
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 24 September 2020
Sec. RNA
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.01025
This article is part of the Research TopicRNA Modification in Human Cancers: Roles and Therapeutic ImplicationsView all 48 articles
 Fayou Wang1,2†
Fayou Wang1,2† Jialiang Yang3,4,5†
Jialiang Yang3,4,5† Huixin Lin4,5
Huixin Lin4,5 Qian Li4,6Zixuan Ye4
Qian Li4,6Zixuan Ye4 Qingqing Lu4,5
Qingqing Lu4,5 Luonan Chen2*Zhidong Tu3*
Luonan Chen2*Zhidong Tu3* Geng Tian4,5*
Geng Tian4,5*Studying transcriptome chronological change from tissues across the whole body can provide valuable information for understanding aging and longevity. Although there has been research on the effect of single-tissue transcriptomes on human aging or aging in mice across multiple tissues, the study of human body-wide multi-tissue transcriptomes on aging is not yet available. In this study, we propose a quantitative model to predict human age by using gene expression data from 46 tissues generated by the Genotype-Tissue Expression (GTEx) project. Specifically, the biological age of a person is first predicted via the gene expression profile of a single tissue. Then, we combine the gene expression profiles from two tissues and compare the predictive accuracy between single and two tissues. The best performance as measured by the root-mean-square error is 3.92 years for single tissue (pituitary), which deceased to 3.6 years when we combined two tissues (pituitary and muscle) together. Different tissues have different potential in predicting chronological age. The prediction accuracy is improved by combining multiple tissues, supporting that aging is a systemic process involving multiple tissues across the human body.
Different people may age at different rates as revealed by recent studies (Li et al., 2009; Horvath, 2013). Some people appear younger than their chronological age, and others appear older. In an extreme case, a 16-year-old girl without any known genetic syndromes or chromosomal abnormalities appeared to stop growing and looked like an infant (Walker et al., 2009). It is a challenge to identify her “actual” age. Many factors, for instance, lifestyle, and environmental factors, can hasten or delay aging (Feldman et al., 1994; Hultsch et al., 1999). Thus, a set of biomarkers that can reliably reflect real age has practical value. There are special cases in which such age biomarkers are particularly useful. For example, people may need to verify an athlete's age in sporting events such as the Olympic Games or to determine a suspect's age in certain forensic cases.
Different types of biomarkers have been proposed to quantify human age (Li et al., 2009). Physical parameters, such as visual acuity, auditory threshold, and maximum work rate, have been used as indicators of aging for more than three decades (Furukawa et al., 1975; Borkan and Norris, 1980). Other criteria, such as gray hair and skin wrinkles, can also reflect chronological age (Van Neste and Tobin, 2004). However, these parameters often do not provide accurate estimation of age and cannot reveal the internal molecular changes of the human body or the underlying aging mechanisms.
With the rapid development of high-throughput technologies, genomic, and epigenetic data are accumulating to an unprecedented status. This provides a new route of estimating aging at the molecular level. Associations between epigenetic variations (e.g., DNA methylation and histone modification) and age have been reported (Fraga and Esteller, 2007). It is manifested that gene expression and the methylation profile of blood (Bocklandt et al., 2011; Hannum et al., 2013; Horvath, 2013), the gene expression profile of brain (Fraser et al., 2005), and telomere length (Harley et al., 1990; Benetos et al., 2001) are good indicators of age in human and other primates. In addition, these biomarkers may also provide candidate targets for intervention to extend the human life span (Baker and Sprott, 1988).
Previous studies on age prediction using gene expression mainly rely on single tissues, such as blood or brain. The predictive ability of different tissues had not been thoroughly studied. Because aging is a concordant process involving multiple tissues (Kujoth et al., 2005), it might be effective to build an age-prediction model with information from multiple tissues. In this study, we built an optimal age prediction model by using the Genotype-Tissue Expression (GTEx) profile among 46 human tissues and then compared the predictive efficiency of a single tissue and combining two tissues.
From the GTEx (V6), the gene expression profiles from 46 tissues were used. A detailed description of sample collection, RNA preparation, RNA sequencing, gene expression estimation, etc., are listed in the GTEx consortium paper (The GTEx Consortium, 2015). We first normalized the original gene expression data from GTEx via quantile normalization.
The genes in each tissue were ranked based on the Pearson correlation of donor age and corresponding gene expression. Then, we picked top genes from 50 to 6400 with multiples of 2 as a model input and tuned it by 10-fold cross-validation (CV).
In this paper, we use root-mean-square error (RMSE) to measure the accuracy of the models. RMSE is a frequently used measure of the differences between values (sample or population values) predicted by a model or between an estimator and the values observed. In the age-prediction models, we use RMSE to measure the quality of the model: the smaller the RMSE, the higher the accuracy of the model—and on the contrary, the lower the accuracy of the model. The RMSE of predicted value ŷ, a regression's dependent variable y, is computed for different predictions as the square root of the mean of the squares of the deviations:
Our age-prediction model is based on the elastic net algorithm (Zhou and Hastile, 2005). The elastic net algorithm has a sparsity property and favors grouping effects so that strongly correlated predictors tend to be in or out of the model together. These properties let the method specifically fit our study because gene expression is highly interrelated, and our prediction model relies on only a small number of genes. The age-prediction process is formulated as follows:
where Agei is the chronological age of the donor of sample i with 1 ≤ i ≤ M, M is the number of samples in a particular tissue, xij is the log2-transformed expression of gene j with 1 ≤ j ≤ N for sample i, N is the number of preselected genes in the tissue, ω0 is the intercept, ωj is the weight of gene j, is the predicted value of ω, 0 ≤ α ≤ 1 is a parameter to balance the L1 (e.g., lasso) and L2 (e.g., ridge regression) penalty, and λ is the lasso parameter. The two parameters α and λ are optimized by a 10-fold CV. After ω0 and ωj (1 ≤ j ≤ N) are determined, the following equation is used to predict age for a new sample y with an expression level known for selected genes:
It is worth noting that the main purpose of this study is to compare the predictive capability of a single tissue with double tissues. Because the main focus is not to identify the “best” predictive models, we do not compare the performance of elastic net with other machine learning methods. However, given the wide application of elastic net in age prediction (Hannum et al., 2013), we consider it to be an appropriate choice to serve the main purpose of this work.
