genet. 82, 207–223. doi: 10.1007/BF02715819
Delprat, A., Guillén, Y., and Ruiz, A. (2019). Computational sequence analysis of inversion breakpoint regions in the cactophilic Drosophila mojavensis lineage. J. Hered. 110, 102–117. doi: 10.1093/jhered/esy057
Deng, W., Maust, B. S., Nickle, D. C., Learn, G. H., Liu, Y., Heath, L., et al. (2010). DIVEIN: a web server to analyze phylogenies, sequence divergence, diversity, and informative sites. Biotechniques 48, 405–408. doi: 10.2144/000113370
Dionne, M. S., Pham, L. N., Shirasu-Hiza, M., and Schneider, D. S. (2006). Akt and foxo dysregulation contribute to infection-induced wasting in Drosophila. Curr. Biol. 16, 1977–1985. doi: 10.1016/j.cub.2006.08.052
Dobzhansky, T. (1947). Genetics of natural populations. XIV. A response of certain gene arrangements in the third chromosome of Drosophila pseudoobscura to natural selection. Genetics 32, 142–160.
Dolezal, T., Krejcova, G., Bajgar, A., Nedbalova, P., and Strasser, P. (2019). Molecular regulations of metabolism during immune response in insects. Insect. Biochem. Mol. Biol. 109, 31–42. doi: 10.1016/j.ibmb.2019.04.005
Dolgova, O. (2013). Genetic and Phenotypic Differentiation in three Chromosomal Arrangements of Drosophila subobscura. Ph.D. thesis, Universitat Autònoma de Barcelona, Barcelona.
Durmaz, E., Rajpurohit, S., Betancourt, N., Fabian, D. K., Kapun, M., Schmidt, P., et al. (2019). A clinal polymorphism in the insulin signaling transcription factor foxo contributes to life-history adaptation in Drosophila. Evolution 73, 1774–1792. doi: 10.1111/evo.13759
Eilbeck, K., Lewis, S., Mungall, C., Yandell, M., Stein, L., Durbin, R., et al. (2005). The sequence ontology: a tool for the unification of genome annotations. Genome Biol. 6:R44. doi: 10.1186/gb-2005-6-5-r44
Ellinghaus, D., Kurtz, S., and Willhoeft, U. (2008). LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9:18. doi: 10.1186/1471-2105-9-18
Fabian, D. K., Kapun, M., Nolte, V., Kofler, R., Schmidt, P. S., Schlötterer, C., et al. (2012). Genome-wide patterns of latitudinal differentiation among populations of Drosophila melanogaster from North America. Mol. Ecol. 21, 4748–4769. doi: 10.1111/j.1365-294X.2012.05731.x
Faria, R., Johannesson, K., Butlin, R. K., and Westram, A. M. (2019). Evolving inversions. Trends Ecol. Evol. 34, 239–248. doi: 10.1016/j.tree.2018.12.005
Farré, M., Robinson, T. J., and Ruiz-Herrera, A. (2015). An integrative breakage model of genome architecture, reshuffling and evolution. Bioessays 37, 479–488. doi: 10.1002/bies.201400174
Finn, R. D., Attwood, T. K., Babbitt, P. C., Bateman, A., Bork, P., Bridge, A. J., et al. (2017). InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199. doi: 10.1093/nar/gkw1107
Finn, R. D., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Mistry, J., Mitchell, A. L., et al. (2016). The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285. doi: 10.1093/nar/gkv1344
Flatt, T., and Partridge, L. (2018). Horizons in the evolution of aging. BMC Biol. 16:93. doi: 10.1186/s12915-018-0562-z
Fontdevila, A., Zapata, C., Alvarez, G., Sanchez, L., Méndez, J., and Enriquez, I. (1983). Genetic coadaptation in the chromosomal polymorphism of Drosophila subobscura. I. Seasonal changes of gametic disequilibrium in a natural population. Genetics 105, 935–955.
