
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet., 24 July 2020
Sec. Genetics of Common and Rare Diseases
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.00833
This article is part of the Research TopicGenetics of Familial Hypercholesterolemia: New InsightView all 17 articles
 Chin-Chou Huang1,2,3,4*
Chin-Chou Huang1,2,3,4* Min-Ji Charng1,2*
Min-Ji Charng1,2*Familial hypercholesterolemia (FH) is a common genetic disease with an incidence of about 1 in 200–500 individuals. Genetic mutations markedly elevate low-density lipoprotein cholesterol and atherosclerotic cardiovascular disease (ASCVD) in FH patients. With advances in clinical diagnosis and genetic testing, more genetic mutations have been detected, including those in low-density lipoprotein receptor (LDLR), apolipoprotein B (APOB), proprotein convertase subtilisin/kexin type 9 (PCSK9), and so on. Globally, most FH patients remain undiagnosed, untreated, or inappropriately treated. Recently, there was a Global Call to Action by the Global Familial Hypercholesterolemia Community to reduce the health burden of FH. Asia, despite being the most populous continent with half of the global population, has low FH detection rates compared to Western countries. Therefore, we aimed to review the current status of FH genetic diagnosis in Asia to understand the gaps in FH diagnosis and management in this region.
Familial hypercholesterolemia (FH) is a common genetic disease with an incidence of about 1 in 200–500 individuals (Marks et al., 2003; Benn et al., 2012). Lipid metabolism changes in FH patients, including reduced low-density lipoprotein receptor (LDLR)-mediated low-density lipoprotein cholesterol (LDL-C) catabolism, impaired apolipoprotein B (APOB)-mediated LDL clearance, and increased proprotein convertase subtilisin/kexin type 9 (PCSK9) levels, mediate post-translational destruction of LDLRs (Chiou and Charng, 2016, Sturm et al., 2018). Genetic mutations in FH patients have been detected in LDLR, APOB, and PCSK9, and are either in a heterozygous (HeFH) or homozygous (HoFH) FH form (Chiou and Charng, 2016; Sturm et al., 2018, Di Taranto et al., 2020; Nawawi et al., 2020). These genetic mutations result in a marked elevation of LDL-C levels and atherosclerotic cardiovascular disease (ASCVD) if these patients are left underdiagnosed and undertreated (Nordestgaard et al., 2013; Gidding et al., 2015, Elkins and Fruh, 2019). For patients harboring FH-causing variants, the risk of ASCVD may be increased by 4.4- to 6.8- fold (Lee et al., 2019). Therefore, early diagnosis of FH is important.
However, globally, the majority of FH patients remain undiagnosed, untreated, or insufficiently treated (Nordestgaard et al., 2013; Khera et al., 2016, Cao et al., 2018; Wang et al., 2019). Based on clinical criteria, FH is not rare in the Asian population (Wang et al., 2019). However, around 42.5% to 62.5% of the FH patients remain underdiagnosed, according to definitions by the Dutch Lipid Clinic Network (DLCN) and Simon Broome (SB). This finding supports the importance of genetic testing for the diagnosis of FH (Cao et al., 2018).
Furthermore, there are great advances in diagnostic technologies, such as next generation sequencing (NGS) (Di Resta and Ferrari, 2018), to improve the genetic diagnosis of FH. In the era of targeted and personalized medicine (Prodan Žitnik et al., 2018), it will be useful to identify different patient types to aid the selection of safer and more effective treatments.
Recently, there was a Global Call to Action by the Global Familial Hypercholesterolemia Community to reduce the health burden of FH (Wilemon et al., 2020). Asia is the most populous continent in the world, with more than half of the world’s population. It is therefore important to better understand the current status of FH diagnosis and management in Asia. Here, we aimed to review the current status of FH genetic diagnosis in Asia.
There are two related studies on FH detection in Asia (Nordestgaard et al., 2013; Pang et al., 2019). Nordestgaard et al. (2013) reported the estimated number and percentage of individuals diagnosed with FH globally. Among roughly 200 countries/territories worldwide, data were only available in 22 countries/territories, including 3 in Asia. The data from Asian countries were derived from the second WHO consultation on FH in 1998. The estimated percentage of individuals diagnosed with FH was only 1% (n = 14,100) in Hong Kong, <1% (n = 46,300) in Taiwan, and <1% (n = 254,800) in Japan (Nordestgaard et al., 2013).
Another study in 2019 compared FH health care in 12 countries, including 9 countries selected in the Asia-Pacific region, and 2 in the Southern Hemisphere and United Kingdom (UK). In the United Kingdom, which was the benchmark, the estimated percentage of individuals diagnosed with FH was 10–20%. In non-Asian countries, the estimated percentage was 5.3% in South Africa, 4% in Australia, 2.9% in Brazil, and 2.5% in New Zealand. In Asian countries, however, the estimated percentage was only 3.8% in Taiwan, 2.2% in Hong Kong, 1.4% in Malaysia, 1% in Japan, <1% in China, <1% in the Philippines, and <1% in Vietnam. Although there is a continuous improvement in FH detection rates, compared to the 2013 data, there are still important gaps in comparison to other non-Asian countries and the United Kingdom (Pang et al., 2019).
The awareness and knowledge about FH have been surveyed among physicians across Asia (Pang et al., 2015, 2017). One anonymous internet-based survey involving 230 physicians was reported in 2015 (Pang et al., 2015). These physicians were selected from Japan, South Korea, and Taiwan. Only a small proportion of them was aware of the heritability (47%), prevalence (27%), risk of FH-related cardiovascular disease (13%), and FH specialists in their geographic area (35%) (Pang et al., 2015).
Another survey was done with 1,078 physicians and reported in 2017 (Pang et al., 2017). These physicians were from 10 different countries/regions, including Australia, Japan, Malaysia, South Korea, Philippines, Hong Kong, China, Vietnam, Taiwan, and the United Kingdom. Self-perceived FH familiarity was only 34% and there were no significant differences among most countries (except in Japan and China, where the value was lower) and the United Kingdom. Physicians from the Asia-Pacific region were significantly worse at selecting the correct FH descriptions (72%), compared to those from the United Kingdom (89%) (P = 0.001). FH guideline awareness was significantly lower in physicians from the Asia-Pacific region (35%), compared to those from the United Kingdom (61%) (P < 0.001). The awareness of lipid specialists for referral or medical advice was also significantly lower in physicians from the Asia-Pacific region (35%), compared to those from the United Kingdom (50%) (P = 0.003) (Pang et al., 2017).
These studies showed that the awareness, knowledge, and perception about FH were still low among Asian physicians, particularly in developing countries (Pang et al., 2015, 2017). More efforts are needed for improvement in these parameters, where extensive education and training programs could be of help.
FH genetic studies in Taiwan are summarized in Table 1.
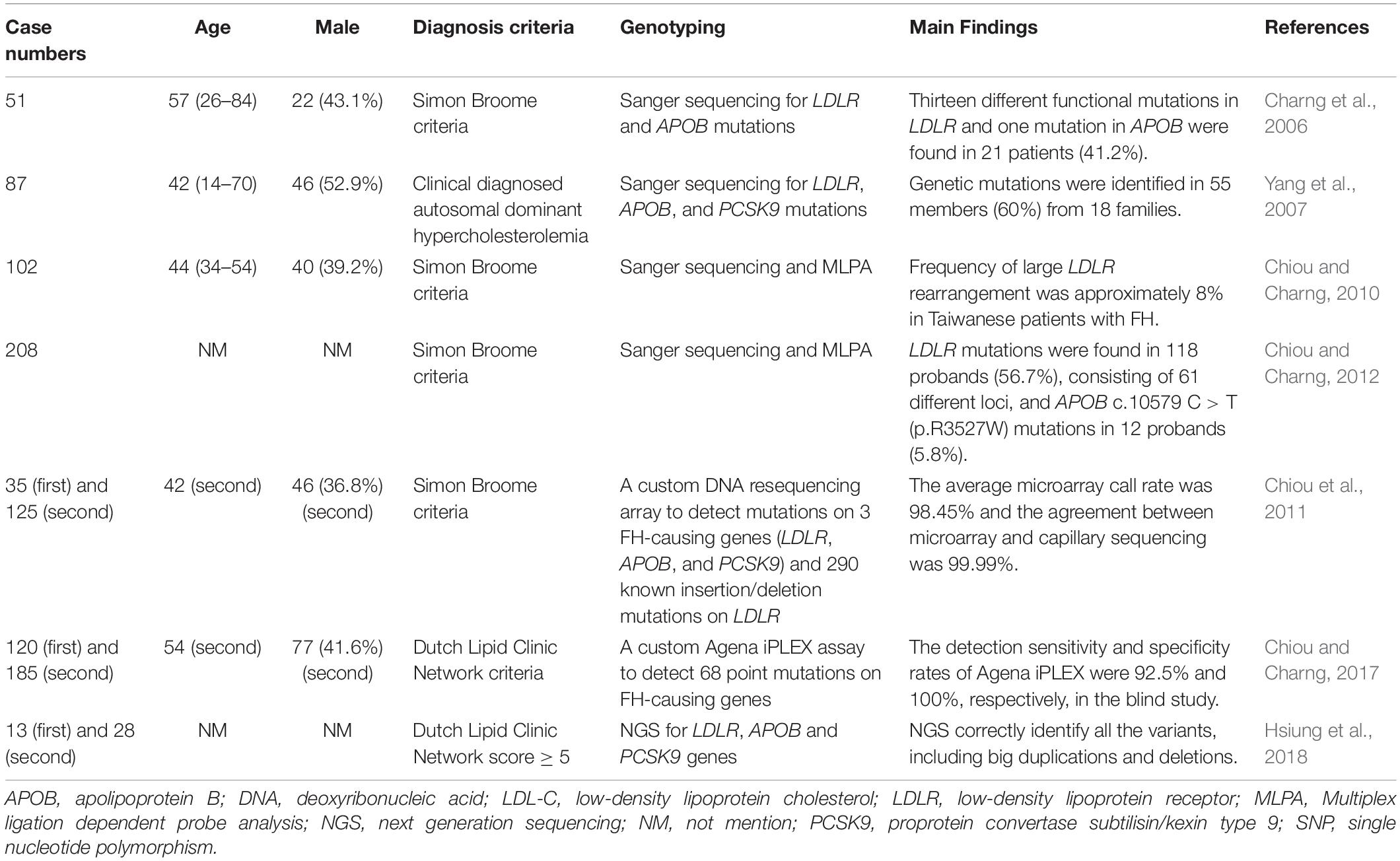
Table 1. Genetic diagnosis of familial hypercholesterolemia in Taiwan.