To identify the best age-prediction model, we applied the 10-fold CV strategy to the analysis. In addition, we bootstrapped the CV process 100 times and averaged the validation RMSE and Pearson correlation coefficient (PCC) to reduce the potential bias that originated from random sampling when splitting the sample into training and testing sets.
As stated above, there are three model parameters, namely the preselection threshold N, parameter α to balance the lasso and ridge regression penalties, and lasso parameter λ. These parameters are tuned by 10-fold CV. Specifically, we let N increase from 50 to 6400 by multiples of 2, α increase from 0 to 1 with a step-wise addition of 0.01, and λ increase from 2−10 to 210 with multiples of 2. The set of parameters yielding the lowest averaged validation RMSE in the 100 bootstrapped, 10-fold, CV runs were chosen as the optimal parameters for single and double tissue. It is of note that we reranked and selected genes (based on the 9 fold training data) in each CV to avoid overfitting.
Because the number of overlapping samples among three tissues are often less than 70, we only analyzed samples that came from two tissues. To balance the contribution of individual tissue, an equal number of top gene expression profiles from each tissue were combined as features in the prediction model. A similar analysis was then applied to tune the model parameters. The performance of each tissue and double tissues were evaluated by RMSE from both validation and testing data.
The DAVID (6.7) (Huang et al., 2009) (https://david.ncifcrf.gov/tools.jsp) bioinformatics resource consists of an integrated biological knowledge base and analytic tools aimed at systematically extracting biological meaning from large gene/protein lists. We can use DAVID, a high-throughput and integrated data-mining environment, to analyze gene functional classification, functional annotation charts, or clustering and functional annotation tables through gene lists derived from our age-prediction models. By following this protocol, investigators are able to gain an in-depth understanding of the aging themes in lists of genes that are enriched in genome-scale studies.
We develop a computational framework to predict donor age depending on the gene expression profile of one single or two tissues generated from GTEx (Version 6). GTEx contains expression profiles of more than 41,298 genes in 46 human tissues. There are 34,443 genes and 8,375 samples that passed the quality control and data processing procedure that was used as the benchmark data in this study. Detailed information on the samples for 46 tissues is provided in Table 1. As can be seen from Table 1, the ages of donors range from 20 to 70, and the number of samples varies from 71 to 430 for each tissue.
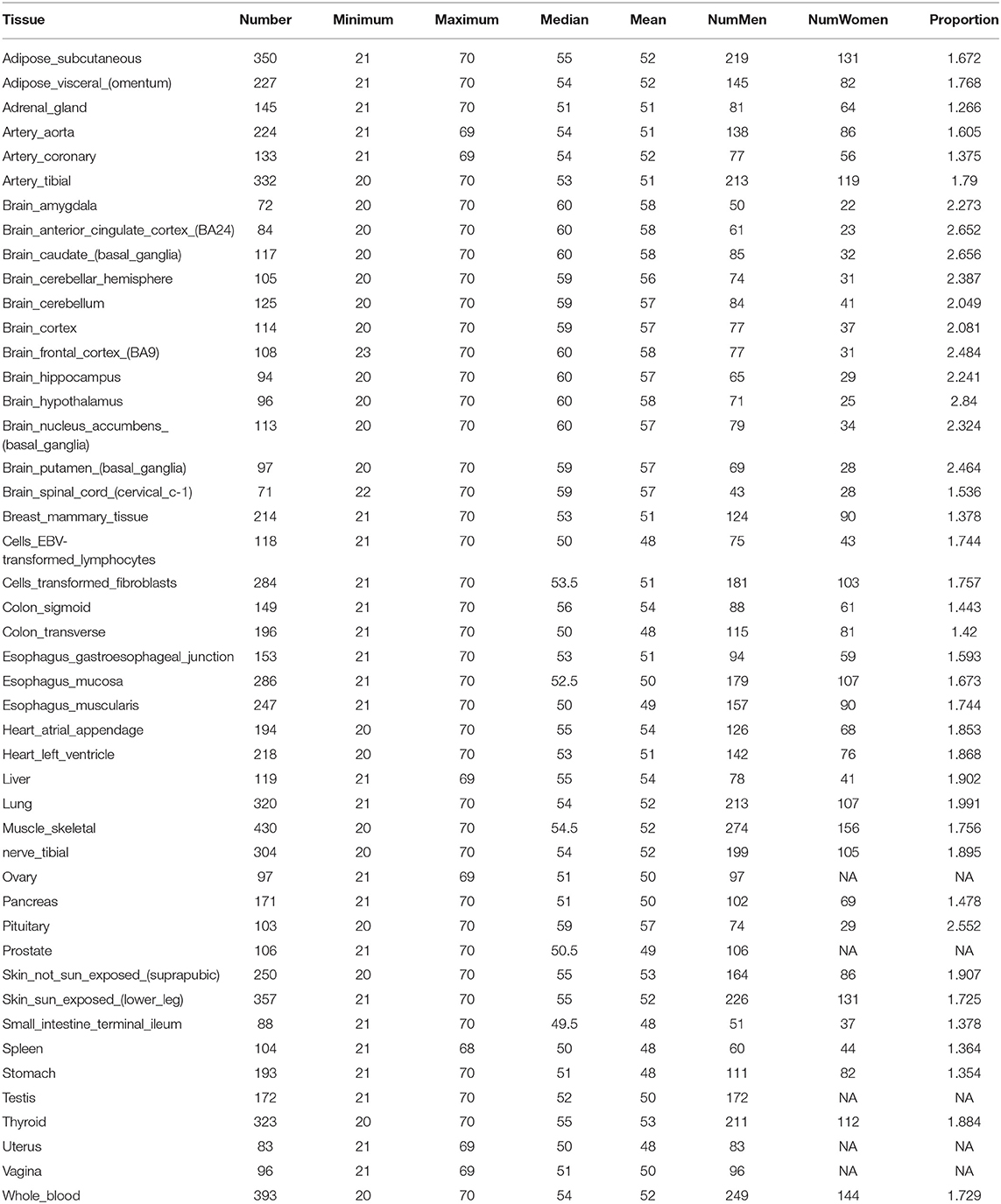
Table 1. Sample Information of 46 tissues in GTEX.
