 Tsung-Ming Hu
Tsung-Ming Hu Ying-Chieh Wang
Ying-Chieh Wang Chia-Liang Wu
Chia-Liang Wu Shih-Hsin Hsu
Shih-Hsin Hsu Hsin-Yao Tsai
Hsin-Yao Tsai Min-Chih Cheng
Min-Chih Cheng- 1Department of Psychiatry, Yuli Branch, Taipei Veterans General Hospital, Hualien, Taiwan
- 2Department of Future Studies and LOHAS Industry, Fo Guang University, Jiaosi, Taiwan
- 3Institute of Medical Sciences, Tzu Chi University, Hualien City, Taiwan
Objective: Schizophrenia is a chronic debilitating neurobiological disorder of aberrant synaptic connectivity and synaptogenesis. Postsynaptic density (PSD)–related proteins in N-methyl-D-aspartate receptor–postsynaptic signaling complexes are crucial to regulating the synaptic transmission and functions of various synaptic receptors. This study examined the role of PSD-related genes in susceptibility to schizophrenia.
Methods: We resequenced 18 genes encoding the disks large-associated protein (DLGAP), HOMER, neuroligin (NLGN), neurexin, and SH3 and multiple ankyrin repeat domains (SHANK) protein families in 98 schizophrenic patients with family psychiatric history using semiconductor sequencing. We analyzed the protein function of the identified rare schizophrenia-associated mutants via immunoblotting and immunocytochemistry.
Results: We identified 50 missense heterozygous mutations in 98 schizophrenic patients with family psychiatric history, and in silico analysis revealed some as damaging or pathological to the protein function. Ten missense mutations were absent from the dbSNP database, the gnomAD (non-neuro) dataset, and 1,517 healthy controls from Taiwan BioBank. Immunoblotting revealed eight missense mutants with altered protein expressions in cultured cells compared with the wild type.
Conclusion: Our findings suggest that PSD-related genes, especially the NLGN, SHANK, and DLGAP families, harbor rare functional mutations that might alter protein expression in some patients with schizophrenia, supporting contributing rare coding variants into the genetic architecture of schizophrenia.
Introduction
Schizophrenia is a chronic debilitating mental illness affecting approximately 1% of the global population and is characterized by overt psychotic (positive) symptoms, various deficits (negative symptoms), and impaired cognitive function (Freedman, 2003). On average, schizophrenia is estimated as approximately 80% hereditable, suggesting that genetic factors are involved in its pathogenesis (Mowry and Gratten, 2013). Studies have demonstrated a reduction of neuronal processes and synaptic dysfunction in schizophrenia (McGlashan and Hoffman, 2000; Hayashi-Takagi and Sawa, 2010), which is regulated by postsynaptic density (PSD)–related proteins in N-methyl-D-aspartate (NMDA) receptor–postsynaptic signaling complexes in various synaptic receptors (Takeuchi et al., 1997; Fukaya and Watanabe, 2000). Moreover, accumulating evidence from genetic, proteomic, and neuroanatomical studies indicates that aberrant synaptic plasticity and NMDA receptor–postsynaptic signaling complexes are involved in the pathophysiology of schizophrenia (Dracheva et al., 2001; Clinton and Meador-Woodruff, 2004; Kristiansen et al., 2007; Kirov et al., 2012; Focking et al., 2015). In vitro functional evidence together with human genetics data strongly suggests that mutations in a variety of postsynaptic scaffolding proteins may contribute to the etiology of schizophrenia (Sun et al., 2011; Peykov et al., 2015; Yu et al., 2018). Therefore, PSD-related genes involved in the formation and functional integrity of synapses can be considered candidate genes for schizophrenia.
In humans, the neuroligin (NLGN) gene family (NLGN1, NLGN2, NLGN3, NLGN4X, and NLGN4Y) encodes postsynaptic cell-adhesion molecules that are vital to the formation of functional synapses via trans-synaptic interactions with presynaptically expressed neurexin (NRXN) gene family and interactions with postsynaptic scaffolding molecules (Missler et al., 1998). Missense mutations in NLGN2 or NLGN3 have been associated with several neuropsychiatric diseases, such as schizophrenia (Sun et al., 2011) and autism (Jamain et al., 2003; Xu et al., 2014; Parente et al., 2017). The SH3 and multiple ankyrin repeat domains (SHANK) protein family comprises three main genes with highly similar domains that function as anchoring or scaffolding proteins to bridge postsynaptic membrane proteins and the cytoskeleton (Naisbitt et al., 1999; Tu et al., 1999). Mutations of the SHANK gene family (SHANK1-3) have been linked to psychiatric disorders such as schizophrenia, autism spectrum disorders, or intellectual disability (Wilson et al., 2003; Durand et al., 2007; Berkel et al., 2010; Sato et al., 2012; Peykov et al., 2015). The disks large-associated protein (DLGAP) family encoded by DLGAP1, DLGAP2, DLGAP3, and DLGAP4 is involved in organizing the postsynaptic signaling complex in glutamatergic synapses and has been linked to a variety of psychiatric disorders (Takeuchi et al., 1997; Rasmussen et al., 2017). The homer protein family of dendritic proteins includes three members encoded by three different genes (HOMER1, HOMER2, and HOMER3) (Shiraishi-Yamaguchi and Furuichi, 2007). Homer proteins act as multimodal adaptors by interacting with several PSD proteins and are involved postsynaptically by regulating glutamatergic receptor trafficking, involving the function of plasma membrane ion channels and intracellular messenger systems (Thomas, 2002).
There has been growing debate that the genetic contribution to individual susceptibility to common complex diseases can be caused either by common disease-common variants with low penetrance or common disease-rare variants with high penetrance (Manolio et al., 2009; Schork et al., 2009; Sullivan et al., 2012). De novo mutations, including copy number variations and single-nucleotide variants (SNVs) of PSD-related proteins’ genes, have been identified in some sporadic patients with schizophrenia (Stefansson et al., 2008; Walsh et al., 2008; Xu et al., 2008, 2011; Grozeva et al., 2012; Fromer et al., 2014; Purcell et al., 2014; Xing et al., 2016). We previously identified several rare [minor allele frequency (MAF) < 0.01] schizophrenia-associated mutations in PSD-associated protein genes, including DLGAP1, DLGAP2, DLGAP3, and NLGN2 (Sun et al., 2011; Li et al., 2013a, b; Li et al., 2014), which may contribute to some of the pathogenic mechanisms of schizophrenia in certain patients. Taken together, resequencing studies have implicated rare pathologic variants as susceptibility genes for schizophrenia.
To identify rare pathogenic SNVs, we resequenced 18 genes for PSD-related proteins, including members of the DLGAP (4 genes), HOMER (3 genes), NLGN (5 genes), NRXN (3 genes), and SHANK gene family (3 genes), in 98 schizophrenic patients with family psychiatric history using semiconductor sequencing technology. We analyzed the protein function of the identified rare schizophrenia-associated mutants via immunoblotting and immunocytochemistry.
Materials and Methods
Subjects and Study Outline
We recruited Han Chinese patients from Taiwan fulfilling the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition diagnostic criteria for schizophrenia. Healthy controls were from a medical center as usual medical check in east Taiwan. We excluded participants with organic brain syndromes, intellectual disability, substance-related psychosis, or mood disorder with psychotic features. The study was performed in two stages. At stage 1, 98 unrelated schizophrenic patients with family psychiatric history (46 males, mean age = 41 ± 9 years; 52 females, mean age = 45 ± 11 years) were screened for rare mutations using semiconductor sequencing. At stage 2, we genotyped the identified rare missense mutations and compared them with an independent sample of 484 nonrelated patients with schizophrenia (262 males, mean age = 48 ± 12 years; 222 females, mean age = 52 ± 10 years) and 533 healthy controls (252 males, mean age = 43 ± 15 years; 281 females, mean age = 45 ± 13 years) using Sanger sequencing.
This study was approved by Antai Medical Care Cooperation Antai-Tian-Sheng Memorial Hospital Institution Review Board (IRB no. 101020) and was performed in accordance with the 1964 World Medical Association’s Declaration of Helsinki and its later amendments. Written informed consent was obtained after the procedures are explained.