There are some studies that investigated the genetic mutations of FH patients and the development of genetic diagnostic methods in Taiwan (Charng et al., 2006; Yang et al., 2007, Chiou and Charng, 2010; Chiou et al., 2011, Chiou and Charng, 2012, 2017, Hsiung et al., 2018).
In a small population of 51 unrelated FH patients in Taiwan, LDLR and APOB mutations were screened using Sanger sequencing. Among these patients, 13 different functional mutations in LDLR and 1 in APOB were found in 21 patients (41.2%). For the LDLR mutations, there were 10 single-point missense mutations, 1 two-point mutation in the same allele, 1 non-sense mutation, and 1 frame-shift mutation. The study discovered 3 novel mutations, including 2 missense mutations [LDLR c.1592 T > A (p.M531K) and LDLR c.1597 T > C (p.W533R)] and 1 frame-shift mutation [LDLR c.1953,1954 del [TA] (p.M652Fs)] (Charng et al., 2006).
In another study, 87 FH patients from 30 unrelated Taiwanese families were screened for LDLR, APOB, and PCSK9 mutations via Sanger sequencing. Genetic mutations were identified in 55 members (60%), including 6 previously reported LDLR mutations [LDLR c.383 G > A (p.C128Y), LDLR c.268 G > A (p.D90N), LDLR c.1216 C > T (p.R406W), LDLR c.1448 G > A (p.W483X), LDLR c.571 C > T (p.G191X), and LDLR c.1285 G > A (p.V429L)], 2 novel LDLR mutations [LDLR c.1954 del AT (p.M631Fs) and LDLR c.1586 + 5 G > C], 1 known [APOB c.10579 C > T (p.R3527W)], and 1 novel missense mutations [APOB c.10718 C > T (p.R3567W)] in the APOB gene. The study found no PCSK9 gene mutations in this Taiwanese patient cohort (Yang et al., 2007).
One hundred two unrelated probands, who fulfilled the FH diagnostic criteria, were tested for mutations using Sanger sequence analysis and multiple ligation-dependent probe amplification (MLPA). Gene mutations were detected in 60 probands (58.8%), including LDLR mutations in 52 probands (51.0%) and the APOB c.10579 C > T (p.R3527W) mutation in 8 probands (7.8%). Among the 42 probands with mutations undetected by exome sequence analysis, 8 had abnormal MLPA patterns, including 2 with exon 6 to 18 deletions, 2 with exon 9 deletions, 1 with exon 6 to 8 deletions, 1 with exon 11 deletions, 1 with exon 3 to 5 duplications, and 1 with exon 7 to 12 duplications. This study found that the frequency of large LDLR rearrangements was approximately 8% in Taiwanese FH patients (Chiou and Charng, 2010).
In 208 Taiwanese patients with clinically diagnosed FH, common FH mutations were investigated using Sanger sequencing and MLPA. The top three most common mutations were LDLR c.986G > A (p.C329Y) (13.1%), c.1747C > T (p.H583Y) (10.8%), and APOB c.10579C > T (p.R3527W) (9.2%). Among these patients, 118 probands (56.7%) had LDLR mutations and 12 (5.8%) had the APOB c.10579 C > T (p.R3527W) mutation. Three novel mutations were discovered, including LDLR c.64 del G (p.A22Fs), LDLR c.1661 C > T (p.S554L), and LDLR c.2099 A > G (p.D700G). One haplotype [CAAGCCCCATGG/(dTA)n-112nt] was associated with LDLR c.1747 C > T (p.H583Y), and two were associated with LDLR c.986 G > A (p.C329Y). In comparison to southern Chinese patients, the same LDLR binding-domain pattern was associated with APOB c.10579 C > T (p.R3527W) in Taiwanese patients. The study found different common FH mutation-associated haplotypes and haplotype homologies in Taiwan and southern China, suggesting multiple historical migrations of Taiwanese and the presence of common ancestors in southern China (Chiou and Charng, 2012).
Some studies have investigated genetic diagnosis techniques in Taiwan. A custom DNA resequencing array was designed to detect mutations on all three FH-causing genes (LDLR, APOB, and PCSK9) and 290 known LDLR insertion/deletion mutations in the Taiwanese patients. In previously sequenced patients, the average call rate was 98.45% and the agreement between microarray and capillary sequencing was 99.99%. In screening patients blindly, the FH array detected at least 1 mutation in 77.5% of patients clinically diagnosed with definite FH and in 52.9% of patients with probable FH. The high-throughput FH resequencing array detected LDLR, APOB, and PCSK9 with high efficiency and accuracy and identifies disease-causing mutations (Chiou et al., 2011).
A mass spectrometric assay (Agena iPLEX assay) was designed to simultaneously diagnose 68 point mutations in FH-causing genes in Taiwanese patients. In the first part of the study, only 1 discrepancy was found between the mass spectrometry and Sanger sequencing data of 180 previously sequenced patients. In the second part, 62 probands with mutations were identified by both techniques out of 185 FH probands, with only 5 mutations detected by Sanger sequencing. The sensitivity and specificity of mass spectrometry were 92.5% and 100%, respectively. This study found great potential for this assay due to its low cost, speed (around 1 day), and flexibility for FH genetic screening in Taiwanese patients (Chiou and Charng, 2017).
Probes for NGS were designed to capture the whole LDLR gene and all coding sequences of APOB and PCSK9 in Taiwanese patients. These probes correctly identified all the variants in 13 DNA samples, including 3 large duplications and 2 large deletions. In a new cohort of 28 unrelated FH patients with a Dutch Lipid Clinic Network score ≥ 5, they identified the causative variants in 21 unrelated probands. Five of them carried the novel splice site variant c.1186 + 2 T > G in LDLR. This study showed that this panel can comprehensively detect disease-causing variants in LDLR, APOB, and PCSK9 in FH patients (Hsiung et al., 2018).
With the advances in molecular diagnostic methods, the detection rate of genetic mutations in clinically diagnosed FH has increased in Taiwan in recent years. However, there were still large proportions of FH patients that did not have genetic diagnoses. There were multiple LDLR and APOB mutations identified in Taiwan, including some novel mutations. However, PCSK9 mutations, which had been reported in China and Japan, have not been identified in Taiwan.
FH genetic studies in China are summarized in Table 2.

Table 2. Genetic diagnosis of familial hypercholesterolemia in China.
In 105 young patients (age ≤ 35 years) with high LDL-C (LDL-C ≥ 3.4 mmol/L), genetic analysis was performed for 9 genes (LDLR, APOB, PCSK9, APOE, STAP1, LIPA, LDLRAP1, ABCG5 and ABCG8). Genetic mutations were detected in 40 patients (38.1%). This study found that the best LDL-C threshold was 4.56 mmol/L for genetically confirmed FH (Cao et al., 2018).
