
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 08 December 2020
Sec. Pharmacogenetics and Pharmacogenomics
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.491895
This article is part of the Research Topic Precision Psychiatry from a Pharmacogenetics Perspective View all 10 articles
 Beatriz Carvalho Henriques1
Beatriz Carvalho Henriques1 Esther H. Yang1,2Diego Lapetina1,2Michael S. Carr1Vasyl Yavorskyy1Joshua Hague1,2
Esther H. Yang1,2Diego Lapetina1,2Michael S. Carr1Vasyl Yavorskyy1Joshua Hague1,2 Katherine J. Aitchison1,2,3*
Katherine J. Aitchison1,2,3*Many genetic variants in drug metabolizing enzymes and transporters have been shown to be relevant for treating psychiatric disorders. Associations are strong enough to feature on drug labels and for prescribing guidelines based on such data. A range of commercial tests are available; however, there is variability in included genetic variants, methodology, and interpretation. We herein provide relevant background for understanding clinical associations with specific variants, other factors that are relevant to consider when interpreting such data (such as age, gender, drug–drug interactions), and summarize the data relevant to clinical utility of pharmacogenetic testing in psychiatry and the available prescribing guidelines. We also highlight areas for future research focus in this field.
Genome-wide association studies (GWAS) and related multi-omic strategies lend themselves well to phenotypes with polygenic modes of inheritance. By contrast, pharmacokinetic genes are associated with traits relevant to response to treatment (such as concentrations of medications and their metabolites) in an oligogenic manner with Mendelian patterns of inheritance and relatively large effect sizes. Many of the genes exhibiting a strength of association strong enough for consensus prescribing recommendations are in drug metabolizing enzymes (CPIC; DPWG; Hiemke et al., 2018; PharmGKB; FDA labels). We herein provide a review of the genetics of drug metabolizing enzymes and transporters relevant for medications prescribed in psychiatry. We searched databases such as PubMed, PharmVar, PharmGKB, CPIC, DPWG, and DrugBank as well as relevant reviews, book chapters, and dissertations with search terms including each drug and drug metabolizing enzyme or transporter; each paper thus retrieved was reviewed by a minimum of two coauthors.
Drug metabolism and transport includes Phase I (addition of a reactive group to the molecule), Phase II (transfer of a polar group to the Phase I metabolite), and Phase III (transport of compounds away from the interior of the cells in an energy-dependent manner, introduced by Ishikawa (1992) (Xu et al., 2005). While the liver and gut are most relevant to phase I metabolism, the above activities occur throughout the body, with many drug metabolizing enzymes being widely expressed (Aitchison et al., 2010). Their activity is subject to mechanisms including competitive and non-competitive inhibition, and induction (Pelkonen et al., 2008; Hisaka et al., 2010; Chen F. et al., 2018; Chen J.T. et al., 2018).
Phase I enzymes catalyze reactions that alter the hydrophobicity, molecular weight, and reactivity of the substrate, occurring through hydrolysis, reduction, and oxidation reactions. Phase I enzymes relevant to psychotropics include the cytochrome P450 (CYP) family of enzymes, flavin-containing monooxygenases, esterases, epoxide hydrolases (EH), and microsomal epoxide hydrolases (mEH).
The CYP superfamily and flavin-containing monooxygenases (FMOs) are oxidoreductases. The most studied of these are the CYPs. Individuals sensitive to the antihypertensive agent debrisoquine and to the anti-arrhythmic agent sparteine gave rise to initial observations regarding variable enzyme activity (Mahgoub et al., 1977; Eichelbaum, 1984; Smith, 1986; reviewed in Johansson and Ingelman-Sundberg, 2011). This led to sequencing efforts that identified the first CYP2D6 loss-of-function mutation (Gough et al., 1990; Hanioka et al., 1990; Kagimoto et al., 1990). Multiple mutations in P450s relevant to psychotropics have since been discovered, with the frequency thereof differing by ethnicity (Aitchison et al., 2000c).
Cytochrome P450 enzyme classification (previously led by the Cytochrome P450 Nomenclature Committee and now transferred to the Pharmacogene Variation Consortium) is as follows: after the letters “CYP” comes a number indicating the CYP family (Cupp and Tracy, 1998). Enzymes within the same family (e.g., CYP1) have a minimum of 36% amino acid sequence homology. The next layer of differentiation is represented by a letter indicating the sub family. Within a subfamily (e.g., CYP1A), there is approximately 70% amino acid homology (Nebert et al., 1987). The final layer is another number representing the isoform (e.g., CYP1A1 and CYP1A2). For all, the enzymes are not italicized, while the corresponding gene names are.
Mutations in the CYP genes can be classified in two different manners: pharmacologically in terms of enzyme function, or genetically in terms such as loss or gain of function (van der Weide and Steijns, 1999). Many CYPs have four distinct levels of enzyme activity: poor, intermediate, normal (previously known as extensive), and ultrarapid (Blake et al., 2013; Caudle et al., 2020). Reduced enzyme–substrate affinity, enzyme stability, or splice site variants leading to lack of functional protein can be a result of mutations (for reviews, see, for example, Ingelman-Sundberg, 2004a, b). Gene duplication (or multiple copies, i.e., multiplication) or single nucleotide polymorphisms (SNPs) affecting transcription, on the other hand, may be associated with increased enzyme activity (ultrarapid metabolizers; UM) (Johansson et al., 1993; Sim et al., 2006; Wang et al., 2014, 2015). Gain-of-function variants may increase medication clearance, consequently reducing the concentration, while loss-of-function mutations reduce clearance, increasing the concentration (Zanger and Schwab, 2013); the opposite is true for prodrugs such as codeine, where loss-of-function mutations lead to lack of production of the pharmacologically active analgesic.
The CYP1 family includes CYP1A and CYP1B. CYP1A1 and CYP1A2 lie in a head–head configuration on chromosome 15, and share a promoter region to which the aryl hydrocarbon receptor binds (at xenobiotic responsive elements) (Jorge-Nebert et al., 2010).
Substrates specific to psychiatric conditions include antipsychotics [e.g., chlorpromazine (CPZ), trifluoperazine, clozapine, olanzapine], tertiary amine tricyclics (e.g., amitriptyline, imipramine, and clomipramine) as well as some selective serotonin reuptake inhibitors (such as fluvoxamine), and zopiclone. Additional substrates include analgesics (paracetamol), anti-inflammatories, cardiovascular agents (e.g., lignocaine), xanthines (caffeine, theophylline, aminophylline), and tacrine (Imaoka and Funae, 1990; Aitchison et al., 2000a, c; Lobo et al., 2008; Turpeinen et al., 2009; Theophylline Pathway; Zanger and Schwab, 2013). CYP1A2 is also involved in toxicity (e.g., bioactivation of arylamines and heterocyclic amines implicated in the formation of colon and bladder cancer, and the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, also known as MPTP) (McManus et al., 1990; Boobis et al., 1994; Eaton et al., 1995; Coleman et al., 1996; Hammons et al., 1997).
The enzyme is inducible by paracetamol, omeprazole, primaquine, carbamazepine, polycyclic aromatic hydrocarbons (e.g., 3-methylcholanthrene), heterocyclic aromatic hydrocarbons (such as 2,3,7,8-tetrachlorodibenzo-p-dioxin), and products of combustion such as cigarette or cannabis smoke (Rost et al., 1994; Parker et al., 1998; reviewed in Aitchison et al., 2000c). Interestingly, a group of phenothiazines represented by perazine and promazine has been shown to induce this enzyme as well, accelerating their own metabolism and that of concomitant medications metabolized by this route (Wójcikowski et al., 2012). It can also be induced by various dietary substances including cruciferous vegetables (Cruciferae: including broccoli, brussels sprouts, cabbage, cauliflower, radishes, and watercress), heterocyclic amines (produced in meat browned at high temperatures), and caffeine (Rost et al., 1994; Parker et al., 1998; Aitchison et al., 2000a, c; Ghotbi et al., 2007; Yoshinari et al., 2008; Dobrinas et al., 2011; Arici and Özhan, 2017). Amine metabolism may be affected by cruciferous vegetables for a significant duration (Murray et al., 2001), which may also affect other enzymes. The inducibility by smoking is of particular relevance to psychiatry, as many patients are smokers (discussed in Fiore et al., 1995; Nakajima et al., 1999; Aitchison et al., 2000c; Ghotbi et al., 2007; Dobrinas et al., 2011). Indeed, a recent study conducted by Lesche et al. (2020) on the impact of genotype of various CYP enzymes (including CYP1A2) and the presence of known inducers and inhibitors demonstrated that, for patients prescribed clozapine, a greater percentage of the variation in plasma concentration of this medication was explained by smoking status than by CYP1A2 genotyping information (in a cohort where 82% of individuals tested positive for the CYP1A2∗1F variant).
Several polymorphisms have been detected in CYP1A2 (Jiang et al., 2005; Browning et al., 2010). Individuals with the CYP1A2∗1F c.−163C>A SNP that confers higher inducibility (Sachse et al., 1998; Chida et al., 1999; Han et al., 2002; Sim, 2013) have higher levels of caffeine metabolism. An initial report in Whites showed a reduction in olanzapine serum concentration in association with this variant (Laika et al., 2010). A subsequent study in Norwegian Whites was not able to replicate this association with olanzapine serum concentration, but in CSF, the ratio of 4’-N-desmethylolanzapine to olanzapine was associated with smoking and CYP1A2 genotype, with the highest ratios being in smokers homozygous for the CYP1A2∗1F (Skogh et al., 2011). A later paper by the same group was also not able to replicate an association between the CYP1A2∗1F and systemic exposure to olanzapine, but did find a relatively modest effect of other variants (rs2472297C>T lying in the intergenic region between CYP1A1 and CYP1A2 and rs4410790C>T upstream of the aryl hydrocarbon receptor locus; Söderberg et al., 2013). Attempts at replication of an association between the CYP1A2∗1F and olanzapine exposure in Asians have also been negative (Shimoda et al., 2002; Obase et al., 2003; Chen et al., 2005; Ghotbi et al., 2007; Nozawa et al., 2008).
There are also loss-of-function variants. The CYP1A2∗6 haplotype containing the c.1291 C>T (previously known as the c.5090 C>T) mutation causes an Arg431Trp amino acid substitution resulting in a complete loss of enzyme function (Chevalier et al., 2001; Zhou et al., 2004). By analogy, owing to the behavioral effects seen on administering clozapine to a CYP1A2 knockout mouse, it is possible that people with this variant could experience more side effects of medications metabolized by CYP1A2, including clozapine and olanzapine (Aitchison et al., 2000b). The CYP1A2∗1C haplotype has a promoter mutation (−3860 G>A) which has been associated with a reduction in caffeine metabolism in Japanese (Nakajima et al., 1999). Other known variants of CYP1A2 with decreased activity include CYP1A2∗1K (characterized by polymorphisms −729C>T, −739T>G, and −163C>A), identified in an Ethiopian population (Aklillu et al., 2003). Likewise, variants CYP1A2∗3 (2116 G>A and 5347 T>C) and CYP1A2∗4 (2499 A>T) are associated with reduced activity and have been identified (Chevalier et al., 2001; Zhou et al., 2004).1 The CYP1A2∗7 has a splice site mutation in the donor site of intron 6 (3533G>A) and was found in heterozygous state in one patient with very high clozapine concentration and plasma caffeine clearance at the lower limit of the normal range, consistent with the mutation leading to no functional CYP1A2 enzyme (Allorge et al., 2003).
Many agents also contribute to the inhibition of CYP1A2, such as: apiaceous vegetables (parsnips, celery, dill, parsley) (Lampe et al., 2000), fluvoxamine (Brosen et al., 1993), grapefruit juice (Fuhr et al., 1993), estrogens (Knutti et al., 1981; Rietveld et al., 1984; Abernethy and Todd, 1985; Vistisen et al., 1992; Le Marchand et al., 1997), quinolone antibiotics (Fuhr et al., 1992), and in smokers, heavy ethanol consumption (Rizzo et al., 1997).
CYP2 genes comprise clusters on different chromosomes (Sezutsu et al., 2013; Zanger and Schwab, 2013).
The CYP2A subfamily includes CYP2A6 and CYP2A13 (Hoffman et al., 2001; Zanger and Schwab, 2013), with CYP2A6 being of relevance to psychiatry. CYP2A6 is mainly expressed in the liver, where it accounts for approximately 4% of total CYP content (Shimada et al., 1994; Haberl et al., 2005). CYP2A13 is expressed at reduced levels in the respiratory tract (Leclerc et al., 2011; Raunio and Rahnasto-Rilla, 2012). CYP2A6 was first recognized as the enzyme responsible for coumarin 7-hydroxylation, and is also the primary nicotine C-oxidase (Pelkonen et al., 2000; Fuhr et al., 2007; Mwenifumbo et al., 2007; Raunio and Rahnasto-Rilla, 2012). In addition to nicotine, CYP2A6 contributes to the metabolism of promazine, valproic acid, disulfiram, and caffeine as well as to other medications and toxins (Crespi et al., 1990; Yamazaki et al., 1992; Gonzalez and Gelboin, 1994; Oscarson et al., 1998; Komatsu et al., 2000; Murai et al., 2009; Tanner and Tyndale, 2017).
Like many of the CYP superfamily, CYP2A6 is a highly polymorphic gene, with many known mutations affecting enzymatic activity (Di et al., 2009; McDonagh et al., 2012). Polymorphisms arise from the occurrence of gene conversion events, deletions, duplications, multiple nucleotide insertions/deletions, and SNPs. The frequency of these events varies by ethnicity, with Asians having the highest frequency of loss-of-function mutations (∼50%), and Whites the least (∼9%) (Nakajima et al., 2006; di Iulio et al., 2009). CYP2A6 expression and activity are also impacted by induction and inhibition effects, age, and interactions with other hepatic enzymes, co-enzymes, and co-factors (Tanner and Tyndale, 2017).
Loss-of-function is often a result of the common CYP2A6∗2 and CYP2A6∗4 alleles. With a frequency of 1–5% in Whites, the CYP2A6∗2 rs1801272 SNP encodes an inactive enzyme due to a Leu160His substitution (Tanner et al., 2017). The CYP2A6∗4 haplotype (and its subtypes such as CYP2A6∗4A and CYP2A6∗4H) denotes a complete gene deletion, where the subtypes represent different genomic mechanisms for the deletion. This deletion is found at higher frequencies in Asians and Blacks [e.g., CYP2A6∗4 has a haplotype frequency of up to 15% in a specific Asian group (Pang et al., 2015)]. Other variants such as CYP2A6∗9 result in a reduced enzyme functionality. Both complete loss-of-function and reduced function variants may result in a reduction of treatment efficacy, with atypical metabolite formation (e.g., switching from coumarin 7-hydroxylation to 3-hydroxylation) (Hadidi et al., 1997; Komatsu et al., 2000; Fujita and Sasaki, 2007). Associations between CYP2A6 variants and smoking cessation have been reported (reviewed by Tanner and Tyndale, 2017).
The CYP2B subfamily members are CYP2B6 and a CYP2B7P (a pseudogene). CYP2B6 is strongly induced by phenobarbital (Faucette et al., 2004). It accounts for ∼1% of total hepatic CYP content (Ward et al., 2003), with variance in inter-individual expression of up to 300-fold (Lang et al., 2001; Lamba et al., 2003; Desta et al., 2007; Hofmann et al., 2008; Wang and Tompkins, 2008; Ohtsuki et al., 2012).
There are 38 different CYP2B6 haplotypes currently described, some of which are associated with defined changes in enzyme function (Lang et al., 2001, 2004; Lamba et al., 2003; Klein et al., 2005; “Pharmacogene Variation Consortium: CYP2B6”; Zukunft et al., 2005). Two of the haplotypes are structural variants representing hybrids whose sequence is partly derived from CYP2B6 and partly from CYP2B7P, and gene duplications have also been identified (Martis et al., 2013). With a frequency of 15–50% across different ethnicities (of which Blacks have the highest), the most common allele is CYP2B6∗6. The mutations c.516G>A and c.785A>G lead to amino acid substitutions Gln172His and Lys262Arg respectively, and are associated with a reduction in enzyme activity (Lang et al., 2001; Tsuchiya et al., 2004; Hofmann et al., 2008; Turpeinen and Zanger, 2012). Those who contain homozygous copies of CYP2B6∗6 show increased plasma concentrations of relevant drugs, which has been linked to increased risk of Adverse Drug Reactions (ADRs) (Haas et al., 2004; Ribaudo et al., 2006; Zanger et al., 2007; King and Aberg, 2008; Lubomirov et al., 2011; Yimer et al., 2012; Zanger and Klein, 2013). The CYP2B6∗4 has a haplotype frequency averaging at 9%, being up to 45% in Africans, 27% in Hispanics, 21% in Europeans, and 19% in Asians (Reference SNP:rs2279343). The enzyme encoded by this variant clears bupropion [relevant for smoking cessation and also used in the treatment of depression and attention deficit hyperactivity disorder (ADHD)] more rapidly than the wild-type (Lang et al., 2001; Kirchheiner et al., 2003; Hesse et al., 2004; Rotger et al., 2007). The CYP2B6∗18 contains the c.983T>C substitution, which leads to Ile328Thr substitution. This haplotype is found in some African populations (e.g., the Bantu) (Jamshidi et al., 2010).
Forming a ∼390 kb cluster at chromosome 10q24, the CYP2C subfamily contains four genes: CYP2C8, CYP2C9, CYP2C18, and CYP2C19 (Goldstein and de Morais, 1994; Nelson et al., 2004). All exhibit extensive homology in both DNA and amino acid sequence, and are thus responsible for the metabolism of partially overlapping subsets of drugs (Coller et al., 2002; Koukouritaki et al., 2004; Rettie and Jones, 2005; Lai et al., 2009; Naraharisetti et al., 2010; Ohtsuki et al., 2012). Their main expression is in the liver, comprising 20% of hepatic CYP content (Shimada et al., 1994). Within the CYP2C subfamily, CYP2C9 is the most abundantly expressed, followed by CYP2C8 and CYP2C19. Psychotropic substrates for CYP2C19 include diazepam (Jung et al., 1997), phenytoin (Bajpai et al., 1996; Mamiya et al., 1998), propranolol (Otton et al., 1990), selective serotonin reuptake inhibitors (SSRIs; Hicks et al., 2015), and tricyclics (Hicks et al., 2017). CYP2C18 is distal to CYP2C19 on chromosome 10 but appears to be expressed only at the mRNA level and not at the protein level (Chen and Goldstein, 2009).
CYP2C8 is the second most important cytochrome after CYP3A4 for the conversion of buprenorphine to its active metabolite, norbuprenorphine (Picard et al., 2005), and its expression is under genetic control. Work in Asian populations has identified variants that are associated with no functional enzyme, specifically the CYP2C8∗5.001, the CYP2C8∗7.001, and the CYP2C8∗11.001 at 0.006, 0.0025, and 0.003 (in Koreans; 0.01 in Vietnamese and 0.0014 in Chinese) frequency, respectively in E. Asian populations tested (Soyama et al., 2002; Hichiya et al., 2005; Yeo et al., 2011).
CYP2C9 metabolizes phenytoin. It is also relevant to drugs prescribed to treat physical comorbidities in those with chronic mental health conditions. These include anti-diabetic agents (such as tolbutamide, glimepiride, and nateglinide), angiotensin II blockers (losartan, valsartan, candesartan, and irbesartan), fluvastatin, warfarin, and nonsteroidal anti-inflammatory drugs including COX2 inhibitors (e.g., celecoxib) (Michaels and Wang, 2014). Of the reduced function variants, CYP2C9∗2 and CYP2C9∗3 are the most common, and have been studied in relation to the metabolism of drugs with a narrow therapeutic index, such as phenytoin, tolbutamide, and warfarin (Lee et al., 2002). The functional mutations in CYP2C9∗2 and CYP2C9∗3 are rs1799853 and rs1057910, leading to Arg144Cys and Ile259Leu substitutions. CYP2C9∗2 is associated with a 10-fold lower Vmax and 2-fold lower Vm for (S)-warfarin hydroxylation. Median daily warfarin dose was in one study 4.0, 2.9, 2.6, and 1 mg for individuals of CYP2C9∗1/∗1, CYP2C9∗1/∗2, CYP2C9∗1/∗3, and CYP2C9 homozygous mutant genotype, respectively (King et al., 2004). Individuals who are affected by two reduced function alleles have a greater chance of ADRs such as gastrointestinal bleeding from NSAIDs (Martinez et al., 2004), hypoglycemia (Holstein et al., 2005), and bleeding from warfarin (Ogg et al., 1999). In a GWAS of response to warfarin, a CYP2C9 marker was separately genotyped in addition to the array-based genomic analysis and was identified as the top signal (Takeuchi et al., 2009). Predictive modeling followed, and included a target of the drug (VKORC1), as well as CYP2C9 (Eriksson and Wadelius, 2012; Maagdenberg et al., 2018); the FDA label summarizes findings of a meta-analysis in which patients carrying at least one copy of the CYP2C9∗2 or CYP2C9∗3 alleles required a mean daily warfarin dose 17 or 37%, respectively less than wild-type individuals (Sanderson et al., 2005).
Like many CYPs (Kalow and Tyndale, 1992), CYP2C19 is also expressed extrahepatically in multiple tissues including in the brain (Aitchison et al., 2010). Substrates of this enzyme include: diazepam and its metabolite desmethyldiazepam, moclobemide (Roh et al., 1996), SSRIs (fluoxetine, sertraline, paroxetine, citalopram, escitalopram), tertiary amine tricyclics (e.g., amitriptyline, imipramine, and clomipramine), as well as clozapine, olanzapine, phenytoin, and propranolol to lesser extents. The SSRIs fluoxetine and fluvoxamine also inhibit CYP2C19 (Preskorn, 1997). Conversely, phenothiazines represented by perazine and promazine have been shown to induce this CYP enzyme (Wójcikowski et al., 2012). Other substrates include the anticoagulant clopidogrel, cyclophosphamide, nelfinavir, proguanil, proton pump inhibitors [omeprazole (Karam et al., 1996) and pantoprazole], thalidomide, and voriconazole (Desta et al., 2002). Of the 35 allelic variants described in the CYP Database, CYP2C19∗2-∗8 are the most common loss of function (poor metabolizer or PM) haplotypes.
There is substantial interethnic variation in the incidence of PMs of CYP2C19, being 2–5% in Whites, 2% in Saudi Arabians, 4% in Black Zimbabweans, 5% in Ethiopians, 13% in Koreans, 15–17% in Chinese, 21% in Indians, and 18–23% in Japanese (Evans et al., 1995; Aitchison et al., 2000c). When the square root of the PM phenotypic frequency (equal to the frequency of PM CYP2C19 alleles) is plotted versus longitude, an increase in this value versus longitude may be seen, with an increment in the value occurring between Saudi Arabia and Bombay (Evans et al., 1995; Saeed and Mayet, 2013). The increasing frequency of PMs is mainly owing to the higher frequencies of the null haplotypes CYP2C19∗2 and CYP2C19∗3. The most common gain-of-function haplotype is the c.−806C>T (rs12248560) defining the CYP2C19∗17 haplotype. Of note, however, this may be found in combination with loss-of-function variants such as the c.1A>G (rs28399504) associated with the CYP2C19∗4 haplotype, or another loss of function variant (c.463G>T) (Scott et al., 2012, 2013; Skierka and Black, 2014). It is therefore necessary to accurately characterize haplotypes with the c.−806C>T. Tables available via PharmGKB2 provides further details on CYP2C19 haplotype frequencies by ethnic group.
The most common PM haplotype is CYP2C19∗2, which accounts for about 86% of all the PMs in the White population and 69–87% in the E. Asian population. The substitution of G681A in exon 5 of the CYP2C19∗2 haplotype creates an aberrant splice site (de Morais et al., 1994). The second most common PM haplotype is CYP2C19∗3, which represents about 13–31% of E. Asian PMs and 1.5% of White PMs. The substitution of G636A mutation in exon 4 of the CYP2C19∗3 creates a premature stop codon. A third variant, CYP2C19∗4 accounts for approximately 3% of White PM alleles and contains an A → G mutation in the initiation codon (i.e., c. 1A>G). CYP2C19∗5 accounts for 1.5% of White PM alleles and is rare in E. Asians. The CYP2C19∗5 haplotype is a result of a c.C1297T mutation in exon 9, in which causes an Arg433Trp change in the heme-binding region. CYP2C19∗6 (a c. G395A base substitution resulting in an Arg132Gln coding change in exon 3) and CYP2C19∗7 (a GT → GA mutation in the donor splice site of intron 5 at c.819 +2) each account for a further 1.5% of White PM alleles. CYP2C19∗8, a T358C substitution in exon 3 that result in a Trp120Arg change, is a less common PM allele. The products of CYP2C19∗6 and CYP2C19∗8 show reduced catalytic activity (2% and 9% of wild-type S-mephenytoin hydroxylase activity, respectively); the others described above are associated with failure to express active CYP2C19. CYP2C19∗2A and CYP2C19∗3 have both been identified in an Ethiopian population and found to account for all the PM alleles in the 114 individuals studied (Persson et al., 1996).
In CYP2C19 PMs, diazepam clearance is significantly lower than in NMs (Bertilsson, 1995). The mean clearance is lower in Chinese compared to Whites. Owing to the relatively high frequency of PMs in E. Asians, there is a greater frequency of individuals carrying one PM haplotype (i.e., heterozygous PMs). Consistent with this, “many Hong Kong physicians routinely prescribe smaller diazepam doses for Chinese than for white Whites” (Kumana et al., 1987). The main variant responsible for this effect is the G681A, which has a gene-dosage association effect on diazepam clearance (Qin et al., 1999). For recent data on antidepressants and CYP2C19, see the relevant section. Dose adjustment by CYP2C19 genotype has been published for amitriptyline, citalopram, clomipramine, imipramine, moclobemide, and trimipramine (Kirchheiner et al., 2001). CYP2C19 PMs show a significantly higher efficacy for triple therapy for Helicobacter pylori (proton pump inhibitor, clarithromycin, and amoxicillin) (Klotz, 2006, 2009).
The CYP2D subfamily consists of a gene cluster comprising CYP2D6, with two pseudogenes, CYP2D7 and CYP2D8 (Yasukochi and Satta, 2011). CYP2D6 accounts for 1.5% of microsomal CYP content in the liver (Michaels and Wang, 2014), and is involved in metabolizing the majority of psychotropic drugs (Bertilsson et al., 2002). It is also expressed in other organs, including the brain (Niznik et al., 1990; Kalow and Tyndale, 1992; Siegle et al., 2001; Aitchison et al., 2010), and has been associated with synthesis of neurotransmitters (Yu et al., 2003; Niwa et al., 2017). The CYP2D6 gene is highly polymorphic, even compared to some of the other CYPs (Pharmacogene Variation Consortium: CYP2D6). It is the most extensively studied genetically variable drug metabolizing enzyme (Bertilsson et al., 2002; Ingelman-Sundberg, 2004b), and has over 110 unique alleles identified (Kertesz et al., 2007).
These studies have revealed that there is significant variation of allelic variants between ethnic groups (Aitchison et al., 2000c)3. For example, the CYP2D6∗4 haplotype (previously known as g.1846G>A, genomic location of NG_008376.3 (Reference SNP (refSNP) Cluster Report: rs3892097) 1847G>A, Gough et al., 1990; Hanioka et al., 1990; Kagimoto et al., 1990) has a frequency of 19% in Whites (approximately 70–90% of all the PM alleles) (Aitchison et al., 1999), and 6% in Africans and 1% in South Asians (Aitchison et al., 2000c; Mammen et al., 2018). The second most frequent PM haplotype in Whites (2–2.5%) is CYP2D6∗5 (Aitchison et al., 1999), which represents a complete gene deletion, and occurs at a frequency of 5.3, 2.9, and 2.9% in Africans, Asians, and Hispanics, respectively (Bradford, 2002; Del Tredici et al., 2018). The CYP2D6∗10 haplotype has key C188T and G4268C base substitutions in exons 1 and 9, respectively, that result in Pro34Ser and Ser486Thr amino acid substitutions (Yokota et al., 1993; Sakuyama et al., 2008). This haplotype is associated with reduced enzymatic activity (Johansson et al., 1994). With an allelic frequency of 0.43, it is very high in East Asians (Aitchison et al., 2000c; Mammen et al., 2018), and similar to other reduced activity metabolizers has been associated with ADRs such as tardive dyskinesia (Ohmori et al., 1998; Puangpetch et al., 2016). However, some of these apparent CYP2D6∗10 alleles may in fact be CYP2D6∗36 hybrid alleles. The CYP2D6∗17 haplotype exhibits a similar reduction in enzymatic activity (Masimirembwa et al., 1996; Oscarson et al., 1997), and is found predominantly in Africans, with frequencies of 34% in Zimbabwe, 28% in Ghana, 17% in Tanzania, and 9% in Ethiopia (Bertilsson et al., 2002). CYP2D6∗41 is the most common reduced activity (IM) haplotype in Whites, a key SNP 2989G>A (genomic position on NG_008376.3 7189G>A) occupying an intronic position leading to a splicing defect (Raimundo et al., 2000, 2004; Rau et al., 2006; Toscano et al., 2006; Hicks et al., 2013, 2016; Wang et al., 2014). Tables available via PharmGKB4 provides details of CYP2D6 haplotype frequencies by ethnic group.
At the opposite end of the activity spectrum are the UM allelic variants, which most commonly have extra functional copies of the CYP2D6 gene in tandem on the chromosome, seen at a frequency of 0.9–4% in Whites (Johansson et al., 1993; Aitchison et al., 1999). An apparently less common mechanism for UM alleles is upregulation of gene expression owing to SNP-related enhancer activity (Wang et al., 2015). Individuals possessing UM alleles were first identified as having lower than expected blood concentration of tricyclic antidepressants such as clomipramine (Bertilsson et al., 1993a, b; Dalen et al., 1998; Roberts et al., 2004). CYP2D6 gene duplication or multiplication events occur at rates up to 29% in Ethiopians (Aklillu et al., 1996) by old techniques such as restriction fragment length polymorphism, and remain to be accurately characterized in terms of frequency using more current approaches.
The diversity in CYP2D6 phenotype has clinical implications (Ingelman-Sundberg, 2005). Individuals with two PM haplotypes have no functional enzyme, are classified as PMs, and are more prone to ADRs for drugs with a narrow therapeutic window (Steimer et al., 2004, 2005). At the other end of the spectrum, UMs may also show more ADRs, such as tardive dyskinesia (Koola et al., 2014) or symptoms of morphine overdose on codeine (Crews et al., 2014), owing to enhanced formation of toxic metabolites (Pinto and Dolan, 2011). Variation in CYP2D6 is highly relevant to psychiatry: for most antidepressants and antipsychotics, there are clinical guidelines that state that pharmacogenomic information for CYP2D6 could or should be used in prescribing (Bousman et al., 2019a). A review with modeling found that for antidepressants metabolized by CYP2D6, normal metabolizers (NMs) would require at least double the dose required by PMs, while cost analyses have associated PM status with not only higher ADRs but also with more drop outs from treatment (Chou et al., 2003; Kirchheiner et al., 2004; Zhou et al., 2004; Tanner et al., 2020).
Haplotype functionality may be used to derive an activity score (Caudle et al., 2020), with resources provided by PharmGKB to assist with this process.3 In the most recent update, the activity score of the CYP2D6∗10 haplotype was adjusted from 0.5 to 0.25, and the phenotype assignment for an activity score of 1 adjusted from NM to IM.
A relatively small number of allelic variants have been identified for CYP2E1, such as CYP2E1∗2, which is associated with reduced enzyme activity (Hu et al., 1997; Mittal et al., 2015). This enzyme is produced primarily in the liver, although it is also found in the brain (García-Suástegui et al., 2017), and is responsible for metabolizing ethanol (into acetaldehyde), paracetamol/acetaminophen, and other substances into reactive intermediates, whose toxicity is enhanced in alcoholics (Cederbaum, 2012). Indeed, CYP2E1 is responsible for 20% of total ethanol metabolism (to which other enzymes such as catalases also contribute) (Heit et al., 2013). Gene transcription is induced by ethanol consumption (a moderate level of intake at 140 g ethanol per week producing an increase in expression of CYP2E1 in the intestine, but not in the liver; Liangpunsakul et al., 2005). Interestingly, a 96-bp insertion polymorphism in the CYP2E1 gene, which is associated with higher activity of the encoded enzyme, has been proposed as a possible protective factor against alcoholism (Cartmell et al., 2005). In addition to its relevance to alcohol use disorders, the role of CYP2E1 in metabolizing ethanol is a potential alcohol–drug interaction site. With occasional alcohol usage, medications such as clozapine at least partly metabolized by CYP2E1 may have their half-life increased owing to competitive inhibition with alcohol. With chronic alcohol use, the induction effect predominates, thus reducing the efficacy of CYP2E1-dependent drugs by decreasing half-life (Cederbaum, 2012).
In mice, tobacco smoke induces CYP2E1 activity in the lungs, liver and kidney (Zevin and Benowitz, 1999). In male smokers, CYPE1 clearance may be increased (Benowitz et al., 1999). There may additionally be a complicated interaction effect of smoking and alcohol at CYP2E1, whereby CYP2E1 activity (as measured by chlorzoxazone metabolism) appears to be enhanced in non-alcoholic female smokers (Girre et al., 1994), while in males (Howard et al., 2003) CYP2E1∗1D has been associated with nicotine and alcohol co-dependence in one study (Howard et al., 2003), which was not replicated in Taiwanese (Huang et al., 2018).
The CYP3 family comprises the CYP3A subfamily of four genes (CYP3A4, CYP3A5, CYP3A7, and CYP3A43) and two pseudogenes (CYP3AP1 and CYP3AP2). CYP3A4 is the most abundant, although CYP3A4 and CYP3A5 have overlapping substrate specificity and in those deficient in CYP3A4, CYP3A5 and other members of the CYP3A family become crucial. The sum of the activity of all CYP3As is the total CYP3A activity, which is responsible for metabolizing ∼50% of all clinically relevant drugs (Guengerich, 1999; Bu, 2006) as well as endogenous and exogenous steroids. They are found mainly in the liver, with lower concentrations found in the intestine, respiratory tract, brain, lung and kidney (Shimada et al., 1994). Owing to their intestinal and hepatic locations, these enzymes play a significant role in the first pass metabolism of all orally administered drugs. Similar substrate specificity is due to high sequence similarity between the enzymes. CYP3A can exhibit substantial interindividual and interethnic variation in its enzymatic activity or expression, partly owing to genetic polymorphism, marked effects of inducers and inhibitors, and epigenetic mechanisms of regulation of gene expression. CYP3A inducers (such as carbamazepine, phenytoin, rifampicin, and phenothiazines such as perazine and promazine) can greatly decrease plasma concentrations of other CYP3A substrates, resulting in reduced efficacy of the substrate (Wójcikowski et al., 2012; Gupta et al., 2018). Conversely, the administration of CYP3A inhibitors (e.g., ketoconazole) can increase the plasma concentration of other substrates, increasing ADRs or even toxicity.
Inhibition/induction effects at the level of the intestine may be more important than those occurring at the hepatic level for certain drugs in some individuals. Indeed, the effects of efflux transporters such as p-glycoprotein can increase exposure of drugs to CYP3A enzymes in the intestine by prolonging transit time across the enterocyte (Wacher et al., 2001). Interestingly, there is broad overlap between substrates for and inhibitors of CYP3A enzymes and p-glycoprotein (Bruyere et al., 2010).
CYP3A4, is the most abundant CYP3A isoform in the intestine and liver (Nebert and Russell, 2002). Up to 30-fold interindividual variation in activity is seen (Ma et al., 2002); however, unlike the distribution of enzymes strongly under genetic control (such as CYP2D6), the distribution is unimodal. Some functional polymorphisms, such as CYP3A4∗22 (a intron 6 SNP, rs35599367, C>T), which is a loss of function mutation associated with 1.7–2.5 decrease in mRNA expression for heterozygous and homozygous carriers, respectively, have been identified in East Asians (who have a lower CYP3A activity) (Elens et al., 2011; Wang et al., 2011; Okubo et al., 2013). Two alleles associated with no active enzyme, CYP3A4∗20 and CYP3A4∗26, have also been identified (Westlind-Johnsson et al., 2006; Werk et al., 2014). Recent screening of over 1000 Han Chinese for mutations in CYP3A4 found seven novel exonic variants (CYP3A4∗28-∗34) (Hu et al., 2017).
Midazolam clearance or an erythromycin breath test may be used in vivo to measure the activity of CYP3A enzyme in both the intestinal epithelium and liver (Goh et al., 2002). Alfentanil is demethylated by CYP3A4 and may be a useful CYP3A probe due to the pupillary response to alfentanil (Baririan et al., 2005; Klees et al., 2005). Other probes for in vivo CYP3A activity include: alprazolam (4-hydroxylation), cortisol (6-β hydroxylation), dextromethorphan (N-demethylation), diazepam (N-demethylation), nifedipine (oxidation), terfenadine (C-hydroxylation), testosterone (6-β hydroxylation), and triazolam (1-hydroxylation) (Jurica and Sulcova, 2012). Itraconazole and ketoconazole are potent CYP3A4 inhibitors (Jurica and Sulcova, 2012). Due to the presence of multiple substrate binding domains within CYP3A4, the use of at least two structurally unrelated probe substrates is recommended when investigating inhibition effects; crystal structures show that multiple substrate/inhibitor molecules may be simultaneously bound (Korzekwa et al., 1998; Schrag and Wienkers, 2001; Tucker et al., 2001; Ekroos and Sjogren, 2006; Foti et al., 2010).
Although various functional genetic variants have been identified as above outlined, these do not account for the degree of phenotypic variation in enzyme activity seen at the population level. The major mechanisms for the regulation of CYP3A expression in fact appear to be epigenetic, including DNA methylation (Dannenberg and Edenberg, 2006), histone acetylation, and miRNA-mediated mechanisms. In the 5’-region of CYP3A4 gene, histone acetylation occurs in response to the pregnane X receptor (PXR) agonist rifampicin (Xie et al., 2009). CYP3A4 is also regulated in the promoter region of the constitutive androstane receptor (CAR) in response to dexamethasone at a lower rate of expression (Assenat et al., 2004). In addition, hepatocyte nuclear factor 4α can regulate the gene expression of PXR and CAR mediated xenobiotic induction of CYP3A4 (Tirona et al., 2003). In regard to miRNA-mediated mechanisms, miR-27b regulates CYP3A4 expression by binding to the 3’untranslated region (UTR) of CYP3A4 mRNA (Pan et al., 2009), miR-148a regulates other liver specific genes by binding to the 3’UTR of PXR mRNA (Takagi et al., 2008), and the vitamin D receptor (VDR, also an indirect modulator of CYP3A) may be downregulated by miR-27b (Li et al., 2015). Targets genes of the PXR are CYP3A4, CYP2B6, MDR1, members of UGT superfamily, multidrug resistance-related protein-3 (MRP3), and organic anion transporting polypeptide-2 (OARP-2) transporters (Klaassen and Slitt, 2005; Tolson and Wang, 2010) in multiple cell types. P-glycoprotein expression at the blood brain barrier is regulated by PXR activation (Bauer et al., 2004). The PXR is also known as the steroid and xenobiotic receptor (SXR); tamoxifen activates both CYP3A4 and MDR1 gene expression through the PXR/SXR in the breast cancer cells (Nagaoka et al., 2006). CAR, PXR, and VDR are members of the nuclear receptor family that also includes FXR, LXR, RXR, and PPARα, which together participate in the complex coordinated regulation of transcription of drug metabolizing enzyme and transporter genes (Czekaj, 2000; Czekaj and Skowronek, 2012). Genetic variants in nuclear receptors contribute to interindividual differences in response to drugs that are metabolized by CYP3A enzymes (Lamba and Schuetz, 2008).