Semiconductor Sequencing
Semiconductor sequencing was performed using the Ion TorrentTM Personal Genome MachineTM System (Thermo Fisher Scientific) according to the manufacturer’s protocol. Briefly, genomic DNA was isolated from peripheral blood cells of the subjects using a Gentra Puregene Blood Kit (QIAGEN) and quantified using the QubitTM dsDNA HS Assay Kit (Thermo Fisher Scientific). Custom amplification primers were designed using Ion AmpliSeqTM Designer software (Thermo Fisher Scientific) to cover the exonic regions of 18 genes (SHANK1, SHANK2, SHANK3, NRXN1, NRXN2, NRXN3, HOMER1, HOMER2, HOMER3, DLGAP1, DLGAP2, DLGAP3, DLGAP4, NLGN1, NLGN2, NLGN3, NLGN4X, and NLGN4Y). Genomic DNA libraries were prepared using two primer pools and an Ion AmpliSeqTM Library Kit 2.0 (Thermo Fisher Scientific). Each library was ligated to a different barcode adapter using an Ion XpressTM Barcode Adapters Kit (Thermo Fisher Scientific) and purified using AgencourtTM AMPureTM XP Reagent (Beckman Coulter). The concentrations of the purified libraries were determined by a quantitative polymerase chain reaction (PCR) with the Ion Library TaqManTM Quantitation Kit (Thermo Fisher Scientific). Emulsion PCR and subsequent Ion SphereTM particle enrichment were performed using the Ion PGMTM Hi-Q View OT2 Kit and Ion PGMTM Enrichment Beads on an Ion OneTouchTM 2 system (Thermo Fisher Scientific). The final product was sequenced on the Ion 318TM Chip using the Ion PGMTM Hi-QTM View Sequencing Kit (Thermo Fisher Scientific). Data analysis, including alignment to the hg19 human reference genome, base calling, and variant calling, was performed using Torrent Suite built-in software. Variants were annotated using Ion ReporterTM Software (Thermo Fisher Scientific).
PCR Reaction and Fluorescence-Based Cycle Sequencing
PCR primer sequences were designed using the Primer3 website1. PCR amplification was performed according to our well-established laboratory protocols. For sequencing, aliquots of PCR products were purified using an IllustraTM ExoProStarTM 1-Step Kit (GE Healthcare Bio-Sciences) and then sequenced using a BigDyeTM Terminator v3.1 Cycle Sequencing Kit on a 3730 DNA Analyzer (Perkin Elmer Applied Biosystems) according to the manufacturer’s protocol.
In silico Analysis
We explored whether the mutations we identified were documented in Taiwan BioBank2, a nationwide research database that collected, stored, and analyzed the biological data necessary for the research designed to trace the common chronic diseases occurring locally in Taiwan. A possible impact of amino acid substitution was analyzed using the publicly available prediction programs: Polyphen-23, SIFT4, PMut5, and PROVEAN6. Evolutionary conservation was assessed using the multiz tool of the University of California Santa Cruz Genome Browser7.
Construction of Expression Plasmids and Organelle Marker Vectors
A pcDNA3.1/C-terminal–green fluorescent protein (GFP) vector containing the coding sequence of human NLGN2 (Sun et al., 2011) was used as a template to generate an NLGN2R309Q mutant variant. Human NLGN3, SHANK2, SHANK3, DLGAP1, and DLGAP3 cDNAs were cloned into expression vectors (tGFP and Myc-DDK tagged) using the PrecisionShuttle vector System (OriGene). Vectors containing the mutant were generated using a QuikChange® Lightning Site-Directed Mutagenesis kit (Agilent Technologies). The authenticity of the cloned sequences was confirmed by qualitative restriction enzyme digestion and Sanger sequencing. Human lymphocyte-specific protein tyrosine kinase (LCK) with a tag vector for plasma membrane marking was purchased from OriGene.
Cell Culture, Transfection, and Immunocytochemistry
HEK-293 and neuro-2a cells were cultured in minimal essential media supplemented with 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 100 U penicillin, 100 μg streptomycin (Invitrogen), and 10% fetal bovine serum. The cells were cultured on 24-well plates (10,000 cells/well) and cotransfected with 450 ng of reporter plasmid and 50 ng of organelle marker vector using LipofectamineTM 3000 (Invitrogen). After 24 h, the cells were washed with cold phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde in PBS (pH 7.4) for 30 min at 37°C. After that, they were washed three times with PBS and permeabilized for 15 min in PBS containing 0.1% Triton X-100. The fixed and permeabilized cells were simultaneously stained with Alexa Fluor® 594 wheat germ agglutinin (WGA) (Invitrogen) and mounted in a medium containing 4′,6-diamidino-2-phenylindole (VECTOR Laboratories). Images were acquired using a fluorescence microscope and analyzed with ZEN 2 software (Zeiss).
Protein Preparation and Immunoblotting Analysis
HEK-293 and neuro-2a cells were transfected in 6-well plates. After 24 h of expression, they were washed twice with cold PBS and resuspended in lysis solution containing 20 mM HEPES (pH 7.6), 7.5 mM NaCl, 2.5 mM MgCl2, 0.1 mM EDTA, 0.1% Triton X-100, 0.1 mM Na3VO4, and 50 mM NaF, and protease inhibitor cocktail tablet (one complete mini tablet/10 mL; Roche Diagnostics GmbH). The homogenates were centrifuged at 13,000 rpm for 30 min at 4°C, and the supernatants were stored at −20°C until use. Membrane proteins were extracted using a Subcellular Protein Fractionation Kit for Cultured Cells (Thermo Fisher Scientific) according to the manufacturer’s protocol.
Immunoblotting was performed according to our standard protocol using primary antibodies (anti-NLGN2 and anti-GFP, Santa Cruz Biotechnology; anti-SHANK3, anti-tGFP, and anti-DDK, OriGene) and secondary antibodies (anti-mouse, Kirkegaard and Perry Laboratories; anti-rabbit, GE Healthcare; anti-goat, Santa Cruz Biotechnology). The chemiluminescence signal was visualized using a Trident ECL plus kit (GeneTex). Band intensities were assessed utilizing the National Institutes of Health ImageJ software8 and normalized to the intensity of the GFP or tGFP band.
Statistical Analysis
In the immunoblotting study, we used the Mann-Whitney U-test to compare the means between two groups (wild and mutant types) and analyze statistical significance. The Kruskal–Wallis H-test was compared among three or more independent groups. p < 0.05 was considered statistically significant. All the calculations were implemented using SPSS 18th version.
Results
Identification of Variants of PSD-Related Genes in Schizophrenia
Our custom-designed panel covering the exon regions of 18 genes allowed the analysis of 267 exons (5-bp exon padding) by the targeted resequencing of 755 amplicons (96.72% coverage). We initially screened for mutations in 98 schizophrenic patients with family psychiatric history. Data generation from semiconductor sequencing was listed in Supplementary Table 1. After the semiconductor sequencing of all the amplicons, 50 heterozygous missense mutations were identified (Table 1). The genotypes of these 50 missense mutations in 98 patients with schizophrenia are listed in Supplementary Table 2. The genotype frequency distributions of the 49 mutants did not deviate from Hardy–Weinberg equilibrium in the patient group, but the genotype distribution of DLGAP2P384Q did significantly deviate from Hardy–Weinberg equilibrium. Five missense mutations (DLGAP2P384Q, NRXN2L81Q, SHANK1D897E, SHANK1V1504A, and DLGAP4R747Q) with MAF greater than 5% were identified. Ten missense mutations (DLGAP3H22N, DLGAP3G352E, DLGAP3S455R, NRXN2P642T, SHANK2L158F, NLGN2R309Q, DLGAP1F70L, SHANK3P209A, NLGN3V346M, and NLGN3Q691R) were absent from the dbSNP database, the gnomAD (non-neuro) dataset, and 1,517 healthy controls from Taiwan BioBank, indicating that they were rare mutations. One patient harbored two rare missense mutations (DLGAP1F70L/NLGN3Q691R), and another a double-missense mutation (NLGN3R195Q/V346M) in the NLGN3 gene.