Fragata, I., Lopes-Cunha, M., Bárbaro, M., Kellen, B., Lima, M., Santos, M. A., et al. (2014). How much can history constrain adaptive evolution? A real-time evolutionary approach of inversion polymorphisms in Drosophila subobscura. J. Evol. Biol. 27, 2727–2738. doi: 10.1111/jeb.12533
Fuller, Z. L., Haynes, G. D., Richards, S., and Schaeffer, S. W. (2016). Genomics of natural populations: How differentially expressed genes shape the evolution of chromosomal inversions in Drosophila pseudoobscura. Genetics 204, 287–301. doi: 10.1534/genetics.116.191429
Fuller, Z. L., Haynes, G. D., Richards, S., and Schaeffer, S. W. (2017). Genomics of natural populations: evolutionary forces that establish and maintain gene arrangements in Drosophila pseudoobscura. Mol. Ecol. 26, 6539–6562. doi: 10.1111/mec.14381
Fuller, Z. L., Koury, S. A., Phadnis, N., and Schaeffer, S. W. (2019). How chromosomal rearrangements shape adaptation and speciation: case studies in Drosophila pseudoobscura and its sibling species Drosophila persimilis. Mol. Ecol. 28, 1283–1301. doi: 10.1111/mec.14923
Gienapp, P., Teplitsky, C., Alho, J. S., Mills, J. A., and Merilä, J. (2008). Climate change and evolution: disentangling environmental and genetic responses. Mol. Ecol. 17, 167–178. doi: 10.1111/j.1365-294X.2007.03413.x
Götz, W. (1965). Beitrag zur Kenntnis der Inversionen, Duplikationen und Strukturtypen von Drosophila subobscura Coll. Z. Vererbungsl. 96, 285–296. doi: 10.1007/BF00896828
Götz, W. (1967). Untersuchungen über den chromosomalen Strukturpolymorphismus in kleinasiatischen und persischen Populationen von Drosophila subobscura Coll. Mol. Gen. Genet. 100, 1–38. doi: 10.1007/BF00425773
Grant, C. E., Bailey, T. L., and Noble, W. S. (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. doi: 10.1093/bioinformatics/btr064
Greig, D. (2007). A screen for recessive speciation genes expressed in the gametes of F1 hybrid yeast. PLoS Genet. 3:e21. doi: 10.1371/journal.pgen.0030021
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximumlikelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Hanson, M. A., and Lemaitre, B. (2020). New insights on Drosophila antimicrobial peptide function in host defense and beyond. Curr. Opin. Immunol. 62, 22–30. doi: 10.1016/j.coi.2019.11.008
Hastings, P. J., Ira, G., and Lupski, J. R. (2009). A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 5:e1000327. doi: 10.1371/journal.pgen.1000327
Hill, T., and Betancourt, A. J. (2018). Extensive exchange of transposable elements in the Drosophila pseudoobscura group. Mob. DNA 9:20.
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q., and Vinh, L. S. (2018). UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522. doi: 10.1093/molbev/msx281
Hoffmann, A. A., and Rieseberg, L. H. (2008). Revisiting the impact of inversions in evolution: from population genetic markers to drivers of adaptive shifts and speciation? Annu. Rev. Ecol. Evol. Syst. 39, 21–42. doi: 10.1146/annurev.ecolsys.39.110707.173532
Holt, C., and Yandell, M. (2011). MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12:491. doi: 10.1186/1471-2105-12-491
Hughes, L. (2000). Biological consequences of global warming: Is the signal already apparent? Trends Ecol. Evol. 15, 56–61.
Hughes, S. E., Miller, D. E., Miller, A. L., and Hawley, R. S. (2018). Female meiosis: synapsis, recombination, and segregation in Drosophila melanogaster. Genetics 208, 875–908. doi: 10.1534/genetics.117.300081
Huson, D. H., and Bryant, I. D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Joyce, E. F., Paul, A., Chen, K. E., Tanneti, N., and McKim, K. S. (2012). Multiple barriers to nonhomologous DNA end joining during meiosis in Drosophila. Genetics 191, 739–746. doi: 10.1534/genetics.112.140996
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Kapun, M., Fabian, D. K., Goudet, J., and Flatt, T. (2016). Genomic evidence for adaptive inversion clines in Drosophila melanogaster. Mol. Biol. Evol. 33, 1317–1336. doi: 10.1093/molbev/msw016
Kapun, M., and Flatt, T. (2018). The adaptive significance of chromosomal inversion polymorphisms in Drosophila melanogaster. Mol. Ecol. 28, 1263–1282. doi: 10.1111/mec.14871
Karageorgiou, C., Gámez-Visairas, V., Tarrío, R., and Rodríguez-Trelles, F. (2019). Long-read based assembly and synteny analysis of a reference Drosophila subobscura genome reveals signatures of structural evolution driven by inversions recombination-suppression effects. BMC Genomics 20:223.
