
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 19 October 2020
Sec. Livestock Genomics
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.550515
This article is part of the Research Topic Epigenetic Variation Influences on Livestock Production and Disease Traits View all 24 articles
 Mengqi Wang1,2,3
Mengqi Wang1,2,3 Yan Liang1
Yan Liang1 Eveline M. Ibeagha-Awemu3
Eveline M. Ibeagha-Awemu3 Mingxun Li1,2
Mingxun Li1,2 Huimin Zhang2
Huimin Zhang2 Zhi Chen2
Zhi Chen2 Yujia Sun2
Yujia Sun2 Niel A. Karrow4
Niel A. Karrow4 Zhangping Yang1,2
Zhangping Yang1,2 Yongjiang Mao1,2*
Yongjiang Mao1,2*Staphylococcus aureus intramammary infection is one of the most common causes of chronic mastitis in dairy cows, whose development may be associated with epigenetic changes in the expression of important host defense genes. This study aimed to construct a genome-wide DNA methylation profile of the mammary gland of Chinese Holstein cows (n = 3) following experimentally induced S. aureus mastitis, and to explore the potential gene regulatory mechanisms affected by DNA methylation during S. aureus mastitis. DNA was extracted from S. aureus-positive (n = 3) and S. aureus-negative (n = 3) mammary gland quarters and subjected to methylation-dependent restriction-site associated DNA sequencing (Methyl-RAD Seq). Results showed that CmCGG/CmCWGG DNA methylation sites were unevenly distributed and concentrated on chromosomes 5, 11, and 19, and within intergenic regions and intron regions of genes. Compared with healthy control quarters, 9,181 significantly differentially methylated (DM) CmCGG sites and 1,790 DM CmCWGG sites were found in the S. aureus-positive quarters (P < 0.05, |log2FC| > 1). Furthermore, 363 CmCGG differently methylated genes (DMGs) and 301 CmCWGG DMGs (adjusted P < 0.05, |log2FC| > 1) were identified. Gene ontology and KEGG enrichment analysis indicated that CmCGG DMGs are involved in immune response pathways, while the CmCWGG DMGs were mainly enriched in gene ontology terms related to metabolism. The mRNAs of 526 differentially methylated CmCGG genes and 124 differentially methylated CmCWGG genes were also significantly differentially expressed (RNA-Seq data) in the same samples, herein denoted differentially methylated and expressed genes (DMEGs) (P < 0.05). Functional enrichment analysis of DMEGs revealed roles related to biological processes, especially the regulation of immune response to diseases. CmCGG DMEGs like IL6R, TNF, BTK, IL1R2, and TNFSF8 enriched in several immune-related GO terms and pathways indicated their important roles in host immune response and their potential as candidate genes for S. aureus mastitis. These results suggest potential regulatory roles for DNA methylation in bovine mammary gland processes during S. aureus mastitis and serves as a reference for future epigenetic regulation and mechanistic studies.
Mastitis, which is defined as inflammation of the mammary gland, is one of the most common production-limiting diseases of the dairy industry (Beniæ et al., 2018). Mastitis not only has significant negative impacts on milk production, including decreased milk yield and milk quality (Detilleux et al., 2015), but also influences the reproductive system (Kumar et al., 2017) as well as increases susceptibility to other diseases (Cha et al., 2013). Mastitis is caused by varied pathogens leading to the development of subclinical/chronic (25–65% incidence worldwide) or clinical (∼5% incidence worldwide) infections (Dufour et al., 2012). Staphylococcus aureus (S. aureus), a Gram positive bacterium, is the major cause of chronic mastitis and accounts for up to 64% of total mastitis cases in Canada (Reksen et al., 2006). S. aureus mastitis is usually asymptomatic, persistent, easily reoccurs and some S. aureus isolates have developed antibiotic resistance (Makovec and Ruegg, 2003; Jensen et al., 2013). Preventive measures to control the occurrence and spread of S. aureus IMI (intramammary infections) are hampered by false-positive bacterial diagnostic tests, sudden outbreaks of IMI and poor therapeutic response to antibiotic treatment (Sampimon et al., 2009).
Mastitis results from the interaction between environment (pathogens) and genetic factors. Attempts to control mastitis through improved farm environment and management practices, and application of antibiotics have not eradicated the disease. The possibility to use genetic selection to improve mastitis resistance was born when heritable genetic variation in the incidence of mastitis was demonstrated (Lush, 1950). However, the use of conventional breeding strategies based on phenotypic selection to eradicate mastitis have shown limited success due to the low heritability of the trait (Emanuelson et al., 1988; Carlén et al., 2004; Koivula et al., 2005; Vallimont et al., 2009). Genomic selection has recently been identified as a complementary or alternative tool for selection to reduce mastitis (Martin et al., 2018; Weigel and Shook, 2018). To support genomic selection, several genome wide association studies (GWAS) have identified genes, markers and quantitative trait loci (QTL) significantly associated with mastitis (Wang X. et al., 2015; Suravajhala and Benso, 2017; Cai et al., 2018; Welderufael et al., 2018; Kurz et al., 2019).
In recent years, attention has been devoted to the study of transcriptome and post-transcriptional regulatory mechanisms of S. aureus mastitis, with aim to find additional biomarkers from different perspectives, for use in genomic selection, molecular diagnostics and for therapeutic purposes (Wang et al., 2016; Luoreng et al., 2018; Han, 2019). Furthermore, growing evidence demonstrates that epigenetics also profoundly influence health, growth, development and production traits of cattle (Ibeagha-Awemu and Zhao, 2015; Reynolds et al., 2017; Sun et al., 2019). Several studies have reported the potential contribution of epigenetic mechanisms like DNA methylation (Song et al., 2016; Zhang et al., 2018), histone modification (He et al., 2016) and non-coding RNA (Li et al., 2015; Pu et al., 2017; Ma et al., 2019) to mastitis risk. As the most characterized epigenetic regulatory mechanism, DNA methylation was reported to regulate mammary gland health (Zhang et al., 2018; Chen et al., 2019; Wang et al., 2019). For example, hypermethylation of CD4 gene promoter was reported to repress its expression during clinical mastitis (Wang et al., 2013; Usman et al., 2016). Genome-wide DNA methylation analysis of peripheral blood lymphocytes identified 1,078 differentially methylated genes between cows with S. aureus-mastitis and healthy cows, as well as the potential of NRG1, MST1, and NAT9 genes to serve as biomarkers of S. aureus mastitis progression (Song et al., 2016). Furthermore, DNA methylation was found to regulate the expression of bovine immune-related genes, including IL6, IL8, and TLR4, in response to lipopolysaccharide-induced mastitis (Korkmaz, 2018). Additionally, abnormal expression of IL6 was revealed to be due to altered DNA methylation rather than genetic mutation during clinical mastitis (Zhang et al., 2018). Moreover, methylation changes in NCKAP5 and transposon MTD were associated with the development of S. aureus mastitis (Wang et al., 2020), while hypomethylation of global DNA methylation induced by S. aureus infection repressed DNA methyltransferase activity in bovine mammary epithelial cells (Wu et al., 2020).
DNA methylation occurs mainly in the form of CpG (addition of methyl group to the fifth position of cytosine base followed by guanosine), and to a lesser extend at non-CpG sites, including CpA, CpT, and CpC. However, the functions of non-CpG sites are not yet elucidated or still controversial (Patil et al., 2014). Methods used for whole genome DNA methylation profiling include whole genome sequencing approaches (e.g., whole genome bisulfite sequencing) and enrichment based immunoprecipitation or enzyme based methods that reduce genome complexity through enzymatic digestion followed by bisulfite treatment and next generation sequencing (Li et al., 2010). Methyl-RAD Seq is one of such enzymatic based methods that uses methylation-dependent restriction enzymes (FspEI, MspJI, etc.) to reduce genome size and focus the analysis on methylation rich regions (Wang S. et al., 2015). Methyl-RAD Seq has been improved in technological ways making it more advantageous than other enzymatic based methods such as reduced representation bisulfite sequencing (RRBS) (Gu et al., 2011), MeDIP-seq (Taiwo et al., 2012), MethylCap-seq (Brinkman et al., 2010) and MethylSeq (Brunner et al., 2009). CmCGG and CmCWGG (W = T or A) are two primary target sites of methylation-dependent restriction enzymes used for some DNA methylation sequencing approaches including Methyl-RAD Seq, providing the data for next-step functional analysis for both CpG and non-CpG sites (Wang S. et al., 2015).
The reports above support the hypothesis that DNA methylation contributes to susceptibility to, or outcome of S. aureus mastitis. However, the global DNA methylation pattern of mammary gland tissue from dairy cows infected with S. aureus mastitis is ambiguous, and related functional reports are scarce. To further test this hypothesis, the aim of the present study was to profile the genome-wide DNA methylation landscape of mammary gland tissue of Holstein cows, and to identify differentially methylated sites and genes and their potential involvement in the host response to S. aureus-induced mastitis. Data from this study is expected to provide reference for further functional studies and mechanistic exploration of epigenetic mechanisms underlying S. aureus mastitis and to contribute to improve management of S. aureus infection in dairy herd.
Three primiparous Holstein cows in mid-lactation (142 ± 25 days) and without a history of disease were selected from Yangzhou University Dairy Farm (Jiangsu, China) for this study. Cows were in their first parity and were under routine farm management and feeding regime. The milk somatic cell counts (SCC) of cows was monitored for 3 weeks prior to the experimental IMI challenge. Furthermore, milk samples were collected weekly and cultured at 37°C for 24 h on Aureus ChromoSelect Agar Base Media (HiMedia, Mumbai, India) to detect the presence of S. aureus. Only cows negative for S. aureus and having a SCC of ≤100,000 cells/mL were used (Rainard et al., 2018). The S. aureus strain Rosenbach ATCC®29213TM (ATCC, Manassas, VA, United States) was used for the IMI challenge as described previously (Li et al., 2015). Briefly, 5 mL S. aureus suspension (1 × 107 CFU, SA treatment) was injected into one mammary gland quarter of each of the three cows, while the same volume of aseptic vehicle phosphate buffer (PBS) solution was injected into another quarter of each of the same three cows (CL treatment). Milk samples were aseptically collected from each quarter just before S. aureus IMI challenge and then at 6, 12, 18, and 24 h post IMI challenge. Quantification of SCC and test for the presence of S. aureus in milk samples were done as described previously with three experimental repeats for each sample from each sampling time (Li et al., 2015). Mammary gland tissues were surgically collected at 24 h post IMI challenge from each quarter and immediately sectioned into small pieces (around 50 mg) and snap frozen and stored in liquid nitrogen. All the above animal experiments followed the guide of animal welfare and use procedures of Yangzhou University and permission to carry out the study was approved by the Institutional Animal Care and Use Committee of Yangzhou University (NO: 201404018).
DNA was extracted from three S. aureus positive mammary tissues (SA treatment) and three control tissues (CL treatment) using QIAamp DNA mini kit (Qiagen Inc., Shanghai, China) according to manufacturer’s protocol. The concentration and purification of DNA samples were measured with a NanoDrop ND-1000 (NanoDrop Technologies, Wilmington, DE, United States) and confirmed by agarose gel.
Preparation of genome-wide DNA methylation sequencing libraries was carried out according to method reported earlier (Wang S. et al., 2015) using Methyl-RAD Seq method. Briefly, DNA was digested with 4 U FspEI (NEB) at 37°C for 4 h followed by adaptor ligation and PCR amplification. Barcodes were introduced through PCR for multiplex sequencing. The sequences of primers, adaptors and barcodes are listed in Supplementary Table S1A. The PCR products were purified using the QIAquick PCR Purification Kit (Qiagen Inc., Shanghai, China), and single-end sequenced (1 × 36 bp) on an Illumina HiSeq 2500 system (Illumina, San Diego, United States). Sequencing was performed to achieve a coverage depth of 10x.
The raw sequences were trimmed with Trimmomatic (v 0.36) (Bolger et al., 2014) to remove adaptor sequences. Reads with more than 8% ambiguous bases, or more than 15% bases with a quality score of less than 30 were also trimmed. Clean reads that passed quality control and with expected restriction sites were defined as enzyme reads and were used for subsequent analyses. Sequences containing CmCGG or CmCWGG (W = T or A) (methylation on second C) with FspEI restriction sites were extracted from the bovine reference genome UMD 3.1.1 (GCF_000003055.6 Bos Taurus UMD 3.1.1) and used as reference for methyl-RAD data analysis. Enzyme reads were mapped to the reference sequences with SOAP (v2.21) (Li et al., 2008) allowing two mismatches (-M 4 -v 2 -r 0). The methylation sites that were found in all SA and/or CL samples and with >3× coverage were retained for further analyses. The normalized read-depth (reads per million, RPM) was used as the unit for relative quantification of level of DNA methylation. SnpEff (v4.3p) (Cingolani et al., 2012) was used to obtain information on untranslated regions (UTR) from the bovine reference genome (UMD 3.1.1). The distribution of mapped enzyme reads to different genic regions {3′ UTR, 5′ UTR, Upstream [2,000 bp upstream of transcription start site (TSS)], exon, intron and downstream [2,000 bp downstream of transcription termination site (TTS)]} and intergenic regions was achieved with bedtools (v 2.25.0) (Quinlan and Hall, 2010).