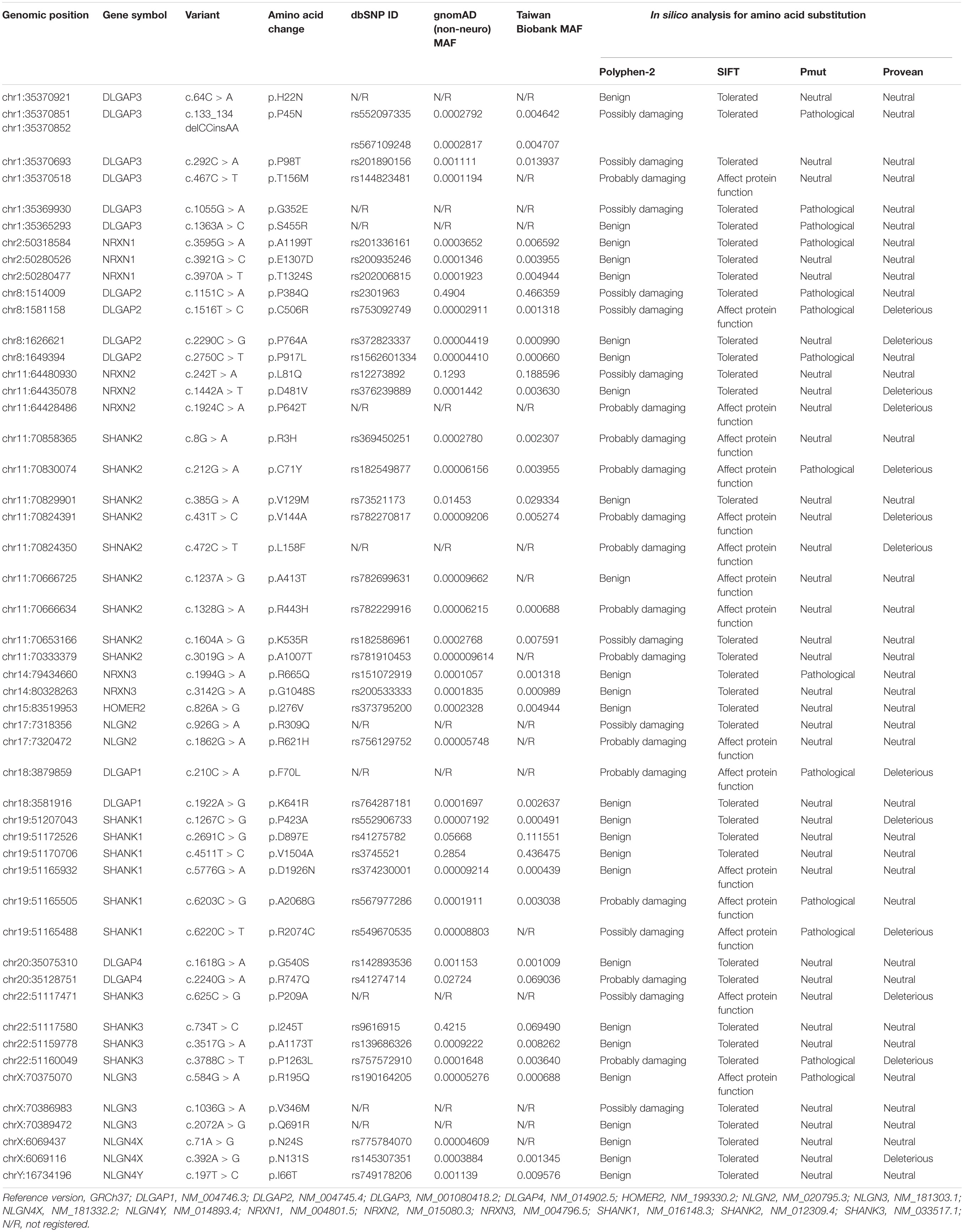
Table 1. Fifty missense mutations identified from 98 patients with schizophrenia in the semiconductor sequencing stage.
Next, we focused on ten missense variants absent from the gnomAD (non-neuro) dataset and Taiwan BioBank database for a genotyping assay in an independent sample set to ascertain whether these mutations were patient-associated in our Taiwan sample. These 10 missense mutations were also absent from a new collection of 468 patients with schizophrenia and 533 healthy controls from Taiwan (Supplementary Table 3). NRXN2P642T is located at presynaptic NRXNs, which form a transsynaptic complex with postsynaptic NLGNs (Boucard et al., 2005). We focused on the 9 rare missense mutations (DLGAP3H22N, DLGAP3G352E, DLGAP3S455R, SHANK2L158F, NLGN2R309Q, DLGAP1F70L, SHANK3P209A, NLGN3V346M, and NLGN3Q691R) located at postsynaptic regions for further in silico analysis and functional assays. The schematic genomic structures of the SHANK, DLGAP, and NLGN gene families and the positions of these 9 patient-associated missense mutations are illustrated in Figure 1. Evolutionary conservation analysis revealed that the amino acids corresponding to the nine rare mutations were highly conserved among eight species (Figure 1). In silico analysis showed that seven mutations (DLGAP3G352E, DLGAP3S455R, SHANK2L158F, NLGN2R309Q, SHANK3P209A, NLGN3V346M, and NLGN3Q691R) were predicted to be pathogenic in at least one prediction programs, whereas DLGAP1F70L was predicted to be pathogenic in all four prediction programs (Table 1).
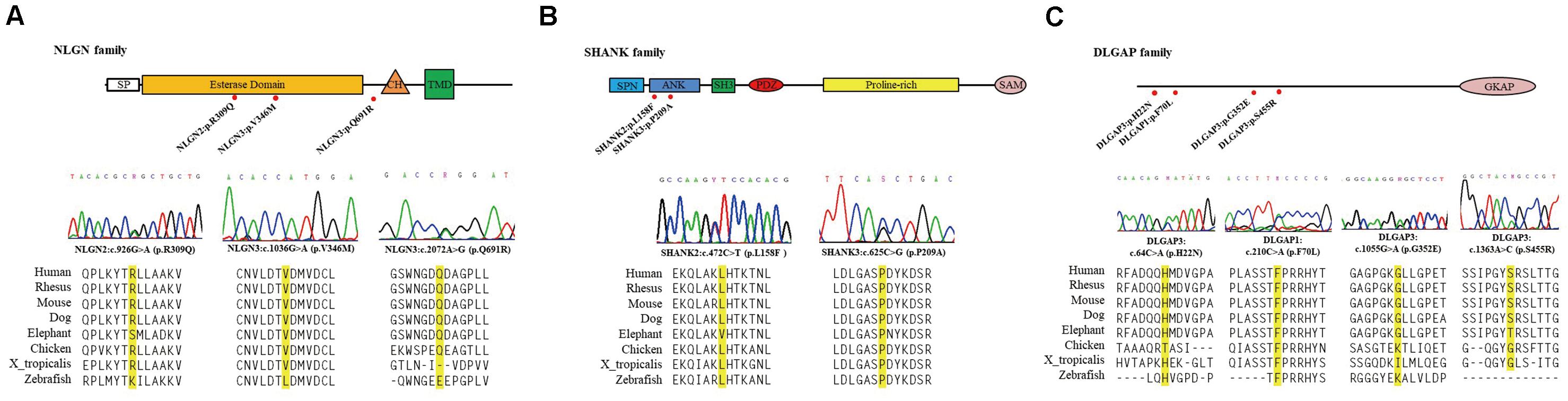
Figure 1. Schematic genomic structures of the NLGN (A), SHANK (B), and DLGAP (C) gene families. The locations and sequence electropherograms of the variants found in patients with schizophrenia are shown. The sequence alignment of eight mammalian species indicates the conservation of the mutated amino acids. SP, signal peptide domain; CH, cholinesterase domain; TMD, transmembrane domain; SPN, shank/ProSAP N-terminal domain; ANK, ankyrin repeats domain; SH3, SRC homology 3 domain; PDZ, PSD-95/D/ZO-1 domain; SAM, sterile α motif; GKAP, guanylate kinase-associated protein.
Immunoblotting Analysis of Gene Mutants in Cultured Cells
We investigated whether 9 rare missense mutations affected protein expression via immunoblotting of HEK-293 or neuro-2a cells after 24 h of transiently expressing wild-type (WT) or mutant protein. NLGN2-GFP fusion protein expression tended to reduce in HEK-293 and neuro-2a cells carrying the NLGN2R309Q mutant compared with NLGN2WT (Figure 2A). The Mann–Whitney U-test was performed between the two conditions each time (HEK-293, p = 0.1; neuro-2a, p = 0.1). NLGN3-DDK fusion protein expression was significantly increased in HEK-293 cells carrying the NLGN3R195Q/V346M mutant, whereas it was significantly decreased in cells carrying the NLGN3Q691R mutant compared with NLGN3WT (Figure 2B). A Kruskal–Wallis H-test was performed between the three conditions (p = 0.027). The SHANK2-tGFP fusion protein with the SHANK2L158F mutant was significantly increased in HEK-293 cells but tended to decrease in neuro-2a cells compared with SHANK2WT (Figure 2C). The Mann–Whitney U-test was performed between the two conditions (HEK-293, p = 0.002; neuro-2a, p = 0.1). The protein expression of the SHANK3-tGFP fusion protein with SHANK3P209A tended to increase in HEK-293 cells but tended to decrease in neuro-2a cells compared with SHANK3WT (Figure 2D). The Mann–Whitney U-test was performed between the two conditions each time (HEK-293, p = 0.1; neuro-2a, p = 0.1). Immunoblotting of cell lysates from the HEK-293 cells carrying DLGAP3WT and DLGAP3H22N, DLGAP3G352E, and DLGAP3S455R revealed significantly decreased levels of DLGAP3-tGFP fusion proteins in mutants relative to WT cells (Figure 2E). A Kruskal–Wallis H-test was performed between the four conditions (p = 0.034). HEK-293 cells with DLGAP1F70L mutant demonstrated similar levels of DLGAP1-tGFP fusion protein expression as the WT cells (Figure 2F). The Mann–Whitney U-test was performed between the two conditions each time (p = 0.7).