As shown in Figure 1, our prediction framework has multiple steps. First, we rank the genes in each tissue based on the PCC of donor age and the corresponding gene expression. Top age-associated genes in one single or two tissues were then used to construct features in an elastic net regularization model, which is a sparse learning model capable of handling data with small sample sizes but numerous features (Zhou and Hastile, 2005). The parameters of the models were tuned through 10-fold CV according to the RMSE. Functions of genes were annotated by the DAVID Tools (see “Methods” for detailed information).
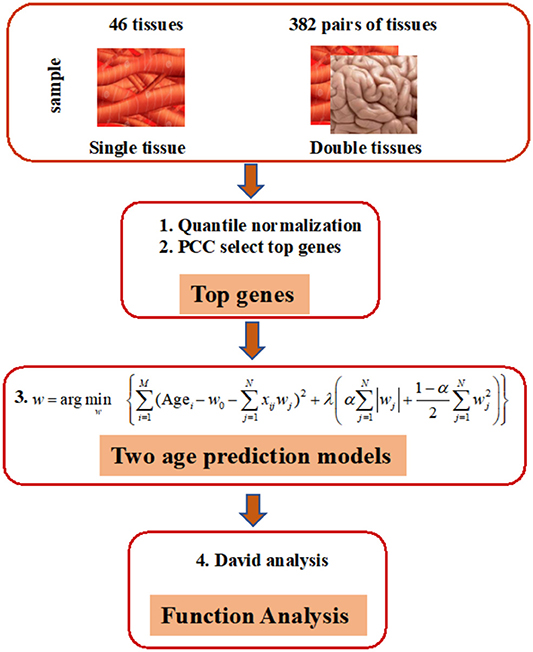
Figure 1. Overview of elastic net method for building age-prediction model. 1. Normalize the original gene expression data from GTEx via quantile normalization. 2. Select the top 50, 100, 200, 400, 600, 800, 1,600, 3,200, and 6,400 genes, obtained via the Pearson correlation of the age and corresponding gene expression, and build the age-prediction model for each of 46 tissues. 3. Construct age-prediction model for multiple tissues as was done for single tissues. Because overlapping samples among three tissues are often less than 70, only two-tissue studies are contained in the current study. 4. Use the selected genes for DAVID analysis.
Our method was first applied to 46 single tissues, respectively. The performance of each tissue is listed in Table 2. As mentioned above, the number of top age-associated genes was taken as a parameter to our model. We selected the top 50, 100, 200, 400, 600, 800, 1,600, 3,200, and 6,400 genes and tested their performances by the 10-fold CV. It turns out that the number of top genes has some influence on prediction accuracy. The lowest RMSE (i.e., 3.8 years) was achieved for pituitary while selecting 600 genes. Pituitary is one of the most studied tissues and is highly associated with human aging (Seeman and Robbins, 1994). Other good tissues for age prediction include small intestine terminal ileum, spleen and testis, and brain/spinal cord. The most accessible tissue, whole blood, seems to be unsuitable for this task. Hannum et al. (2013) applied a blood gene expression profile to predict age based on a much larger sample size (488 in total). However, the RMSE is 7.22 years, which is comparable to our result. We also plotted the RMSEs for all other tissues (using the top 600 genes) in Figure 2A for a better view.
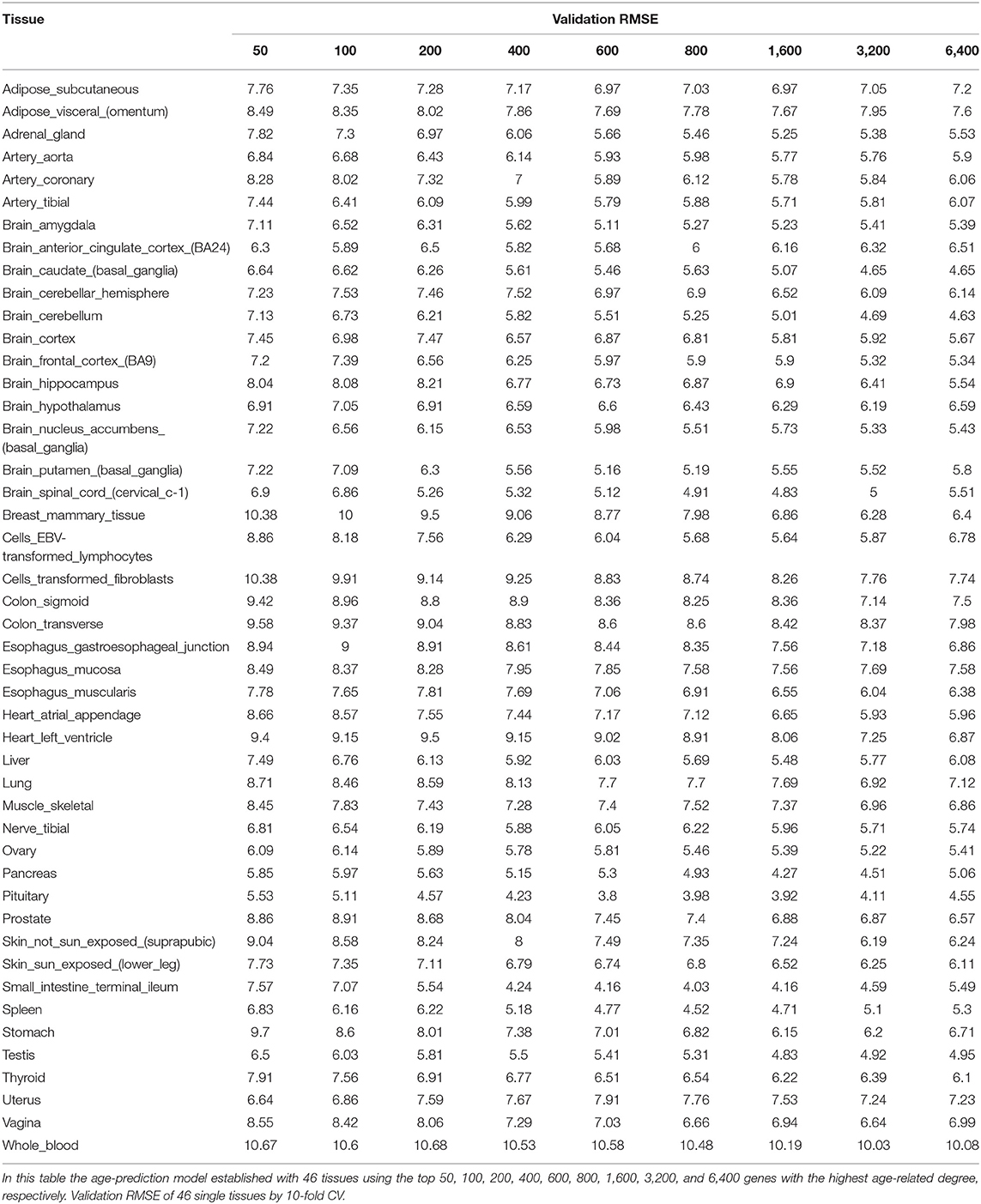
Table 2. Prediction accuracy by using single tissue.
