
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 09 December 2020
Sec. Genomics of Plants and the Phytoecosystem
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.560368
This article is part of the Research Topic Comparative Genomics and Functional Genomics Analyses in Plants View all 37 articles
 Peninah Cheptoo Rono1,2,3†
Peninah Cheptoo Rono1,2,3† Xiang Dong1,2,3†Jia-Xin Yang1,2†Fredrick Munyao Mutie1,2,3Millicent A. Oulo1,2,3Itambo Malombe4
Xiang Dong1,2,3†Jia-Xin Yang1,2†Fredrick Munyao Mutie1,2,3Millicent A. Oulo1,2,3Itambo Malombe4 Paul M. Kirika4
Paul M. Kirika4 Guang-Wan Hu1,2*
Guang-Wan Hu1,2* Qing-Feng Wang1,2
Qing-Feng Wang1,2The genus Alchemilla L., known for its medicinal and ornamental value, is widely distributed in the Holarctic regions with a few species found in Asia and Africa. Delimitation of species within Alchemilla is difficult due to hybridization, autonomous apomixes, and polyploidy, necessitating efficient molecular-based characterization. Herein, we report the initial complete chloroplast (cp) genomes of Alchemilla. The cp genomes of two African (Afromilla) species Alchemilla pedata and Alchemilla argyrophylla were sequenced, and phylogenetic and comparative analyses were conducted in the family Rosaceae. The cp genomes mapped a typical circular quadripartite structure of lengths 152,438 and 152,427 base pairs (bp) in A. pedata and A. argyrophylla, respectively. Alchemilla cp genomes were composed of a pair of inverted repeat regions (IRa/IRb) of length 25,923 and 25,915 bp, separating the small single copy (SSC) region of 17,980 and 17,981 bp and a large single copy (LSC) region of 82,612 and 82,616 bp in A. pedata and A. argyrophylla, respectively. The cp genomes encoded 114 unique genes including 88 protein-coding genes, 37 transfer RNA (tRNA) genes, and 4 ribosomal RNA (rRNA) genes. Additionally, 88 and 95 simple sequence repeats (SSRs) and 37 and 40 tandem repeats were identified in A. pedata and A. argyrophylla, respectively. Significantly, the loss of group II intron in atpF gene in Alchemilla species was detected. Phylogenetic analysis based on 26 whole cp genome sequences and 78 protein-coding gene sequences of 27 Rosaceae species revealed a monophyletic clustering of Alchemilla nested within subfamily Rosoideae. Based on a protein-coding region, negative selective pressure (Ka/Ks < 1) was detected with an average Ka/Ks value of 0.1322 in A. argyrophylla and 0.1418 in A. pedata. The availability of complete cp genome in the genus Alchemilla will contribute to species delineation and further phylogenetic and evolutionary studies in the family Rosaceae.
The genus Alchemilla L. (Rosaceae), composed of more than 1,000 species, is important for its ornamental and medicinal values (Kaya et al., 2012; Boroja et al., 2018). It is widely distributed in the Holarctic region with high species richness in west Eurasia and few species found in montane tropical Asia, Madagascar, South Africa, and East African mountains (Izmailow, 1981; Fröhner, 1995; Gehrke et al., 2008). Alchemilla, together with Aphanes and Lachemilla, is classified under subfamily Rosoideae, tribe Potentilleae and subtribe Alchemillinae (Rothmaler, 1937; Kalkman, 2004; Notov and Kusnetzova, 2004; Soják, 2008; Xiang et al., 2016). Initially, Alchemilla was categorized under tribe Sanguisorbinae by Hutchinson (1964) due to superficial similarity of floral traits but was later reclassified as Potentilleae following Schulze-Menz’s (1964) observational concern on the anther structure and further confirmation through molecular characterization using nuclear ribosomal DNA and trnL/F region of chloroplast (cp) DNA (Eriksson et al., 2003). Morphologically, Alchemilla is distinguished from other Rosaceae genera by the silvery-silky white hair covering on the stems and the leaf surface, achene fruits that are hidden within the calyx tube, inconspicuous individuality, and small but fairly showy inflorescence (Graham, 1960; Faghir et al., 2014; Gehrke et al., 2016). Notwithstanding, circumscription within the genus remain poorly understood due to hybridization, autonomous apomixes, and polyploidization dominant in Alchemilla species (Izmailow, 1982; Czapik, 1996; Gehrke et al., 2008). The genus Alchemilla is commonly used as perfect example of apomictic traits combined with morphological polymorphism in Rosaceae (Czapik, 1996; Bicknell and Koltunow, 2004; Hayirhoglu-Ayaz et al., 2006; Salamone et al., 2013). This has resulted in the existence of diverse micro-species and species complexes with variable indumentum, unstable flower characteristic structure, and heteroblastic plasticity, making Alchemilla a taxonomically difficult group (Hörandl, 2004; Hayirhoglu-Ayaz et al., 2006; Lundberg et al., 2009). This necessitated the need for complete cp genome in the genus Alchemilla to help resolve the taxonomic and phylogenetic uncertainties between its species.
Chloroplast (cp) is found in the cytoplasmic matrix of a plant cell and plays significant roles in photosynthesis, carbon fixation, and synthesis of starch, fatty acids, and amino acids (Daniell et al., 2016). It is a semi-autonomous organelle, similar to the nuclei and the mitochondria, essential in the transfer and expression of the plant’s genetic material (Wolfe et al., 1987). The cp has its own double-stranded circular genome whose size in most terrestrial plants ranges between 120 and 180 kb, encoding about 110–130 different genes in a highly conserved order (Shinozaki et al., 1986; Liu et al., 2017). Based on its genome content and the ultrastructure features, the cp traces its origin to free-living cyanobacteria through a single event of endosymbiosis (Gray, 1989; Keeling, 2004). The cp genome has a characteristic quadripartite structure comprising two identical copies of inverted repeat (IRa/IRb) regions separated by the large single copy (LCS) and small single copy (SSC) regions (Zhao M.-L. et al., 2018). The two IR regions are paramount in defining the size and structure of the cp genome of land plants (Palmer et al., 1988). Variations in genome size could consequently be due to expansion/contraction or loss of one of the IR regions in some species. For instance, the loss of one copy of IR in Taxus chinensis var. mairea resulted in the reduction of its genome size (Zhang et al., 2014). The uniqueness of the cp is evident in its maternal inheritance, small size, conserved sequences, and simple structure (Kress et al., 2005; Parks et al., 2009; Liu et al., 2017). The evolutionary process in angiosperms is dependent on the conserved structure, gene content, and organization of the cp genome (Doyle et al., 1992; Saski et al., 2005). This makes it an appropriate candidate for plant taxonomy, and comparative genomic and evolutionary studies. The advent of the next-generation DNA sequencing technology (Shendure and Ji, 2008) has magnified the rate of cp genome sequencing reports since the process is simpler, cost-effective and fast, resulting in the expansion of the cp genetics and genomics (Daniell et al., 2016). Since the report of the first cp genome sequence of tobacco (Nicotiana tabacum) (Shinozaki et al., 1986), several species in the Rosaceae have had their complete cp genome deposited to the National Center for Biotechnology Information (NCBI) organelle genome database1 (Salamone et al., 2013; Zhao Y. et al., 2018; Wang et al., 2020). However, none of the Alchemilla species genome has been sequenced to date.
In this study, the initial cp genomes of the genus Alchemilla are reported in Alchemilla pedata and Alchemilla argyrophylla. First, we obtained the complete cp genome of the two species and characterized the structure, gene content, and organization of each genome. Second, we establish the codon usage frequencies, simple sequence repeats (SSRs), regions of high sequence divergence, and nucleotide substitution rates. Finally, phylogenetic position was evaluated by comparative analysis based on 24 complete cp genomes and 78 protein-coding gene (PCG) sequences of Rosaceae species. Our results provide a reference for the resolution of Alchemilla species classification and facilitate elucidation of evolutionary and phylogenetic relationships in Rosaceae.
Plant materials of two Alchemilla species of Alchemilla pedata (voucher number SAJIT-001337) and Alchemilla argyrophylla (SAJIT-002399) were collected from Mt. Kenya, Kenya. Young leaves were sampled and immediately preserved using silica gel in plastic bags (Chase and Hills, 1991). The voucher specimens were deposited at the East African Herbarium (EA) in the National Museums of Kenya and at the Herbarium of Wuhan Botanical Garden, CAS (HIB) (China). The total genomic DNA of the two species was extracted from 0.5 g of the silica dried leaves using modified cetyltrimethylammonium bromide (CTAB) protocol (Doyle, 1991). Results were then sequenced based on the Illumina paired end technology platform at the Novogen Company (Beijing, China).
Genome assembly was performed using GetOrganelle v1.6.2d with default parameters (Jin et al., 2020). The GetOrganelle first filtered plastid-like reads, conducted the de novo assembly, purified the assembly, and finally generated the complete plastid genomes (Camacho et al., 2009; Bankevich et al., 2012; Langmead and Salzberg, 2012). K-mer gradients for a mean and maximum 150-bp reads were set to as “-k 21, 45, 65, 85,105” for both species. Bandage (Wick et al., 2015) was used to visualize the final assembly graphs to authenticate the automatically generated plastid genome. The best fit k-mer of 45 was selected for use in a subsequent analysis of the genomes. The quality of the newly assembled genomes was evaluated on read level basis by aligning the trimmed raw reads to the de novo assemblies using Geneious mapper, Geneious version 9.1.4 (Kearse et al., 2012) with medium- to low-sensitivity option and iteration up to five times (Köhler et al., 2020). Gene annotation was conducted using Plastid Genome Annotator (PGA) (Qu et al., 2019) with an annotated plastome Amborella trichopoda (GenBank accession no. GCA_000471905.1) as the initial reference genome. Further annotation confirmation was performed with published genomes of Fragaria virginiana (JN884817) and Fragaria vesca subsp. vesca (KC507760) in the Rosacea family. Geneious was used to manually correct and complement problematic annotations. The whole genome circular map was drawn using Organelle Genome DRAW software (Lohse et al., 2013).
The cp genome sequence of the A. pedata and A. argyrophylla was compared with that of eight other Rosaceae species. This included Dasiphora fruticosa (MF683841), Fragaria iinumae (KC507759), Fragaria nipponica (KY769125), Fragaria pentaphylla (KY434061), Fragaria orientalis (KY769126), Fragaria chiloensis (JN884816), F. virginiana (JN884817), Fragaria mandshurica (KC507760), F. vesca (KC507760), Rosa multiflora (NC_039989), Rosa odorata var. gigantea (KF753637), and Hagenia abyssinica (KX008604) retrieved from GenBank database. The mVISTA (Mayor et al., 2000) program under the shuffle-LAGAN alignment strategy (Frazer et al., 2004) was applied to compare all the complete genomes with A. argyrophylla as reference. The contraction and expansion of the IR boundaries of five Rosaceae species were visualized using the IRscope software (Amiryousefi et al., 2018).
REPuter online program (Kurtz et al., 2001) was used to identify long repeat sequences (forward, reverse, complementary, and palindromic). Repeat sequence locations and sizes in cp genomes were visualized with a minimal criterion of 30 bp, a hamming distance of 3, and less than 90% identity between two repeat copies. Tandem repeat finder (Benson, 1999) was used to identify tandem repeats in the two species A. pedata and A. argyrophylla cp genomes with default alignment parameters of match, mismatch, and insertions and deletions (indels) of 2, 7, and 7, respectively. SSRs were detected using the perl script MicroSAtellite (MISA) (Thiel et al., 2003) with a size motif of one to six nucleotides and a threshold of 10, 5, 5, 3, and 3 for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide, respectively. The codon usage frequency and relative synonymous codon usage (RSCU) of the two species were conducted based on 88 PCGs using MEGA 5 (Tamura et al., 2011).