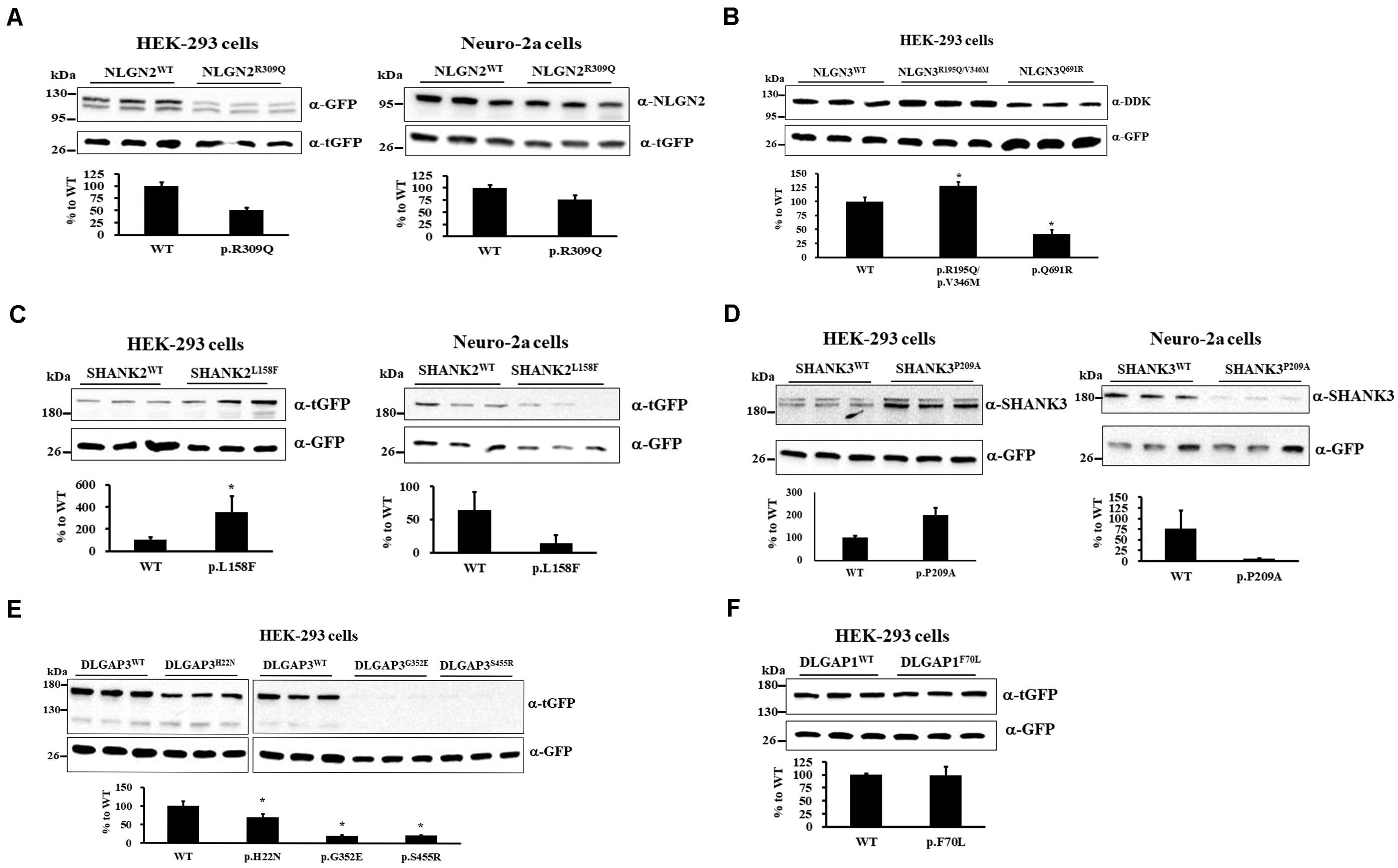
Figure 2. Immunoblotting of gene mutants in cultured cells. (A) Immunoblotting of protein extracts from HEK-293 cells (left) and neuro-2a cells (right) transiently cotransfected with a plasmid expressing either NLGN2WT or NLGN2R309Q cDNAs with pCMV6-AC-tGFP. (B) Immunoblotting of protein extracts from HEK-293 cells transiently cotransfected with a plasmid expressing NLGN3WT, NLGN3R195Q/V346M, or NLGN3Q691R cDNAs with pcDNA3.1-GFP. (C) Immunoblotting of protein extracts from HEK-293 cells (left) and neuro-2a cells (right) transiently cotransfected with a plasmid expressing either SHANK2WT or SHANK2L158F cDNAs with pcDNA3.1-GFP. (D) Immunoblotting of protein extracts from HEK-293 cells (left) and neuro-2a cells (right) transiently cotransfected with a plasmid expressing either SHANK3WT or SHANK3P209A cDNAs with pcDNA3.1-GFP. (E) Immunoblotting of protein extracts from HEK-293 cells transiently cotransfected with a plasmid expressing DLGAP3WT or DLGAP3 mutant cDNAs (DLGAP3H22N, DLGAP3G352E, and DLGAP3S455R) with pcDNA3.1-GFP. (F) Immunoblotting of protein extracts from HEK-293 cells transiently cotransfected with a plasmid expressing either DLGAP1WT or DLGAP1F70L cDNAs with pCMV6-AC-GFP. For normalization, lysates were analyzed in parallel by anti-tGFP or anti-GFP immunoblotting. Results are representative of three independent experiments. WT, wild-type. Graphs represent means ± standard errors. *p < 0.05.
Localization Analysis of Gene Mutants in Cultured Cells
To understand the effects of rare mutations on protein translocation, we examined the protein subcellular localization of the WT and the mutant proteins in HEK-293 and neuro-2a cells. GFP signal was observed in the membrane fraction (WGA) of HEK-293 transfected with NLGN2WT-GFP plasmid but not NLGN2R309Q-GFP plasmid (Figure 3A). NLGN2 WT proteins colocalized to the plasma membrane marker (LCK) in transfected neuro-2a cells, whereas NLGN2R309Q mutant proteins colocalized to LCK at a lower level (Figure 3B). We further examined and quantified the presence of the overexpressed NLGN2-GFP fusion protein in the membrane fraction using an immunoblot assay. The NLGN2-GFP fusion protein was significantly reduced in the membrane fraction of HEK-293 cells carrying NLGN2R309Q mutant but tended to reduce in neuro-2a cells carrying NLGN2R309Q mutant compared with the WT construct (Figures 3C,D). The Mann–Whitney U-test was performed between the two conditions each time (HEK-293, p = 0.015; neuro-2a, p = 0.1). Immunocytochemistry revealed NLGN3WT, NLGN3R195Q/V346M, and NLGN3Q691R proteins colocalized to LCK, whereas SHANK2WT, SHANK2L158F, SHANK3WT, SHANK3P209A, DLGAP3WT, DLGAP3H22N, DLGAP3G352E, and DLGAP3S455R mostly localized in the cytosol (Supplementary Figure 1).