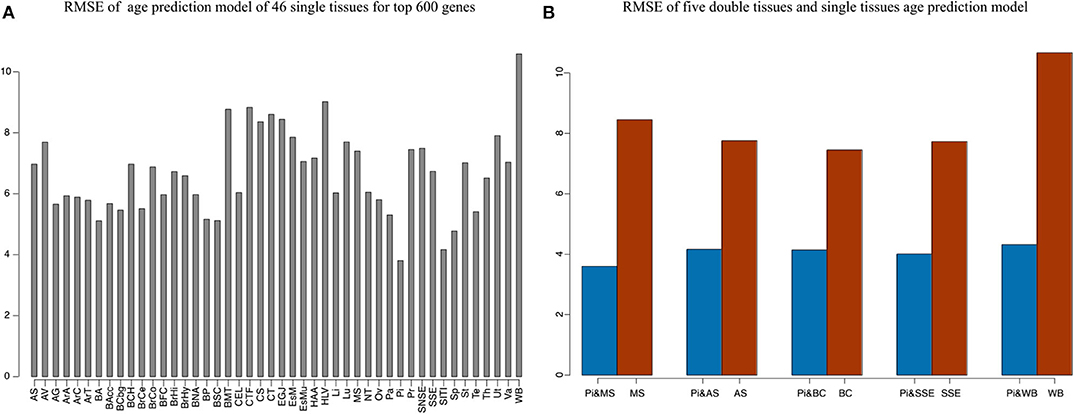
Figure 2. The accuracy of 46 single tissues and five double tissues in age prediction. (A) The RMSE of single tissue age predictors for the top 600 genes. We select the top 50, 100, 200, 400, 600, 800, 1,600, 3,200, and 6,400 genes, which are obtained via Pearson correlation of age and gene expression, and then build the age-prediction model across the 46 single tissues. Because the best predictive model appears in the top 600 genes, here we show the RMSE of the top 600 gene model. As can be seen from the figure, the minimum RMSE is 3.8, which corresponds to the age-prediction model of pituitary tissue. (B) Blue represents the RMSE of the top 600 genes of pituitary and the top 50 genes of muscle, adipose subcutaneous, brain cerebellum, skin sun exposed, and whole blood, and brown represents RMSE of the first 50 genes of muscle, adipose subcutaneous, brain cerebellum, skin sun exposed, and whole blood.
Because aging is a process associated with multiple tissues (Kujoth et al., 2005), it is reasonable to assume that combining multiple tissues can improve age-prediction accuracy. Because there are at least 71 samples in a single tissue, we selected people with at least 70 samples in two tissues for a relatively fair comparison, which derives 382 combinations in total. The combinations were used to train 382 elastic net models (Zhou and Hastile, 2005), whose performances were also evaluated by the 10-fold CV. The results show that it is possible to improve age prediction by combining two tissues. As we mentioned above, the best prediction RMSE for single tissue (3.8 years) was achieved at pituitary with 600 genes. We added 50, 100, 200, and 400 selected genes from one other tissue, including muscle skeletal, adipose subcutaneous, brain cerebellum, skin sun exposed, and whole blood, whose performances are listed in Table 3 and shown in Figure 2B. As can be seen, the validation RMSE decreases to 3.6 by combining 50 genes from muscle skeletal (see also Figures 3A,B). However, the prediction accuracy is worse when adding other tissues, indicating that different tissues might undergo aging at different rates or mechanisms. Generally speaking, the age-prediction accuracy is elevated with the increase of tissue number, which supports that aging is a concordant process involving multiple tissues (Kujoth et al., 2005).
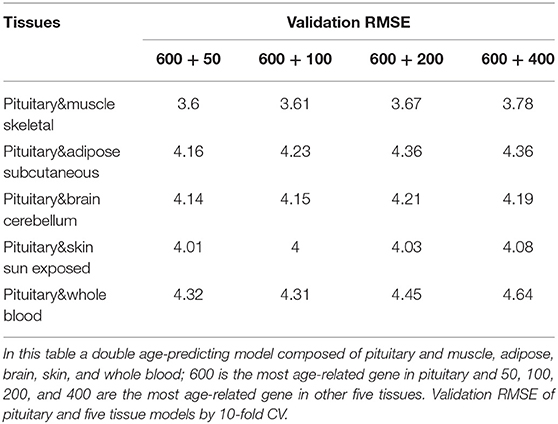
Table 3. Prediction accuracy by combining double tissues.