To evaluate the evolutionary rate variation within the Alchemilla species, 78 protein-coding regions within the cp genomes were explored with F. virginiana as reference species. PCGs were extracted using Geneious var. 9.1.4. Gaps and stop codons were manually removed, and the seven sequences were separately aligned using MAFFT v7. 308 (Katoh and Standley, 2013). The aligned files were converted into AXT format using the parseFastaIntoAXT.pl Perl script2. The non-synonymous (Ka) and synonymous (Ks) substitution rates as well as Ka/Ks ratio of each gene were estimated using the software KaKs_calculator 1.2 using the default model averaging (MA) method (Zhang et al., 2006). Taking into consideration that KaKs_calculator uses model averaging estimates in site selection, we implored the impact of site selection in 78 genes of five species phylogenetically related to Alchemilla. Positive selective pressure within shared genes of the seven species of subfamily Rosoideae was evaluated using PAML v4.7 (Yang, 2007) package implemented in EasyCodeML software (Gao et al., 2019). Non-synonymous (dN) and synonymous substitution (dS) substitution rates, and their ratio (ω = dN/dS) were calculated based on four site-specific models (M0 vs. M3, M1a vs. M2a, M7 vs. M8, and M8a vs. M8) with likelihood ratio test (LRT) threshold of p < 0.05 elucidating adaptation signatures within the genome. The models permit dN/dS variation within sites while keeping the ω ratio fixed within branches. Selective pressure analysis was conducted along ML tree in plain Newick format based on protein-coding sites used in the generation of phylogenetic relationship of the selected seven species. Here, individual coding DNA sequences (CDSs) were aligned in correspondence to their amino acids and their selection evaluated based on both ω and LRT values.
Phylogenetic relationship analysis was done based on two data sets: (1) complete cp genome sequences (genomic tree) and (2) PCGs (CDS tree). The complete cp genomes of A. pedata and A. argyrophylla obtained from this study and other 24 cp genomes of Rosaceae species downloaded from NCBI database were inferred for the genomic tree (Supplementary Table S1). Multiple sequence alignment was performed using MAFFT v7.308 with default parameters setting. Phylogenetic relationship reconstructions were performed based on maximum-likelihood (ML) analysis using the program IQ-Tree v.6.1 (Nguyen et al., 2015) with 1,000 bootstrap replications. The best fit model TVM+I+G4 (Kalyaanamoorthy et al., 2017) was chosen according to Bayesian information criterion (BIC). The CDS tree was constructed by ML, PhyML, and BI methods based on 78 PCGs shared by all the 27 species under comparative evaluation. Gene sequences were extracted and aligned individually using Mega 7 and concatenated into a single file using PhyloSuite (Zhang et al., 2020). The ML CDS tree phylogenies were inferred using IQ-Tree with the best fit model GTR+F+R2 from ModelFinder in accordance to Akaike information criterion (AIC). Bayesian inference (BI) phylogenetic relationship of our taxa was constructed using MrBayes 3.2.6 (Ronquist et al., 2012) in PhyloSiute under GT+F+I+G4 best fit model from ModelFinder (two runs, 200,000 generations) following the AIC. The online program PhyMl vl 3.0 (Guindon et al., 2010) was used to infer phylogenetic relationship following GTR+G+I model selected by sms (Lefort et al., 2017). The constructed tree was visualized using FigTree version 1.4.4 (Rambaut, 2018).
The total number of assembled reads was 3,383,185 and 1,158,253 with an average genome coverage depth of 3,299.6 and 1,133.5 in Alchemilla pedata and Alchemilla argyrophylla, respectively. The complete cp genomes display a typical circular structure with DNA sizes 152,438 and 152,427 bp for A. pedata and A. argyrophylla, respectively (Figure 1A). The quadripartite structure is composed of 82,612 bp in the LSC region, 17,980 bp in the SSC region, and the two IR regions made up of 25,923 bp each in A. pedata. A. argyrophylla has 82,616 bp in the LSC, 17,981 bp in the SSC region, and the two IR regions, each having 25,915 bp in length. The overall guanine–cytosine (GC) content for both Alchemilla species are 37% with 42.7, 34.9, and 30.4% in the IRs, LSC, and SSC, respectively. These are similar to other complete cp genome sequences of Rosaceae species obtained from NCBI database ranging between 36.6 and 37.1% (Table 1). To assess for any mis-assemblies, the raw reads were aligned against the de novo assembled genomes. The high per-base read coverages, plotted against the genomes position for each of the assemblies (Figures 1B,C), reveal the quality of our assembly. The annotated cp genomes were deposited in the GenBank database with the following accession numbers; A. argyrophylla MT382661 and A. pedata MT382662.
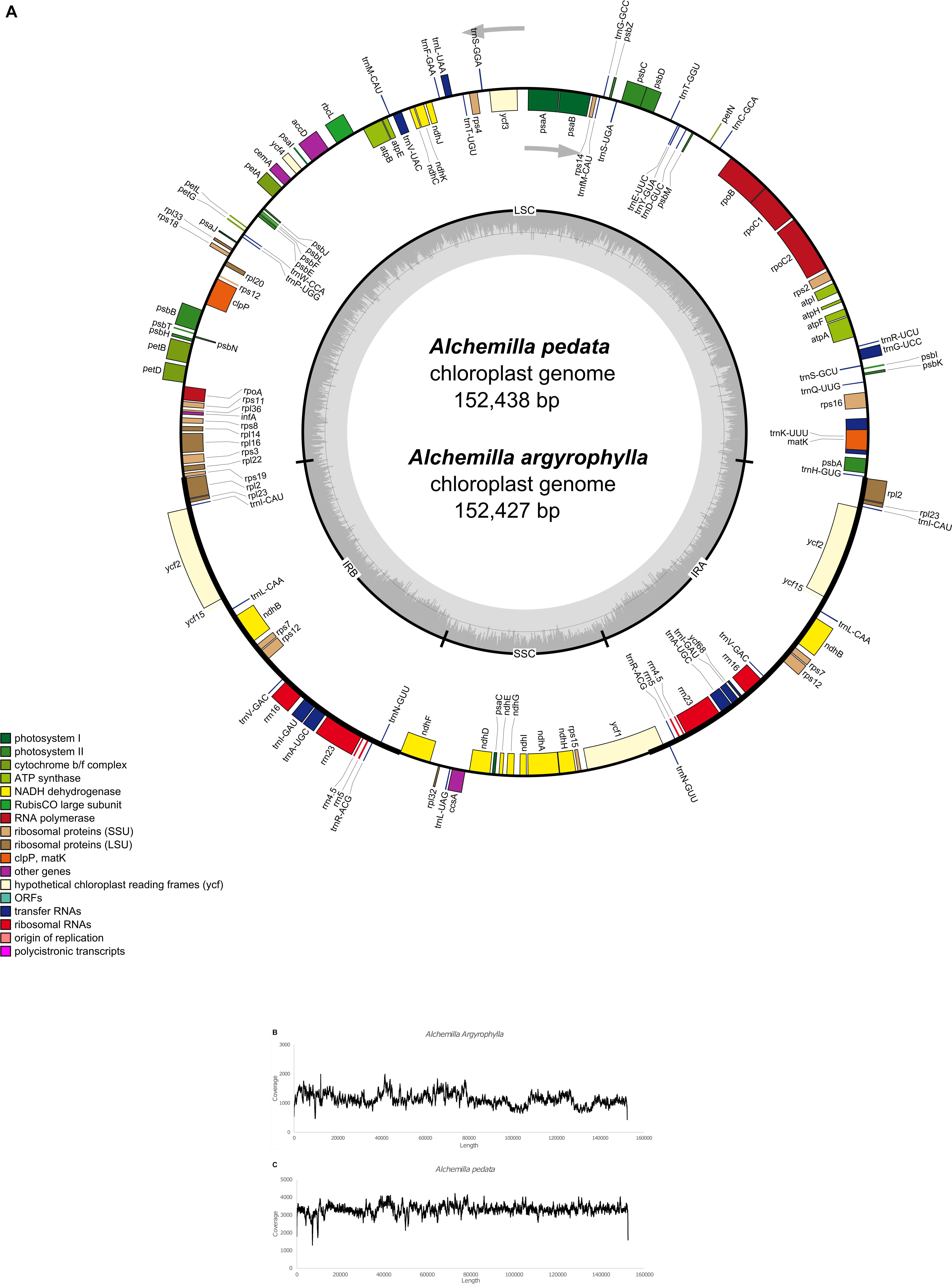
Figure 1. (A) The genome map of Alchemilla pedata and Alchemilla argyrophylla drawn using Organelle Genome DRAW software (Lohse et al., 2013). The genes outside the circle are transcribed in the counterclockwise direction, while those inside are transcribed in the clockwise direction. The colored bars indicate different functional groups. The lighter gray color denotes the AT content, while the gray area in the inner circle corresponds to the guanine–cytosine (GC) content of the genome. IRA, inverted repeat region A; IRB, inverted repeat region B; LSC, large singe copy; SSC, small single copy. (B) A. argyrophylla and (C) A. pedata graphs of read level coverage plots. The de novo assembly reads were trimmed, aligned, and mapped to their chloroplast genome after initial alignment with Bowtie in GetOrganelle using Geneious mapper (Kearse et al., 2012).
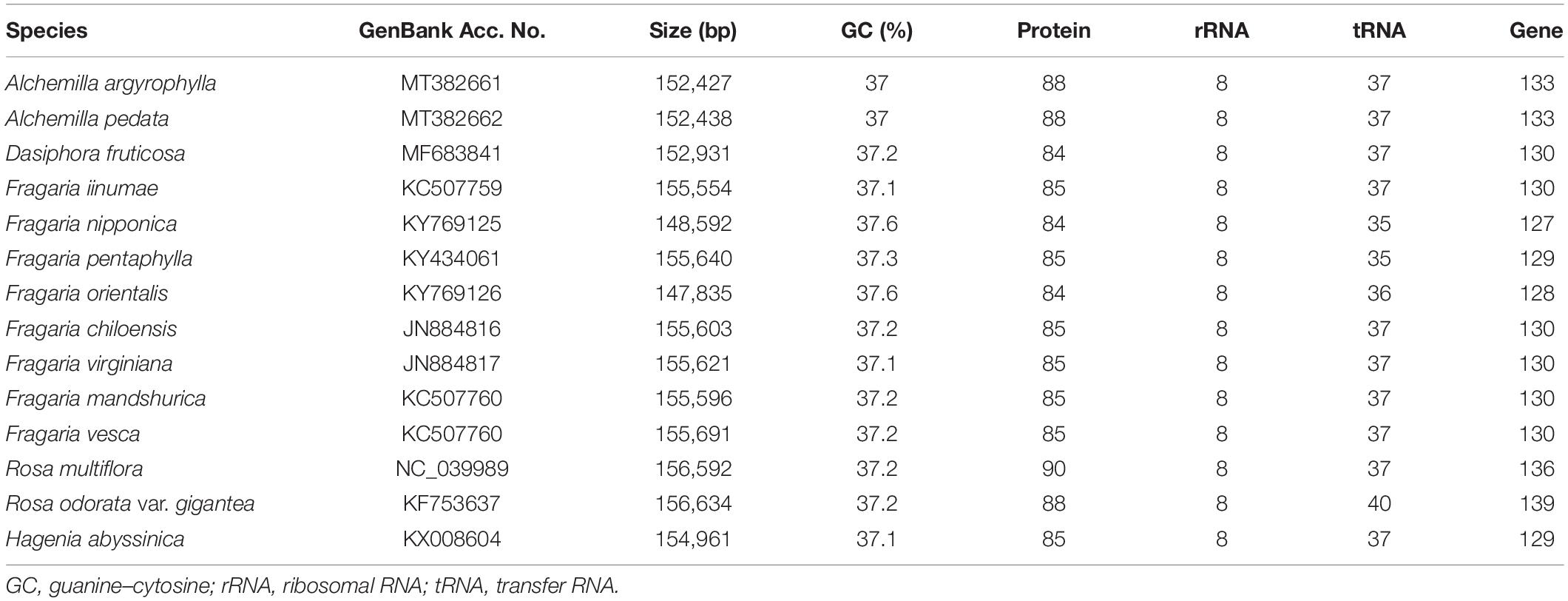
Table 1. Comparison of chloroplast genomes of 14 Rosaceae species.