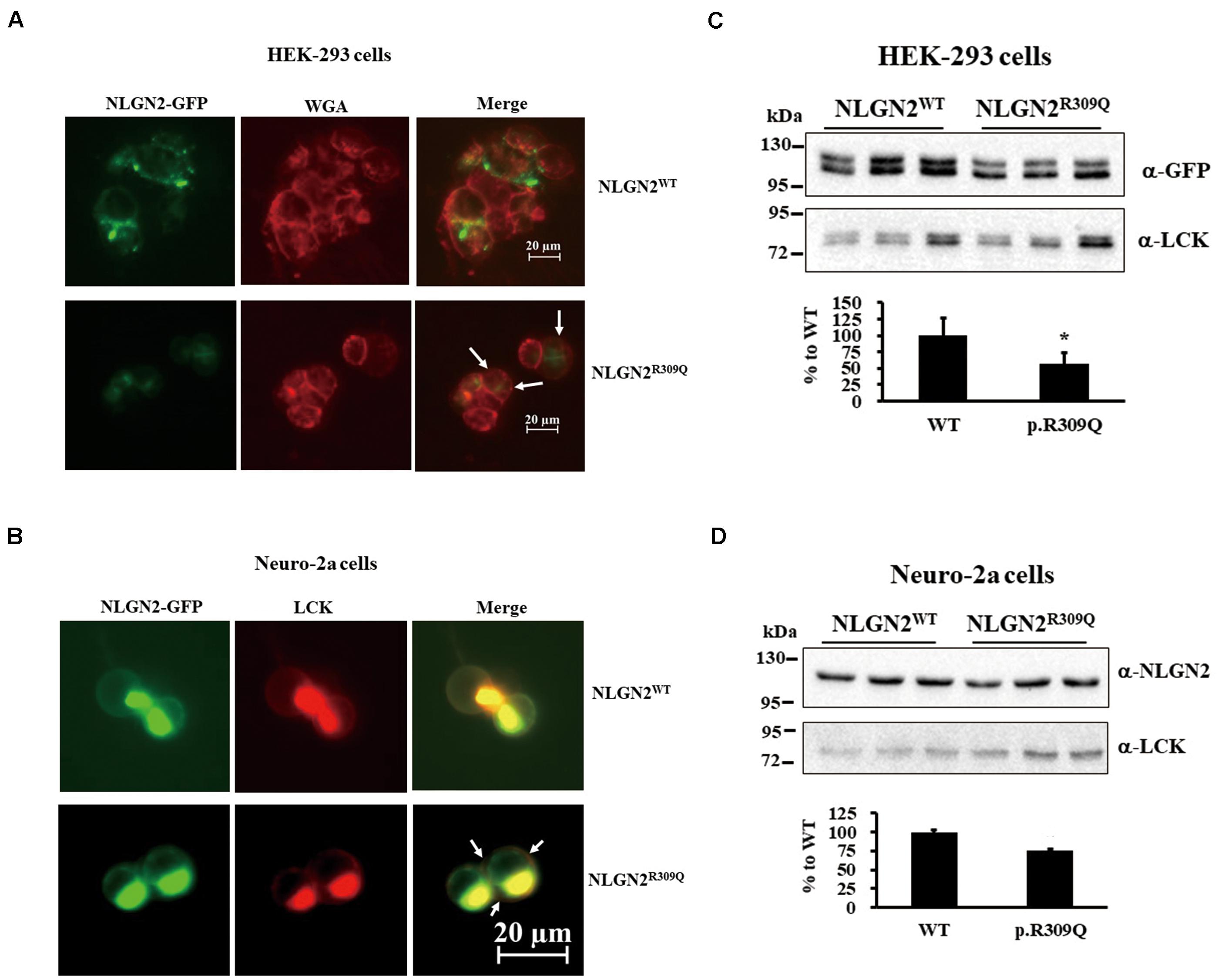
Figure 3. NLGN2-GFP fusion protein expression was reduced in the membrane fraction of cultured cells carrying NLGN2R309Q mutant compared with the WT construct. (A) Subcellular localization of NLGN2 fusion GFP protein in HEK-293 cells transiently cotransfected either with NLGN2WT or NLGN2R309Q plasmids. WT proteins colocalized to the membrane in transfected HEK-293 cells, whereas NLGN2R309Q mutant proteins colocalized to the membrane (WGA) at a lower level (arrow). (B) Subcellular localization of NLGN2 fusion GFP protein in neuro-2a cells transiently cotransfected either with NLGN2WT or NLGN2R309Q plasmids and LCK plasmids. WT proteins colocalized to the plasma membrane marker (LCK) in transfected neuro-2a cells, whereas NLGN2R309Q mutant proteins colocalized to LCK at a lower level (arrow). (C) Immunoblotting of protein extracts from the membrane fraction of HEK-293 overexpressed either with NLGN2WT or NLGN2R309Q plasmid. (D) Immunoblotting of protein extracts from the membrane fraction of neuro-2a cells overexpressed either with NLGN2WT or NLGN2R309Q plasmid. WT, wild-type. WGA and LCK, membrane protein markers wheat germ agglutinin and lymphocyte-specific protein tyrosine kinase, respectively. Graphs represent means ± standard errors. *p < 0.05.
Clinical Findings of the Patients With Rare Missense Mutations
Because eight variants that altered protein expression were absent from the dbSNP database, the gnomAD (non-neuro) dataset, and Taiwan BioBank, we presumed that they existed in the multiple psychosis family. The familial relationships of the patients with schizophrenia with rare coding variants identified in this study are presented in Figure 4. Details regarding patient history are shown in Supplementary Table 4. We did not perform segregation analysis in multiplex pedigrees; we then could not confirm that the identified rare missense mutations were de novo or transmitted from parents to multiple affected members.
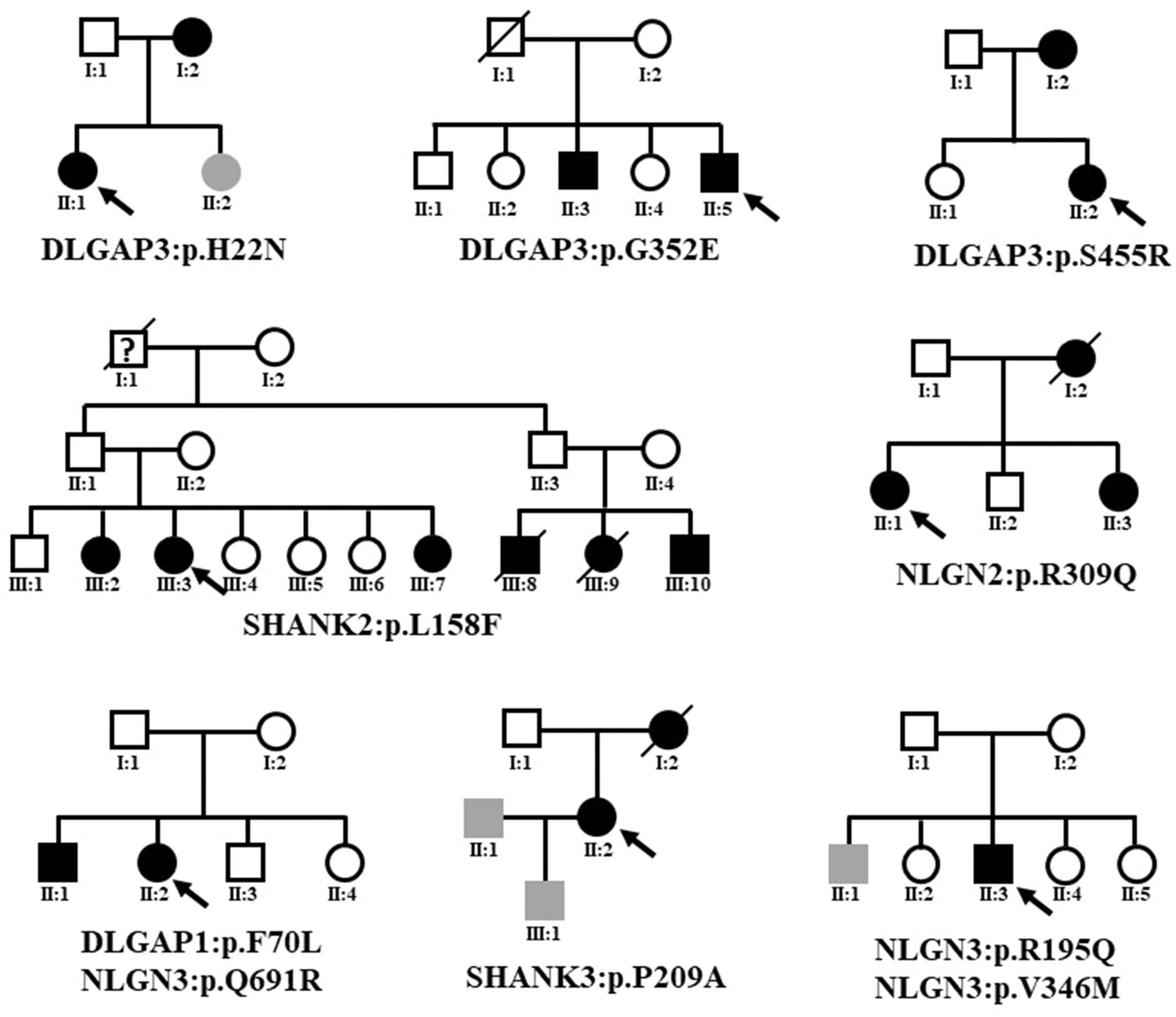
Figure 4. Familial relationships of the patients with schizophrenia with rare coding variants identified in this study. The black symbol indicates schizophrenia. The gray symbol indicates an intellectual disability. The question mark indicates unclear psychosis. Black arrows indicate the index case subjects.
Discussion
Schizophrenia is a disorder involving polygenic inheritance, with rare variants having large effects on risk and common variants having minor impacts (Craddock et al., 2007; McClellan et al., 2007). A genome-wide association study (GWAS) has been used to evaluate an association with common genetic variants or markers (typically with an MAF > 5 %) with schizophrenia (Schizophrenia Working Group of the Psychiatric Genomics Consrotium, 2014). Because GWAS focuses on identifying common variants, it is plausible that low frequency (MAF < 5%) could explain additional trait variability of schizophrenia. Accumulated studies demonstrated that loss-of-function variants and missense variants were enriched in genes with shared similar cellular function, having clinical implications for finding additional drug targets (Lips et al., 2012; Rees et al., 2019). Targeted sequencing focuses on many targeted regions, such as a functional gene group, and identifies sequence variants with high confidence and accuracy (Bewicke-Copley et al., 2019). PSD proteins are essential for protein trafficking in neurons and synaptic plasticity, processes commonly associated with schizophrenia pathogenesis (Pocklington et al., 2014). Here, to understand whether rare mutations in PSD protein-related genes are associated with schizophrenia, we carried out a mutation screening study of 18 PSD protein-related genes in a sample of schizophrenia from Taiwan. Notably, we identified 10 rare missense mutations in the PSD protein-related genes from schizophrenic patients with a family psychiatric history. These 10 missense variants, which were absent from the dbSNP database, the gnomAD (non-neuro) dataset, and Taiwan BioBank, were not detected in the subsequent collection of an independent sample (468 patients with schizophrenia and 533 healthy controls), presuming they might be ultrarare mutations of PSD protein-related genes and exist in the multiplex psychosis family. Our data are also in line with two exome sequencing-based studies that provide strong evidence that de novo mutations from schizophrenia affect genes coding for postsynaptic proteins complex (Fromer et al., 2014; Purcell et al., 2014). In addition, similar resequencing studies reported that rare mutations with loss of function in a variety of postsynaptic scaffolding proteins were present in patients with schizophrenia (Xing et al., 2016; Yu et al., 2018).
