 Na Han1,2
Na Han1,2 Wen Zhang
Wen Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
TECHNOLOGY AND CODE article
Front. Genet. , 31 August 2020
Sec. Statistical Genetics and Methodology
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.01007
Pathogenic microorganism analysis based on next-generation sequencing technology is an important tool for clinical diagnosis, public health surveillance, and outbreak investigation. However, scientific researchers without the relevant background lack the time, training, or infrastructure to use large data sets or install and use command line tools. Therefore, the bioinformatic team at the Chinese Center for Disease Control and Prevention developed the Microbial Data Analysis Cloud Platform (MDACP) as a safe, professional, and efficient pathogen genetic data analysis platform for rapid microbial data mining, such as for candidate pathogen detection, genome typing, and traceability. MDACP is a web service system based on the Docker platform and can be used for data analysis on various operating systems. The platform focuses on pathogen analysis and continuously develops new analysis processes according to the analysis needs of the users. This platform has a friendly user interface and is easy to operate, allowing users to submit data through data pages or graphical clients, flexibly control parameters according to data conditions, and analyze data in parallel with multiple tasks. Researchers can quickly carry out bioinformatic analyses without coding work, promote follow-up research and information mining of projects, and improve the utilization of big data in the field of disease control. MDACP enables research personnel to conduct data analysis and management and assists clinicians and disease control personnel with mining information, such as pathogen identification, classification, and traceability.
Infectious diseases seriously threaten human health and are an important consideration for ensuring national biosafety. Infectious diseases also impact animals and plants, which may have a major effect on animal husbandry and agriculture. Introduction of foreign pathogens can cause human infectious diseases, as well as animal and plant diseases and even ecological disasters. Accurate and rapid detection and identification of pathogens and their resistance phenotypes is key to preventing and controlling infectious diseases. Traditional pathogen detection and identification technologies are mainly based on the immune response or nucleic acid amplification and hybridization, which can only detect one or a few pathogens at a low cost and efficiency and with a long experimental cycle; moreover, very few types of pathogens can be routinely detected. With the rapid development of genomic technology, particularly genome sequencing technology, and the advent of the big data era, it has become possible to detect and identify pathogenic microorganisms (Deurenberg et al., 2017).
Next-generation sequencing (NGS) is a powerful tool in medical microbiology and provides operational information that is difficult or impossible to achieve with traditional microbial technology. This method is widely used in studies of clinical and public health (Motroa and Moran-Gilad, 2017; Besser et al., 2018; Rossen et al., 2018). However, the use of NGS platforms requires sequencers to generate high quality and reliable sequencing data, as well as the means to analyze and interpret the large data sets generated. Analysis of large data sets often requires a combination of bioinformatics skills and high computational resources, making this approach impractical for many diagnostic medical microbiology laboratories (Deurenberg et al., 2017). Additionally, researchers who are unfamiliar with bioinformatic sequence analysis experience difficulty in determining the most appropriate protocol for achieving their research aims, selecting and applying the best bioinformatic tools, and identifying IT resources to access, store, and process large amounts of related to sequence data (Agrawal et al., 2017).
To overcome these challenges, the bioinformatics team at the Chinese Center for Disease Control and Prevention (China CDC) developed the Microbial Data Analysis Cloud Platform (MDACP). MDACP is a secure, professional, and efficient pathogen genetic data analysis platform that performs rapid and professional microbial data mining, such as candidate pathogen detection, for disease system practitioners and clinicians at all levels. MDACP simplifies the transfer, analysis, and visualization of large microbial data sets, integration of users through data pages or graphical clients, and transfer of publicly available data sets into analytics workflows. It also allows users to adjust the parameters set by workflow developers according to their experience levels or data situation, and to use cloud-computing resources to analyze data simultaneously with multi-tasking. MDACP is a pathogenic microbial data analysis cloud platform that does not require bioinformatics, IT technology, and computing resources. China CDC personnel can quickly carry out bioinformatic analyses, promote follow-up research and information mining of projects, and improve the utilization of big data in the field of disease control.