A total of 114 unique genes were annotated including 88 PCGs, 37 transfer RNA (tRNA) genes, and 4 ribosomal RNA (rRNA) genes in both A. argyrophylla and A. pedata (Table 1). Based on their gene functional category, 59 genes were associated with self-replication while 44 genes are responsible for photosynthesis (Table 2). Similar gene order and genome structure were reported in both Alchemilla species (Figure 1A). The IR regions (IRa and IRb) had 18 duplicate genes comprising seven PCGs (rpl2, rpl23, ycf2, ycf15, ndhB, rps7, and rps12), seven tRNA (trnI-CAU, trnL-CAA, trnV-GAC, trnI-GAC, trnA-UGC, trnR-ACG, and trnN-GUU), and four rRNAs (rrn16, rrn23, rrn4.5, and rrn5). The SSC region had 13 genes of which 12 were PCGs and one tRNA, whereas the LSC had 62 PCGs and 22 tRNA (Figure 1A). In total, 15 genes (trnK-UUU, rps16, trnG-UCC, rpoC1, trnL-AUU, trnV-UAC, petB, petD, rpl16, rpl2, ndhB, trnA-UGC, ndhA, trnA-UGC, and trnI-GAU) had one intron with rpl2 and ndhB duplicated in the IR, whereas two genes, clpP and ycf3, had two introns. The rps12 is a trans-spliced gene, with one exon shared between two introns, in which the 3′ exons were duplicated in the IR regions and the 5′ exon end situated in the LSC region (Table 2). Among the 133 genes, three instances of overlapping sequences were detected in Alchemilla. The psbD and psbC genes shared coding regions (53 bp); ycf68 gene was embedded within trnI-GAU in one of the inverted region (IRa); and matK, which has the longest intron (2,523 bp in A. argyrophylla and 2,528 bp in A. pedata), was embedded in trnK-UUU in the SSC region (Figure 1A).
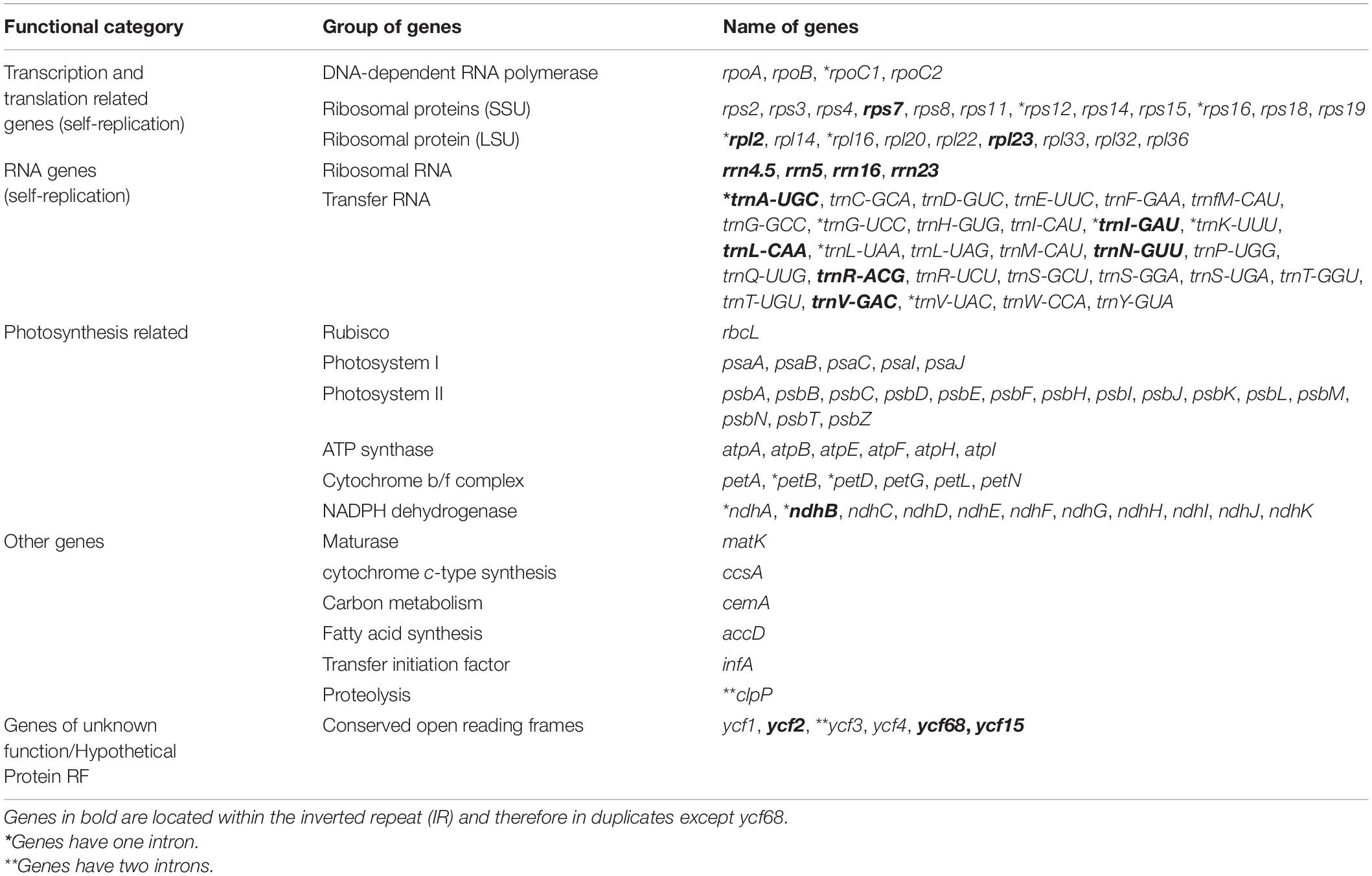
Table 2. Genes present and functional gene category in Alchemilla pedata and Alchemilla argyrophylla chloroplast genome.
The CDSs of the cp genomes were used to estimate the frequency of codon usage of both A. pedata and A. argyrophylla. A total of 22,948 and 22,984 codons encoding 88 genes were detected in A. argyrophylla and A. pedata, respectively. Of all the codons, leucine reported the highest amino acid usage frequency of 10.57% (2,426) in A. argyrophylla and 10.58% (2,432) in A. pedata, whereas cysteine had the lowest amino acid usage frequency of 1.09% (251) in A. argyrophylla and 1.08% (249) in A. pedata (Supplementary Figure S1). The most frequently used codons are AUU (993 and 996), AAA (938 and 940), and GAA (922 and 921) encoding isoleucine, lysine, and glutamic acid in A. argyrophylla and A. pedata, respectively (Supplementary Table S2). Furthermore, RSCU value was estimated in the 88 CDSs in both Alchemilla species. RSCU is the ratio between the expected frequency of use and the actual frequency usage of a particular codon. Codons reporting RSCU value < 1 indicates lower frequency usage than expected, while a score > 1 signifies higher usage frequency (Sharp and Li, 1987; Munyao et al., 2020). In Alchemilla species, apart from the stop codon (UGA), isoleucine (I) codon AUA and leucine (L) codon (CUA) with RSCU value below 1, all the other codons with synonymous codons usage (RSCU > 1) preferred to end with A or U in both A. pedata and A. argyrophylla signifying their preferential codon use. Exceptionally, codon UUG encoding leucine recorded higher bias of RSCU = 1.19 in both species despite ending with G in the third position than did other codons of low frequencies (RSCU < 1) that ended in C or G. Codons AUG (M) and UGG (W) encoding methionine and tryptophan showed no bias (RSCU = 1) (Supplementary Table S2). Our findings are consistent with majority cp genomes of land plants (Cheng et al., 2017). Due to usage frequency variation, RSCU values of the cp genome form a valuable source of evolutionary signature traits resulting from mutation and selection that are essential in studying the evolution of an organism (Morton, 2003; Wang et al., 2018).
Repeat motifs are significant in the computation of phylogenetic and genomic rearrangement (Cavalier-Smith, 2002). In this report, A. pedata reported 36 long repeats comprising four palindromic (P), 14 reverse (R), and 18 forward (F) repeats, whereas A. argyrophylla recorded 42 long repeats composed of 19 P, 8 R, and 15 F repeats (Figure 2). In both species, complementary repeats were not found. This is similar to finding obtained in other Rosaceae species (Gichira et al., 2017). The comparative analysis results revealed that most repeats were between 30 and 40 bp. The longest repeat was a palindromic repeat having 71 bp located in the intergenic spacer (IGS) of the LSC region between trnM-CAU and atpE in both species (Supplementary Table S3). Most repeats were distributed in LSC (non-coding) region, whereas some were found in genes including ndhA, ycf3, ycf1, rpoC1, rpl16, and ndhB. Six repeats were found exclusively in the IR regions, three of them relating to ndhB gene (Supplementary Table S3). The number of tandem repeats was 40 in A. argyrophylla and 37 repeats in A. pedata (Figure 2). In A. argyrophylla, only four repeats were > 30 bp in length, while the rest were between 1 and 28 bp. Of these repeat units, 22 repeats had mismatches, and 15 had indels (Supplementary Table S4a). In A. pedata, two repeats were > 30 bp, and the rest were between 9 and 28 bp in length. Sixteen repeat units reported mismatches, and 11 had indels (Supplementary Table S4b).