Katoh, K., Rozewicki, J., and Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinformatics 20, 1160–1166. doi: 10.1093/bib/bbx108
Kaushal, S., and Freudenreich, C. H. (2019). The role of fork stalling and DNA structures in causing chromosome fragility. Genes Chromosomes Cancer 58, 270–283. doi: 10.1002/gcc.22721
Kehrer-Sawatzki, H., Sandig, C. A., Goidts, V., and Hameister, H. (2005). Breakpoint analysis of the pericentric inversion between chimpanzee chromosome 10 and the homologous chromosome 12 in humans. Cytogenet. Genome Res. 108, 91–97. doi: 10.1159/000080806
Kirkpatrick, M. (2010). How and why chromosome inversions evolve. PLoS Biol. 8:e1000501. doi: 10.1371/journal.pbio.1000501
Kirkpatrick, M., and Barton, N. (2006). Chromosome inversions, local adaptation and speciation. Genetics 173, 419–434. doi: 10.1534/genetics.105.047985
Kolb, J., Chuzhanova, N. A., Högel, J., Vasquez, K. M., Cooper, D. N., Bacolla, A., et al. (2009). Cruciform-forming inverted repeats appear to have mediated many of the microinversions that distinguish the human and chimpanzee genomes. Chromosome Res. 17, 469–483.
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Koske, T., and Maynard-Smith, J. (1954). Genetics and cytology of Drosophila subobscura. X. The fifth linkage group. J. Genet. 52, 521–541. doi: 10.1007/BF02985076
Kramara, J., Osia, B., and Malkova, A. (2018). Break-induced replication: the where, the why, and the how. Trends Genet. 34, 518–531. doi: 10.1016/j.tig.2018.04.002
Krimbas, C. B. (1992). “The inversion polymorphism of Drosophila subobscura,” in Drosophila Inversion Polymorphism, eds C. B. Krimbas and J. R. Powell (Boca Raton, FL: CRC Press), 127–220.
Kulakovskiy, I. V., and Makeev, V. J. (2009). Discovery of DNA motifs recognized by transcription factors through integration of different experimental sources. Biophysics 54, 667–674. doi: 10.1134/S0006350909060013
Kunze-Mühl, E., and Müller, E. (1958). Weitere Untersuchungen uber die chromosomale Struktur und die natürlichen Strukturtypen von Drosophila subobscura Coll. Chromosoma 9, 559–570. doi: 10.1007/BF02568093
Kunze-Mühl, E., and Sperlich, D. (1955). Inversionen und chromosomale Strukturtypen bei Drosophila subobscura Coll. Z. Vererbungsl. 87, 65–84. doi: 10.1007/BF00308333
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Lagesen, K., Hallin, P., Rødland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lazzaro, A., Hilfiker, D., and Zeyer, J. (2015). Structures of microbial communities in alpine soils: seasonal and elevational effects. Front. Microbiol. 6:1330. doi: 10.3389/fmicb.2015.01330
Lazzaro, B. P., and Clark, A. G. (2001). Evidence for recurrent paralogous gene conversion and exceptional allelic divergence in the Attacin genes of Drosophila melanogaster. Genetics 159, 659–671.
Lazzaro, B. P., and Clark, A. G. (2003). Molecular population genetics of inducible antibacterial peptide genes in Drosophila melanogaster. Mol. Biol. Evol. 20, 914–923. doi: 10.1093/molbev/msg109
Lee, J. A., Carvalho, C. M., and Lupski, J. R. (2007). A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131, 1235–1247. doi: 10.1016/j.cell.2007.11.037
Leigh Brown, A. J., and Ish-Horowicz, D. (1981). Evolution of the 87A and 87C heat-shock loci in Drosophila. Nature 290, 677–682.
