 Xin Gao1†
Xin Gao1† Li Zhang2,3†
Li Zhang2,3† Qi Jia1†Liang Tang1
Qi Jia1†Liang Tang1 Wen Guo1,4Tao Wang1Zheyu Wu1,5Wang Zhou1*Zhenxi Li1*
Wen Guo1,4Tao Wang1Zheyu Wu1,5Wang Zhou1*Zhenxi Li1* Jianru Xiao1*
Jianru Xiao1*- 1Orthopedic Oncology Center, Department of Orthopedics, Changzheng Hospital, Second Military Medical University, Shanghai, China
- 2Key Laboratory of Advanced Theory and Application in Statistics and Data Science – MOE, School of Statistics, East China Normal University, Shanghai, China
- 3Center for Bioinformatics and Computational Biology, School of Life Sciences, Institute of Biomedical Sciences, East China Normal University, Shanghai, China
- 4Department of Orthopedics, Taizhou People’s Hospital, Taizhou, China
- 5Department of Orthopedics, Zhongnan Hospital of Wuhan University, Wuhan, China
Spinal schwannoma is the most common primary spinal tumor but its genomic landscape and underlying mechanism driving its initiation remain elusive. The aim of the present study was to gain further insights into the molecular mechanisms of this kind of tumor through whole genome sequencing of nine spinal schwannomas and paired blood samples. The results showed that ATM, CHD4, FAT1, KMT2D, MED12, NF2, and SUFU were the most frequently mutated cancer-related genes. In addition, the somatic copy number alterations (CNA) was potentially associated with spinal schwannoma, among which NF2 was found to be frequently deleted in schwannoma samples. Only a few genes were located within the amplified regions. In contrast, the deleted regions in 15q15.1 and 7q36.1 contained most of these genes. With respect to tumorigenesis, NF2 had the highest variant allele frequency (VAF) than other genes, and homozygous deletion was observed in NF1, NF2, and CDKN2C. Pathway-level analysis suggested that Hippo signaling pathway may be a critical pathway controlling the initiation of spinal schwannoma. Collectively, this systematic analysis of DNA sequencing data revealed that some key genes including NF1, NF2, and CDKN2C and Hippo signaling pathway were associated with spinal schwannoma, which may help improve our understanding about the genomic landscape of spinal schwannoma.
Introduction
Spinal schwannoma is the most common primary spinal tumor, accounting for almost one-third of all spinal tumors (Seppala et al., 1995; Abul-Kasim et al., 2008), with an annual incidence of 0.3 to 0.4 per 100,000 (Seppala et al., 1995). Patients with spinal schwannoma usually have to endure pain, spinal root deficits, pyramidal tract compression, and sphincter disorders, which negatively affect patients’ quality of life (Lenzi et al., 2017). The gold standard treatment for spinal schwannomas is complete surgical resection (Jinnai and Koyama, 2005; Safaee et al., 2017). However, complete tumor removal is associated with a high incidence of complications, in which cutting the involved functionally relevant nerve root is likely to result in a permanent and significant neurological deficit (Celli et al., 2005), with up to 30% of patients developing postoperative neurologic deteriorations (Safaee et al., 2017). Unfortunately, there are no medical therapies available for spinal schwannomas, while a safe and effective treatment for these patients is urgently needed (Campian and Gutmann, 2017). Therefore, it is of great importance to gain a comprehensive insight into the genetic landscape of schwannoma and to identify the potential genes driving its initiation, thereby seeking therapeutic targets.
Schwannomas are derived from Schwann cells, the myelin-producing cells of the peripheral nervous system. Schwannomas can occur either spontaneously or as the hallmark tumor of neurofibromatosis type 2. Previous studies have identified several schwannoma-predisposing mutations. Specifically, mutation of the NF2 gene is the most characteristic genetic risk factor for schwannoma (Rouleau et al., 1993; Trofatter et al., 1993; Lin and Gutmann, 2013; Pathmanaban et al., 2017; Carlson et al., 2018; Roberts et al., 2019). Germline mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas (Piotrowski et al., 2014). Mutation of the SMARCB1 gene was found in 45% of familial and 7% of sporadic schwannomatosis cases (Smith et al., 2012), and it was also reported to play a role in the development of some sporadic spinal schwannomas (Paganini et al., 2018). In order to determine the frequency of these predisposing mutations in solitary schwannomas, Pathmanaban et al. screened 135 schwannoma cases using Sanger sequencing or next-generation sequencing, and the results showed that NF2, LZTR1, or SMARCB1 mutations were found in 54.5% of them (Pathmanaban et al., 2017). Furthermore, in an integrative analysis performed by Agnihotri et al. (2016) whole exome sequencing analysis identified recurrent mutations in NF2, ARID1A, ARID1B, and DDR1, and RNA sequencing identified a recurrent in-frame SH3PXD2A-HTRA1 fusion in 10% cases. Whole exome sequencing was also performed on vestibular schwannomas. NF2, CDC27, and USP8 were identified to be the most common tumor-specific mutations (Havik et al., 2018). Nevertheless, a focused attention on the genomic landscape of schwannomas in the spine is still needed. Furthermore, mutations in non-coding regions, such as introns, regulatory elements, and non-coding RNA, remain widely unexplored.
In this study, whole genome sequencing analysis of nine paired spinal schwannomas (ICD-O of 9560/0) and blood samples was performed to provide some useful information regarding the overall landscape of driver mutations and mutated pathways in schwannomas genomes.
Materials and Methods
DNA Extraction and Whole Genome Sequencing
This study was approved by the Changzheng Hospital medical ethics committee, and genomic DNA samples were obtained from fresh tissues and blood samples after being resected during surgery. All the patients were recruited with written informed consent, in accordance with the Declaration of Helsinki. TIAN amp Blood DNA Kit (TIANGEN Biotech Co., Ltd., Beijing, China) was used for extracting genomic DNA from paired schwannoma and blood samples according to the manufacturer’s instructions. The whole genome DNA were sequenced by Illumina X-Ten platform in Shanghai, China, and 150 bp paired-end reads were generated.
Reads Mapping and Variants Calling and Annotation
The whole genome paired reads of 300bp (150bp at each end) were mapped to human reference genome (UCSC hg19 assembly) using BWA 0.7.12 “mem” mode with default options (Li and Durbin, 2009). The PCR duplicates of the mapped reads and low-quality reads (BaseQ < 20) were then removed by SAMtools “rmdup” with version 0.1.19. The resulting bam files were sorted and indexed by SAMtools sort and index, respectively. Somatic mutations were called by Strelka 2.8.4 software (Saunders et al., 2012) with default options. The somatic mutations should have a minimal of 4 read counts supporting the variant and over 20 reads covering the locus. The somatic copy number alteration (CNA)s were called by SAASCNV 0.3.4 (Zhang and Hao, 2015) with P-value < 0.05. We used the ANNOVAR software for variant annotation (Wang et al., 2010). It should be noted that all the somatic mutations were identified in the tumor tissues but absent in the corresponding normal tissues.