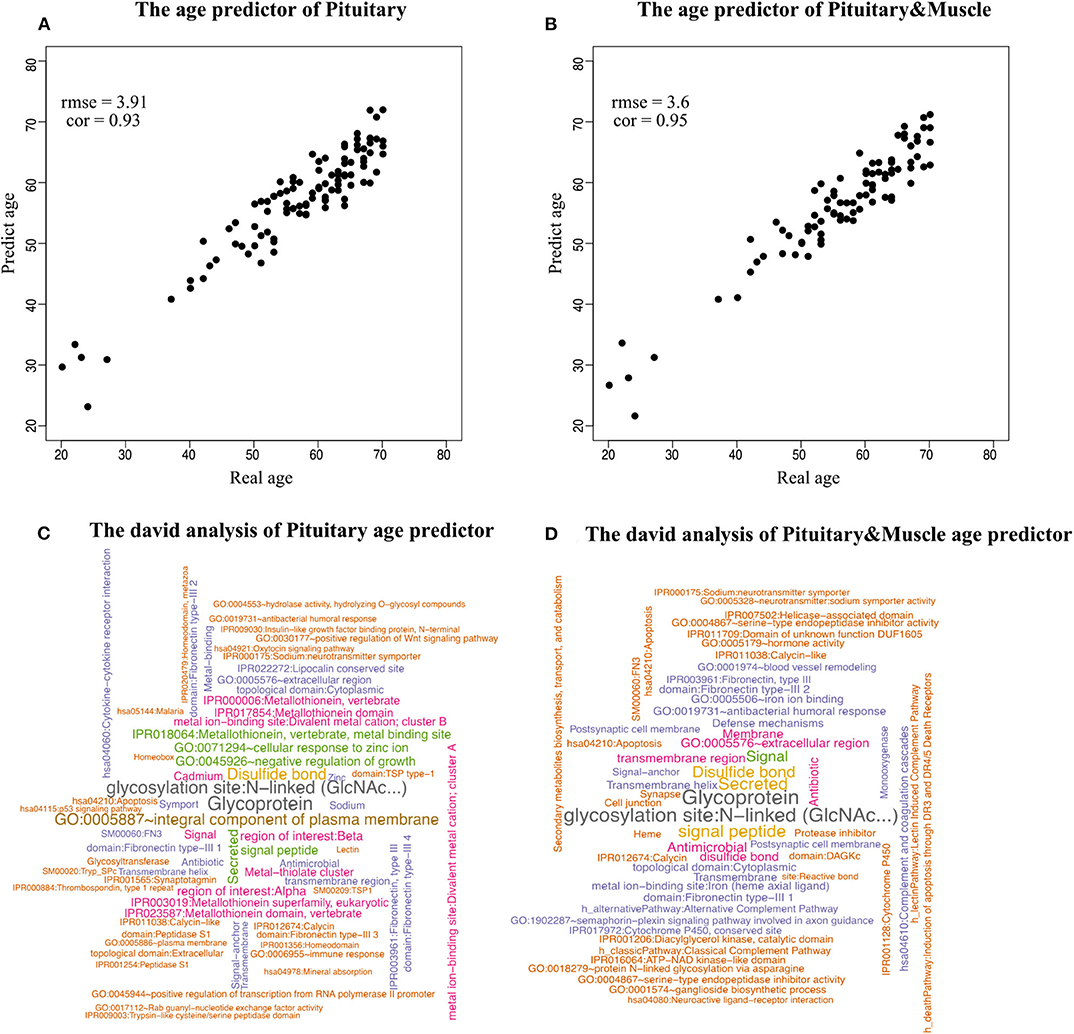
Figure 3. Scatterplot of age prediction and gene functional analysis. (A) Scatterplot of the pituitary age-prediction model for the top 600 genes in 46 single tissues. The RMSE is 3.8, and the PCC of real and predicted age is 0.93. (B) Scatterplot of pituitary for 600 genes and muscle skeletal for 50 genes age-prediction model. The RMSE is 3.6, and the PCC of real and predicted age is 0.95. (C) DAVID analysis of the age-prediction model in pituitary. (D) DAVID analysis of the age-prediction model in pituitary and muscle skeletal.
In our model, we prefilter genes and only allow the top N genes as features to be selected by the elastic net model. There are two elastic net parameters, namely α, which controls the balance between lasso and ridge regression, and λ, the lasso parameter. Because the effects of α and λ have been extensively studied (Zhou and Hastile, 2005), we tested the effect of N on validation error in this study. For most prediction models with a small validation error, the number of genes involved in the model ranges from 300 to 1600. As an indication, only a small or moderate portion of genes are necessary to predict age. This finding is also supported by other studies (Bocklandt et al., 2011; Hannum et al., 2013), in which 200 methylation markers are used to predict the biological age of individuals. The parameters of the best model (e.g., “pituitary&muscle”) are α = 0, λ = 0.5, w0 = 49.1, that is, age = 49.1 − 0.5534609 × RF00019 + 0.4345046 × RASSF8 + 0.4238481 × ALOX15B + …
The model has an intercept of 49.1 years, which is quite close to the mean age of the samples 50.81.
For the best prediction model, we listed the top 50 genes (according to the absolute value of coefficients) and their coefficients in Table 4. Among the top 50 genes, 49 are from pituitary, and only 1 is from muscle (ranked at 15). Interestingly, most of the top genes are age-associated. For example, RASSF8 (ras association domain-containing protein 8), ranks second in the list. RASSF8 encodes a protein that is a member of the transmembrane 4 superfamily and is a lung tumor–suppressor gene candidate. It plays important roles in the regulation of localization, methylation, cell–cell adhesion, cell migration, cell death, response to hypoxia, mitosis, cell growth, wound healing, contact inhibition, and epithelial cell migration (Falvella et al., 2006; Wang et al., 2017; Karthik et al., 2018; Shi L. et al., 2018). Accumulated evidence suggests that RASSF8 is associated with aging (Geigl et al., 2004; Shi Z. et al., 2018; Pagliai et al., 2019). Similarly, ALOX15B (Arachidonate 15-Lipoxygenase Type B), which ranks third on the list, is a protein-coding gene. Diseases associated with ALOX15B include autosomal recessive congenital ichthyosis and prostate cancer (Bhatia et al., 2005; Ginsburg et al., 2016; GeneCards, 2020). This gene is a senescent gene, which can also affect human aging with its expression increasing when prostate epithelial cells become senescent (Bhatia et al., 2005; Alfardan et al., 2019). In addition to age-associated genes, there are also many genes whose association with aging is unknown. For example, no association with aging could be identified in the literature for the top gene RF00019 on the list. In the future, further studies might be needed to elucidate the mechanism for age-dependent functions of RF00019.