Lobachev, K. S., Rattray, A., and Narayanan, V. (2007). Hairpin- and cruciform-mediated chromosome breakage: causes and consequences in eukaryotic cells. Front. Biosci. 12, 4208–4220. doi: 10.2741/2381
Lopes, M., Foiani, M., and Sogo, J. M. (2006). Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell 21, 15–27. doi: 10.1016/j.molcel.2005.11.015
Loukas, M., Krimbas, C. B., and Vergini, Y. (1979). The genetics of Drosophila subobscura populations. IX. Studies on linkage disequilibrium in four natural populations. Genetics 93, 497–523.
Lowe, T. M., and Chan, P. P. (2016). tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57. doi: 10.1093/nar/gkw413
Lowry, D. B., Popovic, D., Brennan, D. J., and Holeski, L. M. (2019). Mechanisms of a locally adaptive shift in allocation among growth, reproduction, and herbivore resistance in Mimulus guttatus. Evolution 73, 1168–1181. doi: 10.1111/evo.13699
Lu, S., Wang, G., Bacolla, A., Zhao, J., Spitser, S., and Vasquez, K. M. (2015). Short inverted repeats are hotspots for genetic instability: relevance to cancer genomes. Cell Rep. 10, 1674–1680. doi: 10.1016/j.celrep.2015.02.039
Mani, R. S., and Chinnaiyan, A. M. (2010). Triggers for genomic rearrangements: insights into genomic, cellular and environmental influences. Nat. Rev. Genet. 11, 819–829. doi: 10.1038/nrg2883
Matzkin, L. M., Merritt, T. J., Zhu, C. T., and Eanes, W. F. (2005). The structure and population genetics of the breakpoints associated with the cosmopolitan chromosomal inversion In(3R)Payne in Drosophila melanogaster. Genetics 170, 1143–1152. doi: 10.1534/genetics.104.038810
Maynard-Smith, J., and Maynard-Smith, S. (1954). Genetics and cytology of Drosophila subobscura. VIII. Heterozygosity, viability and rate of development. J. Genet. 52, 152–164. doi: 10.1007/BF02981496
McBroome, J., Liang, D., and Corbett-Detig, R. (2020). Fine-scale position effects shape the distribution of inversion breakpoints in Drosophila melanogaster. Genome Biol. Evol. doi: 10.1093/gbe/evaa103 [Epub ahead of print].
McKinney, J. A., Wang, G., Mukherjee, A., Christensen, L., Subramanian, S. H. S., Zhao, J., et al. (2020). Distinct DNA repair pathways cause genomic instability at alternative DNA structures. Nat. Commun. 11:236.
Menozzi, P., and Krimbas, C. B. (1992). The inversion polymorphism of D. subobscura revisited: synthetic maps of gene arrangement frequencies and their interpretation. J. Evol. Biol. 5, 625–641. doi: 10.1046/j.1420-9101.1992.5040625.x
Messer, P. W., Ellner, S. P., and Hairston, N. G. Jr. (2016). Can population genetics adapt to rapid evolution? Trends Genet. 32, 408–418. doi: 10.1016/j.tig.2016.04.005
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Orengo, D. J., Puerma, E., Cereijo, U., and Aguadé, M. (2019). The molecular genealogy of sequential overlapping inversions implies both homologous chromosomes of a heterokaryotype in an inversion origin. Sci. Rep. 9:17009.
Orengo, D. J., Puerma, E., Papaceit, M., Segarra, C., and Aguadé, M. A. (2015). Molecular perspective on a complex polymorphic inversion system with cytological evidence of multiply reused breakpoints. Heredity 114, 610–618. doi: 10.1038/hdy.2015
Paaby, A. B., Bergland, A. O., Behrman, E. L., and Schmidt, P. S. (2014). A highly pleiotropic amino acid polymorphism in the Drosophila insulin receptor contributes to life-history adaptation. Evolution 68, 3395–3409. doi: 10.1111/evo.12546
Papaceit, M., Segarra, C., and Aguadé, M. (2012). Structure and population genetics of the breakpoints of a polymorphic inversion in Drosophila subobscura. Evolution 67, 66–79.