In a multi-center study in China, 285 unrelated index cases with clinical FH were analyzed by NGS for LDLR, APOB, and PCSK9 mutations. Genetic mutations were detected in 148 cases (51.9%), including 84 (29.4%) in LDLR, 31 (10.9%) in APOB, and 4 (1.4%) in PCSK9 genes. LDLR c.1448 G > A (p.W483X) was detected in 9 patients, which was the most frequent mutation. There were 8 novel mutations (7 LDLR and 1 APOB), which were considered as pathogenic by in silico analysis. There were 5 true LDLR homozygotes, 16 compound heterozygotes, and 13 double heterozygotes identified. The most severe phenotypes were noted in true LDLR homozygotes (Sun et al., 2018).
There was a review of 163 case reports about Chinese FH patients published before 2018. The mutation frequency was 82% in LDLR, 9% in APOB, and very rare in PCSK9. The study concluded that the epidemiological investigation of FH was not large-scale, FH recognition remained rudimentary, and guidelines for diagnosis and management of FH patients were incomplete in China (Peng et al., 2019).
In a previous systematic review of LDLR mutations in Chinese FH patients, a total of 74 studies were included. Among the 295 probands studied, 131 LDLR mutations were identified. Most of them were located in exon 4, and approximately 60% of them were missense mutations. There were 30 novel mutations and most of them were found pathogenic by in silico analysis. The major LDLR mutations were LDLR c.986 G > A (p.C329Y), LDLR c.1747 C > T (p.H583Y), and LDLR c.1879 G > A (p.A627T) (Jiang et al., 2015).
The majority of FH mutations found in Chinese were in LDLR, there was one in APOB, and were rare in PCSK9. The functionality of these novel mutations was predicted by in silico analysis. Functional testing or familial co-segregation analysis may be required to check the pathogenicity of these mutations.
FH genetic studies in the Hong Kong are summarized in Table 3.
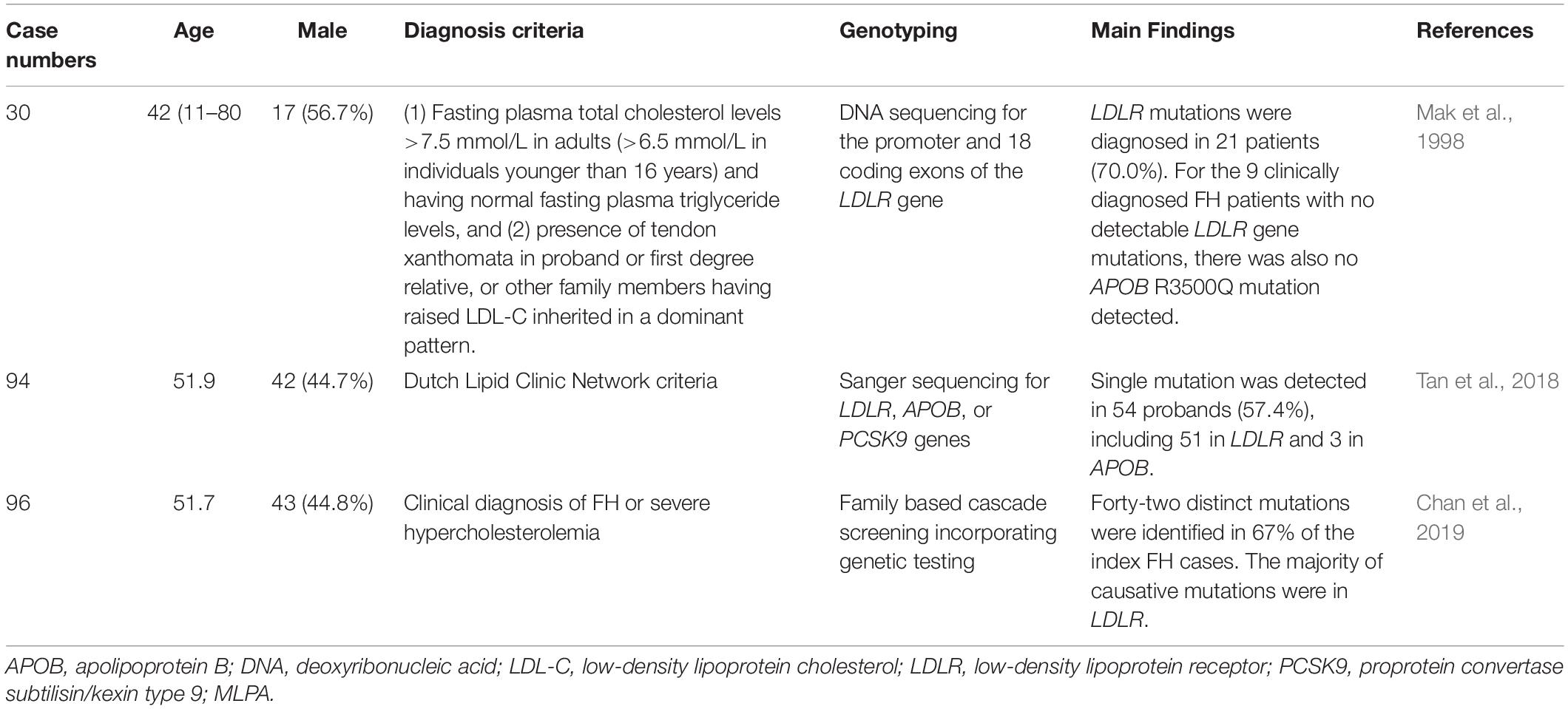
Table 3. Genetic diagnosis of familial hypercholesterolemia in Hong Kong.
Hu et al. reported the epidemiologic features of 252 HeFH patients from the Chinese population in Hong Kong. For patients aged ≥ 18 years, xanthomata was noted in 40.6% of males and 54.8% of females, while corneal arcus was noted in 81.2% of males and 66.9% of females. The incidence of coronary artery disease (CAD) was 9.9% and 8.5% in males and females, respectively. This study found that age and the presence of xanthelasmata were associated with the risk of CAD (Hu et al., 2013, 2016).
In one genetic study from Hong Kong, DNA sequencing was performed for the promoter and coding regions of the 18 exons of the LDLR gene in 30 Chinese FH patients. Overall, LDLR mutations were detected in 21 patients (70.0%). There were 18 mutations in the promoter and 10 exons. Besides, there were 6 polymorphisms with genotypic distributions similar to those in the Japanese population but different from those in the Western populations. For the 9 clinically diagnosed FH patients with no detectable LDLR gene mutations, there was also no APOB R3500Q mutations detected (Mak et al., 1998).
In another genetic study from Hong Kong, a total of 94 unrelated probands with clinical FH received genetic testing for LDLR, APOB, and PCSK9 by Sanger sequencing. Single-gene mutations were detected in 54 probands (57.4%), including 51 (54.2%) in LDLR and 3 (3.2%) in APOB. Heterozygous LDLR mutations were the most common. Compound heterozygous mutations were detected in five cases. Double heterozygous mutations were detected in three cases. A homozygous mutation in exon 10 of LDLR [c.1474G > A (p.D492N)] was detected in one case (Tan et al., 2018).
In a recent study, Chinese subjects with clinical FH received family based cascade screening and genetic testing. Among 96 index cases, 42 distinct mutations were identified in 64 cases (67%). The majority of mutations were in LDLR, and the three most common mutations were LDLR c.1241 T > G (p.L414R), LDLR c.1474G > A (p.D492N), and LDLR c.682G > A (p.E228X). Nine novel LDLR mutations were identified. APOB c.10579 C > T (p.R3527W) was identified in 5% of the total cases (Chan et al., 2019).
FH genetic studies in Japan are summarized in Table 4.