The methylation level of a gene was represented by the sum of the sequencing reads of all its methylated sites. Differential methylated gene (DMG) analysis was accomplished with EdgeR (v 3.26.4) (Robinson et al., 2010). DMGs meeting the thresholds of adjusted P-value < 0.05 and |log2FC| > 1 were considered significant. Significantly DMGs were subjected to gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways functional enrichment analysis with the ClusterProfile (v 3.8) program in R package (Yu et al., 2012).
The DMGs (adjusted P-value < 0.05) were filtered against the mRNA transcriptome data of the same samples (Liao, 2014; Chen et al., 2019, b) to select the differentially methylated genes that were also transcriptionally differentially- expressed (adjusted P-value < 0.05); these will subsequently be referred to as differentially methylated and expressed genes (DMEG). The DMEGs were used for GO and KEGG pathway enrichment with the ClusterProfiler package in R software, and network visualization was performed with ClueGO1 (Bindea et al., 2009).
To validate the reliability of Methyl-RAD Seq data, bisulfite sequencing PCR (BSP) was used to identify the methylation level of three DMEGs (CXCR1, CDH13, and METTL13). The Methyl primer express (v1.0) was used to design the PCR primers (Supplementary Table S1B). Genomic DNA (around 1,000 ng) from each tissue sample was treated with bisulfite sodium using EpiTect Plus Bisulfite kit (Qiagen, Shanghai, China). The BSP reaction was performed in 20 μL, including 1 μL (10 ng) of bisulfite-treated DNA, 1 μL of forward and reverse primer (10 μmoL each), 10 μL of 2 × T5 super PCR MIX (Qingke, Beijing, China) and 7 μL of RNase-free water. The PCR program was as follows: 94°C for 5 min; 40 cycles of 94°C for 5 s, 58°C for 30 s and 72°C for 30 s; and 72°C for 10 min. The PCR product size and purity were confirmed by agarose gel electrophoresis and then cloned into the pMD19-T vector (Shenggong, Shanghai, China). About 20 positive clones for each gene and sample were randomly selected for sequencing (Shenggong, Shanghai, China). The BiO_Analyzer2 was used to analyze the sequence results.
The DNA methylation levels of CmCGG and CmCWGG sites between SA and CL groups were compared with Fisher’s exact Test implemented in EdgeR (v 3.26.4) program. The enrichment score in the gene ontology (GO) analysis in ClusterProfile (v 3.8) was calculated with the following formula:
Where N represents the number of all genes with GO annotation, n is the number of DMGs with GO annotation. M and m are the number of all genes and DMGs annotated in a specific GO term, respectively. Hypergeometric contribution test was used to calculate the significance of gene enrichment in each GO term, and adjusted P-value < 0.05 was considered significant.
As compared with the CL treatment (Figure 1A), the infected mammary gland quarters of the SA treatment showed significant symptoms of inflammation (redness, swelling and warmness to the touch) at 24 h after S. aureus challenge (Figure 1B). Furthermore, cows of the SA treatment showed avoidance behavior and refused to be milked when the infected mammary gland quarter was touched. Histological sections of the infected mammary gland quarters showed atrophic acinar wall and damaged acinar structure with lots of impurities gathered in the acini, that could be exfoliated mammary epithelial cells and inflammatory cells (Figure 1D) as compared to uninfected quarters (Figure 1C).
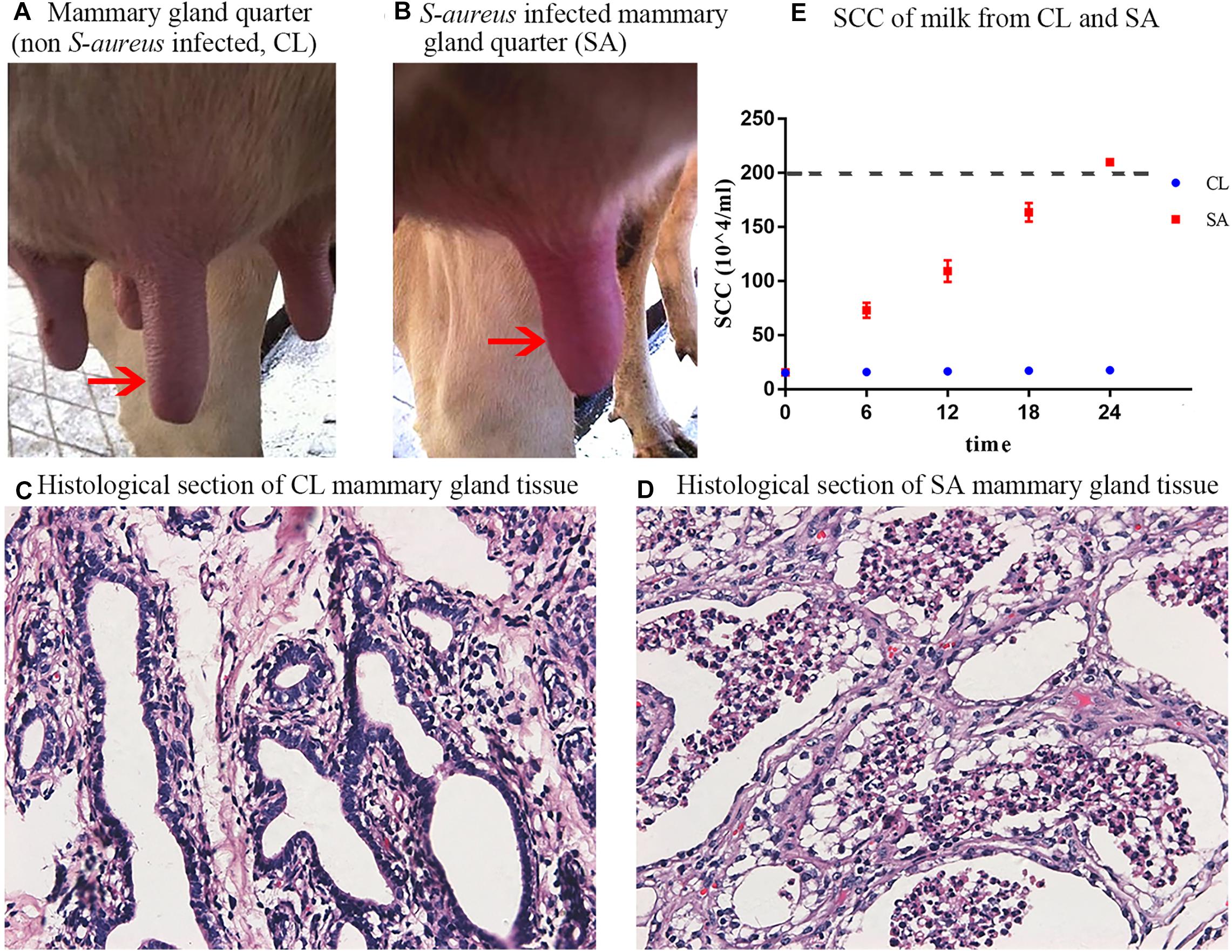
Figure 1. Clinical observation and diagnosis of S. aureus-induced mastitis. (A) Healthy mammary gland quarter; (B) Mammary gland quarter with clinical symptoms: red, swollen and indurated, 24 h after S. aureus induction; (C) Histological section of healthy mammary gland quarter; (D) Histological section of S. aureus-infected mammary gland quarter, showing atrophic acinar wall and damaged acinar structure with lots of magazines gathered in the acini; (E) Individual SCC of milk collected from the healthy (CL group: quarters A1, B1, and C1) and S. aureus-infected (SA group: quarters A2, B2, and C2) mammary gland quarters during the 24 h post S. aureus induction. The red arrows shows the control (A) and challenged (B) mammary gland quarters.
Compared with CL, the milk yield of SA treatment decreased, with flocculent precipitate and increased SCC (P < 0.05) (Figure 1E), indicative of mastitis. Bacterial culture revealed that milk samples collected from all the mammary gland quarters before IMI challenge were S. aureus negative, whereas after IMI, S. aureus was cultured from the SA treatments but not in the CL treatments. In summary, these results confirmed successful induction of S. aureus-induced mastitis in the challenged quarters and a healthy state in control quarters.
The level of genome-wide DNA methylation was analyzed by Methyl-RAD Seq. A total of 81,125,685 raw reads were generated from the six samples, with an average of 13,520,947 raw reads per sample (Table 1). Reads that mapped to multiple positions (mapping rates of 67.7 to 70.13% of clean reads) were removed, only uniquely mapped reads (mapping rates of 26.93 to 29.09%) were kept for further analysis. The CmCGG and CmCWGG (W = A or T) methylation maps were generated to show the genome-wide DNA methylation distribution in the studied samples (Figure 2 and Supplementary Table S2). The CmCGG and CmCWGG methylation sites were unevenly distributed across bovine chromosomes (BTA); BTA with the most CmCGG methylation sites were BTA 11, 5, and 19, while CmCWGG methylation sites were most distributed on BTA 5, 11, and 1. Additionally, CmCGG and CmCWGG methylation sites were more concentrated at the BTA ends.
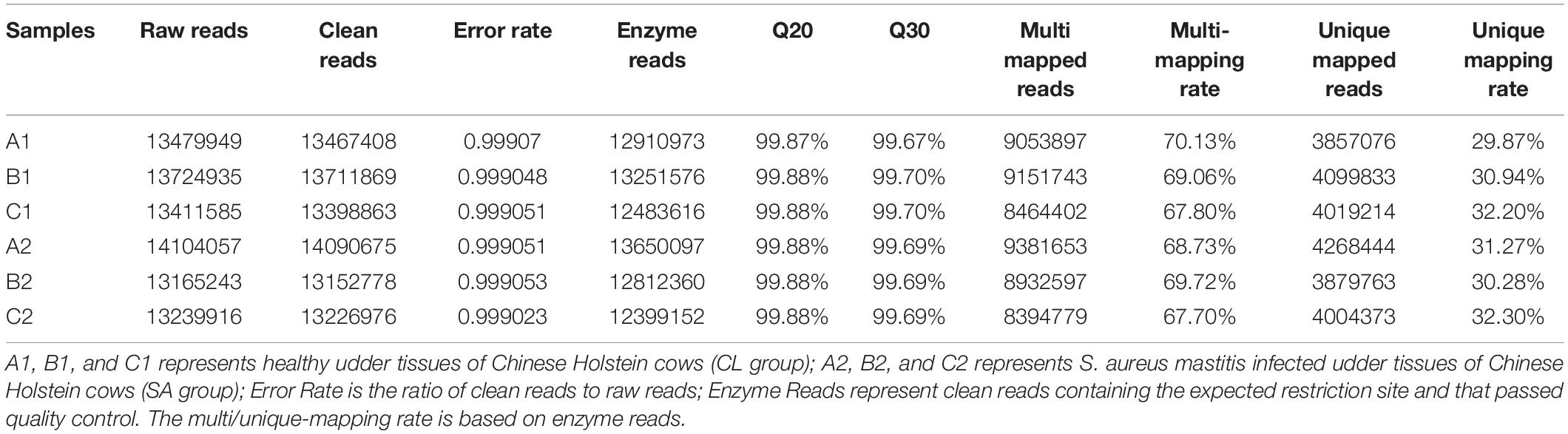
Table 1. Generated reads and mapping statistics of Methyl-RAD Seq sequence data from Staphylococcus aureus induced and healthy udder tissues of Chinese Holstein cows.