The MDACP system uses a browser/server mode to support a cross-platform and horizontal expansion. The web server uses Nginx to handle access control. The platform database is MongoDB, which uses the Flask development framework to handle service access. The system is hosted on a Linux server. The web interface was developed using Python and the graphical client was developed in Java. Tools and workflows were installed with Docker technologies. Currently, MDACP platforms deployed on Alibaba have no restrictions on the number of users when the elastic expansion framework is used for the computing node. The results also show that the system supports 100 users simultaneously running workflows.
MDACP provides two methods for transferring data. Users can upload the data to be analyzed and download the result files individually through the web data page. Alternatively, we provide graphical clients that support Windows and Mac systems to facilitate bulk data transfer between local and cloud storage services. To shorten the data transmission time, the graphical client provides dedicated compression tools and breakpoint retransmissions for large files of raw sequencing data to support data transmission when the network is unstable. We also use data validation during bulk data transfer to ensure the accuracy and security of the user data. Testing results showed that the time required for transmission of 1 Gb data is ~10 min. The transmission speed in different regions also depends on the local network speed.
To protect the safety of personal data, the user must register and log in before using MDACP. The platform data management adopts an access permission isolation mechanism, and the user can only access the data of other users by receiving authorization, thereby further improving the data security for the user. User personal data are safely protected in MDACP by multiple real-time copies, which are retained even if the physical hardware corrupts the data. The system administrator has no access to the user’s data but can generate a new workflow in the MDACP platform to which specific users are granted access.
In contrast to the Seven Bridges and BaseSpace analysis platforms, MDACP analysis has flexible scalability and its analysis function module includes online configuration. Users with privileges can access the process configuration page of the platform and upload new workflows through simple procedures, parameter configuration, and other operations, as shown in Figure 1. The report page of the analysis process can also be customized according to the needs of the user.
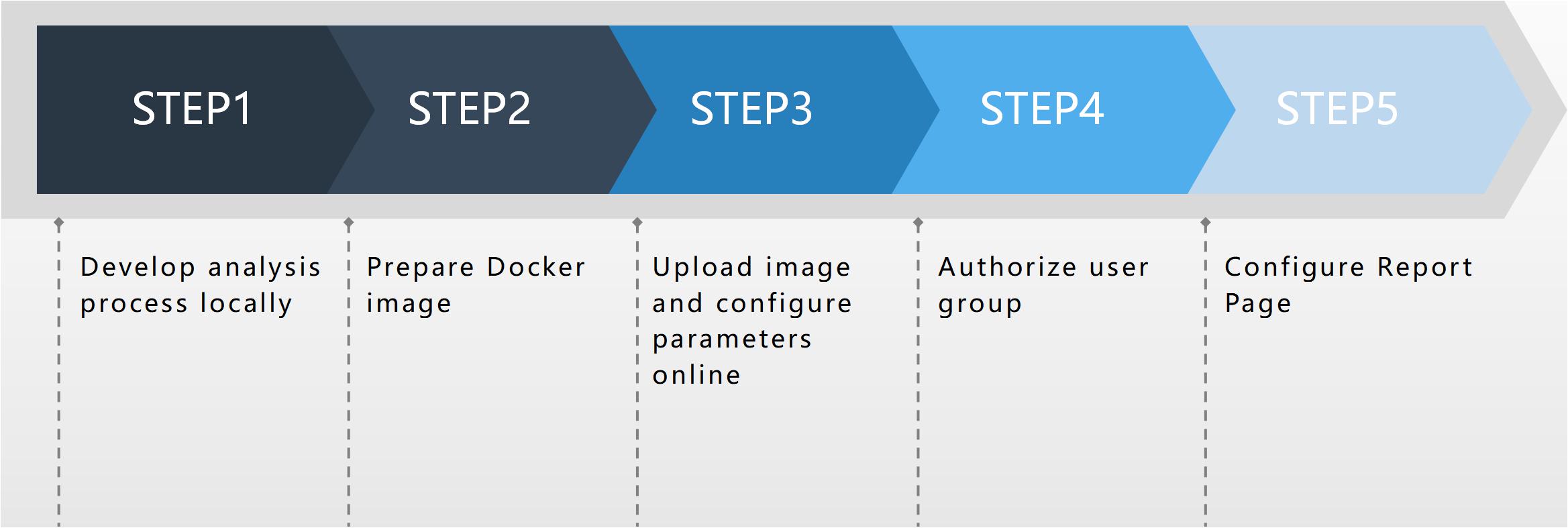
Figure 1. The online deployment process.