Analyses of the Somatic Mutations in Schwannoma
The analyses included identification of potential genes driving schwannoma initiation, and identification of frequently mutated pathways. The potential genes driving schwannoma initiation were identified based on the variant allele frequency (VAF), and the frequently mutated pathways were identified based on the number of mutated genes in the oncogenic pathways. All the analyses were implemented in R maftools package (Mayakonda et al., 2018). The clonality analysis was conducted in R package CLONETv2 with default options (Carreira et al., 2014).
Significantly Amplified and Deleted Regions
The somatic CNAs were first segmented by SAASCNV (Zhang and Hao, 2015). The significantly amplified and deleted regions were identified by GISTIC 2.0 on the Gene Pattern webserver (Mermel et al., 2011). The CNAs with q-value < 0.05 were deemed as the significantly amplified and deleted regions.
Gene Set Enrichment Analysis
The gene sets collected from KEGG pathways were used in the enrichment analysis. The gene set enrichment analysis (GSEA) was conducted by hypergeometric test. The GSEA was implemented in R clusterProfiler package (Yu et al., 2012). The pathways were considered statistically significant if the q-value < 0.05.
Results
Clinical Characteristics and Outcomes of the Patients
The characteristics of 9 patients are shown in Table 1. This cohort was comprised of 4 men and 5 women, with a mean age of 57.6 years (median 63, range 27–69). The most common symptom was pain and hypesthesia. One patient (case 8) was diagnosed with the recurrent schwannoma which was totally resected 15 years ago. Tumor size ranged from 1 to 6.5 cm in the maximum diameter, with 2 of them larger than 5 cm. Schwannomatosis were diagnosed in 3 patients who had multiple spinal schwannomas without bilateral vestibular schwannoma. No pathogenic germline NF2 mutations were found in these patients. Complete resection of the tumor(s) and posterior stabilization of the spine was performed for all patients. According to postoperative pathological examination, the tumor cell percentages of all these samples were above 88% (Range 88–95%). Postoperatively, all patients recovered well without surgical complications. The mean follow-up duration was 42.9 (median 45, range 33–48) months. All patients were alive with no evidence of disease at the last follow-up in July 2019.
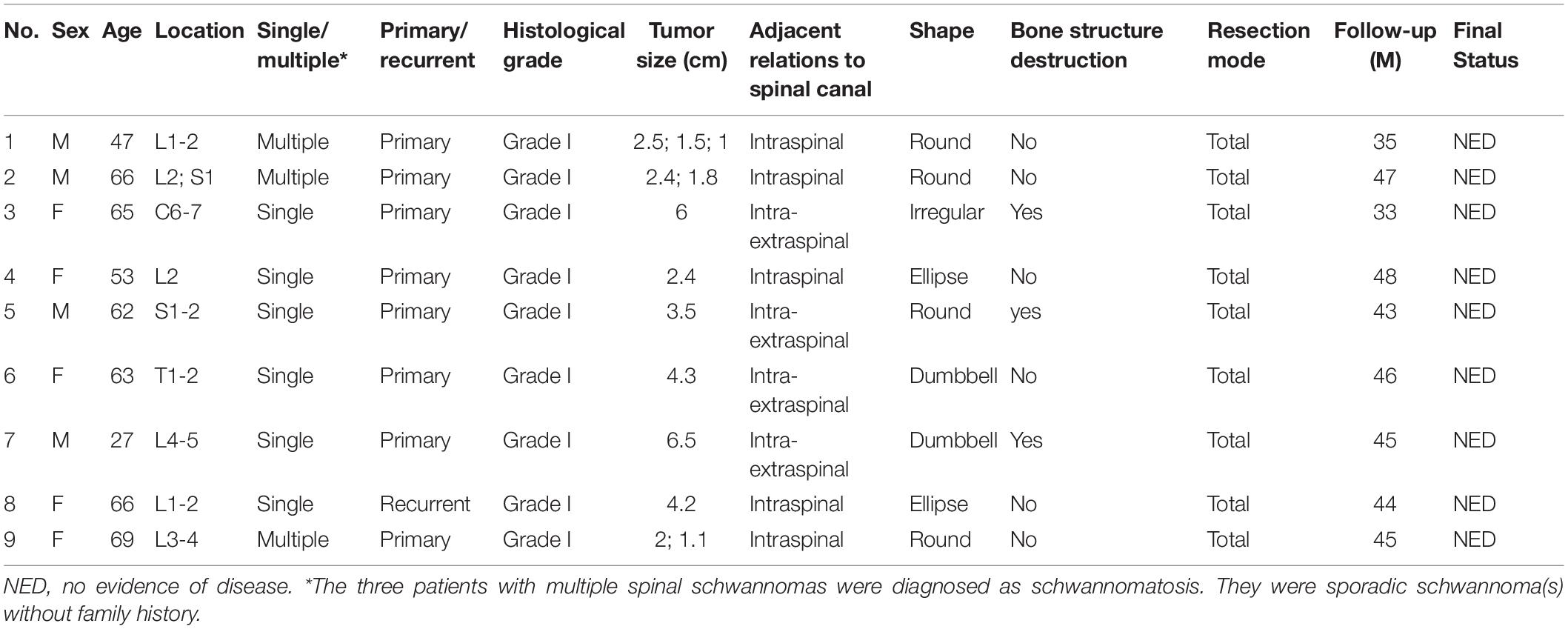
Table 1. Clinical characteristics of nine patients with spinal schwannoma.
Whole Genome Sequencing of Nine Spinal Schwannomas and Paired Blood Samples
To explore the alterations in the genome of spinal schwannoma, we performed whole genome sequencing (WGS) to a median depth of 36.77X (range, 32.34X to 40.59X), and identified 832 somatic mutations, which were present in tumors but absent in paired blood samples, including 763 single nucleotide variants (SNVs) and 69 insertion or deletions (InDels) across the 9 patients (Figures 1A,B), with a median of 87 variants (Figure 1C). Based on the RefSeq gene annotation, we identified TTN, MUC4, FLG2, MUC17, OR2T4, ZNF850, FAM186A, ALMS1, FAM47C, and ATM as the top ten mutated genes (Figure 1D), however, only ATM was previously reported to be implicated in cancer (Kim et al., 2014; Chen et al., 2015; Feng et al., 2015).