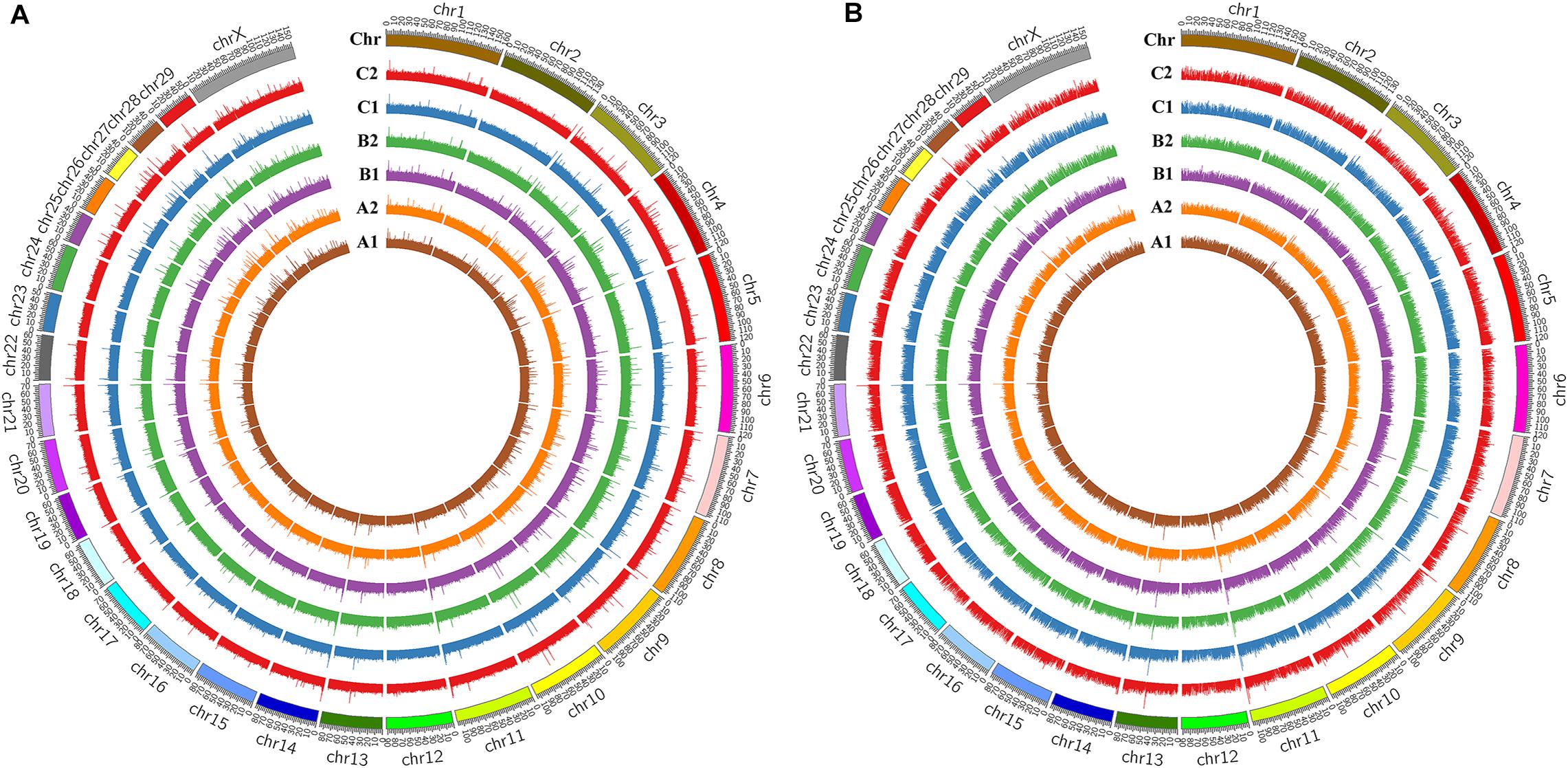
Figure 2. DNA methylation profiles of mammary gland tissues from Chinese Holstein cows. (A) CmCGG methylation map; (B) CmCWGG methylation map (W = A or T); CmCGG means the methylation whose primary target site is CmCGG, while CmCWGG means the methylation with CmCWGG site (W = A/T/H); A1, B1, and C1 indicate the whole genome wide DNA methylation patterns of healthy mammary gland quarters, while A2, B2, and C2 indicate quarters with S. aureus-induced mastitis.
To further understand the distribution of DNA methylation sites in the studied mammary gland tissues, gene body, upstream and downstream of genes were analyzed. As shown in Figure 3, the methylation level in gene bodies was higher than in the regions before the TSS and after the TTS and a sharp increase was observed around the TSS and a decrease around the TTS. Moreover, the distribution of DNA methylation sites in different functional gene elements showed enrichment in intergenic regions and introns, followed by exons and upstream regions, while the 3′UTRs and 5′UTRs had less methylation sites (Figure 4).
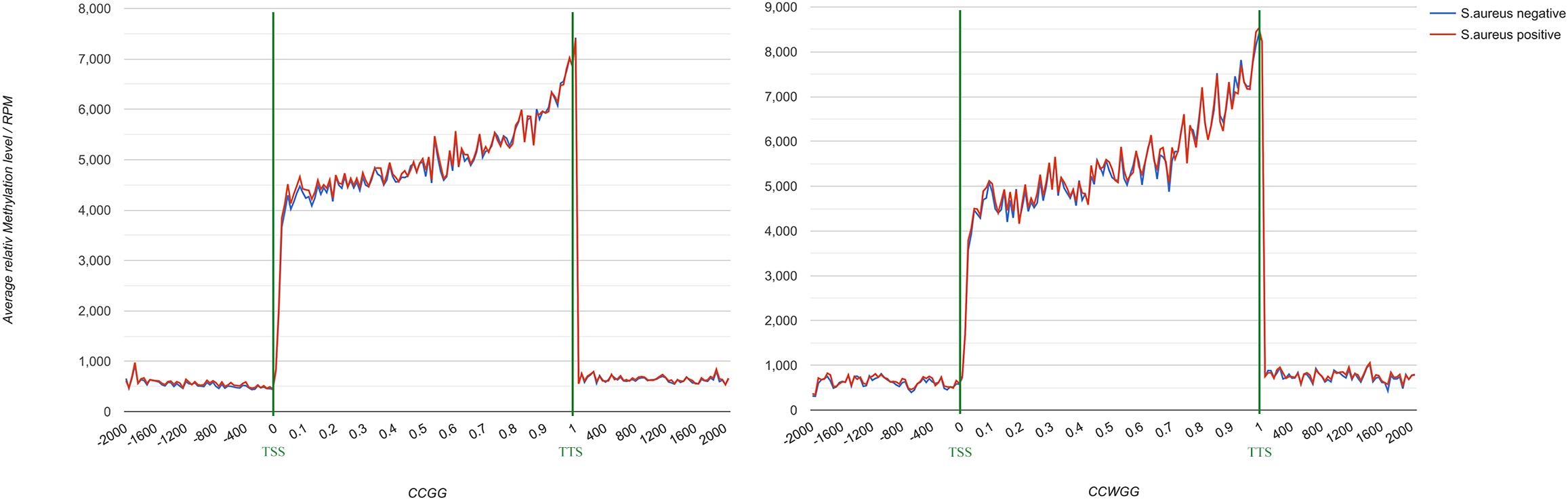
Figure 3. DNA methylation level across gene in mammary gland quarters from Chinese Holstein cows. TSS, transcriptional start site; TTS, transcriptional termination site; Abscissa represents the gene body, expressed as the percentage of gene length (from 0 to 1),the region of 2000 bp before TSS (from –2000 bp to the start of gene body marked as 0), and 2000 bp after TTS [following the end of gene body (marked as 1) until 2000 bp]. Ordinate represents the average relative DNA methylation level (RPM, reads per million) and different colors means different groups.
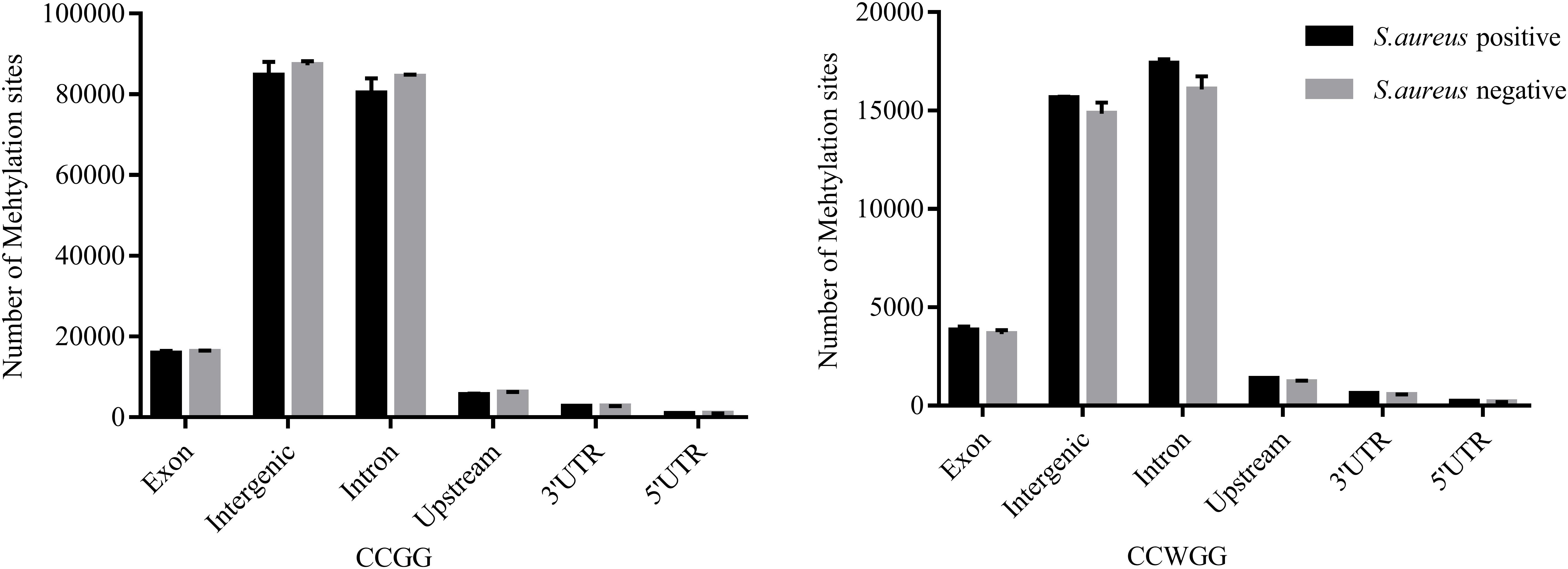
Figure 4. The distribution of DNA methylation sites in functional gene elements. Abscissa represents functional gene elements, and upstream represents 2000 bp region before transcriptional start site. Ordinate represents the number of DNA methylation sites.
Exploration of the DNA methylation variations between S. aureus mastitis and healthy control tissues, showed significant CmCGG/CmCWGG methylation differences between the two groups (Figures 5A,B). A total of 9,181 CmCGG methylation sites and 1,790 CmCWGG methylation sites were differentially methylated between SA group and CL group (Supplementary Table S3). Furthermore, differentially methylated sites were unevenly distributed among chromosomes, with higher density of sites on BTA 5, 11, and 19 (Supplementary Table S4). All the methylation sites were annotated to genes to further assess the DNA methylation variations. A total of 363 CmCGG DMGs and 301 CmCWGG DMGs were obtained (Supplementary Table S5), including 28 DMGs common to the two DNA methylation types, which showed enrichment on BTA 5, 7, 11, 18, and 19 (Figures 5C,D and Supplementary Table S4).