The CYP3A5∗3 (6981A>G) and CYP3A5∗6 (14685G>A) splice site variants are associated with no functional protein (Kuehl et al., 2001; PharmVar CYP3A5 Page, retrieved from https://www.pharmvar.org/gene/CYP3A5). The CYP3A5∗7 variant (27126_27127insT) is also associated with CYP3A5 poor metabolizer status (Hustert et al., 2001).5 The majority (80–85%) of White people are CYP3A5∗3/∗3 genotype, which means they are CYP3A5 poor metabolizers (van Schaik et al., 2002). Owing to this and other factors affecting CYP3A expression (see below), CYP3A5 is expressed more frequently in those of African descent compared to Whites (55% vs 33% in one study of 47 livers Kuehl et al., 2001), However, as the lists of medications metabolized by CYP3A4 and by CYP3A5 overlap with each other and the sum of the activity in both of these enzymes is the total CYP3A activity, for many medications CYP3A4 is able to substitute for CYP3A5 in those who are CYP3A5 poor metabolizers. For those who are CYP3A5 extensive (normal) metabolizers, they require lower than the usual dose (of relevant medications such as tacrolimus) prescribed for Whites (Birdwell et al., 2015). Functional effects of combined CYP3A4 and CYP3A5 enzyme deficiency may be marked (Werk et al., 2014).
Whilst CYP3A7 is mainly found in embryonic, fetal, and newborn liver, it may persist; it metabolizes dehydroepiandrosterone and its sulfate (DHEA-S). Persistent CYP3A7 expression in adults and lower levels of DHEA-S in women with polycystic ovary syndrome has been associated with a promoter variant, CYP3A7∗1C (Goodarzi et al., 2008). Two pseudogenes are found between CYP3A7 and CYP3A5 (CYP3A7-3AP1 and CYP3A7-CYP3AP; Nelson et al., 2004; CYP3A5 RefSeqGene on chromosome 7, 2020, retrieved from https://www.ncbi.nlm.nih.gov/nuccore/NG_007938.2).
Of the total CYP3A hepatic content, CYP3A43 represents a relatively low proportion. Variants in this gene have nonetheless been identified and analyzed for association with clearance of antipsychotics (Variant Annotations). A frameshift mutation is present (c.74delA from the sequence start or c.-30delA from the ATG start, rs61469810), leading to a premature stop codon, a missense mutation (c.1018C>G/P340A, rs680055), and other silent/non-functional mutations. Increased olanzapine clearance in association with rs472660 AA genotype in the CATIE sample was found in an analysis of CYP3A43 markers available on a particular array (the Affymetrix 500K) (Bigos et al., 2011). The A variant appears more frequent in those of African descent; after accounting for CYP3A43 genotype, race was no longer a significant predictor of olanzapine clearance.
There are six human FMOs (Krueger et al., 2002), encoding enzymes FMO1-5 (the sixth gene is a pseudogene). FMO substrates include CPZ, trifluoperazine, prochlorperazine, promazine, promethazine, and other phenothiazines (Lomri et al., 1993), amphetamines, clomipramine, clozapine, desipramine, imipramine, ketoconazole, methamphetamine, moclobemide, olanzapine, ranitidine, and tamoxifen (Beedham, 1997; Motika et al., 2007; Foti and Dalvie, 2016).
FMO1 is expressed in adult kidney, intestine, and fetal liver (Yeung et al., 2000). Lower quantities are found in other organs such as the ovaries, testis, adrenal gland and bladder. Substrates include psychotropics mentioned above, disulfiram, nicotine, and pesticides (Phillips and Shephard, 2017). Some of the variability in FMO1 expression can be accounted for by a promoter SNP (characterizing the FMO1∗6 allele), which has a frequency of 30, 13, and 11% in Hispanics, those of African descent, and Europeans, respectively.
Flavin-containing monooxygenase 2 (FMO2) is expressed in the lungs. The majority of Whites and Asians are homozygous for a non-functional allele: FMO2∗2A (a C>T mutation at position 1414 that results in a premature stop codon). The wild-type (FMO2∗1) haplotype is found in African-Americans (26%), Puerto Ricans (7%) and Mexicans (2%) (Whetstine et al., 2000; Furnes et al., 2003). In some populations in Africa, the frequency approaches 50% (Veeramah et al., 2008). The functional haplotype protects against toxicity caused by organophosphate insecticides, however, it also increases the risk of pulmonary toxicity for chemicals containing thioureas. It can metabolize drugs including nicotine, prochlorperazine, and trifluoperazine (Krueger and Williams, 2005) and is responsible for activating anti-tubercular drugs. Hormones including gonadal hormones (and possibly corticosteroids – a glucocorticoid responsive element has been found in the 5’flanking region of the rabbit FMO2 gene) regulate FMO2 expression.
Flavin-containing monooxygenase 3 (FMO3)is present mainly in the liver; lower concentrations can be found in the lungs, kidneys, small intestine, and brain (Chen et al., 2016). Substrates include amphetamine, chlorpromazine, clozapine, imipramine, methamphetamine, and nicotine. Interindividual and interethnic protein concentration variability can be partially explained by the multiple SNPs that have been identified in the FMO3 gene (Cashman and Zhang, 2002; Krueger et al., 2002). These lead to amino acid substitutions or absence of functional protein, and are associated with the autosomal recessive hereditary condition of trimethylaminuria and milder forms thereof (Mackay et al., 2011). One such variant (Glu158Lys or E158K) may be associated with mild trimethylaminuria and potentially greater neurotoxicity of amphetamine and methamphetamine (which are metabolized to a greater extent to hydroxylamine metabolites by the E158K compared to the wild-type enzyme) (Motika et al., 2007). Trimethylaminuria may be associated with various neuropsychiatric presentations, ranging from depression, anxiety, suicidality, paranoia, addiction (Ayesh et al., 1993) to seizures (McConnell et al., 1997). Flavin-containing monooxygenase 3 converts trimethylamine to trimethylamine N-oxide, which is excreted in the urine, but also appears in the sweat, saliva, breath, and vaginal secretions.
Flavin-containing monooxygenase 3 activity is affected by hormones (the symptoms of trimethylaminuria can be worse in women, especially after puberty, after taking oral contraceptives, and at the time of the menstrual cycle or perimenopause), dietary content (choline, lecithin, tyramine), and intestinal bacterial overgrowth (reducing trimethylamine N-oxide to trimethylamine). Brussel sprout consumption acts as an inhibitor of FMO3, decreasing FMO3 activity, and can worsen the trimethylaminuria condition (Motika et al., 2007). For individuals deficient in FMO3, supplementation with folate and riboflavin is indicated (Motika et al., 2007). Choline and lecithin are found in egg yolk, kidney, liver, legumes, peas, salt-water fish, shellfish, and soybeans. The enzyme is subject to competitive inhibition effects (e.g., by CPZ, and imipramine) (Adali et al., 1998). Methimazole is a potent inhibitor of both FMO1 and FMO3. Recent publications have shown that N-oxidation of nicotine mediated by FMO1 and FMO3 occurs in the brain, and, moreover, that functional variation in FMO3 (rs2266780, E308G) is associated with nicotine dependence (Teitelbaum et al., 2018).
The metabolism of approximately 10% of therapeutic drugs with ester, amide, and thioester functional groups is catalyzed by esterases (Fukami and Yokoi, 2012). A common family of esterases, the B-esterase family, includes cholinesterases such as acetylcholinesterase (AChE). Cholinergic transmissions are regulated by AChE, selectively inactivating acetylcholine released from the presynaptic cleft of neurons of the brain, skeletal muscle, and the autonomic nervous system (Hasin et al., 2005).
Epoxide hydrolases are a family of enzymes that transform reactive epoxide molecules into more stable and more soluble diols (El-Sherbeni and El-Kadi, 2014). EPHX1 encodes mEH. It is a highly polymorphic gene, with over 100 SNPs identified. Enzyme activity is reduced by 40% in the variant with the c.337 T>C SNP, and 25% in the c.416 A>G variant (Caruso et al., 2014). Alcohol dependence has been associated with these SNPs (Bhaskar et al., 2013). Possibly altered response to carbamazepine and warfarin has been associated with genetic variants in EPHX1 (Nakajima et al., 2005; Puranik et al., 2013; Caruso et al., 2014; Daci et al., 2015; Liu et al., 2015).
Phase II enzymes include EH, glutathione S-transferases (GSTs), N-acetyltransferases, sulfotransferases, and UDP-glucuronosyltransferases (UGTs) (Jancova et al., 2010), the actions of which lead to the formation of more hydrophilic molecules for renal or biliary excretion (or further metabolite activation, which may be associated with toxicity). Within these groups, the enzymes most relevant to psychotropic drug metabolism are shown in Figure 1.
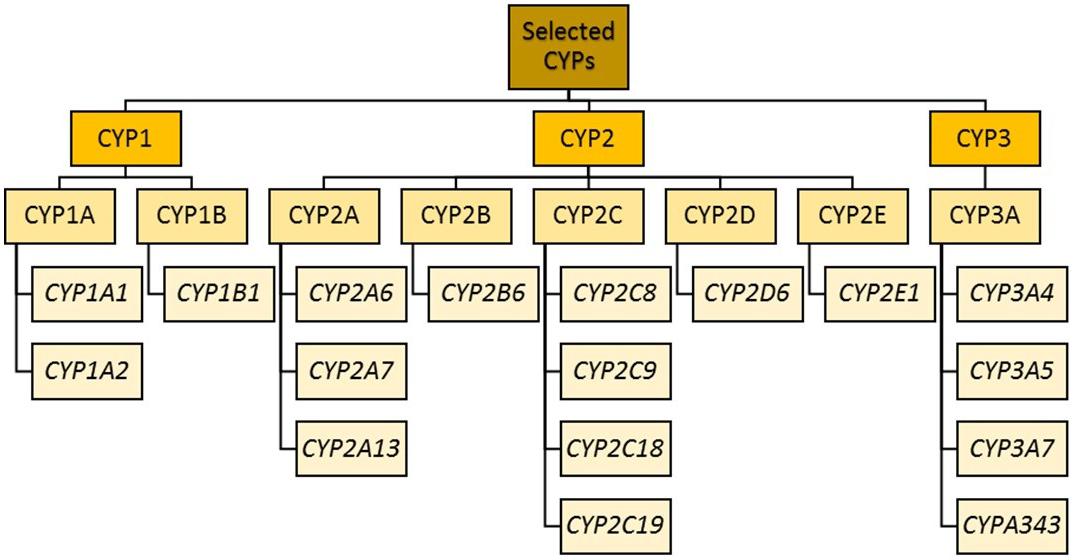
Figure 1. CYP enzyme families most relevant to psychotropic drug metabolism.
Enzymes in this phase can be classified as either type I or type II conjugation. In type I, an activated conjugating agent combines with the substrate to yield a conjugated product through the addition of functional motifs (such as acetate, glutathione, glucuronate, or sulfate), consequently increasing the xenobiotic polarity and hydrophilicity. In type II conjugation, the substrate is activated and then combined with a moiety such as a methyl group or amino acid (Jančová and Šiller, 2012).
Cytosolic enzymes expressed in the liver and intestine are encoded by the genes NAT1 and NAT2. NAT1 shows additional wide tissue distribution (Windmill et al., 2000; Sim et al., 2007, 2014) and is expressed in fetal and neonatal tissue, while NAT2 is not expressed until approximately a year later (Pacifici et al., 1986; Pariente-Khayat et al., 1991). The substrate specificity of NAT1 and NAT2 overlaps. Moreover, genetic variants in one are linked to those in another; they can therefore act in a concerted fashion to “cox and box” against evolutionary selection pressures with mutually compensatory mechanisms.
Glutathione S-transferases are relevant not only to drug metabolism, but also to detoxification of reactive intermediates such as those formed by catecholamine peroxidation (aminochrome, dopachrome, adrenochrome) in the defense against oxidative stress (Jančová and Šiller, 2012). Glucuronosyltransferases are located mainly on the luminal membrane of the endoplasmic reticulum, and act in concert with the CYP enzymes present on the cytosolic surface (Ouzzine et al., 1999; Ishii et al., 2007). Some glucuronidated products are less active; others, such as morphine-6-glucuronide, are pharmacologically active (Gong et al., 1991). UDP-glucuronosyltransferase substrates of relevance to neuropsychiatry include: apomorphine, dopamine, ethanol, lamotrigine, morphine, oxazepam, serotonin, and valproic acid (de Leon, 2003; Ouzzine et al., 2014). In the; brain, UGTs are found in the endothelial cells and astrocytes of the blood–brain barrier, as well as in the pituitary, pineal, neuro-olfactory tissue, and circumventricular organ (Ouzzine et al., 2014).
UDP-glucuronosyltransferase nomenclature is similar to that of the CYPs, with the UGT1 and UGT2 subfamilies being the most relevant for drug metabolism (Mackenzie et al., 2005).6 UDP-glucuronosyltransferase activity is influenced by factors including cigarette smoking, obesity, age, and gender (Liston et al., 2001). Although relevant to the metabolism of both endogenous and exogenous substances, there are to date relatively few studies that have both therapeutic drug monitoring data and UGT enzyme phenotype (Stingl et al., 2014). UGT1A4, UGT1A6, and UGT2B7 are relevant to the clearance of multiple psychotropics including valproic acid, lamotrigine, olanzapine, clozapine, paliperidone, chlorpromazine, and loxapine (Stingl et al., 2014; Mazerska et al., 2016). These enzymes are also expressed in the brain (King et al., 1999; Ghosh et al., 2013). Elevated plasma lamotrigine has been observed when given in combination with valproic acid, which has been attributed to competitive inhibition of UGT1A4 and UGT2B7 metabolism (Gidal et al., 2003; Rowland et al., 2006). Reduced lamotrigine clearance is seen in patients with benign unconjugated hyperbilirubinemia (Gilbert’s syndrome), which is associated with the UGT1A1∗28 haplotype in Whites and the UGT1A1∗6 haplotype in Asians (Akaba et al., 1998; Beutler et al., 1998; Barbarino et al., 2014; UGT1A1 and common exons allele nomenclature). Some associations as yet awaiting replication have been found: elevation of valproic acid clearance in carriers of UGT1A3∗5 (Chu et al., 2012), reduction of lamotrigine clearance by 60% in those homozygous for UGT2B7∗2 (Blanca Sánchez et al., 2010), and a doubling of the clearance in UGT1A4∗3 heterozygotes (Gulcebi et al., 2011). A doubling in the direct glucuronidation of olanzapine is seen in those of UGT1A4∗3/∗3 genotype, with a reduction in those with at least one UGT2B10∗2 variant (Erickson-Ridout et al., 2011). Individuals homozygous for the UGT2B15∗2 haplotype have 50% lower benzodiazepine clearance (lorazepam, and the diazepam CYP metabolite oxazepam) (Chung et al., 2005; He et al., 2009). The 142T>G UGT1A4 polymorphism is associated with reduced clozapine and olanzapine concentrations (Mori et al., 2005; Ghotbi et al., 2010).
The human sulfotransferase superfamily of enzymes contains at least 13 members, with partially overlapping substrate specificity and tissue distributions (Riches et al., 2009). Some sulfo-conjugates are active; however, sulfo-conjugation usually reduces biological activity. For example, pregnenolone sulfate blocks the activation of GABAA receptors (Majewska et al., 1988), although it is a positive allosteric modulator of the NMDA receptor (Wu et al., 1991).
Many exogenous and endogenous compounds can undergo N-, O-, S-, or arsenic-methylation (Feng et al., 2010). The co-factor required is S-adenosylmethionine (SAM), formed from ATP and L-methionine. Catechol O-methyltransferase (COMT) is a magnesium-dependent enzyme (Axelrod, 1957) that has a key role in the modulation of functions such as cardiovascular function, cognition, and pain processing, which are catechol dependent. Catechol O-methyltransferase is involved in the inactivation of catecholamine neurotransmitters (dopamine, noradrenaline), catechol-estrogens and other catechol drugs such as L–DOPA (Weinshilboum et al., 1999). There two forms of COMT: a cytoplasmic soluble form (S-COMT), and a membrane-bound form (MB-COMT), located on the cytosolic side of the endoplasmic reticulum. S-COMT is found in the liver, intestine and kidney (Taskinen et al., 2003), whereas the MB-COMT is more highly expressed in the central nervous system (Tunbridge et al., 2006). The COMT Val158Met (rs4680) polymorphism has been associated with a variety of relevant phenotypes including cognition (Goldman et al., 2009), pain tolerance (Goldman, 2014), and age of onset of psychosis after adolescent cannabis consumption (Caspi et al., 2005; Lodhi et al., 2017).
The final step in drug processing is the export of compounds away from the interior of cells in an energy-dependent manner. Metabolized molecules are transported by the ATP-binding cassette (ABC) superfamily (Hugo Gene Nomeclature Committee, 2020); energy (ATP) is used to transport substances out of the cell against a concentration gradient in multiple different organs including the brain during this phase (Dean et al., 2001; Borst and Elferink, 2002; Doring and Petzinger, 2014).
ABCB1 (previously called MDR1) was the first member to be cloned (Riordan et al., 1985; Roninson et al., 1986; Ueda et al., 1987), with the encoded protein (p-glycoprotein or p-gp) being called multidrug resistance protein owing to the observation that it was overexpressed in tumor cells with resistance to multiple chemotherapeutic agents.
As reviewed by Hodges et al. (2011), this protein has a complex structure. Two homologous halves each contain six transmembrane domains, which surround an aqueous pore within which conserved residues recognize a diverse range of substrates. It can distinguish stereoisomers and bind multiple substrates simultaneously in close proximity to each other, with associated allosteric, competitive and non-competitive inhibition, and cooperativity between substrates. Polymorphisms in ABCB1 and their role in response to antidepressants have been reviewed (Peters et al., 2009; Hodges et al., 2011).
The most commonly studied variant is a triallelic SNP (c.3435T>C, c.3435T>G and c.3435T>A, reverse strand) (rs1045642). The c.3435T>C (or C3435T) is a synonymous SNP that is in linkage disequilibrium with another synonymous SNP (C1236T, rs1128503) and a coding SNP (G2677T, ClinVar database: rs2032582). Haplotypes such as C1236T-G2677T-C3435T that include the C3435T have been associated with reduced inhibition by cyclosporin and verapamil of p-gp mediated substrate (in this case paclitaxel) efflux, with differences being more pronounced at higher levels of p-gp expression (Kimchi-Sarfaty et al., 2007). Sensitivity to rapamycin inhibition was not altered (Kimchi-Sarfaty et al., 2007). The altered sensitivity appeared to be owing to conformational change (as indicated by the use of a conformation sensitive monoclonal antibody, Kimchi-Sarfaty et al., 2007). The 3435C variant frequency varies between 34 and 90% in different ethnic groups (reviewed in Borst and Elferink, 2002; Hodges et al., 2011).
P-gp substrates include many psychotropic drugs (e.g., fluvoxamine, paroxetine, venlafaxine, amitriptyline, desipramine, trimipramine, doxepin, olanzapine, risperidone (RIS), paliperidone, CPZ, diazepam, lamotrigine, carbamazepine, and phenytoin (Uhr and Grauer, 2003; Gunes et al., 2008; Uhr et al., 2008; Aller et al., 2009; O’Brien et al., 2012; de Klerk et al., 2013; Palleria et al., 2013; Lund et al., 2017; UniProtKB). Data on citalopram vary depending on the model system and fluoxetine and mirtazapine are not p-gp substrates (reviewed in Peters et al., 2009). The list of non psychotropic substrates is extensive (reviewed in Hodges et al., 2011).
Overlapping substrate specificity with other ABC transporters is present. P-gp is expressed on the apical membrane of the intestine from the duodenum to the rectum, being coregulated with CYP3A4 in the duodenum and jejunum, and coregulated with CYP3A5 in the rectum and sigmoid colon (von Richter et al., 2004; Ufer et al., 2008; Cascorbi, 2011; Fromm and Kim, 2011). It shares substrate specificity with CYP3A4, and both are regulated by St John’s Wort (Johne et al., 1999), amongst other drugs. High affinity substrates such as verapamil also inhibit p-gp at the blood–brain barrier, causing drugs such as loperamide to affect the central nervous system (an anti-diarrheal medicine that normally has no central nervous system effects) (Elsinga et al., 2004). A review on the topic of p-gp and its relevance to drug–drug interactions (DDI) underlines that data observed in vitro may not always be reflected by that seen in clinical practice in vivo (Lund et al., 2017). In vivo data indicate that carbamazepine and phenytoin are p-gp inducers, while fluvoxamine and paroxetine are p-gp inhibitors (Lund et al., 2017).
In the liver, p-gp levels vary 50-fold. More than 51000 mutations in the ABCB1 gene region including over 137 missense7 variants have been identified. Pharmacogenetic studies to date have often focused on a limited number of SNPs, such as the three described above. Data up to 2009 in regard to associations with response to antidepressants were summarized as equivocal (Peters et al., 2009), with a subsequent pharmacogenetically guided clinical trial (Singh, 2015) and a meta-analysis including this trial concluding in favor of this gene potentially having a role in pharmacogenetically guided treatment (Bousman et al., 2019b). Singh (2015) suggests that ABCB1 should be considered together with ABCC1. Fabbri and Serretti (2015); Lett et al. (2016), and Amare et al. (2017) include data on ABCB1 in their antidepressant response reviews, with a recent study in an E. Asian population reporting an association with response to serotonin noradrenaline reuptake inhibitors (SNRIs; Shan et al., 2019). In a review on clozapine, Krivoy et al. (2016), concluded that ABCB1 genotypes including the C3435T were associated with clozapine concentration and response.
While many studies have focused on the above outlined SNPs, particularly the C3435T, an approach in which haplotypes are linked to transporter phenotypes and systematically cataloged to inform clinical association analyses is surely desirable. For example, using in silico molecular techniques to predict amino acid residues that bind to psychotropics and hence which mutations might be investigated for clinical association analyses could be an informative approach. Further, elucidating mechanisms by which different co-administered medications might interact at p-gp would be helpful.
After initial prescription, psychiatric medicines have a 40–60% failure rate (Correll et al., 2015). Implementation of pharmacogenetics can improve current methods of physician judgment and therapeutic trials. Challenges to data standardization are prevalent (de Leon, 2009; Malhotra et al., 2012; Altman et al., 2013; Bousman and Hopwood, 2016; Bousman et al., 2018). To address this, the Clinical Pharmacogenetics Implementation Consortium (CPIC) was created in 2009 by PharmGKB and the Pharmacogenomics Research Network (Relling and Klein, 2011) to provide prescribing guidelines for genetic variants. CPIC consists of four levels of recommendation concerning drug-gene pairs (Caudle et al., 2016).8 Recommendation levels are denoted based on literature reviews presented to the CPIC writing committee. Evidence classifications include “high,” “moderate,” or “weak,” based on design, quality, and generalizability of the research. Therapeutic recommendations are graded as “strong,” “moderate,” or “optional” (Caudle et al., 2014). Guidelines focus on gene–drug pairs where the prescribing recommendations are actionable (level A or B) (Table 1).9
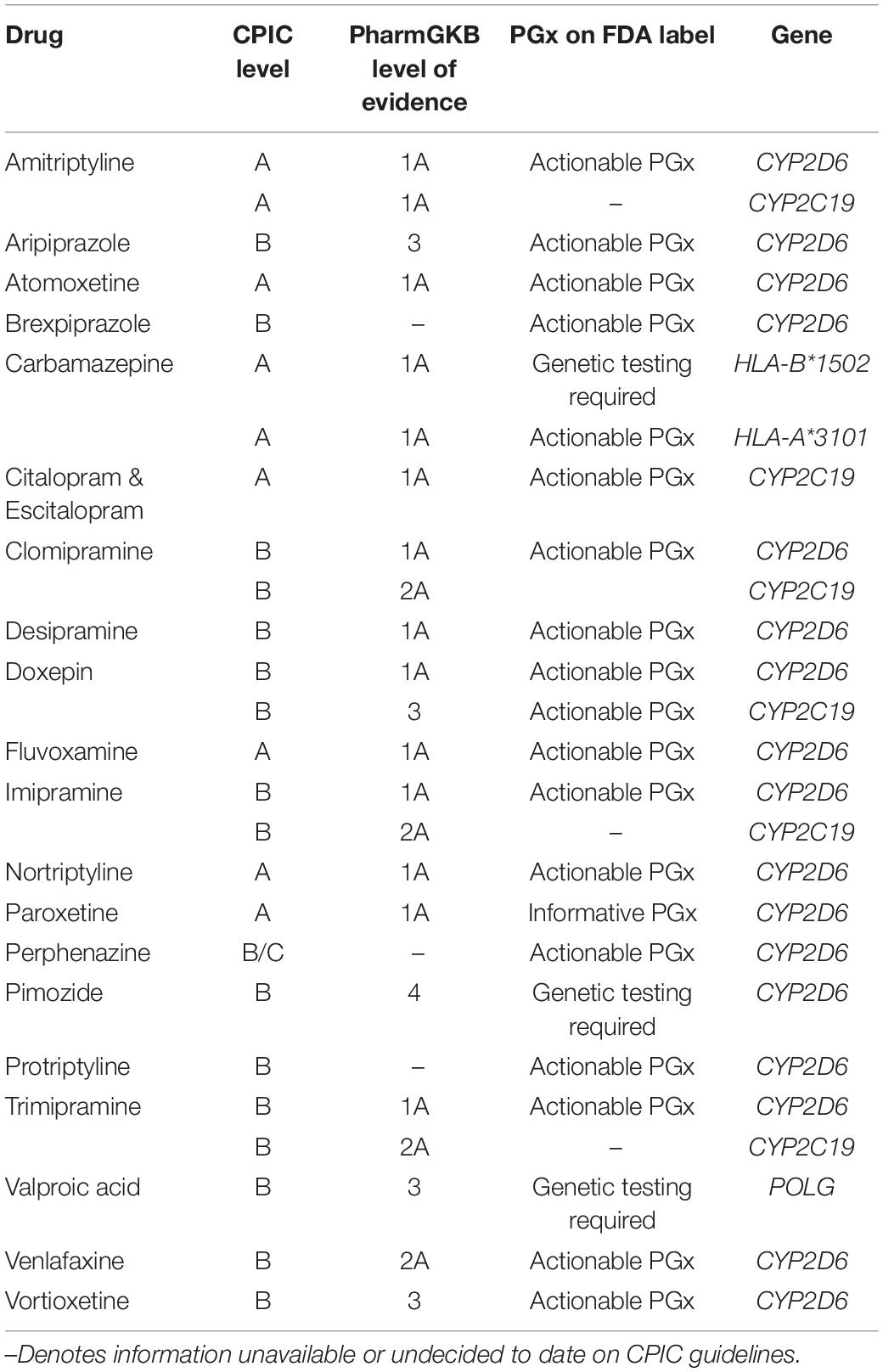
Table 1. Mental health medications: Clinical Pharmacogenetics Implementation Consortium (CPIC) evidence levels, pharmacogenomic FDA label, and associated genes.
Prior to the implementation of CPIC, in 2005, the Royal Dutch Pharmacists Association established a similar body, the Dutch Pharmacogenetics Working Group (DPWG), to provide prescribing guidelines for specific gene–drug pairs to physicians and pharmacists in the Netherlands and now used internationally.10 Similar to CPIC, evidence for strength of a prescribing recommendation (such as to avoid a particular drug in the presence of a specific genotype) is ranked on a 0–4 scale (Bank et al., 2018). While there is significant overlap between the recommendations offered by these two organizations, some differences in therapeutic recommendations can be found (Bank et al., 2018; van Westrhenen et al., 2020).
Below are provided further details for pharmacogenetic associations for specific classes of medications relevant to psychiatry.
There is significant interindividual variation in treatment response and adverse reactions to mood stabilizers (Murru et al., 2015; Tang and Pinsky, 2015; Pisanu et al., 2016). The current CPIC gene–drug pair list includes carbamazepine, oxcarbazepine and valproic acid (Saruwatari et al., 2010; Drozda et al., 2014), with guidelines available for the first two (Relling et al., 2011; Phillips et al., 2018).
Carbamazepine and oxcarbazepine are anticonvulsants approved for treating epilepsy, trigeminal neuralgia, and bipolar disorder (Phillips et al., 2018). Therapeutic drug monitoring for anticonvulsants is well-established. Both share dose-dependent (type A) ADRs including ataxia. Type B ADRs (not predictable from the pharmacology) are potentially lethal and include osteoporosis, aplastic anemia, and Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN).
Genetic variants having actionable levels with carbamazepine and oxcarbazepine are HLA-B∗15:02, HLA-A∗31:01 and SCN1A (Relling and Klein, 2011; Phillips et al., 2018). Associations have been shown in Asians with HLA-A∗31:01 and carbamazepine induced SJS/TEN (Ferrell and McLeod, 2008; Stern and Divito, 2017). A 2004 report in Han Chinese found that the SJS/TEN frequency reduced to 0% after HLA-B∗1502 genotype pre-screening (Chung et al., 2004). East Asians exhibit the highest HLA-B∗15:02 haplotype frequency (∼15%) compared to other populations (>1%). In Hong Kong, Taiwan, and Thailand, testing for this haplotype prior to prescribing carbamazepine and oxcarbazepine is standard practice (Chen et al., 2014; Sukasem and Chantratita, 2016; Lin et al., 2018). However, recent data indicate that the HLA-B∗15:02 frequency in other populations may also be high enough to justify testing in other ethnic groups (Fang et al., 2019). HLA-A∗31:01 haplotype frequency also varies by ethnicity, being up to 15% in most Asian and White groups and infrequent in those of African descent (Fan et al., 2017).
Valproic acid (or its derivative, divalproex sodium, which is converted to valproic acid in the stomach) increases the levels of γ-aminobutyric acid (GABA) in the brain, blocking voltage gated ion channels (particularly calcium and sodium), and inhibiting histone deacetylase enzymes, including HDAC1. Genetic factors are associated with differential efficacy and ADRs (Kasperaviciūte and Sisodiya, 2009; Löscher et al., 2009; Fricke-Galindo et al., 2018). Hepatic metabolism occurs via CYP-mediated oxidation, glucuronidation, and mitochondrial oxidation (Johannessen and Landmark, 2010; Chatzistefanidis et al., 2012; Ghodke-Puranik et al., 2013).
ADRs associated with valproic acid include hepatotoxicity, mitochondrial toxicity, and potentially fatal hyperammonemia encephalopathy, among others (Linnet and Wiborg, 1996; Johannessen and Landmark, 2010; Singh et al., 2015). Valproic acid is contraindicated in patients with disorders secondary to mutations in DNA polymerase gamma (POLG), which replicates mitochondrial DNA. Patients with POLG-related disorders have elevated risk of fatal hyperammonemia encephalopathy. The onset of such may vary from childhood to late adulthood. It is therefore contraindicated in children with clinical suspicion of a hereditary mitochondrial disorder. In those over two years of age with suggestive symptoms (such as migraine with defined types of aura), valproate POLG testing is required,11 and it should be used if the testing is negative, other anticonvulsants have failed, and liver function is monitored.
In this section, pharmacogenetic data available for some specific medications are used to illustrate key applicable principles.
Perphenazine undergoes substantial first-pass hepatic phase I and II metabolism. Serum concentrations vary widely due to polymorphisms in multiple phase I enzymes: up to 30-fold in CYP2D6 NMs (Linnet and Wiborg, 1996). Initial studies showed that after 4–5 weeks, improvement was associated with plasma perphenazine concentrations above 2 nmol/l, while extrapyramidal effects occurred at concentrations above 3 nmol/l (Hansen, 1981; Hansen et al., 1982; Hansen and Larsen, 1983). In a larger study of over 200 patients, a wider therapeutic range (2–6 nmol/l) was suggested (Hansen and Larsen, 1985). Perphenazine binds dopamine D2 and alpha-1/alpha-2 receptors with 70 and 50% antagonism. The main active metabolite, 7-hydroxyperphenazine, binds dopamine D2 and alpha-1/alpha-2 receptors with 70 and 50% the antagonism of perphenazine (Hals et al., 1986). It is formed in a reaction catalyzed by CYP2D6, with other metabolites including N-dealkylated perphenazine (formed in part by other CYPs), and perphenazine sulfoxide (Dahl-Puustinen et al., 1989; Olesen and Linnet, 2000). Compared to perphenazine, the concentration of perphenazine sulfoxide is in the same range, while N-dealkylated perphenazine is approximately three times that of perphenazine (Hansen et al., 1979). At therapeutically relevant concentrations of perphenazine, CYP3A4 accounts for about 40% of the N-dealkylation, with CYP isoforms 1A2, 2C19 and 2D6 contributing 20–25% (Olesen and Linnet, 2000).
The peak serum concentration and the AUC of perphenazine for CYP2D6 PMs is about 3 and 4 times, respectively that of NMs in single dose kinetics (Dahl-Puustinen et al., 1989), and at steady state, the median concentration-to-dose ratio of perphenazine in CYP2D6 PMs is about twice that of NMs, with patients on concomitant inhibitors showing a median concentration in between the two groups (Linnet and Wiborg, 1996). Jerling et al. (1996) conducted a study of patients during treatment; CYP2D6 genotype was shown to significantly predict the oral clearance of perphenazine (patients with two CYP2D6 PM alleles having lower clearance than heterozygote PMs or NMs) (Jerling et al., 1996).
It would be expected that individuals deficient in CYP2D6 or on potent CYP2D6 inhibitors, higher perphenazine concentrations would be found and hence more adverse effects, whilst in CYP2D6 UMs, there would be lower concentrations, with less adverse effects and potentially a lower therapeutic efficacy. Consistent with this, paroxetine, a potent CYP2D6 inhibitor (Lam et al., 2002), increases the AUC of perphenazine 7-fold in NMs, which is associated with increased side effects (Ozdemir et al., 1997).
Since 1984 pimozide has been used to treat Gilles de la Tourette’s syndrome (Pringsheim and Marras, 2009), and also to treat psychotic disorders. Its use has been limited owing to an ADR of prolongation of the QT interval on the electrocardiogram, which is associated with risk for Torsades de Pointes (a type of ventricular fibrillation that may cause sudden cardiac death) (Fulop et al., 1987; Committee on Safety of Medicines-Medicines Control Agency, 1995). In an isolated rabbit heart, this effect was shown to be attributable to pimozide itself, not to metabolites (Flockhart et al., 2000); this is due to an effect of the drug on potassium channels encoded by the human ether-a-go-go-related gene (HERG, otherwise known as KCNH2), which is responsible for the delayed repolarization current in the heart.
It is important to determine which cytochromes might contribute to the pimozide concentration profile. In vitro analyses showed that the formation of the major metabolite, 1,3-dihydro-1-(4-piperidinyl)-2H-benzimidazol-2-one (DHPBI), by N-dealkylation was primarily dependent on CYP3A4, with a lesser contribution by CYP1A2 (Desta et al., 1998). CYP2D6 may also play a role, but due to it being inhibited by pimozide, it was not possible to draw a conclusion regarding this from this in vitro study.
Case reports of interactions between pimozide and CYP2D6 inhibitors such as paroxetine and fluoxetine (Ahmed et al., 1993; Horrigan and Barnhill, 1994), as well as investigation of differential interaction with clarithromycin (an inhibitor of CYP3A) by CYP2D6 status led to recognition that CYP2D6 was a major contributor to the in vivo pharmacokinetics of pimozide (Desta et al., 1998). The effect of a single dose (6 mg) on the QTc interval (QT interval corrected for heart rate) was measured over time, and showed the greatest increase within the first 20 hours, with NMs showing a larger increase (by nearly 20 ms), followed by a reduction from 20 to 50 h, and then an increase at approximately 60–100 h. The late elevation was more significant in CYP2D6 PMs, women, and clarithromycin-treated individuals, and appeared more sustained than the early increase. Owing to the more sustained nature, the late onset elevation may be more relevant to significant QTc prolongation; the early peak in NMs warrants further investigation in UMs. In CYP2D6 PMs, half-life increased from 29 ± 18 h to 36 ± 19 h, while in NMs, the corresponding values were 17 ± 7 and 23 ± 10 h. For subjects with relevant data, the pimozide induced QTc interval changes coincided with the concentration-time course of pimozide. The prescription of CYP3A inhibitors, such as valproate, is now contraindicated with pimozide. In the above study, interestingly, pimozide rapidly increased plasma prolactin concentration, the maximum increase occurring 4 hours post dose, with a sharp reduction thereafter.
Simulated steady-state pharmacokinetic profiling of pimozide in CYP2D6 PMs, IMs, and NMs led to specification in the FDA label in 2011 that CYP2D6 PMs should not be prescribed more than 4 mg, with the maximum recommended dose in CYP2D6 NMs being 10 mg (Rogers et al., 2012). In the simulated data, 4 mg/day in CYP2D6 PMs was the maximum dose that did not result in plasma concentrations in excess of those observed in CYP2D6 NMs receiving 10 mg/day (Desta et al., 1998). Pimozide is commenced at 0.05 mg/kg (Preskorn, 2012), once daily. If the patient is a CYP2D6 NM and is not on a CYP2D6 inhibitor, the dose may be increased every third day to a maximum of 0.2 mg/kg/day, to a maximum of 10 mg/day. If the CYP2D6 status is not known, CYP2D6 genotyping should be done before deciding to increase the dose above 0.05 mg/kg/d, which is the maximum dose for a CYP2D6 PM, or if on a CYP2D6 inhibitor such as paroxetine, fluoxetine, and bupropion. Paroxetine will convert 60% of CYP2D6 NMs to PMs at 20 mg daily, while at 40 mg daily, 95% will be phenocopied to PMs (Preskorn, 2003). Phenoconversion (the conversion of an individual’s genetically defined metabolizer status to another status owing to the effect of a pharmacologically active substance) to CYP2D6 PM status by the action of an enzyme inhibitor has been estimated as being 6 times more common than genetically determined CYP2D6 PM status (Preskorn, 2012, 2013).
First pass metabolism of pimozide includes both the gut and the liver as CYP3A represents 70% and 30% of the total CYP450 in the intestine and the liver, respectively (Kolars et al., 1994; Shimada et al., 1994). Metabolism will be subject to the influence of gut microbiota, diet, and other factors including hormones (CYP3A4 being subject to regulation by the PXR and CAR) (Lamba et al., 2005; Pan et al., 2009).
The drug label does not currently include dosing recommendations for CYP2D6 UMs; further research including genotyping CYP2D6 is required for pimozide, and other CYP2D6 metabolized medications.