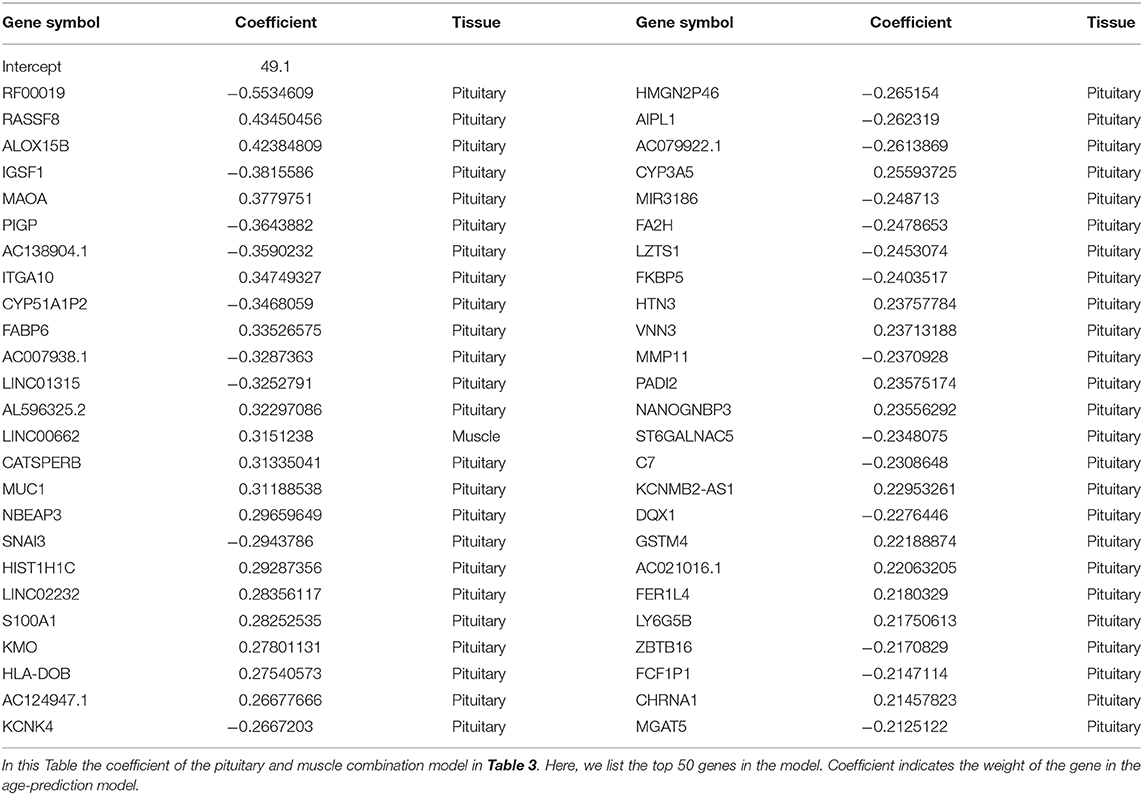
Table 4. Best models for age prediction using pituitary & muscle skeletal tissue.
To identify the biological processes associated with genes in the prediction model, we performed functional annotation analysis using the DAVID tools (Huang et al., 2009), a web-accessible set of tools that allow researchers to infer the biological meaning behind large lists of genes. Because our focus is on enriched functional categories rather than on individual genes, we selected the functional clustering with adjusted P < 0.05. The top cluster is related to glycoprotein (P = 1.79 × 10−8). Histidine-rich glycoprotein (HRG) is present at high levels in plasma, and it is synthesized by parenchymal liver cells and transported as a free protein as well as being stored in α-granules of platelets and released after thrombin stimulation (Blank and Shoenfeld, 2008). Levels of HRG variants in human blood are associated with chronological age and predict mortality (Hong et al., 2019). Also noteworthy were clusters related to age, for instance, GO:0045926~negative regulation of growth (P = 1.08 × 10−4) (Figures 3C,D).
Each human individual has two “ages.” One is the chronological age defined by the time that has passed since birth, and the other is biological age, which describes a shortfall between a population cohort average life expectancy and the perceived life expectancy of an individual of the same age (Jackson et al., 2003). An accurate estimation of biological age is helpful in studying aging, and several approaches have been proposed so far (Borkan and Norris, 1980; Dubina et al., 1983; Hannum et al., 2013). The aging prediction strategy in this study reflects the donor's biological age, effectively providing a possible way to identify key genetics or environmental factors that lead to younger biological age than the chronological age.
By constructing elastic net models, we can predict human age as well as identifying genes strongly associated with human aging. For example, RASSF8 and ALOX15B have been studied to be associated with human aging and age-associated diseases. The function enrichment analysis revealed some common functions, such as glycoprotein and signal peptide in prediction models of multiple tissues, suggesting their general association with aging. In the future, we will identify tissue-common and tissue-specific aging genes and functions.
Our results suggest that the expression level of a small number of genes can reliably predict human age. In the single-tissue model, the predicted age showed a higher deviation from the true chronological age compared to predictions based on two tissues. This reveals that tissues within the same individual have heterogeneous aging rates. The tissue specificity of aging is reported by studies performed in model organisms (Herndon et al., 2002; Libina et al., 2003; Niedernhofer, 2008). On the other hand, aging is a concordant process involving multiple tissues. Different tissues have different potentials for revealing the chronological age of the host, jointly considering that multiple tissues can reduce the variation derived from a single tissue. For instance, our results indicate that blood is a poor choice for age prediction although it is one of the most accessible tissues. In both validation and test data sets, predicted age is more easily deviated from chorological age in blood compared with other tissues. The poor prediction performance of blood is also supported by the other study using the human whole blood transcriptome (Hannum et al., 2013), suggesting that the blood transcriptome fluctuates more due to its frequent interactions with other tissues and environmental factors through circulation (Benetos et al., 1993; Franklin et al., 1997).