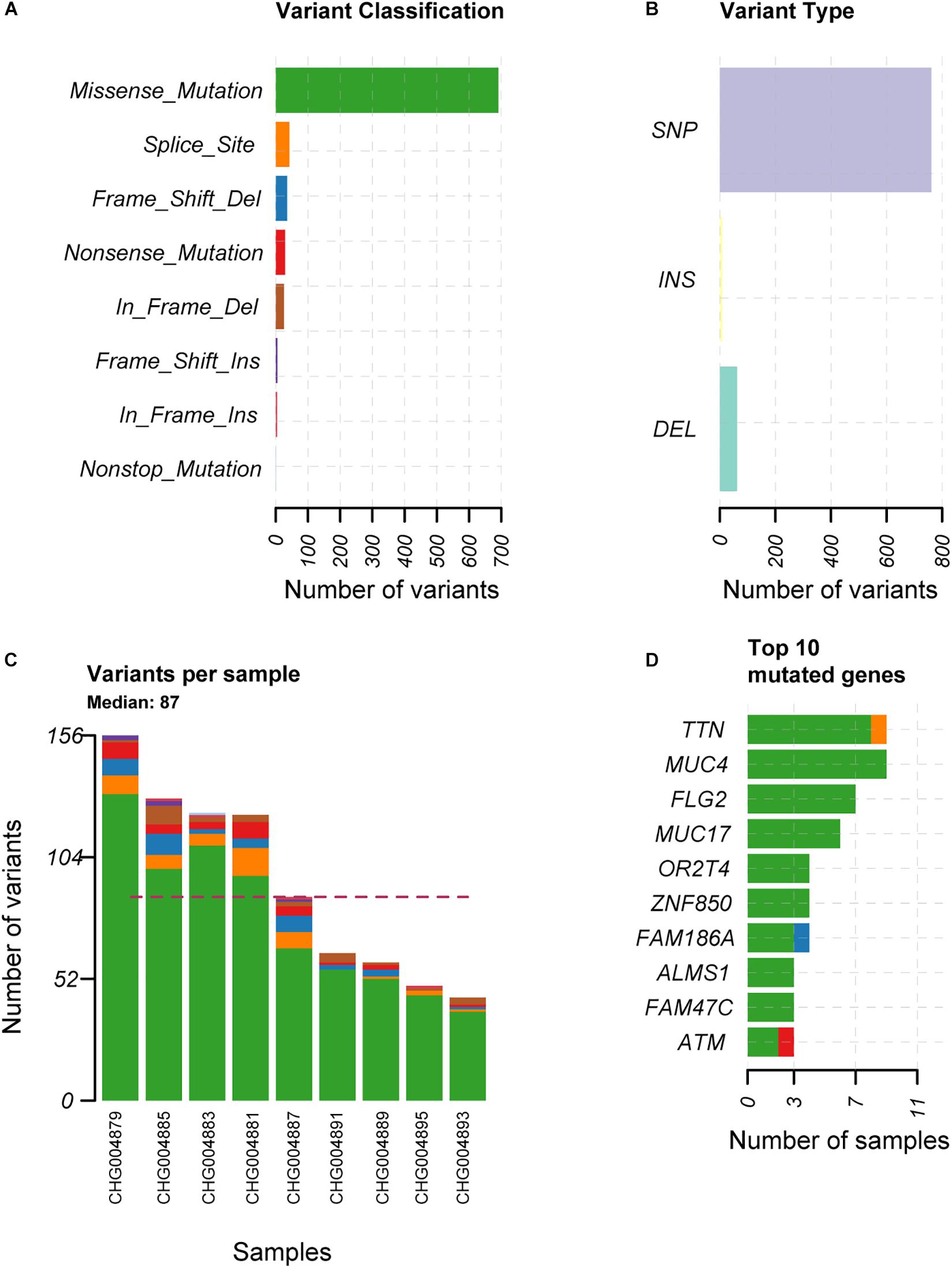
Figure 1. The overview of the somatic mutations in spinal schwannoma. The variant classification, variant type, the number of variants per sample, and the top-ten mutated genes in the nine spinal schwannomas are displayed in (A–D), respectively. The x-axis in (A,B,D) represent the number of mutations or samples. The x-axis in (C) represents the nine samples.
Genetic Landscape of Spinal Schwannoma
To identify the genes responsible for schwannoma, we further investigated whether the mutated genes could be found in the COSMIC Cancer Gene Census database (Forbes et al., 2008), a database curating genes causally implicated in cancer. As shown Figure 2A, ATM, accounting for 33% of the samples, was the most frequently mutated cancer-related gene in schwannoma, followed by CHD4 (22%), FAT1 (22%), KMT2D (22%), MED12 (22%), NF2 (22%), and SUFU (22%). Most of them were predicted as pathogenic by SIFT, PolyPhen-2, or MutationTaster (Table 2). Particularly, NF2 (Neurofibromin 2) has been widely identified as the pathogenic genes for both sporadic and familial schwannoma (Jacoby et al., 1994; Rm et al., 2018).
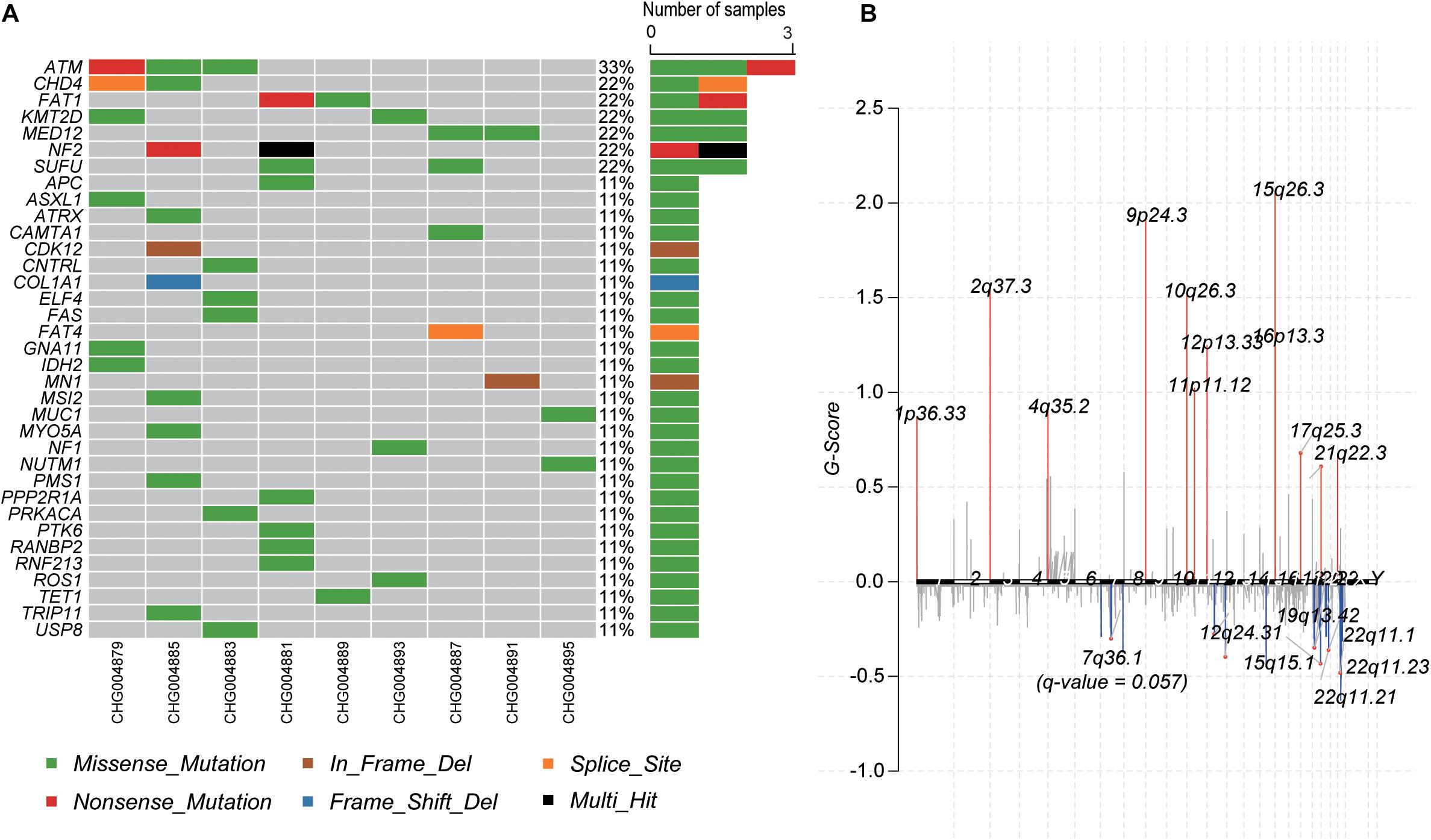
Figure 2. The mutational landscape of spinal schwannoma. (A) The somatic mutations of cancer driver genes across the nine spinal schwannomas. (B) The somatic copy number alterations along the chromosomes. The G-score was to evaluate the significance of the gains and losses.