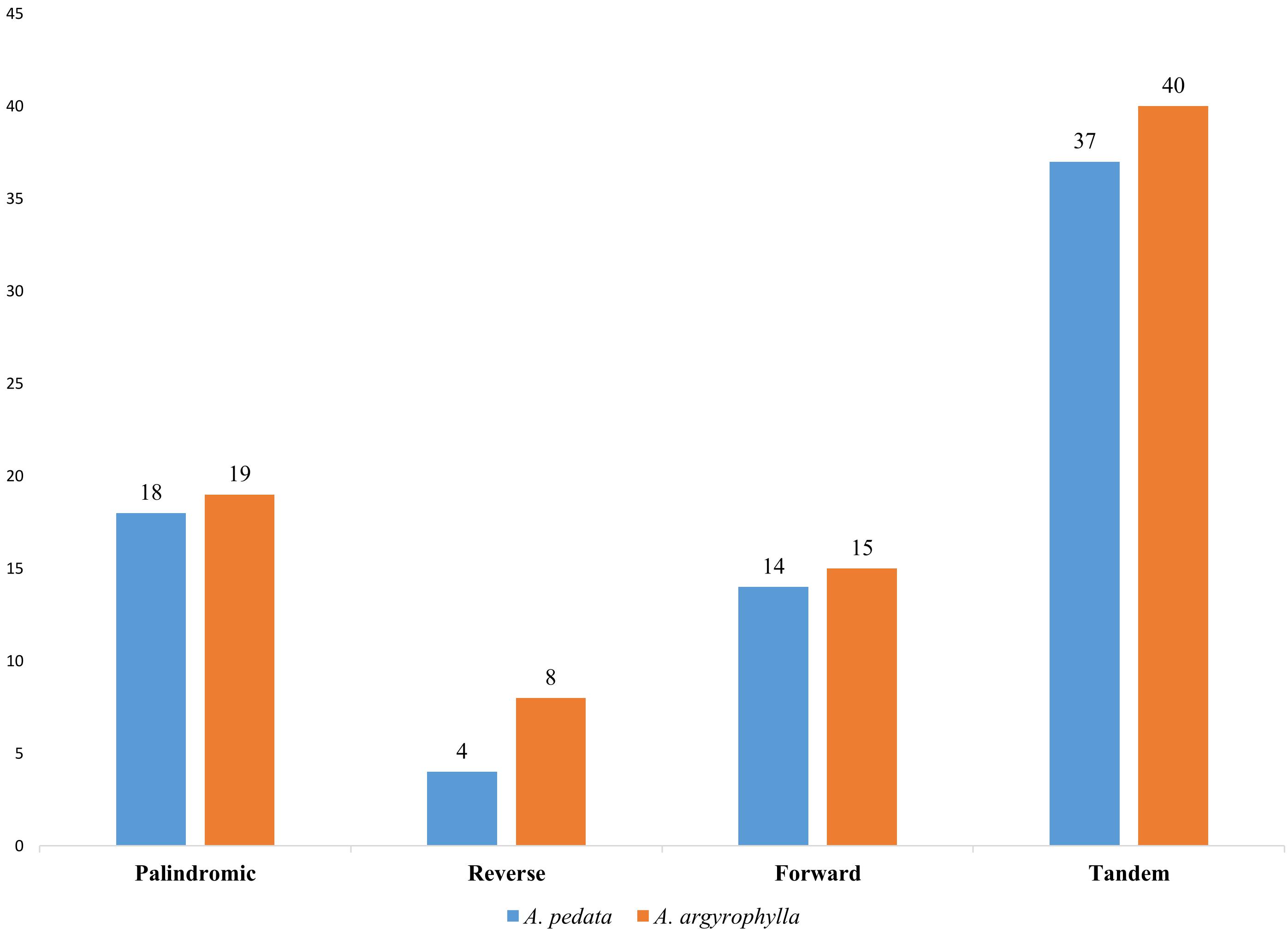
Figure 2. Number of long repeat sequence types in Alchemilla pedata and Alchemilla argyrophylla in the distribution of palindromic, reverse, forward, and tandem repeats.
SSRs, also called short tandem repeats or microsatellites, are repeating sequences of about 1–6 bp that are uniparentally inherited and widely distributed in the whole cp genome (Cheng et al., 2015). SSRs are ideally co-dominant, having the highest degree of intraspecific polymorphism (Weber, 1990), high mutation rates, locus specificity, and multi-allelism (Kuang et al., 2011; Asaf et al., 2017). Thus, the microsatellites are valuable markers ideal for molecular breeding (Rafalski and Tingey, 1993), population genetics (Powell et al., 1995), gene mapping, and genetic linkage analysis (Pugh et al., 2004; Xue et al., 2012). In our study, a total 95 SSRs were identified in A. argyrophylla composed of 70 mononucleotides, 16 dinucleotides, 5 trinucleotides, and four tetranucleotides (Figure 3 and Table 3). Similarly, A. pedata cp genome had 88 SSRs composed of 62 mononucleotides, 17 dinucleotides, 8 trinucleotides, and 3 tetranucleotides (Figure 3 and Table 3). In both species, mononucleotides were the most abundant repeat types (A. argyrophylla 73.68% and A. pedata 70.45%). Pentanucleotides and hexanucleotides were not detected in both species (Figure 3). Apart from one mononucleotide, all the other SSRs were rich in A and T (Table 3). These findings are consistent with contention that SSRs are typically composed of polyadenine (PolyA) and polythyamine (PolyT) repeats in line with previous reports (Cheng et al., 2015; Shen et al., 2016). This perpetually contributes to biasness in base composition of the whole cp genome, where A/T content in the reported Alchemilla species is 62.98% in A. argyrophylla and 62.99% in A. pedata compared with the GC content represented by 37.0% in both species.
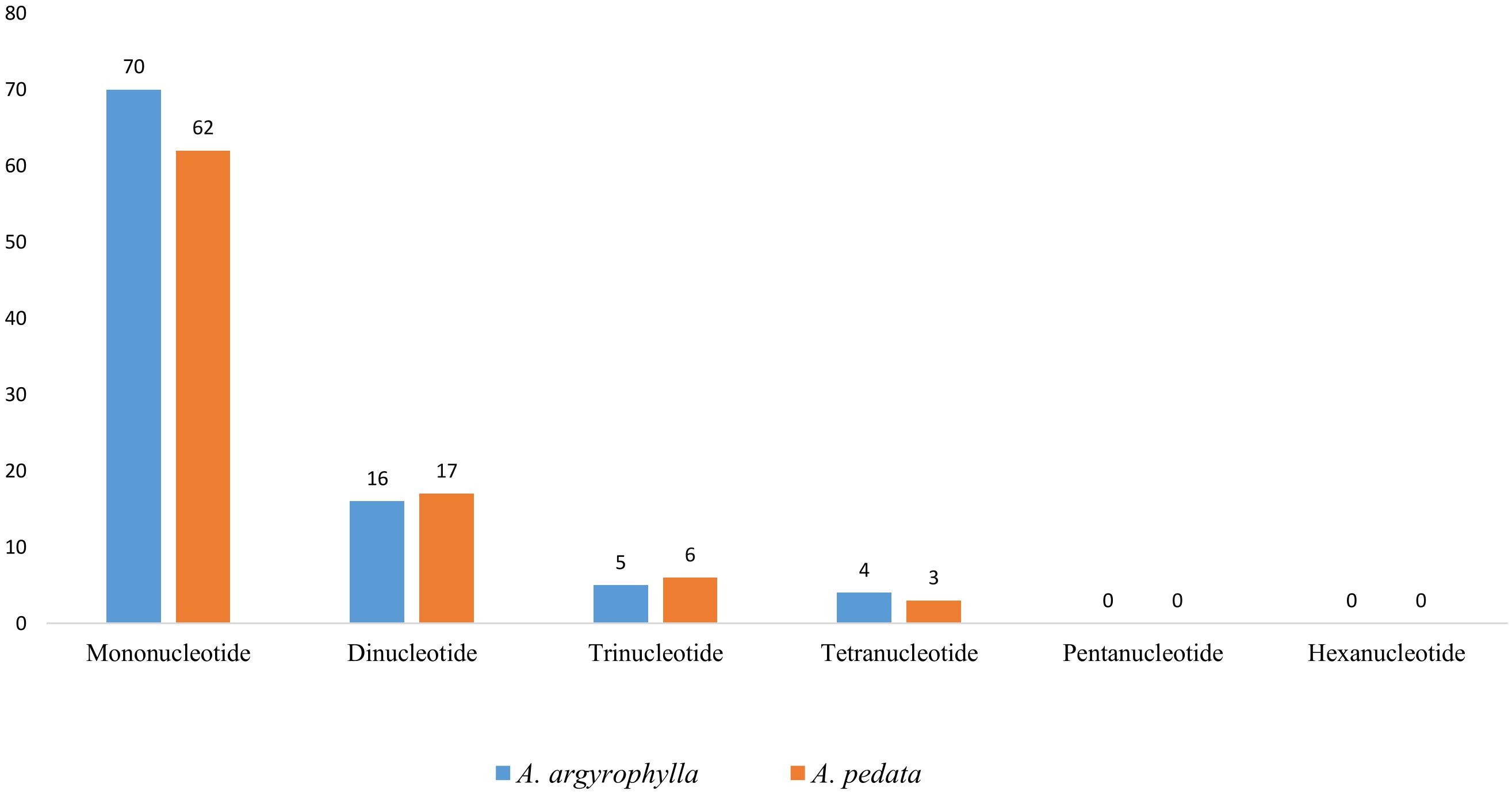
Figure 3. Number of different simple sequence repeat (SSR) units in Alchemilla argyrophylla and Alchemilla pedata comprising mono-, di-, tri, tetra-, penta-, and hexa-nucleotide repeats.

Table 3. Number of different simple sequence repeats (SSRs) in Alchemilla argyrophylla and Alchemilla pedata.
For further analysis of the cp genome of Alchemilla species, 12 whole cp genome sequences of the Rosaceae species were downloaded from NCBI, and the basic genomic characteristics were compared (Table 1). A high similarity was observed in all genome sequences. The genome size ranged from 148,592 bp in Fragaria nipponica to 156,634 in Rosa odorata var. gigantea and was concurrent with the lowest and highest number of genes that ranged from 127 to 139, respectively. The number of PCGs ranged from 84 in Fragaria orientalis to 90 in Rosa multiflora, whereas tRNA genes ranged from 35 in F. nipponica to Fragaria pentaphylla to 40 in R. odorata var. gigantea. The cp genome structure and gene arrangement are conserved in all the species evaluated (Table 1).
To ascertain divergence within the cp genome, multiple alignment analysis of Alchemilla species and seven other Rosacea cp genomes were conducted using mVISTA program. Results showed that IR regions have higher similarity than the SC regions (Supplementary Figure S2). Higher conservation was observed in the coding regions than the non-coding regions, which is a common phenomenon in most angiosperms (Cheng et al., 2017). Significantly, the most conserved regions were observed in the tRNA and rRNA regions across all species. High variation was observed in the IGS regions of trnH-GUG–psbA, trnK-UUU–rps16, rps16–trnQ-UUG, trnR-UCU–atpA, ndhC–trnV-UAC, atpF–atpH, trnC-GCA–petN, petN–psbM, trnD-GUC–trnY-GUA, psaA–ycf3, rps4–trnT-UGU, trnL-UAA–trnF-GAA, ndhC–trnV-UAC, petD–rpoA, rps11–rpl36, trnL-CAA–ndhB, and PetA–psbJ in the LSC. Non-coding regions in ndhF–rpl32, rpl32–trnL-UAG, ccsA–ndhD, and rps15–ycf1 reported high divergence in the SSC. Coding regions with the highest variation include ycf1, ndhF, rpoA, infA, accD, rpoC2, and matK. Divergence was also detected in introns of trnK-UUU, rps16, ycf3, petD, rpl16, clpP, rps12, and ndhA. These are regions of rapid evolutionary changes and therefore are essential sites for the development of molecular markers that could be useful in population genetics and phylogenetic studies. Our results are consistent with findings of other Rosaceae species (Shen et al., 2016; Jian et al., 2018). Generally, A. pedata and A. argyrophylla are highly similar to Dasiphora and Fragaria species and most divergent from Pentactina and Prinsepia species among the seven evaluated species (Supplementary Figure S2).
The IR boundaries of A. pedata and A. argyrophylla were compared with those of five other species of Rosaceae to analyze probable expansion or contraction in the IR (Figure 4). Despite the IRs being the most conserved region of the cp genome, constant variation in the position of the IR/SC boundary and their associated adjacent genes observed in plant lineages has been because of the contraction and expansion of the IR region, which subsequently acts as an evolutionary indicator (Wang et al., 2008). Our results showed that the different species had varied IR sizes, ranging from 25,311 bp in Dasiphora fruticosa to 26,053 bp in R. odorata (Figure 4). The rpl22 and rps19 genes lied exclusively in the LSC region adjacent to the LSC/IRb junction, while the rpl19 shifted away from the LSC/IRb boundary with gap of 4–14 bp. The ndhF gene in the analyzed species was located entirely in the SSC region having varied distances from the IRa/SSC boarder (JBL). However, Hagenia abyssinica had the ndhF gene stretching 21 bp into IRb region. The ycf1 gene stretched through the SSC/IRa boarder (JSA) in all the species at varied lengths. The trnH gene located entirely in the LSC region stretched 3–34 bp away from the IRa/LSC junction in all the analyzed species. Generally, the trnH gene in monocots is located in the IR region, while that in dicots is located in the LSC region (Asano, 2004; Cheng et al., 2017).