Some improvements can be expected to increase the prediction accuracy. First, only two tissues were considered in this study due to sample size limitation. In the future, we may include more tissues. Second, we only use gene expression to predict age. Many other molecular biomarkers have also been reported successfully in predicting human age, for example, methylation (Hannum et al., 2013) and telomere length (Harley et al., 1990; Benetos et al., 2001). Last, there are many choices of machine learning technologies that can be adopted, for example, support vector machine (Cortes and Vapnik, 1995) and neural network (Mcculloch and Pitts, 1990). Combining multiple types of genomics data and data analysis methods will certainly facilitate the prediction efficiency greatly (Dobin et al., 2013).
We have developed a computational framework to predict individual age through age-associated gene expression of single and two tissues. The predicted age is an indicator of biological age reflecting the life span and true functionality of a human body. Although gene expression from a single tissue could be used to estimate individual chronological age, the prediction accuracy is improved by properly combining those with other tissues. Different tissues provide different potential in predicting age, more reliable gene expression–based age markers are obtained in pituitary and skeletal muscle compared with blood.
All datasets generated for this study are included in the article/supplementary material.
ZT, LC, JY, and GT conceived, designed, and managed the study. FW and JY performed the experiments. HL, QLi, ZY, QLu, and GT provided computational support and technical assistance. All authors approved the final manuscript.
This study was partially supported by the Natural Science Foundation of Hunan, China (Grant No. 2018JJ2461) and Talent introduction fund of Zhejiang University Ningbo Institute of Technology (20200709).
JY, HL, QLi, ZY, QLu, and GT were employed by the company Geneis Beijing Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors appreciate those contributors who make the Genotype-Tissue Expression (GTEx). They would also like to thank reviewers for their careful reading and valuable suggestions.
GTEx, The Genotype-Tissue Expression; RMSE, The root-mean-square error; PCC, The Pearson correlation coefficient; CV, Cross-validation.
Alfardan, R., Guo, C., Toth, L. A., and Nie, D. (2019). Impaired recovery from influenza a/x-31(H3N2) infection in mice with 8-lipoxygenase deficiency. Med. Sci. 7:60. doi: 10.3390/medsci7040060
Baker, G. T, III., and Sprott, R. L. (1988). Biomarkers of aging. Exp. Gerontol. 23, 223–239. doi: 10.1016/0531-5565(88)90025-3
Benetos, A., Laurent, S., Hoeks, A. P., Boutouyrie, P. H., and Safar, M. E. (1993). Arterial alterations with aging and high blood pressure. A noninvasive study of carotid and femoral arteries. Arterioscler. Thromb. 13, 90–97. doi: 10.1161/01.ATV.13.1.90
Benetos, A., Okuda, K., Lajemi, M., Kimura, M., Thomas, F., Skurnick, J., et al. (2001). Telomere length as an indicator of biological aging: the gender effect and relation with pulse pressure and pulse wave velocity. Hypertension 37, 381–385. doi: 10.1161/01.HYP.37.2.381
Bhatia, B., Tang, S., Yang, P., Doll, A., Aumueller, G., Newman, R. A., et al. (2005). Cell-autonomous induction of functional tumor suppressor 15-lipoxygenase 2 (15-LOX2) contributes to replicative senescence of human prostate progenitor cells. Oncogene 24, 3583–3595. doi: 10.1038/sj.onc.1208406
Blank, M., and Shoenfeld, Y. (2008). Histidine-rich glycoprotein modulation of immune/autoimmune, vascular, and coagulation systems. Clin. Rev. Allerg. Immunol. 34, 307–312. doi: 10.1007/s12016-007-8058-6
Bocklandt, S., Lin, W., Sehl, M. E., Sanchez, F. J., Sinsheimer, J. S., Horvath, S., et al. (2011). Epigenetic predictor of age. PLoS ONE 6:e14821. doi: 10.1371/journal.pone.0014821
Borkan, G. A., and Norris, A. H. (1980). Assessment of biological age using a profile of physical parameters. J. Gerontol. 35, 177–184. doi: 10.1093/geronj/35.2.177
Cortes, C., and Vapnik, V. (1995). Support-vector networks. Mach. Learn. 20, 273–297. doi: 10.1007/BF00994018
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Dubina, T. L., Dyundikova, V. A., and Zhuk, E. V. (1983). Biological age and its estimation. II. Assessment of biological age of albino rats by multiple regression analysis. Exp. Gerontol. 18, 5–18. doi: 10.1016/0531-5565(83)90046-3
Falvella, F. S., Manenti, G., Spinola, M., Pignatiello, C., Conti, B., Pastorino, U., et al. (2006). Identification of RASSF8 as a candidate lung tumor suppressor gene. Oncogene 25, 3934–3938. doi: 10.1038/sj.onc.1209422
Feldman, H. A., Goldstein, I., Hatzichristou, D. G., Krane, R. J., and McKinlay, J. B. (1994). Impotence and its medical and psychosocial correlates: results of the massachusetts male aging study. J. Urol. 151, 54–61. doi: 10.1016/S0022-5347(17)34871-1
Fraga, M. F., and Esteller, M. (2007). Epigenetics and aging: the targets and the marks. Trends Genet. 23, 413–418. doi: 10.1016/j.tig.2007.05.008
Franklin, S. S., Gustin, W. T, Wong, N. D., Larson, M. G., Weber, M. A., Kannel, W. B., et al. (1997). Hemodynamic patterns of age-related changes in blood pressure. The framingham heart study. Circulation 96, 308–315. doi: 10.1161/01.CIR.96.1.308
Fraser, H. B., Khaitovich, P., Plotkin, J. B., Paabo, S., and Eisen, M. B. (2005). Aging and gene expression in the primate brain. PLoS Biol. 3:e274. doi: 10.1371/journal.pbio.0030274
Furukawa, T., Inoue, M., Kajiya, F., Inada, H., and Takasugi, S. (1975). Assessment of biological age by multiple regression analysis. J. Gerontol. 30, 422–434. doi: 10.1093/geronj/30.4.422