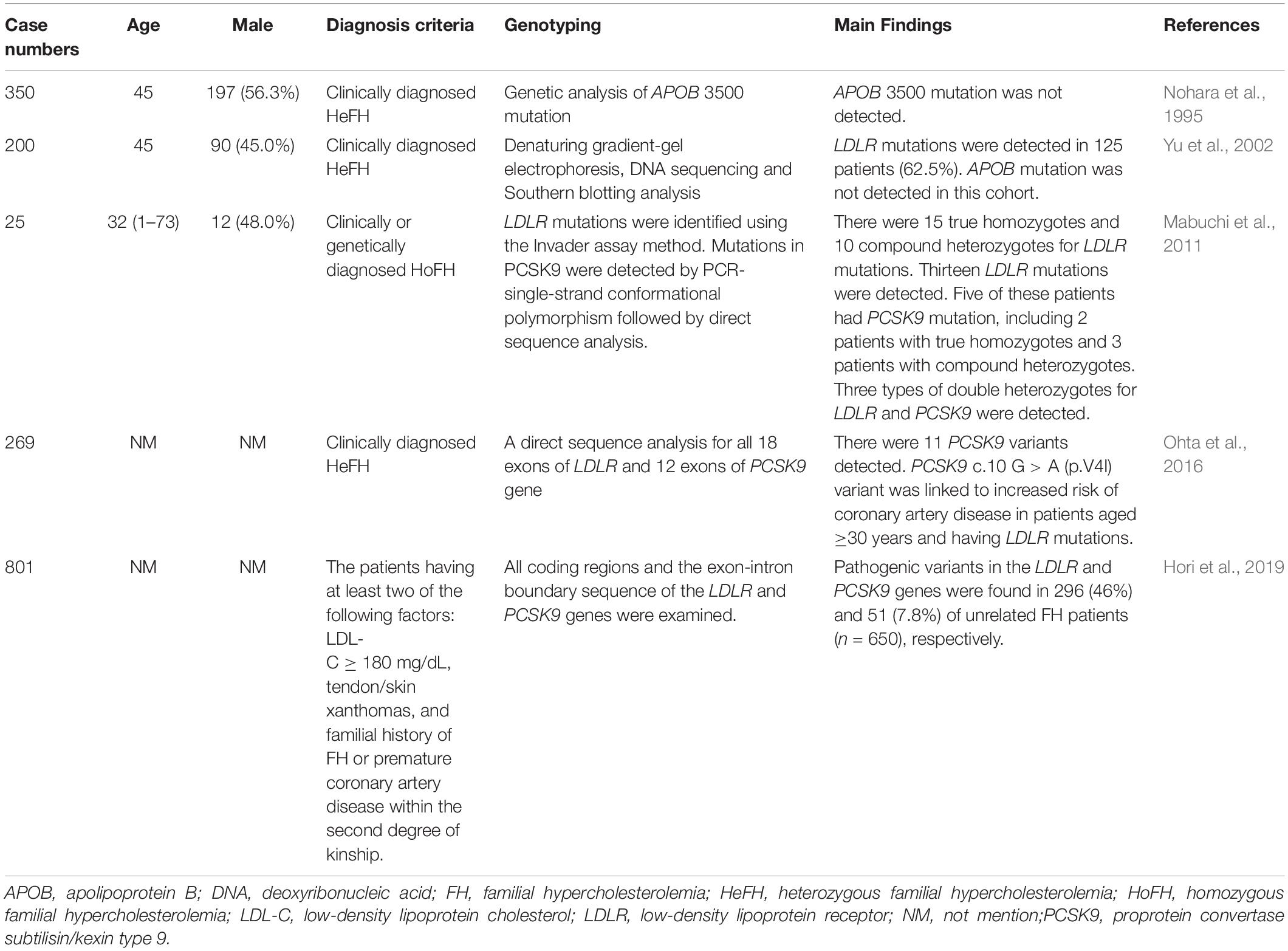
Table 4. Genetic diagnosis of familial hypercholesterolemia in Japan.
In a previous report, 385 clinically diagnosed HeFH patients received genetic analysis for APOB 3500 mutations to estimate the frequency of familial defective apolipoprotein B-100 (FDB) in Japan. The study detected no APOB 3500 mutation in this cohort (Nohara et al., 1995).
In a study conducted in the Hokuriku district of Japan, 200 unrelated FH patients received genetic analysis for LDLR and APOB genes. LDLR mutations were detected in 125 patients (62.5%). There were 37 probable disease-causing mutations, 22 of which were novel. Common LDLR mutations included LDLR c. 2431 C > T (p.K811X) (19.5%), LDLR c. 2054 C > T (p.P685L) (6.0%), FH-Tonami-1 (6.0%), LDLR c.2141-3 C > A (5.5%), and FH-Tonami-2 (4.5%). APOB mutations were not detected in this cohort (Yu et al., 2002).
In another study conducted in the Hokuriku district of Japan, 25 clinical HoFH patients received genetic analysis reports. There were 15 true homozygotes and 10 compound heterozygotes for LDLR mutations. Five of these patients had PCSK9 mutation [PCSK9 c.94G > A (p.E32K)], including 2 true homozygotes and 3 compound heterozygotes (LDLR and PCSK9). APOB mutations were not detected in any FH patient. The study also confirmed a high incidence of HoFH (1/171,167) and HeFH (1/208) in the Hokuriku district of Japan (Mabuchi et al., 2011).
In 269 clinically diagnosed HeFH patients in Japan, genetic analyses were performed for LDLR and PCSK9 gene mutations. Eleven PCSK9 variants were detected. The four most frequent PCSK9 variants were c.63_64insCTG (p.L21_22insL), c.158 C > T (p.A53V), c.10 G > A (p.V4I), and c.94G > A (p.E32K). PCSK9 c.158 C > T (p.A53V) and c.63_64insCTG (p.L21_22insL) were in linkage disequilibrium with each other. Interestingly, the PCSK9 c.10 G > A (p.V4I) variant was linked to an increased risk of CAD in patients aged ≥ 30 years with LDLR mutations. The study suggested that PCSK9 c.10 G > A (p.V4I) might affect LDLR metabolism and modify LDLR mutation phenotypes (Ohta et al., 2016).
Recently, 801 clinically diagnosed Japanese HeFH patients were analyzed for LDLR and PCSK9 mutations. Among the 650 unrelated FH patients, LDLR pathogenic variants were identified in 296 patients (46%) and PCSK9 pathogenic variants were identified in 51 patients (7.8%). PCSK9 c.94G > A (p.E32K) was the most frequently detected pathogenic PCSK9 variant in the Japanese FH population. The five most frequent LDLR pathogenic variants, found in about 17% of this FH cohort, were identified: LDLR c.1845 + 2 T > C, c.1012T > A (p.C338S), c.1297G > C (p.D433H), c.1702C > G (p.L568V), and c.2431A > T (p.K811X). This study found that LDLR and PCSK9 pathogenic variants were common in heterozygous Japanese FH patients (Hori et al., 2019).
APOB mutations, especially APOB c.10579 C > T (p.R3527W), were common mutations in Taiwan (9.2%) (Chiou and Charng, 2012) and China (10.9%) (Sun et al., 2018). However, it was not detected in Japan in the past (Nohara et al., 1995; Yu et al., 2002). The first Japanese case of APOB mutation, APOB c.10580 G > A (p.Arg3527Gln), was just reported recently (Hori et al., 2020). The origin of this APOB mutation was unknown and might be imported from other Asian countries. In contrast, PCSK9 mutations, especially PCSK9 c.94G > A (p.E32K), were quite prevalent in Japan (7.8%) (Hori et al., 2019), but have not been detected in Taiwan and were very rare in other Asian regions.
FH genetic studies in Korea are summarized in Table 5.
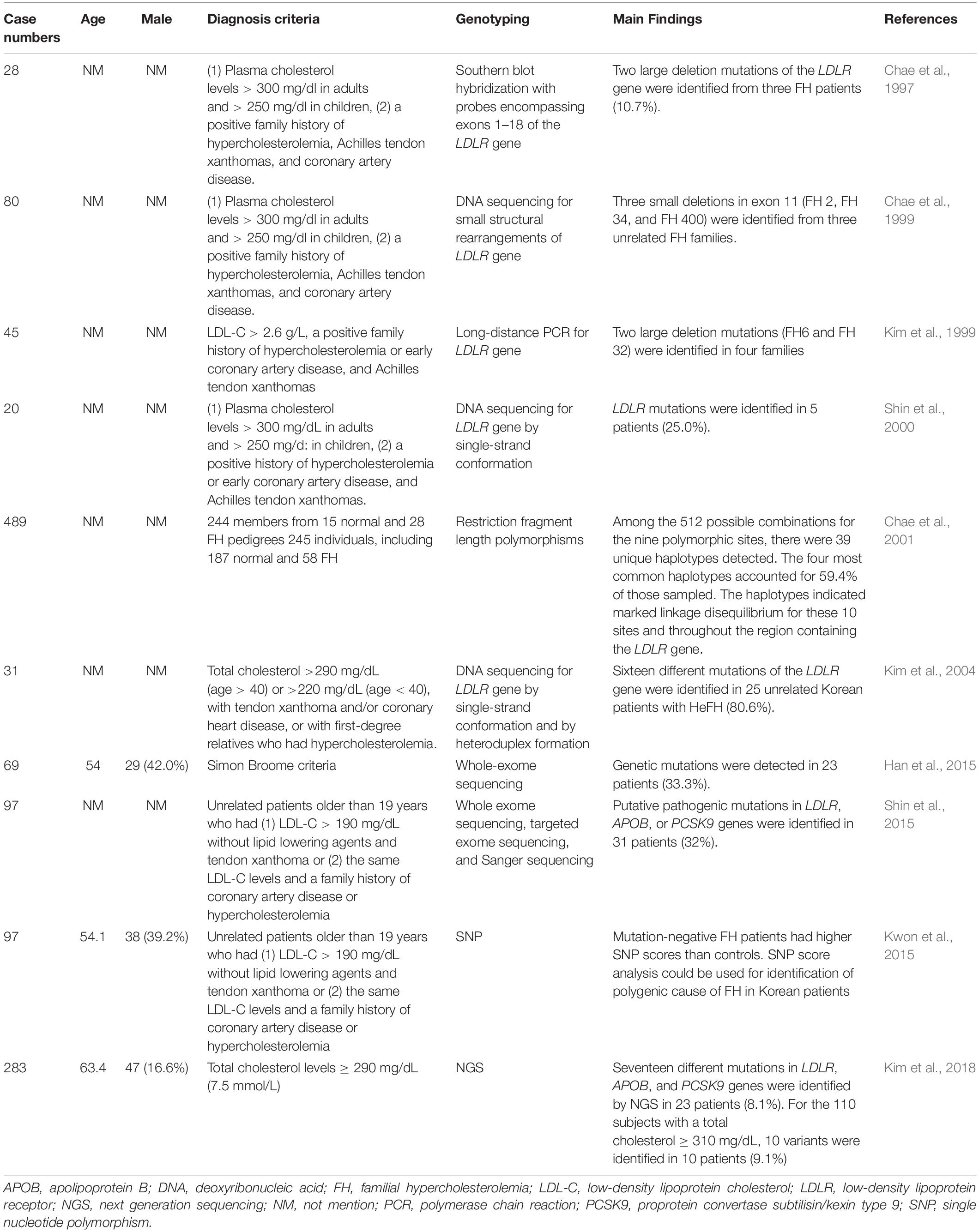
Table 5. Genetic diagnosis of familial hypercholesterolemia in Korea.