MDACP is a web-based cloud platform that allows anyone with access to the Internet to perform pathogenic microbiological analyses without the need for local computing resources and expertise. In addition, MDACP uses Docker technology for analysis process encapsulation, which enables flexible expansion of computing resources, high availability, and good isolation, thereby improving the repeatability and practicability of the analysis process. MDACP has a user-friendly graphical interface and provides convenient pathogenic microbial analysis for inexperienced users, and standardized but configurable options maintain high functionality for experienced researchers.
Data are always saved in MDACP as multiple real-time copies, even if the physical hardware corrupts the data. In addition, the platform data management adopts an access permission isolation mechanism, and the user can only access the data of other users by receiving authorization, further improving data security.
MDACP is a computing environment based on Docker technology that configures complex software tools and workflows, allowing microbiologists or physicians with little experience in programming to quickly perform pathogenic microbiological data analyses in a web interaction mode. Compared with other research methods, and while generating analytical results without deviation, real-time and rapid analyses are important targets for pathogenic microbial data analysis. The development and published analyses of the bioinformatics team are the main deployment targets of the MDACP workflow. MDACP allows collaborative researchers to share their stable and biologically meaningful analysis tools for visual deployment with more users. In addition, the personalized analysis process, jointly developed according to the analysis needs of the user and open source software recommended by most users, is an important part of the MDACP workflow. In the future, MDACP will support user customization and workflow function sharing, and more analysis workflows will be open to users.
Currently, MDACP has opened 35 workflows, allowing disease control personnel and clinicians to perform pathogen identification and screen for drug resistance genes, fractals, and traceability. It also has analysis tools for raw read preprocessing, sequence assembly, gene prediction and annotation, specific analysis, and graphical transformation of results. We provide detailed functional descriptions, usage instructions, and referenced open source software for each workflow to help users quickly apply MDACP to perform data analysis and cite results.
According to the MDACP workflow, deployment conditions can be divided into the following three parts: (1) workflow developed and published by ourselves, including the 16SPIP workflow of pathogen identification for metagenomic samples (Miao et al., 2017), predictive process Effector search for secretory system effector proteins of pathogen genome type iii (Zhang and Bergelson, 2012), core genotyping of Streptococcus suis (Chen et al., 2013), and process of microbial genome evolution analysis based on genetic similarity ANItools (Zhang et al., 2014) and (2) cooperative researchers share the co-deployed analytical workflow, such as the metagenomic resistance gene detection process ARGs-OAP v2.0 (Yin et al., 2018). We then simplified the analysis steps of the process and supported simultaneous analysis of multiple samples to facilitate the determination of difference in resistance genes between samples; (3) several widely used bioinformatics tools, such as Prokka (Seemann, 2014) and Centrifuge (Kim et al., 2016). The MDACP platform connects these existing microbial genomic analysis tools into workflows on a point-and-click interface, which will be easier for user to use.
Previously, users needed Unix command line or high computing resources to run this bioinformatics software. Through our platform, users without programming experience or with limited computing resources can perform various steps of pathogenic microbial data analysis with various types of online MDACP workflows (Table 1) (Kent, 2002; Segata et al., 2011; Luo et al., 2012; Page et al., 2015; Wick et al., 2017).