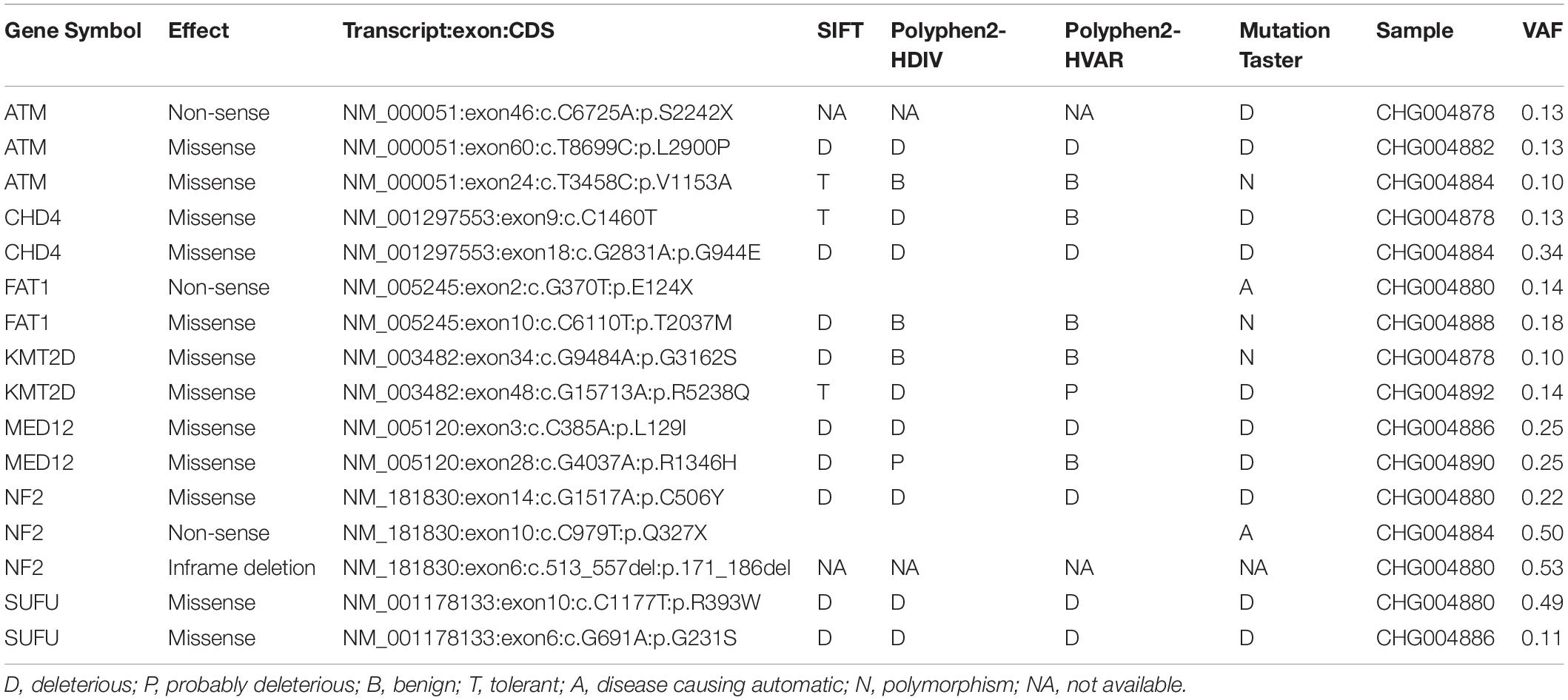
Table 2. The pathogenicity of the somatic mutations in spinal schwannoma by SIFT, PolyPhen-2, and MutationTaster.
Moreover, we also profiled the somatic CNA in schwannoma based on the GISTIC algorithm (q-value < 0.05). The identified significantly amplified regions were 1p36.33, 2q37.3, 4q35.2, 9p24.3, 10q26.3, 11p11.12, 12p13.33, 15q26.3, 16p13.3, and 17q25.3, while the significantly deleted regions were 12q24.31, 15q15.1, 19q13.42, 22q11.1, 22q11.21, and 22q11.23 (Figure 2B). In accordance with the somatic mutations and InDels, NF2, harbored in the cytoband 22q11, was also identified to be frequently deleted in the samples of schwannoma (Supplementary Figure S1). These results indicated that NF2 played a key role in the initiation of schwannoma.
Furthermore, we also investigated the genes within these CNA regions potentially involved in spinal schwannoma. Only a few genes were located within the amplified regions. whereas the deleted regions contained most of these genes (Supplementary Tables S1, S2). Specifically, ITPKA, LTK, CHP, OIP5, RTF1, RPAP1, NDUFAF1, NUSAP1, INO80, EXD1, and OIP5-AS1, fell within the cytoband of 15q15.1 (n = 6, 66.7%). The gene set enrichment analysis revealed that these genes might participate in the maintenance of chromosomal structure, such as DNA conformation change, chromosome segregation, and nucleosome organization (q-value < 0.05). In addition, we observed that XRCC2 and MLL3/KMT2C were located within 7q36.1 (n = 5, 55.6%), which had a q-value 0.057, slightly higher than the threshold 0.05, however, the two genes were involved in the tumorigenesis or progression of several malignant tumors (Figure 2B and Supplementary Table S1), suggesting that these genes might play key roles in schwannoma.
The Potential Genes Driving Schwannoma Initiation
With the whole genome sequencing data, we aimed to identify the genes potentially driving schwannoma initiation. In tumorigenesis, the more the VAF was close to 50%, the earlier the mutation may occur. We then ranked the genes based on the median of VAF across the samples (Figure 3A). Among the genes mutated in more than one sample, NF2 had a higher VAF than other genes, followed by DEPDC5, which were involved in mTOR signaling pathway. Furthermore, we also conducted the subclonality analysis, and identified that NF2 gene mutations were present in all subclones of CHG004880 and CHG004884, suggesting that NF2 was a candidate driver in spinal schwannoma (Supplementary Figure S2). Particularly, SOX10 and CHD4 also showed relatively high VAF among those genes (VAF > 20%). In addition, we also examined the five samples without NF2 gene mutations. Interestingly, the spinal schwannoma samples of CHG004882 and CHG004886 harbored homozygous deletion in NF1 (Figure 3B). Homozygous deletion of CDKN2C was observed in CHG004890 (Figure 3C). These results indicated that the CNV loss in NF1 and CDKN2C might also contribute to tumorigenesis of spinal schwannoma.