It is suggested that CYP3A4 also be genotyped for pimozide treatment, given its association with sudden cardiac death. It has a less clear genotype–phenotype relationship (with no updated data on PharmVar), and thus has not yet been introduced into clinical guidelines. In the absence of genotyping, probe drugs such as nifedipine may be utilized to test the activity of multiple CYPs (de Andrés et al., 2014); however, such estimation of CYP3A4 phenotype is influenced by any concomitant medication and/or dietary effects.
Haloperidol (HAL) is a butyrophenone and first-generation antipsychotic (FGA) drug that acts as a dopaminergic antagonist in the mesolimbic system. It is used to treat a variety of psychiatric conditions, including psychoses (e.g., schizophrenia, schizoaffective disorder, bipolar disorder with mania or psychotic symptoms, substance-induced psychotic disorder) and other conditions with hallucinations (e.g., alcohol withdrawal, delirium, Lewy body dementia). Adverse effects may include tardive dyskinesia, neuroleptic malignant syndrome, and a prolonged QTc interval. Two major routes of metabolism, N-glucuronidation and O-glucuronidation, are effected by UGT enzymes, specifically the former by UGT1A4, and the latter by UGT1A4, UGT1A9, and UGT2B7 (Kato et al., 2012). Various CYP isoenzymes contribute to the metabolic pathways of this medication, most notably CYP3A4, and, to a lesser extent, CYP2D6. Cytosolic carbonyl reductase catalyzes the formation of reduced HAL, which retains 10–20% of the activity of the parent compound. Reduced HAL can be further metabolized by CYP3A4 to a tetrahydropyridine. The reduced drug can also be back-oxidized by CYP3A4 to HAL (Pan et al., 1998; Kudo and Ishizaki, 1999; Tateishi et al., 2000; discussed in Aitchison et al., 1999). Owing to its lipophilicity, HAL is extensively metabolized in humans, with large interindividual variations in pharmacokinetics arising. With a proposed therapeutic range of 5.6–16.9 μg/l in serum (Ulrich et al., 1998), being able to appreciably predict pharmacokinetic parameters in individuals is of utmost importance to optimize efficacy and safety. At lower doses, CYP2D6 contributes to HAL metabolism significantly, but with higher doses, and longer term treatments, CYP3A4 back-oxidation and N-dealkylation considerably outweigh the contributions of CYP2D6 (Fang et al., 1997; Pan et al., 1998; Zhou et al., 2009). Nyberg et al. (1995) showed that CYP2D6 PMs exhibited higher plasma concentrations of HAL over a 4-week treatment period with HAL decanoate, as compared to seven NMs in the study. However, Ohnuma et al. (2003) showed that, in a large number of Japanese patients, the presence of neither an enzyme activity-reducing mutation (CYP2D6∗10A) nor activity-increasing mutations (duplications) in CYP2D6 alone could appreciably predict HAL concentrations.
Haloperidol is a medication that is CPIC level B (for CYP2D6),12 with a guideline currently in progressguidelines. Further, in the DPWG guidelines, there is a recommendation for initial dose to be reduced to 50% in PMs or for selection of an alternative medication based on a metabolic pathway different than CYP2D6. Possible dose adjustments are also mentioned for UMs.13 In a study of 70 patients in which the most commonly prescribed medication was HAL, the risk of tardive dyskinesia increased with increasing number of functional CYP2D6 genes, with UMs showing the highest risk (Koola et al., 2014).
Enzyme induction effects are also relevant for HAL metabolism. First, there is the effect of smoking. From a relevant review (Desai et al., 2001), it may be deduced that smoking increases the clearance of oral HAL (via effects including on CYP1A2), especially at doses lower than 0.5 mg/kg/day. Carbamazepine (which induces several CYPs including the CYP3As) reduces plasma HAL concentration (Hesslinger et al., 1999).
Chlorpromazine is a phenothiazine that was the first antipsychotic to be introduced (reviewed in Basu et al., 2007). Its biotransformation includes hydroxylation (by CYP2D6 and CYP1A2), N-methylation, N-N-didemethylation, N-oxidation, S-oxidation, and glutathione conjugation. The hydroxylated metabolite can undergo further oxidation leading to formation of an electrophilic quinone imine intermediate, which is capable of mediating toxic effects (by reacting with cellular proteins and DNA) or underdoing glutathione conjugation (Wen and Zhou, 2009). Muralidharan et al. (1996) confirmed the contribution of CYP2D6 to the hydroxylation pathway using quinidine, whilst also showing that CYP2D6 genetic polymorphism was not the major contributor to inter-individual variability in plasma concentrations. The latter finding was confirmed by Yoshii and colleagues (Yoshii et al., 2000), whose microsomal inhibition studies of chlorpromazine 7-hydroxylation showed that CYP1A2 may play a more important role in the hydroxylation reaction for individuals deficient in CYP2D6. Indeed, Gill et al. (1997) reported that an individual with schizophrenia who was homozygous for the CYP2D6∗4 variant (then known as the “B”) and therefore a PM and had been intolerant and non-compliant with multiple medications settled on a very low dose (50 mg) of chlorpromazine (Gill et al., 1997).
Zuclopenthixol is a thioxanthene derivative used to treat schizophrenia, having high affinity for both D2 and D1 dopamine receptors (Kumar and Strech, 2005). Its metabolic pathways include sulfoxidation, N-dealkylation, and glucuronidation (Zhou et al., 2009), with metabolites not known to have antipsychotic activity.
Dahl et al. (1991) showed that clearance of zuclopenthixol was associated with debrisoquine hydroxylation, and further studies confirmed the role of CYP2D6 in zuclopenthixol metabolism (Zhou et al., 2009). Moreover, PMs had a 1.9-fold higher AUC of zuclopenthixol compared to NMs (n = 6 for each group) after a single 6 or 10 mg dose (Dahl et al., 1991). Linnet and Wiborg (1996) found similar results: investigation of phenotypic relationships to zuclopenthixol concentration showed that, in 12 psychiatric patients, CYP2D6 PMs had 60% greater concentrations than NMs, but were similar to NMs who were co-administered CYP2D6 inhibiting drugs.
Furthermore, in another study, psychiatric patients treated with zuclopenthixol who experienced adverse neurological events (tardive dyskinesia, parkinsonism) tended to have a higher frequency of non-functional CYP2D6∗3 and ∗4 alleles, but these results did not attain statistical significance (Jaanson et al., 2002).
Zuclopenthixol is CPIC level B,14 with a guideline in progress (and also not yet available from DPWG). One review suggests considering dose adjustment (58 and 88% for CYP2D6 PMs and IMs, respectively) or selecting an alternative medication (Stingl et al., 2013).
Aripiprazole was marketed as the first antipsychotic with dopamine and serotonin partial agonism. In Europe, aripiprazole is indicated for use in the treatment of schizophrenia and treatment of moderate to severe manic and episodes associated with bipolar I disorder and for the prevention of new manic episodes in those whose manic episodes respond to aripiprazole (Koskinen et al., 2010; Abilify 10 mg tablets; Abilify; Summary of Product Information, EMA). Other licensed indications include adjunctive treatment of major depressive disorder, Tourette’s syndrome, and irritability in autism spectrum disorder (Mailman and Murthy, 2010). Global therapeutic efficacy has been measured versus aripiprazole and dehydroaripiprazole serum concentrations, with a reported 68% response rate in those with concentrations of 150–300 ng/ml of aripiprazole, and a 57 and 50% response rate with concentrations less than 150 ng/ml or above 300 ng/ml, respectively (Kirschbaum et al., 2008). Therapeutic drug monitoring (TDM) has “recommended” (level 2 evidence) for aripiprazole by the interdisciplinary TDM group of the Arbeitsgemeinschaft für Neuropsychopharmakologie und Pharmakopsychiatrie (AGNP), with a therapeutic target range of 100–350 ng/ml for aripiprazole, or 150–500 for aripiprazole and dehydroaripiprazole (Hiemke et al., 2018).
Aripiprazole undergoes substantial first pass metabolism in the liver unless administered in a long-acting injectable (LAI) form. It is metabolized by dehydrogenation, hydroxylation, and N-dealkylation. In vitro studies show that CYP3A4 and CYP2D6 conduct the dehydrogenation and hydroxylation of aripiprazole, with CYP3A4 additionally catalyzing the N-dealkylation. Although a substrate for these enzymes, it does not appear to inhibit the activity of these enzymes. In clinical studies, 10–30 mg/day doses of aripiprazole had no significant effect on the metabolism of substrates of CYP2D6 or CYP3A4 activity as indexed by dextromethorphan; it does not appear to be an inhibitor of CYP2C9, CYP2C19, or CYP1A2 (Hjorthoj et al., 2015), nor a substrate for CYP1A enzymes, and hence no dose adjustment is required in smokers.
In a large pharmacokinetic study (N = 1288), CYP2D6 PMs and IMs had a 1.4 times increase in exposure to the active moiety compared to NMs, leading to a 15% decrease in medication dosage of aripiprazole. Switch in medication from aripiprazole was not, however, significantly associated with CYP2D6 status (Jukic et al., 2019).
The active dehydro-aripiprazole metabolite has a similar affinity as aripiprazole for dopamine D2 receptors; at steady state it represents about 40% of the plasma concentration of aripiprazole (area under the curve or AUC; Hjorthoj et al., 2015), after oral administration or 29–33% after administration in the form of the LAI Abilify Maintena, and is therefore thought to contribute to the sustained pharmacologic effect of aripiprazole. Both aripiprazole and dehydro-aripiprazole are highly bound to plasma protein, mainly to albumin (reviewed in DeLeon et al., 2004). The average elimination half-life is oral aripiprazole is ∼75 h, but in CYP2D6 PMs, the average half-life extends to ∼146 h (Hjorthoj et al., 2015). The half-life of oral aripiprazole in CYP2D6 IMs (75.2 h) was significantly longer than that in CYP2D6 NMs (45.8 h); the systemic clearance of aripiprazole in IMs is approximately 60% that of NM subjects, with the maximum concentration being the same in IMs as in NMs (Kubo et al., 2007). At steady state, PMs have a significantly lower concentration to dose ratio than NMs, while in one report, IMs did not differ (van der Weide and van der Weide, 2015). However, van der Weide and van der Weide (2015) included in their IM group individuals who were heterozygous NMs (NM/PM genotype). In another report (Hendset et al., 2014), median serum concentrations were 1.6-fold or 1.8-fold higher in individuals of CYP2D6 PM/IM or IM/IM genotype, respectively than in those who were heterozygous NMs.
For patients who are known CYP2D6 PMs, FDA recommends administration of half of the usual dose of aripiprazole, and the DPWG guidelines recommend reducing maximum daily dose to 10 mg/day or 300 mg/month, i.e., 67–75% of the standard maximum dose.15 Given that at a dose as low as 2 mg, D2 receptor occupancy is ∼70% (71.6 ± 5.5%, Kegeles et al., 2008), and the recommendation by consensus guidelines of doses of aripiprazole lower than those used in the initial marketing phase of the drug (Aitchison et al., 2009), it may well be recommendable to start at the lowest dose (2 mg) and to go no higher than 5 mg in CYP2D6 poor metabolizers. While there are as yet no guidelines for other CYP2D6 phenotypic groups, in the case of IMs, the Japanese data would suggest that a cautious dosing in the 2–5 mg range should be appropriate.
In addition, packaging information for aripiprazole offers additional guidelines should the medication be taken with known CYP inducers or inhibitors (Abilify - aripiprazole tablet, 2016). In the case of co-prescription of CYP3A4 or CYP2D6 inhibitors, dosage is recommended to be reduced (by 50% in the case of strong inhibitors such as ketoconazole and fluoxetine, respectively). Likewise, should aripiprazole be taken with known CYP3A4 inducers, dosage increase is recommended (doubling in the case of carbamazepine). On cessation of any inhibitors/inducers, the dose should be readjusted accordingly (Abilify).16
It has been noted that aripiprazole and 2,3-(dichlorophenyl) piperazine (2,3-DCPP), one of its metabolites, affect cholesterol biosynthesis by inhibiting 7-dehydrocholesterol reductase (DHCR7), the enzyme that converts 7-dehydrocholesterol (7-DHC) to cholesterol (Korade et al., 2010, 2016; Kim et al., 2016; Genaro-Mattos et al., 2018). Cholesterol is of critical importance to brain development; mutations in DHCR7 gene leads to Smith-Lemli-Opitz Syndrome, a neurodevelopmental condition, and exposure to DHCR7 inhibitors during the first trimester of pregnancy is associated with increased rates of fetal malformations, intrauterine death, and spontaneous abortions (Boland and Tatonetti, 2016). Thus, aripiprazole should be contraindicated during the first trimester of pregnancy; the Summary of Product Characteristics states17 “this medicinal product should not be used in pregnancy unless the expected benefit clearly justifies the potential risk to the fetus.” The most critical period for formation of the neural tube is the first six weeks of gestation, when many women do not realize they are pregnant. Therefore, it is recommended that women receiving aripiprazole in reproductive years should have a discussion of whether the woman is sexually active and of methods of contraception.
In a multiple-dose study, the mean terminal-phase elimination half-life of aripiprazole was 29.9 days and 46.5 days after 4-week injection of LAI 300 mg dose and 400 mg dose, respectively (Mallikaarjun et al., 2013). Data regarding differential half-life of the LAI by CYP2D6 genotype and/or CYP3A activity are not available. Aripiprazole lauroxil is a prodrug that undergoes bioactivation by hydroxylation and can be administered once every 6 weeks; it is similarly lacking pharmacogenetic data thus far.
Risperidone is an atypical antipsychotic used for treating schizophrenia, acting mainly on 5-HT2A and D2 receptors (Zhou et al., 2009); it is converted to 9-hydroxyrisperidone (9-OH-RIS) by CYP2D6, with the latter being excreted in the urine and feces. In a meta-analysis conducted by Stingl and Viviani (2015), they estimated dose adjustment of RIS to be 56 and 146% in CYP2D6 PMs and UMs, respectively, mentioning increased risk of toxicity in PMs. The DPWG note increased risk of treatment failure in CYP2D6 PMs and UMs and recommend using 67% of the standard dose in the former, and choosing an alternative drug or titrating the dose according to the maximum for the active metabolite (12 mg/day of paliperidone) in the latter. In a recent review by van Westrhenen et al. (2020), these recommendations were updated to suggest reducing the maximum dose by 33% (to 4 mg/day) in IMs as well as in PMs. For UMs, it was suggested to select an alternative medication or use therapeutic drug monitoring. It is worth noting that Cui et al. (2020) have found significant differences in recommendations of RIS dosage according to ethnicity. Specifically, adjustment in titration of this medication should be reduced in people of Asian ethnicity compared to Whites.
In a Norwegian population (Mannheimer et al., 2014, 2016), it was found that the metabolic ratio (MR) for RIS, expressed as RIS/9-OH-RIS, was, not surprisingly, associated with CYP2D6 PM status: an MR threshold of >1 predicted PM status with 91% accuracy (Mannheimer et al., 2016).
RIS metabolism by CYP2D6 is inhibited by the phenothiazine drug perazine when the two are co-administered (Paulzen et al., 2017), resulting in an increase in RIS and (RIS + 9-OH-RIS) concentrations and a reduction in the 9-OH-RIS/RIS ratio. Animal models have previously shown the role of phenothiazines in inhibiting the CYP2D family (Daniel et al., 2005).
In a study focusing on the relationship between genetic and epigenetic variation and response to RIS, three CpG sites in CYP2D6 and two to three CpG sites in CYP3A4 appeared to be more methylated in poor responders (Shi et al., 2017).
The effect of CYP2D6 genotype on RIS metabolism has been studied in young Thai autistic spectrum individuals (Nuntamool et al., 2017). Genotypes CYP2D6∗5/∗10, ∗10/∗10 and ∗10/∗41 showed reduced RIS metabolism, with significantly higher RIS plasma concentrations. While such an association was not seen in the CYP2D6∗4/∗10 genotype group, this was likely owing to the relatively low frequency of the CYP2D6∗4 variant in this ethnicity, and the wide spread of the data in the small subgroup of CYP2D6∗4/∗10 genotype. The CYP2D6∗10 variant was also associated with higher MR of RIS/9-OH-RIS. Hendset et al. (2014) genotyped for CYP2D6 ∗3, ∗4,∗ 5, ∗6, ∗9, ∗10 and ∗41, and classified as “∗1/def” (heterozygous for normal and deficient function) or “def/red” (heterozygous for deficient and reduced function); RIS serum concentration was 4.5 times higher in the def/red group compared to ∗1/def. In addition, a 3 to 4-fold increase in the serum concentration of RIS was shown in the red/red group.
In addition to variation in metabolic activity and treatment response, Molden et al. (2016) found evidence of a relationship between genotype and discontinuation of treatment. Individuals classified as PMs for CYP2D6 had active moiety (RIS + 9-OH-RIS) concentration 1.5 times higher than NMs. Consequently, there was an over-representation of adverse events and discontinuation of treatment for PMs. Conversely, a similar study with Croatian psychiatric patients receiving RIS injections found individuals classified as UM with concentrations of RIS active moiety (RIS + 9-OH-RIS) not reaching the threshold recommended for therapeutic range (Ganoci et al., 2016). Kaur et al. (2017) reported an association between the CYP2D6∗4 PM haplotype and treatment dropout due to poor response.
In the largest study of RIS and CYP2D6 to date (1288 patients), approximately 1.4 and 1.6-fold RIS exposure increase was observed in CYP2D6 IMs and PMs, respectively (Jukic et al., 2019). A higher incidence of RIS-associated ADRs (de Leon et al., 2005) and treatment failure (Jukic et al., 2019) is observed in CYP2D6 PMs compared with NMs, with increased treatment failure rate also being observed in CYP2D6 UMs (Jukic et al., 2019). It is possible that the latter may be exposed to subtherapeutic drug concentrations, and also possible the effect of CYP2D6 on normally minor synthesis pathways for serotonin and dopamine may at least partly relate to such associations. Recent systematic reviews and meta-analyses support the need for dosage adjustment of RIS based on CYP2D6 genotype (Cui et al., 2020; Zhang et al., 2020).
The relationship between CYP2D6 and hyperprolactinemia (a possible adverse effect of RIS) appears to be U-shaped, with a tendency (though not consistently replicated) for both extremes of CYP2D6 metabolic phenotype (i.e., PMs and UMs) to show an association with hyperprolactinemia (Troost et al., 2007; Roke et al., 2013; Youngster et al., 2014; Sukasem et al., 2016). Hyperprolactinemia has also been associated with the DRD2 Taq1A variant (Sukasem et al., 2018).
Interaction of known CYP2D6 inhibitors such as fluoxetine, bupropion, lamotrigine, sertraline, and citalopram are strongly correlated with the concentration of RIS in young male patients, compared to the available concentration of its metabolites (Calarge and Miller, 2011). A similar relationship has been described for thioridazine, and levomepromazine (Mannheimer et al., 2008). The same relationship was not found for duloxetine, another known CYP2D6 inhibitor (Hendset et al., 2010). Ishak et al. (2008) have described an association between RIS discontinuation caused by DDI from CYP2D6 inhibitors.
To a lesser extent, RIS metabolism is also mediated by CYP3A4 and DDI with inducers of this CYP enzyme are supported by the literature. Co-medication with armodafinil results in a decrease in plasma concentration of both RIS and 9OH-RIS (Darwish et al., 2015). The same relationship is true for rifampin (Mahatthanatrakul et al., 2007; Kim et al., 2008).
Olanzapine is an antipsychotic licensed for use in schizophrenia and related psychotic disorders and bipolar disorder. The main circulating metabolites are desmethylolanzapine and olanzapine-10-glucuronide (Callaghan et al., 1999; Erickson-Ridout et al., 2011; Lu et al., 2016). The conversion to desmethylolanzapine is predominantly catalyzed by CYP1A2, with lesser roles for CYP2D6, CYP2C8, and CYP2C19 (Ereshefsky, 1996; Callaghan et al., 1999; Korprasertthaworn et al., 2015; Okubo et al., 2016). Okubo and colleagues investigated the role of CYP1A2, CYP2D6, and FMO3 in individuals of varying CYP2D6 and FMO3 genotype (Okubo et al., 2016). Olanzapine N-demethylation and N-oxygenation were found to be catalyzed by CYP1A2 and CYP2D6, and by CYP2D6 and FMO3, respectively, in experiments using liver microsomes and recombinant enzymes. The effects on olanzapine oxidation activities of furafylline (which inhibits CYP1A2), quinidine (inhibits CYP2D6), and heat treatment (inhibits FMO3-mediated activities) were investigated. Each, and the combination of all three treatments suppressed the metabolic clearances of olanzapine by 28, 33, 25, and 85%, respectively. Using recombinant CYP2D6 enzymes CYP2D6.1 and CYP2D6.10, only the wild-type variant was capable of the 2-hydroxylation conversion of olanzapine into 2-hydroxymethyl-olanzapine; CYP2D6 appears to be the only enzyme catalyzing olanzapine 2-hydroxylation. Direct glucuronidation (at the 10 and 4 positions) is conducted by UGT1A4 and UGT2B10, with the UGT1A4∗3 and UGT2B10∗2 haplotypes being associated with increased and decreased glucuronidation, respectively (Erickson-Ridout et al., 2011).
Drug-drug (Drug Interactions Flockhart TableTM; DrugBank) interactions are of importance in the prescription of olanzapine. CYP1A2 is induced by smoking; the plasma concentration to dose ratio of olanzapine is therefore lower in smokers (Tsuda et al., 2014). Inhibition of CYP1A2 by fluvoxamine also increases the concentration to dose ratio (Chiu et al., 2004). CYP1A2 is also inhibited by estrogens; as a result, gender (clearance is reduced in women) and body fat content influence the metabolism of olanzapine (Ereshefsky, 1996; Callaghan et al., 1999). Valproic acid co-prescription results in a decrease in olanzapine concentration (reviewed by Vella and Mifsud, 2014). In contrast, protease inhibitors used in the treatment of HIV such as ritonavir in combination with fosamprenavir induce olanzapine metabolism (via CYP1A2 and/or UGT), leading to a recommendation to increase olanzapine dose by 50% when prescribed with such (Jacobs et al., 2014).
Quetiapine is another commonly prescribed antipsychotic. While literature supports CYP3A4 being the main enzyme in the quetiapine metabolic pathway, CYP2D6 is involved in the further metabolism of its principal metabolite, N-desalkylquetiapine. In an analysis of therapeutic drug monitoring data, patients from a Norwegian psychiatric hospital were genotyped for CYP2D6, CYP3A5, and ABCB1 (3435C>T) and the associations with dose-corrected serum concentrations of quetiapine and N-desalkylquetiapine were analyzed (Bakken et al., 2015). The mean dose-corrected serum concentration (C/D) of N-desalkylquetiapine was estimated to be 33 and 22% higher in CYP2D6 PMs (P = 0.03) and heterozygous CYP2D6 NMs (P = 0.001), respectively, compared with CYP2D6 NMs. There was no significant association with ABCB1 3435C>T polymorphism or CYP3A5 genotype.
Quetiapine has, however, been observed to have a serum level 2.5 times higher in those either heterozygous or homozygous for CYP3A4∗22 compared to those of CYP3A4 wild-type (van der Weide and van der Weide, 2014), with concentration to dose ratios that were 67% higher. The percentage of patients who had levels of quetiapine above the therapeutic range was also about five times higher in the ∗22 carrier group (16.1 versus 2.9%). Quetiapine serum levels based on reduced CYP3A4 metabolic activity were comparable to results found with coadministered CYP3A4 inhibitors, such as ketoconazole. The frequency of the CYP3A4∗22 haplotype is up to 10%.
In terms of DDI, valproate coadministration with quetiapine appears to result in a variable degree of increase in quetiapine plasma levels (Aichhorn et al., 2006; Winter et al., 2007), which may result in toxicity on occasion (Anderson and Fritz, 2000). In a review of therapeutic monitoring data from more than 2000 patients, it was reported that the following factors were associated with an increase in quetiapine concentration: age of at least 70 years, comedication with clozapine, fluvoxamine, and to a lesser extent citalopram/escitalopram, while, conversely, the following were associated with reduced quetiapine concentration: age under 18 years and comedication with carbamazepine or oxazepam, and to a lesser extent levomepromazine or lamotrigine (Castberg et al., 2007). The largest effect sizes were seen with fluvoxamine (+159%), clozapine (+82%), age at least 70 years (+67%), and carbamazepine (−86%). Another study found that dose-corrected quetiapine concentrations were approximately 60% lower in patients co-medicated with lamotrigine (Andersson et al., 2011).
Ziprasidone is a less commonly prescribed antipsychotic. Data indicate ziprasidone is mainly metabolized by glutathione and enzymatic reduction by aldehyde oxidase, followed by S-methylation to S-methyl-dihydroziprasidone by thiolmethyltransferase (Obach and Walsky, 2005). Approximately one third of its clearance is thought to be CYP3A4-dependent (Beedham et al., 2003). It is therefore subject to CYP3A-mediated induction (e.g., by carbamazepine, Miceli et al., 2000) and inhibition effects. As this medication has been associated with increases in the QTc interval (Aronow and Shamliyan, 2018), inhibition effects (e.g., by fluvoxamine or ketoconazole) should be avoided. Ziprasidone is also contraindicated in the presence of other medications that also prolong QTc (Kutcher et al., 2005; Table 4 in Hicks et al., 2017).
Tricyclic antidepressants (TCAs) and SSRIs both undergo first pass metabolism in the liver, with the CYP enzymes playing a prominent role in this. The cytochromes involved include CYP2D6, CYP2C19, CYP2C18, the CYP3A family, CYP1A2, CYP2C9, and CYP2B6, with the first two enzymes having a higher affinity for most antidepressants than the rest of the enzymes (Brosen, 1993; Koyama et al., 1997; Jann and Cohen, 2000).
Imipramine, the first TCA was derived from a phenothiazine, showing improvement without serious side effects in 500 patients with severe depression (Kuhn, 1958; Hillhouse and Porter, 2015). Although TCAs are still used (e.g., second-line or with somatic symptoms) to treat depression (Uher et al., 2009b; American Psychiatric Association, 2010; Kennedy et al., 2016), the treatment of pain (e.g., migraine, neuropathic, cancer-associated) is now their more common therapeutic use (Watson, 2000; Laird et al., 2008; Baltenberger et al., 2015).
Tricyclics include tertiary and secondary amines. A tertiary amine has a nitrogen bonded to three carbons, while in the case of a secondary amine, the nitrogen is bonded to only two carbons. The tricyclics amitriptyline, clomipramine, imipramine, trimipramine, doxepin, and dothiepin are tertiary amines. Tertiary amines are demethylated to secondary amines mainly by CYP2C19, but also by CYP1A2, CYP2C9, and CYP3A4, while both tertiary and secondary undergo parallel hydroxylation reactions mainly by CYP2D6, with CYP2C19 making a lesser contribution (Bertilsson et al., 2002; Aitchison, 2003; Bertilsson, 2007). The secondary amine metabolites of amitriptyline and imipramine are nortriptyline and desipramine, respectively, each also available as licensed medications.
Using hepatic microsomes of varying CYP2C19 activity and recombinant CYPs, Koyama et al. (1997) demonstrated that imipramine N-demethylation was catalyzed by CYP2C19 and CYP1A2 (high affinity and low affinity components, respectively), imipramine 2-hydroxylation was mediated by CYP2D6 and CYP2C19 (high affinity and low affinity components, respectively), and that in individuals deficient in CYP2C19, CYP1A2, and CYP2D6 play a major role in imipramine N-demethylation and 2-hydroxylation respectively. Among the recombinant human CYPs, CYP2C19, 2C18, 2D6, 1A2, 3A4, and 2B6 in rank order catalyzed the N-demethylation, whereas CYP2D6, 2C19, 1A2, 2C18, and 3A4 catalyzed the 2-hydroxylation. In a monoclonal antibody inhibition, Yang et al. (1999) concluded similarly that imipramine was metabolized to 2-hydroxyimipramine by 2C19 and 2D6, and to desipramine by 1A2, 2C18, 2C19, and 2D6, with the contributions of the isoforms to desipramine formation varying for 2C19 (13–50%), 1A2 (23–41%), and 3A4 (8–26%).
Tricyclic antidepressants inhibit presynaptic noradrenaline (also known as norepinephrine) and serotonin reuptake via the noradrenaline and serotonin transporters, respectively, with the tertiary amines having a greater affinity for the serotonin transporter than the secondary amines, which are relatively selective for the noradrenaline transporter (Owens, 1996). The tertiary amines are therefore dual SNRIs, while the secondary amines are noradrenaline reuptake inhibitors (or NARIs). There are also contrasts in their CYP inhibition. Tertiary amines TCAs (e.g., amitriptyline, imipramine) inhibit CYP2C19 (estimated Ki of 37.7 and 56.8 μM, respectively). By contrast, the secondary amines show negligible CYP2C19 inhibition activity (Shin et al., 2002) but inhibit CYP2D6 slightly more than tertiary amine TCAs; for example, estimated Ki values for the tertiary amine TCAs amitriptyline and imipramine are 31.0, 28.6 μM, respectively, with Ki s for nortriptyline and desipramine being 7.9 and 12.5 (Shin et al., 2002). Although therapeutic plasma concentrations are less than 1 μM (Yau et al., 2007; Aitchison et al., 2010), cerebral concentrations may be higher (Weigmann et al., 2000; Aitchison et al., 2010). Further, this differential may be affected by factors such as p-gp expression at the blood–brain barrier. It is therefore possible that with repeated dosing, as the concentration of a tertiary amine TCA increases in the brain, that the level of CYP2C19 inhibition increases, and that this leads to reduction in the demethylation reaction centrally. This would be expected to be associated with a greater degree of dual reuptake inhibition and may at least partly explain the clinical observation of time for antidepressant effect to maximize. This hypothesis is consistent with a report of an inverse relationship between CYP2C19 activity and response to TCAs (mainly tertiary amines, Aitchison et al., under revision).
There are also contrasts between the tertiary and secondary amines and side effect/adverse drug reaction potential. The side effects are associated with antagonism at the following receptors: adrenergic α1 and α2 receptors, muscarinic (cholinergic) receptors, and histamine H1 receptors (Cusack et al., 1994; Owens et al., 1997; Sanchez and Hyttel, 1999). Specifically, blockade of muscarinic receptors in the parasympathetic nervous system is associated with dry mouth, blurred vision, constipation, urinary retention, and if at toxic levels, delirium; alpha adrenergic receptor antagonism is associated with orthostatic hypotension; and histamine H1 receptor blockade with sedation and weight gain. Other side effects (such as palpitations, vertigo, sweating, tremors, and interference with sexual function) may also occur (Asberg et al., 1970; Ziegler et al., 1978; Uher et al., 2009a; Hodgson et al., 2015) and may represent more than one pharmacodynamic mechanism. The tertiary amine TCAs have greater cholinergic receptor binding than the secondary amines, which in turn have greater affinity than the hydroxylated metabolites (Rudorfer and Potter, 1999). Some effects may be related to specific metabolites (e.g., N-methyl quaternary ammonium derivatives of amitriptyline, doxepin, and imipramine are antagonists at both central nervous system and cardiac muscarinic receptors) (Ehlert et al., 1990). Hydroxylated metabolite concentration has been associated with increases QTc interval (Schneider et al., 1988; Stern et al., 1991). It is therefore possible that CYP2D6 UM might have elevated hydroxy-metabolite plasma concentrations (Bertilsson et al., 1985) resulting in an increased risk of cardiotoxicity. Moreover, therapeutic drug monitoring does not usually include measuring hydroxylated metabolite plasma concentrations. In the case of a combination of CYP2C19 poor metabolizer status and CYP2D6 UM status, it might be advisable to avoid TCA prescription; this is in fact the CPIC recommendation for this combination [Table 4 in Hicks et al. (2017)].
The association between CYP2D6 and CYP2C19 genotype and clinical response to TCAs (treatment efficacy and/or side effects) has been reviewed (Hicks et al., 2017). In brief, studies support the existence of a concentration–effect relationship for TCAs and/or their active metabolites (Ziegler et al., 1977; Sjöqvist et al., 1980; Gram et al., 1984; Perry et al., 1987) (Preskorn and Jerkovich, 1990). In an early report, high concentrations of nortriptyline were linked to adverse effects, with decreased antidepressant effect (Asberg et al., 1970). Concentration-dependent side effects have been observed in individuals deficient in CYP2D6 when treated with usual doses of TCAs from accumulation of the parent drug and/or active metabolites (Bertilsson et al., 1981; Sjöqvist and Bertilsson, 1984; Balant-Gorgia et al., 1989). Ethnic groups with a higher frequency of CYP2D6 IM alleles achieve higher levels of TCAs than Whites and have a faster rate of recovery from depressive episodes (Raskin and Crook, 1975; Ziegler and Biggs, 1977; Rudorfer and Robins, 1982). An excess of CYP2D6 PM alleles has been found in amongst patients with a history of adverse reactions to TCAs and relevant SSRIs (Chen et al., 1996). CYP2D6 PMs have high levels of desipramine, associated with adverse effects necessitating dose reduction (Spina et al., 1997). An inverse correlation between the frequency of adverse drug events and number of functional CYP2D6 genes has been found, including patients on TCAs (Chou et al., 2003). Studies published since the Hicks et al. (2017) review include that of Hodgson et al. (2015).
There are guidelines by both CPIC and DPWG (for a comparison, see Bank et al., 2018). CPIC guidelines for CYP2D6 are as follows: for both PMs and UMs, it is suggested to avoid use of TCAs due to possible side effects or subthreshold concentrations, respectively. In both cases, when TCAs are still prescribed, therapeutic drug monitoring is recommended, with PMs starting at 50% regular dosage and for UMs consideration being given to use TDM to titrate up to a higher target dose. The DPWG provide specific suggested increases in the starting dosages for amitriptyline, clomipramine, doxepin, imipramine, and nortriptyline of 125, 150, 200, 170%, respectively followed by TDM (Bank et al., 2018). For CYP2D6 IMs, a 25% reduction in the initial dose is recommended by CPIC. For many drugs, evidence is still accumulating, and therefore implementation of the recommendations is “optional,” or at prescriber discretion.
In regard to CYP2C19 status, there are CPIC guidelines for the tertiary amines amitriptyline, clomipramine, doxepin, imipramine, and trimipramine. For CYP2D6 UMs, RMs, or PMs, CPIC provides an optional recommendation to substitute with medications not metabolized by CYP2C19, such as nortriptyline or desipramine. In the case of CYPC19 PMs, a 50% decrease in initiation dose is suggested, with TDM to guide titration (Hicks et al., 2017).
There are also CPIC guidelines for amitriptyline where both CYP2D6 and CYP2C19 data are available. If an individual is a CYP2D6 or CYP2C19 PM and a NM for the other enzyme, it is recommended to avoid the medication or, if warranted, consider a 50% decrease in initiation dose; for a CYP2D6 UM and CYP2C19 NM, it is recommended to avoid the medication or, if warranted, consider titrating to a higher target dose (compared to CYP2D6 NMs); and for a CYP2D6 IM and CYP2C19 NM, to consider a 25% decrease in initiation dose ((Hicks et al., 2017). No adjustments in dosage is necessary for those who are NMs for CYP2D6 and CYP2C19, or an NM for CYP2D6 and an IM for CYP2C19 (Hicks et al., 2017).
Mirtazapine acts as antagonist at adrenergic α2-autoreceptors and α2-heteroreceptors as well as at 5-HT2 and 5-HT3 receptors (Anttila and Leinonen, 2001). The α2-autoreceptor blockade leads to increased release of noradrenaline while the blockade of α2-heteroreceptor on serotonergic neurons increases serotonin release. Owing to antagonism of 5-HT2 and 5-HT3, transmission is enhanced at only 5-HT1A (and related receptors). It is a racemic mixture of R(−) and S(+)-enantiomers, with effects on heart rate and blood pressure correlating more strongly with R (−) than with S (+) concentration, and sedation being associated with both enantiomers (Brockmöller et al., 2007). The main metabolic pathway for mirtazapine is 8-hydroxylation, catalyzed by mainly by CYP2D6 (65%) at low concentrations, reducing to 20% at higher concentrations, where CYP1A2 (50%), CYP3A4 (20%), and CYP2C9 (10%) contribute more (Dahl et al., 1997; Störmer et al., 2000). Other metabolic pathways are N-demethylation and N-oxidation. The former is conducted by mainly by CYP3A4 (50–70%), with CYP1A2 (50% at low concentrations, 5% at high concentrations), CYP2C8 (<20%), and CYP2C9 (<5%) also contributing. N-oxidation is catalyzed by CYP1A2 and CYP3A4, with the former playing a larger role (80%) at low concentrations and the latter being responsible for a greater proportion (85%) of the reaction at higher drug concentrations (Dahl et al., 1997; Störmer et al., 2000). Enzyme polymorphism may additionally affect the relative contributions of these CYPs.
The maximum concentration and area under the curve are greater in females as compared to males (Timmer et al., 2000; Sennef et al., 2003). In non-smokers and at lower concentrations of mirtazapine, CYP2D6 genotype affects the plasma levels and clearance of the S-enantiomer and its metabolites (Störmer et al., 2000; Brockmöller et al., 2007; Sirot et al., 2012; Hayashi et al., 2015). At higher concentrations (250 μM), CYP3A4 contributes to about 70%, while CYP2D6, CYP2C8, CYP2C9, and CYP1A2 each account for less than 15% of its metabolism (Störmer et al., 2000). Unlike the tricyclics, there is no clear relationship between mirtazapine plasma concentration and its efficacy. While an increase in the maximal serum concentration for coadministered amitriptyline has been described (Sennef et al., 2003), the overall inhibitory effect of mirtazapine on CYPs is not thought to be clinically significant (Anttila and Leinonen, 2001; Spina et al., 2008). In the Sennef et al. (2003) study, coadministered amitriptyline increased the maximum concentration of mirtazapine (by 36%) in only males. S-hydroxymirtazapine concentration has been reported as being elevated in individuals of CYP2B6∗6/∗6 genotype (Sirot et al., 2012). As yet, there are no DPWG or CPIC guidelines for mirtazapine based on genotype.
The second SSRI to be synthesized, fluoxetine was the first SSRI to enter widespread use (Wong et al., 1974, 1975). Selective serotonin reuptake inhibitors are now widely used to treat depression, escitalopram having the highest affinity for the serotonin transporter (Owens et al., 2001) and being an allosteric modulator, one molecule increasing the binding of a second at this target.
In brief, SSRIs are partly metabolized by CYP2D6 (Brosen, 1993). Demethylenation is the initial step of paroxetine metabolism (an SSRI), primarily conducted by CYP2D6 (a high affinity saturable process,Bloomer et al., 1992). Further conjugation of paroxetine results in glucuronide and sulfate conjugated metabolites (Haddock et al., 1989). Paroxetine is a potent competitive inhibitor of CYP2D6 (Bourin et al., 2001) nonetheless, differences in steady-state plasma concentration of paroxetine by CYP2D6 phenotype are seen (Gex-Fabry et al., 2008). Higher doses of paroxetine (e.g., 30 mg) are associated with a greater degree of CYP2D6 inhibition (Findling et al., 2006). In diabetic neuropathy, paroxetine has an analgesic effect, plasma concentrations greater than 300–400 nmol/l being associated with optimal response (Sindrup et al., 1990, 1991).