In 28 clinically diagnosed HeFH patients, genetic analysis was performed for LDLR gene by Southern blot hybridization. Three of them (10.7%) were diagnosed with two large LDLR deletion mutations (FH29 and FH110) (Chae et al., 1997).
In 80 clinically diagnosed FH patients, genetic analysis was performed to detect small structural rearrangements of the LDLR gene, which were undetected by southern blot hybridization. Three novel small deletions were identified (FH 2, FH 34 and FH 400) (Chae et al., 1999).
In 45 unrelated HeFH patients, genetic analysis was performed to assess the LDLR gene mutations by long-distance PCR. Two different large deletion mutations were identified (FH6 and FH 32) (Kim et al., 1999).
In 20 unrelated FH patients, genetic analysis was performed for the LDLR gene by single-strand conformation polymorphism. LDLR mutations were detected in five patients (25%). There were four novel point mutations, including one non-sense mutation and three missense mutations (Shin et al., 2000).
To study the role of the LDLR gene in polygenic hypercholesterolemia, a study was conducted in 244 members of 43 different pedigrees (15 normal and 28 FH pedigrees) and 245 individuals (187 normal and 58 FH) for 9 different restriction fragment length polymorphisms (RFLPs) (TaqI, StuI, HincII, BstEII, AvaII, PvuII, MspIA, MspIB, and NcoI) and a sequence variation at Arg450. Frequencies of these polymorphisms were similar between FH patients and controls. Among the 512 possible combinations for the nine polymorphic sites, 39 unique haplotypes were detected. The four most common haplotypes accounted for 59.4% of the total patients sampled. The study found that the haplotypes indicated marked linkage disequilibrium for these 10 sites and throughout the region containing the LDLR gene (Chae et al., 2001).
In 31 clinically diagnosed HeFH patients, genetic analysis was performed for the LDLR gene by single-strand conformation and heteroduplex formation. LDLR gene mutations were detected in 25 patients (80.6%). There were sixteen different LDLR mutations. The study identified one mutation that had only been reported in a Korean FH patient, the in-frame 36-bp deletion (1591del36) in exon 11. Five novel mutations were identified, including LDLR c.311G > A (C104Y), c.661del17, c.1705insCTAG, c.2088C > A (C696X), and c.941-1G > A (Kim et al., 2004).
In 69 clinically diagnosed FH cases, whole-exome sequencing analysis was applied to identify genetic mutations in the 3 known FH-related genes (LDLR, APOB, and PCSK9 genes). Genetic mutations were detected in 23 patients (33.3%). The most common genetic mutation was in the LDLR gene, which was identified in 19 patients (82.6%), included 2 copy number deletions and 17 mutations. There was also one APOB gene mutation and one PCSK9 gene mutation. Two frameshift deletions in the LDLR gene and one PCSK9 mutation were novel causative mutations. By co-segregation in their relatives, one copy number deletion and novel mutation were validated (Han et al., 2015).
One study enrolled patients with >190 mg/dL LDL-C and xanthoma or a family history of CAD or hypercholesterolemia. Among 97 patients, putative pathogenic mutations in LDLR, APOB, or PCSK9 genes were identified in 31 patients (32%). When comparing the four clinical diagnosis criteria of FH (Simon Broome, Dutch, MEDPED, and Japanese), mutation detection rates were higher using the MEDPED criteria (67–75%) and lower in the Simon Broome or Dutch criteria (35%–37%). The study found that 225 mg/dL was the best LDL-C threshold for putative mutations in Korean FH patients (Shin et al., 2015).
The polygenic cause of FH in Korean patients was investigated in 97 FH patients and 2,274 controls from the Korean Health Examinee (HEXA) shared control study. These patients received genotyping for 12 single nucleotide polymorphisms (SNPs) used in prior studies to assess the polygenic causes of FH in Caucasian patients and 4 SNPs associated with LDL-C levels in East Asians. The study found that mutation-negative FH patients had higher SNP scores than controls. The study also showed that SNP score analysis could be used for the identification of the polygenic cause of FH in Korean patients (Kwon et al., 2015).
A total of 283 subjects with total cholesterol levels ≥ 290 mg/dL (7.5 mmol/L) were selected from the Namwon and Dong-gu Studies. Seventeen different mutations in LDLR, APOB, and PCSK9 genes were identified by NGS in 23 patients (8.1%). For the 110 subjects with a total cholesterol ≥ 310 mg/dL, 10 variants were identified in 10 patients (9.1%). There were two novel LDLR variants, including LDLR c.2038 (p.L680V), and c.2201 (p.T734F) (Kim et al., 2018).
FH genetic studies in Malaysia are summarized in Table 6.
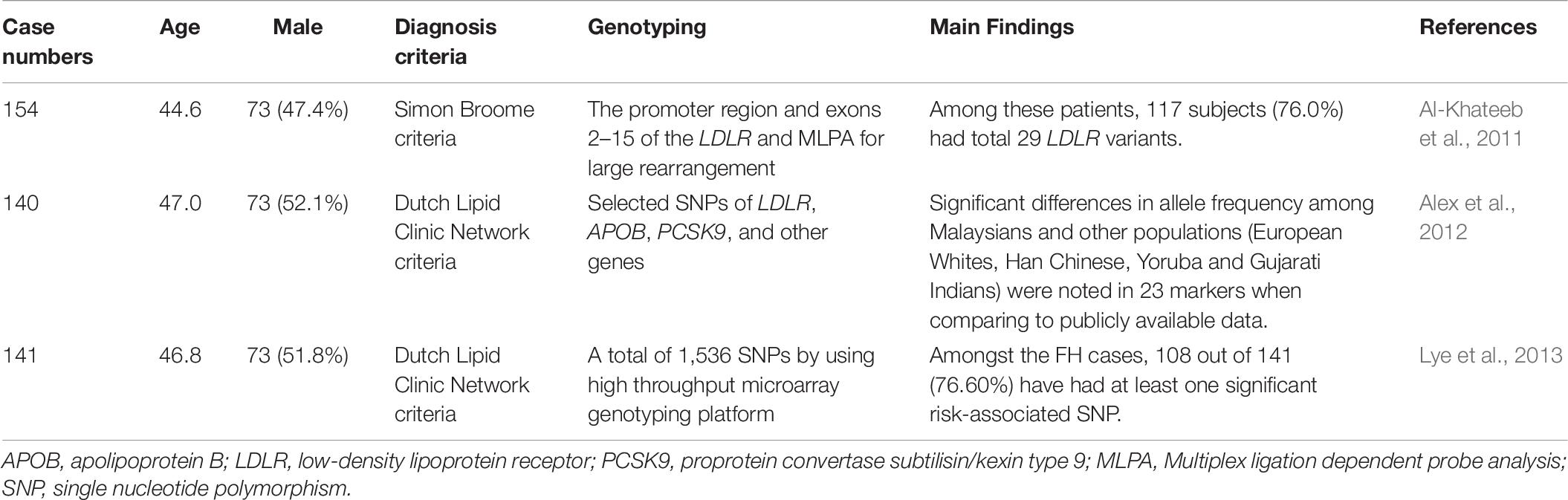
Table 6. Genetic diagnosis of familial hypercholesterolemia in Malaysia.