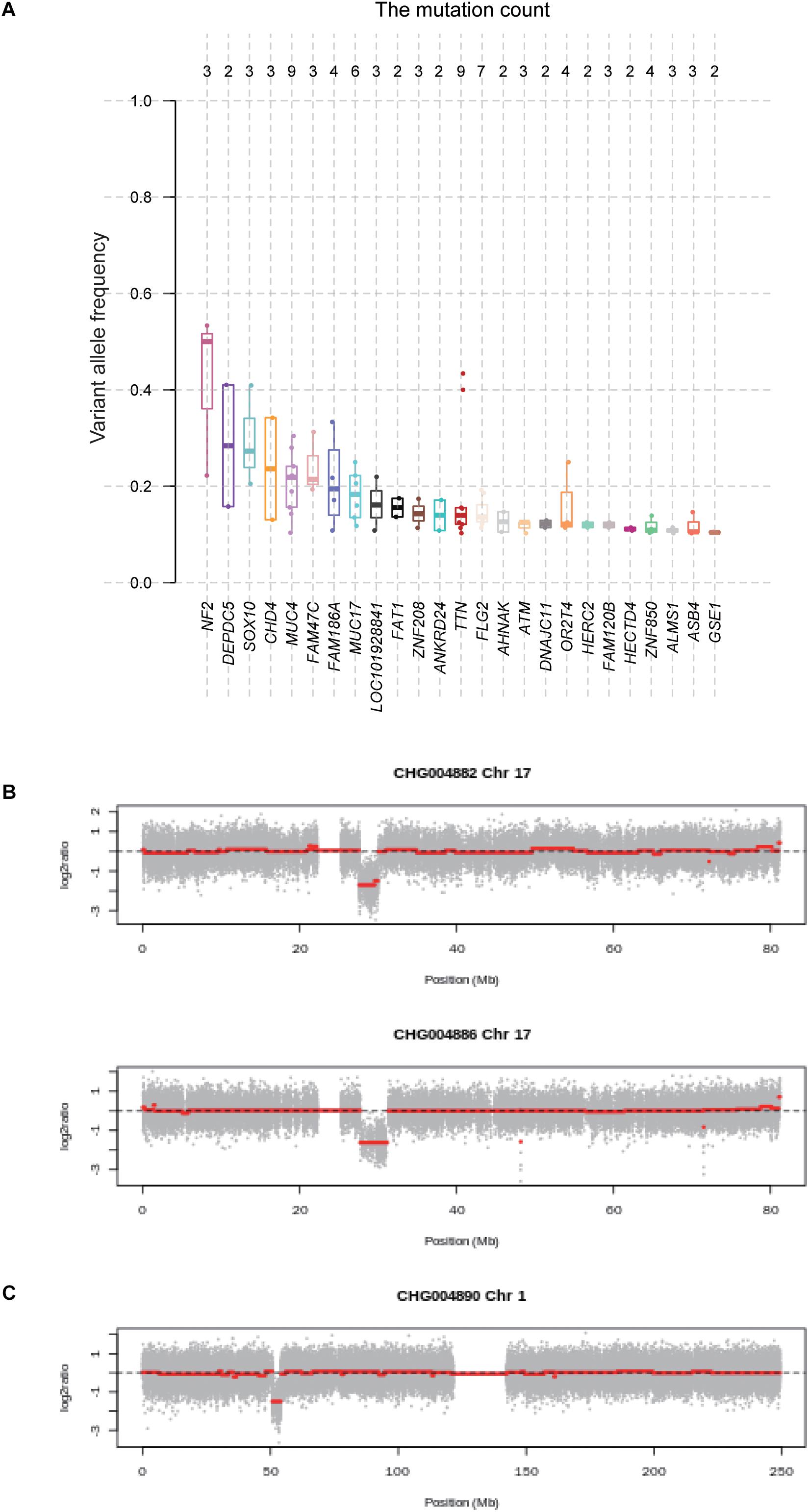
Figure 3. The variant allele frequency (VAF) of the genes mutated in spinal schwannomas. (A) The numbers on the top represent the number of mutations. The genes are ordered by the median of VAF across the mutations. (B,C) The log2 copy number ratio of chromosomes 17 and 1 in the spinal schwannoma samples.
Identification of Frequently Mutated Pathways in Schwannoma
To further investigate the critical pathways that controlled the initiation of schwannoma, we mapped the genes with somatic mutations to the oncogenic pathways. The Hippo signaling pathway was observed to be frequently mutated in schwannoma (Figure 4A). Combining the somatic mutations with CNAs, we found that NF2, SAV1, LLGL1/2, and CSNK1D/E were key regulators with mutations in Hippo signaling pathway (Figure 4B). Particularly, NF2, SAV1, and LLGL1/2, the upstream regulators of YAP/TAZ transcription factors, were frequently deleted or mutated with loss-of-function patterns, which may promote the transcription of YAP/TAZ target genes (Figure 4B). Among the 9 samples, 6 were detected to have mutations in the Hippo signaling pathway (Figure 4C), accounting for 67% of the samples. These results suggested that the Hippo signaling pathway was a critical pathway for schwannoma.