Fluoxetine is a racemic mixture of S(+) and R(−)-fluoxetine, with the former being metabolized predominantly by CYP2D6 to S-norfluoxetine and the latter by CYP2D6 and CYP2C9 to R-norfluoxetine; CYP3A4 and CYP2C19 make minor contributions to this demethylation reaction (Hicks et al., 2015). R/S-fluoxetine and S-norfluoxetine are all potent SSRIs, with R-norfluoxetine being 20 times less potent (Hicks et al., 2015). The strength of CYP2D6 inhibition for SSRIs is as follows in reducing order: paroxetine, fluoxetine, norfluoxetine, desmethylcitalopram, fluvoxamine, sertraline, citalopram (Baumann and Rochat, 1995). Although fluoxetine is less potent as an inhibitor of CYP2D6 than paroxetine, owing to its substantially longer half-life – 1–3 days and 4–6 days after acute and chronic administration, respectively, with the corresponding values being 4 and 16 days for norfluoxetine,18 inhibition effects may endure for weeks to months after multiple dosing (Liston et al., 2002). Fluoxetine and sertraline also inhibit CYP2C19 (Bertilsson and Dahl, 1996) while norfluoxetine is a moderate CYP3A4 inhibitor (Hemeryck and Belpaire, 2002).
The primary route of metabolism for citalopram (a racemic mixture of the R- and S-enantiomers of citalopram) and escitalopram (S-citalopram) is N-demethylation by CYP2C19, CYP2D6, and CYP3A4 (Sindrup et al., 1993; Kobayashi et al., 1997; Rochat et al., 1997; von Moltke et al., 2001). CYP2D6 then conducts the N-demethylation of N-desmethylescitalopram to N-didesmethylescitalopram (von Moltke et al., 2001). The medication and its metabolites may inhibit enzymes: citalopram and R- or S-desmethylcitalopram are weak inhibitors of CYP2C19, while R- and S-didesmethylcitalopram are moderate inhibitors, with mean IC50 values of 18.7 and 12.1 μM, respectively. S-citalopram and S-desmethylcitalopram are weak inhibitors of CYP2D6 (IC50 = 70–80 μM); R-desmethylcitalopram shows stronger inhibition at this enzyme (IC50 25.5 ± 2.1 μM) (von Moltke et al., 2001). Fluvoxamine is predominantly a CYP1A2 inhibitor (Christensen et al., 2002) but also inhibits other CYPs including the CYP3As (Hemeryck and Belpaire, 2002).
SSRIs are more 20 to 1500-fold more selective for inhibiting serotonin than noradrenaline. They do not stimulate the release of serotonin or norepinephrine presynaptically (Rothman et al., 2001) and have weak/no-direct pharmacological action at postsynaptic serotonin receptors (e.g., 5-HT1A, 5-HT2A, and 5-HT2C) (Thomas et al., 1987; Owens et al., 1997; Sanchez and Hyttel, 1999), and minimal binding affinity for other postsynaptic receptors (adrenergic α1, α2, and β, histamine H1, muscarinic, and dopamine D2 receptors) (Thomas et al., 1987; Owens et al., 1997).
Associations between SSRI phenotypes (concentrations, efficacy, tolerability) and CYP2D6 and CYP2C19 genotypes are provided in Supplementary Tables S7–S11 in Hicks et al. (2015). In a meta-analysis of the main functional CYP2C19 variants in Whites (the CYP2C19∗2 and the CYP2C19∗17, plus wild-type by exclusion of these) for individuals treated with citalopram or escitalopram (in the GENDEP, STAR∗D, GenPod, and PGRN-AMPS studies), CYP2C19 PMs had greater symptom improvement and higher remission rates compared to NMs (Fabbri et al., 2018). This is consistent with earlier data indicating that CYP2C19 PMs respond better to escitalopram if treatment is tolerated (Mrazek et al., 2011). At weeks 2–4, PMs showed increased risk of side effects (Fabbri et al., 2018). In a retrospective analysis of data from 2087 patients treated with escitalopram and genotyped for CYP2C19, PMs had an increase in exposure and a higher rate of treatment dropout compared with CYP2C19 NMs (Jukic et al., 2018). Conversely, the CYP2C19∗17 haplotype was associated with an increase in CYP2C19 activity by approximately 20%, with those of CYP2C19∗1/∗17 and CYP2C19∗17/∗17 genotype showing a 50% increase in treatment failure rate compared with NMs (Jukic et al., 2018). Moreover, replicated findings that CYP2C19 UMs treated with escitalopram exhibit increased suicidal ideation (Jukic et al., 2017; Rahikainen et al., 2019) indicates that distinguishing between CYP2C19 NMs and UMs is clinically relevant for the escitalopram treatment. Shelton et al. (2020) using a combinatorial PGx algorithm (covering several different genes) reported a significant association with variation in the metabolism of escitalopram/citalopram.
The CPIC guidelines for SSRIs (Hicks et al., 2015) cover two medications for CYP2D6: paroxetine and fluvoxamine. The recommendation for paroxetine in the case of CYP2D6 UMs is to select an alternative drug and likewise for PMs, with implementation being optional for the latter. For fluvoxamine, in the case of CYP2D6 UMs there was insufficient data for a recommendation, with an optional recommendation to consider a 25–50% reduction in the starting dose for CYP2D6 PMs, and titrate to response, or consider using an alternative medication not metabolized by CYP2D6.
Three medications are included in the CPIC SSRI guidelines in regard to CYP2C19: citalopram, escitalopram and sertraline. A 50% reduction of the standard dosage for the three drugs is recommended for PM status, with and titration to response, or considering using an alternative medication not metabolized by CYP2D6 (strength of the recommendation being moderate for citalopram and escitalopram and moderate for sertraline). For CYP2C19 UMs, for citalopram and escitalopram, selection of a medication not metabolized by CYP2C19 is recommended, while for sertraline, initiation at the normal dose may be tried, with substitution being considered if patients do not respond to treatment. The recommendations are classified as moderate for citalopram/escitalopram and optional for sertraline (Hicks et al., 2015).
In addition to the CPIC guidelines, Stingl et al. (2013) suggest that in the case of fluoxetine (not included in the guidelines above), due to its role as both substrate and inhibitor of CYP2D6, physicians should be careful if co-prescribing it with other CYP2D6 substrates.
Venlafaxine is a SNRI, which means that like the tertiary amine tricyclics, it inhibits neurotransmitter reuptake at both the serotonin and the noradrenaline (also known as epinephrine) transporters The major metabolic route for venlafaxine is O-demethylation, which is mediated very specifically by CYP2D6 to an active metabolite, O-desmethylvenlafaxine (Otton et al., 1996). The N-demethylation is conducted by CYP3A4 and CYP2C19 (Sangkuhl et al., 2014). This means that the ratio of the O- and N-demethylated metabolites of venlafaxine may in fact be used as a biomarker of CYP2D6 activity, predicting CYP2D6 poor metabolizers with a specificity and sensitivity of >85% (Mannheimer et al., 2016). In in vitro studies, venlafaxine is a weaker inhibitor of CYP2D6 than are the SSRIs paroxetine, fluoxetine, fluvoxamine, and sertraline, and has minimal or no effect on CYP1A2, CYP2C9, and CYP3A4 (Ball et al., 1997; von Moltke et al., 1997). In a study of 1003 Scandinavians (mostly White), it was found that CYP2D6 metabolism measured as the O/N-desmethylvenlafaxine ratio was significantly lower in carriers of CYP2D6∗41 vs. CYP2D6∗9–10 (Jukic et al., 2019). The annotated DPWG guideline states that for CYP2D6 poor (PM) and intermediate metabolizers (IM), select an alternative to venlafaxine or reduce the dose and monitor patient’s plasma metabolite level; for CYP2D6 ultrarapid metabolizers (UM), increase dose to 150% of the normal dose or select an alternative to venlafaxine.19
Duloxetine acts as a serotonin and noradrenaline reuptake inhibitor, and a weak dopamine reuptake inhibitor (e.g., in the frontal cortex).20 CYP1A2 and to a lesser extent CYP2D6 convert duloxetine into its main metabolites 4-hydroxy and 5-hydroxy duloxetine; activity (Knadler et al., 2011). CYP1A2 inducers including cigarette smoke therefore result in a reduction in duloxetine concentration (Augustin et al., 2018).
Atomoxetine is a noradrenaline reuptake inhibitor used as second-line in the treatment of ADHD. It is metabolized mainly by CYP2D6 to 4-hydroxyatomoxetine and by CYP2C19 to N-desmethylatomoxetine, which is subsequently metabolized via CYP2D6 to N-desmethyl-4-hydroxyatomoxetine (Atomoxetine). Other enzymes (CYP1A2, CYP2B6, CYP2C19, CYP3A4, and CYP2E1) also contribute to the hydroxylation, with glucuronidation occurring subsequently (Yu et al., 2016).21 Atomoxetine may take 2-4 weeks for its full effect to be seen (Savill et al., 2015); peak concentrations have been associated with treatment efficacy and CYP2D6 genotype has been associated with both peak concentration and half-life (e.g., exposure is on average 10-fold greater in CYP2D6 PMs; reviewed in the CPIC atomoxetine guidelines, Brown et al., 2019). Individuals homozygous for the CYP2D6∗10 haplotype show a 5-fold higher peak atomoxetine concentration compared with individuals with at least one normal function haplotype; individuals heterozygous for the CYP2D6∗10 also had higher atomoxetine exposure compared with CYP2D6 NMs (Cui et al., 2007; Matsui et al., 2012; Byeon et al., 2015).
The initiation dose for children and adolescents 0.5mg/kg/day. For UMs, NMs and IMs without a CYP2D6∗10, after three days an increase in dose to 1.2mg/kg/day is recommended. At the two week point, if there is neither efficacy nor adverse events, measurement of peak concentration 1–2 h after dose should be considered, and should this be less than 200 ng/ml, the dose may be increased until the concentration reaches 400 ng/ml. For those with an activity score of 0 (PMs), 0.5–1.0 (IMs including those with a CYP2D6∗10) the recommendation is that if there is neither efficacy nor adverse events by two weeks, to consider measuring plasma concentration 2–4 h (4 h for PMs) after dosing; if response is inadequate and the concentration is less than 200 ng/ml, the dose may be increased to approach 400 ng/ml; while if unacceptable side effects are present at any time, a reduction in dose should be considered. Of note, while the strength of the evidence for IMs, NMs, and UMs is moderate, for PMs, it is strong (Brown et al., 2019).
For adults, the starting dose is 40 mg/day. For UMs, NMs and IMs without a CYP2D6∗10, the dose should be increased to 80 mg/day after three days; if there is neither efficacy nor adverse events at two weeks, it is recommended to consider increasing the dose to 100 mg/day. After a further two weeks, if there is no clinical response, measurement of peak concentration 1–2 h after dose should be considered, and should this be less than 200 ng/ml, the dose may be increased until the concentration reaches 400 ng/ml. Doses above 100 mg/day may be needed to achieve target concentrations. For those with an activity score of 0, or 0.5–1.0 (IMs including those with a CYP2D6∗10), the recommendation is that if there is neither efficacy nor adverse events, at two weeks increase the dose to 80 mg/day. If resultant efficacy is inadequate, consideration should be given to measuring plasma concentration 2–4 h (4 h for PMs) after dosing; if the concentration is less than 200 ng/ml, the dose may be increased to approach 400 ng/ml; while if unacceptable side effects are present at any time, a reduction in dose should be considered.
To date one paper shows an association between CYP2C19 and atomoxetine pharmacokinetics, with PMs showing higher atomoxetine concentration and half-life, and with correspondingly lower values for N-desmethylatomoxetine (Choi et al., 2014). Replication of this is required before any guidelines result.
Many genetic variants in drug metabolizing enzymes and transporters have been shown to be relevant for psychiatry. Associations are strong enough to feature on drug labels and for prescribing guidelines based on such data (CPIC; DPWG). The International Society of Psychiatric Genetics recommends HLA-A and HLA-B testing prior to use of carbamazepine and oxcarbazepine, and suggests that genetic information for CYP2C19 and CYP2D6 would be likely to be most beneficial for individuals who have experienced insufficient efficacy or an adverse reaction to a previously tried antidepressant or antipsychotic (International Society for Psychiatric Genetics [ISPG], 2019). A range of (non-validated) commercial tests are currently available; however, there is variability in included genetic variants, methodology, and interpretation. This variability presents challenges for clinicians and other end users. Maruf et al. (2020) suggest the following should be considered: (a) whether or not the lab is accredited; (b) the relevance of the genetic variants to the medications of interest; (c) test logistics (such as turnaround time). With genes such as CYP2D6 that are particularly challenging, a pragmatic approach may need to be taken: balancing a desire for a fast turnaround in a clinically accredited laboratory with a comprehensive coverage of all relevant functional variants.
While considerable progress has been made in determining reference samples by Gaedigk et al. (2019), what is still required is a consensus regarding the minimum set of informative variants in relevant genes that should be genotyped, methodologies for genotyping these efficiently and in a validated manner, and standardized interpretation with reporting algorithms and decision-support tools that can be integrated into electronic medical records. In addition, there has been relatively little work to date clinical associations with genetic variants in more than one gene (amitriptyline in Hicks et al., 2017; Greden et al., 2019, Aitchison et al., in submission). Depression was predicted to be responsible for the greatest global burden of disease by 2030 (Malhi and Mann, 2018) and in fact, given the evidence of increasing prevalence in association with the current viral pandemic (Holmes et al., 2020; Galea et al., 2020), this may be an underestimate. Depression is the mental health condition with the most prescribing guidelines in association with gene–drug pairs. Pharmacogenetically informed care has the potential to enhance treatment efficacy and reduce ADRs for this common disorder associated with not only the type of health burdens previously measured but also with a negative impact on outcomes from other health conditions ranging from cardiovascular to infectious diseases. Further, pharmacogenetics may not only reduce the risk of undesirable drug-drug interactions but may also in fact inform the utility of drug-drug interactions that may have a beneficial therapeutic effect – such as the induction of expression of ABCB1 (Tian et al., 2005) (which may be associated with viral resistance). Significant ground in this area has been covered to date, but much remains to be covered. For example, ABCB1 would appear to be the CYP2D6 equivalent of drug transporters and is largely uncharted territory in terms of specific genotype–phenotype relationships by substrate binding including cooperativity, inhibition, and induction.
All authors contributed to manuscript drafting (each citation being reviewed by at least two authors), and approved the final version for publication.
MC was funded by a University of Alberta Office of the Provost and VP (Academic) Summer Student Award). BCH, VY, and JH were funded by an Alberta Innovates Strategic Research Project: SRP51_PRIME – Pharmacogenomics for the Prevention of Adverse Drug Reactions in mental health (PI KJA, Co-PI Chad Bousman), grant agreement number G2018000868.
KA is a member of the Pharmacogene Variation Consortium, Clinical Pharmacogenetics Implementation Consortium, has received two research grants in the last two years from Janssen Inc., Canada (fellowship grants for trainees) and provided consultancy services (unpaid) for HLS Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Glen Baker for his helpful comments on some draft text for one section of the manuscript and Keanna Wallace for some proofreading and referencing.
Abernethy, D. R., and Todd, E. L. (1985). Impairment of caffeine clearance by chronic use of low-dose oestrogen-containing oral contraceptives. Eur. J. Clin. Pharmacol. 28, 425–428. doi: 10.1007/bf00544361
Abilify - aripiprazole tablet (2016). Abilify - aripiprazole tablet. URL: https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=c040bd1d-45b7-49f2-93ea-aed7220b30ac
Abilify 10 mg tablets. Electronic Medicines Compendium. Retrieved from https://www.medicines.org.uk/emc/product/7969/smpc
Abilify. Summary of Product Characteristics, European Medicines Agency Retrieved from https://www.ema.europa.eu/documents/product-information/abilify-epar-product-information_en.pdf
Adali, O., Carver, G. C., and Philpot, R. M. (1998). Modulation of human flavin-containing monooxygenase 3 activity by tricyclic antidepressants and other agents: importance of residue 428. Arch. Biochem. Biophys. 358, 92–97. doi: 10.1006/abbi.1998.0835
Ahmed, I., Dagincourt, P. G., Miller, L. G., and Shader, R. I. (1993). Possible interaction between fluoxetine and pimozide causing sinus bradycardia. Can. J. Psychiatry 38, 62–63. doi: 10.1177/070674379303800116
Aichhorn, W., Marksteiner, J., Walch, T., Zernig, G., Saria, A., and Kemmler, G. (2006). Influence of age, gender, body weight and valproate comedication on quetiapine plasma concentrations. Int. Clin. Psychopharmacol. 21, 81–85. doi: 10.1097/01.yic.0000188213.46667.f1
Aitchison, K. J. (2003). Pharmacogenetic studies of CYP2D6, CYP2C19, and CYP1A2, and investigation of their role in clinical response to antipsychotics and antidepressants. Ph. D Thesis (London, UK: King’s College London).
Aitchison, K. J., Bienroth, M., Cookson, J., Gray, R., Haddad, P. M., Moore, B., et al. (2009). A UK consensus on the administration of aripiprazole for the treatment of mania. J. Psychopharmacol. 23, 231–240. doi: 10.1177/0269881108098820
Aitchison, K. J., Datla, K., Rooprai, H., Fernando, J., and Dexter, D. (2010). Regional distribution of clomipramine and desmethylclomipramine in rat brain and peripheral organs on chronic clomipramine administration. J. Psychopharmacol. 24, 1261–1268. doi: 10.1177/0269881109105789
Aitchison, K. J., Gonzalez, F. J., Quattrochi, L. C., Sapone, A., Zhao, J. H., Zaher, H., et al. (2000a). Identification of novel polymorphisms in the 5’ flanking region of CYP1A2, characterization of interethnic variability, and investigation of their functional significance. Pharmacogenetics 10, 695–704. doi: 10.1097/00008571-200011000-00004
Aitchison, K. J., Jann, M. W., Zhao, J. H., Sakai, T., Zaher, H., Wolff, K., et al. (2000b). Clozapine pharmacokinetics and pharmacodynamics studied with Cyp1A2-null mice. J. Psychopharmacol. 14, 353–359. doi: 10.1177/026988110001400403
Aitchison, K. J., Jordan, B. D., and Sharma, T. (2000c). The relevance of ethnic influences on pharmacogenetics to the treatment of psychosis. Drug Metabol. Drug Interact. 16, 15–38.
Aitchison, K. J., Munro, J., Wright, P., Smith, S., Makoff, A. J., Sachse, C., et al. (1999). Failure to respond to treatment with typical antipsychotics is not associated with CYP2D6 ultrarapid hydroxylation. Br. J. Clin. Pharmacol. 48, 388–394. doi: 10.1046/j.1365-2125.1999.00006.x
Akaba, K., Kimura, T., Sasaki, A., Tanabe, S., Ikegami, T., Hashimoto, M., et al. (1998). Neonatal hyperbilirubinemia and mutation of the bilirubin uridine diphosphate-glucuronosyltransferase gene: a common missense mutation among Japanese, Koreans and Chinese. Biochem. Mole. Biol. Int. 46, 21–26. doi: 10.1080/15216549800203512
Aklillu, E., Carrillo, J. A., Makonnen, E., Bertilsson, L., and Ingelman-Sundberg, M. (2003). Xanthine oxidase activity is influenced by environmental factors in Ethiopians. Eur. J. Clin. Pharmacol. 59, 533–536. doi: 10.1007/s00228-003-0653-8
Aklillu, E., Persson, I., Bertilsson, L., Johansson, I., Rodrigues, F., and Ingelman-Sundberg, M. (1996). Frequent distribution of ultrarapid metabolizers of debrisoquine in an ethiopian population carrying duplicated and multiduplicated functional CYP2D6 alleles. J. Pharmacol. Exp. Ther. 278, 441–446.
Aller, S. G., Yu, J., Ward, A., Weng, Y., Chittaboina, S., Zhuo, R., et al. (2009). Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722. doi: 10.1126/science.1168750
Allorge, D., Chevalier, D., Lo-Guidice, J. M., Cauffiez, C., Suard, F., Baumann, P., et al. (2003). Identification of a novel splice-site mutation in the CYP1A2 gene. Br. J. Clin. Pharmacol. 56, 341–344. doi: 10.1046/j.1365-2125.2003.01858.x
Altman, R. B., Whirl-Carrillo, M., and Klein, T. E. (2013). Challenges in the pharmacogenomic annotation of whole genomes. Clin. Pharmacol. Ther. 94, 211–213. doi: 10.1038/clpt.2013.111
Amare, A. T., Schubert, K. O., and Baune, B. T. (2017). Pharmacogenomics in the treatment of mood disorders: Strategies and Opportunities for personalized psychiatry. EPMA J. 8, 211–227. doi: 10.1007/s13167-017-0112-8
American Psychiatric Association. (2010). Practice Guideline for the Treatment of Patients With Major Depressive Disorder 3rd edn. Washington: American Psychiatric Association.
Anderson, D. T., and Fritz, K. L. (2000). Quetiapine (Seroquel) concentrations in seven postmortem cases. J. Analyt. Toxicol. 24, 300–304. doi: 10.1093/jat/24.4.300
Andersson, M. L., Björkhem-Bergman, L., and Lindh, J. D. (2011). Possible drug-drug interaction between quetiapine and lamotrigine–evidence from a Swedish TDM database. Br. J. Clin. Pharmacol. 72, 153–156. doi: 10.1111/j.1365-2125.2011.03941.x
Anttila, S. A., and Leinonen, E. V. (2001). A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev. 7, 249–264. doi: 10.1111/j.1527-3458.2001.tb00198.x
Arici, M., and Özhan, G. (2017). The genetic profiles of CYP1A1, CYP1A2 and CYP2E1 enzymes as susceptibility factor in xenobiotic toxicity in Turkish population. Saudi Pharm. J. 25, 294–297. doi: 10.1016/j.jsps.2016.06.001
Aronow, W. S., and Shamliyan, T. A. (2018). Effects of atypical antipsychotic drugs on QT interval in patients with mental disorders. Ann. Transl. Med. 6:147. doi: 10.21037/atm.2018.03.17
Asberg, M., Cronholm, B., Sjöqvist, F., and Tuck, D. (1970). Correlation of subjective side effects with plasma concentrations of nortriptyline. Br. Med. J. 4, 18–21. doi: 10.1136/bmj.4.5726.18
Assenat, E., Gerbal-Chaloin, S., Larrey, D., Saric, J., Fabre, J. M., Maurel, P., et al. (2004). Interleukin 1beta inhibits CAR-induced expression of hepatic genes involved in drug and bilirubin clearance. Hepatology 40, 951–960. doi: 10.1002/hep.20387
Atomoxetine. Retrieved from https://www.pharmgkb.org/chemical/PA134688071
Augustin, M., Schoretsanitis, G., Hiemke, C., Gründer, G., Haen, E., and Paulzen, M. (2018). Differences in Duloxetine Dosing Strategies in Smoking and Nonsmoking Patients: Therapeutic Drug Monitoring Uncovers the Impact on Drug Metabolism. J. Clin. Psychiatry 79:17m12086. doi: 10.4088/JCP.17m12086
Axelrod, J. (1957). O-Methylation of Epinephrine and Other Catechols Invitro and Invivo. Science 126, 400–401. doi: 10.1007/Bf01227268
Ayesh, R., Mitchell, S. C., Zhang, A., and Smith, R. L. (1993). The fish odour syndrome: biochemical, familial, and clinical aspects. BMJ 307, 655–657. doi: 10.1136/bmj.307.6905.655
Bajpai, M., Roskos, L. K., Shen, D. D., and Levy, R. H. (1996). Roles of cytochrome P4502C9 and cytochrome P4502C19 in the stereoselective metabolism of phenytoin to its major metabolite. Drug Metab. Dispos. 24, 1401–1403.
Bakken, G. V., Molden, E., and Hermann, M. (2015). Impact of genetic variability in CYP2D6, CYP3A5, and ABCB1 on serum concentrations of quetiapine and N-desalkylquetiapine in psychiatric patients. Ther. Drug Monit. 37, 256–261. doi: 10.1097/FTD.0000000000000135
Balant-Gorgia, A. E., Balant, L. P., and Garrone, G. (1989). High blood concentrations of imipramine or clomipramine and therapeutic failure: a case report study using drug monitoring data. Ther. Drug Monit. 11, 415–420. doi: 10.1097/00007691-198911040-00008
Ball, S. E., Ahern, D., Scatina, J., and Kao, J. (1997). Venlafaxine: in vitro inhibition of CYP2D6 dependent imipramine and desipramine metabolism; comparative studies with selected SSRIs, and effects on human hepatic CYP3A4, CYP2C9 and CYP1A2. Br. J. Clin. Pharmacol. 43, 619–626. doi: 10.1046/j.1365-2125.1997.00591.x
Baltenberger, E. P., Buterbaugh, W. M., Martin, S., and Thomas, A. J. (2015). Review of antidepressants in the treatment of neuropathic pain. Mental Health Clin. 5, 123–133. doi: 10.9740/mhc.2015.05.123
Bank, P. C. D., Caudle, K. E., Swen, J. J., Gammal, R. S., Whirl-Carrillo, M., Klein, T. E., et al. (2018). Comparison of the Guidelines of the Clinical Pharmacogenetics Implementation Consortium and the Dutch Pharmacogenetics Working Group. Clin. Pharmacol. Ther. 103, 599–618. doi: 10.1002/cpt.762
Barbarino, J. M., Haidar, C. E., Klein, T. E., and Altman, R. B. (2014). PharmGKB summary: very important pharmacogene information for UGT1A1. Pharmacogen. Genom. 24, 177–183. doi: 10.1097/FPC.0000000000000024
Baririan, N., Horsmans, Y., Desager, J. P., Verbeeck, R., Vanbinst, R., Wallemacq, P., et al. (2005). Alfentanil-induced miosis clearance as a liver CYP3A4 and 3A5 activity measure in healthy volunteers: improvement of experimental conditions. J. Clin. Pharmacol. 45, 1434–1441. doi: 10.1177/0091270005282629
Basu, A., Pereira, J., and Aitchison, K. J. (2007). “The pharmacological management of schizophrenia,” in College Seminar Series in General Adult Psychiatry (2nd Edition ed, ed. G. Stein (London: Gaskell), 238–294.
Bauer, B., Hartz, A. M., Fricker, G., and Miller, D. S. (2004). Pregnane X receptor up-regulation of P-glycoprotein expression and transport function at the blood-brain barrier. Mol. Pharmacol. 66, 413–419. doi: 10.1124/mol.66.3
Baumann, P., and Rochat, B. (1995). Comparative pharmacokinetics of selective serotonin reuptake inhibitors: a look behind the mirror. Int. Clin. Psychopharmacol. 10, 15–21. doi: 10.1097/00004850-199503001-00004
Beedham, C. (1997). The role of non-P450 enzymes in drug oxidation. Pharm. World Sci. 19, 255–263. doi: 10.1201/9781420028485.ch8
Beedham, C., Miceli, J. J., and Obach, R. S. (2003). Ziprasidone metabolism, aldehyde oxidase, and clinical implications. J. Clin. Psychopharmacol. 23, 229–232. doi: 10.1097/01.jcp.0000084028.22282.f2
Benowitz, N. L., Jacob, I. I. I., Saunders, S., and Gourlay, S. (1999). Carbon monoxide, cigarette smoking and CYP2E1 activity. Clin. Pharmacol. Therapeut. 65, 154–154. doi: 10.1016/S0009-9236(99)80148-9
Bertilsson, L. (1995). Geographical/interracial differences in polymorphic drug oxidation. Current state of knowledge of cytochromes P450 (CYP) 2D6 and 2C19. Clin. Pharmacokinet 29, 192–209. doi: 10.2165/00003088-199529030-00005
Bertilsson, L. (2007). Metabolism of antidepressant and neuroleptic drugs by cytochrome p450s: clinical and interethnic aspects. Clin. Pharmacol. Therapeut. 82, 606–609. doi: 10.1038/sj.clpt.6100358
Bertilsson, L., Aberg-Wistedt, A., Gustafsson, L. L., and Nordin, C. (1985). Extremely rapid hydroxylation of debrisoquine: a case report with implication for treatment with nortriptyline and other tricyclic antidepressants. Ther. Drug Monit. 7, 478–480. doi: 10.1097/00007691-198512000-00021
Bertilsson, L., and Dahl, M. L. (1996). Polymorphic drug oxidation - Relevance to the treatment of psychiatric disorders. CNS Drugs 5, 200–223. doi: 10.2165/00023210-199605030-00006
Bertilsson, L., Dahl, M. L., Dalen, P., and Al-Shurbaji, A. (2002). Molecular genetics of CYP2D6: clinical relevance with focus on psychotropic drugs. Br. J. Clin. Pharmacol. 53, 111–122. doi: 10.1046/j.0306-5251.2001.01548.x
Bertilsson, L., Dahl, M. L., Ekqvist, B., and Llerena, A. (1993a). Disposition of the neuroleptics perphenazine, zuclopenthixol, and haloperidol cosegregates with polymorphic debrisoquine hydroxylation. Psychopharmacol. Ser. 10, 230–237. doi: 10.1007/978-3-642-78010-3_21
Bertilsson, L., Dahl, M. L., Sjoqvist, F., Aberg-Wistedt, A., Humble, M., Johansson, I., et al. (1993b). Molecular basis for rational megaprescribing in ultrarapid hydroxylators of debrisoquine. Lancet 63:341.
Bertilsson, L., Mellstrom, B., Sjokvist, F., Martenson, B., and Asberg, M. (1981). Slow hydroxylation of nortriptyline and concomitant poor debrisoquine hydroxylation: clinical implications. Lancet 1, 560–561. doi: 10.1016/s0140-6736(81)92894-4
Beutler, E., Gelbart, T., and Demina, A. (1998). Racial variability in the UDP-glucuronosyltransferase 1 (UGT1A1) promoter: a balanced polymorphism for regulation of bilirubin metabolism? Proc. Natl. Acad. Sci. U S A. 95, 8170–8174. doi: 10.1073/pnas.95.14.8170
Bhaskar, L. V., Thangaraj, K., Patel, M., Shah, A. M., Gopal, K., Saikrishna, L., et al. (2013). EPHX1 gene polymorphisms in alcohol dependence and their distribution among the Indian populations. Am. J. Drug Alcohol Abuse 39, 16–22. doi: 10.3109/00952990.2011.643991
Bigos, K. L., Bies, R. R., Pollock, B. G., Lowy, J. J., Zhang, F., and Weinberger, D. R. (2011). Genetic variation in CYP3A43 explains racial difference in olanzapine clearance. Mol. Psychiatry 16, 620–625. doi: 10.1038/mp.2011.38
Birdwell, K. A., Decker, B., Barbarino, J. M., Peterson, J. F., Stein, C. M., Sadee, W., et al. (2015). Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines for CYP3A5 Genotype and Tacrolimus Dosing. Clin. Pharmacol. Ther. 98, 19–24. doi: 10.1002/cpt.113
Blake, C. M., Kharasch, E. D., Schwab, M., and Nagele, P. (2013). A meta-analysis of CYP2D6 metabolizer phenotype and metoprolol pharmacokinetics. Clin. Pharmacol. Ther. 94, 394–399. doi: 10.1038/clpt.2013.96
Blanca Sánchez, M., Herranz, J. L., Leno, C., Arteaga, R., Oterino, A., Valdizán, E. M., et al. (2010). UGT2B7_-161C>T polymorphism is associated with lamotrigine concentration-to-dose ratio in a multivariate study. Therapeu. Drug Monit. 32, 177–184. doi: 10.1097/FTD.0b013e3181ceecc6
Bloomer, J. C., Woods, F. R., Haddock, R. E., Lennard, M. S., and Tucker, G. T. (1992). The role of cytochrome P4502D6 in the metabolism of paroxetine by human liver microsomes. Br. J. Clin. Pharmacol. 33, 521–523. doi: 10.1111/j.1365-2125.1992.tb04082.x
Boland, M. R., and Tatonetti, N. P. (2016). Investigation of 7-dehydrocholesterol reductase pathway to elucidate off-target prenatal effects of pharmaceuticals: a systematic review. Pharmacogen. J. 16, 411–429. doi: 10.1038/tpj.2016.48
Boobis, A. R., Lynch, A. M., Murray, S., de la Torre, R., Solans, A., Farre, M., et al. (1994). CYP1A2-catalyzed conversion of dietary heterocyclic amines to their proximate carcinogens is their major route of metabolism in humans. Cancer Res. 54, 89–94.
Borst, P., and Elferink, R. O. (2002). Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 71, 537–592. doi: 10.1146/annurev.biochem.71.102301.093055
Bourin, M., Chue, P., and Guillon, Y. (2001). Paroxetine: a review. CNS Drug Rev. 7, 25–47. doi: 10.1111/j.1527-3458.2001.tb00189.x
Bousman, C. A., and Hopwood, M. (2016). Commercial pharmacogenetic-based decision-support tools in psychiatry. Lancet Psychiatry 3, 585–590. doi: 10.1016/S2215-0366(16)00017-1
Bousman, C., Allen, J., and Eyre, H. A. (2018). Pharmacogenetic Tests in Psychiatry. Am. J. Psychiatry 175:189. doi: 10.1176/appi.ajp.2017.17101086
Bousman, C., Maruf, A. A., and Muller, D. J. (2019a). Towards the integration of pharmacogenetics in psychiatry: a minimum, evidence-based genetic testing panel. Curr. Opin. Psychiatry 32, 7–15. doi: 10.1097/YCO.0000000000000465
Bousman, C. A., Arandjelovic, K., Mancuso, S. G., Eyre, H. A., and Dunlop, B. W. (2019b). Pharmacogenetic tests and depressive symptom remission: a meta-analysis of randomized controlled trials. Pharmacogenomics 20, 37–47. doi: 10.2217/pgs-2018-0142
Bradford, L. D. (2002). CYP2D6 allele frequency in European Caucasians, Asians, Africans and their descendants. Pharmacogenomics 3, 229–243. doi: 10.1517/14622416.3.2.229
Brockmöller, J., Meineke, I., and Kirchheiner, J. (2007). Pharmacokinetics of mirtazapine: enantioselective effects of the CYP2D6 ultra rapid metabolizer genotype and correlation with adverse effects. Clin. Pharmacol. Ther. 81, 699–707. doi: 10.1038/sj.clpt.6100116
Brosen, K. (1993). The pharmacogenetics of the selective serotonin reuptake inhibitors. Clin. Invest. 71, 1002–1009.
Brosen, K., Skjelbo, E., Rasmussen, B. B., Poulsen, H. E., and Loft, S. (1993). Fluvoxamine is a potent inhibitor of cytochrome P4501A2. Biochem. Pharmacol. 45, 1211–1214. doi: 10.1016/0006-2952(93)90272-x
Brown, J. T., Bishop, J. R., Sangkuhl, K., Nurmi, E. L., Mueller, D. J., Dinh, J. C., et al. (2019). Clinical Pharmacogenetics Implementation Consortium Guideline for Cytochrome P450 (CYP)2D6 Genotype and Atomoxetine Therapy. Clin. Pharmacol. Ther. 106, 94–102. doi: 10.1002/cpt.1409
Browning, S. L., Tarekegn, A., Bekele, E., Bradman, N., and Thomas, M. G. (2010). CYP1A2 is more variable than previously thought: a genomic biography of the gene behind the human drug-metabolizing enzyme. Pharmacogenet Genom. 20, 647–664. doi: 10.1097/FPC.0b013e32833e90eb
Bruyere, A., Decleves, X., Bouzom, F., Ball, K., Marques, C., Treton, X., et al. (2010). Effect of variations in the amounts of P-glycoprotein (ABCB1), BCRP (ABCG2) and CYP3A4 along the human small intestine on PBPK models for predicting intestinal first pass. Mol. Pharm. 7, 1596–1607. doi: 10.1021/mp100015x
Bu, H. Z. (2006). A literature review of enzyme kinetic parameters for CYP3A4-mediated metabolic reactions of 113 drugs in human liver microsomes: structure-kinetics relationship assessment. Curr. Drug Metab. 7, 231–249. doi: 10.2174/138920006776359329
Byeon, J. Y., Kim, Y. H., Na, H. S., Jang, J. H., Kim, S. H., Lee, Y. J., et al. (2015). Effects of the CYP2D6∗10 allele on the pharmacokinetics of atomoxetine and its metabolites. Arch. Pharm. Res. 38, 2083–2091. doi: 10.1007/s12272-015-0646-z
Calarge, C. A., and Miller, D. (2011). Predictors of risperidone and 9-hydroxyrisperidone serum concentration in children and adolescents. J. Child Adolesc. Psychopharmacol. 21, 163–169. doi: 10.1089/cap.2010.0038
Callaghan, J. T., Bergstrom, R. F., Ptak, L. R., and Beasley, C. M. (1999). Olanzapine. Pharmacokinetic and pharmacodynamic profile. Clin. Pharmac. 37, 177–193. doi: 10.2165/00003088-199937030-00001
Cartmell, M. T., Schulz, H. U., O’Reilly, D. A., Yang, B. M., Kielstein, V., Dunlop, S. P., et al. (2005). Cytochrome P450 2E1 high activity polymorphism in alcohol abuse and end-organ disease. World J. Gastroenterol. 11, 6445–6449. doi: 10.3748/wjg.v11.i41.6445
Caruso, A., Bellia, C., Pivetti, A., Agnello, L., Bazza, F., Scazzone, C., et al. (2014). Effects of EPHX1 and CYP3A4 polymorphisms on carbamazepine metabolism in epileptic patients. Pharmgenom. Pers. Med. 7, 117–120. doi: 10.2147/PGPM.S55548
Cascorbi, I. (2011). P-glycoprotein: tissue distribution, substrates, and functional consequences of genetic variations. Handb. Exp. Pharmacol. 201, 261–283. doi: 10.1007/978-3-642-14541-4_6
Cashman, J. R., and Zhang, J. (2002). Interindividual differences of human flavin-containing monooxygenase 3: genetic polymorphisms and functional variation. Drug Metab. Dispos. 30, 1043–1052. doi: 10.1124/dmd.30.10.1043
Caspi, A., Moffitt, T. E., Cannon, M., McClay, J., Murray, R., Harrington, H., et al. (2005). Moderation of the effect of adolescent-onset cannabis use on adult psychosis by a functional polymorphism in the catechol-O-methyltransferase gene: longitudinal evidence of a gene X environment interaction. Biol. Psychiatry 57, 1117–1127. doi: 10.1016/j.biopsych.2005.01.026
Castberg, I., Skogvoll, E., and Spigset, O. (2007). Quetiapine and drug interactions: evidence from a routine therapeutic drug monitoring service. J. Clin. Psychiatry 68, 1540–1545. doi: 10.4088/jcp.v68n1011
Caudle, K. E., Gammal, R. S., Whirl-Carrillo, M., Hoffman, J. M., Relling, M. V., and Klein, T. E. (2016). Evidence and resources to implement pharmacogenetic knowledge for precision medicine. Am. J. Health Syst. Pharm. 73, 1977–1985. doi: 10.2146/ajhp150977
Caudle, K. E., Klein, T. E., Hoffman, J. M., Muller, D. J., Whirl-Carrillo, M., Gong, L., et al. (2014). Incorporation of pharmacogenomics into routine clinical practice: the Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline development process. Curr. Drug Metab. 15, 209–217. doi: 10.2174/1389200215666140130124910
Caudle, K. E., Sangkuhl, K., Whirl-Carrillo, M., Swen, J. J., Haidar, C. E., Klein, T. E., et al. (2020). Standardizing CYP2D6 Genotype to Phenotype Translation: Consensus Recommendations from the Clinical Pharmacogenetics Implementation Consortium and Dutch Pharmacogenetics Working Group. Clin. Transl. Sci. 13, 116–124. doi: 10.1111/cts.12692
Cederbaum, A. I. (2012). Alcohol metabolism. Clin. Liver Dis. 16, 667–685. doi: 10.1016/j.cld.2012.08.002
Chatzistefanidis, D., Georgiou, I., Kyritsis, A. P., and Markoula, S. (2012). Functional impact and prevalence of polymorphisms involved in the hepatic glucuronidation of valproic acid. Pharmacogenomics 13, 1055–1071. doi: 10.2217/pgs.12.78
Chen, F., Zhang, B., Parker, R. B., & Laizure, S. C. (2018). Clinical implications of genetic variation in carboxylesterase drug metabolism. Expert Opin Drug Metab Toxicol, 14(2), 131-142. doi: 10.1080/17425255.2018.1420164
Chen, J.-T., Wei, L., Chen, T.-L., Huang, C.-J., and Chen, R.-M. (2018). Regulation of cytochrome P450 gene expression by ketamine: a review. Exp. Opin. Drug Metab. Toxicol. 14, 709–720.