Parmesan, C. (2006). Ecological and evolutionary responses to recent climate change. Annu. Rev. Ecol. Evol. Syst. 37, 637–669. doi: 10.1146/annurev.ecolsys.37.091305.110100
Pegueroles, C., Aquadro, C. F., Mestres, F., and Pascual, M. (2013). Gene flow and gene flux shape evolutionary patterns of variation in Drosophila subobscura. Heredity 110, 520–529. doi: 10.1038/hdy.2012.118
Pegueroles, C., Araúz, P. A., Pascual, M., and Mestres, F. (2010a). A recombination survey using microsatellites: the O chromosome of Drosophila subobscura. Genetica 138, 795–804.
Pegueroles, C., Ferrés-Coy, A., Martí-Solano, M., Aquadro, C. F., Pascual, M., and Mestres, F. (2016). Inversions and adaptation to the plant toxin ouabain shape DNA sequence variation within and between chromosomal inversions of Drosophila subobscura. Sci. Rep. 6:23754. doi: 10.1038/srep23754
Pegueroles, C., Ordoñez, V., Mestres, F., and Pascual, M. (2010b). Recombination and selection in the maintenance of the adaptive value of inversions. J. Evol. Biol. 23, 2709–2717. doi: 10.1111/j.1420-9101.2010.02136.x
Powell, J. R. (1997). Progress and Prospects in Evolutionary Biology: The Drosophila Model. Oxford: Oxford University Press.
Prazeres da Costa, O., González, J., and Ruiz, A. (2009). Cloning and sequencing of the breakpoint regions of inversion 5g fixed in Drosophila buzzatii. Chromosoma 118, 349–360. doi: 10.1007/s00412-008-0201-5
Prevosti, A., Ribo, G., Serra, L., Aguadé, M., Balaña, J., Monclus, M., et al. (1988). Colonization of America by Drosophila subobscura: experiment in natural populations that supports the adaptive role of chromosomal-inversion polymorphism. Proc. Natl. Acad. Sci. U.S.A. 85, 5597–5600. doi: 10.1073/pnas.85.15.5597
Puerma, E., Orengo, D. J., and Aguadé, M. (2016a). Multiple and diverse structural changes affect the breakpoint regions of polymorphic inversions across the Drosophila genus. Sci. Rep. 6:36248. doi: 10.1038/srep36248
Puerma, E., Orengo, D. J., and Aguadé, M. (2016b). The origin of chromosomal inversions as a source of segmental duplications in the Sophophora subgenus of Drosophila. Sci. Rep. 6:30715. doi: 10.1038/srep30715
Puerma, E., Orengo, D. J., and Aguadé, M. (2017). Inversion evolutionary rates might limit the experimental identification of inversion breakpoints in non-model species. Sci. Rep. 7:17281.
Puerma, E., Orengo, D. J., Cruz, F., Gómez-Garrido, J., Librado, P., Salguero, D., et al. (2018). The high-quality genome sequence of the oceanic island endemic species Drosophila guanche reveals signals of adaptive evolution in genes related to flight and genome stability. Genome Biol. Evol. 10, 1956–1969. doi: 10.1093/gbe/evy135
Puerma, E., Orengo, D. J., Salguero, D., Papaceit, M., Segarra, C., and Aguadé, M. (2014). Characterization of the breakpoints of a polymorphic inversion complex detects strict and broad breakpoint reuse at the molecular level. Mol. Biol. Evol. 31, 2331–2341. doi: 10.1093/molbev/msu177
Puig-Giribets, M., Guerreiro, M. P. G., Santos, M., Ayala, F. J., Tarrío, R., and Rodríguez-Trelles, F. (2019). Chromosomal inversions promote genomic islands of concerted evolution of Hsp70 genes in the Drosophila subobscura species subgroup. Mol. Ecol. 28, 1316–1332. doi: 10.1111/mec.14511
Ranz, J. M., Maurin, D., Chan, Y. S., von Grotthuss, M., Hillier, L. W., Roote, J., et al. (2007). Principles of genome evolution in the Drosophila melanogaster species group. PLoS Biol. 5:e152. doi: 10.1371/journal.pbio.0050152
Reese, M. G. (2001). Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput. Chem. 26, 51–56.