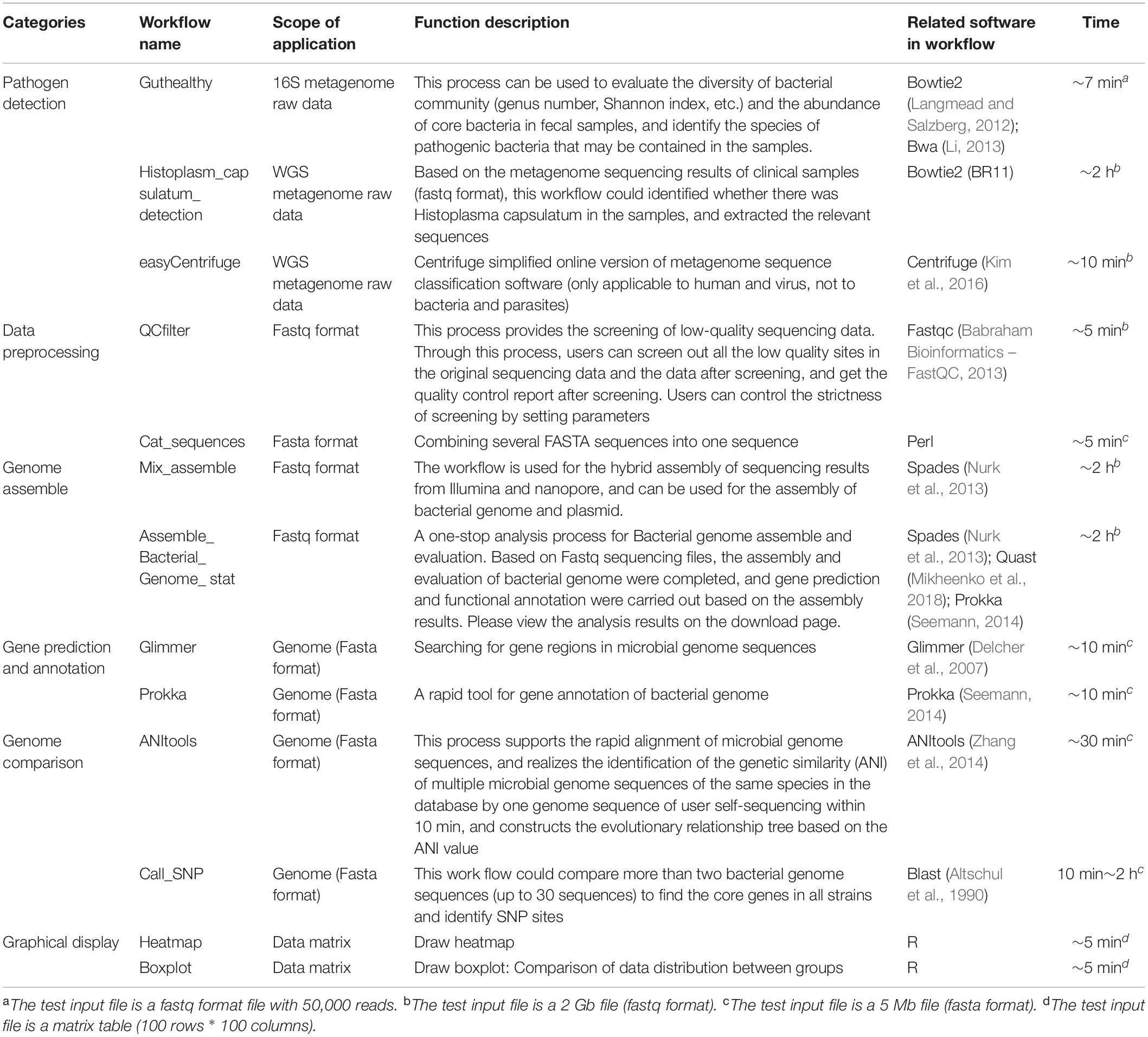
Table 1. MDACP workflow.