Chen, S., Chou, W. H., Blouin, R. A., Mao, Z., Humphries, L. L., Meek, Q. C., et al. (1996). The cytochrome P450 2D6 (CYP2D6) enzyme polymorphism: screening costs and influence on clinical outcomes in psychiatry. Clin. Pharmacol. Ther. 60, 522–534. doi: 10.1016/S0009-9236(96)90148-4
Chen, X., Wang, L., Zhi, L., Zhou, G., Wang, H., Zhang, X., et al. (2005). The G-113A polymorphism in CYP1A2 affects the caffeine metabolic ratio in a Chinese population. Clin. Pharmacol. Ther. 78, 249–259. doi: 10.1016/j.clpt.2005.05.012
Chen, Y., and Goldstein, J. A. (2009). The transcriptional regulation of the human CYP2C genes. Curr. Drug Metabol. 10, 567–578. doi: 10.2174/138920009789375397
Chen, Y., Zane, N. R., Thakker, D. R., and Wang, M. Z. (2016). Quantification of Flavin-containing Monooxygenases 1, 3, and 5 in Human Liver Microsomes by UPLC-MRM-Based Targeted Quantitative Proteomics and Its Application to the Study of Ontogeny. Drug Metab. Dispos. 44, 975–983. doi: 10.1124/dmd.115.067538
Chen, Z., Liew, D., and Kwan, P. (2014). Effects of a HLA-B∗15:02 screening policy on antiepileptic drug use and severe skin reactions. Neurology 83, 2077–2084. doi: 10.1212/WNL.0000000000001034
Chevalier, D., Cauffiez, C., Allorge, D., Lo-Guidice, J. M., Lhermitte, M., Lafitte, J. J., et al. (2001). Five novel natural allelic variants-951A>C, 1042G>A (D348N), 1156A>T (I386F), 1217G>A (C406Y) and 1291C>T (C431Y)-of the human CYP1A2 gene in a French Caucasian population. Hum. Mutat. 17, 355–356. doi: 10.1002/humu.49
Chida, M., Yokoi, T., Fukui, T., Kinoshita, M., Yokota, J., and Kamataki, T. (1999). Detection of three genetic polymorphisms in the 5’-flanking region and intron 1 of human CYP1A2 in the Japanese population. Jap. J. Cancer Res. 90, 899–902. doi: 10.1111/j.1349-7006.1999.tb00832.x
Chiu, C. C., Lane, H. Y., Huang, M. C., Liu, H. C., Jann, M. W., Hon, Y. Y., et al. (2004). Dose-dependent alternations in the pharmacokinetics of olanzapine during coadministration of fluvoxamine in patients with schizophrenia. J. Clin. Pharmacol. 44, 1385–1390. doi: 10.1177/0091270004270291
Choi, C. I., Bae, J. W., Lee, Y. J., Lee, H. I., Jang, C. G., and Lee, S. Y. (2014). Effects of CYP2C19 genetic polymorphisms on atomoxetine pharmacokinetics. J. Clin. Psychopharmacol. 34, 139–142. doi: 10.1097/JCP.0b013e3182a608a2
Chou, W. H., Yan, F. X., Robbins-Weilert, D. K., Ryder, T. B., Liu, W. W., Perbost, C., et al. (2003). Comparison of two CYP2D6 genotyping methods and assessment of genotype-phenotype relationships. Clin. Chem. 49, 542–551. doi: 10.1373/49.4.542
Christensen, M., Tybring, G., Mihara, K., Yasui-Furokori, N., Carrillo, J. A., Ramos, S. I., et al. (2002). Low daily 10-mg and 20-mg doses of fluvoxamine inhibit the metabolism of both caffeine (cytochrome P4501A2) and omeprazole (cytochrome P4502C19). Clin. Pharmacol. Ther. 71, 141–152. doi: 10.1067/mcp.2002.121788
Chu, X. M., Zhang, L. F., Wang, G. J., Zhang, S. N., Zhou, J. H., and Hao, H. P. (2012). Influence of UDP-glucuronosyltransferase polymorphisms on valproic acid pharmacokinetics in Chinese epilepsy patients. Eur. J. Clin. Pharmacol. 68, 1395–1401. doi: 10.1007/s00228-012-1277-7
Chung, J. Y., Cho, J. Y., Yu, K. S., Kim, J. R., Jung, H. R., Lim, K. S., et al. (2005). Effect of the UGT2B15 genotype on the pharmacokinetics, pharmacodynamics, and drug interactions of intravenous lorazepam in healthy volunteers. Clin. Pharmacol. Therapeu. 77, 486–494. doi: 10.1016/j.clpt.2005.02.006
Chung, W. H., Hung, S. I., Hong, H. S., Hsih, M. S., Yang, L. C., Ho, H. C., et al. (2004). Medical genetics: a marker for Stevens-Johnson syndrome. Nature 428:486. doi: 10.1038/428486a
ClinVar database: rs2032582. Retrieved from https://www.ncbi.nlm.nih.gov/clinvar/?term==rs2032582
Coleman, T., Ellis, S. W., Martin, I. J., Lennard, M. S., and Tucker, G. T. (1996). 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is N-demethylated by cytochromes P450 2D6, 1A2 and 3A4–implications for susceptibility to Parkinson’s disease. J. Pharmacol. Exp. Ther. 277, 685–690.
Coller, J. K., Krebsfaenger, N., Klein, K., Endrizzi, K., Wolbold, R., Lang, T., et al. (2002). The influence of CYP2B6, CYP2C9 and CYP2D6 genotypes on the formation of the potent antioestrogen Z-4-hydroxy-tamoxifen in human liver. Br. J. Clin. Pharmacol. 54, 157–167. doi: 10.1046/j.1365-2125.2002.01614.x
Committee on Safety of Medicines-Medicines Control Agency. (1995). Cardiac arrhythmias with pimozide (Orap). Current Problems in Pharmacovigilance, London, UK: Committee on Safety of Medicines-Medicines Control Agency 21.
Correll, C. U., Detraux, J., De Lepeleire, J., and De Hert, M. (2015). Effects of antipsychotics, antidepressants and mood stabilizers on risk for physical diseases in people with schizophrenia, depression and bipolar disorder. World Psychiatry 14, 119–136. doi: 10.1002/wps.20204
Crespi, C. L., Penman, B. W., Leakey, J. A. E., Arlotto, M. P., Stark, A., Parkinson, A., et al. (1990). Human cytochrome P450IIA3: cDNA sequence role of the enzyme in the metabolic of promutagens comparison to nitrosamine activation by human cytochrome P450IIE1. Carcinogenesis 11, 1293–1300. doi: 10.1093/carcin/11.8.1293
Crews, K. R., Gaedigk, A., Dunnenberger, H. M., Leeder, J. S., Klein, T. E., Caudle, K. E., et al. (2014). Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450 2D6 genotype and codeine therapy: 2014 update. Clin. Pharmacol. Ther. 95, 376–382. doi: 10.1038/clpt.2013.254
Cui, Y. M., Teng, C. H., Pan, A. X., Yuen, E., Yeo, K. P., Zhou, Y., et al. (2007). Atomoxetine pharmacokinetics in healthy Chinese subjects and effect of the CYP2D6∗10 allele. Br. J. Clin. Pharmacol. 64, 445–449. doi: 10.1111/j.1365-2125.2007.02912.x
Cui, Y., Yan, H., Su, Y., Wang, L., Lu, T., Zhang, D., et al. (2020). CYP2D6 Genotype-Based Dose Recommendations for Risperidone in Asian People. Front. Pharmacol. 11:936. doi: 10.3389/fphar.2020.00936
Cupp, M. J., and Tracy, T. S. (1998). Cytochrome P450: new nomenclature and clinical implications. Am. Fam. Physician. 57, 107–116.
Cusack, B., Nelson, A., and Richelson, E. (1994). Binding of antidepressants to human brain receptors: focus on newer generation compounds. Psychopharmacology 114, 559–565. doi: 10.1007/BF02244985
CYP3A4. Retrieved from https://www.pharmvar.org/gene/CYP3A4
CYP3A5 RefSeqGene on chromosome 7. (2020). URL: https://www.ncbi.nlm.nih.gov/nuccore/NG_007938.2
Czekaj, P. (2000). Phenobarbital-induced expression of cytochrome P450 genes. Acta Biochim. Pol. 47, 1093–1105. doi: 10.18388/abp.2000_3962
Czekaj, P., and Skowronek, R. (2012). “Transcription Factors Potentially Involved in Regulation of Cytochrome P450 Gene Expression,” in Topics on Drug Metabolism, ed. J. Paxton. (IntechOpen).
Daci, A., Beretta, G., Vllasaliu, D., Shala, A., Govori, V., Norata, G. D., et al. (2015). Polymorphic Variants of SCN1A and EPHX1 Influence Plasma Carbamazepine Concentration, Metabolism and Pharmacoresistance in a Population of Kosovar Albanian Epileptic Patients. PLoS One 10:e0142408. doi: 10.1371/journal.pone.0142408
Dahl, M. L., Ekqvist, B., Widén, J., and Bertilsson, L. (1991). Disposition of the neuroleptic zuclopenthixol cosegregates with the polymorphic hydroxylation of debrisoquine in humans. Acta Psychiatr. Scand. 84, 99–102. doi: 10.1111/j.1600-0447.1991.tb01428.x
Dahl, M. L., Voortman, G., Alm, C., Elwin, C. E., Delbressine, L., Vos, R., et al. (1997). In Vitro and In Vivo Studies on the Disposition of Mirtazapine in Humans. Clin. Drug Invest. 13, 37–46. doi: 10.2165/00044011-199713010-00005
Dahl-Puustinen, M.-L., Lidén, A., Alm, C., Nordin, C., and Bertilsson, L. (1989). Disposition of perphenazine is related to polymorphic debrisoquin hydroxylation in luman beings. Clin. Pharmacol. Therapeut. 46, 78–81. doi: 10.1038/clpt.1989.109
Dalen, P., Dahl, M. L., Bernal Ruiz, M. L., Nordin, J., and Bertilsson, L. (1998). 10-Hydroxylation of nortriptyline in white persons with 0, 1, 2, 3, and 13 functional CYP2D6 genes. Clin. Pharmacol. Ther. 63, 444–452. doi: 10.1016/S0009-9236(98)90040-6
Daniel, W. A., Haduch, A., and Wójcikowski, J. (2005). Inhibition of rat liver CYP2D in vitro and after 1-day and long-term exposure to neuroleptics in vivo-possible involvement of different mechanisms. Eur. Neuropsychopharmacol. 15, 103–110. doi: 10.1016/j.euroneuro.2004.05.008
Dannenberg, L. O., and Edenberg, H. J. (2006). Epigenetics of gene expression in human hepatoma cells: expression profiling the response to inhibition of DNA methylation and histone deacetylation. BMC Genomics 7:181. doi: 10.1186/1471-2164-7-181
Darwish, M., Bond, M., Yang, R., Hellriegel, E. T., and Robertson, P. (2015). Evaluation of potential pharmacokinetic drug-drug interaction between armodafinil and risperidone in healthy adults. Clin. Drug Investig. 35, 725–733. doi: 10.1007/s40261-015-0330-6
de Andrés, F., Sosa-Macías, M., and Llerena, A. (2014). A rapid and simple LC-MS/MS method for the simultaneous evaluation of CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 hydroxylation capacity. Bioanalysis 6, 683–696. doi: 10.4155/bio.14.20
de Klerk, O. L., Nolte, I. M., Bet, P. M., Bosker, F. J., Snieder, H., den Boer, J. A., et al. (2013). ABCB1 gene variants influence tolerance to selective serotonin reuptake inhibitors in a large sample of Dutch cases with major depressive disorder. Pharmacogenom. J. 13, 349–353. doi: 10.1038/tpj.2012.16
de Leon, J. (2003). Glucuronidation enzymes, genes and psychiatry. Int. J. Neuropsychopharmacol. 6, 57–72. doi: 10.1017/S1461145703003249
de Leon, J. (2009). The future (or lack of future) of personalized prescription in psychiatry. Pharmacol. Res. 59, 81–89. doi: 10.1016/j.phrs.2008.10.002
de Leon, J., Susce, M. T., Pan, R. M., Fairchild, M., Koch, W. H., and Wedlund, P. J. (2005). The CYP2D6 poor metabolizer phenotype may be associated with risperidone adverse drug reactions and discontinuation. J. Clin. Psychiatry 66, 15–27. doi: 10.4088/jcp.v66n0103
de Morais, S. M., Wilkinson, G. R., Blaisdell, J., Nakamura, K., Meyer, U. A., and Goldstein, J. A. (1994). The major genetic defect responsible for the polymorphism of S-mephenytoin metabolism in humans. J. Biol. Chem. 269, 15419–15422.
Dean, M., Hamon, Y., and Chimini, G. (2001). The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res. 42, 1007–1017.
Del Tredici, A. L., Malhotra, A., Dedek, M., Espin, F., Roach, D., Zhu, G. D., et al. (2018). Frequency of CYP2D6 Alleles Including Structural Variants in the United States. Front. Pharmacol. 9:305. doi: 10.3389/fphar.2018.00305
DeLeon, A., Patel, N. C., and Crismon, M. L. (2004). Aripiprazole: a comprehensive review of its pharmacology, clinical efficacy, and tolerability. Clin. Ther. 26, 649–666. doi: 10.1016/s0149-2918(04)90066-5
Desai, H. D., Seabolt, J., and Jann, M. W. (2001). Smoking in patients receiving psychotropic medications: a pharmacokinetic perspective. CNS Drugs 15, 469–494. doi: 10.2165/00023210-200115060-00005
Desta, Z., Kerbusch, T., Soukhova, N., Richard, E., Ko, J. W., and Flockhart, D. A. (1998). Identification and characterization of human cytochrome P450 isoforms interacting with pimozide. J. Pharmacol. Exp. Ther. 285, 428–437.
Desta, Z., Saussele, T., Ward, B., Blievernicht, J., Li, L., Klein, K., et al. (2007). Impact of CYP2B6 polymorphism on hepatic efavirenz metabolism in vitro. Pharmacogenomics 8, 547–558. doi: 10.2217/14622416.8.6.547
Desta, Z., Zhao, X., Shin, J. G., and Flockhart, D. A. (2002). Clinical significance of the cytochrome P450 2C19 genetic polymorphism. Clin. Pharmacokinet 41, 913–958. doi: 10.2165/00003088-200241120-00002
di Iulio, J., Fayet, A., Arab-Alameddine, M., Rotger, M., Lubomirov, R., Cavassini, M., et al. (2009). In vivo analysis of efavirenz metabolism in individuals with impaired CYP2A6 function. Pharmacog. Genom. 19, 300–309. doi: 10.1097/FPC.0b013e328328d577
Di, Y. M., Chow, V. D., Yang, L. P., and Zhou, S. F. (2009). Structure, function, regulation and polymorphism of human cytochrome P450 2A6. Curr. Drug Metab. 10, 754–780. doi: 10.2174/138920009789895507
Dobrinas, M., Cornuz, J., Oneda, B., Kohler Serra, M., Puhl, M., and Eap, C. B. (2011). Impact of smoking, smoking cessation, and genetic polymorphisms on CYP1A2 activity and inducibility. Clin. Pharmacol. Ther. 90, 117–125. doi: 10.1038/clpt.2011.70
Doring, B., and Petzinger, E. (2014). Phase 0 and phase III transport in various organs: combined concept of phases in xenobiotic transport and metabolism. Drug Metab. Rev. 46, 261–282. doi: 10.3109/03602532.2014.882353
Drozda, K., Muller, D. J., and Bishop, J. R. (2014). Pharmacogenomic testing for neuropsychiatric drugs: current status of drug labeling, guidelines for using genetic information, and test options. Pharmacotherapy 34, 166–184. doi: 10.1002/phar.1398
Drug Interactions Flockhart TableTM. Retrieved from http://medicine.iupui.edu/clinpharm/ddis/table.aspx
DrugBank. Olanazapine. https://go.drugbank.com/drugs/DB00334
Eaton, D. L., Gallagher, E. P., Bammler, T. K., and Kunze, K. L. (1995). Role of cytochrome P4501A2 in chemical carcinogenesis: implications for human variability in expression and enzyme activity. Pharmacogenetics 5, 259–274. doi: 10.1097/00008571-199510000-00001
Ehlert, F. J., Delen, F. M., Yun, S. H., and Liem, H. A. (1990). The interaction of amitriptyline, doxepin, imipramine and their N-methyl quaternary ammonium derivatives with subtypes of muscarinic receptors in brain and heart. J. Pharmacol. Exp. Ther. 253, 13–19. doi: 10.1007/bf03297258
Ekroos, M., and Sjogren, T. (2006). Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. U S A 103, 13682–13687. doi: 10.1073/pnas.0603236103
El-Sherbeni, A. A., and El-Kadi, A. O. (2014). The role of epoxide hydrolases in health and disease. Arch. Toxicol. 88, 2013–2032. doi: 10.1007/s00204-014-1371-y
Elens, L., Becker, M. L., Haufroid, V., Hofman, A., Visser, L. E., Uitterlinden, A. G., et al. (2011). Novel CYP3A4 intron 6 single nucleotide polymorphism is associated with simvastatin-mediated cholesterol reduction in the Rotterdam study. Pharmacogenet. Genomics 21, 861–866. doi: 10.1097/FPC.0b013e32834c6edb
Elsinga, P. H., Hendrikse, N. H., Bart, J., Vaalburg, W., and van Waarde, A. (2004). PET Studies on P-glycoprotein function in the blood-brain barrier: how it affects uptake and binding of drugs within the CNS. Curr. Pharm. Des. 10, 1493–1503. doi: 10.2174/1381612043384736
Ereshefsky, L. (1996). Pharmacokinetics and drug interactions: update for new antipsychotics. J. Clin. Psychiatry 57, 12–25.
Erickson-Ridout, K. K., Zhu, J., and Lazarus, P. (2011). Olanzapine metabolism and the significance of UGT1A448V and UGT2B1067Y variants. Pharmacog. Genom. 21, 539–551. doi: 10.1097/FPC.0b013e328348c76b
Eriksson, N., and Wadelius, M. (2012). Prediction of warfarin dose: why, when and how? Pharmacogenomics 13, 429–440. doi: 10.2217/pgs.11.184
Evans, D. A., Krahn, P., and Narayanan, N. (1995). The mephenytoin (cytochrome P450 2C 19) and dextromethorphan (cytochrome P450 2D6) polymorphisms in Saudi Arabians and Filipinos. Pharmacogenetics 5, 64–71. doi: 10.1097/00008571-199504000-00002
Fabbri, C., and Serretti, A. (2015). Pharmacogenetics of major depressive disorder: top genes and pathways toward clinical applications. Curr. Psychiatry Rep. 17:50. doi: 10.1007/s11920-015-0594-9
Fabbri, C., Tansey, K. E., Perlis, R. H., Hauser, J., Henigsberg, N., Maier, W., et al. (2018). Effect of cytochrome CYP2C19 metabolizing activity on antidepressant response and side effects: Meta-analysis of data from genome-wide association studies. Eur. Neuropsychopharmacol. 28, 945–954. doi: 10.1016/j.euroneuro.2018.05.009
Fan, W. L., Shiao, M. S., Hui, R. C., Su, S. C., Wang, C. W., Chang, Y. C., et al. (2017). HLA Association with Drug-Induced Adverse Reactions. J. Immunol. Res. 2017:3186328. doi: 10.1155/2017/3186328
Fang, H., Xu, X., Kaur, K., Dedek, M., Zhu, G. D., Riley, B. J., et al. (2019). A Screening Test for HLA-B∗15:02 in a Large United States Patient Cohort Identifies Broader Risk of Carbamazepine-Induced Adverse Events. Front. Pharmacol. 10:149. doi: 10.3389/fphar.2019.00149
Fang, J., Baker, G. B., Silverstone, P. H., and Coutts, R. T. (1997). Involvement of CYP3A4 and CYP2D6 in the metabolism of haloperidol. Cell. Mole. Neurobiol. 17, 227–233. doi: 10.1023/a:1026317929335
Faucette, S. R., Wang, H., Hamilton, G. A., Jolley, S. L., Gilbert, D., Lindley, C., et al. (2004). Regulation of CYP2B6 in primary human hepatocytes by prototypical inducers. Drug Metab. Dispos. 32, 348–358. doi: 10.1124/dmd.32.3.348
Feng, J., Sun, J., Wang, M. Z., Zhang, Z., Kim, S. T., Zhu, Y., et al. (2010). Compilation of a comprehensive gene panel for systematic assessment of genes that govern an individual’s drug responses. Pharmacogenomics 11, 1403–1425. doi: 10.2217/pgs.10.99
Ferrell, P. B., and McLeod, H. L. (2008). Carbamazepine, HLA-B∗1502 and risk of Stevens-Johnson syndrome and toxic epidermal necrolysis: US FDA recommendations. Pharmacogenomics 9, 1543–1546. doi: 10.2217/14622416.9.10.1543
Findling, R. L., Nucci, G., Piergies, A. A., Gomeni, R., Bartolic, E. I., Fong, R., et al. (2006). Multiple dose pharmacokinetics of paroxetine in children and adolescents with major depressive disorder or obsessive-compulsive disorder. Neuropsychopharmacology 31, 1274–1285. doi: 10.1038/sj.npp.1300960
Fiore, M. C., Jorenby, D. E., Schensky, A. E., Smith, S. S., Bauer, R. R., and Baker, T. B. (1995). Smoking status as the new vital sign: effect on assessment and intervention in patients who smoke. Mayo Clin. Proc. 70, 209–213. doi: 10.1016/S0025-6196(11)64939-2
Flockhart, D. A., Drici, M. D., Kerbusch, T., Soukhova, N., Richard, E., Pearle, P. L., et al. (2000). Studies on the mechanism of a fatal clarithromycin-pimozide interaction in a patient with Tourette syndrome. J. Clin. Psychopharmacol. 20, 317–324. doi: 10.1097/00004714-200006000-00005
Foti, R. S., and Dalvie, D. K. (2016). Cytochrome P450 and Non-Cytochrome P450 Oxidative Metabolism: Contributions to the Pharmacokinetics, Safety, and Efficacy of Xenobiotics. Drug Metab. Dispos. 44, 1229–1245. doi: 10.1124/dmd.116.071753
Foti, R. S., Wienkers, L. C., and Wahlstrom, J. L. (2010). Application of cytochrome P450 drug interaction screening in drug discovery. Comb. Chem. High Throughput Screen 13, 145–158. doi: 10.2174/138620710790596718
Fricke-Galindo, I., LLerena, A., Jung-Cook, H., and López-López, M. (2018). Carbamazepine adverse drug reactions. Exp. Rev. Clin. Pharmacol. 11, 705–718. doi: 10.1080/17512433.2018.1486707
Fromm, M. F., and Kim, R. B. (2011). Drug transportersHandbook of experimental. Pharmacology. 201:454.
Fuhr, U., Anders, E. M., Mahr, G., Sorgel, F., and Staib, A. H. (1992). Inhibitory potency of quinolone antibacterial agents against cytochrome P450IA2 activity in vivo and in vitro. Antimicrob. Agents Chemother. 36, 942–948. doi: 10.1128/aac.36.5.942
Fuhr, U., Jetter, A., and Kirchheiner, J. (2007). Appropriate Phenotyping Procedures for Drug Metabolizing Enzymes and Transporters in Humans and Their Simultaneous Use in the “Cocktail” Approach. Clin. Pharmacol. Ther. 81, 270–83. doi: 10.1038/sj.clpt.6100050
Fuhr, U., Klittich, K., and Staib, A. H. (1993). Inhibitory effect of grapefruit juice and its bitter principal, naringenin, on CYP1A2 dependent metabolism of caffeine in man. Br. J. Clin. Pharmacol. 35, 431–436. doi: 10.1111/j.1365-2125.1993.tb04162.x
Fujita, K., and Sasaki, Y. (2007). Pharmacogenomics in drug-metabolizing enzymes catalyzing anticancer drugs for personalized cancer chemotherapy. Curr. Drug Metab. 8, 554–562. doi: 10.2174/138920007781368890
Fukami, T., and Yokoi, T. (2012). The emerging role of human esterases. Drug Metab. Pharmacokinet 27, 466–477. doi: 10.2133/dmpk.dmpk-12-rv-042
Fulop, G., Phillips, R. A., Shapiro, A. K., Gomes, J. A., Shapiro, E., and Nordlie, J. W. (1987). ECG changes during haloperidol and pimozide treatment of Tourette’s disorder. Am. J. Psychiatry 144, 673–675. doi: 10.1176/ajp.144.5.673
Furnes, B., Feng, J., Sommer, S. S., and Schlenk, D. (2003). Identification of novel variants of the flavin-containing monooxygenase gene family in African Americans. Drug Metab. Dispos. 31, 187–193. doi: 10.1124/dmd.31.2.187
Gaedigk, A., Turner, A., Everts, R. E., Scott, S. A., Aggarwal, P., Broeckel, U., et al. (2019). Characterization of Reference Materials for Genetic Testing of CYP2D6 Alleles: A GeT-RM Collaborative Project. J. Mol. Diagn. 21, 1034–1052. doi: 10.1016/j.jmoldx.2019.06.007
Galea, S., Merchant, R. M., and Lurie, N. (2020). The Mental Health Consequences of COVID-19 and Physical Distancing: The Need for Prevention and Early Intervention. JAMA Internal Med. 180, 817–818. doi: 10.1001/jamainternmed.2020.1562
Ganoci, L., Lovrić, M., Živković, M., Šagud, M., Klarica Domjanović, I., and Božina, N. (2016). The Role Of Cyp2d6, Cyp3a4/5, And Abcb1 Polymorphisms In Patients Using Long-Acting Injectable Risperidone. Clin. Therapeu. 38, e10–e11. doi: 10.1016/j.clinthera.2016.07.110
García-Suástegui, W. A., Ramos-Chávez, L. A., Rubio-Osornio, M., Calvillo-Velasco, M., Atzin-Méndez, J. A., Guevara, J., et al. (2017). The Role of CYP2E1 in the Drug Metabolism or Bioactivation in the Brain. Oxid. Med. Cell Longev. 2017:4680732. doi: 10.1155/2017/4680732
Genaro-Mattos, T. C., Tallman, K. A., Allen, L. B., Anderson, A., Mirnics, K., Korade, Z., et al. (2018). Dichlorophenyl piperazines, including a recently-approved atypical antipsychotic, are potent inhibitors of DHCR7, the last enzyme in cholesterol biosynthesis. Toxicol. Appl. Pharmacol. 349, 21–28. doi: 10.1016/j.taap.2018.04.029
Gex-Fabry, M., Eap, C. B., Oneda, B., Gervasoni, N., Aubry, J. M., Bondolfi, G., et al. (2008). CYP2D6 and ABCB1 genetic variability: influence on paroxetine plasma level and therapeutic response. Ther. Drug Monit. 30, 474–482. doi: 10.1097/FTD.0b013e31817d6f5d
Ghodke-Puranik, Y., Thorn, C. F., Lamba, J. K., Leeder, J. S., Song, W., Birnbaum, A. K., et al. (2013). Valproic acid pathway: pharmacokinetics and pharmacodynamics. Pharmacogenet Genom. 23, 236–241. doi: 10.1097/FPC.0b013e32835ea0b2
Ghosh, C., Hossain, M., Puvenna, V., Martinez-Gonzalez, J., Alexopolous, A., Janigro, D., et al. (2013). Expression and functional relevance of UGT1A4 in a cohort of human drug-resistant epileptic brains. Epilepsia 54, 1562–1570. doi: 10.1111/epi.12318
Ghotbi, R., Christensen, M., Roh, H. K., Ingelman-Sundberg, M., Aklillu, E., and Bertilsson, L. (2007). Comparisons of CYP1A2 genetic polymorphisms, enzyme activity and the genotype-phenotype relationship in Swedes and Koreans. Eur. J. Clin. Pharmacol. 63, 537–546. doi: 10.1007/s00228-007-0288-2
Ghotbi, R., Mannheimer, B., Aklillu, E., Suda, A., Bertilsson, L., Eliasson, E., et al. (2010). Carriers of the UGT1A4 142T >>G gene variant are predisposed to reduced olanzapine exposure–an impact similar to male gender or smoking in schizophrenic patients. Eur. J. Clin. Pharmacol. 66, 465–474. doi: 10.1007/s00228-009-0783-8
Gidal, B. E., Sheth, R., Parnell, J., Maloney, K., and Sale, M. (2003). Evaluation of VPA dose and concentration effects on lamotrigine pharmacokinetics: implications for conversion to lamotrigine monotherapy. Epilepsy Res. 57, 85–93. doi: 10.1016/j.eplepsyres.2003.09.008
Gill, M., Hawi, Z., and Webb, M. (1997). Homozygous mutation at cytochrome P4502D6 in an individual with schizophrenia: Implications for antipsychotic drugs, side effects and compliance. Irish J. Psychol. Med. 14, 38–39. doi: 10.1017/S0790966700002925
Girre, C., Lucas, D., Hispard, E., Menez, C., Dally, S., and Menez, J. F. (1994). Assessment of cytochrome P4502E1 induction in alcoholic patients by chlorzoxazone pharmacokinetics. Biochem. Pharmacol. 47, 1503–1508. doi: 10.1016/0006-2952(94)90524-x
Goh, B. C., Lee, S. C., Wang, L. Z., Fan, L., Guo, J. Y., Lamba, J., et al. (2002). Explaining interindividual variability of docetaxel pharmacokinetics and pharmacodynamics in Asians through phenotyping and genotyping strategies. J. Clin. Oncol. 20, 3683–3690. doi: 10.1200/JCO.2002.01.025
Goldman, D. (2014). Roles of COMT, NPY and GCH1 in acute and chronic pain/stress response. Mol. Pain 10:O5.
Goldman, D., Weinberger, D. R., Malhotra, A. K., and Goldberg, T. E. (2009). The role of COMT Val158Met in cognition. Biol. Psychiatry 65, e1–e2. doi: 10.1016/j.biopsych.2008.07.032
Goldstein, J. A., and de Morais, S. M. (1994). Biochemistry and molecular biology of the human CYP2C subfamily. Pharmacogenetics 4, 285–299. doi: 10.1097/00008571-199412000-00001
Gong, Q. L., Hedner, T., Hedner, J., Bjorkman, R., and Nordberg, G. (1991). Antinociceptive and ventilatory effects of the morphine metabolites: morphine-6-glucuronide and morphine-3-glucuronide. Eur. J. Pharmacol. 193, 47–56. doi: 10.1016/0014-2999(91)90199-z
Gonzalez, F. J., and Gelboin, H. V. (1994). Role of Human Cytochromes P450 in the Metabolic Activation of Chemical Carcinogens and Toxins. Drug Metab. Rev. 26, 165–183. doi: 10.3109/03602539409029789
Goodarzi, M. O., Xu, N., and Azziz, R. (2008). Association of CYP3A7∗1C and serum dehydroepiandrosterone sulfate levels in women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 93, 2909–2912. doi: 10.1210/jc.2008-0403
Gough, A. C., Miles, J. S., Spurr, N. K., Moss, J. E., Gaedigk, A., Eichelbaum, M., et al. (1990). Identification of the primary gene defect at the cytochrome P450 CYP2D locus. Nature 347, 773–776. doi: 10.1038/347773a0
Gram, L. F., Kragh-Sorensen, P., Kristensen, C. B., Moller, M., Pedersen, O. L., and Thayssen, P. (1984). Plasma level monitoring of antidepressants: theoretical basis and clinical application. Adv. Biochem. Psychopharmacol. 39, 399–411.
Greden, J. F., Parikh, S. V., Rothschild, A. J., Thase, M. E., Dunlop, B. W., and DeBattista, C. (2019). Impact of pharmacogenomics on clinical outcomes in major depressive disorder in the GUIDED trial: A large, patient- and rater-blinded, randomized, controlled study. J. Psychiatr. Res. 111, 59–67. doi: 10.1016/j.jpsychires.2019.01.003
Guengerich, F. P. (1999). Cytochrome P-450 3A4: regulation and role in drug metabolism. Annu. Rev. Pharmacol. Toxicol. 39, 1–17. doi: 10.1146/annurev.pharmtox.39.1.1
Gulcebi, M. I., Ozkaynakcı, A., Goren, M. Z., Aker, R. G., Ozkara, C., and Onat, F. Y. (2011). The relationship between UGT1A4 polymorphism and serum concentration of lamotrigine in patients with epilepsy. Epilepsy Res. 95, 1–8. doi: 10.1016/j.eplepsyres.2011.01.016
Gunes, A., Spina, E., Dahl, M. L., and Scordo, M. G. (2008). ABCB1 polymorphisms influence steady-state plasma levels of 9-hydroxyrisperidone and risperidone active moiety. Ther. Drug Monit. 30, 628–633. doi: 10.1097/FTD.0b013e3181858ca9
Gupta, N., Hanley, M. J., Venkatakrishnan, K., Bessudo, A., Rasco, D. W., Sharma, S., et al. (2018). Effects of Strong CYP3A Inhibition and Induction on the Pharmacokinetics of Ixazomib, an Oral Proteasome Inhibitor: Results of Drug-Drug Interaction Studies in Patients With Advanced Solid Tumors or Lymphoma and a Physiologically Based Pharmacokinetic Analysis. J. Clin. Pharmacol. 58, 180–192. doi: 10.1002/jcph.988
Haas, D. W., Ribaudo, H. J., Kim, R. B., Tierney, C., Wilkinson, G. R., Gulick, R. M., et al. (2004). Pharmacogenetics of efavirenz and central nervous system side effects: an Adult AIDS Clinical Trials Group study. AIDS 18, 2391–2400.
Haberl, M., Anwald, B., Klein, K., Weil, R., Fuss, C., Gepdiremen, A., et al. (2005). Three haplotypes associated with CYP2A6 phenotypes in Caucasians. Pharmacog. Genom. 15, 609–624. doi: 10.1097/01.fpc.0000171517.22258.f1
Haddock, R. E., Johnson, A. M., Langley, P. F., Nelson, D. R., Pope, J. A., Thomas, D. R., et al. (1989). Metabolic pathway of paroxetine in animals and man and the comparative pharmacological properties of its metabolites. Acta psychiatrica Scandinavica. Supplementum 350, 24–26. doi: 10.1111/j.1600-0447.1989.tb07163.x
Hadidi, H., Zahlsen, K., Idle, J. R., and Cholerton, S. (1997). A single amino acid substitution (Leu160His) in cytochrome P450 CYP2A6 causes switching from 7-hydroxylation to 3-hydroxylation of coumarin. Food Chem. Toxicol. 35, 903–907. doi: 10.1016/s0278-6915(97)00066-5
Hals, P.-A., Hall, H., and Dahl, S. G. (1986). Phenothiazine drug metabolites: Dopamine D2 receptor, -α1-and -α2-adrenoceptor binding. Eur. J. Pharmacol. 125, 373–381. doi: 10.1016/0014-2999(86)90793-4
Hammons, G. J., Milton, D., Stepps, K., Guengerich, F. P., Tukey, R. H., and Kadlubar, F. F. (1997). Metabolism of carcinogenic heterocyclic and aromatic amines by recombinant human cytochrome P450 enzymes. Carcinogenesis 18, 851–854. doi: 10.1093/carcin/18.4.851
Han, X. M., Ouyang, D. S., Chen, X. P., Shu, Y., Jiang, C. H., Tan, Z. R., et al. (2002). Inducibility of CYP1A2 by omeprazole in vivo related to the genetic polymorphism of CYP1A2. Br. J. Clin. Pharmacol. 54, 540–543. doi: 10.1046/j.1365-2125.2002.01686.x
Hanioka, N., Kimura, S., Meyer, U. A., and Gonzalez, F. J. (1990). The human CYP2D locus associated with a common genetic defect in drug oxidation: a G1934—-A base change in intron 3 of a mutant CYP2D6 allele results in an aberrant 3’ splice recognition site. Am. J. Hum. Genetics 47, 994–1001.