Figure 4. Comparative distance between the boundaries of the two inverted repeats (IRa/IRb), small single copy (SSC), and large single copy (LSC) regions and adjacent genes among the chloroplast genomes of seven Rosaceae species using IRscope software (Amiryousefi et al., 2018). The species name and their corresponding genome lengths are shown on the left side of the figure. JLB, JSB, JSA, and JLA correspond to LSC/IRb, IRb/SSC, SSC/IRa, and IRa/LSC junctions, respectively. Genes drawn above the track indicate direct transcription, and genes below the track indicate complement transcription. The arrows indicate the distance from the beginning or ending coordinate of the specific gene from the corresponding junction; AT bar above or below the gene extending into two regions shows to what extent in base pairs it has stretched. The figure is not drawn to scale based on sequence length but only shows the relative change near or at the IR/SC junctions.
Non-synonymous (Ka) and synonymous (Ks) substitutions and their proportional ratios (Ka/Ks) similarly referred to as (dN/dS) have been used to evaluate nucleotide’s natural selection pressure and evolution rates (Ninio, 1984; Yang and Nielsen, 2000). In most protein-coding regions, occurrence of synonymous substitutions has been reported more frequently than occurrence of non-synonymous substitutions (Makałowski and Boguski, 1998). The synonymous substitutions normally do not alter the amino acid chain unlike the non-synonymous substitutions that change the amino acid sequence. In this study, Ka and Ks values were estimated in 78 genes of the A. pedata and A. argyrophylla, computed against a close relative Fragaria virginiana (Supplementary Table S5). In our evaluation, none of the genes reported Ka value above 1 of which ycf1 PCG reported the highest value (Ka = 0.0416) in A. pedata and (Ka = 0.0411) in A. argyrophylla. On the other hand, the highest Ks value was recorded in photosynthesis gene psbD (Ks = 1.2010) in A. pedata and petL (Ks = 0.2642) in A. argyrophylla.
The Ka/Ks value indicates the intensity of selective pressure imposed on a particular gene. Neutral selection is denoted by a Ka/Ks value of 1, Ka/Ks ratio < 1 signifies negative (purifying) selection, and Ka/Ks ratio > 1 indicates positive (adaptive) selection (Nei and Kumar, 2000). Purifying selection is common in many protein-coding regions (Nielsen, 2005). In this study, most of the genes had Ka/Ks ratio of less than 0.5, accounting for over 90% of the analyzed genes. However, the high Ka/Ks values were noted in rps7 (Ka/Ks = 50), rpl23 (Ka/Ks = 50), and psbJ (Ka/Ks = 47) in A. argyrophylla and rpl32 (Ka/Ks = 50) and psbJ (Ka/Ks = 47) in A. pedata due to very low Ks value < 0.001 implying 0 synonymous changes in the genes. This means that there was very low or no substitutions (NA) between the aligned gene sequences (Mo et al., 2020). We therefore replaced the high Ka/Ks values in these genes with 0. The average Ka/Ks value was found to be 0.1322 in A. argyrophylla and 0.1418 in A. pedata, signifying an overall negative selection pressure of the genes (Supplementary Table S5). Genes with Ka/Ks > 0.5 included petN, psbL, and psbN in A. argyrophylla and petN, psbD, psbL, and PsbN in A. pedata. In both Alchemilla species, the least Ka/Ks value (0.0010) was recorded in photosynthesis-related genes (atpH, ndhI, petD, petD, petG, petL, psaC, psbA, psbH, psbI, psbM, and psbT) and self-replicating genes (rps2, rps19, and rps36), indicating significant purifying selection (Supplementary Table S5). The same functional protein-coding sequences in seven Rosoideae species were used to detect sites of positive selection. Among the four models, comparative LRT of M7 vs. M8 was positive in determining p-value of chi square < 0.05 and the selection strength. Bayes empirical Bayes (BEB) (Yang et al., 2005) and naïve empirical Bayes (NEB) analyses were implemented in model M8. In the BEB method, three sites were detected as site of positive selection, which represented one photosynthesis-related gene ndhB, self-replication gene rpoC1, and hypothetical gene ycf2 (Table 4). NEB method on the other hand detected 59 sites that coded for 17 genes under selective pressure. Among the genes, rpoc2, ycf2, and ndhB had p > 0.99%. The generally slow evolutionary rates and subsequent low Ka/Ks ratio observed in Alchemilla species is a common attribute of the cp genome. The varying results of Ka/Ks ratio obtained in our study give evidence that evolutionary rates of cp genomes vary among genes. Similar conclusions were drawn by Menezes et al. (2018) in the cp genome analysis of Malpighiales.
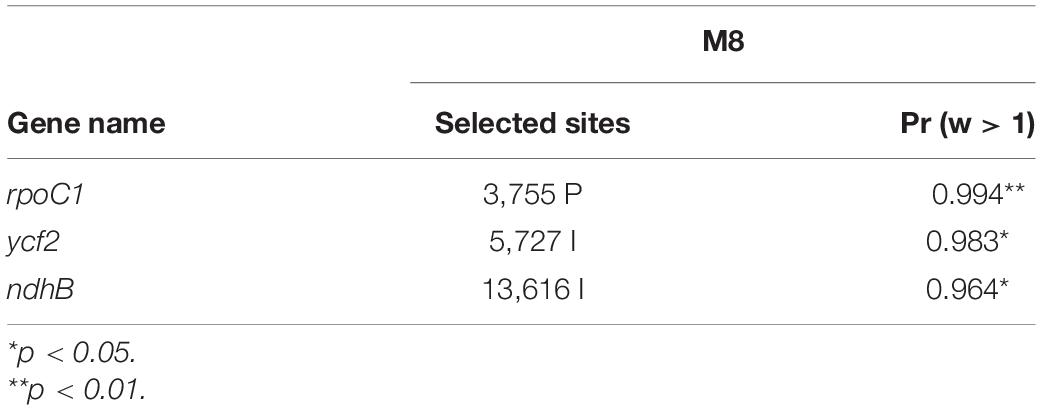
Table 4. Positively selected sites detected in the chloroplast genome of subfamily Rosoideae based of Bayes empirical Bayes (BEB) method.
In order to understand the evolutionary relationship among Rosaceae species, complete cp genome sequences and 78 PCGs in 27 species of subfamily Rosoideae and Amygdaloideae were used to infer the phylogenetic position of A. argyrophylla and A. pedata with Elaeagnus macrophylla (Elaeagnaceae) and Morus indica (Moraceae) as outgroups. The cp genome sequences and its PCGs provide precise and systematic genomic information for phylogenetic and evolutionary relationship reconstruction (Yang X. et al., 2020). Phylogenetic analysis was conducted using ML, BI, and PhyML methods (Figures 5,6). A comparison between the CDS tree and the genome tree revealed an overall similar topology with few incongruences observed in subfamily Amygdaloideae (Figure 6). In both trees, Alchemilla species were found to be closely related to species of genus Fragaria and Dasiphora with strong bootstrap support in the subfamily Rosoideae. The Alchemilla species clustered together, assuming a monophyletic clade. Our findings are consistent with previous phylogenies reconstructed including representatives of the genus Alchemilla using molecular markers (Eriksson et al., 2003; Xiang et al., 2016; Zhang et al., 2017).
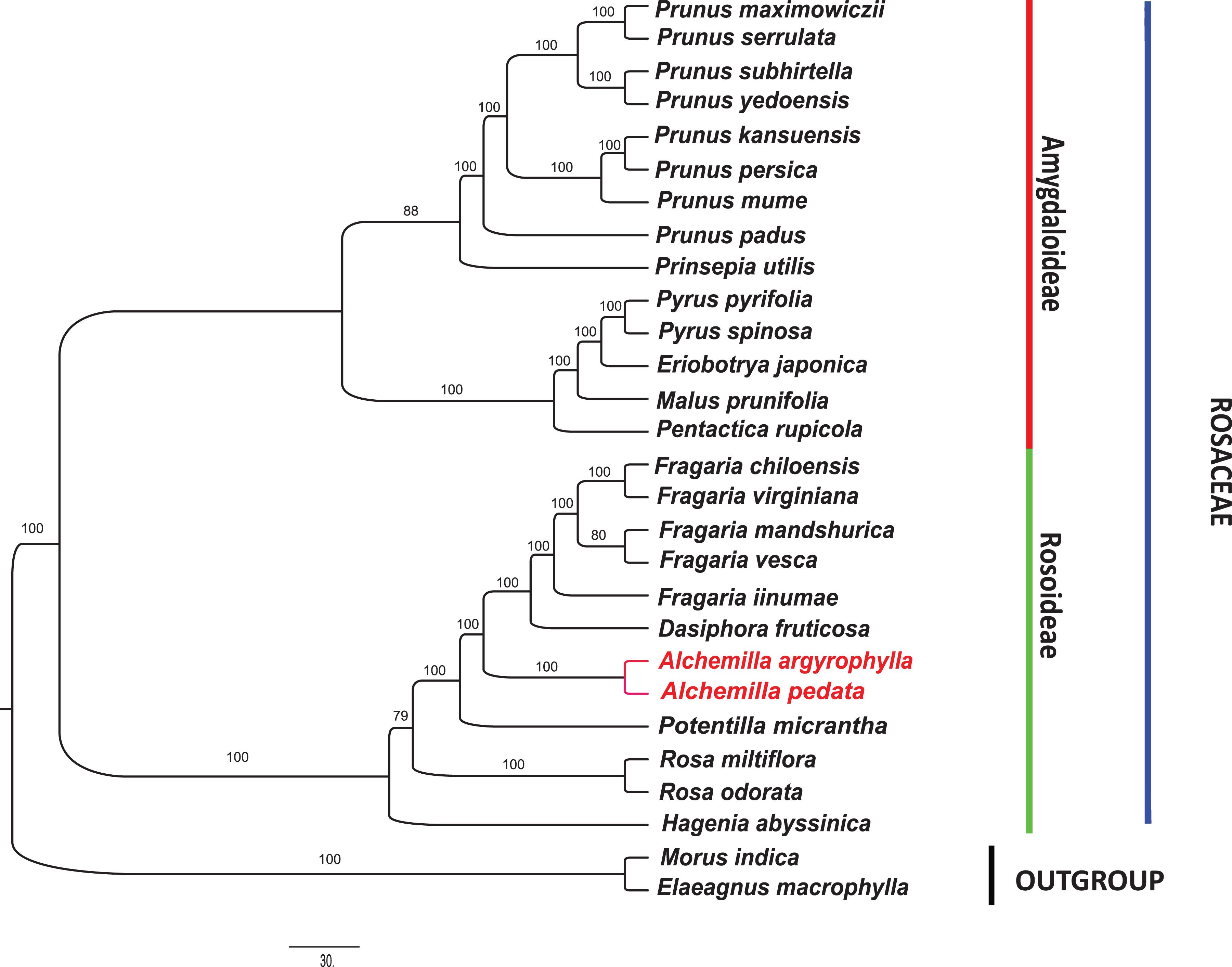
Figure 5. Phylogenetic tree reconstruction of Rosaceae species based on maximum-likelihood (ML) analysis using the program IQ-Tree v.6.1 (Nguyen et al., 2015) with 1,000 bootstrap replications in 26 complete chloroplast genome sequences. Morus indica and Elaeagnus macrophylla were used as outgroups.