A total of 154 unrelated FH patients from Malaysia (Kelantan) were analyzed for LDLR gene mutations. Among these patients, 117 subjects (76.0%) had a total of 29 LDLR variants. The most frequent variant was LDLR c.1060 + 7 T > C (n = 18, 11.7%). The second most frequent variant was c.1411A > G (p.R471G) (n = 17, 11.0%) in LDLR. Eight of them [LDLR c.300 C > T (p.D100D), c.415G > C (p.D139H), c.1411A > G (p.R471G), c.1705 + 117 T > G, c.1186 + 41T > A, 1705 + 112C > G, Dup exon 12, and c.1966_2010del17 (p.W666Fs)] were reported for the first time. The study also found that patients with pathogenic mutations had higher LDL-C levels and a higher incidence of tendon xanthoma and cardiovascular diseases than those with non-pathogenic variants (Al-Khateeb et al., 2011).
A total of 140 clinically diagnosed autosomal dominant hypercholesterolemia (ADH) and 111 healthy control subjects were analyzed for selected SNPs in LDLR, APOB, PCSK9, and other genes. The ADH patients included Malays (60.0%), Indians (25.0%), and Chinese (15.0%). A total of 310 markers were examined, including 73 from LDLR, 130 from APOB, and 107 from PCSK9. Significant differences in allele frequency among Malaysians and other populations (European Whites, Han Chinese, Yoruba, and Gujarati Indians) were noted in 23 markers when compared to publicly available data. These markers included 2 on LDLR, 17 on APOB, and 4 on PCSK9 (Alex et al., 2012).
In another study, 141 patients with clinically diagnosed FH and 111 unrelated control subjects were genetically analyzed via a high throughput microarray genotyping platform. Among the FH cases, 108 (76.60%) had at least one SNP that was associated with FH risk. Eleven of the 14 SNPs were significantly associated with an increased risk of FH, including 1 SNP in LDLR, 7 in APOB, and 3 in PCSK9. The study showed that APOB rs12720762 was associated with the highest risk of FH (odds ratio 14.78, p < 0.001) (Lye et al., 2013).
Two independent studies from the same team in Malaysia used SNPs for genetic analysis (Alex et al., 2012; Lye et al., 2013). Although they found SNPs with different allele frequencies between Malaysians, European Whites, Han Chinese, Yoruba, and Gujarati Indians, the functional significance of these SNPs requires further investigation.
FH genetic studies in Singapore are summarized in Table 7.

Table 7. Genetic diagnosis of familial hypercholesterolemia in Singapore.
In Singapore, 96 clinically suspected FH patients were genetically analyzed via NGS of 26 lipid-related genes. The cohort included Chinese (78.1%), Malays (13.5%), Indians (5.2%), and other ethnicities. A total of 50 patients (52.1%) had LDLR mutations and 4 (4.2%) had APOB mutations. No PCSK9 mutations were detected in this cohort. There were 15 novel LDLR mutations. The study concluded that the mutation distribution was similar to other Asian countries, but the spectrum was different locally (Pek et al., 2018).
FH genetic studies in Philippines are summarized in Table 8.

Table 8. Genetic diagnosis of familial hypercholesterolemia in Philippines.
In the Philippines, 60 suspected FH patients were analyzed for LDLR mutations. LDLR mutations were detected in 12 patients (20%), and six novel mutations were identified. The study found that LDLR mutations were significantly associated with LDL-C levels, FH scores, and a family history of dyslipidemia (Punzalan et al., 2005).
In addition to LDLR mutations, further studies are still needed to identify relevant mutations in APOB, PCSK9, and other FH-related genes in this population.
FH genetic studies in Sri Lanka are summarized in Table 9.
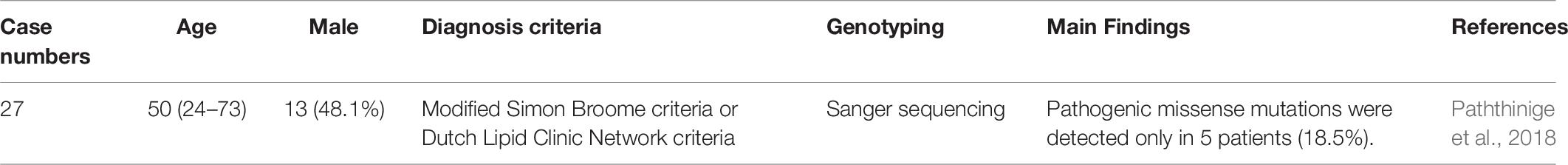
Table 9. Genetic diagnosis of familial hypercholesterolemia in Sri Lankan.
In Sri Lanka, a total of 27 clinically diagnosed FH patients were analyzed for LDLR mutations using Sanger sequencing. Among these patients, pathogenic missense mutations were detected in only five (18.5%). Four patients were heterozygous for LDLR mutations, including LDLR c.682 G > C (p.E228Q) in 2 patients, LDLR c.1720C > A (p.R574S) in 1, and LDLR c.1855 T > A (p.F619L) in 1. One patient was compound heterozygous for LDLR c.2289 G > T (p.E763D) and LDLR c.1670 C > G (p.T557S). The study found that LDLR mutations were markedly low in this population (Paththinige et al., 2018).
This is the first report on LDLR mutations in this population. However, the detection rate of genetic mutations was low. Furthermore, the study did not screen for other FH mutations, including those in APOB, PCSK9, and LDLRAP1.
FH genetic studies in India are summarized in Table 10.

Table 10. Genetic diagnosis of familial hypercholesterolemia in India.
In one Indian study, a total of 100 potential FH cases were genetically analyzed using Sanger sequencing, MLPA, and NGS. Genetic mutations were identified in 47 cases (47.0%). Based on modified Dutch Lipid Clinic Network criteria, mutations were detected in 91.4% of definite FH cases, in 40% of probable FH cases, and in 18.8% of possible FH cases. A total of 38 pathogenic variants were identified, including 33 LDLR mutations, 3 APOB mutations, and 2 PCSK9 mutations. There were ten novel pathogenic variants. Interestingly, a likely founder mutation in intron 10 (c.1587-1 G > A) of the LDLR gene was observed in 6 North Indian families. However, the conventional pathogenic variants in previously reported LDLR, APOB, and PCSK9 mutations were not detected. This study found different genetic variants between the Indian population and Western populations (Setia et al., 2020).
In addition, there were other genetic studies as abstracts from India. However, we did not report them in this review article.
Among the genetic studies in Asia, the detection rates of genetic mutations varied. The differences may be due to either the diagnostic methods used or the ethnicities sampled. With the advances in genetic diagnostic methods, detection rates will be further improved. Most of the genetic studies in Asia were confined to one ethnicity. Only a few studies enrolled FH patients with different ethnicities. Data from Singapore revealed a similar distribution of mutations as compared to other Asian countries (Pek et al., 2018). Data from Malaysia revealed significant differences in the allele frequency of markers for LDLR, APOB, and PCSK9 genes compared to publicly available data from other populations (Alex et al., 2012). Until now, there is limited literature discussing the genetic differences in FH between Asian and non-Asian populations. Further studies are still needed to compare the genetic differences in FH between different ethnicities.
Although there has been a continuous improvement in FH detection rates, there are still important gaps to fill in Asian countries in comparison to other countries and the United Kingdom. FH awareness, knowledge, and perception remain low in Asian physicians, particularly in less economically developed countries. More efforts are required to close this gap, such as extensive education and training programs. Although there have been some FH genetic studies in the Asian population, each utilizing different techniques, further studies are required to understand the different genetic backgrounds of FH in Asia.
C-CH and M-JC contributed to conception and design of the study, manuscript revision, read, and approved the submitted version. C-CH wrote the first draft of the manuscript.
This work was funded by Taiwan’s National Ministry of Science and Technology (Grant Number, MOST108-2314-B-075-063-MY2) and by Taipei Veterans General Hospital (Grant Number, V109C-071).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Hsing-Yi Liu for her excellent technical assistance.