MDACP simplifies and accelerates the generation of microbiological data analysis results, and the user-friendly interface saves time. The user can upload sequencing data to the system through the web data page or graphical client. When the data are verified and available, the user can run the workflow on the web workflow page and submit the analysis task. This platform allows the user to submit multiple analysis tasks at the same time, analyze the data simultaneously, and view the analysis report on the web task report page. For example, users can assemble their bacterial genome using the “Assemble_Bacteria_Genome_Stat” tool in three steps: (1) upload the fastq format sequencing files to MDACP; (2) choose the “Assemble_Bacteria_Genome_Stat” tool and select the input files; and (3) click the “Run” button and download the assembled genome after ~10 min. The report files are shown on the automatically refreshed page. MDACP has a user-friendly interface allowing users to submit data through data pages or graphical clients, flexibly control parameters according to data conditions, and analyze data simultaneously with multiple tasks. To date, 417 China CDC users have submitted more than 4,600 analysis tasks on this platform, which will continue to increase.
In contrast to other web-based platforms, such as Seven Bridges and BaseSpace, MDACP focuses on the microbiology field and supports various types of free analysis workflows based on the needs of clinicians and for communicable disease control. Compared with the MicrobiomeAnalyst (Chong et al., 2020) and MetaCoMET1 platforms, which support online analysis of biome tables generated by metagenomic analysis, the MDACP platform supports workflows involving direct analysis of raw sequencing data and can easily be applied in resource-limited situations as well as in clinical laboratories and public health lab settings where bioinformatics expertise is lacking. With our MDACP platform, users can easily perform several types of bioinformatics analysis on a point-and-click interface in 5 min–2 h (Table 1). The run time depends on the workflow chosen by the user.
This MDACP platform can not only facilitate data analysis and management for scientific research, but also assist clinicians and disease control personnel with information mining such as pathogen identification, classification, and traceability. The platform is free and available for research users at https://analysis.mypathogen.org.
Publicly available datasets were analyzed in this study. This data can be found here: https://analysis.mypathogen.org.
WZ designed this study and wrote this manuscript. WZ, NH, JM, XP, TZ, YQ, and XL contributed to data analysis and platform establishment. All authors reviewed the manuscript.
This study was supported by grants from National Key Research and Development Program of China (2018YFC1200100), the Major Infectious Diseases Such as AIDS and Viral Hepatitis Prevention and Control Technology Major Projects (Grant Nos: 2018ZX10712-001, 2018ZX10303-402, 2018ZX10714-002, and 2018ZX10305-410), and the National Natural Science Foundation of China (Grant Nos: 81301402 and 81700016).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
China CDC, Chinese Center for Disease Control and Prevention; MDACP, Microbial Data Analysis Cloud Platform; NGS, Next-generation sequencing.
Agrawal, S., Arze, C., Adkins, R. S., Crabtree, J., Riley, D., Vangala, M., et al. (2017). CloVR-Comparative: automated, cloud-enabled comparative microbial genome sequence analysis pipeline. BMC Genom. 18:332. doi: 10.1186/s12864-017-3717-3
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool (BLAST). J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Andrews, S. (2013). Babraham Bioinformatics – Fastqc a Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Besser, J., Carleton, H. A., Gerner-Smidt, P., Lindsey, R. L., and Trees, E. (2018). Next-generation sequencing technologies and their application to the study and control of bacterial infections. Clin. Microbiol. Infect. 24, 335–341. doi: 10.1016/j.cmi.2017.10.013
Chen, C., Zhang, W., Zheng, H., Lan, R., Wang, H., Du, P., et al. (2013). Minimum core genome sequence typing of bacterial pathogens: a unified approach for clinical and public health microbiology. J. Clin. Microbiol. 52, 2582–2591. doi: 10.1128/jcm.00535-13
Chong, J., Liu, P., Zhou, G., and Xia, J. (2020). Using microbiome analyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 15, 799–821. doi: 10.1038/s41596-019-0264-1
Delcher, A. L., Bratke, K. A., Powers, E. C., and Salzberg, S. L. (2007). Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23, 673–679. doi: 10.1093/bioinformatics/btm009
Deurenberg, R. H., Bathoorn, E., Chlebowicz, M. A., Couto, N., Ferdous, M., García-Cobos, S., et al. (2017). Application of next generation sequencing in clinical microbiology and infection prevention. J. Biotechnol. 243, 16–24. doi: 10.1016/j.jbiotec.2016.12.022
Kent, W. J. (2002). BLAT–the BLAST-like alignment tool. Genome Res. 12, 656–664. doi: 10.1101/gr.229202.