NLGN2 and NLGN3 are similar in structure and sequence to NLGN1 and share an N-terminal signal peptide, a large esterase homology domain, a single transmembrane region, and a C-terminal cytoplasmic tail (Ichtchenko et al., 1996). We identified three schizophrenia-associated mutants where NLGN2R309Q and NLGN3V346M were located at the esterase domain of the NLGN protein, and NLGN3Q691R was located between the esterase and cholinesterase domain. Notably, one putative damaging missense mutation, NLGN2R309Q, was absent in the 503 controls, 1,517 healthy controls from Taiwan BioBank, the dbSNP database, and the gnomAD (non-neuro) dataset. To understand the effects of NLGN2R309Q, NLGN3V346M, and NLGN3Q691R on protein expression and translocation, we examined the protein expression and the subcellular localization of the WT and the mutant proteins in HEK-293 and neuro-2a cells. NLGN2 expression were significantly reduced by NLGN2R309Q in HEK-293 and neuro-2a cells. In addition, NLGN2R309Q mutant protein was significantly reduced in the membrane fraction of HEK-293 cells and tended to reduce in neuro-2a cells, suggesting that NLGN2R309Q affects NLGN2 trafficking. Our functional analysis identified the NLGN2R309Q mutant as a loss-of-function, supporting our previous findings of the association of rare missense mutations in NLGN2 with schizophrenia (Sun et al., 2011). NLGN3 expression was significantly increased by NLGN3R195Q/V346M and reduced by NLGN3Q691R in HEK-293 cells. However, we did not observe the differential location of NLGN3 protein between NLGN3WT and two mutants (NLGN3R195Q/V346M and NLGN3Q691R), concluding that the NLGN3 mutants significantly reduced NLGN3 expression but did not affect NLGN3 trafficking. The effect of the NLGN3R195Q/V346M and NLGN3Q691R on schizophrenia pathophysiology requires further elucidation. Taken together, these findings suggest that NLGN2 and NLGN3 mutations are part of the genetic underpinnings of schizophrenia and support the implication of synaptic dysfunction in schizophrenia pathogenesis.
SHANK family members share several central domain regions, including a Shank/ProSAP N-terminal domain (SPN), multiple ankyrin (ANK) repeat domains, an SRC homology 3 (SH3) domain, a PSD-05/D/ZO-1 (PDZ) domain, a proline-rich region, and a C-terminal sterile α motif domain (Kursula, 2019). We identified two heterozygous missense mutations (SHANK2L158F and SHANK3P209A) located at the ANK domain of the SHANK family protein in two unrelated patients with schizophrenia. We speculated that these two mutations might affect the binding ability of SHANK proteins with their interactions with other PSD proteins. Assuming that the SHANK gene mutations affected protein expression, we observed that cells carrying SHANK2L158F and SHANK3P209A mutants altered the levels of SHANK2 and SHANK3 proteins, respectively, relative to WT cells. Immunocytochemistry revealed these two mutants and WT proteins mostly localized in the cytosol. Thus, we suggest that these two mutants significantly alter protein levels but do not affect protein trafficking. However, we found that these two SHANK mutant proteins (SHANK2L158F and SHANK3P209A) were increased in HEK-293 cells but decreased in neuro-2a. Thus, the effect of these mutants on schizophrenia pathophysiology requires further clarification. Peykov and colleagues identified a rare causative SHANK2 variant (SHANK2A1731S) in schizophrenia (Peykov et al., 2015), and several groups have found missense mutations in SHANK3 in schizophrenia (Gauthier et al., 2010; de Sena Cortabitarte et al., 2017). Gauthier and colleagues identified a nonsense mutation of SHANK3 changing an arginine to a stop codon (SHANK3R1117X) in three brothers diagnosed with schizophrenia spectrum disorders (Gauthier et al., 2010). However, increasing evidence is linking SHANK3 gene mutations as a cause of autism (Durand et al., 2007; Waga et al., 2011). Schizophrenia and autism have overlapping symptoms, especially the negative symptoms, suggesting that they share some common biological basis in their pathogenesis. These observations reflect that rare risk mutations in the SHANK gene family might underlie some of the cognitive and social dysfunction present in schizophrenia and autism. Taken together, this suggests that the rare missense variant burden is increased in patients with schizophrenia and those with autism compared with healthy controls.
We previously detected three rare schizophrenia-associated missense mutations (DLGAP3G381S, DLGAP3G587R, and DLGAP3R770L) in patients with schizophrenia (Li et al., 2013a) and speculated that they might affect the posttranslational modification of DLGAP3 protein via kinases phosphorylation. In this study, we identified three additional schizophrenia-associated DLGAP3 missense mutants (DLGAP3H22N, DLGAP3G352E, DLGAP3S455R) in an independent sample of patients with schizophrenia. Notably, the DLGAP3S455R, DLGAP3G587R, and DLGAP3R770L mutants were located at the C-terminal of the DLGAP3 gene, which interacts with focal adhesion kinase and proline-rich tyrosine kinase 2 (Bongiorno-Borbone et al., 2005), presuming the DLGAP3S455R mutant affects the anchoring of specific protein kinases in the PSD region. We observed that DLGAP3H22N, DLGAP3G352E, and DLGAP3S455R significantly reduced DLGAP3 protein expression in HEK-293 cells; however, DLGAP3 was expressed in the cytosol in all three mutants and the WT. Thus, we suggest that these mutants significantly reduce DLGAP3 activity but do not affect protein trafficking. Wan and colleagues suggested that DLGAP3 plays a role in inhibiting mGluR5 activity and the downstream triggering of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor endocytosis (Wan et al., 2011). Chen and colleagues identified a role for SAPAP3 in regulating postsynaptic mGluRs and endocannabinoid-mediated synaptic plasticity (Chen et al., 2011). We speculated that DLGAP3H22N, DLGAP3G352E, and DLGAP3S455R mutants might affect the binding ability of DLGAP3 with proteins associated with metabotropic glutamate receptors. However, these rare DLGAP3 mutants were not located in a known functional domain but were predicted as highly conserved among different species. Furthermore, we identified a DLGAP1 missense mutant (DLGAP1F70L) in 1 of 98 patients with schizophrenia, but not in a subsequent collection of 468 patients and 533 healthy controls, suggesting that DLGAP1F70L might be a patient-specific rare mutation. This missense mutation is located at conserved sequences, presuming that this mutant may impair critical biological functions of DLGAP1. However, the in silico analysis and functional analysis by immunoblotting denied this possibility. Thus, the relationship between rare mutations in the DLGAP gene family and schizophrenia pathogenesis needs to be elucidated.
This study has several weaknesses. First, we identified 50 missense mutations in the PSD protein-related genes from our sample. However, we cannot rule out the possibility that common SNPs within these genes may be associated with schizophrenia. Besides, several potentially valuable regions were not sequenced, such as introns and noncoding regions. We also did not detect insertions and deletions variants because of the technical limitation of semiconductor sequencing (Loman et al., 2012). Second, because we were not able to obtain samples from family members of probands, we did not confirm that the identified rare missense mutations were segregated with schizophrenia. Third, our functional assays were conducted using HEK-293 and neuro-2a cells, not primary neuronal cells. Although HEK-293 and neuro-2a cells are straightforward to grow in culture and suitably used as hosts for gene expression, primary neuronal cells are widely used to study synaptic function, morphology, neurotoxicity, neurotransmitter release, and disease modeling. Thus, the findings in the present study must be interpreted cautiously. Fourth, we found that DLGAP3 and DLGAP1 protein were not well detected in neuro-2a cells (data not shown), while the SHANK proteins with the SHANK mutants were increased in HEK-293 cells but decreased in neuro-2a. The discrepant results may be due to cell line–specific expressions and the differences in the genetic and biochemical processes regulating genes or genome activity in human and mouse cells (Zhang et al., 2019). Also, in vitro experiments may not fully or accurately predict the effects on a living organism. Further, in vivo studies using animal models carrying these mutants should be needed to validate our in vitro results and to understand the functional consequence of these mutants in vivo. Besides, we were not able to directly evaluate expression changes of PSD-related proteins from patients. Thus, the clinical relevance of the mutations we identified should be interpreted with caution. Finally, a larger sample size is required to have sufficient statistical power to implicate rare pathologic variants with high confidence.