Hansen, L. B. (1981). “The clinical significance of measuring perphenazine in plasma during oral antipsychotic treatment,” in Clinical pharmacology in psychiatry - neuroleptic and antidepressant research, E. Usdin (London: Macmillan), 211–216. doi: 10.1007/978-1-349-05929-4_18
Hansen, L. B., and Larsen, N. E. (1983). “Plasma levels of perphenazine related to clinical effect and extrapyramidal side-effects,” in Clinical pharmacology in psychiatry - bridging the experimental-therapeutic gap, L.F. Gram E. Usdin S.G. Dahl (London: Macmillan), 175–181. doi: 10.1007/978-1-349-06671-1_16
Hansen, L. B., and Larsen, N. E. (1985). Therapeutic advantages of monitoring plasma concentrations of perphenazine in clinical practice. Psychopharmacology 87, 16–19. doi: 10.1007/bf00431770
Hansen, L. B., Elley, J., Christensen, T. R., Larsen, N. E., Naestoft, J., and Hvidberg, E. F. (1979). Plasma levels of perphenazine and its major metabolites during simultaneous treatment with anticholinergic drugs. Br. J. Clin. Pharmacol. 7, 75–80. doi: 10.1111/j.1365-2125.1979.tb00900.x
Hansen, L. B., Larsen, N. E., and Gulmann, N. (1982). Dose-response relationships of perphenazine in the treatment of acute psychoses. Psychopharmacology 78, 112–115. doi: 10.1007/bf00432245
Hasin, Y., Avidan, N., Bercovich, D., Korczyn, A. D., Silman, I., Beckmann, J. S., et al. (2005). Analysis of genetic polymorphisms in acetylcholinesterase as reflected in different populations. Curr. Alzheimer Res. 2, 207–218. doi: 10.2174/1567205053585909
Hayashi, Y., Watanabe, T., Aoki, A., Ishiguro, S., Ueda, M., Akiyama, K., et al. (2015). Factors Affecting Steady-state Plasma Concentrations of Enantiomeric Mirtazapine and its Desmethylated Metabolites in Japanese Psychiatric Patients. Pharmacopsychiatry 48, 279–285. doi: 10.1055/s-0035-1565069
He, X., Hesse, L. M., Hazarika, S., Masse, G., Harmatz, J. S., Greenblatt, D. J., et al. (2009). Evidence for oxazepam as an in vivo probe of UGT2B15: oxazepam clearance is reduced by UGT2B15 D85Y polymorphism but unaffected by UGT2B17 deletion. Br. J. Clin. Pharmacol. 68, 721–730. doi: 10.1111/j.1365-2125.2009.03519.x
Heit, C., Dong, H., Chen, Y., Thompson, D. C., Deitrich, R. A., and Vasiliou, V. K. (2013). The role of CYP2E1 in alcohol metabolism and sensitivity in the central nervous system. Subcell Biochem. 67, 235–247. doi: 10.1007/978-94-007-5881-0_8
Hemeryck, A., and Belpaire, F. M. (2002). Selective serotonin reuptake inhibitors and cytochrome P-450 mediated drug-drug interactions: an update. Curr. Drug Metab. 3, 13–37. doi: 10.2174/1389200023338017
Hendset, M., Molden, E., Enoksen, T. B., Refsum, H., and Hermann, M. (2010). The effect of coadministration of duloxetine on steady-state serum concentration of risperidone and aripiprazole: a study based on therapeutic drug monitoring data. Ther. Drug Monit. 32, 787–790. doi: 10.1097/FTD.0b013e3181fc50d5
Hendset, M., Molden, E., Knape, M., and Hermann, M. (2014). Serum concentrations of risperidone and aripiprazole in subgroups encoding CYP2D6 intermediate metabolizer phenotype. Ther. Drug. Monit. 36, 80–85. doi: 10.1097/FTD.0000000000000018
Hesse, L. M., He, P., Krishnaswamy, S., Hao, Q., Hogan, K., von Moltke, L. L., et al. (2004). Pharmacogenetic determinants of interindividual variability in bupropion hydroxylation by cytochrome P450 2B6 in human liver microsomes. Pharmacogenetics 14, 225–238. doi: 10.1097/00008571-200404000-00002
Hesslinger, B., Normann, C., Langosch, J. M., Klose, P., Berger, M., and Walden, J. (1999). Effects of carbamazepine and valproate on haloperidol plasma levels and on psychopathologic outcome in schizophrenic patients. J. Clin. Psychopharmacol. 19, 310–315. doi: 10.1097/00004714-199908000-00005
Hichiya, H., Tanaka-Kagawa, T., Soyama, A., Jinno, H., Koyano, S., Katori, N., et al. (2005). Functional characterization of five novel CYP2C8 variants, G171S, R186X, R186G, K247R, and K383N, found in a Japanese population. Drug Metab. Dispos. 33, 630–636. doi: 10.1124/dmd.105.003830
Hicks, J. K., Bishop, J. R., Sangkuhl, K., Müller, D. J., Ji, Y., Leckband, S. G., et al. (2015). Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and CYP2C19 Genotypes and Dosing of Selective Serotonin Reuptake Inhibitors. Clin. Pharmacol. Ther. 98, 127–134. doi: 10.1002/cpt.147
Hicks, J. K., Sangkuhl, K., Swen, J. J., Ellingrod, V. L., Müller, D. J., Shimoda, K., et al. (2016). Clinical pharmacogenetics implementation consortium guideline (CPIC) for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants: 2016 update. Clin. Pharmacol. Ther. 102, 37–44. doi: 10.1002/cpt.597
Hicks, J. K., Sangkuhl, K., Swen, J. J., Ellingrod, V. L., Müller, D. J., Shimoda, K., et al. (2017). Clinical pharmacogenetics implementation consortium guideline (CPIC) for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants: 2016 update. Clin. Pharmacol. Ther. 102, 37–44. doi: 10.1002/cpt.597
Hicks, J. K., Swen, J. J., Thorn, C. F., Sangkuhl, K., Kharasch, E. D., Ellingrod, V. L., et al. (2013). Clinical Pharmacogenetics Implementation Consortium guideline for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants. Clin. Pharmacol. Ther. 93, 402–408. doi: 10.1038/clpt.2013.2
Hiemke, C., Bergemann, N., Clement, H. W., Conca, A., Deckert, J., Domschke, K., et al. (2018). Consensus Guidelines for Therapeutic Drug Monitoring in Neuropsychopharmacology: Update 2017. Pharmacopsychiatry 51:e1. doi: 10.1055/s-0037-1600991
Hillhouse, T. M., and Porter, J. H. (2015). A brief history of the development of antidepressant drugs: from monoamines to glutamate. Exp. Clin. Psychopharmacol. 23, 1–21. doi: 10.1037/a0038550
Hisaka, A., Ohno, Y., Yamamoto, T., and Suzuki, H. (2010). Prediction of pharmacokinetic drug-drug interaction caused by changes in cytochrome P450 activity using in vivo information. Pharmacol. Ther. 125, 230–248. doi: 10.1016/j.pharmthera.2009.10.011
Hjorthoj, C., Ostergaard, M. L. D., Benros, M. E., Toftdahl, N. G., Erlangsen, A., Andersen, J. T., et al. (2015). Association between alcohol and substance use disorders and all-cause and cause-specific mortality in schizophrenia, bipolar disorder, and unipolar depression: a nationwide, prospective, register-based study. Lancet Psychiatry 2, 801–808. doi: 10.1016/S2215-0366(15)00207-2
Hodges, L. M., Markova, S. M., Chinn, L. W., Gow, J. M., Kroetz, D. L., Klein, T. E., et al. (2011). Very important pharmacogene summary: ABCB1 (MDR1. P-glycoprotein). Pharmacogenet Genomics 21, 152–161. doi: 10.1097/FPC.0b013e3283385a1c
Hodgson, K., Tansey, K. E., Uher, R., Dernovšek, M. Z., Mors, O., Hauser, J., et al. (2015). Exploring the role of drug-metabolising enzymes in antidepressant side effects. Psychopharmacology 232, 2609–2617. doi: 10.1007/s00213-015-3898-x
Hoffman, S. M., Nelson, D. R., and Keeney, D. S. (2001). Organization, structure and evolution of the CYP2 gene cluster on human chromosome 19. Pharmacogenetics 11, 687–698. doi: 10.1097/00008571-200111000-00007
Hofmann, M. H., Blievernicht, J. K., Klein, K., Saussele, T., Schaeffeler, E., Schwab, M., et al. (2008). Aberrant splicing caused by single nucleotide polymorphism c.516G >T [Q172H], a marker of CYP2B6∗6, is responsible for decreased expression and activity of CYP2B6 in liver. J. Pharmacol. Exp. Ther. 325, 284–292. doi: 10.1124/jpet.107.133306
Holmes, E. A., O’Connor, R. C., Perry, V. H., Tracey, I., Wessely, S., Arseneault, L., et al. (2020). Multidisciplinary research priorities for the COVID-19 pandemic: a call for action for mental health science. Lancet Psychiatry 7, 547–560. doi: 10.1016/S2215-0366(20)30168-1
Holstein, A., Plaschke, A., Ptak, M., Egberts, E. H., El-Din, J., Brockmöller, J., et al. (2005). Association between CYP2C9 slow metabolizer genotypes and severe hypoglycaemia on medication with sulphonylurea hypoglycaemic agents. Br. J. Clin. Pharmacol. 60, 103–106. doi: 10.1111/j.1365-2125.2005.02379.x
Horrigan, J. P., and Barnhill, L. J. (1994). Paroxetine-pimozide drug interaction. J. Am. Acad. Child Adolesc. Psychiatry 33, 1060–1061. doi: 10.1097/00004583-199409000-00022
Howard, L. A., Ahluwalia, J. S., Lin, S. K., Sellers, E. M., and Tyndale, R. F. (2003). CYP2E1∗1D regulatory polymorphism: association with alcohol and nicotine dependence. Pharmacogenetics 13, 321–328. doi: 10.1097/01.fpc.0000054090.48725.a2
Hu, G. X., Dai, D. P., Wang, H., Huang, X. X., Zhou, X. Y., Cai, J., et al. (2017). Systematic screening for CYP3A4 genetic polymorphisms in a Han Chinese population. Pharmacogenomics 18, 369–379. doi: 10.2217/pgs-2016-0179
Hu, Y., Oscarson, M., Johansson, I., Yue, Q. Y., Dahl, M. L., Tabone, M., et al. (1997). Genetic polymorphism of human CYP2E1: characterization of two variant alleles. Mol. Pharmacol. 51, 370–376.
Huang, C. Y., Kao, C. F., Chen, C. C., Kuo, P. H., and Huang, M. C. (2018). No association of CYP2E1 genetic polymorphisms with alcohol dependence in Han Taiwanese population. J. Formosan Med. Assoc. 117, 646–649. doi: 10.1016/j.jfma.2018.03.007
Hugo Gene Nomeclature Committee (2020). Atp Binding Cassette Transporters (Abc). Retrieved from https://www.genenames.org/data/genegroup/#!/group/417 (accessed on 24 August 2020) doi: 10.1016/j.jfma.2018.03.007
Hustert, E., Haberl, M., Burk, O., Wolbold, R., He, Y. Q., Klein, K., et al. (2001). The genetic determinants of the CYP3A5 polymorphism. Pharmacogenetics 11, 773–779. doi: 10.1097/00008571-200112000-00005
Imaoka, S., and Funae, Y. (1990). Purification and characterization of rat pulmonary cytochrome P-450. J. Biochem. 108, 33–36. doi: 10.1093/oxfordjournals.jbchem.a123157
Ingelman-Sundberg, M. (2004a). Human drug metabolising cytochrome P450 enzymes: properties and polymorphisms. Naunyn Schmiedebergs Arch. Pharmacol. 369, 89–104. doi: 10.1007/s00210-003-0819-z
Ingelman-Sundberg, M. (2004b). Pharmacogenetics of cytochrome P450 and its applications in drug therapy: the past, present and future. Trends Pharmacol. Sci. 25, 193–200. doi: 10.1016/j.tips.2004.02.007
Ingelman-Sundberg, M. (2005). Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J. 5, 6–13. doi: 10.1038/sj.tpj.6500285
International Society for Psychiatric Genetics [ISPG]. (2019). Genetic testing statement: genetic testing and psychiatric disorders: a statement from the International Society of Psychiatric Genetics. Retrieved from http://ispg.net/genetic-testing-statement/
Ishak, K. J., Tan, Y., Glass, J., Luong, D., and Caro, J. J. (2008). Risk of discontinuation of risperidone after exposure to potentially interacting drugs: a nested case-control study in patients with schizophrenia. Clin. Ther. 30, 1251–1263. doi: 10.1016/s0149-2918(08)80049-5
Ishii, Y., Iwanaga, M., Nishimura, Y., Takeda, S., Ikushiro, S., Nagata, K., et al. (2007). Protein-protein interactions between rat hepatic cytochromes P450 (P450s) and UDP-glucuronosyltransferases (UGTs): evidence for the functionally active UGT in P450-UGT complex. Drug Metab. Pharmacokinet 22, 367–376. doi: 10.2133/dmpk.22.367
Ishikawa, T. (1992). The ATP-dependent glutathione S-conjugate export pump. Trends Biochem. Sci. 17, 463–468. doi: 10.1016/0968-0004(92)90489-v
Jaanson, P., Marandi, T., Kiivet, R. A., Vasar, V., Vään, S., Svensson, J. O., et al. (2002). Maintenance therapy with zuclopenthixol decanoate: associations between plasma concentrations, neurological side effects and CYP2D6 genotype. Psychopharmacology 162, 67–73. doi: 10.1007/s00213-002-1059-5
Jacobs, B. S., Colbers, A. P., Velthoven-Graafland, K., Schouwenberg, B. J., and Burger, D. M. (2014). Effect of fosamprenavir/ritonavir on the pharmacokinetics of single-dose olanzapine in healthy volunteers. Int. J. Antimicrob. Agents 44, 173–177. doi: 10.1016/j.ijantimicag.2014.03.014
Jamshidi, Y., Moreton, M., McKeown, D. A., Andrews, S., Nithiyananthan, T., Tinworth, L., et al. (2010). Tribal ethnicity and CYP2B6 genetics in Ugandan and Zimbabwean populations in the UK: implications for efavirenz dosing in HIV infection. J. Antimicrob. Chemother. 65, 2614–2619. doi: 10.1093/jac/dkq369
Jančová, P., and Šiller, M. (2012). “Phase II Drug Metabolism,” in Topics on Drug Metabolism, ed. J. Paxton (Croatia InTech), 35–60.
Jancova, P., Anzenbacher, P., and Anzenbacherova, E. (2010). Phase II drug metabolizing enzymes. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 154, 103–116.
Jann, M. W., and Cohen, L. J. (2000). The influence of ethnicity and antidepressant pharmacogenetics in the treatment of depression. Drug Metabol. Drug Interact. 16, 39–67. doi: 10.1515/dmdi.2000.16.1.39
Jaquenoud Sirot, E., Harenberg, S., Vandel, P., Lima, C. A., Perrenoud, P., Kemmerling, K., et al. (2012). Multicenter study on the clinical effectiveness, pharmacokinetics, and pharmacogenetics of mirtazapine in depression. J. Clin. Psychopharmacol. 32, 622–629. doi: 10.1097/JCP.0b013e3182664d98
Jerling, M., Dahl, M. L., Aberg-Wistedt, A., Liljenberg, B., Landell, N. E., Bertilsson, L., et al. (1996). The CYP2D6 genotype predicts the oral clearance of the neuroleptic agents perphenazine and zuclopenthixol. Clin. Pharmacol. Ther. 59, 423–428. doi: 10.1016/S0009-9236(96)90111-3
Jiang, Z., Dalton, T. P., Jin, L., Wang, B., Tsuneoka, Y., Shertzer, H. G., et al. (2005). Toward the evaluation of function in genetic variability: characterizing human SNP frequencies and establishing BAC-transgenic mice carrying the human CYP1A1_CYP1A2 locus. Hum. Mutat. 25, 196–206. doi: 10.1002/humu.20134
Johannessen, S. I., and Landmark, C. J. (2010). Antiepileptic drug interactions - principles and clinical implications. Curr. Neuropharmacol. 8, 254–267. doi: 10.2174/157015910792246254
Johansson, I., and Ingelman-Sundberg, M. (2011). Genetic polymorphism and toxicology–with emphasis on cytochrome p450. Toxicol. Sci. 120, 1–13. doi: 10.1093/toxsci/kfq374
Johansson, I., Lundqvist, E., Bertilsson, L., Dahl, M. L., Sjoqvist, F., and Ingelman-Sundberg, M. (1993). Inherited amplification of an active gene in the cytochrome P450 CYP2D locus as a cause of ultrarapid metabolism of debrisoquine. Proc. Natl. Acad. Sci. U S A 90, 11825–11829. doi: 10.1073/pnas.90.24.11825
Johansson, I., Oscarson, M., Yue, Q. Y., Bertilsson, L., Sjoqvist, F., and Ingelmansundberg, M. (1994). Genetic-Analysis of the Chinese Cytochrome P4502d Locus - Characterization of Variant Cyp2d6 Genes Present in Subjects with Diminished Capacity for Debrisoquine Hydroxylation. Mole. Pharmacol. 46, 452–459.
Johne, A., Brockmoller, J., Bauer, S., Maurer, A., Langheinrich, M., and Roots, I. (1999). Pharmacokinetic interaction of digoxin with an herbal extract from St John’s wort (Hypericum perforatum). Clin. Pharmacol. Ther. 66, 338–345. doi: 10.1053/cp.1999.v66.a101944
Jorge-Nebert, L. F., Jiang, Z., Chakraborty, R., Watson, J., Jin, L., McGarvey, S. T., et al. (2010). Analysis of human CYP1A1 and CYP1A2 genes and their shared bidirectional promoter in eight world populations. Hum. Mutat. 31, 27–40. doi: 10.1002/humu.21132
Jukić, M. M., Haslemo, T., Molden, E., and Ingelman-Sundberg, M. (2018). Impact of CYP2C19 Genotype on Escitalopram Exposure and Therapeutic Failure: A Retrospective Study Based on 2,087 Patients. Am. J. Psychiatry 175, 463–470. doi: 10.1176/appi.ajp.2017.17050550
Jukić, M. M., Opel, N., Ström, J., Carrillo-Roa, T., Miksys, S., Novalen, M., et al. (2017). Elevated CYP2C19 expression is associated with depressive symptoms and hippocampal homeostasis impairment. Mol. Psychiatry 22:1224. doi: 10.1038/mp.2017.93
Jukic, M. M., Smith, R. L., Haslemo, T., Molden, E., and Ingelman-Sundberg, M. (2019). Effect of CYP2D6 genotype on exposure and efficacy of risperidone and aripiprazole: a retrospective, cohort study. Lancet Psychiatry 6, 418–426. doi: 10.1016/S2215-0366(19)30088-4
Jung, F., Richardson, T. H., Raucy, J. L., and Johnson, E. F. (1997). Diazepam metabolism by cDNA-expressed human 2C P450s: identification of P4502C18 and P4502C19 as low K(M) diazepam N-demethylases. Drug Metab. Dispos. 25, 133–139.
Jurica, J., and Sulcova, A. (2012). “Determination of Cytochrome P450 Metabolic Activity Using Selective Markers,” in Topics on Drug Metabolism, ed. J. Paxton. (IntechOpen).
Kagimoto, M., Heim, M., Kagimoto, K., Zeugin, T., and Meyer, U. A. (1990). Multiple mutations of the human cytochrome P450IID6 gene (CYP2D6) in poor metabolizers of debrisoquine. Study of the functional significance of individual mutations by expression of chimeric genes. J. Biol. Chem. 265, 17209–17214.
Kalow, W., and Tyndale, R. (1992). “Debrisoquine/sparteine monooxygenase and other P450s in brain,” in Pharmacogenetics of drug metabolism, ed. W. Kalow (Pregamon Press), 649–656.
Karam, W. G., Goldstein, J. A., Lasker, J. M., and Ghanayem, B. I. (1996). Human CYP2C19 is a major omeprazole 5-hydroxylase, as demonstrated with recombinant cytochrome P450 enzymes. Drug Metab. Dispos. 24, 1081–1087.
Kasperaviciūte, D., and Sisodiya, S. M. (2009). Epilepsy pharmacogenetics. Pharmacogenomics 10, 817–836. doi: 10.2217/pgs.09.34
Kato, Y., Nakajima, M., Oda, S., Fukami, T., and Yokoi, T. (2012). Human UDP-glucuronosyltransferase isoforms involved in haloperidol glucuronidation and quantitative estimation of their contribution. Drug Metab. Dispos. 40, 240–248. doi: 10.1124/dmd.111.042150
Kaur, G., Gupta, D., Chavan, B. S., Sinhmar, V., Prasad, R., Tripathi, A., et al. (2017). Identification of genetic correlates of response to Risperidone: Findings of a multicentric schizophrenia study from India. Asian J. Psychiatry 29, 174–182. doi: 10.1016/j.ajp.2017.07.026
Kegeles, L. S., Slifstein, M., Frankle, W. G., Xu, X., Hackett, E., Bae, S. A., et al. (2008). Dose-occupancy study of striatal and extrastriatal dopamine D2 receptors by aripiprazole in schizophrenia with PET and [18F]fallypride. Neuropsychopharmacology 33, 3111–3125. doi: 10.1038/npp.2008.33
Kennedy, S. H., Lam, R. W., McIntyre, R. S., Tourjman, S. V., Bhat, V., Blier, P., et al. (2016). Canadian Network for Mood and Anxiety Treatments (CANMAT) 2016 Clinical Guidelines for the Management of Adults with Major Depressive Disorder: Section 3. Pharmacological Treatments. Can. J. Psychiatry 61, 540–560. doi: 10.1177/0706743716659417
Kertesz, S. G., Pletcher, M. J., Safford, M., Halanych, J., Kirk, K., Schumacher, J., et al. (2007). Illicit drug use in young adults and subsequent decline in general health: The coronary artery risk development in young adults (CARDIA) study. Drug Alcohol. Depend. 88, 224–233. doi: 10.1016/j.drugalcdep.2006.10.017
Kim, K. A., Park, P. W., Liu, K. H., Kim, K. B., Lee, H. J., Shin, J. G., et al. (2008). Effect of rifampin, an inducer of CYP3A and P-glycoprotein, on the pharmacokinetics of risperidone. J. Clin. Pharmacol. 48, 66–72. doi: 10.1177/0091270007309888
Kim, H. Y., Korade, Z., Tallman, K. A., Liu, W., Weaver, C. D., Mirnics, K., et al. (2016). Inhibitors of 7-Dehydrocholesterol Reductase: Screening of a Collection of Pharmacologically Active Compounds in Neuro2a Cells. Chem. Res. Toxicol. 29, 892–900. doi: 10.1021/acs.chemrestox.6b00054
Kimchi-Sarfaty, C., Oh, J. M., Kim, I. W., Sauna, Z. E., Calcagno, A. M., Ambudkar, S. V., et al. (2007). A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 315, 525–528. doi: 10.1126/science.1135308
King, B. P., Khan, T. I., Aithal, G. P., Kamali, F., and Daly, A. K. (2004). Upstream and coding region CYP2C9 polymorphisms: correlation with warfarin dose and metabolism. Pharmacogenetics 14, 813–822. doi: 10.1097/00008571-200412000-00004
King, C. D., Rios, G. R., Assouline, J. A., and Tephly, T. R. (1999). Expression of UDP-glucuronosyltransferases (UGTs) 2B7 and 1A6 in the human brain and identification of 5-hydroxytryptamine as a substrate. Arch. Biochem. Biophys. 365, 156–162. doi: 10.1006/abbi.1999.1155
King, J., and Aberg, J. A. (2008). Clinical impact of patient population differences and genomic variation in efavirenz therapy. AIDS 22, 1709–1717. doi: 10.1097/QAD.0b013e32830163ad
Kirchheiner, J., Brøsen, K., Dahl, M. L., Gram, L. F., Kasper, S., Roots, I., et al. (2001). CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr. Scand. 104, 173–192. doi: 10.1034/j.1600-0447.2001.00299.x
Kirchheiner, J., Klein, C., Meineke, I., Sasse, J., Zanger, U. M., Murdter, T. E., et al. (2003). Bupropion and 4-OH-bupropion pharmacokinetics in relation to genetic polymorphisms in CYP2B6. Pharmacogenetics 13, 619–626. doi: 10.1097/01.fpc.0000054125.14659.d0
Kirchheiner, J., Nickchen, K., Bauer, M., Wong, M. L., Licinio, J., Roots, I., et al. (2004). Pharmacogenetics of antidepressants and antipsychotics: the contribution of allelic variations to the phenotype of drug response. Mol. Psychiatry 9, 442–473. doi: 10.1038/sj.mp.4001494
Kirschbaum, K. M., Müller, M. J., Malevani, J., Mobascher, A., Burchardt, C., Piel, M., et al. (2008). Serum levels of aripiprazole and dehydroaripiprazole, clinical response and side effects. World J. Biol. Psychiatry 9, 212–218. doi: 10.1080/15622970701361255
Klaassen, C. D., and Slitt, A. L. (2005). Regulation of hepatic transporters by xenobiotic receptors. Curr. Drug Metab. 6, 309–328. doi: 10.2174/1389200054633826
Klees, T. M., Sheffels, P., Dale, O., and Kharasch, E. D. (2005). Metabolism of alfentanil by cytochrome p4503a (cyp3a) enzymes. Drug Metab. Dispos. 33, 303–311. doi: 10.1124/dmd.104.002709
Klein, K., Lang, T., Saussele, T., Barbosa-Sicard, E., Schunck, W. H., Eichelbaum, M., et al. (2005). Genetic variability of CYP2B6 in populations of African and Asian origin: allele frequencies, novel functional variants, and possible implications for anti-HIV therapy with efavirenz. Pharmacog. Genom. 15, 861–873. doi: 10.1097/01213011-200512000-00004
Klotz, U. (2006). Clinical impact of CYP2C19 polymorphism on the action of proton pump inhibitors: a review of a special problem. Int. J. Clin. Pharmacol. Ther. 44, 297–302. doi: 10.5414/cpp44297
Klotz, U. (2009). Pharmacokinetics and drug metabolism in the elderly. Drug Metab. Rev. 41, 67–76. doi: 10.1080/03602530902722679
Knadler, M. P., Lobo, E., Chappell, J., and Bergstrom, R. (2011). Duloxetine: clinical pharmacokinetics and drug interactions. Clin. Pharmacokinet 50, 281–294. doi: 10.2165/11539240-000000000-00000
Knutti, R., Rothweiler, H., and Schlatter, C. (1981). Effect of pregnancy on the pharmacokinetics of caffeine. Eur. J. Clin. Pharmacol. 21, 121–126. doi: 10.1007/bf00637512
Kobayashi, K., Chiba, K., Yagi, T., Shimada, N., Taniguchi, T., Horie, T., et al. (1997). Identification of cytochrome P450 isoforms involved in citalopram N-demethylation by human liver microsomes. J. Pharmacol. Exp. Ther. 280, 927–933.
Kolars, J. C., Lown, K. S., Schmiedlin-Ren, P., Ghosh, M., Fang, C., Wrighton, S. A., et al. (1994). CYP3A gene expression in human gut epithelium. Pharmacogenetics 4, 247–259. doi: 10.1097/00008571-199410000-00003
Komatsu, T., Yamazaki, H., Shimada, N., Nakajima, M., and Yokoi, T. (2000). Roles of cytochromes P450 1A2, 2A6, and 2C8 in 5-fluorouracil formation from tegafur, an anticancer prodrug, in human liver microsomes. Drug Metab. Dispos. 28, 1457–1463.
Koola, M. M., Tsapakis, E. M., Wright, P., Smith, S., Kerwin Rip, R. W., Nugent, K. L., et al. (2014). Association of tardive dyskinesia with variation in CYP2D6: Is there a role for active metabolites? J. Psychopharmacol. 28, 665–670. doi: 10.1177/0269881114523861
Korade, Z., Kim, H. Y., Tallman, K. A., Liu, W., Koczok, K., Balogh, I., et al. (2016). The Effect of Small Molecules on Sterol Homeostasis: Measuring 7-Dehydrocholesterol in Dhcr7-Deficient Neuro2a Cells and Human Fibroblasts. J. Med. Chem. 59, 1102–1115. doi: 10.1021/acs.jmedchem.5b01696
Korade, Z., Xu, L., Shelton, R., and Porter, N. A. (2010). Biological activities of 7-dehydrocholesterol-derived oxysterols: implications for Smith-Lemli-Opitz syndrome. J. Lipid Res. 51, 3259–3269. doi: 10.1194/jlr.m009365
Korprasertthaworn, P., Polasek, T. M., Sorich, M. J., McLachlan, A. J., Miners, J. O., Tucker, G. T., et al. (2015). In Vitro Characterization of the Human Liver Microsomal Kinetics and Reaction Phenotyping of Olanzapine Metabolism. Drug Metab. Disposit. 43, 1806–1814. doi: 10.1124/dmd.115.064790
Korzekwa, K. R., Krishnamachary, N., Shou, M., Ogai, A., Parise, R. A., Rettie, A. E., et al. (1998). Evaluation of atypical cytochrome P450 kinetics with two-substrate models: evidence that multiple substrates can simultaneously bind to cytochrome P450 active sites. Biochemistry 37, 4137–4147. doi: 10.1021/bi9715627
Koskinen, J., Lohonen, J., Koponen, H., Isohanni, M., and Miettunen, J. (2010). Rate of Cannabis Use Disorders in Clinical Samples of Patients With Schizophrenia: A Meta-analysis. Schizophrenia Bull. 36, 1115–1130. doi: 10.1093/schbul/sbp031
Koukouritaki, S. B., Manro, J. R., Marsh, S. A., Stevens, J. C., Rettie, A. E., McCarver, D. G., et al. (2004). Developmental expression of human hepatic CYP2C9 and CYP2C19. J. Pharmacol. Exp. Ther. 308, 965–974. doi: 10.1124/jpet.103.060137
Koyama, E., Chiba, K., Tani, M., and Ishizaki, T. (1997). Reappraisal of human CYP isoforms involved in imipramine N-demethylation and 2-hydroxylation: a study using microsomes obtained from putative extensive and poor metabolizers of S-mephenytoin and eleven recombinant human CYPs. J. Pharmacol. Exp. Ther. 281, 1199–1210.
Krivoy, A., Gaughran, F., Weizman, A., Breen, G., and MacCabe, J. H. (2016). Gene polymorphisms potentially related to the pharmacokinetics of clozapine: a systematic review. Int. Clin. Psychopharmacol. 31, 179–184. doi: 10.1097/YIC.0000000000000065
Krueger, S. K., and Williams, D. E. (2005). Mammalian flavin-containing monooxygenases: structure/function, genetic polymorphisms and role in drug metabolism. Pharmacol. Ther. 106, 357–387. doi: 10.1016/j.pharmthera.2005.01.001
Krueger, S. K., Williams, D. E., Yueh, M. F., Martin, S. R., Hines, R. N., Raucy, J. L., et al. (2002). Genetic polymorphisms of flavin-containing monooxygenase (FMO). Drug Metab. Rev. 34, 523–532. doi: 10.1081/DMR-120005653
Kubo, M., Koue, T., Maune, H., Fukuda, T., and Azuma, J. (2007). Pharmacokinetics of aripiprazole, a new antipsychotic, following oral dosing in healthy adult Japanese volunteers: influence of CYP2D6 polymorphism. Drug Metab. Pharmacokinet 22, 358–366. doi: 10.2133/dmpk.22.358
Kudo, S., and Ishizaki, T. (1999). Pharmacokinetics of haloperidol: an update. Clin. Pharmacokinetics 37, 435–456. doi: 10.2165/00003088-199937060-00001
Kuehl, P., Zhang, J., Lin, Y., Lamba, J., Assem, M., Schuetz, J., et al. (2001). Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat. Genet 27, 383–391. doi: 10.1038/86882
Kuhn, R. (1958). The treatment of depressive states with G 22355 (imipramine hydrochloride). Am. J. Psychiatry 115, 459–464. doi: 10.1176/ajp.115.5.459
Kumana, C. R., Lauder, I. J., Chan, M., Ko, W., and Lin, H. J. (1987). Differences in diazepam pharmacokinetics in Chinese and white Caucasians–relation to body lipid stores. Eur. J. Clin. Pharmacol. 32, 211–215. doi: 10.1007/bf00542199
Kumar, A., and Strech, D. (2005). Zuclopenthixol dihydrochloride for schizophrenia. Cochrane Data. System. Rev. 4:CD005474. doi: 10.1002/14651858.CD005474
Kutcher, S., Brooks, S. J., Gardner, D. M., Honer, B., Kopala, L., Labelle, A., et al. (2005). Expert Canadian consensus suggestions on the rational, clinical use of ziprasidone in the treatment of schizophrenia and related psychotic disorders. Neuropsychiatr. Dis. Treat. 1, 89–108. doi: 10.2147/nedt.1.2.89.61042
Lai, X. S., Yang, L. P., Li, X. T., Liu, J. P., Zhou, Z. W., and Zhou, S. F. (2009). Human CYP2C8: structure, substrate specificity, inhibitor selectivity, inducers and polymorphisms. Curr. Drug Metab. 10, 1009–1047. doi: 10.2174/138920009790711832
Laika, B., Leucht, S., Heres, S., Schneider, H., and Steimer, W. (2010). Pharmacogenetics and olanzapine treatment: CYP1A2∗1F and serotonergic polymorphisms influence therapeutic outcome. Pharmacogenom. J. 10, 20–29. doi: 10.1038/tpj.2009.32
Laird, B., Colvin, L., and Fallon, M. (2008). Management of cancer pain: basic principles and neuropathic cancer pain. Eur. J. Cancer 44, 1078–1082. doi: 10.1016/j.ejca.2008.03.022
Lam, Y. W., Gaedigk, A., Ereshefsky, L., Alfaro, C. L., and Simpson, J. (2002). CYP2D6 inhibition by selective serotonin reuptake inhibitors: analysis of achievable steady-state plasma concentrations and the effect of ultrarapid metabolism at CYP2D6. Pharmacotherapy 22, 1001–1006. doi: 10.1592/phco.22.12.1001.33603
Lamba, J., and Schuetz, E. (2008). Genetic Variants of Xenobiotic Receptors and their Implications in Drug Metabolism and Pharmacogenetics. Nuclear Recept. Drug Metab. 2008, 241–273. doi: 10.1002/9780470409107.ch9
Lamba, J., Lamba, V., and Schuetz, E. (2005). Genetic variants of PXR (NR1I2) and CAR (NR1I3) and their implications in drug metabolism and pharmacogenetics. Curr. Drug Metab. 6, 369–383. doi: 10.2174/1389200054633880
Lamba, V., Lamba, J., Yasuda, K., Strom, S., Davila, J., Hancock, M. L., et al. (2003). Hepatic CYP2B6 expression: gender and ethnic differences and relationship to CYP2B6 genotype and CAR (constitutive androstane receptor) expression. J. Pharmacol. Exp. Ther. 307, 906–922. doi: 10.1124/jpet.103.054866
Lampe, J. W., King, I. B., Li, S., Grate, M. T., Barale, K. V., Chen, C., et al. (2000). Brassica vegetables increase and apiaceous vegetables decrease cytochrome P450 1A2 activity in humans: changes in caffeine metabolite ratios in response to controlled vegetable diets. Carcinogenesis 21, 1157–1162. doi: 10.1093/carcin/21.5.157
Lang, T., Klein, K., Fischer, J., Nüssler, A. K., Neuhaus, P., Hofmann, U., et al. (2001). Extensive genetic polymorphism in the human CYP2B6 gene with impact on expression and function in human liver. Pharmacogenetics 11, 399–415.
Lang, T., Klein, K., Richter, T., Zibat, A., Kerb, R., Eichelbaum, M., et al. (2004). Multiple novel nonsynonymous CYP2B6 gene polymorphisms in Caucasians: demonstration of phenotypic null alleles. J. Pharmacol Exp. Ther. 311, 34–43. doi: 10.1124/jpet.104.068973
Le Marchand, L., Franke, A. A., Custer, L., Wilkens, L. R., and Cooney, R. V. (1997). Lifestyle and nutritional correlates of cytochrome CYP1A2 activity: inverse associations with plasma lutein and alpha-tocopherol. Pharmacogenetics 7, 11–19. doi: 10.1097/00008571-199702000-00002
Leclerc, J., Courcot-Ngoubo Ngangue, E., Cauffiez, C., Allorge, D., Pottier, N., Lafitte, J. J., et al. (2011). Xenobiotic metabolism and disposition in human lung: transcript profiling in non-tumoral and tumoral tissues. Biochimie 93, 1012–1027. doi: 10.1016/j.biochi.2011.02.012
Lee, C. R., Goldstein, J. A., and Pieper, J. A. (2002). Cytochrome P450 2C9 polymorphisms: a comprehensive review of the in-vitro and human data. Pharmacogenetics 12, 251–263. doi: 10.1097/00008571-200204000-00010
Lesche, D., Mostafa, S., Everall, I., Pantelis, C., and Bousman, C. A. (2020). Impact of CYP1A2, CYP2C19, and CYP2D6 genotype- and phenoconversion-predicted enzyme activity on clozapine exposure and symptom severity. Pharmacogen. J. 20, 192–201. doi: 10.1038/s41397-019-0108-y
Lett, T. A., Walter, H., and Brandl, E. J. (2016). Pharmacogenetics and Imaging-Pharmacogenetics of Antidepressant Response: Towards Translational Strategies. CNS Drugs 30, 1169–1189. doi: 10.1007/s40263-016-0385-9
Li, F., Zhang, A., Shi, Y., Ma, Y., and Du, Y. (2015). 1alpha,25-Dihydroxyvitamin D3 prevents the differentiation of human lung fibroblasts via microRNA-27b targeting the vitamin D receptor. Int. J. Mol. Med. 36, 967–974. doi: 10.3892/ijmm.2015.2318
Liangpunsakul, S., Kolwankar, D., Pinto, A., Gorski, J. C., Hall, S. D., and Chalasani, N. (2005). Activity of CYP2E1 and CYP3A enzymes in adults with moderate alcohol consumption: a comparison with nonalcoholics. Hepatology 41, 1144–1150. doi: 10.1002/hep.20673
Lin, C. W., Huang, W. I., Chao, P. H., Chen, W. W., and Hsiao, F. Y. (2018). Temporal trends and patterns in carbamazepine use, related severe cutaneous adverse reactions, and HLA-B∗15:02 screening: A nationwide study. Epilepsia 59, 2325–2339. doi: 10.1111/epi.14599
Linnet, K., and Wiborg, O. (1996). Steady-state serum concentrations of the neuroleptic perphenazine in relation to CYP2D6 genetic polymorphism. Clin. Pharmacol. Ther. 60, 41–47. doi: 10.1016/S0009-9236(96)90165-4
Liston, H. L., DeVane, C. L., Boulton, D. W., Risch, S. C., Markowitz, J. S., and Goldman, J. (2002). Differential time course of cytochrome P450 2D6 enzyme inhibition by fluoxetine, sertraline, and paroxetine in healthy volunteers. J. Clin. Psychopharmacol. 22, 169–173. doi: 10.1097/00004714-200204000-00010
Liston, H. L., Markowitz, J. S., and DeVane, C. L. (2001). Drug glucuronidation in clinical psychopharmacology. J. Clin. Psychopharmacol. 21, 500–515. doi: 10.1097/00004714-200110000-00008
Liu, H. Q., Zhang, C. P., Zhang, C. Z., Liu, X. C., and Liu, Z. J. (2015). Influence of two common polymorphisms in the EPHX1 gene on warfarin maintenance dosage: a meta-analysis. Biomed. Res. Int. 2015:564149. doi: 10.1155/2015/564149
Lobo, E. D., Bergstrom, R. F., Reddy, S., Quinlan, T., Chappell, J., Hong, Q., et al. (2008). In vitro and in vivo evaluations of cytochrome P450 1A2 interactions with duloxetine. Clin. Pharmacokinet 47, 191–202. doi: 10.2165/00003088-200847030-00005
Lodhi, R. J., Wang, Y., Rossolatos, D., MacIntyre, G., Bowker, A., Crocker, C., et al. (2017). Investigation of the COMT Val158Met variant association with age of onset of psychosis, adjusting for cannabis use. Brain Behav. 7:e00850. doi: 10.1002/brb3.850
Lomri, N., Yang, Z., and Cashman, J. R. (1993). Expression in Escherichia coli of the flavin-containing monooxygenase D (form II) from adult human liver: determination of a distinct tertiary amine substrate specificity. Chem. Res. Toxicol. 6, 425–429. doi: 10.1021/tx00034a006
Löscher, W., Klotz, U., Zimprich, F., and Schmidt, D. (2009). The clinical impact of pharmacogenetics on the treatment of epilepsy. Epilepsia 50, 1–23. doi: 10.1111/j.1528-1167.2008.01716.x
Lu, M. L., Wu, Y. X., Chen, C. H., Kuo, P. T., Chen, Y. H., Lin, C. H., et al. (2016). Application of plasma levels of olanzapine and N-desmethyl-olanzapine to monitor clinical efficacy in patients with schizophrenia. PloS one 11:e0148539. doi: 10.1371/journal.pone.0148539
Lubomirov, R., Colombo, S., di Iulio, J., Ledergerber, B., Martinez, R., Cavassini, M., et al. (2011). Association of pharmacogenetic markers with premature discontinuation of first-line anti-HIV therapy: an observational cohort study. J. Infect. Dis. 203, 246–257. doi: 10.1093/infdis/jiq043
Lund, M., Petersen, T. S., and Dalhoff, K. P. (2017). Clinical Implications of P-Glycoprotein Modulation in Drug-Drug Interactions. Drugs 77, 859–883. doi: 10.1007/s40265-017-0729-x
Ma, M. K., Woo, M. H., and McLeod, H. L. (2002). Genetic basis of drug metabolism. Am. J. Health Sys. 59, 2061–2069. doi: 10.1093/ajhp/59.21.2061
Maagdenberg, H., Bierings, M. B., van Ommen, C. H., van der Meer, F. J., Appel, I. M., Tamminga, R. Y., et al. (2018). Effects of age and genetic variations in VKORC1, CYP2C9 and CYP3A4 on the phenprocoumon dose in pediatric patients. Pharmacogenomics 19, 1195–1202. doi: 10.2217/pgs-2018-0095
Mackay, R. J., McEntyre, C. J., Henderson, C., Lever, M., and George, P. M. (2011). Trimethylaminuria: causes and diagnosis of a socially distressing condition. Clin. Biochem. Rev. 32, 33–43.