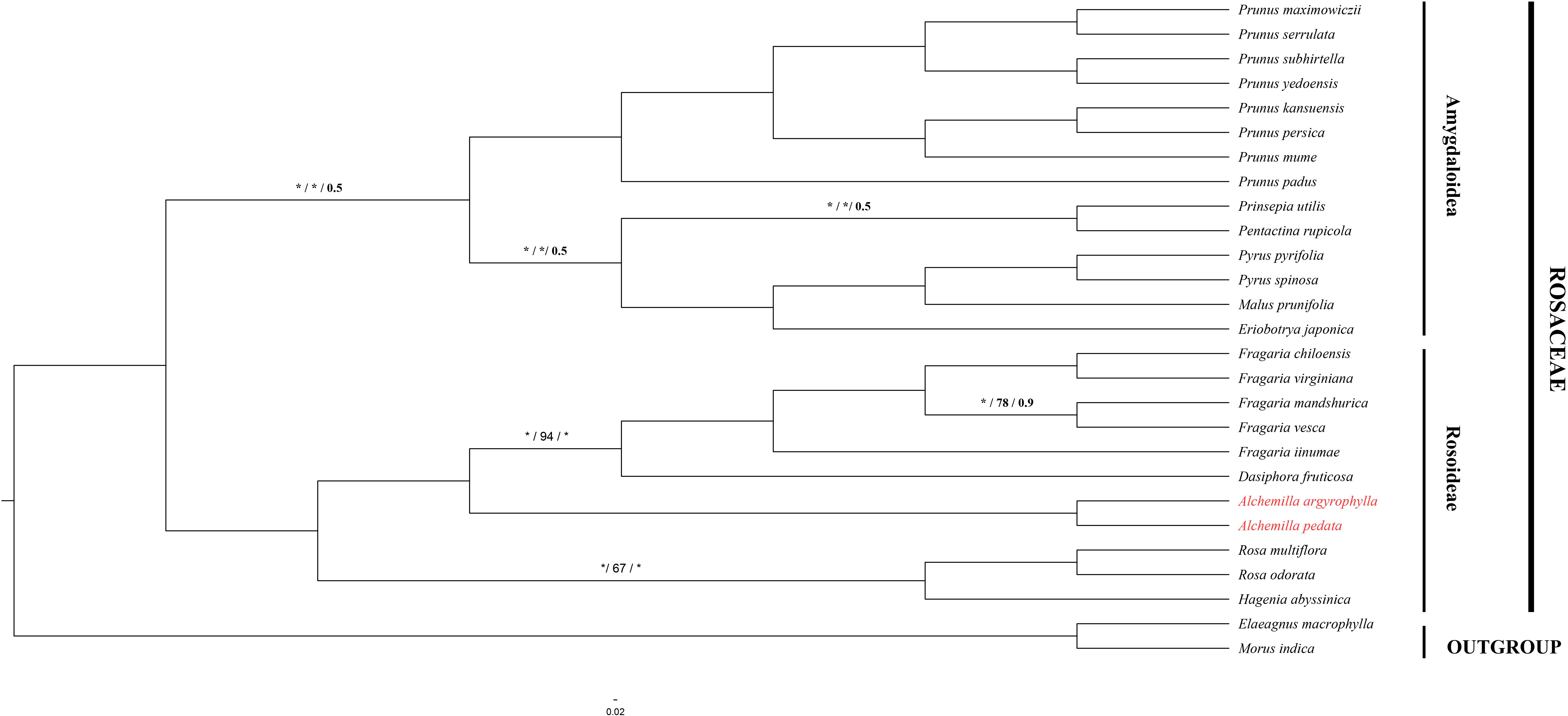
Figure 6. Phylogenetic tree reconstruction based on maximum-likelihood IQ-Tree in PhyloSuite (Zhang et al., 2020) with 1,000 replications, PhyML (Lefort et al., 2017) and BI based on MrBayers (Ronquist et al., 2012) in 200,000 generation using 78 protein-coding genes common in 27 Rosaceae species. The numbers above the branch represent bootstrap support value for ML/PhyML/BI methods, where the asterisk signifies maximum support value of 100 in IQ and 1 BI. Blank branches signify 100% support value.
Our study is the initial report and analysis of the complete cp genome of the Alchemilla species. They are represented by Alchemilla pedata and Alchemilla argyrophylla, a herb and a shrub, respectively, of the Afromilla clade found in the cooler mountainous regions of East Africa at altitudes of 2,250–4,500 m above sea level (Graham, 1960). The sequenced cp genome comprises 114 unique genes with slight variation in genome size of 152,427 bp in A. argyrophylla and 152, 438 bp in A. pedata (Figure 1A). Compared with other species, the size variation could be a result of the expansion and contraction of the IR region (Palmer et al., 1988). Similar to other cp genomes of higher plants, the genome annotation of Alchemilla species revealed an LSC-IR–SSC-IR arrangement as well as a systemic gene category and functional classification. Comparative analysis with other Rosaceae species reveals the conserved structural and organizational nature with slight variation in gene content and genome length (Table 1). Furthermore, the hypothetical ycf68 embedded within trnI-GUA, previously not annotated in the most Rosacea species, was detected in the two annotated Alchemilla species (Figure 1A). Nucleotide substitution rates in the cp genome of angiosperms are considered lower than those of the nuclear genome (Wolfe et al., 1987). Therefore, the low gene substitution rate reported by the PCGs in Alchemilla is consistent with other cp genomes of higher plants. Concomitantly, the low rate of nucleotide substitutions in the PCG could be accentuated by recombination between the IRs. This is primarily due to the recurrent intra-chromosomal recombination event interplay between the two identical IR regions of the cp genome. As a result, selective constraints are imposed on both the structural stability and the sequence homogeneity (Wolfe et al., 1987; Palmer et al., 1988). The ratios between non-synonymous and synonymous substitutions (Ka/Ks) are fundamental in elucidating natural selection pressure (Nei and Kumar, 2000). In Alchemilla, petN, psbL, and psbN with Ka/Ks > 0.5 and rpoC2, ycf2, and ndhB with p > 0.99% are essential in unfolding evolutionary history of the genus (Table 4 and Supplementary Table S5). The genomic information in this study will be fundamental in the phylogenetic studies as well as the generation of molecular markers of not only the African Alchemilla clade but also genus Alchemilla.
Introns are generally conserved regions among land plants; and therefore, instances of intron loss or gain in the cp genome could signify an evolutionary event (Daniell et al., 2016). In the annotated Alchemilla cp genomes, we report the absence of an intron in atpF belonging to group II introns. This is a rare phenomenon in land plant besides Euphorbiaceae and Malpighiales (Daniell et al., 2008). A similar observation was made in Fragaria vesca for the first time in Rosoideae (Shulaev et al., 2011), which we found to be closely related to our species based on phylogenetic analysis (Figure 5). Subsequent absence of the atpF intron has been evident in species of Rosa, Potentilla, Rubus, and Fragaria (Yang J. et al., 2020). However, a comparison with relative species of subfamily Amygdaloidea indicates the retention of introns within atpF genes (Supplementary Table S6). Based on the present phylogenetic framework, the loss of intron within atpF genes seems to have taken place once within the Rosoideae subfamily. The loss of intron in atpF genes is yet to be determined in other species of subfamily Rosoideae, genera in the Rosaceae, and the broader families of Rosids (Yang J. et al., 2020). Introns are broadly classified as either group I or group II and are considered as the mobile genetic elements in the cp genome. The cp genome of land plants has 17 to 20 introns classified under group II within tRNA and PCGs (Daniell et al., 2008). The only group I intron present within trnL-AUU gene is considered most ancient since it is also found in cyanobacteria other than in land plants and algae’s cp genome (Vogel et al., 1999). The splicing function of trnL and other RNA transcripts for genes such as trnA, rpl2, rps12, and atpF is fully dependent on the function of maturases. The matK is the only maturase encoded protein present in the cp genome of land plants, yet it lacks the reverse transcriptase domain and hence cannot promote intron mobility (Hao et al., 2010; Liu et al., 2016). This causes the splicing of group II introns, including atpF intron, to be dependent on the host encoded splicing factors through lariat formation (Barkan, 2004). The loss of atpF intron among Alchemilla species and its close relatives could be used as a new classification basis of structural change within Rosaceae. Furthermore, the presence or absence of introns maybe insightful in understanding phylogenetic relationships as well as providing a potentially resourceful marker for evolutionary lineages in angiosperms.
Over the previous century, the taxonomic treatment of Rosaceae has yielded varied results pertaining to its family position among the angiosperms (Potter et al., 2007). Furthermore, classification within the family has always differed depending on the treatment imposed (Hutchinson, 1964; Schulze-Menz, 1964; Kalkman, 2004; Potter et al., 2007). Morgan et al. (1994) resolved that Rosaceae be composed of monophyletic groups based on phylogenetic analysis of rbcL sequences across the family. This acclamation was later supported by phylogenetic studies based on ndhF, matK, and trnL-trnF sequences (Potter et al., 2002; Zhang et al., 2017). However, there exist uncertainties within genera resulting from unresolved tree portions having weak support (Potter et al., 2007). Notably in the genus Alchemilla, the distinction between the African clade (Afromilla) and the Eurasian (Eualchemilla) clade is geographically significant but remain unresolved (Gehrke et al., 2016). The contention requires the separation of the Alchemilla into Eualchemilla and Afromilla clade, which has been restrained due to lack of substantial morphological borderlines despite extensive and significant studies (Notov and Kusnetzova, 2004; Gehrke et al., 2008; Lundberg et al., 2009). The emergence and rapid development in cp genome sequencing technologies have been essential in providing resourceful genomic information that has been fundamental for the reconstruction of lower and higher plant phylogenies as well as evolutionary trends (Daniell et al., 2016; Yang X. et al., 2020). Significantly, the cp genome sequences have been proven effective in eliminating phylogenetic incongruences arising from incomplete lineage sorting (ILS) and hybridization (Lundberg et al., 2009; Morales-Briones et al., 2018). In this study, A. pedata and A. argyrophylla nested into a monophyletic clade with 100% bootstrap support. They share a recent ancestry with Dasiphora and Fragaria species (Figure 5). There is need for more complete cp genomes of Alchemilla species for higher precision in phylogenetic conclusion. This study is therefore resourceful for further delimitation of species in Alchemilla and phylogenetic studies of the genus.
The assembled chloroplast genome sequences have been uploaded to and deposited in GenBank and we have received the accession numbers which are shown in Table 1 in the manuscript.
PR and XD performed the experiment. PR, XD, and J-XY performed the data analysis. PR drafted the manuscript. FM, MO, IM, and PK revised the manuscript. G-WH and Q-FW designed and supervised the experiment. All the authors contributed to and approved the final manuscript.
This work was supported by the National Natural Science Foundation of China (31970211) and Sino-Africa Joint Research Center, CAS (SAJC201614).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.560368/full#supplementary-material
Amiryousefi, A., Hyvönen, J., and Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Asaf, S., Khan, A. L., Khan, M. A., Waqas, M., Kang, S.-M., Yun, B.-W., et al. (2017). Chloroplast genomes of Arabidopsis halleri ssp. gemmifera and Arabidopsis lyrata ssp. petraea: structures and comparative analysis. Sci. Rep. 7:7556. doi: 10.1038/s41598-017-07891-5
Asano, T. (2004). Complete nucleotide sequence of the sugarcane (Saccharum Officinarum) chloroplast genome: a comparative analysis of four monocot chloroplast genomes. DNA Res. 11, 93–99. doi: 10.1093/dnares/11.2.93
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Barkan, A. (2004). “Intron splicing in plant organelles,” in Molecular Biology and Biotechnology of Plant Organelles, eds H. Daniell and C. D. Chase (Dordrecht: Springer), 295–322.