Regan, J. C., Froy, H., Walling, C. A., Moatt, J. P., and Nussey, D. H. (2020). Dietary restriction and insulin-like signalling pathways as adaptive plasticity: a synthesis and re-evaluation. Funct. Ecol. 34, 107–128. doi: 10.1111/1365-2435.13418
Rego, C., Balanyà, J., Fragata, I., Matos, M., Rezende, E. L., and Santos, M. (2010). Clinal patterns of chromosomal inversion polymorphisms in Drosophila subobscura are partly associated with thermal preferences and heat stress resistance. Evolution 64, 385–397. doi: 10.1111/j.1558-5646.2009.00835.x
Rezende, E. L., Balanyà, J., Rodríguez-Trelles, F., Rego, C., Fragata, I., Matos, M., et al. (2010). Climate change and chromosomal inversions in Drosophila subobscura. Clim. Res. 43, 103–114. doi: 10.1007/s10709-018-0035-x
Richards, S., Liu, Y., Bettencourt, B. R., Hradecky, P., Letovsky, S., and Nielsen, R. (2005). Comparative genome sequencing of Drosophila pseudoobscura: chromosomal, gene, and cis-element evolution. Genome Res. 15, 1–18. doi: 10.1101/gr.3059305
Rivera, J., Keränen, S. V. E., Gallo, S. M., and Halfon, M. S. (2019). REDfly: the transcriptional regulatory element database for Drosophila. Nucleic Acids Res. 47, D828–D834. doi: 10.1093/nar/gky957
Rodríguez-Trelles, F. (2003). Seasonal cycles of allozyme-by-chromosomal-inversion gametic disequilibrium in Drosophila subobscura. Evolution 57, 839–848. doi: 10.1111/j.0014-3820.2003.tb00295.x
Rodríguez-Trelles, F., Alvarez, G., and Zapata, C. (1996). Time-series analysis of seasonal changes of the O inversion polymorphism of Drosophila subobscura. Genetics 142, 179–187.
Rodríguez-Trelles, F., and Rodríguez, M. A. (1998). Rapid micro-evolution and loss of chromosomal diversity in Drosophila in response to climate warming. Evol. Ecol. 12, 829–838. doi: 10.1023/A:1006546616462
Rodríguez-Trelles, F., and Rodríguez, M. A. (2007). Comment on ‘Global genetic change tracks global climate warming in Drosophila subobscura’. Science 315:1497. doi: 10.1126/science.1136298
Rodríguez-Trelles, F., and Rodríguez, M. A. (2010). Measuring evolutionary responses to global warming: cautionary lessons from Drosophila. Insect Conserv. Divers. 3, 44–50. doi: 10.1111/j.1752-4598.2009.00071.x
Rodríguez-Trelles, F., Tarrío, R., and Santos, M. (2013). Genome-wide evolutionary response to a heat wave in Drosophila. Biol. Lett. 9:20130228. doi: 10.1098/rsbl.2013.0228
Romero-Olivares, A. L., Allison, S. D., and Treseder, K. K. (2017). Soil microbes and their response to experimental warming over time: a meta-analysis of field studies. Soil Biol. Biochem. 107, 32–40. doi: 10.1016/j.soilbio.2016.12.026
Said, I., Byrne, A., Serrano, V., Cardeno, C., Vollmers, C., and Corbett-Detig, R. B. (2018). Linked genetic variation and not genome structure causes widespread differential expression associated with chromosomal inversions. Proc. Natl. Acad. Sci. U.S.A. 115, 5492–5497. doi: 10.1073/pnas.1721275115
Sakofsky, C. J., Ayyar, S., Deem, A. K., Chung, W. H., Ira, G., and Malkova, A. (2015). Translesion polymerases drive microhomology-mediated break-induced replication leading to complex chromosomal rearrangements. Mol. Cell 60, 860–872. doi: 10.1016/j.molcel.2015.10.041
Santos, J., Serra, L., Solé, E., and Pascual, M. (2010). FISH mapping of microsatellite loci from Drosophila subobscura and its comparison to related species. Chromosome Res. 18, 213–226.