Mackenzie, P. I., Bock, K. W., Burchell, B., Guillemette, C., Ikushiro, S., Iyanagi, T., et al. (2005). Nomenclature update for the mammalian UDP glycosyltransferase (UGT) gene superfamily. Pharmacog. Genom. 15, 677–685. doi: 10.1097/01.fpc.0000173483.13689.56
Mahatthanatrakul, W., Nontaput, T., Ridtitid, W., Wongnawa, M., and Sunbhanich, M. (2007). Rifampin, a cytochrome P450 3A inducer, decreases plasma concentrations of antipsychotic risperidone in healthy volunteers. J. Clin. Pharm. Ther. 32, 161–167. doi: 10.1111/j.1365-2710.2007.00811.x
Mahgoub, A., Idle, J. R., Dring, L. G., Lancaster, R., and Smith, R. L. (1977). Polymorphic hydroxylation of Debrisoquine in man. Lancet 2, 584–586. doi: 10.1016/s0140-6736(77)91430-1
Mailman, R. B., and Murthy, V. (2010). Third generation antipsychotic drugs: partial agonism or receptor functional selectivity? Curr. Pharm. Des. 16, 488–501. doi: 10.2174/138161210790361461
Majewska, M. D., Mienville, J. M., and Vicini, S. (1988). Neurosteroid pregnenolone sulfate antagonizes electrophysiological responses to GABA in neurons. Neurosci. Lett. 90, 279–284. doi: 10.1016/0304-3940(88)90202-9
Malhi, G. S., and Mann, J. J. (2018). Depression. Lancet 392, 2299–2312. doi: 10.1016/S0140-6736(18)31948-2
Malhotra, A. K., Zhang, J. P., and Lencz, T. (2012). Pharmacogenetics in psychiatry: translating research into clinical practice. Mol. Psychiatry 17, 760–769. doi: 10.1038/mp.2011.146
Mallikaarjun, S., Kane, J. M., Bricmont, P., McQuade, R., Carson, W., Sanchez, R., et al. (2013). Pharmacokinetics, tolerability and safety of aripiprazole once-monthly in adult schizophrenia: an open-label, parallel-arm, multiple-dose study. Schizophrenia Res. 150, 281–288. doi: 10.1016/j.schres.2013.06.041
Mamiya, K., Ieiri, I., Shimamoto, J., Yukawa, E., Imai, J., Ninomiya, H., et al. (1998). The effects of genetic polymorphisms of CYP2C9 and CYP2C19 on phenytoin metabolism in Japanese adult patients with epilepsy: studies in stereoselective hydroxylation and population pharmacokinetics. Epilepsia 39, 1317–1323. doi: 10.1111/j.1528-1157.1998.tb01330.x
Mammen, G., Rueda, S., Roerecke, M., Bonato, S., Lev-Ran, S., and Rehm, J. (2018). Association of Cannabis With Long-Term Clinical Symptoms in Anxiety and Mood Disorders: A Systematic Review of Prospective Studies. J. Clin. Psychiatry 79, :17r11839. doi: 10.4088/JCP.17r11839
Mannheimer, B., Haslemo, T., Lindh, J. D., Eliasson, E., and Molden, E. (2016). Risperidone and Venlafaxine Metabolic Ratios Strongly Predict a CYP2D6 Poor Metabolizing Genotype. Therapeut. Drug Monit. 38, 127–134. doi: 10.1097/FTD.0000000000000251
Mannheimer, B., Holm, J., Koukel, L., Bertilsson, L., Osby, U., and Eliasson, E. (2014). Risperidone metabolic ratio as a biomarker of individual CYP2D6 genotype in schizophrenic patients. Eur. J. Clin. Pharmacol. 70, 695–699. doi: 10.1007/s00228-014-1664-3
Mannheimer, B., von Bahr, C., Pettersson, H., and Eliasson, E. (2008). Impact of multiple inhibitors or substrates of cytochrome P450 2D6 on plasma risperidone levels in patients on polypharmacy. Ther. Drug Monit. 30, 565–569. doi: 10.1097/FTD.0b013e31818679c9
Martinez, C., Blanco, G., Ladero, J. M., Garcia-Martin, E., Taxonera, C., Gamito, F. G., et al. (2004). Genetic predisposition to acute gastrointestinal bleeding after NSAIDs use. Br. J. Pharmacol. 141, 205–208. doi: 10.1038/sj.bjp.0705623
Martis, S., Mei, H., Vijzelaar, R., Edelmann, L., Desnick, R. J., and Scott, S. A. (2013). Multi-ethnic cytochrome-P450 copy number profiling: novel pharmacogenetic alleles and mechanism of copy number variation formation. Pharmacog. J. 13, 558–566. doi: 10.1038/tpj.2012.48
Maruf, A. A., Fan, M., Arnold, P. D., Müller, D. J., Aitchison, K. J., and Bousman, C. A. (2020). Pharmacogenetic Testing Options Relevant to Psychiatry in Canada: Options de tests pharmacogénétiques pertinents en psychiatrie au Canada. Canadian journal of psychiatry. Revue Can. De Psychiatrie 65, 521–530. doi: 10.1177/0706743720904820
Masimirembwa, C., Persson, I., Bertilsson, L., Hasler, J., and Ingelman-Sundberg, M. (1996). A novel mutant variant of the CYP2D6 gene (CYP2D6∗17) common in a black African population: association with diminished debrisoquine hydroxylase activity. Br. J. Clin. Pharmacol. 42, 713–719. doi: 10.1046/j.1365-2125.1996.00489.x
Matsui, A., Azuma, J., Witcher, J. W., Long, A. J., Sauer, J. M., Smith, B. P., et al. (2012). Pharmacokinetics, safety, and tolerability of atomoxetine and effect of CYP2D6∗10/∗10 genotype in healthy Japanese men. J. Clin. Pharmacol. 52, 388–403. doi: 10.1177/0091270011398657
Mazerska, Z., Mroz, A., Pawlowska, M., and Augustin, E. (2016). The role of glucuronidation in drug resistance. Pharmacol. Ther. 159, 35–55. doi: 10.1016/j.pharmthera.2016.01.009
McConnell, H. W., Mitchell, S. C., Smith, R. L., and Brewster, M. (1997). Trimethylaminuria associated with seizures and behavioural disturbance: a case report. Seizure 6, 317–321. doi: 10.1016/s1059-1311(97)80080-5
McDonagh, E. M., Wassenaar, C., David, S. P., Tyndale, R. F., Altman, R. B., Whirl-Carrillo, M., et al. (2012). PharmGKB summary: very important pharmacogene information for cytochrome P-450, family 2, subfamily A, polypeptide 6. Pharmacogen. Genom. 22, 695–708. doi: 10.1097/FPC.0b013e3283540217
McManus, M. E., Burgess, W. M., Veronese, M. E., Huggett, A., Quattrochi, L. C., and Tukey, R. H. (1990). Metabolism of 2-acetylaminofluorene and benzo(a)pyrene and activation of food-derived heterocyclic amine mutagens by human cytochromes P-450. Cancer Res. 50, 3367–3376.
Miceli, J. J., Anziano, R. J., Robarge, L., Hansen, R. A., and Laurent, A. (2000). The effect of carbamazepine on the steady-state pharmacokinetics of ziprasidone in healthy volunteers. Br. J. Clin. Pharmacol. 49, 65s–70s. doi: 10.1046/j.1365-2125.2000.00157.x
Michaels, S., and Wang, M. Z. (2014). The revised human liver cytochrome P450 “Pie”: absolute protein quantification of CYP4F and CYP3A enzymes using targeted quantitative proteomics. Drug Metab. Dispos. 42, 1241–1251. doi: 10.1124/dmd.114.058040
Mittal, B., Tulsyan, S., Kumar, S., Mittal, R. D., Agarwal, G., and Makowski, G. S. (2015). Chapter Four - Cytochrome P450 in Cancer Susceptibility and Treatment, New Jersey: Elsevier 71, 77–139.
Molden, E., Waade, R. B., Hoff, M., and Haslemo, T. (2016). Impact of Ageing on Serum Concentrations of Risperidone and Its Active Metabolite in Patients with Known CYP2D6 Genotype. Basic Clin. Pharmacol. Toxicol. 119, 470–475. doi: 10.1111/bcpt.12614
Mori, A., Maruo, Y., Iwai, M., Sato, H., and Takeuchi, Y. (2005). UDP-glucuronosyltransferase 1A4 polymorphisms in a Japanese population and kinetics of clozapine glucuronidation. Drug Metab. Dispos. Biol. Fate Chem. 33, 672–675. doi: 10.1124/dmd.104.002576
Motika, M. S., Zhang, J., and Cashman, J. R. (2007). Flavin-containing monooxygenase 3 and human disease. Exp. Opin. Drug Metab. Toxicol. 3, 831–845. doi: 10.1517/17425255.3.6.831
Mrazek, D. A., Biernacka, J. M., O’Kane, D. J., Black, J. L., Cunningham, J. M., Drews, M. S., et al. (2011). CYP2C19 variation and citalopram response. Pharmacogen. Genom. 21, 1–9. doi: 10.1097/fpc.0b013e328340bc5a
Murai, K., Yamazaki, H., Nakagawa, K., Kawai, R., and Kamataki, T. (2009). Deactivation of anti-cancer drug letrozole to a carbinol metabolite by polymorphic cytochrome P450 2A6 in human liver microsomes. Xenobiotica 39, 795–802. doi: 10.3109/00498250903171395
Muralidharan, G., Cooper, J. K., Hawes, E. M., Korchinski, E. D., and Midha, K. K. (1996). Quinidine inhibits the 7-hydroxylation of chlorpromazine in extensive metabolisers of debrisoquine. Eur. J. Clin. Pharmacol. 50, 121–128. doi: 10.1007/s002280050079
Murray, G. I., Melvin, W. T., Greenlee, W. F., and Burke, M. D. (2001). Regulation, function, and tissue-specific expression of cytochrome P450 CYP1B1. Annu. Rev. Pharmacol. Toxicol. 41, 297–316. doi: 10.1146/annurev.pharmtox.41.1.297
Murru, A., Popovic, D., Pacchiarotti, I., Hidalgo, D., León-Caballero, J., and Vieta, E. (2015). Management of adverse effects of mood stabilizers. Curr. Psychiatry Rep. 17:603. doi: 10.1007/s11920-015-0603-z
Mwenifumbo, J. C., Sellers, E. M., and Tyndale, R. F. (2007). Nicotine metabolism and CYP2A6 activity in a population of black African descent: impact of gender and light smoking. Drug Alcohol. Depend. 89, 24–33. doi: 10.1016/j.drugalcdep.2006.11.012
Nagaoka, R., Iwasaki, T., Rokutanda, N., Takeshita, A., Koibuchi, Y., Horiguchi, J., et al. (2006). Tamoxifen activates CYP3A4 and MDR1 genes through steroid and xenobiotic receptor in breast cancer cells. Endocrine 30, 261–268. doi: 10.1007/s12020-006-0003-6
Nakajima, M., Fukami, T., Yamanaka, H., Higashi, E., Sakai, H., Yoshida, R., et al. (2006). Comprehensive evaluation of variability in nicotine metabolism and CYP2A6 polymorphic alleles in four ethnic populations. Clin. Pharmacol. Ther. 80, 282–297. doi: 10.1016/j.clpt.2006.05.012
Nakajima, M., Yokoi, T., Mizutani, M., Kinoshita, M., Funayama, M., and Kamataki, T. (1999). Genetic polymorphism in the 5’-flanking region of human CYP1A2 gene: effect on the CYP1A2 inducibility in humans. J. Biochem. 125, 803–808. doi: 10.1093/oxfordjournals.jbchem.a022352
Nakajima, Y., Saito, Y., Shiseki, K., Fukushima-Uesaka, H., Hasegawa, R., Ozawa, S., et al. (2005). Haplotype structures of EPHX1 and their effects on the metabolism of carbamazepine-10,11-epoxide in Japanese epileptic patients. Eur. J. Clin. Pharmacol. 61, 25–34. doi: 10.1007/s00228-004-0878-1
Naraharisetti, S. B., Lin, Y. S., Rieder, M. J., Marciante, K. D., Psaty, B. M., Thummel, K. E., et al. (2010). Human liver expression of CYP2C8: gender, age, and genotype effects. Drug Metab. Dispos. 38, 889–893. doi: 10.1124/dmd.109.031542
Nebert, D. W., Adesnik, M., Coon, M. J., Estabrook, R. W., Gonzalez, F. J., Guengerich, F. P., et al. (1987). The P450 gene superfamily: recommended nomenclature. DNA 6, 1–11. doi: 10.1089/dna.1987.6.1
Nebert, D. W., and Russell, D. W. (2002). Clinical importance of the cytochromes P450. Lancet 360, 1155–1162. doi: 10.1016/S0140-6736(02)11203-7
Nelson, D. R., Zeldin, D. C., Hoffman, S. M., Maltais, L. J., Wain, H. M., and Nebert, D. W. (2004). Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics 14, 1–18. doi: 10.1097/00008571-200401000-00001
Niwa, T., Shizuku, M., and Yamano, K. (2017). Effect of genetic polymorphism on the inhibition of dopamine formation from p-tyramine catalyzed by brain cytochrome P450 2D6. Arch. Biochem. Biophys. 620, 23–27. doi: 10.1016/j.abb.2017.03.009
Niznik, H. B., Tyndale, R. F., Sallee, F. R., Gonzalez, F. J., Hardwick, J. P., Inaba, T., et al. (1990). The dopamine transporter and cytochrome P45OIID1 (debrisoquine 4-hydroxylase) in brain: resolution and identification of two distinct [3H]GBR-12935 binding proteins. Arch. Biochem. Biophys. 276, 424–432. doi: 10.1016/0003-9861(90)90741-g
Nozawa, M., Ohnuma, T., Matsubara, Y., Sakai, Y., Hatano, T., Hanzawa, R., et al. (2008). The relationship between the response of clinical symptoms and plasma olanzapine concentration, based on pharmacogenetics: Juntendo University Schizophrenia Projects (JUSP). Ther. Drug Monit. 30, 35–40. doi: 10.1097/FTD.0b013e31816336fd
Nuntamool, N., Ngamsamut, N., Vanwong, N., Puangpetch, A., Chamnanphon, M., Hongkaew, Y., et al. (2017). Pharmacogenomics and Efficacy of Risperidone Long-Term Treatment in Thai Autistic Children and Adolescents. Basic Clin. Pharmacol. Toxicol. 121, 316–324. doi: 10.1111/bcpt.12803
Nyberg, S., Farde, L., Halldin, C., Dahl, M. L., and Bertilsson, L. (1995). D2 dopamine receptor occupancy during low-dose treatment with haloperidol decanoate. Am. J. Psychiatry 152, 173–178. doi: 10.1176/ajp.152.2.173
Obach, R. S., and Walsky, R. L. (2005). Drugs that inhibit oxidation reactions catalyzed by aldehyde oxidase do not inhibit the reductive metabolism of ziprasidone to its major metabolite. J. Clin. Psychopharmacol. 25, 605–608. doi: 10.1097/01.jcp.0000186740.22395.50
Obase, Y., Shimoda, T., Kawano, T., Saeki, S., Tomari, S. Y., Mitsuta-Izaki, K., et al. (2003). Polymorphisms in the CYP1A2 gene and theophylline metabolism in patients with asthma. Clin. Pharmacol. Ther. 73, 468–474. doi: 10.1016/s0009-9236(03)00013-4
O’Brien, F. E., Dinan, T. G., Griffin, B. T., and Cryan, J. F. (2012). Interactions between antidepressants and P-glycoprotein at the blood-brain barrier: clinical significance of in vitro and in vivo findings. Br. J. Pharmacol. 165, 289–312. doi: 10.1111/j.1476-5381.2011.01557.x
Ogg, M. S., Brennan, P., Meade, T., and Humphries, S. E. (1999). CYP2C9∗3 allelic variant and bleeding complications. Lancet 354:1124. doi: 10.1016/S0140-6736(05)76918-X
Ohmori, O., Suzuki, T., Kojima, H., Shinkai, T., Terao, T., Mita, T., et al. (1998). Tardive dyskinesia and debrisoquine 4-hydroxylase (CYP2D6) genotype in Japanese schizophrenics. Schizophr. Res. 32, 107–113. doi: 10.1016/s0920-9964(98)00018-8
Ohnuma, T., Shibata, N., Matsubara, Y., and Arai, H. (2003). Haloperidol plasma concentration in Japanese psychiatric subjects with gene duplication of CYP2D6. Br. J. Clin. Pharmacol. 56, 315–320. doi: 10.1046/j.0306-5251.2003.01872.x
Ohtsuki, S., Schaefer, O., Kawakami, H., Inoue, T., Liehner, S., Saito, A., et al. (2012). Simultaneous absolute protein quantification of transporters, cytochromes P450, and UDP-glucuronosyltransferases as a novel approach for the characterization of individual human liver: comparison with mRNA levels and activities. Drug Metab. Dispos. 40, 83–92. doi: 10.1124/dmd.111.042259
Okubo, M., Murayama, N., Shimizu, M., Shimada, T., Guengerich, F. P., and Yamazaki, H. (2013). CYP3A4 intron 6 C >>T polymorphism (CYP3A4∗22) is associated with reduced CYP3A4 protein level and function in human liver microsomes. J. Toxicol. Sci. 38, 349–354. doi: 10.2131/jts.38.349
Okubo, M., Narita, M., Murayama, N., Akimoto, Y., Goto, A., and Yamazaki, H. (2016). Individual differences in in vitro and in vivo metabolic clearances of the antipsychotic drug olanzapine from non-smoking and smoking Japanese subjects genotyped for cytochrome P4502D6 and flavincontaining monooxygenase 3. Hum. Psychopharmacol. 31, 83–92. doi: 10.1002/hup.2515
Olesen, O. V., and Linnet, K. (2000). Identification of the human cytochrome P450 isoforms mediating in vitro N-dealkylation of perphenazine. Br. J. Clin. Pharmacol. 50, 563–571. doi: 10.1046/j.1365-2125.2000.00298.x
Oscarson, M., Gullsten, H., Rautio, A., Bernal, M. L., Sinues, B., Dahl, M. L., et al. (1998). Genotyping of human cytochrome P450 2A6 (CYP2A6), a nicotine C-oxidase. FEBS Lett. 438, 201–205. doi: 10.1016/s0014-5793(98)01297-6
Oscarson, M., Hidestrand, M., Johansson, I., and Ingelman-Sundberg, M. (1997). A combination of mutations in the CYP2D6∗17 (CYP2D6Z) allele causes alterations in enzyme function. Mol. Pharmacol. 52, 1034–1040. doi: 10.1124/mol.52.6.1034
Otton, S. V., Ball, S. E., Cheung, S. W., Inaba, T., Rudolph, R. L., and Sellers, E. M. (1996). Venlafaxine oxidation in vitro is catalysed by CYP2D6. Br. J. Clin. Pharmacol. 41, 149–156. doi: 10.1111/j.1365-2125.1996.tb00173.x
Otton, S. V., Gillam, E. M., Lennard, M. S., Tucker, G. T., and Woods, H. F. (1990). Propranolol oxidation by human liver microsomes–the use of cumene hydroperoxide to probe isoenzyme specificity and regio- and stereoselectivity. Br. J. Clin. Pharmacol. 30, 751–760. doi: 10.1111/j.1365-2125.1990.tb03846.x
Ouzzine, M., Gulberti, S., Ramalanjaona, N., Magdalou, J., and Fournel-Gigleux, S. (2014). The UDP-glucuronosyltransferases of the blood-brain barrier: their role in drug metabolism and detoxication. Front. Cell Neurosci. 8:349. doi: 10.3389/fncel.2014.00349
Ouzzine, M., Magdalou, J., Burchell, B., and Fournel-Gigleux, S. (1999). An internal signal sequence mediates the targeting and retention of the human UDP-glucuronosyltransferase 1A6 to the endoplasmic reticulum. J. Biol. Chem. 274, 31401–31409. doi: 10.1074/jbc.274.44.31401
Owens, M. J. (1996). Molecular and cellular mechanisms of antidepressant drugs. Depress Anxiety 4, 153–159. doi: 10.1002/(SICI)1520-6394(1996)4
Owens, M. J., Knight, D. L., and Nemeroff, C. B. (2001). Second-generation SSRIs: human monoamine transporter binding profile of escitalopram and R-fluoxetine. Biol. Psychiatry 50, 345–350. doi: 10.1016/s0006-3223(01)01145-3
Owens, M. J., Morgan, W. N., Plott, S. J., and Nemeroff, C. B. (1997). Neurotransmitter receptor and transporter binding profile of antidepressants and their metabolites. J. Pharmacol. Exp. Ther. 283, 1305–1322.
Ozdemir, V., Naranjo, C. A., Herrmann, N., Reed, K., Sellers, E. M., and Kalow, W. (1997). Paroxetine potentiates the central nervous system side effects of perphenazine: contribution of cytochrome P4502D6 inhibition in vivo. Clin. Pharmacol. Ther. 62, 334–347. doi: 10.1016/S0009-9236(97)90037-0
Pacifici, G. M., Bencini, C., and Rane, A. (1986). Acetyltransferase in humans: development and tissue distribution. Pharmacology 32, 283–291. doi: 10.1159/000138181
Palleria, C., Di Paolo, A., Giofrè, C., Caglioti, C., Leuzzi, G., Siniscalchi, A., et al. (2013). Pharmacokinetic drug-drug interaction and their implication in clinical management. J. Res. Med. Sci. 18, 601–610.
Pan, L. P., De Vriendt, C., and Belpaire, F. M. (1998). In-vitro characterization of the cytochrome P450 isoenzymes involved in the back oxidation and N-dealkylation of reduced haloperidol. Pharmacogenetics 8, 383–389. doi: 10.1097/00008571-199810000-00003
Pan, Y. Z., Gao, W., and Yu, A. M. (2009). MicroRNAs regulate CYP3A4 expression via direct and indirect targeting. Drug Metab. Dispos. 37, 2112–2117. doi: 10.1124/dmd.109.027680
Pang, C., Liu, J. H., Xu, Y. S., Chen, C., and Dai, P. G. (2015). The allele frequency of CYP2A6∗4 in four ethnic groups of China. Exp. Mole. Pathol. 98, 546–548. doi: 10.1016/j.yexmp.2015.03.040
Pariente-Khayat, A., Pons, G., Rey, E., Richard, M. O., D’Athis, P., Moran, C., et al. (1991). Caffeine acetylator phenotyping during maturation in infants. Pediatr. Res. 29, 492–495. doi: 10.1203/00006450-199105010-00015
Parker, A. C., Pritchard, P., Preston, T., and Choonara, I. (1998). Induction of CYP1A2 activity by carbamazepine in children using the caffeine breath test. Br. J. Clin. Pharmacol. 45, 176–178. doi: 10.1046/j.1365-2125.1998.00684.x
Paulzen, M., Haen, E., Hiemke, C., Stegmann, B., Lammertz, S. E., Gründer, G., et al. (2017). Cytochrome P450-mediated interaction between perazine and risperidone: implications for antipsychotic polypharmacy. Br. J. Clin. Pharmacol. 83, 1668–1675. doi: 10.1111/bcp.13255
Pelkonen, O., Rautio, A., Raunio, H., and Pasanen, M. (2000). CYP2A6: a human coumarin 7-hydroxylase. Toxicology 144, 139–147. doi: 10.1016/s0300-483x(99)00200-0
Pelkonen, O., Turpeinen, M., Hakkola, J., Honkakoski, P., Hukkanen, J., and Raunio, H. (2008). Inhibition and induction of human cytochrome P450 enzymes: current status. Arch. Toxicol. 82, 667–715. doi: 10.1007/s00204-008-0332-8
Perry, P. J., Pfohl, B. M., and Holstad, S. G. (1987). The relationship between antidepressant response and tricyclic antidepressant plasma concentrations. A retrospective analysis of the literature using logistic regression analysis. Clin. Pharmacokinet 13, 381–392. doi: 10.2165/00003088-198713060-00003
Persson, I., Aklillu, E., Rodrigues, F., Bertilsson, L., and Ingelman-Sundberg, M. (1996). S-mephenytoin hydroxylation phenotype and CYP2C19 genotype among Ethiopians. Pharmacogenetics 6, 521–526. doi: 10.1097/00008571-199612000-00005
Peters, E. J., Reus, V., and Hamilton, S. P. (2009). The ABCB1 transporter gene and antidepressant response. F1000 Biol. Rep. 1:23. doi: 10.3410/B1-23
Pharmacogene Variation Consortium: CYP2D6. Retrieved from https://www.pharmvar.org/gene/CYP2D6
PharmVar CYP3A5 Page. Retrieved from https://www.pharmvar.org/gene/CYP3A5
Phillips, E. J., Sukasem, C., Whirl-Carrillo, M., Müller, D. J., Dunnenberger, H. M., Chantratita, W., et al. (2018). Clinical Pharmacogenetics Implementation Consortium Guideline for HLA Genotype and Use of Carbamazepine and Oxcarbazepine: 2017 Update. Clin. Pharmacol. Ther. 103, 574–581. doi: 10.1002/cpt.1004
Phillips, I. R., and Shephard, E. A. (2017). Drug metabolism by flavin-containing monooxygenases of human and mouse. Exp. Opin. Drug Metab. Toxicol. 13, 167–181. doi: 10.1080/17425255.2017.1239718
Picard, N., Cresteil, T., Djebli, N., and Marquet, P. (2005). In vitro metabolism study of buprenorphine: evidence for new metabolic pathways. Drug Metab. Dispos. 33, 689–695. doi: 10.1124/dmd.105.003681
Pinto, N., and Dolan, M. E. (2011). Clinically relevant genetic variations in drug metabolizing enzymes. Curr. Drug Metab. 12, 487–497. doi: 10.2174/138920011795495321
Pisanu, C., Melis, C., and Squassina, A. (2016). Lithium Pharmacogenetics: Where Do We Stand? Drug Dev. Res. 77, 368–373. doi: 10.1002/ddr.21341
Preskorn, S. H. (1997). Clinically relevant pharmacology of selective serotonin reuptake inhibitors. An overview with emphasis on pharmacokinetics and effects on oxidative drug metabolism. Clin. Pharmacokinet 32, 1–21. doi: 10.2165/00003088-199700321-00003
Preskorn, S. H. (2003). Reproducibility of the in vivo effect of the selective serotonin reuptake inhibitors on the in vivo function of cytochrome P450 2D6: an update (part I). J. Psychiatr. Pract. 9, 150–158. doi: 10.1097/00131746-200303000-00006
Preskorn, S. H. (2012). Changes in the product label for pimozide illustrate both the promises and the challenges of personalized medicine. J. Clin. Psychiatry 73, 1191–1193. doi: 10.4088/JCP.12com07963
Preskorn, S. H. (2013). Complexities of personalized medicine: how genes, drug-drug interactions, dosing schedules, and other factors can combine to produce clinically meaningful differences in a drug’s effect. J. Psychiatr. Pract. 19, 397–405. doi: 10.1097/01.pra.0000435038.91049.cb
Preskorn, S. H., and Jerkovich, G. S. (1990). Central nervous system toxicity of tricyclic antidepressants: phenomenology, course, risk factors, and role of therapeutic drug monitoring. J. Clin. Psychopharmacol. 10, 88–95. doi: 10.1097/00004714-199004000-00003
Pringsheim, T., and Marras, C. (2009). Pimozide for tics in Tourette’s syndrome. Cochrane Database Syst. Rev. 2:CD006996. doi: 10.1002/14651858.CD006996.pub2
Puangpetch, A., Vanwong, N., Nuntamool, N., Hongkaew, Y., Chamnanphon, M., and Sukasem, C. (2016). CYP2D6 polymorphisms and their influence on risperidone treatment. Pharmgenom. Pers. Med. 9, 131–147. doi: 10.2147/PGPM.S107772
Puranik, Y. G., Birnbaum, A. K., Marino, S. E., Ahmed, G., Cloyd, J. C., Remmel, R. P., et al. (2013). Association of carbamazepine major metabolism and transport pathway gene polymorphisms and pharmacokinetics in patients with epilepsy. Pharmacogenomics 14, 35–45. doi: 10.2217/pgs.12.180
Qin, X. P., Xie, H. G., Wang, W., He, N., Huang, S. L., Xu, Z. H., et al. (1999). Effect of the gene dosage of CgammaP2C19 on diazepam metabolism in Chinese subjects. Clin. Pharmacol. Ther. 66, 642–646. doi: 10.1016/S0009-9236(99)90075-9
Rahikainen, A. L., Vauhkonen, P., Pett, H., Palo, J. U., Haukka, J., Ojanperä, I., et al. (2019). Completed suicides of citalopram users-the role of CYP genotypes and adverse drug interactions. Int. J. Legal. Med. 133, 353–363. doi: 10.1007/s00414-018-1927-0
Raimundo, S., Fischer, J., Eichelbaum, M., Griese, E. U., Schwab, M., and Zanger, U. M. (2000). Elucidation of the genetic basis of the common ‘intermediate metabolizer’ phenotype for drug oxidation by CYP2D6. Pharmacogenetics 10, 577–581. doi: 10.1097/00008571-200010000-00001
Raimundo, S., Toscano, C., Klein, K., Fischer, J., Griese, E. U., Eichelbaum, M., et al. (2004). A novel intronic mutation, 2988G >A, with high predictivity for impaired function of cytochrome P450 2D6 in white subjects. Clin. Pharmacol. Ther. 76, 128–138. doi: 10.1016/j.clpt.2004.04.009
Raskin, A., and Crook, T. H. (1975). Antidepressants in black and white inpatients. Differential response to a controlled trial of chlorpromazine and imipramine. Arch. Gen. Psychiatry 32, 643–649. doi: 10.1001/archpsyc.1975.01760230109008
Rau, T., Diepenbruck, S., Diepenbruck, I., and Eschenhagen, T. (2006). The 2988G - >A polymorphism affects splicing of a CYP2D6 minigene. Clin. Pharmacol. Ther. 80, 555–558. doi: 10.1016/j.clpt.2006.08.008
Raunio, H., and Rahnasto-Rilla, M. (2012). CYP2A6: genetics, structure, regulation, and function. Drug Metabol. Drug Interact. 27, 73–88. doi: 10.1515/dmdi-2012-0001
Reference SNP (refSNP) Cluster Report: rs3892097. Retrieved from https://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?rs==3892097
Reference SNP:rs2279343. Retrieved from https://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?do_not_redirect&rs==rs2279343
Relling, M. V., and Klein, T. E. (2011). CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin. Pharmacol. Ther. 89, 464–467. doi: 10.1038/clpt.2010.279
Relling, M. V., Gardner, E. E., Sandborn, W. J., Schmiegelow, K., Pui, C. H., Yee, S. W., et al. (2011). Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin. Pharmacol. Ther. 89, 387–391. doi: 10.1038/clpt.2010.320
Rettie, A. E., and Jones, J. P. (2005). Clinical and toxicological relevance of CYP2C9: drug-drug interactions and pharmacogenetics. Annu. Rev. Pharmacol. Toxicol. 45, 477–494. doi: 10.1146/annurev.pharmtox.45.120403.095821
Ribaudo, H. J., Haas, D. W., Tierney, C., Kim, R. B., Wilkinson, G. R., Gulick, R. M., et al. (2006). Pharmacogenetics of plasma efavirenz exposure after treatment discontinuation: an Adult AIDS Clinical Trials Group Study. Clin. Infect. Dis. 42, 401–407. doi: 10.1086/499364
Riches, Z., Stanley, E. L., Bloomer, J. C., and Coughtrie, M. W. (2009). Quantitative evaluation of the expression and activity of five major sulfotransferases (SULTs) in human tissues: the SULT “pie”. Drug Metab. Dispos. 37, 2255–2261. doi: 10.1124/dmd.109.028399
Rietveld, E. C., Broekman, M. M., Houben, J. J., Eskes, T. K., and van Rossum, J. M. (1984). Rapid onset of an increase in caffeine residence time in young women due to oral contraceptive steroids. Eur. J. Clin. Pharmacol. 26, 371–373. doi: 10.1007/bf00548769
Riordan, J. R., Deuchars, K., Kartner, N., Alon, N., Trent, J., and Ling, V. (1985). Amplification of P-glycoprotein genes in multidrug-resistant mammalian cell lines. Nature 316, 817–819. doi: 10.1038/316817a0
Rizzo, N., Hispard, E., Dolbeault, S., Dally, S., Leverge, R., and Girre, C. (1997). Impact of long-term ethanol consumption on CYP1A2 activity. Clin. Pharmacol. Ther. 62, 505–509. doi: 10.1016/S0009-9236(97)90045-X
Roberts, R. L., Mulder, R. T., Joyce, P. R., Luty, S. E., and Kennedy, M. A. (2004). No evidence of increased adverse drug reactions in cytochrome P450 CYP2D6 poor metabolizers treated with fluoxetine or nortriptyline. Hum. Psychopharmacol. 19, 17–23. doi: 10.1002/hup.539
Rochat, B., Amey, M., Gillet, M., Meyer, U. A., and Baumann, P. (1997). Identification of three cytochrome P450 isozymes involved in N-demethylation of citalopram enantiomers in human liver microsomes. Pharmacogenetics 7, 1–10. doi: 10.1097/00008571-199702000-00001
Rogers, H. L., Bhattaram, A., Zineh, I., Gobburu, J., Mathis, M., Laughren, T. P., et al. (2012). CYP2D6 genotype information to guide pimozide treatment in adult and pediatric patients: basis for the U.S. Food and Drug Administration’s new dosing recommendations. J. Clin. Psychiatry 73, 1187–1190. doi: 10.4088/JCP.11m07572
Roh, H. K., Dahl, M. L., Tybring, G., Yamada, H., Cha, Y. N., and Bertilsson, L. (1996). CYP2C19 genotype and phenotype determined by omeprazole in a Korean population. Pharmacogenetics 6, 547–551. doi: 10.1097/00008571-199612000-00008
Roke, Y., van Harten, P. N., Franke, B., Galesloot, T. E., Boot, A. M., and Buitelaar, J. K. (2013). The effect of the Taq1A variant in the dopamine D2 receptor gene and common CYP2D6 alleles on prolactin levels in risperidone-treated boys. Pharmacogen. Genom. 23, 487–493. doi: 10.1097/FPC.0b013e3283647c33
Roninson, I. B., Chin, J. E., Choi, K. G., Gros, P., Housman, D. E., Fojo, A., et al. (1986). Isolation of human mdr DNA sequences amplified in multidrug-resistant KB carcinoma cells. Proc. Natl. Acad. Sci. U S A 83, 4538–4542. doi: 10.1073/pnas.83.12.4538
Rost, K. L., Brosicke, H., Heinemeyer, G., and Roots, I. (1994). Specific and dose-dependent enzyme induction by omeprazole in human beings. Hepatology 20, 1204–1212. doi: 10.1002/hep.1840200516
Rotger, M., Tegude, H., Colombo, S., Cavassini, M., Furrer, H., Decosterd, L., et al. (2007). Predictive value of known and novel alleles of CYP2B6 for efavirenz plasma concentrations in HIV-infected individuals. Clin. Pharmacol. Ther. 81, 557–566. doi: 10.1038/sj.clpt.6100072
Rothman, R. B., Baumann, M. H., Dersch, C. M., Romero, D. V., Rice, K. C., Carroll, F. I., et al. (2001). Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse 39, 32–41. doi: 10.1002/1098-2396(20010101)39
Rowland, A., Elliot, D. J., Williams, J. A., Mackenzie, P. I., Dickinson, R. G., and Miners, J. O. (2006). In vitro characterization of lamotrigine N2-glucuronidation and the lamotrigine-valproic acid interaction. Drug Metab. Disp. 34, 1055–1062. doi: 10.1124/dmd.106.009340
Rudorfer, M. V., and Robins, E. (1982). Amitriptyline overdose: clinical effects on tricyclic antidepressant plasma levels. J. Clin. Psychiatry 43, 457–460.
Rudorfer, M. V., and Potter, W. Z. (1999). Metabolism of tricyclic antidepressants. Cell. Mole. Neurobiol. 19, 373–409. doi: 10.1023/a:1006949816036
Sachse, C., Brockmoller, J., Hildebrand, M., Muller, K., and Roots, I. (1998). Correctness of prediction of the CYP2D6 phenotype confirmed by genotyping 47 intermediate and poor metabolizers of debrisoquine. Pharmacogenetics 8, 181–185.
Saeed, L. H., and Mayet, A. Y. (2013). Genotype-phenotype analysis of CYP2C19 in healthy saudi individuals and its potential clinical implication in drug therapy. Int. J. Med. Sci. 10, 1497–1502. doi: 10.7150/ijms.6795
Sakuyama, K., Sasaki, T., Ujiie, S., Obata, K., Mizugaki, M., Ishikawa, M., et al. (2008). Functional characterization of 17 CYP2D6 allelic variants (CYP2D6.2, 10, 14A-B, 18, 27, 36, 39, 47-51, 53-55, and 57). Drug Metab. Dispos. 36, 2460–2467. doi: 10.1124/dmd.108.023242
Sanchez, C., and Hyttel, J. (1999). Comparison of the effects of antidepressants and their metabolites on reuptake of biogenic amines and on receptor binding. Cell Mol. Neurobiol. 19, 467–489.