Alex, L., Chahil, J. K., Lye, S. H., Bagali, P., and Ler, L. W. (2012). Differences in allele frequencies of autosomal dominant hypercholesterolemia SNPs in the Malaysian population. J. Hum. Genet. 57, 358–362. doi: 10.1038/jhg.2012.34
Al-Khateeb, A., Zahri, M. K., Mohamed, M. S., Sasongko, T. H., Ibrahim, S., Yusof, Z., et al. (2011). Analysis of sequence variations in low-density lipoprotein receptor gene among Malaysian patients with familial hypercholesterolemia. BMC Med. Genet. 12:40. doi: 10.1186/1471-2350-12-40
Benn, M., Watts, G. F., Tybjaerg-Hansen, A., and Nordestgaard, B. G. (2012). Familial hypercholesterolemia in the danish general population: prevalence, coronary artery disease, and cholesterol-lowering medication. J. Clin. Endocrinol. Metab. 97, 3956–3964. doi: 10.1210/jc.2012-1563
Cao, Y. X., Wu, N. Q., Sun, D., Liu, H. H., Jin, J. L., Li, S., et al. (2018). Application of expanded genetic analysis in the diagnosis of familial hypercholesterolemia in patients with very early-onset coronary artery disease. J. Transl. Med. 16:345. doi: 10.1186/s12967-018-1737-7
Chae, J. J., Kim, S. H., Kim, U. K., Han, K. H., Kim, H. S., Kastner, D. L., et al. (1999). Three novel small deletion mutations of the LDL receptor gene in Korean patients with familial hypercholesterolemia. Clin. Genet. 55, 325–331. doi: 10.1034/j.1399-0004.1999.550505.x
Chae, J. J., Kim, S. H., Kim, U. K., Hong, S. S., Kim, Y. S., Namkoong, Y., et al. (2001). Polymorphic DNA haplotypes at the human low-density lipoprotein receptor gene locus in Koreans. Hum. Biol. 73, 105–119. doi: 10.1353/hub.2001.0004
Chae, J. J., Park, Y. B., Kim, S. H., Hong, S. S., Song, G. J., Han, K. H., et al. (1997). Two partial deletion mutations involving the same Alu sequence within intron 8 of the LDL receptor gene in Korean patients with familial hypercholesterolemia. Hum. Genet. 99, 155–163. doi: 10.1007/s004390050331
Chan, M. L., Cheung, C. L., Lee, A. C., Yeung, C. Y., Siu, C. W., Leung, J. Y., et al. (2019). Genetic variations in familial hypercholesterolemia and cascade screening in East Asians. Mol. Genet. Genomic. Med. 7:e00520. doi: 10.1002/mgg3.520
Charng, M. J., Chiou, K. R., Chang, H. M., Cheng, H. M., Ye, Z. X., and Lin, S. J. (2006). Identification and characterization of novel low-density lipoprotein receptor mutations of familial hypercholesterolaemia patients in Taiwan. Eur. J. Clin. Invest. 36, 866–874. doi: 10.1111/j.1365-2362.2006.01735.x
Chiou, K. R., and Charng, M. J. (2010). Detection of mutations and large rearrangements of the low-density lipoprotein receptor gene in Taiwanese patients with familial hypercholesterolemia. Am. J. Cardiol. 105, 1752–1758. doi: 10.1016/j.amjcard.2010.01.356
Chiou, K. R., and Charng, M. J. (2012). Common mutations of familial hypercholesterolemia patients in Taiwan: characteristics and implications of migrations from southeast China. Gene 498, 100–106. doi: 10.1016/j.gene2012.01.092
Chiou, K. R., and Charng, M. J. (2016). Genetic diagnosis of familial hypercholesterolemia in Han Chinese. J. Clin. Lipidol. 10, 490–496. doi: 10.1016/j.jacl.2016.01.009
Chiou, K. R., and Charng, M. J. (2017). Detection of common sequence variations of familial hypercholesterolemia in Taiwan using DNA mass spectrometry. J. Clin. Lipidol. 11, 386–393. doi: 10.1016/j.jacl.2016.12.014
Chiou, K. R., Charng, M. J., and Chang, H. M. (2011). Array-based resequencing for mutations causing familial hypercholesterolemia. Atherosclerosis 216, 383–389. doi: 10.1016/j.atherosclerosis.2011.02.006
Di Resta, C., and Ferrari, M. (2018). Next generation sequencing: from research area to clinical practice. EJIFCC 29, 215–220.
Di Taranto, M. D., Giacobbe, C., and Fortunato, G. (2020). Familial hypercholesterolemia: a complex genetic disease with variable phenotypes. Eur. J. Med. Genet. 63:103831. doi: 10.1016/j.ejmg.2019.103831
Elkins, J. C., and Fruh, S. (2019). Early diagnosis and treatment of familial hypercholesterolemia. Nurse Pract. 44, 18–24. doi: 10.1097/01.NPR.0000552677.31028.57
Gidding, S. S., Champagne, M. A., de Ferranti, S. D., Defesche, J., Ito, M. K., Knowles, J. W., et al. (2015). The agenda for familial hypercholesterolemia: a scientific statement from the american heart association. Circulation 132, 2167–2192. doi: 10.1161/CIR.0000000000000297
Han, S. M., Hwang, B., Park, T. G., Kim, D. I., Rhee, M. Y., Lee, B. K., et al. (2015). Genetic testing of Korean familial hypercholesterolemia using whole-exome sequencing. PLoS One 10:e0126706. doi: 10.1371/journal.pone.0126706
Hori, M., Ohta, N., Takahashi, A., Masuda, H., Isoda, R., Yamamoto, S., et al. (2019). Impact of LDLR and PCSK9 pathogenic variants in Japanese heterozygous familial hypercholesterolemia patients. Atherosclerosis 289, 101–108. doi: 10.1016/j.atherosclerosis.2019.08.004
Hori, M., Takahashi, A., Son, C., Ogura, M., and Harada-Shiba, M. (2020). The first Japanese cases of familial hypercholesterolemia due to a known pathogenic APOB gene variant, c.10580 G>A: p.(Arg3527Gln). J. Clin. Lipidol. doi: 10.1016/j.jacl.2020.05.007 [Epub ahead of print].
Hsiung, Y. C., Lin, P. C., Chen, C. S., Tung, Y. C., Yang, W. S., Chen, P. L., et al. (2018). Identification of a novel LDLR disease-causing variant using capture-based next-generation sequencing screening of familial hypercholesterolemia patients in Taiwan. Atherosclerosis 277, 440–447. doi: 10.1016/j.atherosclerosis.2018.08.022
Hu, M., Hooper, A. J., Bockxmeer, F. M., Watts, G. F., Chan, J. C., and Tomlinson, B. (2016). Management of Familial Hypercholesterolemia in Hong Kong. J. Atheroscler. Thromb. 23, 520–531. doi: 10.5551/jat.34314
Hu, M., Lan, W., Lam, C. W., Mak, Y. T., Pang, C. P., and Tomlinson, B. (2013). Heterozygous familial hypercholesterolemia in Hong Kong Chinese. Study of 252 cases. Int. J. Cardiol. 167, 762–767. doi: 10.1016/j.ijcard.2012.03.048
Jiang, L., Sun, L. Y., Dai, Y. F., Yang, S. W., Zhang, F., and Wang, L. Y. (2015). The distribution and characteristics of LDL receptor mutations in China: a systematic review. Sci. Rep. 5:17272. doi: 10.1038/srep17272
Khera, A. V., Won, H. H., Peloso, G. M., Lawson, K. S., Bartz, T. M., Deng, X., et al. (2016). Diagnostic yield and clinical utility of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia. J. Am. Coll. Cardiol. 67, 2578–2589. doi: 10.1016/j.jacc.2016.03.520
Kim, H. N., Kweon, S. S., and Shin, M. H. (2018). Detection of familial hypercholesterolemia using next generation sequencing in two population-based cohorts. Chonnam. Med. J. 54, 31–35. doi: 10.4068/cmj.2018.54.1.31
Kim, J. H., Choi, H. K., Lee, H., Park, H. Y., Kim, J. H., Kim, J. W., et al. (2004). Novel and recurrent mutations of the LDL receptor gene in Korean patients with familial hypercholesterolemia. Mol. Cells. 18, 63–70.