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bicknell, R. A., and Koltunow, A. M. (2004). Understanding apomixis: recent advances and remaining conundrums. Plant Cell 16(Suppl. 1), S228–S245. doi: 10.1105/tpc.017921
Boroja, T., Mihailović, V., Katanić, J., Pan, S. P., Nikles, S., Imbimbo, P., et al. (2018). The biological activities of roots and aerial parts of Alchemilla vulgaris L. S. Afr. J. Bot. 116, 175–184. doi: 10.1016/j.sajb.2018.03.007
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Cavalier-Smith, T. (2002). Chloroplast evolution: secondary symbiogenesis and multiple losses. Curr. Biol. 12, R62–R64. doi: 10.1016/S0960-9822(01)00675-3
Chase, M. W., and Hills, H. H. (1991). Silica gel: an ideal material for field preservation of leaf samples for DNA studies. Taxon 40, 215–220. doi: 10.2307/1222975
Cheng, B., Zheng, Y., and Sun, Q. (2015). Genetic diversity and population structure of Taxus cuspidata in the Changbai Mountains assessed by chloroplast DNA sequences and microsatellite markers. Biochem. Syst. Ecol. 63, 157–164. doi: 10.1016/j.bse.2015.10.009
Cheng, H., Li, J., Zhang, H., Cai, B., Gao, Z., Qiao, Y., et al. (2017). The complete chloroplast genome sequence of strawberry (Fragaria × ananassa Duch.) and comparison with related species of Rosaceae. PeerJ 5:e3919. doi: 10.7717/peerj.3919
Czapik, R. (1996). Problems of apomictic reproduction in the FamiliesCompositae andRosaceae. Folia Geobot. Phytotaxonom. 31, 381–387. doi: 10.1007/BF02815382
Daniell, H., Lin, C.-S., Yu, M., and Chang, W.-J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17:134. doi: 10.1186/s13059-016-1004-2
Daniell, H., Wurdack, K. J., Kanagaraj, A., Lee, S.-B., Saski, C., and Jansen, R. K. (2008). The complete nucleotide sequence of the cassava (Manihot esculenta) chloroplast genome and the evolution of atpF in Malpighiales: RNA editing and multiple losses of a group II intron. Theor. Appl. Genet. 116, 723–737. doi: 10.1007/s00122-007-0706-y
Doyle, J. (1991). “DNA protocols for plants,” in Molecular Techniques in Taxonomy, eds G. M. Hewitt, A. W. B. Johnston, and J. P. W. Young (Berlin: Springer), 283–293. doi: 10.1007/978-3-642-83962-7_18
Doyle, J. J., Davis, J. I., Soreng, R. J., Garvin, D., and Anderson, M. J. (1992). Chloroplast DNA inversions and the origin of the grass family (Poaceae). Proc. Natl. Acad. Sci. U.S.A. 89, 7722–7726. doi: 10.1073/pnas.89.16.7722
Eriksson, T., Hibbs, M. S., Yoder, A. D., Delwiche, C. F., and Donoghue, M. J. (2003). The Phylogeny of Rosoideae (Rosaceae) based on sequences of the internal transcribed spacers (ITS) of nuclear ribosomal DNA and the trnL/F region of chloroplast DNA. Int. J. Plant Sci. 164, 197–211. doi: 10.1086/346163
Faghir, M. B., Chaichi, K. K., and Shahvon, R. S. (2014). Foliar epidermis micromorphology of the genus Alchemilla (Rosaceae) in Iran. Phytol. Balcanica 20, 215–225.
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Fröhner, S. E. (1995). “Alchemilla,” in Hegi: Illustrierte Flora von Mitteleuropa, Vol. 4 Teil 2B, eds H. Scholz, H. J. Conert, E. J. Jäger, J. W. Kadereit, W. Schultze-Motel, G. Wagenitz, et al. (Berlin: Verlag Paul Parey), 13–242.
Gao, F., Chen, C., Arab, D. A., Du, Z., He, Y., and Ho, S. Y. W. (2019). EasyCodeML: a visual tool for analysis of selection using CodeML. Ecol. Evol. 9, 3891–3898. doi: 10.1002/ece3.5015
Gehrke, B., Bräuchler, C., Romoleroux, K., Lundberg, M., Heubl, G., and Eriksson, T. (2008). Molecular phylogenetics of Alchemilla, Aphanes and Lachemilla (Rosaceae) inferred from plastid and nuclear intron and spacer DNA sequences, with comments on generic classification. Mol. Phylogenet. Evol. 47, 1030–1044. doi: 10.1016/j.ympev.2008.03.004
Gehrke, B., Kandziora, M., and Pirie, M. D. (2016). The evolution of dwarf shrubs in alpine environments: a case study of Alchemilla in Africa. Ann. Bot. 117, 121–131. doi: 10.1093/aob/mcv159
Gichira, A. W., Li, Z., Saina, J. K., Long, Z., Hu, G., Gituru, R. W., et al. (2017). The complete chloroplast genome sequence of an endemic monotypic genus Hagenia (Rosaceae): structural comparative analysis, gene content and microsatellite detection. PeerJ 5:e2846. doi: 10.7717/peerj.2846
Graham, R. A. (1960). “Rosaceae,” in Flora of Tropical East Africa, eds C. E. Hubbard and E. Milne-Redhead (London: Crown Agents), 1–61.
Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Hao, D. C., Chen, S. L., and Xiao, P. G. (2010). Molecular evolution and positive Darwinian selection of the chloroplast maturase matK. J. Plant Res. 123, 241–247. doi: 10.1007/s10265-009-0261-5
Hayirhoglu-Ayaz, S., İnceer, H., and Frost-Olsen, P. (2006). Chromosome counts in the genus Alchemilla (Rosaceae) from SW Europe. Folia Geobot. 41, 335–344.
Hörandl, E. (2004). Comparative analysis of genetic divergence among sexual ancestors of apomictic complexes using isozyme data. Int. J. Plant Sci. 165, 615–622. doi: 10.1086/386557
Hutchinson, J. (1964). The Genera of Flowering Plants. Dicotyledons, Vol. 1. (London: Oxford University Press), 195–208.
Izmailow, R. (1981). Karyological studies in species of Alchemilla L. from the series Calycinae Bus. (Section Brevicaulon Rothm.). Acta Biol. Cracov. Ser. Bot. 23, 117–130.
Izmailow, R. (1982). Further Karyological studies in species of Alchemilla L. from the series Calycinae Bus.(section Brevicaulon Rothm.). Acta Biol. Cracov. Ser. Bot. 24, 117–130.
Jian, H.-Y., Zhang, Y.-H., Yan, H.-J., Qiu, X.-Q., Wang, Q.-G., Li, S.-B., et al. (2018). The complete chloroplast genome of a key ancestor of modern roses, Rosa chinensis var. Spontanea, and a Comparison with Congeneric Species. Molecules 23:389. doi: 10.3390/molecules23020389
Jin, J.-J., Yu, W.-B., Yang, J.-B., Song, Y., dePamphilis, C. W., Yi, T.-S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21:241. doi: 10.1186/s13059-020-02154-5
Kalkman, C. (2004). “Rosaceae,” in The Families and Genera of Vascular Plants, vol. 6, Flowering Plants - Dicotyledons: Celastrales, Oxalidales, Rosales, Cornales, Ericales, ed. K. Kubitzki (Berlin: Springer), 343–386. doi: 10.1007/978-3-662-07257-8_39
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kaya, B., Menemen, Y., and Saltan, F. (2012). Flavonoid compounds identified in Alchemilla l. species collected in the North-eastern Black sea region of Turkey. Afr. J. Trad. Complement. Altern. Med. 9, 418–425. doi: 10.4314/ajtcam.v9i3.18
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Keeling, P. J. (2004). Diversity and evolutionary history of plastids and their hosts. Am. J. Bot. 91, 1481–1493. doi: 10.3732/ajb.91.10.1481
Köhler, M., Reginato, M., Souza-Chies, T. T., and Majure, L. C. (2020). Insights into chloroplast genome evolution across opuntioideae (Cactaceae) reveals robust yet sometimes conflicting phylogenetic topologies. Front. Plant Sci. 11:729. doi: 10.3389/fpls.2020.00729
Kress, W. J., Wurdack, K. J., Zimmer, E. A., Weigt, L. A., and Janzen, D. H. (2005). Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. U.S.A. 102, 8369–8374. doi: 10.1073/pnas.0503123102
Kuang, D.-Y., Wu, H., Wang, Y.-L., Gao, L.-M., Zhang, S.-Z., and Lu, L. (2011). Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): implication for DNA barcoding and population genetics. Genome 54, 663–673. doi: 10.1139/g11-026
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lefort, V., Longueville, J.-E., and Gascuel, O. (2017). SMS: smart model selection in PhyML. Mol. Biol. Evol. 34, 2422–2424. doi: 10.1093/molbev/msx149
Liu, C., Zhu, H., Xing, Y., Tan, J., Chen, X., Zhang, J., et al. (2016). Albino Leaf 2 is involved in the splicing of chloroplast group I and II introns in rice. J. Exp. Bot. 67, 5339–5347. doi: 10.1093/jxb/erw296
Liu, L.-X., Li, R., Worth, J. R. P., Li, X., Li, P., Cameron, K. M., et al. (2017). The complete chloroplast genome of chinese bayberry (Morella rubra, Myricaceae): implications for understanding the evolution of Fagales. Front. Plant Sci. 8:968. doi: 10.3389/fpls.2017.00968
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). OrganellarGenomeDRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581. doi: 10.1093/nar/gkt289
Lundberg, M., Töpel, M., Eriksen, B., Nylander, J. A. A., and Eriksson, T. (2009). Allopolyploidy in Fragariinae (Rosaceae): comparing four DNA sequence regions, with comments on classification. Mol. Phylogenet. Evol. 51, 269–280. doi: 10.1016/j.ympev.2009.02.020
Makałowski, W., and Boguski, M. S. (1998). Evolutionary parameters of the transcribed mammalian genome: An analysis of 2,820 orthologous rodent and human sequences. Proc. Natl. Acad. Sci. U.S.A. 95, 9407–9412. doi: 10.1073/pnas.95.16.9407
Mayor, C., Brudno, M., Schwartz, J. R., Poliakov, A., Rubin, E. M., Frazer, K. A., et al. (2000). VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 16, 1046–1047. doi: 10.1093/bioinformatics/16.11.1046
Menezes, A. P. A., Resende-Moreira, L. C., Buzatti, R. S. O., Nazareno, A. G., Carlsen, M., Lobo, F. P., et al. (2018). Chloroplast genomes of Byrsonima species (Malpighiaceae): comparative analysis and screening of high divergence sequences. Sci. Rep. 8:2210. doi: 10.1038/s41598-018-20189-4
Mo, Z., Lou, W., Chen, Y., Jia, X., Zhai, M., Guo, Z., et al. (2020). The chloroplast genome of Carya illinoinensis: genome structure, adaptive evolution, and phylogenetic analysis. Forests 11:207. doi: 10.3390/f11020207
Morales-Briones, D. F., Romoleroux, K., Kolář, F., and Tank, D. C. (2018). Phylogeny and evolution of the neotropical radiation of Lachemilla (Rosaceae): uncovering a history of reticulate evolution and implications for infrageneric classification. Syst. Bot. 43, 17–34. doi: 10.1600/036364418X696897
Morgan, D. R., Soltis, D. E., and Robertson, K. R. (1994). Systematic and evolutionary implications of rbc L sequence variation in Rosaceae. Am. J. Bot. 81, 890–903. doi: 10.1002/j.1537-2197.1994.tb15570.x
Morton, B. R. (2003). The role of context-dependent mutations in generating compositional and codon usage bias in grass chloroplast DNA. J. Mol. Evol. 56, 616–629. doi: 10.1007/s00239-002-2430-1
Munyao, J. N., Dong, X., Yang, J.-X., Mbandi, E. M., Wanga, V. O., Oulo, M. A., et al. (2020). Complete Chloroplast Genomes of Chlorophytum comosum and Chlorophytum gallabatense: genome structures, comparative and phylogenetic analysis. Plants 9:296. doi: 10.3390/plants9030296
Nei, M., and Kumar, S. (2000). Molecular Evolution and Phylogenetics. Oxford: Oxford university press.