Sanderson, S., Emery, J., and Higgins, J. (2005). CYP2C9 gene variants, drug dose, and bleeding risk in warfarin-treated patients: a HuGEnet systematic review and meta-analysis. Genet Med. 7, 97–104. doi: 10.1097/01.gim.0000153664.65759.cf
Sangkuhl, K., Stingl, J. C., Turpeinen, M., Altman, R. B., and Klein, T. E. (2014). PharmGKB summary: venlafaxine pathway. Pharmacog. Genom. 24, 62–72. doi: 10.1097/fpc.0000000000000003
Saruwatari, J., Ishitsu, T., and Nakagawa, K. (2010). Update on the Genetic Polymorphisms of Drug-Metabolizing Enzymes in Antiepileptic Drug Therapy. Pharmaceuticals 3, 2709–2732. doi: 10.3390/ph3082709
Savill, N. C., Buitelaar, J. K., Anand, E., Day, K. A., Treuer, T., Upadhyaya, H. P., et al. (2015). The efficacy of atomoxetine for the treatment of children and adolescents with attention-deficit/hyperactivity disorder: a comprehensive review of over a decade of clinical research. CNS Drugs 29, 131–151. doi: 10.1007/s40263-014-0224-9
Schneider, L. S., Cooper, T. B., Severson, J. A., Zemplenyi, T., and Sloane, R. B. (1988). Electrocardiographic changes with nortriptyline and 10-hydroxynortriptyline in elderly depressed outpatients. J. Clin. Psychopharmacol. 8, 402–408.
Schrag, M. L., and Wienkers, L. C. (2001). Covalent alteration of the CYP3A4 active site: evidence for multiple substrate binding domains. Arch. Biochem. Biophys. 391, 49–55. doi: 10.1006/abbi.2001.2401
Scott, S. A., Martis, S., Peter, I., Kasai, Y., Kornreich, R., and Desnick, R. J. (2012). Identification of CYP2C19∗4B: pharmacogenetic implications for drug metabolism including clopidogrel responsiveness. Pharmacogenom. J. 12, 297–305. doi: 10.1038/tpj.2011.5
Scott, S. A., Tan, Q., Baber, U., Yang, Y., Martis, S., Bander, J., et al. (2013). An allele-specific PCR system for rapid detection and discrimination of the CYP2C19∗4A, ∗4B, and ∗17 alleles: implications for clopidogrel response testing. J. Mole. Diagn 15, 783–789. doi: 10.1016/j.jmoldx.2013.06.004
Sennef, C., Timmer, C. J., and Sitsen, J. M. (2003). Mirtazapine in combination with amitriptyline: a drug-drug interaction study in healthy subjects. Hum. Psychopharmacol. 18, 91–101. doi: 10.1002/hup.441
Sezutsu, H., Le Goff, G., and Feyereisen, R. (2013). Origins of P450 diversity. Philosophic. Transact. Soc. B 368:20120428. doi: 10.1098/rstb.2012.0428
Shan, X. X., Qiu, Y., Xie, W. W., Wu, R. R., Yu, Y., Wu, H. S., et al. (2019). ABCB1 Gene Is Associated With Clinical Response to SNRIs in a Local Chinese Han Population. Front. Pharmacol. 10:761. doi: 10.3389/fphar.2019.00761
Shelton, R. C., Parikh, S. V., Law, R. A., Rothschild, A. J., Thase, M. E., Dunlop, B. W., et al. (2020). Combinatorial Pharmacogenomic Algorithm is Predictive of Citalopram and Escitalopram Metabolism in Patients with Major Depressive Disorder. Psychiatry Res. 290:113017. doi: 10.1016/j.psychres.2020.113017
Shi, Y., Li, M., Song, C., Xu, Q., Huo, R., Shen, L., et al. (2017). Combined study of genetic and epigenetic biomarker risperidone treatment efficacy in Chinese Han schizophrenia patients. Transl. Psychiatry 7:e1170. doi: 10.1038/tp.2017.143
Shih, J. C., and Chen, K. (2004). Regulation of MAO-A and MAO-B gene expression. Curr. Med. Chem. 11, 1995–2005. doi: 10.2174/0929867043364757
Shimada, T., Yamazaki, H., Mimura, M., Inui, Y., and Guengerich, F. P. (1994). Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J. Pharmacol. Exp. Ther. 270, 414–423.
Shimoda, K., Someya, T., Morita, S., Hirokane, G., Yokono, A., and Takahashi, S. (2002). Lack of impact of CYP1A2 genetic polymorphism (C/A polymorphism at position 734 in intron 1 and G/A polymorphism at position – 2964 in the 50-flanking region of CYP1A2) on the plasma concentration of haloperidol in smoking male Japanese with schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 26, 261–265. doi: 10.1016/s0278-5846(01)00263-9
Shin, J. G., Park, J. Y., Kim, M. J., Shon, J. H., Yoon, Y. R., Cha, I. J., et al. (2002). Inhibitory effects of tricyclic antidepressants (TCAs) on human cytochrome P450 enzymes in vitro: mechanism of drug interaction between TCAs and phenytoin. Drug Metab. Dispos. 30, 1102–1107. doi: 10.1124/dmd.30.10.1102
Siegle, I., Fritz, P., Eckhardt, K., Zanger, U. M., and Eichelbaum, M. (2001). Cellular localization and regional distribution of CYP2D6 mRNA and protein expression in human brain. Pharmacogenetics 11, 237–245. doi: 10.1097/00008571-200104000-00007
Sim, S. C. (2013). CYP1A2*1E contains the −163C>A substitution and is highly inducible. Pharmacogenet. Genomics 23, 104–105. doi: 10.1097/FPC.0b013e32835ccc76
Sim, E., Abuhammad, A., and Ryan, A. (2014). Arylamine N-acetyltransferases: from drug metabolism and pharmacogenetics to drug discovery. Br. J. Pharmacol. 171, 2705–2725. doi: 10.1111/bph.12598
Sim, E., Westwood, I., and Fullam, E. (2007). Arylamine N-acetyltransferases. Exp. Opin. Drug Metab. Toxicol. 3, 169–184. doi: 10.1517/17425255.3.2.169
Sim, S. C., Risinger, C., Dahl, M. L., Aklillu, E., Christensen, M., Bertilsson, L., et al. (2006). A common novel CYP2C19 gene variant causes ultrarapid drug metabolism relevant for the drug response to proton pump inhibitors and antidepressants. Clin. Pharmacol. Ther. 79, 103–113. doi: 10.1016/j.clpt.2005.10.002
Sindrup, S. H., Brøsen, K., Hansen, M. G., Aaes-Jørgensen, T., Overø, K. F., and Gram, L. F. (1993). Pharmacokinetics of citalopram in relation to the sparteine and the mephenytoin oxidation polymorphisms. Ther. Drug Monit. 15, 11–17. doi: 10.1097/00007691-199302000-00002
Sindrup, S. H., Gram, L. F., Brosen, K., Eshoj, O., and Mogensen, E. F. (1990). The selective serotonin reuptake inhibitor paroxetine is effective in the treatment of diabetic neuropathy symptoms. Pain 42, 135–144. doi: 10.1016/0304-3959(90)91157-e
Sindrup, S. H., Grodum, E., Gram, L. F., and Beck-Nielsen, H. (1991). Concentration-response relationship in paroxetine treatment of diabetic neuropathy symptoms: a patient-blinded dose-escalation study. Ther. Drug Monit. 13, 408–414. doi: 10.1097/00007691-199109000-00003
Singh, A. B. (2015). Improved antidepressant remission in major depression via a pharmacokinetic pathway polygene pharmacogenetic report. Clin. Psychopharmacol. Neurosci. 13, 150–156. doi: 10.9758/cpn.2015.13.2.150
Singh, D., Cho, W. C., and Upadhyay, G. (2015). Drug-Induced Liver Toxicity and Prevention by Herbal Antioxidants: An Overview. Front. Physiol. 6:363. doi: 10.3389/fphys.2015.00363
Sjöqvist, F., and Bertilsson, L. (1984). Clinical pharmacology of antidepressant drugs: pharmacogenetics. Adv. Biochem. Psychopharmacol. 39, 359–372.
Sjöqvist, F., Bertilsson, L., and Asberg, M. (1980). Monitoring tricyclic antidepressants. Therapeu. Drug Monit. 2, 85–93. doi: 10.1097/00007691-198001000-00010
Skierka, J. M., and Black, J. L. III (2014). Analysis of compound heterozygous CYP2C19 genotypes to determine cis and trans configurations. Pharmacogenomics 15, 1197–1205. doi: 10.2217/pgs.14.72
Skogh, E., Sjödin, I., Josefsson, M., and Dahl, M. L. (2011). High correlation between serum and cerebrospinal fluid olanzapine concentrations in patients with schizophrenia or schizoaffective disorder medicating with oral olanzapine as the only antipsychotic drug. J. Clin. Psychopharmacol. 31, 4–9. doi: 10.1097/JCP.0b013e318204d9e2
Smith, R. L. (1986). Special Issue - Human Genetic Variations in Oxidative Drug-Metabolism - Introduction. Xenobiotica 16, 361–365. doi: 10.3109/00498258609050244
Söderberg, M. M., Haslemo, T., Molden, E., and Dahl, M. L. (2013). Influence of CYP1A1/CYP1A2 and AHR polymorphisms on systemic olanzapine exposure. Pharmacog. Genom. 23, 279–285. doi: 10.1097/FPC.0b013e3283602876
Soyama, A., Saito, Y., Komamura, K., Ueno, K., Kamakura, S., Ozawa, S., et al. (2002). Five novel single nucleotide polymorphisms in the CYP2C8 gene, one of which induces a frame-shift. Drug Metab. Pharmacokinet 17, 374–377. doi: 10.2133/dmpk.17.374
Spina, E., Gitto, C., Avenoso, A., Campo, G. M., Caputi, A. P., and Perucca, E. (1997). Relationship between plasma desipramine levels, CYP2D6 phenotype and clinical response to desipramine: a prospective study. Eur. J. Clin. Pharmacol. 51, 395–398. doi: 10.1007/s002280050220
Spina, E., Santoro, V., and D’Arrigo, C. (2008). Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an update. Clin. Ther. 30, 1206–1227. doi: 10.1016/s0149-2918(08)80047-1
Steimer, W., Zopf, K., von Amelunxen, S., Pfeiffer, H., Bachofer, J., Popp, J., et al. (2005). Amitriptyline or not, that is the question: pharmacogenetic testing of CYP2D6 and CYP2C19 identifies patients with low or high risk for side effects in amitriptyline therapy. Clin. Chem. 51, 376–385. doi: 10.1373/clinchem.2004.041327
Steimer, W., Zöpf, K., von Amelunxen, S., Pfeiffer, H., Bachofer, J., Popp, J., et al. (2004). Allele-specific change of concentration and functional gene dose for the prediction of steady-state serum concentrations of amitriptyline and nortriptyline in CYP2C19 and CYP2D6 extensive and intermediate metabolizers. Clin. Chem. 50, 1623–1633. doi: 10.1373/clinchem.2003.030825
Stern, R. S., and Divito, S. J. (2017). Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis: Associations, Outcomes, and Pathobiology-Thirty Years of Progress but Still Much to Be Done. J. Invest. Dermatol. 137, 1004–1008. doi: 10.1016/j.jid.2017.01.003
Stern, S. L., Ribner, H. S., Cooper, T. B., Nelson, L. D., Johnson, M. H., and Suckow, R. F. (1991). 2-Hydroxydesipramine and desipramine plasma levels and electrocardiographic effects in depressed younger adults. J. Clin. Psychopharmacol. 11, 93–98.
Stingl, J. C., Bartels, H., Viviani, R., Lehmann, M. L., and Brockmoller, J. (2014). Relevance of UDP-glucuronosyltransferase polymorphisms for drug dosing: A quantitative systematic review. Pharmacol. Ther. 141, 92–116. doi: 10.1016/j.pharmthera.2013.09.002
Stingl, J. C., Brockmöller, J., and Viviani, R. (2013). Genetic variability of drug-metabolizing enzymes: the dual impact on psychiatric therapy and regulation of brain function. Mole. Psychiatry 18, 273–287. doi: 10.1038/mp.2012.42
Stingl, J., and Viviani, R. (2015). Polymorphism in CYP2D6 and CYP2C19, members of the cytochrome P450 mixed-function oxidase system, in the metabolism of psychotropic drugs. J. Int. Med. 277, 167–177. doi: 10.1111/joim.12317
Störmer, E., von Moltke, L. L., Shader, R. I., and Greenblatt, D. J. (2000). Metabolism of the antidepressant mirtazapine in vitro: contribution of cytochromes P-450 1A2, 2D6, and 3A4. Drug Metab. Dispos. 28, 1168–1175.
Sukasem, C., and Chantratita, W. (2016). A success story in pharmacogenomics: genetic ID card for SJS/TEN. Pharmacogenomics 17, 455–458. doi: 10.2217/pgs-2015-0009
Sukasem, C., Chaichan, C., Nakkrut, T., Satapornpong, P., Jaruthamsophon, K., Jantararoungtong, T., et al. (2018). Association between HLA-B Alleles and Carbamazepine-Induced Maculopapular Exanthema and Severe Cutaneous Reactions in Thai Patients. J. Immunol. Res. 2018:2780272. doi: 10.1155/2018/2780272
Sukasem, C., Hongkaew, Y., Ngamsamut, N., Puangpetch, A., Vanwong, N., Chamnanphon, M., et al. (2016). Impact of Pharmacogenetic Markers of CYP2D6 and DRD2 on Prolactin Response in Risperidone-Treated Thai Children and Adolescents With Autism Spectrum Disorders. J. Clin. Psychopharmacol. 36, 141–146. doi: 10.1097/JCP.0000000000000474
Takagi, S., Nakajima, M., Mohri, T., and Yokoi, T. (2008). Post-transcriptional regulation of human pregnane X receptor by micro-RNA affects the expression of cytochrome P450 3A4. J. Biol. Chem. 283, 9674–9680. doi: 10.1074/jbc.M709382200
Takeuchi, F., McGinnis, R., Bourgeois, S., Barnes, C., Eriksson, N., Soranzo, N., et al. (2009). A genome-wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLoS Genet 5:e1000433. doi: 10.1371/journal.pgen.1000433
Tang, M. H., and Pinsky, E. G. (2015). Mood and affect disorders. Pediatr. Rev. 36, 52–60. doi: 10.1542/pir.36-2-52
Tanner, J. A., and Tyndale, R. F. (2017). Variation in CYP2A6 Activity and Personalized Medicine. J. Personal. Med. 7:18. doi: 10.3390/jpm7040018
Tanner, J. A., Davies, P. E., Overall, C. C., Grima, D., Nam, J., and Dechairo, B. M. (2020). Cost-effectiveness of combinatorial pharmacogenomic testing for depression from the Canadian public payer perspective. Pharmacogenomics 21, 521–531. doi: 10.2217/pgs-2020-0012
Tanner, J. A., Prasad, B., Claw, K. G., Stapleton, P., Chaudhry, A., Schuetz, E. G., et al. (2017). Predictors of Variation in CYP2A6 mRNA, Protein, and Enzyme Activity in a Human Liver Bank: Influence of Genetic and Nongenetic Factors. J. Pharmacol. Exp. Ther. 360, 129–139. doi: 10.1124/jpet.116.237594
Taskinen, J., Ethell, B. T., Pihlavisto, P., Hood, A. M., Burchell, B., and Coughtrie, M. W. (2003). Conjugation of catechols by recombinant human sulfotransferases, UDP-glucuronosyltransferases, and soluble catechol O-methyltransferase: structure-conjugation relationships and predictive models. Drug Metab. Dispos. 31, 1187–1197. doi: 10.1124/dmd.31.9.1187
Tateishi, T., Watanabe, M., Kumai, T., Tanaka, M., Moriya, H., Yamaguchi, S., et al. (2000). CYP3A is responsible for N-dealkylation of haloperidol and bromperidol and oxidation of their reduced forms by human liver microsomes. Life Sci. 67, 2913–2920. doi: 10.1016/S0024-3205(00)00874-2
Teitelbaum, A. M., Murphy, S. E., Akk, G., Baker, T. B., Germann, A., von Weymarn, L. B., et al. (2018). Nicotine dependence is associated with functional variation in FMO3, an enzyme that metabolizes nicotine in the brain. Pharmacogenomics J. 18, 136–143. doi: 10.1038/tpj.2016.92
Theophylline Pathway. Retrieved from https://www.pharmgkb.org/pathway/PA165958541
Thomas, D. R., Nelson, D. R., and Johnson, A. M. (1987). Biochemical effects of the antidepressant paroxetine, a specific 5-hydroxytryptamine uptake inhibitor. Psychopharmacology 93, 193–200.
Tian, R., Koyabu, N., Morimoto, S., Shoyama, Y., Ohtani, H., and Sawada, Y. (2005). Functional induction and de-induction of P-glycoprotein by St. John’s wort and its ingredients in a human colon adenocarcinoma cell line. Drug Metab. Disp. 33, 547–554. doi: 10.1124/dmd.104.002485
Timmer, C. J., Sitsen, J. M., and Delbressine, L. P. (2000). Clinical pharmacokinetics of mirtazapine. Clin. Pharmacokinet 38, 461–474. doi: 10.2165/00003088-200038060-00001
Tirona, R. G., Lee, W., Leake, B. F., Lan, L. B., Cline, C. B., Lamba, V., et al. (2003). The orphan nuclear receptor HNF4alpha determines PXR- and CAR-mediated xenobiotic induction of CYP3A4. Nat. Med. 9, 220–224. doi: 10.1038/nm815
Tolson, A. H., and Wang, H. (2010). Regulation of drug-metabolizing enzymes by xenobiotic receptors: PXR and CAR. Adv. Drug Deliv. Rev. 62, 1238–1249. doi: 10.1016/j.addr.2010.08.006
Toscano, C., Klein, K., Blievernicht, J., Schaeffeler, E., Saussele, T., Raimundo, S., et al. (2006). Impaired expression of CYP2D6 in intermediate metabolizers carrying the ∗41 allele caused by the intronic SNP 2988G >>A: evidence for modulation of splicing events. Pharmacogenet Genom. 16, 755–766. doi: 10.1097/01.fpc.0000230112.96086.e0
Troost, P. W., Lahuis, B. E., Hermans, M. H., Buitelaar, J. K., van Engeland, H., Scahill, L., et al. (2007). Prolactin release in children treated with risperidone: impact and role of CYP2D6 metabolism. J. Clin. Psychopharmacol. 27, 52–57. doi: 10.1097/JCP.0b013e31802e68d5
Tsuchiya, K., Gatanaga, H., Tachikawa, N., Teruya, K., Kikuchi, Y., Yoshino, M., et al. (2004). Homozygous CYP2B6 ∗6 (Q172H and K262R) correlates with high plasma efavirenz concentrations in HIV-1 patients treated with standard efavirenz-containing regimens. Biochem. Biophys. Res. Commun. 319, 1322–1326. doi: 10.1016/j.bbrc.2004.05.116
Tsuda, Y., Saruwatari, J., and Yasui-Furukori, N. (2014). Meta-analysis: the effects of smoking on the disposition of two commonly used antipsychotic agents, olanzapine and clozapine. BMJ Open 4:e004216. doi: 10.1136/bmjopen-2013-004216
Tucker, G. T., Houston, J. B., and Huang, S. M. (2001). Optimizing drug development: strategies to assess drug metabolism/transporter interaction potential–towards a consensus. Br. J. Clin. Pharmacol. 52, 107–117. doi: 10.1046/j.0306-5251.2001.temp.1441.x
Tunbridge, E. M., Harrison, P. J., and Weinberger, D. R. (2006). Catechol-o-methyltransferase, cognition, and psychosis: Val158Met and beyond. Biol. Psychiatry 60, 141–151. doi: 10.1016/j.biopsych.2005.10.024
Turpeinen, M., and Zanger, U. M. (2012). Cytochrome P450 2B6: function, genetics, and clinical relevance. Drug Metabol. Drug Interact. 27, 185–197. doi: 10.1515/dmdi-2012-0027
Turpeinen, M., Tolonen, A., Chesne, C., Guillouzo, A., Uusitalo, J., and Pelkonen, O. (2009). Functional expression, inhibition and induction of CYP enzymes in HepaRG cells. Toxicol. In Vitro 23, 748–753. doi: 10.1016/j.tiv.2009.03.008
Ueda, K., Clark, D. P., Chen, C. J., Roninson, I. B., Gottesman, M. M., and Pastan, I. (1987). The human multidrug resistance (mdr1) gene. cDNA cloning and transcription initiation. J. Biol. Chem. 262, 505–508.
Ufer, M., Dilger, K., Leschhorn, L., Daufresne, L., Mosyagin, I., Rosenstiel, P., et al. (2008). Influence of CYP3A4, CYP3A5, and ABCB1 genotype and expression on budesonide pharmacokinetics: a possible role of intestinal CYP3A4 expression. Clin. Pharmacol. Ther. 84, 43–46. doi: 10.1038/sj.clpt.6100505
UGT1A1 and common exons allele nomenclature. Retrieved from https://www.pharmacogenomics.pha.ulaval.ca/wp-content/uploads/2015/04/UGT1A1-allele-nomenclature.html
Uher, R., Farmer, A., Henigsberg, N., Rietschel, M., and Aitchison, K. (2009b). Adverse Reactions to Antidepressants. Br. J. Psychiatry 195, 202–210.
Uher, R., Maier, W., Hauser, J., Marusic, A., Schmael, C., Mors, O., et al. (2009a). Differential efficacy of escitalopram and nortriptyline on dimensional measures of depression. Br. J. Psychiatry 194, 252–259. doi: 10.1192/bjp.bp.108.057554
Uhr, M., and Grauer, M. T. (2003). abcb1ab P-glycoprotein is involved in the uptake of citalopram and trimipramine into the brain of mice. J. Psychiatr. Res. 37, 179–185. doi: 10.1016/s0022-3956(03)00022-0
Uhr, M., Tontsch, A., Namendorf, C., Ripke, S., Lucae, S., Ising, M., et al. (2008). Polymorphisms in the drug transporter gene ABCB1 predict antidepressant treatment response in depression. Neuron 57, 203–209. doi: 10.1016/j.neuron.2007.11.017
Ulrich, S., Wurthmann, C., Brosz, M., and Meyer, F. P. (1998). The relationship between serum concentration and therapeutic effect of haloperidol in patients with acute schizophrenia. Clin. Pharmacokinetics 34, 227–263. doi: 10.2165/00003088-199834030-00005
van der Weide, J., and Steijns, L. S. W. (1999). Cytochrome P450 Enzyme System: Genetic Polymorphisms and Impact on Clinical Pharmacology. Ann. Clin. Biochem. 36, 722–729. doi: 10.1177/000456329903600604
van der Weide, K., and van der Weide, J. (2014). The influence of the CYP3A4∗22 polymorphism on serum concentration of quetiapine in psychiatric patients. J. Clin. Psychopharmacol. 34, 256–260. doi: 10.1097/JCP.0000000000000070
van der Weide, K., and van der Weide, J. (2015). The Influence of the CYP3A4∗22 Polymorphism and CYP2D6 Polymorphisms on Serum Concentrations of Aripiprazole, Haloperidol, Pimozide, and Risperidone in Psychiatric Patients. J. Clin. Psychopharmacol. 35, 228–236. doi: 10.1097/JCP.0000000000000319
van Schaik, R. H., van der Heiden, I. P., van den Anker, J. N., and Lindemans, J. (2002). CYP3A5 variant allele frequencies in Dutch Caucasians. Clin. Chem. 48, 1668–1671. doi: 10.1093/clinchem/48.10.1668
van Westrhenen, R., Aitchison, K. J., Ingelman-Sundberg, M., and Jukic, M. M. (2020). Pharmacogenomics of Antidepressant and Antipsychotic Treatment: How Far Have We Got and Where Are We Going? Front. Psychiatry 11:94. doi: 10.3389/fpsyt.2020.00094
Variant Annotations. Retrieved from https://www.pharmgkb.org/combination/PA427,PA452233/variantAnnotation
Veeramah, K. R., Thomas, M. G., Weale, M. E., Zeitlyn, D., Tarekegn, A., Bekele, E., et al. (2008). The potentially deleterious functional variant flavin-containing monooxygenase 2∗1 is at high frequency throughout sub-Saharan Africa. Pharmacog. Genom. 18, 877–886. doi: 10.1097/FPC.0b013e3283097311
Vella, T., and Mifsud, J. (2014). Interactions between valproic acid and quetiapine/olanzapine in the treatment of bipolar disorder and the role of therapeutic drug monitoring. J. Pharm. Pharmacol. 66, 747–759. doi: 10.1111/jphp.12209
Vistisen, K., Poulsen, H. E., and Loft, S. (1992). Foreign compound metabolism capacity in man measured from metabolites of dietary caffeine. Carcinogenesis 13, 1561–1568. doi: 10.1093/carcin/13.9.1561
von Moltke, L. L., Duan, S. X., Greenblatt, D. J., Fogelman, S. M., Schmider, J., Harmatz, J. S., et al. (1997). Venlafaxine and metabolites are very weak inhibitors of human cytochrome P450-3A isoforms. Biol. Psychiatry 41, 377–380. doi: 10.1016/s0006-3223(96)00406-4
von Moltke, L. L., Greenblatt, D. J., Giancarlo, G. M., Granda, B. W., Harmatz, J. S., and Shader, R. I. (2001). Escitalopram (S-citalopram) and its metabolites in vitro: cytochromes mediating biotransformation, inhibitory effects, and comparison to R-citalopram. Drug Metab. Dispos. 29, 1102–1109.
von Richter, O., Burk, O., Fromm, M. F., Thon, K. P., Eichelbaum, M., and Kivisto, K. T. (2004). Cytochrome P450 3A4 and P-glycoprotein expression in human small intestinal enterocytes and hepatocytes: a comparative analysis in paired tissue specimens. Clin. Pharmacol. Ther. 75, 172–183. doi: 10.1016/j.clpt.2003.10.008
Wacher, V. J., Salphati, L., and Benet, L. Z. (2001). Active secretion and enterocytic drug metabolism barriers to drug absorption. Adv. Drug Deliv. Rev. 46, 89–102. doi: 10.1016/s0169-409x(00)00126-5
Wang, D., Guo, Y., Wrighton, S. A., Cooke, G. E., and Sadee, W. (2011). Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenomics J. 11, 274–286. doi: 10.1038/tpj.2010.28
Wang, D., Papp, A. C., and Sun, X. (2015). Functional characterization of CYP2D6 enhancer polymorphisms. Hum. Mol. Genet 24, 1556–1562. doi: 10.1093/hmg/ddu566
Wang, D., Poi, M. J., Sun, X., Gaedigk, A., Leeder, J. S., and Sadee, W. (2014). Common CYP2D6 polymorphisms affecting alternative splicing and transcription: long-range haplotypes with two regulatory variants modulate CYP2D6 activity. Hum. Mol. Genet 23, 268–278. doi: 10.1093/hmg/ddt417
Wang, H., and Tompkins, L. M. (2008). CYP2B6: new insights into a historically overlooked cytochrome P450 isozyme. Curr. Drug Metab. 9, 598–610. doi: 10.2174/138920008785821710
Ward, B. A., Gorski, J. C., Jones, D. R., Hall, S. D., Flockhart, D. A., and Desta, Z. (2003). The cytochrome P450 2B6 (CYP2B6) is the main catalyst of efavirenz primary and secondary metabolism: implication for HIV/AIDS therapy and utility of efavirenz as a substrate marker of CYP2B6 catalytic activity. J. Pharmacol. Exp. Ther. 306, 287–300. doi: 10.1124/jpet.103.049601
Watson, C. P. (2000). The treatment of neuropathic pain: antidepressants and opioids. Clin. J. Pain 2, S49–S55.
Weigmann, H., Härtter, S., Bagli, M., and Hiemke, C. (2000). Steady state concentrations of clomipramine and its major metabolite desmethylclomipramine in rat brain and serum after oral administration of clomipramine. Eur. Neuropsychopharmacol. 10, 401–405. doi: 10.1016/s0924-977x(00)00098-5
Weinshilboum, R. M., Otterness, D. M., and Szumlanski, C. L. (1999). Methylation pharmacogenetics: catechol O-methyltransferase, thiopurine methyltransferase, and histamine N-methyltransferase. Annu. Rev. Pharmacol. Toxicol. 39, 19–52. doi: 10.1146/annurev.pharmtox.39.1.19
Wen, B., and Zhou, M. (2009). Metabolic activation of the phenothiazine antipsychotics chlorpromazine and thioridazine to electrophilic iminoquinone species in human liver microsomes and recombinant P450s. Chemico Biol. Interact. 181, 220–226. doi: 10.1016/j.cbi.2009.05.014
Werk, A. N., Lefeldt, S., Bruckmueller, H., Hemmrich-Stanisak, G., Franke, A., Roos, M., et al. (2014). Identification and characterization of a defective CYP3A4 genotype in a kidney transplant patient with severely diminished tacrolimus clearance. Clin. Pharmacol. Ther. 95, 416–422. doi: 10.1038/clpt.2013.210
Westlind-Johnsson, A., Hermann, R., Huennemeyer, A., Hauns, B., Lahu, G., Nassr, N., et al. (2006). Identification and characterization of CYP3A4∗20, a novel rare CYP3A4 allele without functional activity. Clin. Pharmacol. Therapeut. 79, 339–349. doi: 10.1016/j.clpt.2005.11.015
Whetstine, J. R., Yueh, M. F., McCarver, D. G., Williams, D. E., Park, C. S., Kang, J. H., et al. (2000). Ethnic differences in human flavin-containing monooxygenase 2 (FMO2) polymorphisms: detection of expressed protein in African-Americans. Toxicol. Appl. Pharmacol. 168, 216–224. doi: 10.1006/taap.2000.9050
Windmill, K. F., Gaedigk, A., Hall, P. M., Samaratunga, H., Grant, D. M., and McManus, M. E. (2000). Localization of N-acetyltransferases NAT1 and NAT2 in human tissues. Toxicol. Sci. 54, 19–29. doi: 10.1093/toxsci/54.1.19
Winter, H. R., DeVane, C. L., Figueroa, C., Ennis, D. J., Hamer-Maansson, J. E., Davis, P. C., et al. (2007). Open-label steady-state pharmacokinetic drug interaction study on co-administered quetiapine fumarate and divalproex sodium in patients with schizophrenia, schizoaffective disorder, or bipolar disorder. Hum. Psychopharmacol. 22, 469–476. doi: 10.1002/hup.869
Wójcikowski, J., Maurel, P., and Daniel, W. A. (2012). Autoinduction of the metabolism of phenothiazine neuroleptics in a primary culture of human hepatocytes. PR 64, 1578–1583. doi: 10.1016/s1734-1140(12)70957-x
Wong, D. T., Bymaster, F. P., Horng, J. S., and Molloy, B. B. (1975). A new selective inhibitor for uptake of serotonin into synaptosomes of rat brain: 3-(p-trifluoromethylphenoxy). N-methyl-3-phenylpropylamine. J. Pharmacol. Exp. Ther. 193, 804–811.
Wong, D. T., Horng, J. S., Bymaster, F. P., Hauser, K. L., and Molloy, B. B. (1974). A selective inhibitor of serotonin uptake: Lilly 110140, 3-(p-trifluoromethylphenoxy)-N-methyl-3-phenylpropylamine. Life Sci. 15, 471–479. doi: 10.1016/0024-3205(74)90345-2
Wu, F. S., Gibbs, T. T., and Farb, D. H. (1991). Pregnenolone sulfate: a positive allosteric modulator at the N-methyl-D-aspartate receptor. Mol. Pharmacol. 40, 333–336.
Xie, Y., Ke, S., Ouyang, N., He, J., Xie, W., Bedford, M. T., et al. (2009). Epigenetic regulation of transcriptional activity of pregnane X receptor by protein arginine methyltransferase 1. J. Biol. Chem. 284, 9199–9205. doi: 10.1074/jbc.M806193200
Xu, C., Li, C. Y., and Kong, A. N. (2005). Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch. Pharm. Res. 28, 249–268. doi: 10.1007/bf02977789
Yamazaki, H., Inui, Y., Yun, C.-H., Guengerich, F. P., and Shimada, T. (1992). Cytochrome P450 2E1 and 2A6 enzymes as major catalysts for metabolic activation of N-nitrosodialkylamines and tobacco-related nitrosamines in human liver microsomes. Carcinogenesis 13, 1789–1794. doi: 10.1093/carcin/13.10.1789
Yang, T. J., Krausz, K. W., Sai, Y., Gonzalez, F. J., and Gelboin, H. V. (1999). Eight inhibitory monoclonal antibodies define the role of individual P-450s in human liver microsomal diazepam, 7-ethoxycoumarin, and imipramine metabolism. Drug Metab. Dispos. 27, 102–109.
Yasukochi, Y., and Satta, Y. (2011). Evolution of the CYP2D gene cluster in humans and four non-human primates. Genes Genet Syst. 86, 109–116. doi: 10.1266/ggs.86.109
Yau, J. L., Noble, J., Thomas, S., Kerwin, R., Morgan, P. E., Lightman, S., et al. (2007). The antidepressant desipramine requires the ABCB1 (Mdr1)-type p-glycoprotein to upregulate the glucocorticoid receptor in mice. Neuropsychopharmacology 32, 2520–2529. doi: 10.1038/sj.npp.1301389
Yeo, C. W., Lee, S. J., Lee, S. S., Bae, S. K., Kim, E. Y., Shon, J. H., et al. (2011). Discovery of a novel allelic variant of CYP2C8, CYP2C8∗11, in Asian populations and its clinical effect on the rosiglitazone disposition in vivo. Drug Metab. Dispos. 39, 711–716. doi: 10.1124/dmd.110.035899
Yeung, C. K., Lang, D. H., Thummel, K. E., and Rettie, A. E. (2000). Immunoquantitation of FMO1 in human liver, kidney, and intestine. Drug Metab. Dispos. 28, 1107–1111. doi: 10.3109/00498259109039550
Yimer, G., Amogne, W., Habtewold, A., Makonnen, E., Ueda, N., Suda, A., et al. (2012). High plasma efavirenz level and CYP2B6∗6 are associated with efavirenz-based HAART-induced liver injury in the treatment of naive HIV patients from Ethiopia: a prospective cohort study. Pharmacogenomics J. 12, 499–506. doi: 10.1038/tpj.2011.34
Yokota, H., Tamura, S., Furuya, H., Kimura, S., Watanabe, M., Kanazawa, I., et al. (1993). Evidence for a new variant CYP2D6 allele CYP2D6J in a Japanese population associated with lower in vivo rates of sparteine metabolism. Pharmacogenetics 3, 256–263. doi: 10.1097/00008571-199310000-00005
Yoshii, K., Kobayashi, K., Tsumuji, M., Tani, M., Shimada, N., and Chiba, K. (2000). Identification of human cytochrome P450 isoforms involved in the 7-hydroxylation of chlorpromazine by human liver microsomes. Life Sci. 67, 175–184. doi: 10.1016/s0024-3205(00)00613-5
Yoshinari, K., Ueda, R., Kusano, K., Yoshimura, T., Nagata, K., and Yamazoe, Y. (2008). Omeprazole transactivates human CYP1A1 and CYP1A2 expression through the common regulatory region containing multiple xenobiotic-responsive elements. Biochem. Pharmacol. 76, 139–145. doi: 10.1016/j.bcp.2008.04.005
Youngster, I., Zachor, D. A., Gabis, L. V., Bar-Chaim, A., Benveniste-Levkovitz, P., Britzi, M., et al. (2014). CYP2D6 genotyping in paediatric patients with autism treated with risperidone: a preliminary cohort study. Devel. Med. Child Neurol. 56, 990–994. doi: 10.1111/dmcn.12470
Yu, A. M., Idle, J. R., Herraiz, T., Kupfer, A., and Gonzalez, F. J. (2003). Screening for endogenous substrates reveals that CYP2D6 is a 5-methoxyindolethylamine O-demethylase. Pharmacogenetics 13, 307–319. doi: 10.1097/01.fpc.0000054094.48725.b7
Yu, G., Li, G. F., and Markowitz, J. S. (2016). Atomoxetine: A Review of Its Pharmacokinetics and Pharmacogenomics Relative to Drug Disposition. J. Child Adolesc. Psychopharmacol. 26, 314–326. doi: 10.1089/cap.2015.0137
Zanger, U. M., and Klein, K. (2013). Pharmacogenetics of cytochrome P450 2B6 (CYP2B6): advances on polymorphisms, mechanisms, and clinical relevance. Front. Genetics 4:24.
Zanger, U. M., and Schwab, M. (2013). Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 138, 103–141. doi: 10.1016/j.pharmthera.2012.12.007
Zanger, U. M., Klein, K., Saussele, T., Blievernicht, J., Hofmann, M. H., and Schwab, M. (2007). Polymorphic CYP2B6: molecular mechanisms and emerging clinical significance. Pharmacogenomics 8, 743–759. doi: 10.2217/14622416.8.7.743
Zevin, S., and Benowitz, N. L. (1999). Drug interactions with tobacco smoking. Clin. Pharmacokinet 36, 425–438. doi: 10.2165/00003088-199936060-00004
Zhang, L., Brown, S. J., Shan, Y., Lee, A. M., Allen, J. D., Eum, S., et al. (2020). CYP2D6 Genetic Polymorphisms and Risperidone Pharmacokinetics: A Systematic Review and Meta-analysis. Pharmacotherapy 40, 632–647. doi: 10.1002/phar.2434
Zhou, H., Josephy, P. D., Kim, D., and Guengerich, F. P. (2004). Functional characterization of four allelic variants of human cytochrome P450 1A2. Arch. Biochem. Biophys 422, 23–30. doi: 10.1016/j.abb.2003.11.019
Zhou, S.-F., Liu, J.-P., and Chowbay, B. (2009). Polymorphism of human cytochrome P450 enzymes and its clinical impact. Drug Metab. Rev. 41, 89–295. doi: 10.1080/03602530902843483
Ziegler, V. E., and Biggs, J. T. (1977). Tricyclic plasma levels. Effect of age, race, sex, and smoking. JAMA 238, 2167–2169. doi: 10.1001/jama.1977.03280210059023
Ziegler, V. E., Clayton, P. J., and Biggs, J. T. (1977). A comparison study of amitriptyline and nortriptyline with plasma levels. Arch. Gen. Psychiatry 34, 607–612. doi: 10.1001/archpsyc.1977.01770170117012
Ziegler, V., Taylor, J., Wetzel, R., and Biggs, J. (1978). Nortriptyline Plasma Levels and Subjective Side Effects. Br. J. Psychiatry 132, 55–60. doi: 10.1192/S0007125000282974
Keywords: pharmacogenomics (PGx), drug metabolism, drug transporters, cytochrome P450 enzymes, psychotropic drugs
Citation: Carvalho Henriques B, Yang EH, Lapetina D, Carr MS, Yavorskyy V, Hague J and Aitchison KJ (2020) How Can Drug Metabolism and Transporter Genetics Inform Psychotropic Prescribing? Front. Genet. 11:491895. doi: 10.3389/fgene.2020.491895
Received: 16 August 2019; Accepted: 25 September 2020;
Published: 08 December 2020.
Edited by:
Edoardo Spina, University of Messina, ItalyReviewed by:
Alessandro Serretti, University of Bologna, ItalyCopyright © 2020 Carvalho Henriques, Yang, Lapetina, Carr, Yavorskyy, Hague and Aitchison. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katherine J. Aitchison, a2FpdGNoaXNAdWFsYmVydGEuY2E=; orcid.org/0000-0002-1107-3024
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.