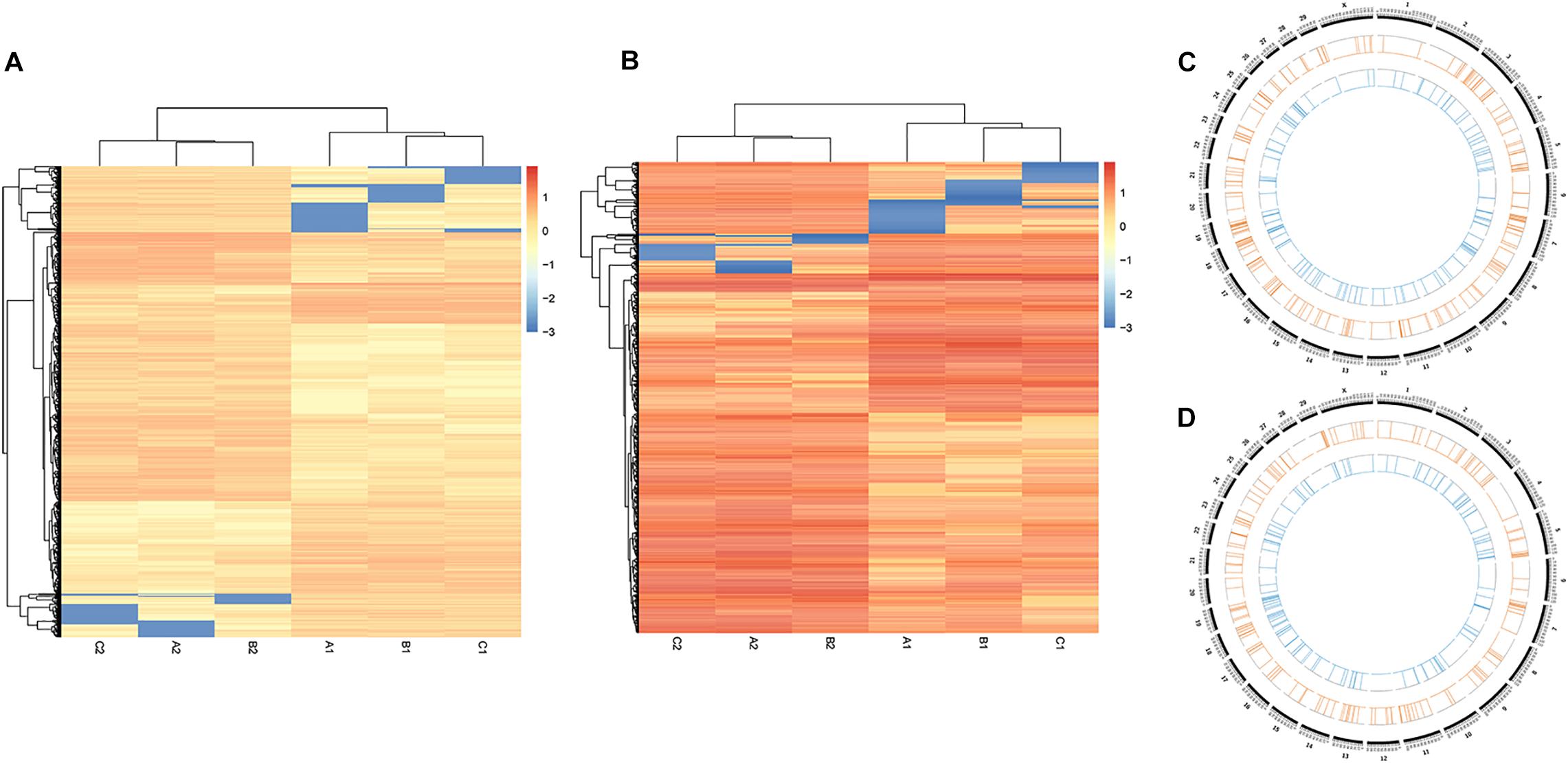
Figure 5. Cluster analysis of DNA methylation levels in healthy and S. aureus-induced mammary gland tissues. (A,B) are the heat maps for CmCGG and CmCWGG methylation, respectively. The colors and intensity indicate the methylation level in the heat map. For example, highly methylated sites are in red color and hypomethylated sites are in blue color. (C) The distribution of CmCGG differently methylated genes across chromosomes, while (D) shows the distribution of CmCWGG differently methylated genes. Blue lines in the inner circle represent genes with significantly down-regulated methylation levels, while the orange lines in the next circle represent genes with significantly up-regulated methylation levels.
Investigation of the potential functions of DMGs through GO and KEGG pathways enrichment analyses revealed that 580 GO terms were significantly enriched by CmCGG DMGs, while CmCWGG DMGs were significantly enriched in 460 GO terms (adjusted P-value < 0.05, Supplementary Table S6). For the CmCGG DMGs, the most significant biological process (BP), cellular component (CC) and molecular function (MF) GO terms were “hematopoietic progenitor cell differentiation” (GO:0002244), and “cytoplasm” (GO:0005737) and “sequence-specific DNA binding,” respectively (Figure 6A). “Kidney development” (GO:0001822), “integral component of endoplasmic reticulum membrane” (GO:0030176) and “actin binding” (GO:0003779) were the most significant BP, CC and MF GO terms enriched by CmCWGG DMGs, respectively (Figure 6B).
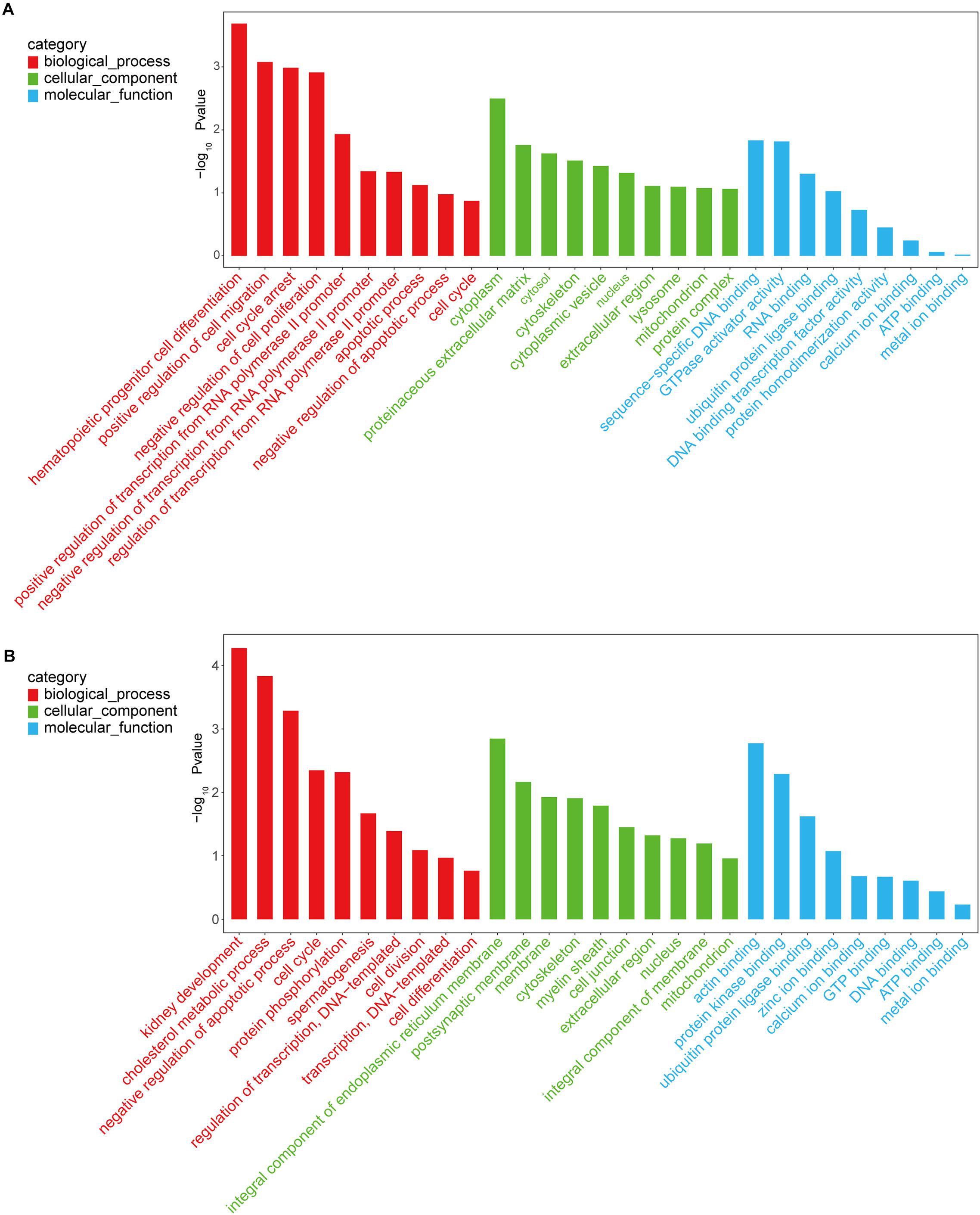
Figure 6. Histogram of gene ontology terms enriched by differently methylated genes. (A) Histogram of CmCGG differently methylated genes; (B) Histogram of CmCWGG differently methylated genes. Ordinate represents the adjusted P-value (-log10 P-value). Abscissa represents the GO terms. Only the top 10 GO terms of each category are listed in ascending order of adjusted P-value.
KEGG pathways enrichment analysis identified 40 pathways for the CmCGG DMGs and 64 pathways for the CmCWGG DMGs (adjusted P-value < 0.05) (Supplementary Table S7). The top 20 significantly enriched pathways in each category are shown in Figure 7. KEGG pathways enriched by CmCGG DMGs were mainly related to diseases and the immune response. The most significantly enriched pathways for CmCGG DMGs were “asthma” (bta05310), “transcriptional mis-regulation in cancer” (bta05202) and “leishmaniasis” (bta05140). Meanwhile, the CmCWGG DMGs enriched KEGG pathways were more related to biosynthesis and metabolism. “N-Glycan biosynthesis” (bta00510), “glycosphingolipid biosynthesis” (bta00601) and “galactose metabolism” (bta00052) were the top most significantly enriched pathways for CmCWGG DMGs.
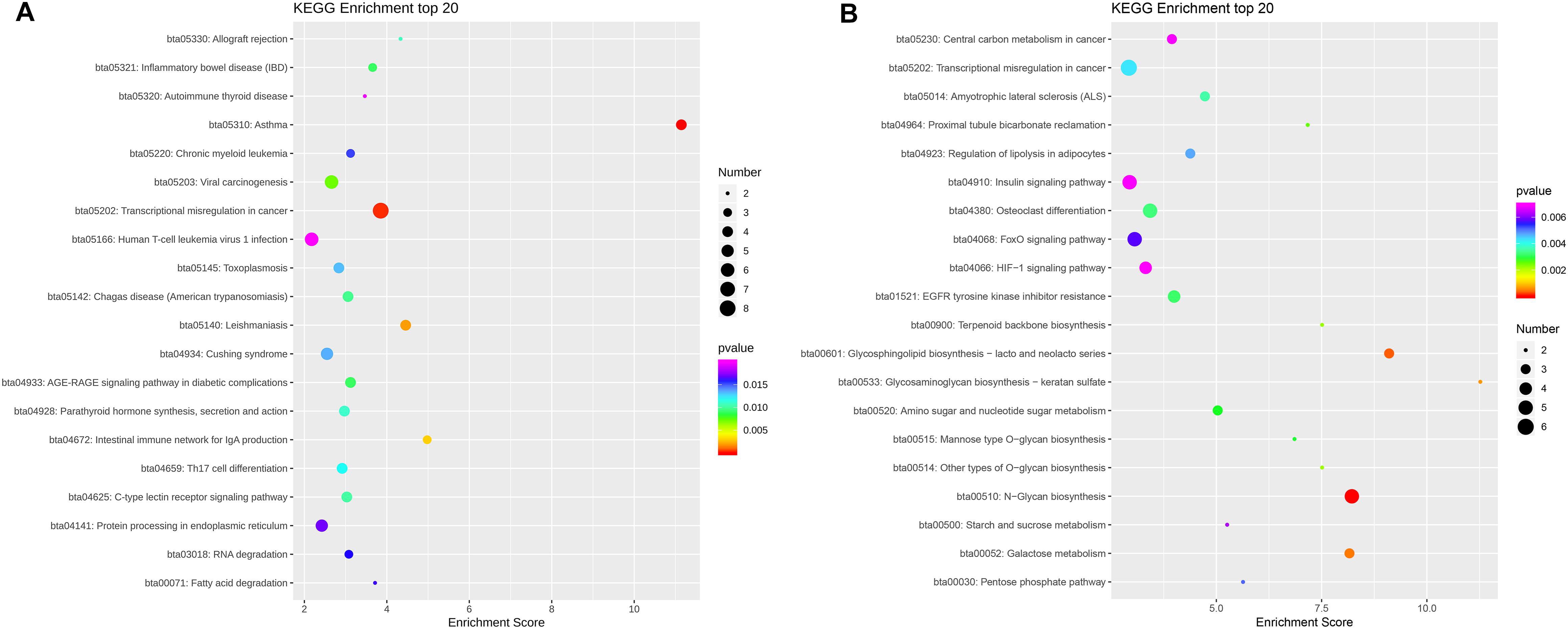
Figure 7. Scatter plot of KEGG pathways enriched by differently methylated genes. (A) Scatter plot of CmCGG differently methylated genes; (B) Scatter plot of CmCWGG differently methylated genes. Ordinate represents the significantly enriched KEGG pathways. The abscissa represents the corresponding enrichment score. The color of the spot indicates the adjusted P-value of each pathways, while the size of the spot indicates the number of differently methylated genes enriched in that pathway. A bigger enrichment score means better enrichment. Only the top 20 KEGG pathways are listed in ascending order of adjusted P-value.
The next step was to identify DMEGs, this was achieved by cross-comparing the above DMGs with those that were also previously found to be differentially transcriptionally expressed (Liao, 2014; Chen et al., 2019, b). As shown in Figure 8, 526 CmCGG and 124 CmCWGG DMEGs were identified. Functional enrichment of the CmCWGG DMEGs indicated that only two GO terms [endochondral bone growth (GO:0003416) and calcium ion transmembrane import into cytosol (GO:0097553)] were significantly enriched by seven DMEGs. There were 430 CmCGG DMEGs that were significantly enriched in 480 GO terms (adjusted P < 0.05, Supplementary Table S8A). The top three most significantly enriched GO terms were “cell adhesion” (GO:0007155), “cell activation” (GO:0001775) and “leukocyte cell-cell adhesion” (GO:0007159). Several enriched (adjusted P < 0.05) GO terms were related to cell proliferation, differentiation, migration and adhesion of immune-related cells, such as T cell, lymphocyte and leukocyte, etc (Table 2 and Supplementary Table S8A). Furthermore, GO terms related to the immune response and regulation processes were also enriched by CmCGG DMEGs, including “regulation of immune system process” (GO:0002682), “myeloid cell activation involved in immune response” (GO:0002275), etc. Some CmCGG DMEGs such as BTK, TNF, TNFAIP8L2, IL6R, and IL18R1, were enriched in multiple immune processes related GO terms (Supplementary Table S9). Meanwhile, 44 KEGG pathways were significantly enriched (adjusted P < 0.05) for CmCGG DMEGs. “Focal adhesion” (KEGG:04510) was the most significantly enriched pathway (adjusted P = 2.52 × 10–5) (Figure 9 and Supplementary Table S8B). The “T cell receptor signaling pathway” (KEGG:04660), “primary immunodeficiency” (KEGG:05340) and other pathways related to cancers and other diseases were enriched (adjusted P < 0.05). A total of 102 immune-related CmCGG DMEGs enriched in 82 GO terms and 3 KEGG pathways related to immune response, including IL6R, TNF, BTK, IL1R2, and TNFSF8, are listed in Supplementary Tables S9A,B.