Kim, D., Song, L., Breitwieser, F. P., and Salzberg, S. L. (2016). Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 26, 1721–1729. doi: 10.1101/gr.210641.116
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv [Preprint], Available at: https://www.scienceopen.com/document?vid=e623e045-f570-42c5-80c8-ef0aea06629c
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1:18. doi: 10.1186/2047-217X-1-18
Miao, J., Han, N., Qiang, Y., Zhang, T., Li, X., and Zhang, W. (2017). 16SPIP: a comprehensive analysis pipeline for rapid pathogen detection in clinical samples based on 16S metagenomic sequencing. BMC Bioinform. 18:568. doi: 10.1186/s12859-017-1975-3
Mikheenko, A., Prjibelski, A., Saveliev, S., Antipov, D., and Gurevich, G. (2018). Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 34, i142–i150. doi: 10.1093/bioinformatics/bty266
Motroa, Y., and Moran-Gilad, J. (2017). Next-generation sequencing applications in clinical bacteriology. Biomol. Detect. Q. 14, 1–6. doi: 10.1016/j.bdq.2017.10.002
Nurk, S., Bankevich, A., Antipov, D., Gurevich, A., Korobeynikov, A., Lapidus, A., et al. (2013). “Assembling genomes and mini-metagenomes from highly chimeric reads,” in Research in Computational Molecular Biology. RECOMB 2013. Lecture Notes in Computer Science, eds M. Deng, R. Jiang, F. Sun, and X. Zhang (Berlin: Springer), doi: 10.1007/978-3-642-37195-0_13 7821.
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Rossen, J. W. A., Friedrich, A. W., Moran-Gilad, J., and ESCMID Study Group for Genomic and Molecular Diagnostics [ESGMD], (2018). Practical issues in implementing whole-genome-sequencing in routine diagnostic microbiology. Clin. Microbiol. Infect. 24, 355–360. doi: 10.1016/j.cmi.2017.11.001
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Yin, X., Jiang, X. T., Chai, B., Li, L., Yang, Y., Cole, J. R., et al. (2018). ARGs-OAP v2.0 with an expanded SARG database and hidden markov models for enhancement characterization and quantification of antibiotic resistance genes in environmental metagenomes. Bioinformatics 34, 2263–2270. doi: 10.1093/bioinformatics/bty053
Zhang, W., and Bergelson, J. (2012). EFFECTORSEARCH: software for identifying effectors of T3SS in bacterial species. Chin. J. Zoon. 28, 528–535.
Keywords: microorganism, genome, pathogen, web resource, analysis cloud platform
Citation: Han N, Miao J, Zhang T, Qiang Y, Peng X, Li X and Zhang W (2020) MDACP: A Pathogen Genome and Metagenome Analysis Cloud Platform. Front. Genet. 11:1007. doi: 10.3389/fgene.2020.01007
Received: 14 May 2020; Accepted: 07 August 2020;
Published: 31 August 2020.
Edited by:
Sebastian Zöllner, University of Michigan, United StatesReviewed by:
Lixin Zhang, Michigan State University, United StatesCopyright © 2020 Han, Miao, Zhang, Qiang, Peng, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen Zhang, emhhbmd3ZW5AaWNkYy5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.