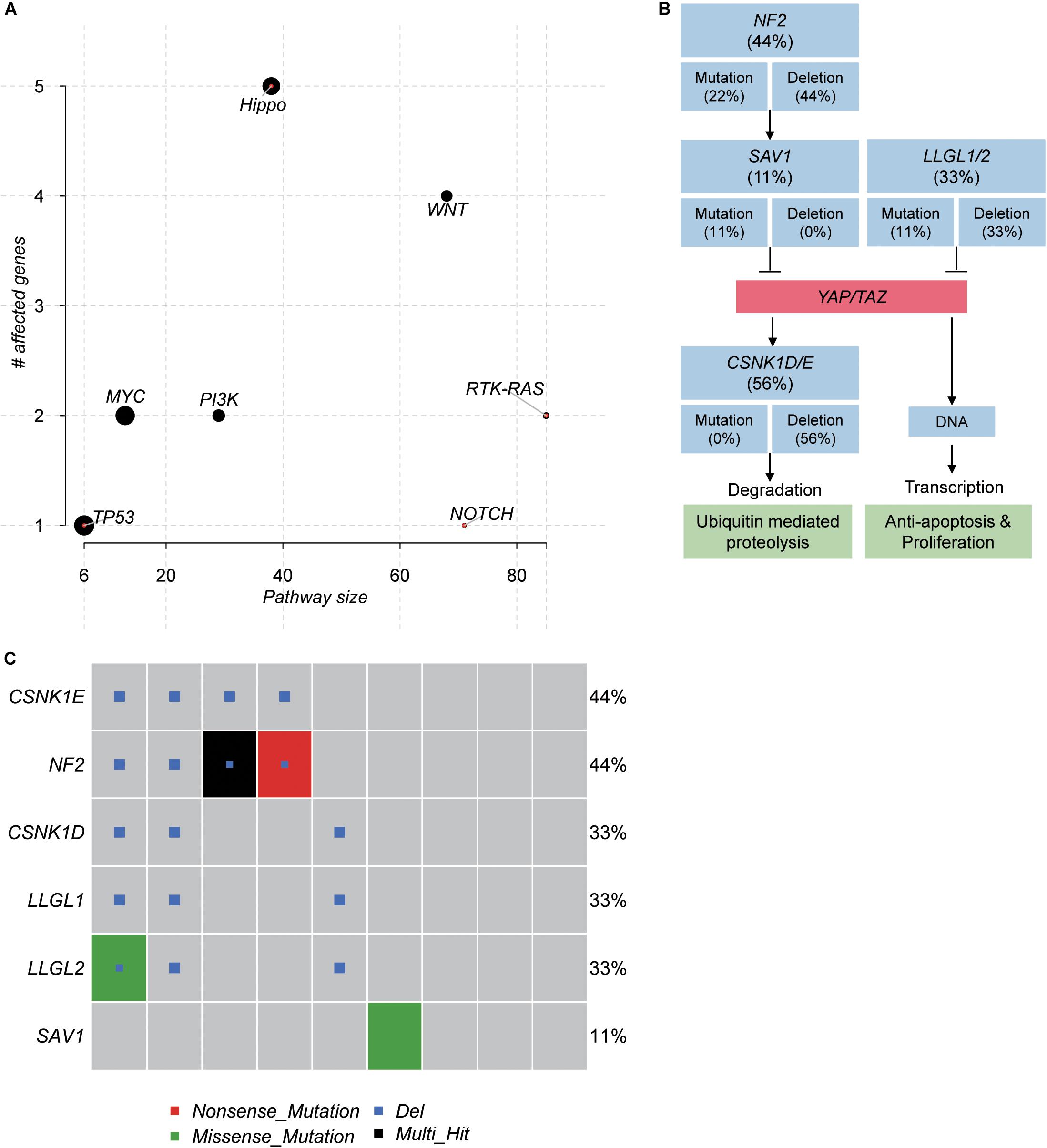
Figure 4. The pathways frequently mutated in spinal schwannoma. (A) The significance of the pathways frequently mutated in spinal schwannoma. The y-axis and x-axis represent the total number of genes and the number of mutated genes in the pathways. (B) The mutated genes and frequency in Hippo signaling pathway. (C) The number of mutated genes in Hippo signaling pathway across the nine spinal schwannomas. The x-axis represents the spinal schwannoma samples. The Multi_Hit indicates multiple variants of a gene were identified in the same sample.
Discussion
Spinal schwannoma is the most common primary spinal tumor, typically arising from spinal nerve roots. To investigate the genomic landscape of the spinal tumor, we performed whole genome sequencing of nine tumors and paired blood samples with high coverage (Supplementary Table S3). Specifically, we identified TTN, MUC4, FLG2, MUC17, OR2T4, ZNF850, FAM186A, ALMS1, FAM47C, and ATM as the top ten mutated genes. However, further analysis of those frequently mutated genes revealed that most of them were long genes and had lower VAF, indicating that most of these genes were passengers following the driver genes. Particularly, ATM, accounting for 33% of the samples, was the most frequently mutated cancer-related gene in schwannoma, followed by CHD4, FAT1, KMT2D, MED12, NF2, and SUFU (>20%). Notably, SUFU was reported to be responsible for meningioma (Aavikko et al., 2012), a type of tumor from central nervous system. In accordance with previous studies of sporadic schwannoma by next generation sequencing (Agnihotri et al., 2016; Havik et al., 2018), NF2 was identified as the pathogenic gene in both spinal and vestibular schwannomas. As the other genes like CHD4, FAT1, KMT2D, and MED12 were not detected in vestibular schwannoma, we thus speculated that some of them might be specific in spinal schwannoma.
Moreover, we also profiled the somatic CNA in schwannoma. NF2 gene, harbored in the cytoband 22q11, was also identified to be frequently deleted in the samples of spinal schwannoma. The integrative analysis of the NF2 point mutations, InDels and copy number deletions revealed that CHG004880 and CHG004884 had bi-allelic mutations in NF2, resulting in bi-allelic NF2 inactivation. Consistently, the occurrence of both somatic mutations and CNAs in NF2 has been reported to result in schwannoma by previous studies (Jacoby et al., 1994; Rm et al., 2018).
Furthermore, we also investigated the genes within these CNAs in spinal schwannoma. We found that there were only a few genes located within the amplified regions. In contrast, the deleted regions contained a majority of genes (Supplementary Tables S1, S2). The higher frequency in deletions than gains suggested that the tumorigenesis of spinal schwannoma might be caused by these deletions. Particularly, losses of XRCC2 and MLL3/KMT2C, which were located within 7q36.1, were also observed in the spinal schwannomas. XRCC2 and MLL3/KMT2C were involved in homologous recombination repair and histone modification, suggesting that loss of DNA damage repair and epigenetic alterations might also play key roles in schwannoma.
Besides, among the somatic mutations and CNAs in the nine spinal schwannomas, SMARCB1, SMARCE1 or LZTR1 were absent in these spinal schwannoma tissues. As the familial spinal schwannoma usually have germline mutations in SMARCB1, SMARCE1 or LZTR1, to our knowledge, these mutations are not prevalent in sporadic spinal schwannoma patients.
In addition, to identify genes potentially initiating the schwannoma, we ranked the genes with somatic mutations by the median of VAF across the samples (Figure 3A). Combined with the clonality analysis, NF2 gene was a candidate driver in spinal schwannoma. Besides NF2, SOX10 and CHD4 also showed relatively high VAF among those genes (VAF > 20%). Notably, a previous study reported that loss of SOX10 function contributed to the phenotype of human Merlin-null schwannoma cells, indicating that the mutation of SOX10 might be associated with the initiation of schwannoma (Doddrell et al., 2013). Moreover, the different location of CHD4 staining has been reported to be used as a potential biomarker to differentiate cellular schwannoma from malignant peripheral sheath tumor (MPNST) (Wu et al., 2018). In addition, NF1 and CDKN2C were also found to be homozygously deleted in spinal schwannoma. To our knowledge, NF1 mutations were rarely reported in spinal schwannoma. Exceptionally, lack of NF1 expression has been observed in a sporadic schwannoma from a patient without neurofibromatosis (Gutmann et al., 1995), suggesting that NF1 might be a novel driver in spinal schwannoma. Similarly, loss of CDKN2C, a cell growth regulator that controls cell cycle G1 progression, might also contribute to spinal schwannoma by the manner of CDKN2A (Koutsimpelas et al., 2011; Rohrich et al., 2016). Moreover, the pathway-level analysis revealed that Hippo signaling pathway was one of the frequently mutated pathways (6/9, 67%), in which, NF2, SAV1, and LLGL1/2 were frequently deleted or mutated with loss-of-function patterns. SAV1 and LLGL1/2 have not been reported to cause the tumorigenesis of spinal schwannoma. However, the Hippo signaling pathway has been widely reported to be implicated in schwannoma by previous studies (Nikuseva-Martic et al., 2007; Oh et al., 2015; Brodhun et al., 2017; Zhao et al., 2018). SAV1 promotes activation of MST-LATS kinase cascade in Hippo signaling, which suppresses the activities of YAP/TAZ by phosphorylation (Bae et al., 2017). LLGL1/2 have been recognized as direct negative regulators of YAP/TAZ via phosphorylation (Cordenonsi et al., 2011). The loss of these tumor suppressors might lead to lower phosphorylation of YAP/TAZ, thereby promoting YAP/TAZ to translocate to the cell nucleus.