Santos, M., Céspedes, W., Balanyà, J., Trotta, V., Calboli, F. C., Fontdevila, A., et al. (2005). Temperature-related genetic changes in laboratory populations of Drosophila subobscura: evidence against simple climatic-based explanations for latitudinal clines. Am. Nat. 165, 258–273. doi: 10.1086/427093
Savage, J. R. (1976). Classification and relationships of induced chromosomal structual changes. J. Med. Genet. 13, 103–122. doi: 10.1136/jmg.13.2.103
Schwenke, R. A., Lazzaro, B. P., and Wolfner, M. F. (2016). Reproduction-immunity trade-offs in insects. Annu. Rev. Entomol. 61, 239–256.
Scully, R., Panday, A., Elango, R., and Willis, N. A. (2019). DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 20, 698–714.
Sebastián, A., and Contreras-Moreira, B. (2014). FootprintDB: a database of transcription factors with annotated cis elements and binding interfaces. Bioinformatics 30, 258–265. doi: 10.1093/bioinformatics/btt663
Senger, K., Armstrong, G. W., Rowell, W. J., Kwan, J. M., Markstein, M., and Levine, M. (2004). Immunity regulatory DNAs share common organizational features in Drosophila. Mol. Cell 13, 19–32.
Seppey, M., Manni, M., and Zdobnov, E. M. (2019). BUSCO: assessing genome assembly and annotation completeness. Methods Mol. Biol. 1962, 227–245. doi: 10.1007/978-1-4939-9173-0_14
Shigyo, N., Umeki, K., and Hirao, T. (2019). Seasonal dynamics of soil fungal and bacterial communities in cool-temperate montane forests. Front. Microbiol. 10:1944. doi: 10.3389/fmicb.2019.01944
Shtraizent, N., DeRossi, C., Nayar, S., Sachidanandam, R., Katz, L. S., Prince, A., et al. (2017). MPI depletion enhances O-GlcNAcylation of p53 and suppresses the Warburg effect. eLife 6:e22477. doi: 10.7554/eLife.22477
Slade, J. D., and Staveley, B. E. (2016). Enhanced survival of Drosophila Akt1 hypomorphs during amino-acid starvation requires foxo. Genome 59, 87–93.
Smit, A. F. A., Hubley, R., and Green, P. (2013/2015). RepeatMasker Open-4.0. Available online at: http://www.repeatmasker.org (accessed November 15, 2019).
Smith, C., Llorente, B., and Symington, L. (2007). Template switching during break-induced replication. Nature 447, 102–105. doi: 10.1038/nature05723
Soderlund, C., Bomhoff, M., and Nelson, W. M. (2011). SyMAP v3.4: a turnkey synteny system with application to plant genomes. Nucleic Acids Res. 39:e68. doi: 10.1093/nar/gkr123
Sogo, J. M., Lopes, M., and Foiani, M. (2002). Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297, 599–602. doi: 10.1126/science.1074023
Solé, E., Balanyà, J., Sperlich, D., and Serra, L. (2002). Long-term changes in the chromosomal inversion polymorphism of Drosophila subobscura. I. Mediterranean populations from southwestern Europe. Evolution 56, 830–835. doi: 10.1111/j.0014-3820.2002.tb01393.x
Sperlich, D., Feuerbach-Mravlag, H., Lange, P., Michaelidis, A., and Pentzos-Daponte, A. (1977). Genetic load and viability distribution in central and marginal populations of Drosophila subobscura. Genetics 86, 835–848.