Kim, S. H., Bae, J. H., Chae, J. J., Kim, U. K., Choe, S. J., Namkoong, Y., et al. (1999). Long-distance PCR-based screening for large rearrangements of the LDL receptor gene in Korean patients with familial hypercholesterolemia. Clin. Chem. 45, 1424–1430. doi: 10.1093/clinchem/45.9.1424
Kwon, M., Han, S. M., Kim, D. I., Rhee, M. Y., Lee, B. K., Ahn, Y. K., et al. (2015). Evaluation of polygenic cause in Korean patients with familial hypercholesterolemia - a study supported by korean society of lipidology and atherosclerosis. Atherosclerosis 242, 8–12. doi: 10.1016/j.atherosclerosis.2015.06.053
Lee, S., Akioyamen, L. E., Aljenedil, S., Rivière, J. B., Ruel, I., and Genest, J. (2019). Genetic testing for familial hypercholesterolemia: impact on diagnosis, treatment and cardiovascular risk. Eur. J. Prev. Cardiol. 26, 1262–1270. doi: 10.1177/2047487319829746
Lye, S. H., Chahil, J. K., Bagali, P., Alex, L., Vadivelu, J., Ahmad, W. A., et al. (2013). Genetic polymorphisms in LDLR, APOB, PCSK9 and other lipid related genes associated with familial hypercholesterolemia in Malaysia. PLoS One 8:e60729. doi: 10.1371/journal.pone.0060729
Mabuchi, H., Nohara, A., Noguchi, T., Kobayashi, J., Kawashiri, M. A., Tada, H., et al. (2011). Molecular genetic epidemiology of homozygous familial hypercholesterolemia in the Hokuriku district of Japan. Atherosclerosis 214, 404–407. doi: 10.1016/j.atherosclerosis.2010.11.005
Mak, Y. T., Pang, C. P., Tomlinson, B., Zhang, J., Chan, Y. S., Mak, T. W., et al. (1998). Mutations in the low-density lipoprotein receptor gene in Chinese familial hypercholesterolemia patients. Arterioscler. Thromb. Vasc. Biol. 18, 1600–1605. doi: 10.1161/01.atv.18.10.1600
Marks, D., Thorogood, M., Neil, H. A., and Humphries, S. E. (2003). A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis 168, 1–14. doi: 10.1016/s0021-9150(02)00330-1
Nawawi, H. M., Chua, Y. A., and Watts, G. F. (2020). The brave new world of genetic testing in the management of the dyslipidaemias. Curr. Opin. Cardiol. 35, 226–233. doi: 10.1097/HCO.0000000000000721
Nohara, A., Yagi, K., Inazu, A., Kajinami, K., Koizumi, J., and Mabuchi, H. (1995). Absence of familial defective apolipoprotein B-100 in Japanese patients with familial hypercholesterolaemia. Lancet 345:1438. doi: 10.1016/s0140-6736(95)92627-5
Nordestgaard, B. G., Chapman, M. J., Humphries, S. E., Ginsberg, H. N., Masana, L., Descamps, O. S., et al. (2013). Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur. Heart J. 34, 3478a–3490a. doi: 10.1093/eurheartj/eht273
Ohta, N., Hori, M., Takahashi, A., Ogura, M., Makino, H., Tamanaha, T., et al. (2016). Proprotein convertase subtilisin/kexin 9 V4I variant with LDLR mutations modifies the phenotype of familial hypercholesterolemia. J. Clin. Lipidol. 10, 547–555. doi: 10.1016/j.jacl.2015.12.024
Pang, J., Chan, D. C., Hu, M., Muir, L. A., Kwok, S., Charng, M. J., et al. (2019). Comparative aspects of the care of familial hypercholesterolemia in the “Ten Countries Study”. J. Clin. Lipidol. 13, 287–300. doi: 10.1016/j.jacl.2019.01.009
Pang, J., Hu, M., Lin, J., Miida, T., Nawawi, H. M., Park, J. E., et al. (2017). An enquiry based on a standardised questionnaire into knowledge, awareness and preferences concerning the care of familial hypercholesterolaemia among primary care physicians in the Asia-Pacific region: the “Ten Countries Study”. BMJ Open 7:e017817. doi: 10.1136/bmjopen-2017-017817
Pang, J., Sullivan, D. R., Harada-Shiba, M., Ding, P. Y., Selvey, S., Ali, S., et al. (2015). Significant gaps in awareness of familial hypercholesterolemia among physicians in selected Asia-Pacific countries: a pilot study. J. Clin. Lipidol. 9, 42–48. doi: 10.1016/j.jacl.2014.09.011
Paththinige, C. S., Rajapakse, J. R. D. K., Constantine, G. R., Sem, K. P., Singaraja, R. R., Jayasekara, R. W., et al. (2018). Spectrum of low-density lipoprotein receptor (LDLR) mutations in a cohort of Sri Lankan patients with familial hypercholesterolemia - a preliminary report. Lipids Health Dis. 17:100. doi: 10.1186/s12944-018-0763-z
Pek, S. L. T., Dissanayake, S., Fong, J. C. W., Lin, M. X., Chan, E. Z. L., Tang, J. I., et al. (2018). Spectrum of mutations in index patients with familial hypercholesterolemia in Singapore: single center study. Atherosclerosis 269, 106–116. doi: 10.1016/j.atherosclerosis.2017.12.028
Peng, J., Wu, X., Wang, S., Zhang, S., Wang, X., Liu, Z., et al. (2019). Familial hypercholesterolemia in China half a century: a review of published literature. Atheroscler. Suppl. 36, 12–18. doi: 10.1016/j.atherosclerosissup.2019.01.003
Prodan Žitnik, I., Černe, D., Mancini, I., Simi, L., Pazzagli, M., Di Resta, M., et al. (2018). Personalized laboratory medicine: a patient-centered future approach. Clin. Chem. Lab. Med. 56, 1981–1991. doi: 10.1515/cclm-2018-0181
Punzalan, F. E., Sy, R. G., Santos, R. S., Cutiongco, E. M., Gosiengfiao, S., Fadriguilan, E., et al. (2005). Low density lipoprotein–receptor (LDL-R) gene mutations among Filipinos with familial hypercholesterolemia. J. Atheroscler. Thromb. 12, 276–283. doi: 10.5551/jat.12.276
Setia, N., Movva, S., Balakrishnan, P., Biji, I. K., Sawhney, J. P. S., Puri, R., et al. (2020). Genetic analysis of familial hypercholesterolemia in Asian Indians: a single-center study. J. Clin. Lipidol. 14, 35–45. doi: 10.1016/j.jacl.2019.12.010
Shin, D. G., Han, S. M., Kim, D. I., Rhee, M. Y., Lee, B. K., Ahn, Y. K., et al. (2015). Clinical features of familial hypercholesterolemia in Korea: predictors of pathogenic mutations and coronary artery disease - a study supported by the Korean Society of Lipidology and Atherosclerosis. Atherosclerosis 243, 53–58. doi: 10.1016/j.atherosclerosis.2015.08.033
Shin, J. A., Kim, S. H., Kim, U. K., Chae, J. J., Choe, S. J., Namkoong, Y., et al. (2000). Identification of four novel mutations of the low-density lipoprotein receptor gene in Korean patients with familial hypercholesterolemia. Clin. Genet. 57, 225–229. doi: 10.1034/j.1399-0004.2000.570309.x
Sturm, A. C., Knowles, J. W., Gidding, S. S., Ahmad, Z. S., Ahmed, C. D., Ballantyne, C. M., et al. (2018). Clinical genetic testing for familial hypercholesterolemia: JACC scientific expert panel. J. Am. Coll. Cardiol. 72, 662–680. doi: 10.1016/j.jacc.2018.05.044
Sun, D., Zhou, B. Y., Li, S., Sun, N. L., Hua, Q., Wu, S. L., et al. (2018). Genetic basis of index patients with familial hypercholesterolemia in Chinese population: mutation spectrum and genotype-phenotype correlation. Lipids Health Dis. 17:252. doi: 10.1186/s12944-018-0900-8
Tan, K., Cheung, C. L., Yeung, C. Y., Siu, D., Leung, J., and Pang, H. K. (2018). Genetic screening for familial hypercholesterolaemia in Hong Kong. Hong Kong Med. J. 3, 7–10.
Wang, Y., Li, Y., Liu, X., Tu, R., Zhang, H., Qian, X., et al. (2019). The prevalence and related factors of familial hypercholesterolemia in rural population of China using chinese modified dutch lipid clinic network definition. BMC Public Health 19:837. doi: 10.1186/s12889-019-7212-4
Wilemon, K. A., Patel, J., Aguilar-Salinas, C., Ahmed, C. D., Alkhnifsawi, M., Almahmeed, W., et al. (2020). Reducing the clinical and public health burden of familial hypercholesterolemia: a global call to action. JAMA Cardiol. [Epub ahead of print] doi: 10.1001/jamacardio.2019.5173,
Yang, K. C., Su, Y. N., Shew, J. Y., Yang, K. Y., Tseng, W. K., Wu, C. C., et al. (2007). LDLR and ApoB are major genetic causes of autosomal dominant hypercholesterolemia in a Taiwanese population. J. Formos. Med. Assoc. 106, 799–807. doi: 10.1016/S0929-6646(08)60044-3
Keywords: apolipoprotein B, Asia, diagnosis, familial hypercholesterolemia, gene, low-density lipoprotein receptor, proprotein convertase subtilisin/kexin type 9, mutation
Citation: Huang C-C and Charng M-J (2020) Genetic Diagnosis of Familial Hypercholesterolemia in Asia. Front. Genet. 11:833. doi: 10.3389/fgene.2020.00833
Received: 13 April 2020; Accepted: 09 July 2020;
Published: 24 July 2020.
Edited by:
Alpo Juhani Vuorio, University of Helsinki, FinlandReviewed by:
Chiara Di Resta, Vita-Salute San Raffaele University, ItalyCopyright © 2020 Huang and Charng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chin-Chou Huang, Y2NodWFuZzRAdmdodHBlLmdvdi50dw==; Min-Ji Charng, bWpjaGFybmdAdmdodHBlLmdvdi50dw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.