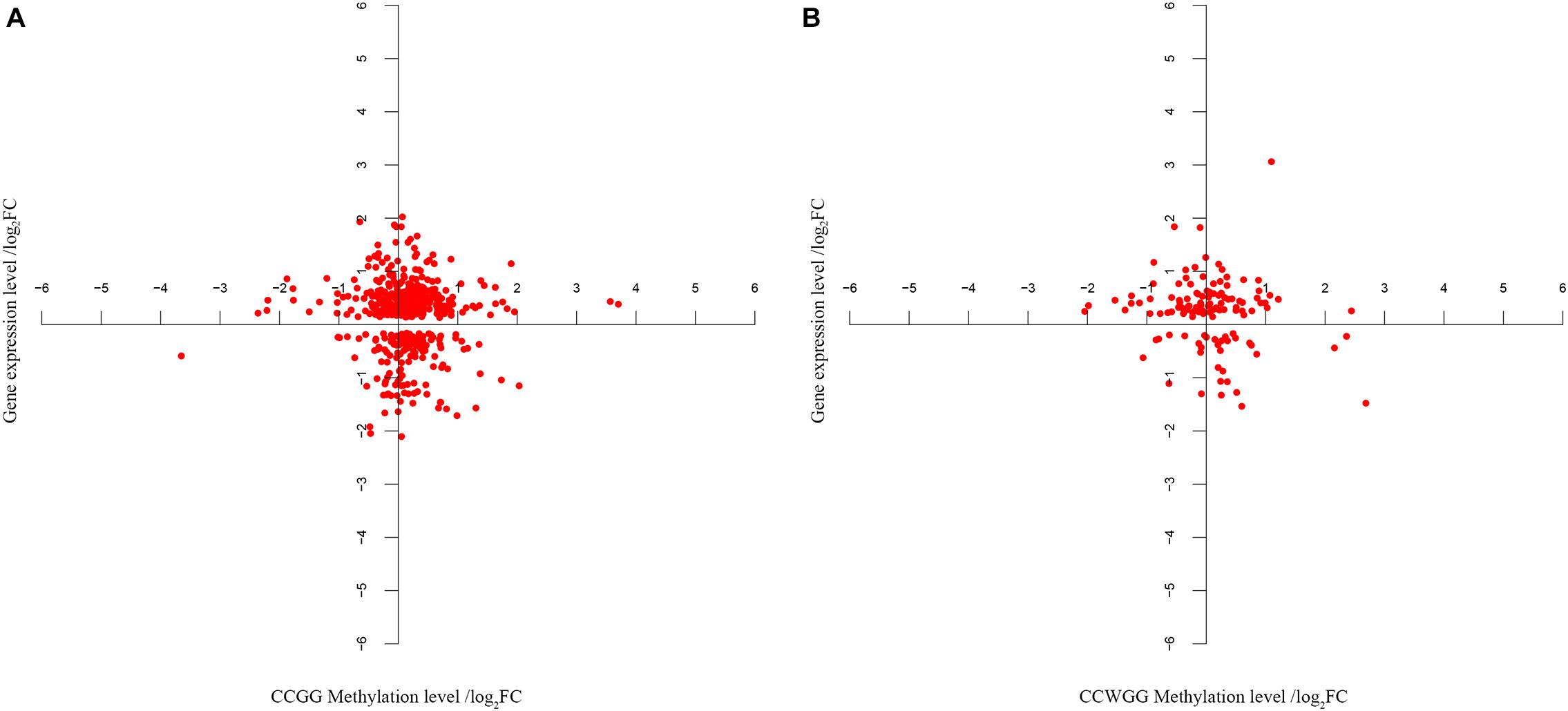
Figure 8. (A,B) The co-expression analysis of gene expression and DNA methylation level for DMEGs. Each point in the figure represents one DMEG, the differently methylated gene whose gene expression level is also significantly different in the same tissues. Abscissa indicates the CmCGG or CmCWGG methylation level, represented by adjusted fold change (log2FC). Ordinate indicates the gene expression level (log2FC).
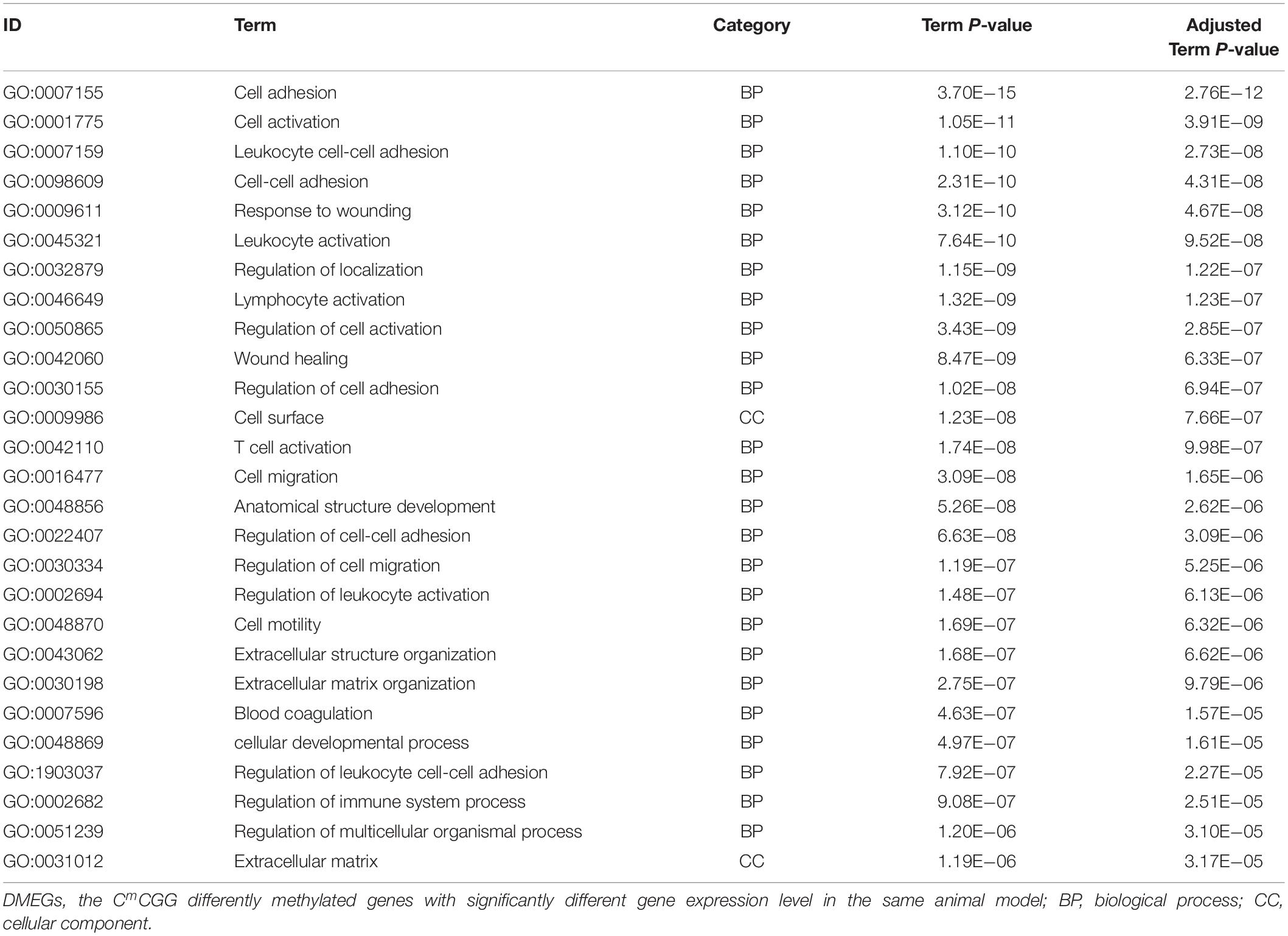
Table 2. Top 30 significantly enriched gene ontology terms for CmCGG DMEGs.
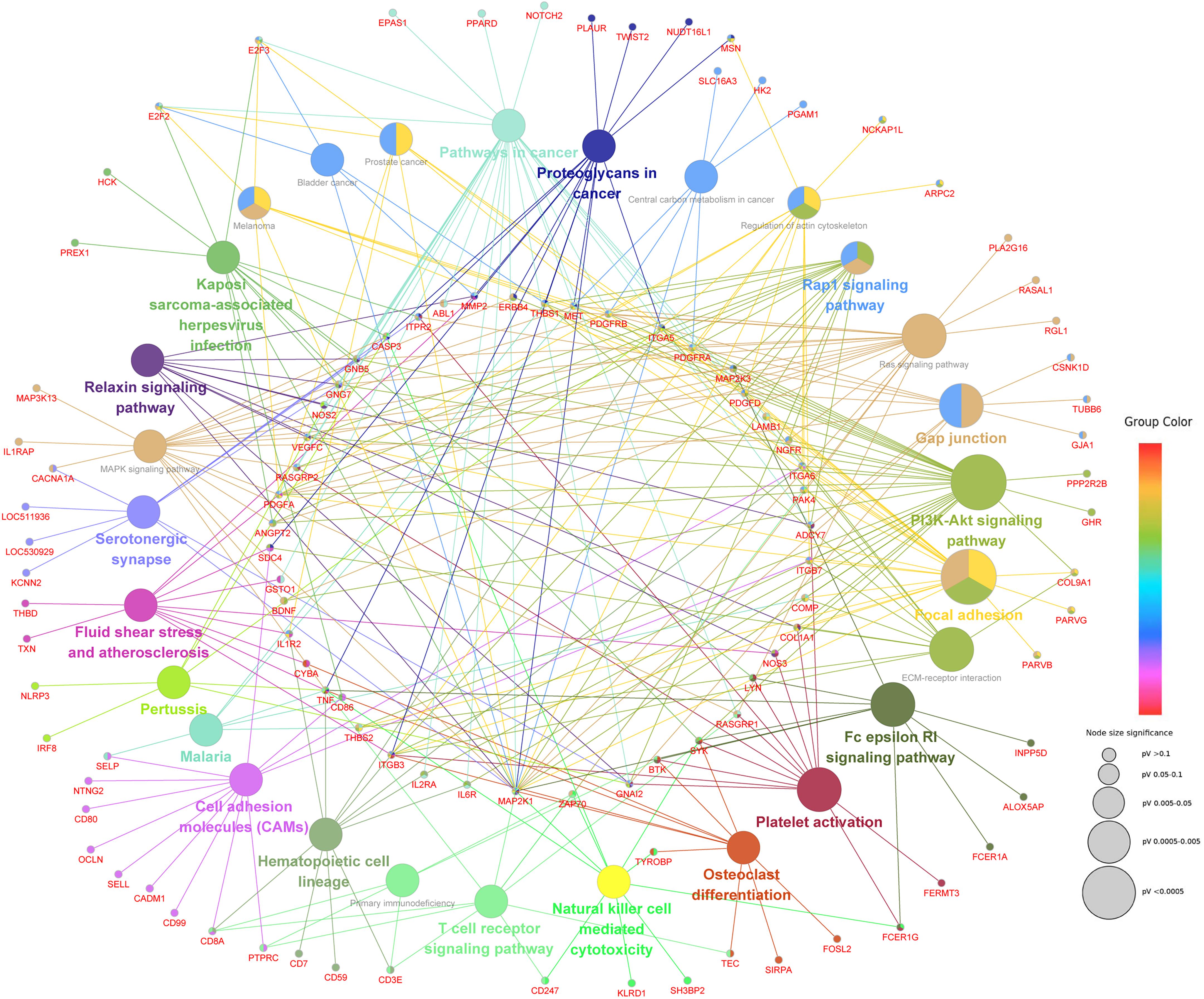
Figure 9. The network of KEGG pathways significantly enriched by CmCGG DMEGs. Each node represents a KEGG pathway or a CmCGG DMEG enriched in the pathway. The color of the node indicates the cluster of KEGG pathways, that more similar colors mean closer clusters with more similar functions. The size of node indicates the adjusted P-value of the corresponding pathway, that a bigger size of the node means more significant enrichment.