Sun, X., Qun, Y., and Xia, X. (2013). An improved implementation of effective number of codons (Nc). Mol. Biol. Evol. 30, 191–196. doi: 10.1093/molbev/mss201
Sunder, S., and Wilson, E. T. (2019). Frequency of DNA end joining in trans is not determined by the predamage spatial proximity of double-strand breaks in yeast. Proc. Natl. Acad. Sci. U.S.A. 116, 9481–9490. doi: 10.1073/pnas.1818595116
Tajima, F. (1993). Simple methods for testing the molecular evolutionary clock hypothesis. Genetics 135, 599–607.
Teleman, A. A. (2010). Molecular mechanisms of metabolic regulation by insulin in Drosophila. Biochem. J. 425, 13–26.
The Gene Ontology Consortium. (2017). Expansion of the gene ontology knowledgebase and resources. Nucleic Acids Res. 45, D331–D338. doi: 10.1093/nar/gkw1108
Tremblay-Belzile, S., Lepage, É., Zampini, É., and Brisson, N. (2015). Short-range inversions: rethinking organelle genome stability: template switching events during DNA replication destabilize organelle genomes. Bioessays 37, 1086–1094. doi: 10.1002/bies.201500064
Voineagu, I., Narayanan, V., Lobachev, K. S., and Mirkin, S. M. (2008). Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proc. Natl. Acad. Sci. U.S.A. 105, 9936–9941. doi: 10.1073/pnas.0804510105
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, G., Christensen, L. A., and Vasquez, K. M. (2006). Z-DNA-forming sequences generate large-scale deletions in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 103, 2677–2682. doi: 10.1073/pnas.0511084103
Wang, G., and Vasquez, K. M. (2006). Non-B DNA structure-induced genetic instability. Mutat. Res. 598, 103–119.
Weirauch, M. T., Yang, A., Albu, M., Cote, A. G., Montenegro-Montero, A., Drewe, P., et al. (2014). Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443. doi: 10.1016/j.cell.2014.08.009
Wellenreuther, M., and Bernatchez, L. (2018). Eco-evolutionary genomics of chromosomal inversions. Trends Ecol. Evol. 33, 427–440. doi: 10.1016/j.tree.2018.04.002
Wesley, C. S., and Eanes, W. F. (1994). Isolation and analysis of the breakpoint sequences of chromosome inversion In(3L)Payne in Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 91, 3132–3136. doi: 10.1073/pnas.91.8.3132
Zhang, F., Khajavi, M., Connolly, A. M., Towne, C. F., Batish, S. D., and Lupski, J. R. (2009). The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat. Genet. 41, 849–853. doi: 10.1038/ng.399
Zhao, J., Bacolla, A., Wang, G., and Vasquez, K. M. (2010). Non-B DNA structure-induced genetic instability and evolution. Cell. Mol. Life Sci. 67, 43–62.
Zollinger, E. (1950). Ein strukturell homozygoter Stamm von Drosophila subobscura aus einer Wild-population. Arch. Klaus-Stiftg 25, 33–35.
Keywords: non-B DNA, genome fragility, foxo (forkhead box subgroup O), Akt1 (serine/threonine–protein kinase B), Attacin antibacterial genes, immunometabolism, thermal adaptation, seasonal selection
Citation: Karageorgiou C, Tarrío R and Rodríguez-Trelles F (2020) The Cyclically Seasonal Drosophila subobscura Inversion O7 Originated From Fragile Genomic Sites and Relocated Immunity and Metabolic Genes. Front. Genet. 11:565836. doi: 10.3389/fgene.2020.565836
Received: 26 May 2020; Accepted: 09 September 2020;
Published: 09 October 2020.
Edited by:
Pedro Simões, University of Lisbon, PortugalReviewed by:
Stephen Wade Schaeffer, Pennsylvania State University (PSU), United StatesCopyright © 2020 Karageorgiou, Tarrío and Rodríguez-Trelles. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charikleia Karageorgiou, Y2hhcmlrbGVpYS5rYXJhZ2Vvcmdpb3VAdWFiLmNhdA==; Rosa Tarrío, cm9zYW1hcmlhLnRhcnJpb0B1YWIuY2F0; Francisco Rodríguez-Trelles, ZnJhbmNpc2Nvam9zZS5yb2RyaWd1ZXp0cmVsbGVzQHVhYi5jYXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.