Admittedly, the present study still had some limitations. First, the sample size is small, which limited the accuracy of the conclusions. Second, the pathogenicity of the mutated genes in schwannoma need to be further validated. Third, the consequences of these potentially pathogenic genes and the underlying mechanisms should be further investigated. However, we aimed to identify the potentially pathogenic genes responsible for schwannoma, and the systematic analysis of DNA sequencing data improved our understanding of the genomic landscape in spinal schwannoma.
Data Availability Statement
The raw data have been uploaded to the National Omics Data Encyclopedia (NODE) database (https://www.biosino.org/node/) with accession number OEP000894.
Author Contributions
XG, WZ, ZL, and JX conceived and designed the study. LZ conducted the analyses. XG and QJ drafted the manuscript. LT, WG, TW, and ZW assisted with the analysis and the interpretation of the results. All authors read and approved the manuscript before submission.
Funding
This work was supported by Key Project funding in the Basic Research Field of the Shanghai Municipal Science and Technology Commission (17JC1400903), National Natural Science Foundation of China (82002838), and Shanghai Youth Science and Technology Talent Sailing Program (20YF1449100).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.507816/full#supplementary-material
Supplementary Figure 1 | The coverage and B allele frequency of chromosome 22 in samples of CHG004878, CHG004880, CHG004884, and CHG004888. The whole chromosome loss in chr22 was observed in samples CHG004878, CHG004880, and CHG004884, but the CHG004888 was found to have focal deletion adjacent to NF2. The position of NF2 in chr22 was highlighted by blue boxes.
Supplementary Figure 2 | The read alignment of NF2 mutations in CHG004880 and CHG004884 by integrative genomic viewer (IGV).
Supplementary Figure 3 | The high resolution version of Figure 3B.
References
Aavikko, M., Li, S. P., Saarinen, S., Alhopuro, P., Kaasinen, E., Morgunova, E., et al. (2012). Loss of SUFU function in familial multiple meningioma. Am. J. Hum. Genet. 91, 520–526. doi: 10.1016/j.ajhg.2012.07.015
Abul-Kasim, K., Thurnher, M. M., McKeever, P., and Sundgren, P. C. (2008). Intradural spinal tumors: current classification and MRI features. Neuroradiology 50, 301–314. doi: 10.1007/s00234-007-0345-7
Agnihotri, S., Jalali, S., Wilson, M. R., Danesh, A., Li, M., Klironomos, G., et al. (2016). The genomic landscape of schwannoma. Nat. Genet. 48, 1339–1348.
Bae, S. J., Ni, L., Osinski, A., Tomchick, D. R., Brautigam, C. A., and Luo, X. (2017). SAV1 promotes Hippo kinase activation through antagonizing the PP2A phosphatase STRIPAK. Elife 6:e30278.
Brodhun, M., Stahn, V., and Harder, A. (2017). Pathogenesis and molecular pathology of vestibular schwannoma. HNO 65, 362–372.
Campian, J., and Gutmann, D. H. (2017). CNS Tumors in Neurofibromatosis. J. Clin. Oncol. 35, 2378–2385. doi: 10.1200/jco.2016.71.7199
Carlson, M. L., Smadbeck, J. B., Link, M. J., Klee, E. W., Vasmatzis, G., and Schimmenti, L. A. (2018). Next Generation Sequencing of Sporadic Vestibular Schwannoma: Necessity of Biallelic NF2 Inactivation and Implications of Accessory Non-NF2 Variants. Otol. Neurotol. 39, 860–871e.
Carreira, S., Romanel, A., Goodall, J., Grist, E., Ferraldeschi, R., Miranda, S., et al. (2014). Tumor clone dynamics in lethal prostate cancer. Sci. Transl. Med. 6:254ra125. doi: 10.1126/scitranslmed.3009448
Celli, P., Trillo, G., and Ferrante, L. (2005). Spinal extradural schwannoma. J. Neurosurg. Spine 2, 447–456. doi: 10.3171/spi.2005.2.4.0447
Chen, W. T., Ebelt, N. D., Stracker, T. H., Xhemalce, B., Van Den, Berg CL, and Miller, K. M. (2015). ATM regulation of IL-8 links oxidative stress to cancer cell migration and invasion. Elife 4:e07270.
Cordenonsi, M., Zanconato, F., Azzolin, L., Forcato, M., Rosato, A., Frasson, C., et al. (2011). The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 147, 759–772. doi: 10.1016/j.cell.2011.09.048
Doddrell, R. D., Dun, X. P., Shivane, A., Feltri, M. L., Wrabetz, L., Wegner, M., et al. (2013). Loss of SOX10 function contributes to the phenotype of human Merlin-null schwannoma cells. Brain 136(Pt 2), 549–563. doi: 10.1093/brain/aws353
Feng, X., Li, H., Dean, M., Wilson, H. E., Kornaga, E., Enwere, E. K., et al. (2015). Low ATM protein expression in malignant tumor as well as cancer-associated stroma are independent prognostic factors in a retrospective study of early-stage hormone-negative breast cancer. Breast Cancer Res. 17:65.
Forbes, S. A., Bhamra, G., Bamford, S., Dawson, E., Kok, C., Clements, J., et al. (2008). The Catalogue of Somatic Mutations in Cancer (COSMIC). Curr. Protoc. Hum. Genet. 2008:18428421
Gutmann, D. H., Silos-Santiago, I., Geist, R. T., Daras, M., and Rutkowski, J. L. (1995). Lack of NF1 expression in a sporadic schwannoma from a patient without neurofibromatosis. J. Neurooncol. 25, 103–111. doi: 10.1007/bf01057754
Havik, A. L., Bruland, O., Myrseth, E., Miletic, H., Aarhus, M., Knappskog, P. M., et al. (2018). Genetic landscape of sporadic vestibular schwannoma. J. Neurosurg. 128, 911–922. doi: 10.3171/2016.10.jns161384
Jacoby, L. B., MacCollin, M., Louis, D. N., Mohney, T., Rubio, M. P., Pulaski, K., et al. (1994). Exon scanning for mutation of the NF2 gene in schwannomas. Hum. Mol. Genet. 3, 413–419. doi: 10.1093/hmg/3.3.413
Jinnai, T., and Koyama, T. (2005). Clinical characteristics of spinal nerve sheath tumors: analysis of 149 cases. Neurosurgery 56, 510–515. doi: 10.1227/01.neu.0000153752.59565.bb
Kim, H., Saka, B., Knight, S., Borges, M., Childs, E., Klein, A., et al. (2014). Having pancreatic cancer with tumoral loss of ATM and normal TP53 protein expression is associated with a poorer prognosis. Clin. Cancer Res. 20, 1865–1872. doi: 10.1158/1078-0432.ccr-13-1239
Koutsimpelas, D., Felmeden, U., Mann, W. J., and Brieger, J. (2011). Analysis of cytogenetic aberrations in sporadic vestibular schwannoma by comparative genomic hybridization. J. Neurooncol. 103, 437–443. doi: 10.1007/s11060-010-0412-5
Lenzi, J., Anichini, G., Landi, A., Piciocchi, A., Passacantilli, E., Pedace, F., et al. (2017). Spinal Nerves Schwannomas: Experience on 367 Cases-Historic Overview on How Clinical, Radiological, and Surgical Practices Have Changed over a Course of 60 Years. Neurol. Res. Int. 2017:3568359.