Geigl, J. B., Langer, S., Barwisch, S., Pfleghaar, K., Lederer, G., and Speicher, M. R. (2004). Analysis of gene expression patterns and chromosomal changes associated with aging. Cancer Res. 64, 8550–8557. doi: 10.1158/0008-5472.CAN-04-2151
GeneCards (2020). The Human Gene Database. Available online at: https://www.genecards.org/cgi-bin/carddisp.pl?gene=ALOX15B&search=linoleic%20acid%20metabolic%20process
Ginsburg, K., Dyson, G., Bollig-Fischer, A., and Powell, I. (2016). Elevated expression of 15-lipoxygenase-2 (Alox15b) is associated with nonaggressive prostate cancer and confers a survival benefits. J. Urology. 195, E1096–E1097. doi: 10.1016/j.juro.2016.02.2243
Hannum, G., Guinney, J., Zhao, L., Zhang, L., Hughes, G., Sadda, S., et al. (2013). Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 49, 359–367. doi: 10.1016/j.molcel.2012.10.016
Harley, C. B., Futcher, A. B., and Greider, C. W. (1990). Telomeres shorten during ageing of human fibroblasts. Nature 345, 458–460. doi: 10.1038/345458a0
Herndon, L. A., Schmeissner, P. J., Dudaronek, J. M., Brown, P. A., Listner, K. M., Sakano, Y., et al. (2002). Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature 419, 808–814. doi: 10.1038/nature01135
Hong, M. G., Dodig-Crnković, T, Chen, X., Drobin, K., Lee, W., Wang, Y., et al. (2019). Levels of histidine-rich glycoprotein variants in human blood are associated to chronological age and predict mortality. bioRxiv. 3:464909. doi: 10.1101/464909
Horvath, S. (2013). DNA methylation age of human tissues and cell types. Genome Biol. 14:3156. doi: 10.1186/gb-2013-14-10-r115
Huang, D. W., Sherman, B. T., and Lempicki, R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. doi: 10.1038/nprot.2008.211
Hultsch, D. F., Hertzog, C., Small, B. J., and Dixon, R. A. (1999). Use it or lose it: engaged lifestyle as a buffer of cognitive decline in aging? Psychol. Aging 14, 245–263. doi: 10.1037/0882-7974.14.2.245
Jackson, S. H., Weale, M. R., and Weale, R. A. (2003). Biological age—what is it and can it be measured? Arch. Gerontol. Geriatr. 36, 103–115. doi: 10.1016/S0167-4943(02)00060-2
Karthik, I. P., Desai, P., Sukumar, S., Dimitrijevic, A., Rajalingam, K., and Mahalingam, S. (2018). E4BP4/NFIL3 modulates the epigenetically repressed RAS effector RASSF8 function through histone methyltransferases. J. Biol. Chem. 293, 5624–5635. doi: 10.1074/jbc.RA117.000623
Kujoth, GC, Hiona, A, Pugh, TD, Someya, S, Panzer, K, and Wohlgemuth, SE. (2005). Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 309, 481–484. doi: 10.1126/science.1112125
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Libina, N., Berman, J. R., and Kenyon, C. (2003). Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell 115, 489–502. doi: 10.1016/S0092-8674(03)00889-4
Mcculloch, W. S., and Pitts, W. (1990). A logical calculus of the ideas immanent in nervous activity. Bull. Math. Biol. 52, 99–115. doi: 10.1007/BF02478259
Niedernhofer, L. J. (2008). Tissue-specific accelerated aging in nucleotide excision repair deficiency. Mech. Ageing Dev. 129, 408–415. doi: 10.1016/j.mad.2008.04.010
Pagliai, G., Sofi, F., Dinu, M., Sticchi, E., Vannetti, F., Lova, R. M., et al. (2019). CLOCK gene polymorphisms and quality of aging in a cohort of nonagenarians - the MUGELLO study. Sci Rep. 9:1472. doi: 10.1038/s41598-018-37992-8
Seeman, T. E., and Robbins, R. J. (1994). Aging and hypothalamic-pituitary-adrenal response to challenge in humans. Endocr. Rev. 15, 233–260. doi: 10.1210/er.15.2.233
Shi, L., Middleton, J., Jeon, Y. J., Magee, P., Veneziano, D., Lagana, A., et al. (2018). KRAS induces lung tumorigenesis through microRNAs modulation. Cell Death Dis. 9:219. doi: 10.1038/s41419-017-0243-9
Shi, Z., Geng, Y., Liu, J., Zhang, H., Zhou, L., Lin, Q., et al. (2018). Single-cell transcriptomics reveals gene signatures and alterations associated with aging in distinct neural stem/progenitor cell subpopulations. Protein Cell 9, 351–364. doi: 10.1007/s13238-017-0450-2
The GTEx Consortium (2015). The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 348, 648–660. doi: 10.1126/science.1262110
Van Neste, D., and Tobin, D. J. (2004). Hair cycle and hair pigmentation: dynamic interactions and changes associated with aging. Micron 35, 193–200. doi: 10.1016/j.micron.2003.11.006
Walker, R. F., Pakula, L. C., Sutcliffe, M. J., Kruk, P. A., Graakjaer, J., and Shay, J. W. (2009). A case study of “disorganized development” and its possible relevance to genetic determinants of aging. Mech. Ageing Dev. 130, 350–356. doi: 10.1016/j.mad.2009.02.003
Wang, L., Liu, W., Zhang, Y. P., and Huang, X. R. (2017). The miR-224 promotes non-small cell lung cancer cell proliferation by directly targeting RASSF8. Eur. Rev. Med. Pharmaco. 21, 3223–3231.
Keywords: age prediction, aging, gene expression, RNA sequencing, genotype-tissue expression (GTEx)
Citation: Wang F, Yang J, Lin H, Li Q, Ye Z, Lu Q, Chen L, Tu Z and Tian G (2020) Improved Human Age Prediction by Using Gene Expression Profiles From Multiple Tissues. Front. Genet. 11:1025. doi: 10.3389/fgene.2020.01025
Received: 25 June 2020; Accepted: 11 August 2020;
Published: 24 September 2020.
Edited by:
You Zhou, First People's Hospital of Changzhou, ChinaReviewed by:
Leiming Wang, Baylor College of Medicine, United StatesCopyright © 2020 Wang, Yang, Lin, Li, Ye, Lu, Chen, Tu and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luonan Chen, bG5jaGVuQHNpYnMuYWMuY24=; Zhidong Tu, emhpZG9uZy50dUBtc3NtLmVkdQ==; Geng Tian, dGlhbmdAZ2VuZWlzLmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.