To assess the reliability and accuracy of the Methyl-RAD Seq data, the methylation levels in promoter regions of three candidate genes were validated by BSP in the SA and CL treatments. CXCR1, CDH13, and METTL13 with high, low and zero methylation levels, respectively, were selected. In general, the BSP results were in good agreement with the Methyl-RAD Seq results. For example, the CXCR1 gene showed high methylation levels (Figure 10A), and the CDH13 gene showed low methylation levels (Figure 10B), while the METTL13 gene was almost completely unmethylated (Supplementary Figure S1) according to both methods.

Figure 10. Bisulfite sequencing PCR results. Columns represent the CpG sites and rows represent the clones. Black and white points represent methylated and unmethylated CpG sites, respectively. The blank region indicate that the CpG site was not detected by BSP. The distribution of sequencing reads detected by Methyl-RAD Seq for two genes in the mammary gland quarters of SA group (A2, B2, and C2) and CL group (A1, B1, and C1) are to the right of each figure. The blue boxes represent two genes, CXCR1 [(A), high methylation level] and CDH13 [(B), low methylation level].
Experimental induction of mastitis by inoculating S. aureus into mammary gland quarters has been used to study the transcriptional and post-transcriptional regulatory immune response mechanisms during IMI (Fang et al., 2016; Enger et al., 2019). This strategy was used in this study whereby one mammary gland quarter was inoculated with S. aureus to induce mastitis (SA treatment), while one quarter of the same animal was inoculated with PBS and served as the control (CL treatment). Typical clinical symptoms of mastitis were observed in the quarters inoculated with S. aureus, such as swelling, redness, warm feeling to the touch, incomplete anatomical structure of acini with exfoliated epithelial cells, as well as significantly increased SCC (>2 million cells/mL) as compared to the healthy PBS quarters, indicating that induction of S. aureus mastitis in the SA treatment was successful.
Methyl-RAD Seq is an improved enzymatic based genome-wide DNA methylation detection method when compared with similar techniques (RRBS, MeDIP Seq, and MethyCap Seq) (Brunner et al., 2009; Taiwo et al., 2012; Huang et al., 2013; Wang S. et al., 2015). Firstly, the process of Methyl-RAD Seq eliminates some enzymatic treatments and purification steps inherent in other methods (RRBS, MeDIP Seq, and MethyCap Seq), which allows application to a wide-range of samples due to it very low DNA input acquirement (about 1 ng). Particularly, the gel purification after DNA digestion is omitted by Methyl-RAD technique, thereby avoiding the denaturation of short fragments (23 or 24 bp) with low melting point in the process of gel electrophoresis (Sanderson et al., 2014). Furthermore, the Methyl-RAD Seq technique is easier and faster, and can be done in a 96-well PCR plate in just 2 days, thus making it an ideal protocol for large-scale methylation detection of numerous samples. Additionally, Methyl-RAD Seq is flexible and users can adjust the label density to meet their specific requirements. Moreover, Methyl-RAD Seq has high specificity, sensitivity and reproducibility with unique abilities to perform qualitative DNA methylation analysis (Huang et al., 2013).
In this study, Methyl-RAD Seq was used for genome-wide DNA methylation profiling of mammary gland tissues of Chinese Holstein cows. Both CmCGG and CmCWGG methylation sites were unevenly distributed among chromosomes with higher density on BTA 5 and 11 in the mammary gland tissues. A previous study also reported that S. aureus-positive cattle had significantly more methylation sites on BTA 11 than S. aureus-negative controls (Song et al., 2016), implying a potential regulatory role of DNA methylation in response to S. aureus mastitis. We observed the intergenic and intron regions had more methylation sites (adjusted P < 0.05) compared with other functional gene elements, which is in agreement with previous studies in other cattle breeds (Song et al., 2016; Fang et al., 2017; Chen et al., 2019, a). Similar to results obtained with other mammalian species (Gim et al., 2015; Song et al., 2016; Zhang X. et al., 2017; Zhang Y. et al., 2017), the methylation level was significantly higher in the gene body than its up- and down-stream regions.
DNA methylation plays an important regulatory role in gene expression, which is also influenced by environmental factors and age (Horvath, 2013; Kanzleiter et al., 2015). It has been noted that knowledge on the spatial and temporal regulation of DNA methylation is necessary for understanding the biology of disease (Bergman and Cedar, 2013; Shin et al., 2015). A total of 9,181 CmCGG and 1,790 CmCWGG methylation sites were identified as differently methylated sites in this study, and were annotated to 363 CmCGG and 301 CmCWGG DMGs (adjusted P-value < 0.05 and |log2FC| > 1). The DNA methylation data in this study was compared with the mRNA transcriptome data generated from these same tissues (Liao, 2014; Chen et al., 2019, b) to explore the effects of DNA methylation on gene expression in response to S. aureus-mastitis. We found that a total of 526 CmCGG and 124 CmCWGG DMGs in this study were also reported as significantly differentially expressed in response to S. aureus-induced mastitis (adjusted P-value < 0.05) (Chen et al., 2019, 2019b), suggesting that DNA methylation may be one of the regulatory mechanisms of gene expression during S. aureus IMI.
DNA methylation of some immune-related genes has been associated with mastitis, such as CXCR1 and CDH13. CXCR1 gene polymorphism was shown to impact milk production and sensitivity to mastitis, and was selected as a candidate marker for improving bovine resistance to mastitis (Verbeke et al., 2015; Pokorska et al., 2016; Siebert et al., 2017). In addition, CpG sites in the promoter region of bovine CXCR1 were found to potentially affect its gene expression during S. aureus-induced mastitis (Mao et al., 2015). Consistent with Mao et al. (2015) the CXCR1 gene DNA methylation level in this study was higher in the CL treatment than in the SA treatment. This is supported by its upregulated expression in the SA treatment (Liao, 2014), suggesting an important role in response to S. aureus mastitis. In contrast, CDH13 was reported to have important regulatory roles in human cancers based on a significant correlation of DNA methylation in the CDH13 promoter with the occurrence, development and prognosis of disease (Yang et al., 2016; Ye et al., 2017; Li Y. et al., 2018). Furthermore, the level DNA methylation in the promoter of CDH13 is often high during cancer processes (Xu et al., 2016; Wang et al., 2018). In this study, the level of DNA methylation in CDH13 was higher in SA treatment than in the CL treatment, suggesting a potential regulatory role during S. aureus mastitis.
In addition to CXCR1 and CDH13, DNA methylation levels of other immune-related genes, including IL6R, IL10, IL17, and NFKBIA, were also significantly different between SA and CL treatments. Moreover, 102 CmCGG DMEGs, including IL6R, TNF, BTK, IL1R2, and TNFSF8 were enriched in several immune-related GO terms, indicating their important roles in host immune response, and their potential as candidate genes for improving resistance to S. aureus mastitis. A higher level of DNA methylation in IL6R was also previously found in mastitis-infected cows; and it was found to regulate the expression of IL6R in part by promoting the inclusion of its alternatively spliced exon 2 (Zhang et al., 2018). In contrast, hypomethylation of IL10 has been reported to promote the incidence of diseases, including Escherichia coli-induced mastitis (Modak et al., 2012) and systemic lupus erythematosus (Lin et al., 2012).
The GO functional enrichment revealed the potential roles of CmCWGG DMGs in biological processes related to cholesterol metabolic process and protein phosphorylation, as well as molecular functions related to protein kinase binding and ubiquitin ligase binding (Figure 8B). Meanwhile, CmCGG DMGs were enriched in several cellular biological processes including cell proliferation, cell differentiation, cell migration, etc., (Figure 8A), suggesting their potential regulatory roles in cell development and diverse biological processes. The CmCGG DMGs were significantly enriched in KEGG pathways related to infectious diseases, including Leishmaniasis, Toxoplasmosis and American trypanosomiasis (Figure 9). S. aureus mastitis is a common infectious disease of dairy cows, and enrichment of these disease pathways in this study suggests regulatory roles of CmCGG DMGs in the host response to S. aureus-induced mastitis. Furthermore, CmCGG DMGs were also enriched in KEGG pathways related to the immune response, such as “Intestinal immune network for IgA production,” “Th17 cell differentiation,” and “C-type lectin receptor signaling pathway.” C-type lectin is important for the regulation of innate immune and inflammatory processes (Del Fresno et al., 2018), and is known to influence host antimicrobial immunity (Dambuza and Brown, 2015). Th17 cell differentiation plays an important role in immune regulation of T cell during specific immune processes (Sordillo, 2011). It was previously reported that Th17 cells showed specific differentiation in S. aureus-mastitis (Zhao et al., 2015), and the diseases and immune-related KEGG pathways in the present study further support the importance of Th17 cells in the host immune response to S. aureus mastitis and the regulatory roles of CmCGG DMGs.
Interestingly, several GO terms and KEGG pathways related to cell adhesion were enriched by CmCGG DMEGs, indicating the important impacts of CmCGG methylation on the process of cell adhesion. Cell adhesion has been noted to influence immune cell function impacting cell proliferation, differentiation and migration as well as host-pathogen interaction (Samanta and Almo, 2015; Li J. et al., 2018). The CmCGG DMEGs were enriched in GO terms related to inflammatory response, immune system development, regulation of immune response as well as other immune response-regulating signaling pathways indicating their potential regulatory roles in response to S. aureus-infection. In addition, we found that CmCGG DMEGs were significantly enriched in pathways related to activities of immune cells, such as leukocyte, lymphocyte and T cell, which are important effector cells of the immune system, thus suggesting important roles for these methylated genes in the host immune response to S. aureus-mastitis. Lymphocytes are important members of the acquired immune system (Hoekstra et al., 2015) and lymphocyte proliferation and viability are detrimentally affected by S. aureus (Nonnecke and Harp, 1988; Hu et al., 2001). T cells activated by monocyte-derived dendritic cells play key roles in immune response to S. aureus (Bharathan, 2016). He et al. (2016) also reported that differently expressed genes due to subclinical S. aureus mastitis in cattle are involved in the T cell receptor-signaling pathway. These reports are supported by the enriched GO terms related to the immune response regulation and immune system, and KEGG disease and immune related pathways observed in this study.
This study constructed the landscape of genome wide DNA methylation of healthy and S. aureus infected mammary gland tissues of Holstein dairy cows, providing basic functional prediction of altered methylation sites or genes in relation to S. aureus mastitis. However, the relatively small sample size (n = 3) limits the reliability of the data. But a major advantage of the data is the fact that infected and health mammary gland quarters where from the same cows which provided room for valid comparisons of methylated sites and also eliminated the bias due to different genetic constitution of infected and control cows if a different set of challenged and control cows were used. Although the sequencing technology (Methyl-RAD) employed for this study had many advantages, such as higher sensitivity and accuracy, the reliability and reference value, but is still limited because of the short-read output, which normally limits the success of unique mapping. This study has provided a general overview and functional prediction of DNA methylation alterations due to S. aureus bacteria but further in-depth analyses are needed. For example, the methylation distribution and alteration at the promotor regions, which has significant impacts on gene expression, deserves more detailed analysis. Furthermore, functional analysis and validation of DMGs should be expand and deepen. More cows and a more reliable sequencing method such as whole genome bisulfite sequencing is expected to further enhance the reliability and reference value of detected methylation sites due to S. aureus bacteria.
The genome-wide DNA methylation profile of mammary gland tissues of Chinese Holstein cows with S. aureus-induced mastitis was generated by the method of Methyl-RAD Seq, indicating the significantly difference of DNA methylation level between S. aureus-challenged and non-challenged quarters. A total of 9,181 CmCGG sites and 1,790 DM CmCWGG sites were significantly DM between SA and CL treatments. Furthermore, potential functions of 363 CmCGG DMGs and 301 CmCWGG DMGs were related to the regulation of immune response and metabolism during S. aureus-infection, respectively. The functional enrichment of 526 CmCGG DMEGs further revealed the regulatory roles of altered methylation of 102 immune-related CmCGG DMEGs involved in immune processes and regulation in response to S. aureus infection, suggesting their potential as candidate genes for S. aureus mastitis. Therefore, these results show that DNA methylation has potential regulatory roles in bovine mammary gland processes during S. aureus mastitis and also serves as a reference for future mechanistic studies aiming to better understand the epigenetic regulation of host response to S. aureus-induced mastitis.
Our sequence data have submitted to NCBI (https://dataview.ncbi.nlm.nih.gov/?search=SUB7329711) and accession number is SUB7329711.
The animal use protocol was reviewed and approved by the animal use and care committee of Yangzhou University.