In summary, we identified 10 rare missense mutations in the PSD protein-related genes from schizophrenia, characterized the functional consequences of nine mutations in cultured cells, and demonstrated eight variants altered protein expression and might be associated with schizophrenia. These findings demonstrate the importance of the identified PSD-related gene mutations, especially in the NLGN, SHANK, and DLGAP family, and shed light on future functional genomic investigations of the genes and related biological pathways of schizophrenia. The in vitro and in vivo impacts of these rare pathological mutations on the pathophysiology of schizophrenia are worthy of future investigation.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by the Antai Medical Care Cooperation Antai-Tian-Sheng Memorial Hospital Institution Review Board (IRB Number: 101020). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
M-CC designed the study and wrote the protocol and draft. T-MH, Y-CW, and C-LW helped recruit and evaluate the patients. M-CC, S-HH, and H-YT conducted the experimental works and analyzed the data. All authors reviewed the article and approved its publication.
Funding
Funding for this study was provided by the Ministry of Science and Technology, Taiwan (grant MOST104-2314-B-480-001-MY3) and Yuli Branch, Taipei Veterans General Hospital, Taiwan (grant VHYL-105-04, VHYL-106-01, VHYL-107-01, and VHYL-107-02).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors sincerely thank Dr. Shaw-Ji Chen, the faculty of the department of psychiatry, Mackay Memorial Hospital, Taitung Branch, Taiwan, for his assistance with the statistics used in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.524258/full#supplementary-material
Footnotes
- ^ http://bioinfo.ut.ee/primer3-0.4.0/primer3/
- ^ https://taiwanview.twbiobank.org.tw
- ^ http://genetics.bwh.harvard.edu/pph2/
- ^ http://sift.bii.a-star.edu.sg/
- ^ http://mmb.pcb.ub.es/PMut/
- ^ http://provean.jcvi.org/seq_submit.php
- ^ http://genome.ucsc.edu/index.html
- ^ http://rsb.info.nih.gov/nih-image/
References
Berkel, S., Marshall, C. R., Weiss, B., Howe, J., Roeth, R., Moog, U., et al. (2010). Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet. 42, 489–491. doi: 10.1038/ng.589
Bewicke-Copley, F., Arjun Kumar, E., Palladino, G., Korfi, K., and Wang, J. (2019). Applications and analysis of targeted genomic sequencing in cancer studies. Comput. Struct. Biotechnol. J. 17, 1348–1359. doi: 10.1016/j.csbj.2019.10.004
Bongiorno-Borbone, L., Kadaré, G., Benfenati, F., and Girault, J.-A. (2005). FAK and PYK2 interact with SAP90/PSD-95-associated protein-3. Biochem. Biophys. Res. Commun. 337, 641–646. doi: 10.1016/j.bbrc.2005.09.099
Boucard, A. A., Chubykin, A. A., Comoletti, D., Taylor, P., and Sudhof, T. C. (2005). A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha- and beta-neurexins. Neuron 48, 229–236. doi: 10.1016/j.neuron.2005.08.026
Chen, M., Wan, Y., Ade, K., Ting, J., Feng, G., and Calakos, N. (2011). Sapap3 deletion anomalously activates short-term endocannabinoid-mediated synaptic plasticity. J. Neurosci. 31, 9563–9573. doi: 10.1523/jneurosci.1701-11.2011
Clinton, S. M., and Meador-Woodruff, J. H. (2004). Abnormalities of the NMDA receptor and associated intracellular molecules in the thalamus in schizophrenia and bipolar disorder. Neuropsychopharmacology 29, 1353–1362. doi: 10.1038/sj.npp.1300451
Craddock, N., O’Donovan, M. C., and Owen, M. J. (2007). Phenotypic and genetic complexity of psychosis. Invited commentary on. Schizophrenia: a common disease caused by multiple rare alleles. Br. J. Psychiatry 190, 200–203. doi: 10.1192/bjp.bp.106.033761
de Sena Cortabitarte, A., Degenhardt, F., Strohmaier, J., Lang, M., Weiss, B., Roeth, R., et al. (2017). Investigation of SHANK3 in schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 174, 390–398. doi: 10.1002/ajmg.b.32528
Dracheva, S., Marras, S. A. E., Elhakem, S. L., Kramer, F. R., Davis, K. L., and Haroutunian, V. (2001). N-Methyl-D-Aspartic acid receptor expression in the dorsolateral prefrontal cortex of elderly patients with schizophrenia. Am. J. Psychiatry 158, 1400–1410. doi: 10.1176/appi.ajp.158.9.1400
Durand, C. M., Betancur, C., Boeckers, T. M., Bockmann, J., Chaste, P., Fauchereau, F., et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27. doi: 10.1038/ng1933
Focking, M., Lopez, L. M., English, J. A., Dicker, P., Wolff, A., Brindley, E., et al. (2015). Proteomic and genomic evidence implicates the postsynaptic density in schizophrenia. Mol. Psychiatry 20, 424–432. doi: 10.1038/mp.2014.63
Fromer, M., Pocklington, A. J., Kavanagh, D. H., Williams, H. J., Dwyer, S., Gormley, P., et al. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184. doi: 10.1038/nature12929
Fukaya, M., and Watanabe, M. (2000). Improved immunohistochemical detection of postsynaptically located PSD-95/SAP90 protein family by protease section pretreatment: a study in the adult mouse brain. J. Comp. Neurol. 426, 572–586. doi: 10.1002/1096-9861(20001030)426:4<572::aid-cne6<3.0.co;2-9
Gauthier, J., Champagne, N., Lafrenière, R. G., Xiong, L., Spiegelman, D., Brustein, E., et al. (2010). De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 107, 7863–7868. doi: 10.1073/pnas.0906232107
Grozeva, D., Conrad, D. F., Barnes, C. P., Hurles, M., Owen, M. J., O’Donovan, M. C., et al. (2012). Independent estimation of the frequency of rare CNVs in the UK population confirms their role in schizophrenia. Schizophr. Res. 135, 1–7. doi: 10.1016/j.schres.2011.11.004
Hayashi-Takagi, A., and Sawa, A. (2010). Disturbed synaptic connectivity in schizophrenia: convergence of genetic risk factors during neurodevelopment. Brain Res. Bull. 83, 140–146. doi: 10.1016/j.brainresbull.2010.04.007
Ichtchenko, K., Nguyen, T., and Sudhof, T. C. (1996). Structures, alternative splicing, and neurexin binding of multiple neuroligins. J. Biol. Chem. 271, 2676–2682. doi: 10.1074/jbc.271.5.2676
Jamain, S., Quach, H., Betancur, C., Rastam, M., Colineaux, C., Gillberg, I. C., et al. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29. doi: 10.1038/ng1136
Kirov, G., Pocklington, A. J., Holmans, P., Ivanov, D., Ikeda, M., Ruderfer, D., et al. (2012). De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 17, 142–153. doi: 10.1038/mp.2011.154
Kristiansen, L. V., Huerta, I., Beneyto, M., and Meador-Woodruff, J. H. (2007). NMDA receptors and schizophrenia. Curr. Opin. Pharmacol. 7, 48–55. doi: 10.1016/j.coph.2006.08.013
Kursula, P. (2019). Shanks – multidomain molecular scaffolds of the postsynaptic density. Curr. Opin. Struct. Biol. 54, 122–128. doi: 10.1016/j.sbi.2019.01.007
Li, J. M., Lu, C. L., Cheng, M. C., Luu, S. U., Hsu, S. H., and Chen, C. H. (2013a). Exonic resequencing of the DLGAP3 gene as a candidate gene for schizophrenia. Psychiatry Res. 208, 84–87. doi: 10.1016/j.psychres.2012.12.015
Li, J. M., Lu, C. L., Cheng, M. C., Luu, S. U., Hsu, S. H., and Chen, C. H. (2013b). Genetic analysis of the DLGAP1 gene as a candidate gene for schizophrenia. Psychiatry Res. 205, 13–17. doi: 10.1016/j.psychres.2012.08.014
Li, J. M., Lu, C. L., Cheng, M. C., Luu, S. U., Hsu, S. H., Hu, T. M., et al. (2014). Role of the DLGAP2 gene encoding the SAP90/PSD-95-associated protein 2 in schizophrenia. PLoS One 9:e85373. doi: 10.1371/journal.pone.0085373
Lips, E. S., Cornelisse, L. N., Toonen, R. F., Min, J. L., Hultman, C. M., Holmans, P. A., et al. (2012). Functional gene group analysis identifies synaptic gene groups as risk factor for schizophrenia. Mol. Psychiatry 17, 996–1006. doi: 10.1038/mp.2011.117