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nielsen, R. (2005). Molecular signatures of natural selection. Annu. Rev. Genet. 39, 197–218. doi: 10.1146/annurev.genet.39.073003.112420
Ninio, J. (1984). The neutral theory of molecular evolution: edited by Mooto Kimura Cambridge University Press. Cambridge, 1983 366 pages. FEBS Lett. 170, 210–211. doi: 10.1016/0014-5793(84)81411-8
Notov, A. A., and Kusnetzova, T. V. (2004). Architectural units, axiality and their taxonomic implications in Alchemillinae. Wulfenia 11, 85–130.
Palmer, J. D., Jansen, R. K., Michaels, H. J., Chase, M. W., and Manhart, J. R. (1988). Chloroplast DNA variation and plant phylogeny. Ann. Mi. Bot. Gard. 75:1180. doi: 10.2307/2399279
Parks, M., Cronn, R., and Liston, A. (2009). Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 7:84. doi: 10.1186/1741-7007-7-84
Potter, D., Eriksson, T., Evans, R. C., Oh, S., Smedmark, J. E. E., Morgan, D. R., et al. (2007). Phylogeny and classification of Rosaceae. Plant Syst. Evol. 266, 5–43. doi: 10.1007/s00606-007-0539-9
Potter, D., Gao, F., Bortiri, P. E., Oh, S.-H., and Baggett, S. (2002). Phylogenetic relationships in Rosaceae inferred from chloroplast mat K and trn L- trn F nucleotide sequence data. Plant Syst. Evol. 231, 77–89. doi: 10.1007/s006060200012
Powell, W., Morgante, M., McDevitt, R., Vendramin, G. G., and Rafalski, J. A. (1995). Polymorphic simple sequence repeat regions in chloroplast genomes: Applications to the population genetics of pines. Proc. Natl. Acad. Sci. U.S.A. 92, 7759–7763. doi: 10.1073/pnas.92.17.7759
Pugh, T., Fouet, O., Risterucci, A. M., Brottier, P., Abouladze, M., Deletrez, C., et al. (2004). A new cacao linkage map based on codominant markers: development and integration of 201 new microsatellite markers. Theor. Appl. Genet. 108, 1151–1161. doi: 10.1007/s00122-003-1533-4
Qu, X.-J., Moore, M. J., Li, D.-Z., and Yi, T.-S. (2019). PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 15:50. doi: 10.1186/s13007-019-0435-7
Rafalski, J. A., and Tingey, S. V. (1993). Genetic diagnostics in plant breeding: RAPDs, microsatellites and machines. Trends Genet. 9, 275–280. doi: 10.1016/0168-9525(93)90013-8
Rambaut, A. (2018). FigTree v1.4.4. Institute of Evolutionary Biology. Edinburgh: University of Edinburgh.
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rothmaler, W. (1937). Systematische Vorarbeiten zu einer Monographie der Gattung Alchemilla (L.) Scop. VII. Aufteilung der Gattung und Nomenklatur. Repertorium Novarum Specierum Regni Vegetabilis 42, 164–173. doi: 10.1002/fedr.19370421106
Salamone, I., Govindarajulu, R., Falk, S., Parks, M., Liston, A., and Ashman, T.-L. (2013). Bioclimatic, ecological, and phenotypic intermediacy and high genetic admixture in a natural hybrid of octoploid strawberries. Am. J. Bot. 100, 939–950. doi: 10.3732/ajb.1200624
Saski, C., Lee, S.-B., Daniell, H., Wood, T. C., Tomkins, J., Kim, H.-G., et al. (2005). Complete chloroplast genome sequence of Glycine max and comparative analyses with other legume genomes. Plant Mol. Biol. 59, 309–322. doi: 10.1007/s11103-005-8882-0
Schulze-Menz, G. K. (1964). “Rosaceae,” in A. Engler’s Syllabus der Pflanzenfamilien, Vol. II, ed. H. Melchoir (Berlin: Borntraeger), 209–218.
Sharp, P. M., and Li, W.-H. (1987). The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15, 1281–1295. doi: 10.1093/nar/15.3.1281
Shen, L., Guan, Q., Amin, A., Zhu, W., Li, M., Li, X., et al. (2016). Complete plastid genome of Eriobotrya japonica (Thunb.) Lindl and comparative analysis in Rosaceae. Springerplus 5:2036. doi: 10.1186/s40064-016-3702-3
Shendure, J., and Ji, H. (2008). Next-generation DNA sequencing. Nat. Biotechnol. 26, 1135–1145. doi: 10.1038/nbt1486
Shinozaki, K., Ohme, M., Tanaka, M., Wakasugi, T., Hayashida, N., Matsubayashi, T., et al. (1986). The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. EMBO J. 5, 2043–2049. doi: 10.1002/j.1460-2075.1986.tb04464.x
Shulaev, V., Sargent, D. J., Crowhurst, R. N., Mockler, T. C., Folkerts, O., Delcher, A. L., et al. (2011). The genome of woodland strawberry (Fragaria vesca). Nat. Genet. 43, 109–116. doi: 10.1038/ng.740
Soják, J. (2008). Notes on Potentilla XXI. A new division of the tribe Potentilleae (Rosaceae) and notes on generic delimitations. Bot. Jahrbücher 127, 349–358.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Thiel, T., Michalek, W., Varshney, R., and Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106, 411–422. doi: 10.1007/s00122-002-1031-0
Vogel, J., Börner, T., and Hess, W. R. (1999). Comparative analysis of splicing of the complete set of chloroplast group II introns in three higher plant mutants. Nucleic Acids Res. 27, 3866–3874. doi: 10.1093/nar/27.19.3866
Wang, L., Xing, H., Yuan, Y., Wang, X., Saeed, M., Tao, J., et al. (2018). Genome-wide analysis of codon usage bias in four sequenced cotton species. PLoS One 13:e0194372. doi: 10.1371/journal.pone.0194372
Wang, Q., Niu, Z., Li, J., Zhu, K., and Chen, X. (2020). The complete chloroplast genome sequence of the Chinese endemic species Sorbus setschwanensis (Rosaceae) and its phylogenetic analysis. Nord. J. Bot. 38:njb.02532. doi: 10.1111/njb.02532
Wang, R.-J., Cheng, C.-L., Chang, C.-C., Wu, C.-L., Su, T.-M., and Chaw, S.-M. (2008). Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 8:36. doi: 10.1186/1471-2148-8-36
Weber, J. L. (1990). Human DNA polymorphisms and methods of analysis. Curr. Opin. Biotechnol. 1, 166–171. doi: 10.1016/0958-1669(90)90026-H
Wick, R. R., Schultz, M. B., Zobel, J., and Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies: Fig. 1. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wolfe, K. H., Li, W. H., and Sharp, P. M. (1987). Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. U.S.A. 84, 9054–9058. doi: 10.1073/pnas.84.24.9054
Xiang, Y., Huang, C.-H., Hu, Y., Wen, J., Li, S., Yi, T., et al. (2016). Evolution of Rosaceae fruit types based on nuclear phylogeny in the context of geological times and genome duplication. Mol. Biol. Evol. 34, 262–281. doi: 10.1093/molbev/msw242
Xue, J., Wang, S., and Zhou, S.-L. (2012). Polymorphic chloroplast microsatellite loci in Nelumbo (Nelumbonaceae). Am. J. Bot. 99, e240–e244. doi: 10.3732/ajb.1100547
Yang, J., Kang, G.-H., Pak, J.-H., and Kim, S.-C. (2020). Characterization and comparison of two complete plastomes of Rosaceae Species (Potentilla dickinsii var. Glabrata and Spiraea insularis) Endemic to Ulleung Island, Korea. Int. J. Mol. Sci. 21:4933. doi: 10.3390/ijms21144933
Yang, X., Xie, D.-F., Chen, J.-P., Zhou, S.-D., Yu, Y., and He, X.-J. (2020). Comparative analysis of the complete chloroplast genomes in Allium Subgenus Cyathophora (Amaryllidaceae): phylogenetic relationship and adaptive evolution. Biomed Res. Int. 2020:1732586. doi: 10.1155/2020/1732586
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yang, Z., and Nielsen, R. (2000). Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 17, 32–43. doi: 10.1093/oxfordjournals.molbev.a026236
Yang, Z., Wong, W. S., and Nielsen, R. (2005). Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 22, 1107–1118. doi: 10.1093/molbev/msi097
Zhang, D., Gao, F., Jakovlić, I., Zou, H., Zhang, J., Li, W. X., et al. (2020). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Zhang, S.-D., Jin, J.-J., Chen, S.-Y., Chase, M. W., Soltis, D. E., Li, H.-T., et al. (2017). Diversification of Rosaceae since the late cretaceous based on plastid phylogenomics. New Phytol. 214, 1355–1367. doi: 10.1111/nph.14461
Zhang, Y., Ma, J., Yang, B., Li, R., Zhu, W., Sun, L., et al. (2014). The complete chloroplast genome sequence of Taxus chinensis var. mairei (Taxaceae): loss of an inverted repeat region and comparative analysis with related species. Gene 540, 201–209. doi: 10.1016/j.gene.2014.02.037
Zhang, Z., Li, J., Zhao, X.-Q., Wang, J., Wong, G. K.-S., and Yu, J. (2006). KaKs_Calculator: calculating Ka and Ks through model selection and model averaging. Genomics Proteomics Bioinformatics 4, 259–263. doi: 10.1016/S1672-0229(07)60007-2
Zhao, M.-L., Song, Y., Ni, J., Yao, X., Tan, Y.-H., and Xu, Z.-F. (2018). Comparative chloroplast genomics and phylogenetics of nine Lindera species (Lauraceae). Sci. Rep. 8:8844. doi: 10.1038/s41598-018-27090-0
Keywords: Alchemilla argyrophylla, A. pedata, chloroplast genome, phylogenetic, Rosaceae
Citation: Rono PC, Dong X, Yang J-X, Mutie FM, Oulo MA, Malombe I, Kirika PM, Hu G-W and Wang Q-F (2020) Initial Complete Chloroplast Genomes of Alchemilla (Rosaceae): Comparative Analysis and Phylogenetic Relationships. Front. Genet. 11:560368. doi: 10.3389/fgene.2020.560368
Received: 08 May 2020; Accepted: 15 October 2020;
Published: 09 December 2020.
Edited by:
Zhiyong Liu, Shenyang Agricultural University, ChinaReviewed by:
Joel Sharbrough, New Mexico Institute of Mining and Technology, United StatesCopyright © 2020 Rono, Dong, Yang, Mutie, Oulo, Malombe, Kirika, Hu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guang-Wan Hu, SHVndWFuZ3dhbmh1QHdiZ2Nhcy5jbg==; Z3Vhbmd3YW5odUBzb2h1LmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.