MW conducted the experiments, analyzed the data, and drafted the manuscript. ZY and YM designed the experiments and guided the experimental implementation. YL and YS handled the samples and prepared libraries for sequencing. ML, HZ, and ZC were involved in data processing and visualization. EI-A provided inputs in data interpretation and critically reviewed the initial manuscript draft. NK provided inputs in the discussion. All authors reviewed and approved the final draft.
The research was financially supported by the National Natural Science Foundation of China (31972555 and 31372286), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (18KJA230003), the Six Talent Peaks Project in Jiangsu Province (NY-093), the Key Project Foundation of Joint International Research Laboratory of Agriculture and Agri-Product Safety, the Ministry of Education of China (JRK20180014), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.550515/full#supplementary-material
Beniæ, M., Maæešiæ, N., Cvetniæ, L., Habrun, B., Cvetniæ, Ž, Turk, R., et al. (2018). Bovine mastitis: a persistent and evolving problem requiring novel approaches for its control-a review. Vet. Arh. 88, 535–557. doi: 10.24099/vet.arhiv.0116
Bergman, Y., and Cedar, H. (2013). DNA methylation dynamics in health and disease. Nat. Struct. Mole. Biol. 20, 274–281.
Bharathan, M. (2016). Monocyte Derived Dendritic Cells: Sentinels and Translators of Immune Response to Staphylococcus aureus. Dissertation, Virginia: Virginia Polytechnic Institute and State University.
Bindea, G., Mlecnik, B., Hackl, H., Charoentong, P., Tosolini, M., Kirilovsky, A., et al. (2009). ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093. doi: 10.1093/bioinformatics/btp101
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brinkman, A. B., Simmer, F., Ma, K., Kaan, A., Zhu, J., and Stunnenberg, H. G. (2010). Whole-genome DNA methylation profiling using MethylCap-seq. Methods 52, 232–236. doi: 10.1016/j.ymeth.2010.06.012
Brunner, A. L., Johnson, D. S., Kim, S. W., Valouev, A., Reddy, T. E., Neff, N. F., et al. (2009). Distinct DNA methylation patterns characterize differentiated human embryonic stem cells and developing human fetal liver. Genome Res. 19, 1044–1056. doi: 10.1101/gr.088773.108
Cai, Z., Guldbrandtsen, B., Lund, M. S., and Sahana, G. (2018). Prioritizing candidate genes post-GWAS using multiple sources of data for mastitis resistance in dairy cattle. BMC Genom. 19:656. doi: 10.1186/s12864-018-5050-x
Carlén, E., Strandberg, E., and Roth, A. (2004). Genetic parameters for clinical mastitis, somatic cell score, and production in the first three lactations of Swedish Holstein cows. J. Dairy Sci. 87, 3062–3070. doi: 10.3168/jds.s0022-0302(04)73439-6
Cha, E., Hertl, J., Schukken, Y., Tauer, L., Welcome, F., and Gröhn, Y. (2013). The effect of repeated episodes of bacteria-specific clinical mastitis on mortality and culling in Holstein dairy cows. J. Dairy Sci. 96, 4993–5007. doi: 10.3168/jds.2012-6232
Chen, J., Wu., Y., Sun, Y., Dong, X., Wang, Z., Zhang, Z., et al. (2019). Bacterial Lipopolysaccharide Induced Alterations of Genome-Wide DNA Methylation and Promoter Methylation of Lactation-Related Genes in Bovine Mammary Epithelial Cells. Toxins 11:298. doi: 10.3390/toxins11050298
Chen, Z., Chu, S., Xu, X., Jiang, J., and Yang, Z. (2019a). Analysis of longissimus muscle quality characteristics and associations with DNA methylation status in cattle. Genes Genom. 41, 1147–1163. doi: 10.1007/s13258-019-00844-4
Chen, Z., Zhou, J., Wang, X., Zhang, Y., Lu, X., Fan, Y., et al. (2019b). Screening candidate microR-15a-IRAK2 regulatory pairs for predicting the response to Staphylococcus aureus-induced mastitis in dairy cows. J. Dairy Res. 86, 425–431. doi: 10.1017/s0022029919000785
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. doi: 10.4161/fly.19695
Dambuza, I. M., and Brown, G. D. (2015). C-type lectins in immunity: recent developments. Curr. Opin. Immunol. 32, 21–27. doi: 10.1016/j.coi.2014.12.002
Del Fresno, C., Iborra, S., Saz-Leal, P., Martínez-López, M., and Sancho, D. (2018). Flexible signaling of myeloid C-type lectin receptors in immunity and inflammation. Front. Immunol. 9:804. doi: 10.3389/fimmu.2018.00804
Detilleux, J., Kastelic, J. P., and Barkema, H. W. (2015). Mediation analysis to estimate direct and indirect milk losses due to clinical mastitis in dairy cattle. Prevent. Vet. Med. 118, 449–456. doi: 10.1016/j.prevetmed.2015.01.009
Dufour, S., Dohoo, I. R., Barkema, H. W., DesCôteaux, L., DeVries, T., Reyher, K. K., et al. (2012). Manageable risk factors associated with the lactational incidence, elimination, and prevalence of Staphylococcus aureus intramammary infections in dairy cows. J. Dairy Sci. 95, 1283–1300. doi: 10.3168/jds.2011-4711
Emanuelson, U., Danell, B., and Philipsson, J. (1988). Genetic parameters for clinical mastitis, somatic cell counts, and milk production estimated by multiple-trait restricted maximum likelihood. J. Dairy Sci. 71, 467–476. doi: 10.3168/jds.s0022-0302(88)79576-4
Enger, B., Tucker, H., Nickerson, S., Parsons, C., and Akers, R. (2019). Effects of Staphylococcus aureus intramammary infection on the expression of estrogen receptor α and progesterone receptor in mammary glands of nonlactating cows administered estradiol and progesterone to stimulate mammary growth. J. Dairy Sci. 102, 2607–2617. doi: 10.3168/jds.2018-15499
Fang, L., Hou, Y., An, J., Li, B., Song, M., Wang, X., et al. (2016). Genome-wide transcriptional and post-transcriptional regulation of innate immune and defense responses of bovine mammary gland to Staphylococcus aureus. Front. Cell. Infect. Microbiol. 6:193. doi: 10.3389/fcimb.2016.00193
Fang, X., Zhao, Z., Yu, H., Li, G., Jiang, P., Yang, Y., et al. (2017). Comparative genome-wide methylation analysis of longissimus dorsi muscles between Japanese black (Wagyu) and Chinese Red Steppes cattle. PLoS One 12:e0182492. doi: 10.1371/journal.pone.0182492
Gim, J.-A., Hong, C. P., Kim, D.-S., Moon, J.-W., Choi, Y., Eo, J., et al. (2015). Genome-wide analysis of DNA methylation before-and after exercise in the thoroughbred horse with MeDIP-Seq. Mole. Cells 38:210. doi: 10.14348/molcells.2015.2138
Gu, H., Smith, Z. D., Bock, C., Boyle, P., Gnirke, A., and Meissner, A. (2011). Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat. Proto. 6:468. doi: 10.1038/nprot.2010.190
Han, H. (2019). Identification of several key genes by microarray data analysis of bovine mammary gland epithelial cells challenged with Escherichia coli and Staphylococcus aureus. Gene 683, 123–132. doi: 10.1016/j.gene.2018.10.004
He, Y., Song, M., Zhang, Y., Li, X., Song, J., Zhang, Y., et al. (2016). Whole-genome regulation analysis of histone H3 lysin 27 trimethylation in subclinical mastitis cows infected by Staphylococcus aureus. BMC Genom. 17:565. doi: 10.1186/s12864-016-2947-0
Hoekstra, M. E., Dijkgraaf, F. E., Schumacher, T. N., and Rohr, J. C. (2015). Assessing T lymphocyte function and differentiation by genetically encoded reporter systems. Trends Immunol. 36, 392–400. doi: 10.1016/j.it.2015.05.008
Hu, S., Concha, C., Johannisson, A., Meglia, G., and Waller, K. P. (2001). Effect of Subcutaneous Injection of Ginseng on Cows with Subclinical Staphylococcus aureus Mastitis. J. Vet. Med. B Infect. Dis. Vet. Publ. Health 48, 519–528. doi: 10.1111/j.1439-0450.2001.00470.x
Huang, X., Lu, H., Wang, J.-W., Xu, L., Liu, S., Sun, J., et al. (2013). High-throughput sequencing of methylated cytosine enriched by modification-dependent restriction endonuclease MspJI. BMC Genet. 14:56. doi: 10.1186/1471-2156-14-56
Ibeagha-Awemu, E. M., and Zhao, X. (2015). Epigenetic marks: regulators of livestock phenotypes and conceivable sources of missing variation in livestock improvement programs. Front. Genet. 6:302. doi: 10.3389/fgene.2015.00302
Jensen, K., Günther, J., Talbot, R., Petzl, W., Zerbe, H., Schuberth, H.-J., et al. (2013). Escherichia coli-and Staphylococcus aureus-induced mastitis differentially modulate transcriptional responses in neighbouring uninfected bovine mammary gland quarters. BMC Genom. 14:36. doi: 10.1186/1471-2164-14-36
Kanzleiter, T., Jähnert, M., Schulze, G., Selbig, J., Hallahan, N., Schwenk, R. W., et al. (2015). Exercise training alters DNA methylation patterns in genes related to muscle growth and differentiation in mice. Am. J. Physiol. Endocrinol. Metabol. 308, E912–E920.
Koivula, M., Mäntysaari, E., Negussie, E., and Serenius, T. (2005). Genetic and phenotypic relationships among milk yield and somatic cell count before and after clinical mastitis. J. Dairy Sci. 88, 827–833. doi: 10.3168/jds.s0022-0302(05)72747-8
Korkmaz, F. (2018). An Analysis of Between-Cow Variation in Innate Immunity in Relation to Mastitis Severity. Graduate College Dissertations Burlington: The University of Vermont.
Kumar, N., Manimaran, A., Kumaresan, A., Jeyakumar, S., Sreela, L., Mooventhan, P., et al. (2017). Mastitis effects on reproductive performance in dairy cattle: a review. Trop. Anim. Health Product. 49, 663–673. doi: 10.1007/s11250-017-1253-4
Kurz, J. P., Yang, Z., Weiss, R. B., Wilson, D. J., Rood, K. A., Liu, G. E., et al. (2019). A genome-wide association study for mastitis resistance in phenotypically well-characterized Holstein dairy cattle using a selective genotyping approach. Immunogenetics 71, 35–47. doi: 10.1007/s00251-018-1088-9
Li, J., Hsu, H.-C., Mountz, J. D., and Allen, J. G. (2018). Unmasking fucosylation: from cell adhesion to immune system regulation and diseases. Cell Chem. Biol. 25, 499–512. doi: 10.1016/j.chembiol.2018.02.005
Li, N., Ye, M., Li, Y., Yan, Z., Butcher, L. M., Sun, J., et al. (2010). Whole genome DNA methylation analysis based on high throughput sequencing technology. Methods 52, 203–212. doi: 10.1016/j.ymeth.2010.04.009
Li, R., Li, Y., Kristiansen, K., and Wang, J. (2008). SOAP: short oligonucleotide alignment program. Bioinformatics 24, 713–714. doi: 10.1093/bioinformatics/btn025
Li, R., Zhang, C. L., Liao, X. X., Chen, D., Wang, W. Q., Zhu, Y. H., et al. (2015). Transcriptome microRNA profiling of bovine mammary glands infected with Staphylococcus aureus. Int. J. Mol. Sci. 16, 4997–5013. doi: 10.3390/ijms16034997
Li, Y., Li, C., Ma, Q., Zhang, Y., Yao, Y., Liu, S., et al. (2018). Genetic variation in CDH13 gene was associated with non-small cell lung cancer (NSCLC): A population-based case-control study. Oncotarget 9:881. doi: 10.18632/oncotarget.22971
Liao, X. (2014). Gene expression profile of Staphylococcus aureus induced mastitis in dairy cows. China: Yangzhou University.
Lin, S., Hsieh, S., Lin, Y., Lee, C., Tsai, M., Lai, L., et al. (2012). A whole genome methylation analysis of systemic lupus erythematosus: hypomethylation of the IL10 and IL1R2 promoters is associated with disease activity. Genes Immun. 13, 214–220. doi: 10.1038/gene.2011.74
Luoreng, Z.-M., Wang, X.-P., Mei, C.-G., and Zan, L.-S. (2018). Comparison of microRNA Profiles between Bovine Mammary Glands Infected with Staphylococcus aureus and Escherichia coli. Int. J. Biol. Sci. 14:87. doi: 10.7150/ijbs.22498
Lush, J. L. (1950). Inheritance of susceptibility to mastitis. J. Dairy Sci. 33, 121–125. doi: 10.3168/jds.s0022-0302(50)91876-5
Ma, S., Tong, C., Ibeagha-Awemu, E. M., and Zhao, X. (2019). Identification and characterization of differentially expressed exosomal microRNAs in bovine milk infected with Staphylococcus aureus. BMC Genom. 20:934. doi: 10.1186/s12864-019-6338-1
Makovec, J. A., and Ruegg, D. P. L. (2003). Antimicrobial resistance of bacteria isolated from dairy cow milk samples submitted for bacterial culture: 8,905 samples (1994–2001). J. Am. Vet. Med. Assoc. 222, 1582–1589. doi: 10.2460/javma.2003.222.1582
Mao, Y., Zhu, X., Li, R., Chen, D., Xin, S., Zhu, Y., et al. (2015). Methylation analysis of CXCR1 in mammary gland tissue of cows with mastitis induced by Staphylococcus aureus. Genet. Mole. Res. 14, 12606–12615. doi: 10.4238/2015.october.19.4
Martin, P., Barkema, H., Brito, L., Narayana, S., and Miglior, F. (2018). Symposium review: novel strategies to genetically improve mastitis resistance in dairy cattle. J. Dairy Sci. 101, 2724–2736. doi: 10.3168/jds.2017-13554
Modak, R., Das Mitra, S., Krishnamoorthy, P., Bhat, A., Banerjee, A., Gowsica, B., et al. (2012). Histone H3K14 and H4K8 hyperacetylation is associated with Escherichia coli-induced mastitis in mice. Epigenetics 7, 492–501. doi: 10.4161/epi.19742
Nonnecke, B. J., and Harp, J. A. (1988). Effects of Staphylococcus aureus on Bovine Mononuclear Leukocyte Proliferation and Viability: Modulation by Phagocytic Leukocytes. J. Dairy Sci. 71, 835–842. doi: 10.3168/jds.s0022-0302(88)79624-1
Patil, V., Ward, R. L., and Hesson, L. B. (2014). The evidence for functional non-CpG methylation in mammalian cells. Epigenetics 9, 823–828. doi: 10.4161/epi.28741
Pokorska, J., Dusza, M., Kułaj, D., Żukowski, K., and Makulska, J. (2016). Single nucleotide polymorphisms in the CXCR1 gene and its association with clinical mastitis incidence in Polish Holstein-Friesian cows. Genet. Mole. Res. GMR 15:15027247
Pu, J., Li, R., Zhang, C., Chen, D., Liao, X., Zhu, Y., et al. (2017). Expression profiles of miRNAs from bovine mammary glands in response to Streptococcus agalactiae-induced mastitis. J. Dairy Res. 84, 300–308. doi: 10.1017/S0022029917000437
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinform 26, 841–842. doi: 10.1093/bioinformatics/btq033
Rainard, P., Foucras, G., Boichard, D., and Rupp, R. (2018). Invited review: low milk somatic cell count and susceptibility to mastitis. J. Dairy Sci. 101, 6703–6714. doi: 10.3168/jds.2018-14593
Reksen, O., Sølverød, L., Branscum, A., and Østerås, O. (2006). Relationships between milk culture results and treatment for clinical mastitis or culling in Norwegian dairy cattle. J. Dairy Sci. 89, 2928–2937. doi: 10.3168/jds.s0022-0302(06)72565-6
Reynolds, L. P., Ward, A. K., and Caton, J. S. (2017). Epigenetics and Developmental Programming in Ruminants: Long-Term Impacts on Growth and Development, Biology of domestic animals. Boca: CRC Press, 85–121.
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Samanta, D., and Almo, S. C. (2015). Nectin family of cell-adhesion molecules: structural and molecular aspects of function and specificity. Cell. Mole. Life Sci. 72, 645–658. doi: 10.1007/s00018-014-1763-4
Sampimon, O., Barkema, H. W., Berends, I., Sol, J., and Lam, T. (2009). Prevalence of intramammary infection in Dutch dairy herds. J. Dairy Res. 76, 129–136. doi: 10.1017/s0022029908003762
Sanderson, B. A., Araki, N., Lilley, J. L., Guerrero, G., and Lewis, L. K. (2014). Modification of gel architecture and TBE/TAE buffer composition to minimize heating during agarose gel electrophoresis. Anal. Biochem. 454, 44–52. doi: 10.1016/j.ab.2014.03.003
Shin, J., Bourdon, C., Bernard, M., Wilson, M. D., Reischl, E., Waldenberger, M., et al. (2015). Layered genetic control of DNA methylation and gene expression: a locus of multiple sclerosis in healthy individuals. Hum Mole. Genet. 24, 5733–5745. doi: 10.1093/hmg/ddv294
Siebert, L., Staton, M., Oliver, S., and Pighetti, G. (2017). Identifying genome associations with unique mastitis phenotypes in response to intramammary Streptococcus uberis challenge, Doctoral Dissertations Knoxville: The University of Tennessee, 86.
Song, M., He, Y., Zhou, H., Zhang, Y., Li, X., and Yu, Y. (2016). Combined analysis of DNA methylome and transcriptome reveal novel candidate genes with susceptibility to bovine Staphylococcus aureus subclinical mastitis. Sci. Rep. 6:29390.
Sordillo, L. M. (2011). New Concepts in the Causes and Control of Mastitis. J. Mammary Gland Biol. Neopl. 16, 271–273. doi: 10.1007/s10911-011-9239-8
Sun, X., Li, J., Fu, L., Jiang, J., Zhao, J., and Wang, G. (2019). The Epigenetic Modification in Mammals under Heat Stress. World J. Vet. Sci. 1:1005.
Suravajhala, P., and Benso, A. (2017). Prioritizing single-nucleotide polymorphisms and variants associated with clinical mastitis. Adv. Appl. Bioinform. Chem. AABC 10:57. doi: 10.2147/aabc.s123604
Taiwo, O., Wilson, G. A., Morris, T., Seisenberger, S., Reik, W., Pearce, D., et al. (2012). Methylome analysis using MeDIP-seq with low DNA concentrations. Nat. Prot. 7:617. doi: 10.1038/nprot.2012.012
Usman, T., Yu, Y., and Wang, Y. (2016). CD4 promoter hyper methylation is associated with lower gene expression in clinical mastitis cows and vice versa in the healthy controls. J. Anim. Sci. 94, 38–38. doi: 10.2527/jas2016.94supplement438x
Vallimont, J., Dechow, C. D., Sattler, C., and Clay, J. (2009). Heritability estimates associated with alternative definitions of mastitis and correlations with somatic cell score and yield. J. Dairy Sci. 92, 3402–3410. doi: 10.3168/jds.2008-1229
Verbeke, J., Piccart, K., Piepers, S., Van Poucke, M., Peelman, L., De Visscher, A., et al. (2015). Somatic cell count and milk neutrophil viability of dairy heifers with specific CXCR1 genotypes following experimental intramammary infection with Staphylococcus chromogenes originating from milk. Vet J. 204, 322–326. doi: 10.1016/j.tvjl.2015.04.010
Wang, D., Wei, Y., Shi, L., Khan, M. Z., Fan, L., Wang, Y., et al. (2019). Genome-wide DNA methylation pattern in a mouse model reveals two novel genes associated with Staphylococcus aureus mastitis. Asian Austr. J. Anim. Sci. 33, 203–211. doi: 10.5713/ajas.18.0858
Wang, D., Wei, Y., Shi, L., Khan, M. Z., Fan, L., Wang, Y., et al. (2020). Genome-wide DNA methylation pattern in a mouse model reveals two novel genes associated with Staphylococcus aureus mastitis. Asian Austr. J. Anim. Sci 33:203. doi: 10.5713/ajas.18.0858
Wang, H.-Q., Yang, C.-Y., Wang, S.-Y., Wang, T., Han, J.-L., Wei, K., et al. (2018). Cell-free plasma hypermethylated CASZ1, CDH13 and ING2 are promising biomarkers of esophageal cancer. J. Biomed. Res. 32, 424–433.
Wang, S., Lv, J., Zhang, L., Dou, J., Sun, Y., Li, X., et al. (2015). MethylRAD: a simple and scalable method for genome-wide DNA methylation profiling using methylation-dependent restriction enzymes. Open Biol. 5:150130. doi: 10.1098/rsob.150130
Wang, X. G., Ju, Z. H., Hou, M. H., Jiang, Q., Yang, C. H., Zhang, Y., et al. (2016). Deciphering transcriptome and complex alternative splicing transcripts in mammary gland tissues from cows naturally infected with Staphylococcus aureus mastitis. PLoS One 11:e0159719. doi: 10.1371/journal.pone.0159719
Wang, X., Ma, P., Liu, J., Zhang, Q., Zhang, Y., Ding, X., et al. (2015). Genome-wide association study in Chinese Holstein cows reveal two candidate genes for somatic cell score as an indicator for mastitis susceptibility. BMC Genet. 16:111. doi: 10.1186/s12863-015-0263-3
Wang, X., Zhang, Y., He, Y., Ma, P., Fan, L., Wang, Y., et al. (2013). Aberrant promoter methylation of the CD4 gene in peripheral blood cells of mastitic dairy cows. Genet. Mol. Res. 12, 6228–6239. doi: 10.4238/2013.december.4.10
Weigel, K. A., and Shook, G. E. (2018). Genetic selection for mastitis resistance. Vet. Clin. Food Anim. Pract. 34, 457–472. doi: 10.1016/j.cvfa.2018.07.001
Welderufael, B. G., Løvendahl, P., De Koning, D.-J., Janss, L. L., and Fikse, W. (2018). Genome-wide association study for susceptibility to and recoverability from mastitis in Danish Holstein cows. Front. Genet. 9:141. doi: 10.3389/fgene.2018.00141
Wu, Y., Chen, J., Sun, Y., Dong, X., Wang, Z., Chen, J., et al. (2020). PGN and LTA from Staphylococcus aureus Induced Inflammation and Decreased Lactation through Regulating DNA Methylation and Histone H3 Acetylation in Bovine Mammary Epithelial Cells. Toxins 12:238. doi: 10.3390/toxins12040238
Xu, Y., Li, X., Wang, H., Xie, P., Yan, X., Bai, Y., et al. (2016). Hypermethylation of CDH13, DKK3 and FOXL2 promoters and the expression of EZH2 in ovary granulosa cell tumors. Mole. Med. Rep. 14, 2739–2745. doi: 10.3892/mmr.2016.5521
Yang, J., Niu, H., Huang, Y., and Yang, K. (2016). A systematic analysis of the relationship of CDH13 promoter methylation and breast cancer risk and prognosis. PLoS One 11:e0149185. doi: 10.1371/journal.pone.0149185
Ye, M., Huang, T., Li, J., Zhou, C., Yang, P., Ni, C., et al. (2017). Role of CDH13 promoter methylation in the carcinogenesis, progression, and prognosis of colorectal cancer: a systematic meta-analysis under PRISMA guidelines. Medicine 96:e5956. doi: 10.1097/md.0000000000005956
Yu, G., Wang, L.-G., Han, Y., and He, Q.-Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS A J. Integr. Biol. 16, 284–287. doi: 10.1089/omi.2011.0118
Zhang, X., Zhang, S., Ma, L., Jiang, E., Xu, H., Chen, R., et al. (2017). Reduced representation bisulfite sequencing (RRBS) of dairy goat mammary glands reveals DNA methylation profiles of integrated genome-wide and critical milk-related genes. Oncotarget 8:115326. doi: 10.18632/oncotarget.23260
Zhang, Y., Li, F., Feng, X., Yang, H., Zhu, A., Pang, J., et al. (2017). Genome-wide analysis of DNA Methylation profiles on sheep ovaries associated with prolificacy using whole-genome Bisulfite sequencing. BMC Genom. 18:759. doi: 10.1186/s12864-017-4068-9
Zhang, Y., Wang, X., Jiang, Q., Hao, H., Ju, Z., Yang, C., et al. (2018). DNA methylation rather than single nucleotide polymorphisms regulates the production of an aberrant splice variant of IL6R in mastitic cows. Cell Stress Chap. 23, 617–628. doi: 10.1007/s12192-017-0871-0
Keywords: genome-wide DNA methylation, mastitis, Staphylococcus aureus, mammary gland tissue, Chinese Holstein
Citation: Wang M, Liang Y, Ibeagha-Awemu EM, Li M, Zhang H, Chen Z, Sun Y, Karrow NA, Yang Z and Mao Y (2020) Genome-Wide DNA Methylation Analysis of Mammary Gland Tissues From Chinese Holstein Cows With Staphylococcus aureus Induced Mastitis. Front. Genet. 11:550515. doi: 10.3389/fgene.2020.550515
Received: 09 April 2020; Accepted: 29 September 2020;
Published: 19 October 2020.
Edited by:
Göran Andersson, Swedish University of Agricultural Sciences, SwedenReviewed by:
Ying Yu, China Agricultural University, ChinaCopyright © 2020 Wang, Liang, Ibeagha-Awemu, Li, Zhang, Chen, Sun, Karrow, Yang and Mao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongjiang Mao, Y2F0dGxlQHl6dS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.