Loman, N. J., Misra, R. V., Dallman, T. J., Constantinidou, C., Gharbia, S. E., Wain, J., et al. (2012). Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 30, 434–439. doi: 10.1038/nbt.2198
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., et al. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753. doi: 10.1038/nature08494
McClellan, J. M., Susser, E., and King, M.-C. (2007). Schizophrenia: a common disease caused by multiple rare alleles. Br. J. Psychiatry 190, 194–199. doi: 10.1192/bjp.bp.106.025585
McGlashan, T. H., and Hoffman, R. E. (2000). Schizophrenia as a disorder of developmentally reduced synaptic connectivity. Arch. Gen. Psychiatry 57, 637–648. doi: 10.1001/archpsyc.57.7.637
Missler, M., Fernandez-Chacon, R., and Sudhof, T. C. (1998). The making of Neurexins. J. Neurochem. 71, 1339–1347. doi: 10.1046/j.1471-4159.1998.71041339.x
Mowry, B. J., and Gratten, J. (2013). The emerging spectrum of allelic variation in schizophrenia: current evidence and strategies for the identification and functional characterization of common and rare variants. Mol. Psychiatry 18, 38–52. doi: 10.1038/mp.2012.34
Naisbitt, S., Kim, E., Tu, J. C., Xiao, B., Sala, C., Valtschanoff, J., et al. (1999). Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 23, 569–582. doi: 10.1016/s0896-6273(00)80809-0
Parente, D. J., Garriga, C., Baskin, B., Douglas, G., Cho, M. T., Araujo, G. C., et al. (2017). Neuroligin 2 nonsense variant associated with anxiety, autism, intellectual disability, hyperphagia, and obesity. Am. J. Med. Genet. A 173, 213–216. doi: 10.1002/ajmg.a.37977
Peykov, S., Berkel, S., Schoen, M., Weiss, K., Degenhardt, F., Strohmaier, J., et al. (2015). Identification and functional characterization of rare SHANK2 variants in schizophrenia. Mol. Psychiatry 20, 1489–1498. doi: 10.1038/mp.2014.172
Pocklington, A. J., O’Donovan, M., and Owen, M. J. (2014). The synapse in schizophrenia. Eur. J. Neurosci. 39, 1059–1067. doi: 10.1111/ejn.12489
Purcell, S. M., Moran, J. L., Fromer, M., Ruderfer, D., Solovieff, N., Roussos, P., et al. (2014). A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190. doi: 10.1038/nature12975
Rasmussen, A. H., Rasmussen, H. B., and Silahtaroglu, A. (2017). The DLGAP family: neuronal expression, function and role in brain disorders. Mol. Brain 10:43. doi: 10.1186/s13041-017-0324-9
Rees, E., Carrera, N., Morgan, J., Hambridge, K., Escott-Price, V., Pocklington, A. J., et al. (2019). Targeted sequencing of 10,198 samples confirms abnormalities in neuronal activity and implicates voltage-gated sodium channels in schizophrenia pathogenesis. Biol. Psychiatry 85, 554–562. doi: 10.1016/j.biopsych.2018.08.022
Sato, D., Lionel, A. C., Leblond, C. S., Prasad, A., Pinto, D., Walker, S., et al. (2012). SHANK1 deletions in males with autism spectrum disorder. Am. J. Hum. Genet. 90, 879–887. doi: 10.1016/j.ajhg.2012.03.017
Schizophrenia Working Group of the Psychiatric Genomics Consrotium (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. doi: 10.1038/nature13595
Schork, N. J., Murray, S. S., Frazer, K. A., and Topol, E. J. (2009). Common vs. rare allele hypotheses for complex diseases. Curr. Opin. Genet. Dev. 19, 212–219. doi: 10.1016/j.gde.2009.04.010
Shiraishi-Yamaguchi, Y., and Furuichi, T. (2007). The Homer family proteins. Genome Biol. 8:206. doi: 10.1186/gb-2007-8-2-206
Stefansson, H., Rujescu, D., Cichon, S., Pietilainen, O. P., Ingason, A., Steinberg, S., et al. (2008). Large recurrent microdeletions associated with schizophrenia. Nature 455, 232–236. doi: 10.1038/nature07229
Sullivan, P. F., Daly, M. J., and O’Donovan, M. (2012). Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat. Rev. Genet. 13, 537–551. doi: 10.1038/nrg3240
Sun, C., Cheng, M. C., Qin, R., Liao, D. L., Chen, T. T., Koong, F. J., et al. (2011). Identification and functional characterization of rare mutations of the neuroligin-2 gene (NLGN2) associated with schizophrenia. Hum. Mol. Genet. 20, 3042–3051. doi: 10.1093/hmg/ddr208
Takeuchi, M., Hata, Y., Hirao, K., Toyoda, A., Irie, M., and Takai, Y. (1997). SAPAPs. A family of PSD-95/SAP90-associated proteins localized at postsynaptic density. J. Biol. Chem. 272, 11943–11951. doi: 10.1074/jbc.272.18.11943
Thomas, U. (2002). Modulation of synaptic signalling complexes by Homer proteins. J. Neurochem. 81, 407–413. doi: 10.1046/j.1471-4159.2002.00869.x
Tu, J. C., Xiao, B., Naisbitt, S., Yuan, J. P., Petralia, R. S., Brakeman, P., et al. (1999). Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 23, 583–592. doi: 10.1016/s0896-6273(00)80810-7
Waga, C., Okamoto, N., Ondo, Y., Fukumura-Kato, R., Goto, Y., Kohsaka, S., et al. (2011). Novel variants of the SHANK3 gene in Japanese autistic patients with severe delayed speech development. Psychiatr. Genet. 21, 208–211. doi: 10.1097/YPG.0b013e328341e069
Walsh, T., McClellan, J. M., McCarthy, S. E., Addington, A. M., Pierce, S. B., Cooper, G. M., et al. (2008). Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320, 539–543. doi: 10.1126/science.1155174
Wan, Y., Feng, G., and Calakos, N. (2011). Sapap3 deletion causes mGluR5-dependent silencing of AMPAR synapses. J. Neurosci. 31, 16685–16691. doi: 10.1523/jneurosci.2533-11.2011
Wilson, H. L., Wong, A. C., Shaw, S. R., Tse, W. Y., Stapleton, G. A., Phelan, M. C., et al. (2003). Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J. Med. Genet. 40, 575–584. doi: 10.1136/jmg.40.8.575
Xing, J., Kimura, H., Wang, C., Ishizuka, K., Kushima, I., Arioka, Y., et al. (2016). Resequencing and association analysis of six PSD-95-related genes as possible susceptibility genes for schizophrenia and autism spectrum disorders. Sci. Rep. 6:27491. doi: 10.1038/srep27491
Xu, B., Roos, J. L., Dexheimer, P., Boone, B., Plummer, B., Levy, S., et al. (2011). Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat. Genet. 43, 864–868. doi: 10.1038/ng.902
Xu, B., Roos, J. L., Levy, S., van Rensburg, E. J., Gogos, J. A., and Karayiorgou, M. (2008). Strong association of de novo copy number mutations with sporadic schizophrenia. Nat. Genet. 40, 880–885. doi: 10.1038/ng.162
Xu, X., Xiong, Z., Zhang, L., Liu, Y., Lu, L., Peng, Y., et al. (2014). Variations analysis of NLGN3 and NLGN4X gene in Chinese autism patients. Mol. Biol. Rep. 41, 4133–4140. doi: 10.1007/s11033-014-3284-5
Yu, Y., Lin, Y., Takasaki, Y., Wang, C., Kimura, H., Xing, J., et al. (2018). Rare loss of function mutations in N-methyl-D-aspartate glutamate receptors and their contributions to schizophrenia susceptibility. Transl. Psychiatry 8:12. doi: 10.1038/s41398-017-0061-y
Keywords: schizophrenia, PSD gene, resequencing, rare mutation, in vitro
Citation: Hu T-M, Wang Y-C, Wu C-L, Hsu S-H, Tsai H-Y and Cheng M-C (2020) Multiple Rare Risk Coding Variants in Postsynaptic Density-Related Genes Associated With Schizophrenia Susceptibility. Front. Genet. 11:524258. doi: 10.3389/fgene.2020.524258
Received: 03 January 2020; Accepted: 09 November 2020;
Published: 04 December 2020.
Edited by:
Claudio Toma, Severo Ochoa Molecular Biology Center (CSIC-UAM), SpainReviewed by:
Gabriel R. Fries, University of Texas Health Science Center at Houston, United StatesAmalia Martinez-Mir, Institute of Biomedicine of Seville (IBIS), Spain
Copyright © 2020 Hu, Wang, Wu, Hsu, Tsai and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min-Chih Cheng, dW5jbGUwNTVAeWFob28uY29t; Y21jQG1haWwudmh5bC5nb3YudHc=
†These authors have contributed equally to this work