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Lin, A. L., and Gutmann, D. H. (2013). Advances in the treatment of neurofibromatosis-associated tumours. Nat. Rev. Clin. Oncol. 10, 616–624. doi: 10.1038/nrclinonc.2013.144
Mayakonda, A., Lin, D. C., Assenov, Y., Plass, C., and Koeffler, H. P. (2018). Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome. Res. 28, 1747–1756. doi: 10.1101/gr.239244.118
Mermel, C. H., Schumacher, S. E., Hill, B., Meyerson, M. L., Beroukhim, R., and Getz, G. (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome. Biol. 12:R41.
Nikuseva-Martic, T., Beros, V., Pecina-Slaus, N., Pecina, H. I., and Bulic-Jakus, F. (2007). Genetic changes of CDH1, APC, and CTNNB1 found in human brain tumors. Pathol. Res. Pract. 203, 779–787. doi: 10.1016/j.prp.2007.07.009
Oh, J. E., Ohta, T., Satomi, K., Foll, M., Durand, G., McKay, J., et al. (2015). Alterations in the NF2/LATS1/LATS2/YAP Pathway in Schwannomas. J. Neuropathol. Exp. Neurol. 74, 952–959. doi: 10.1097/nen.0000000000000238
Paganini, I., Capone, G. L., Vitte, J., Sestini, R., Putignano, A. L., Giovannini, M., et al. (2018). Double somatic SMARCB1 and NF2 mutations in sporadic spinal schwannoma. J. Neurooncol. 137, 33–38. doi: 10.1007/s11060-017-2711-6
Pathmanaban, O. N., Sadler, K. V., Kamaly-Asl, I. D., King, A. T., Rutherford, S. A., Hammerbeck-Ward, C., et al. (2017). Association of Genetic Predisposition With Solitary Schwannoma or Meningioma in Children and Young Adults. JAMA Neurol. 74, 1123–1129. doi: 10.1001/jamaneurol.2017.1406
Piotrowski, A., Xie, J., Liu, Y. F., Poplawski, A. B., Gomes, A. R., Madanecki, P., et al. (2014). Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat. Genet. 46, 182–187. doi: 10.1038/ng.2855
Rm, D. E. C., Decsa, C., Pinto, G. R., Paschoal, E. H. A., Tuji, F. M., Donb, B., et al. (2018). Frequency of the Loss of Heterozygosity of the NF2 Gene in Sporadic Spinal Schwannomas. Anticancer Res. 38, 2149–2154.
Roberts, D. S., Maurya, R., Takemon, Y., Vitte, J., Gong, L., Zhao, J., et al. (2019). Linked-read Sequencing Analysis Reveals Tumor-specific Genome Variation Landscapes in Neurofibromatosis Type 2 (NF2) Patients. Otol. Neurotol. 40, 150–159e.
Rohrich, M., Koelsche, C., Schrimpf, D., Capper, D., Sahm, F., Kratz, A., et al. (2016). Methylation-based classification of benign and malignant peripheral nerve sheath tumors. Acta Neuropathol. 131, 877–887.
Rouleau, G. A., Merel, P., Lutchman, M., Sanson, M., Zucman, J., Marineau, C., et al. (1993). Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature 363, 515–521. doi: 10.1038/363515a0
Safaee, M. M., Lyon, R., Barbaro, N. M., Chou, D., Mummaneni, P. V., Weinstein, P. R., et al. (2017). Neurological outcomes and surgical complications in 221 spinal nerve sheath tumors. J. Neurosurg. Spine 26, 103–111. doi: 10.3171/2016.5.spine15974
Saunders, C. T., Wong, W. S., Swamy, S., Becq, J., Murray, L. J., and Cheetham, R. K. (2012). Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817. doi: 10.1093/bioinformatics/bts271
Seppala, M. T., Haltia, M. J., Sankila, R. J., Jaaskelainen, J. E., and Heiskanen, O. (1995). Long-term outcome after removal of spinal schwannoma: a clinicopathological study of 187 cases. J. Neurosurg. 83, 621–626. doi: 10.3171/jns.1995.83.4.0621
Smith, M. J., Wallace, A. J., Bowers, N. L., Rustad, C. F., Woods, C. G., Leschziner, G. D., et al. (2012). Frequency of SMARCB1 mutations in familial and sporadic schwannomatosis. Neurogenetics 13, 141–145. doi: 10.1007/s10048-012-0319-8
Trofatter, J. A., MacCollin, M. M., Rutter, J. L., Murrell, J. R., Duyao, M. P., Parry, D. M., et al. (1993). A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell 72, 791–800. doi: 10.1016/0092-8674(93)90406-g
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucl. Acids Res. 38:e164. doi: 10.1093/nar/gkq603
Wu, C. C., Pan, M. R., Wei, Y. C., Lin, C. H., Yang, S. F., Tsai, H. P., et al. (2018). CHD4 as a Potential Biomarker in Differentiating Between Cellular Schwannoma and Malignant Peripheral Nerve Sheath Tumor. Appl. Immunohistochem. Mol. Morphol. 26, 775–780.
Yu, G., Wang, L. G., Han, Y., and He, Q. Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. doi: 10.1089/omi.2011.0118
Zhang, Z., and Hao, K. (2015). SAAS-CNV: A Joint Segmentation Approach on Aggregated and Allele Specific Signals for the Identification of Somatic Copy Number Alterations with Next-Generation Sequencing Data. PLoS Comput. Biol. 11:e1004618. doi: 10.1371/journal.pcbi.1004618
Keywords: hippo signaling pathway, copy number alterations, frequently mutated genes, whole genome sequencing, spinal schwannoma
Citation: Gao X, Zhang L, Jia Q, Tang L, Guo W, Wang T, Wu Z, Zhou W, Li Z and Xiao J (2020) Whole Genome Sequencing Identifies Key Genes in Spinal Schwannoma. Front. Genet. 11:507816. doi: 10.3389/fgene.2020.507816
Received: 28 October 2019; Accepted: 10 September 2020;
Published: 30 October 2020.
Edited by:
Ira Ida Skvortsova, Medical University of Innsbruck, AustriaReviewed by:
Miguel Torres-Martin, Mount Sinai Medical Center, United StatesBárbara Meléndez, Hospital Virgen de la Salud, Spain
Netta Mäkinen, Dana–Farber Cancer Institute, United States
Nives Pecina-Slaus, University of Zagreb, Croatia
Copyright © 2020 Gao, Zhang, Jia, Tang, Guo, Wang, Wu, Zhou, Li and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wang Zhou, YnJpbGxpYW50MjEyQDE2My5jb20=; Zhenxi Li, emhlbnhpbGkuZWNudUBnbWFpbC5jb20=; Jianru Xiao, eGlhb2ppYW5ydXZ2aXBAMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship