 Chenmiao Liu
Chenmiao Liu Huiling Chen
Huiling Chen Zhanjun Ren
Zhanjun Ren
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 30 July 2020
Sec. Livestock Genomics
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.00797
This article is part of the Research Topic High-Throughput Phenotyping in the Genomic Improvement of Livestock View all 17 articles
The domestic Bactrian camel is indispensable to agricultural production in the desertification area of China owning to its endurance to hunger and thirst, cold resistance, drought resistance, and good long-distance transportation. Therefore, it is necessary to investigate the genetic diversity, genetic structure, and genes with important roles in the evolution of this species. In this study, 1,568,087 SNPs were identified in 47 domestic Bactrian camels inhabiting four regions of China, namely Inner Mongolia, Gansu, Qinghai, and Xinjiang, by restriction site associated DNA sequencing (RAD-seq). The SNP data were used for nucleotide diversity analysis (π) and linkage disequilibrium (LD) attenuation analysis to elucidate the genetic diversity of the domestic Bactrian camel in the four regions studied. Results showed that Xinjiang camels had the highest nucleotide diversity and the fastest decay rate of the LD coefficient; therefore, Xinjiang camels had the highest genetic diversity. Structure analysis, principal component analysis (PCA), and phylogenetic tree construction by the neighbor-joining (NJ) method showed that Qinghai camels clustered separately, at a larger phylogenetic distance from camels in the other regions. Through analyses of selection signals, it was found that the number of selected genes shared by Inner Mongolia camels, Qinghai camels, Xinjiang camels, and Gansu camels was 7, 24, 25, and 113, respectively. The shared selected genes of the domestic Bactrian camel in the four regions were further analyzed, and three shared genes (GRIA3, XIAP, and THOC2) of the domestic Bactrian camel in China were identified. Gene Ontology (GO) classification and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis were performed on the shared selected genes of the domestic Bactrian camel in all four regions studied. Across all regions, genes involved in the cellular process were the most abundant subcategory under biological process. Cell and cell part represented the main proportion of genes under cellular component. Binding represented the main molecular function. In addition, the shared selected genes of the domestic Bactrian camel in the four regions of China were significantly enriched in the long-term depression pathway. The research should enable further study of the genetic resources of the domestic Bactrian camel, as well as the conservation of these resources.
The domestic Bactrian camel plays an important role in economic activities and trade, as well as in national defense, and has cultural significance; therefore, this animal represents one of the most valuable livestock resources in China (Chen et al., 2018). With the impact of modern civilization, such as the growth of the automobile transportation industry and the destruction of the natural environment, the number of domestic Bactrian camels has been greatly reduced, from 618,600 in 1981 to 380,000 in present-day China. Therefore, it is essential to study the genetic diversity of the domestic Bactrian camel and conserve its germplasm resources (Ming et al., 2017). To date, a variety of studies have been conducted on the origin, domestication, and genetic diversity of the domestic Bactrian camel worldwide. Molecular clock analysis based on the complete mitochondrial genome sequences shows that the wild camel was not the direct ancestor of the domestic Bactrian camel. The domestic Bactrian camel is considered to be monophyletic in terms of its evolutionary origin, and to originate from a single wild population (Ji et al., 2009). The earliest evidence on the domestication of Bactrian camels is mainly derived from the northeast of Ilang and the south of neighboring Turkmenistan, especially in the Kopet Daghmountain region. Bactrian camel skeletons were found in the second cultural layer of the Anau ruins on the northern edge of the mountain range, dating back to 3500–3000 BC (Meadow and Zeder, 1978). Studies of the genetic diversity of the domestic Bactrian camel through analysis of mitochondrial sequence variation found that there were no significant genetic differences between populations in China, Russia, and Mongolia, indicating that a strong gene flow occurred due to the extensive movement of the domestic Bactrian camel (Ming et al., 2017). However, the use of RAD-seq to identify whole-genome single nucleotide polymorphisms (SNPs) for genetic analysis of the domestic Bactrian camel in the four regions of China has not been reported to date.
In the past few decades, the use of SNPs for studying species genetic diversity and population structure has become widespread (Ren et al., 2019). The rapid development of high-throughput sequencing technology has led to the use of restriction site-associated DNA sequencing (RAD-seq) as a relatively cost-effective way to identify a large number of SNPs throughout the genome (Miller et al., 2007; Davey et al., 2011). So far, RAD-seq has been widely applied to population genetics studies of a variety of species (Zhang et al., 2016). For instance, Díaz-Arce et al. (2016) used RAD-seq data to infer the phylogeny of tuna based on whole-genome nuclear markers. Ren et al. (2019) used the RAD-seq method to identify genome-wide SNPs, and studied the genetic diversity and population structure of four Chinese rabbit breeds.
In this study, we discovered genome-wide SNPs in the domestic Bactrian camel using the RAD-seq method. We then investigated the genetic diversity, genetic structure, and genes with important roles in the evolution of this species in four regions of China (Inner Mongolia, Gansu, Qinghai, and Xinjiang). The study clarified the phylogenetic relationship of the domestic Bactrian camel in China and explored the genomic resources of this species. And the SNP resources generated in this study offer a valuable tool for future genetics and genomics research of the domestic Bactrian camel.
Forty-seven venous blood samples were obtained from seven populations of domestic Bactrian agricultural camels from four regions of China (Inner Mongolia, Gansu, Qinghai, and Xinjiang) (Table 1) to ensure that each sample was derived from a different family, and there was no kinship between individuals. All methods and experimental protocols of this study were performed in accordance with guidelines and regulations of the animal ethics committee of Northwest A and F University (China) and the National Natural Science Foundation of China (31172178) Animal Care and Use Committee. DNA samples were extracted following a standard phenol-chloroform extraction procedure (Sambrook and Russell, 2002) and were diluted to 20 ng/μL.
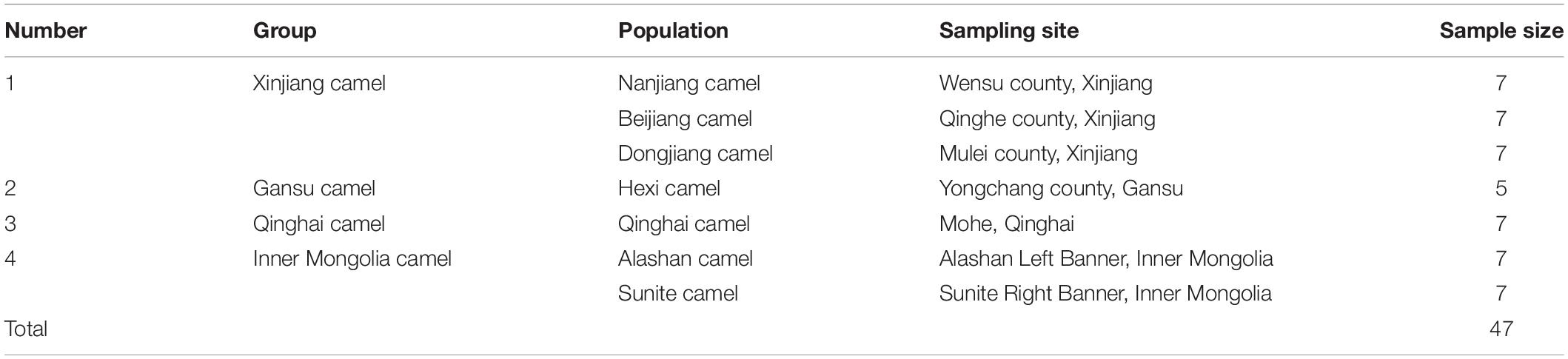
Table 1. Information for domestic Bactrian camels in the four sampled regions of China.
RAD-seq libraries were constructed in accordance with the modified protocol (Baird et al., 2008). In short, EcoRI (New England Biolabs) was used to digest genomic DNA (0.1–1 μg; from a single sample or pooled samples), and P1 adaptors were connected at the cutting site. Then, the samples were pooled, randomly sheared, and size-selected in sequential steps. After the second adaptors (P2) were added, the sequencing libraries were constructed using DNA fragments of 300–700 bp in length. Finally, the constructed libraries were sequenced using the Illumina HiSeq3000 platform, and 100 bp paired-end reads were generated.
Quality trimming generated using fastp is an indispensable step to ensure high confidence of variant calling (Chen et al., 2018). Applying three strict filtering criteria, raw reads were processed to obtain high-quality clean reads: (i) removing reads with ≥10% unidentified nucleotides (N); (ii) removing reads with >50% bases having phred quality scores of ≤20; and (iii) removing reads aligned to the barcode adapter.
The Burrows-Wheeler Aligner (BWA) was used to align the clean reads of each sample with the reference genome1 with the settings “mem 4 -k 32 -M”, where -k is the minimum seed length, and -M being an option used to mark shorter split alignment hits as secondary alignments (Li and Durbin, 2009). GATK’s Unified Genotyper was used to conduct variant calling on all samples (DePristo et al., 2011). GATK’s Variant Filtration with proper standards was used to filter SNPs (-Window 4, -filter “QD < 2.0 | | FS > 60.0 | | MQ < 40.0”,-G_filter “GQ < 20”).
First, we used the VCFtools software suite to study the overall read depth and chromosome distribution of all SNPs (Danecek et al., 2011). The minimum read coverage for a SNP to be called is 3×, and all non-completely missing polymorphic loci (-max-missing 1e-06-non-ref-af 1e-06) were used for counting. The nucleotide diversity (π) of the domestic Bactrian camel in four regions of China was calculated using the PopGenome software package2 (Pfeifer et al., 2014). In addition, we used the 1,568,087 identified SNPs to estimate the linkage disequilibrium (LD) attenuation trend by calculating the LD coefficient (r2) between two points in a range of sequence (typically <5 Mb). The more rapid the decay of r2, the higher the genetic diversity of population is; r2 values in the range of 0,1 represent the correlation between two points: if r2 is 0, there is no correlation between the two loci, whereas if r2 is 1, the two loci are completely correlated. “LD attenuation distance” was used to evaluate the speed of LD attenuation (Guo, 2012).
The genetic structure was studied by phylogenetic tree construction, principal component analysis, and analysis of population structure. Following the identification of SNPs, 1,568,087 SNPs were used to calculate the phylogenetic distance between populations. The phylogenetic tree was constructed using a neighbor-joining (NJ) method with the software Treebest (version 1.9.2) to determine the evolutionary relationship between populations. Bootstrap values were generated from 1,000 replications. After removing sites with a missing rate of 50% or more, the remaining 865,774 loci were used for PCA, which was carried out using the GCTA software in R to further study the population genetic structure between regions (Yang et al., 2011). The STRUCTURE program3 (in order to ensure the independence of SNP marks, we used PLINK software to filter 865,774 SNPs according to LD intensity, the remaining 12,046 loci were used for structure analysis, -indep-pairwise 250 10 0.1, 250 kb window, the step size of 10 SNPs, r2 is <1) was used for analysis of population structure. We predefined the number of genetic clusters from K = 2 to K = 6 (BURNIN = 5,000 times, NUMREPS = 100,000), and repeated each K-value three times (Liu et al., 2019). Next, we used the POPHELPER software4 to calculate the value of ΔK; then, we used the CLUMPP software5 to combine the results of three repetitions (Schraiber and Akey, 2015). The fixation index (Fst) was calculated according to the statistical function of Fst in the PopGenome software package to study the genetic diversity between different regions (Pfeifer et al., 2014). Fst can also be used to infer the genetic distance between different regions. After removing the site with a missing rate of 50% or more, the remaining 865,774 loci were used for Fst analysis. PLINK 1.9 was used to calculate the inbreeding coefficient (Fis) of the domestic Bactrian camel in four regions6. After obtaining the Fis value for each sample, the average value in the region was determined.
The top 5% region was selected based on the interception of two different parameters, namely nucleotide diversity (π) (Nei and Li, 1979) and population differentiation index (FST) (Danecek et al., 2011). Using the 50 kb sliding window method with a step size of 25 kb, the −log10 transform of Nei’s π was used to select the lower end of diversity windows, and these parameters were quantified by internal PERL Scripts. All related graphs were drawn using R scripts (R Core Team, 2013). With Gansu camels as the control group and Inner Mongolia camels as the selection group, the genes of Inner Mongolia camels under selection pressure were identified. With Qinghai camels as the control group and Inner Mongolia camels as the selection group, the genes of Inner Mongolia camels under selection pressure were identified. With Xinjiang camels as the control group and Inner Mongolia camels as the selection group, the genes of Inner Mongolia camels under selection pressure were identified. Venn diagrams were then used to determine the common genes under selection pressure in Inner Mongolia camels; these genes played a crucial role in the evolution of this group. Using the same comparison method, the common genes under selection pressure were identified in Gansu camels, Qinghai camels, and Xinjiang camels.
In order to further systematically elucidate the complex biological functions of the genes, the common genes of the domestic Bactrian camel under selection pressure, in the four regions of China investigated in this study, were mapped against both GO and KEGG databases. GO enrichment analysis was performed with WEGO software, and gene numbers were calculated for every term (Ye et al., 2006). KEGG enrichment analysis using the KEGG database7 and KOBAS software were performed to determine the statistical enrichment in KEGG pathways of the common genes of the domestic Bactrian camel under selection pressure in the four regions (Mao et al., 2005). The calculated p-value was subject to false discovery rate (FDR) correction, applying FDR ≤ 0.05 as the threshold. Pathways meeting this condition were defined as significantly enriched pathways in the abovementioned genes.
RAD sequencing produced a total of 131.13G of raw data for 47 normal sequenced individuals prior to quality filtering, with an average of 2.79G per sample, ranging from 1.36 to 4.57G. After quality filtering of the sequence data, 129.24G of clean data (1.34G to 4.30G for each sample, with an average of 2.75G) were retained, representing an average effective mapping rate of 98.57%. We mapped 1,568,087 regions, and the average spacing of RAD regions on the draft camel genome assembly was 1201.39 bp. The percentage of high-quality clean reads was above 97.73%, and the number of reads on the alignment was mostly above 97.39%. Of the clean reads retained, an average of 19.01 million reads were retained for each sample. In short, our sequencing data showed a high phred quality (Q20 > 94%, Q30 > 87%), and the GC content was stable, at between 40.71 and 45.13% (Supplementary Tables S1–S4).
In this study, 1,568,087 SNPs were generated by RAD-seq. There were 548,830 loci (35%) in the transversion and 1,019,257 loci (65%) in the transition. The ratio of transition to transversion was close to 2:1. There were differences in the number of SNPs of the domestic Bactrian camel between the four regions, and the number of SNPs was in the following order: Xinjiang camels > Inner Mongolia camels > Qinghai camels > Gansu camels. The LD attenuation analysis of the domestic Bactrian camels from the four regions showed that the attenuation rate of the LD coefficient differed between the four regions, and that the attenuation rate was Xinjiang camels > Inner Mongolia camels > Qinghai camels > Gansu camels (Figure 1). Genome-wide nucleotide diversity was estimated from the SNP data. Because nucleotide diversity represents genetic diversity to an extent, it can be concluded from the data in Table 2 that the nucleotide diversity (π) of Xinjiang camels was the highest.
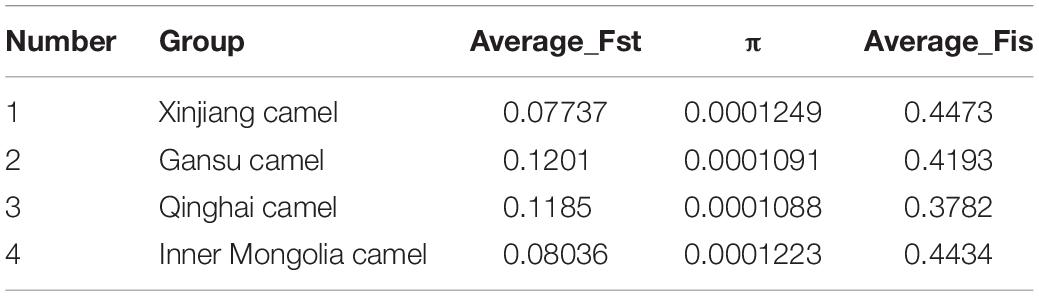
Table 2. Fst, π, and Fis values for domestic Bactrian camels in the four regions of China.
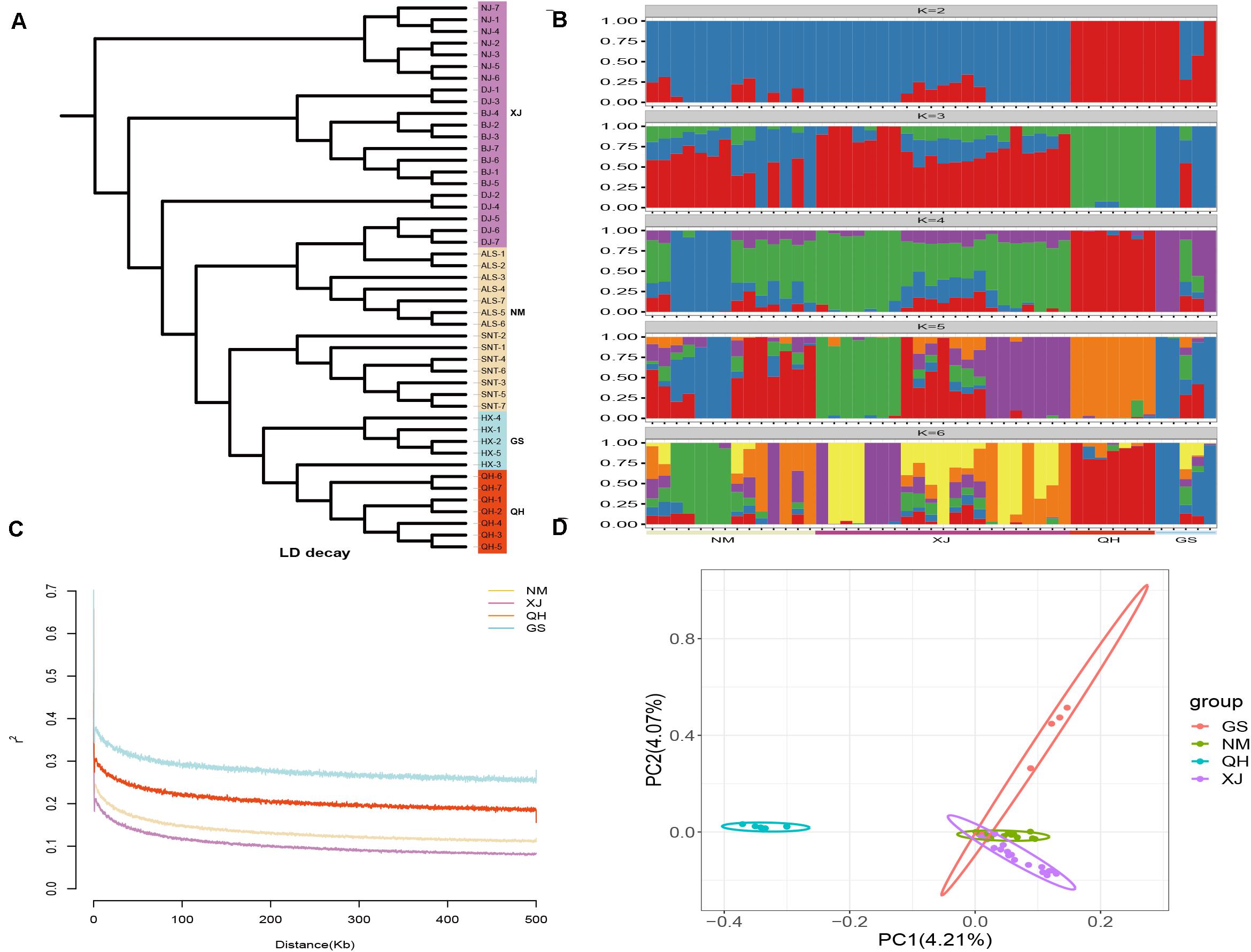
Figure 1. Structure analyses of the domestic Bactrian camel in four regions of China. (A) Phylogenetic tree construction with the neighbor-joining (NJ) method. (B) Groups structure clustering figure of the domestic Bactrian camel in four regions of China. (C) LD attenuation map of the domestic Bactrian camel in four regions of China. (D) Principal components analysis of the domestic Bactrian camel in four regions of China.
Genetic analysis of population structure using STRUCTURE software and PCA showed similar patterns. Cross-validation with K = 5 was the most suitable for the true differentiation history of the domestic Bactrian camel. At K = 5, the ancestral background of Qinghai camels was relatively pure, with a major genetic ancestor. Although the Inner Mongolia camels, Xinjiang camels, and Gansu camels had multiple genetic ancestors, a major genetic ancestor was evident. At K = 2, Qinghai camels were obviously separated from the camels in other regions, indicating that this group was phylogenetically distant from the camels in other regions. The PCA map showed that the Qinghai camels clustered together separately, and were phylogenetically distant from the camels in other regions. Inner Mongolia camels and Xinjiang camels gathered together, indicating that their genetic relationship was relatively close. The phylogenetic tree constructed by the NJ method showed that the domestic Bactrian camels of the four regions gathered together, and the branches of the tree were obvious. Xinjiang camels and Inner Mongolia camels gathered together (Figure 1). The Fst values were calculated to study the genetic distance between different regions. As shown in Table 2, it can be concluded that the average Fst between the Qinghai camels and the domestic Bactrian camels in other regions was 0.1185, second only to Gansu camels (0.1201), indicating a large genetic distance between Qinghai camels and other domestic Bactrian camels. It should be noted that the farther the kinship, the smaller the inbreeding coefficient. The average Fis between Qinghai camels and domestic Bactrian camels in other regions was the lowest, indicating a large genetic distance from camels in the other regions; these findings were consistent with the results of PCA and structure analysis.
The top 5% regions were selected by combining the π and the FST. With Gansu camels as the control group and Inner Mongolia camels as the selection group, 238 selected genes were obtained. With Qinghai camels as the control group and Inner Mongolia camels as the selection group, 365 selected genes were obtained. With Xinjiang camels as the control group and Inner Mongolia camels as the selection group, 287 selected genes were obtained. Among them, 7 selected genes shared by Inner Mongolia camels were identified between all three comparisons (Figure 2 and Supplementary Tables S5–S7). Using the same comparison method, it was found that the number of selected genes shared by Qinghai camels, Xinjiang camels, and Gansu camels was 24, 25, and 113, respectively (Supplementary Figures S1–S3 and Supplementary Tables S8–S16). Venn diagrams were used to further analyze the shared selected genes of the domestic Bactrian camel between the four regions in China, and three shared genes (GRIA3, XIAP, THOC2) were identified in the domestic Bactrian camel across all four regions (Figure 7).
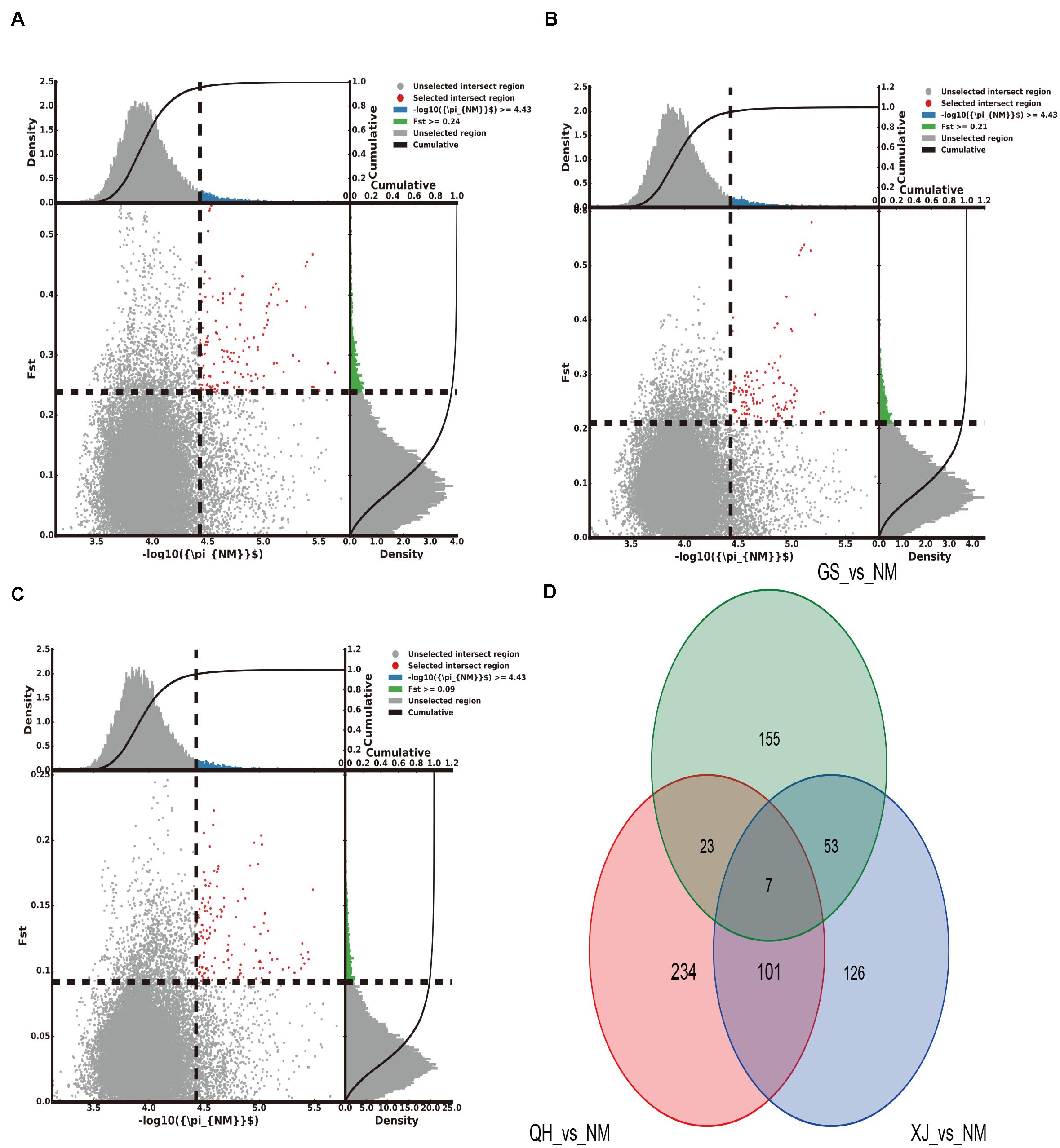
Figure 2. The number of selected genes shared by the Inner Mongolia group of camels. (A) Gansu camels were the control group and Inner Mongolia camels were the selection group; 238 selected genes were obtained. (B) Qinghai camels were the control group and Inner Mongolia camels were the selection group; 365 selected genes were obtained. (C) Xinjiang camels were the control group and Inner Mongolia camels were the selection group; 287 selected genes were obtained. (D) Seven selected genes were shared by Inner Mongolia camels.
GO classification was carried out on the shared selected genes of the domestic Bactrian camel in the four regions. For Inner Mongolia camels, the genes involved in the single-organism process (GO: 0044699) were the most abundant under biological process. Cell (GO: 0005623), cell part (GO: 0044464), and organelle (GO: 0043226) represented the main proportion of the cellular component. Binding (GO: 0005488) accounted for a high proportion of the molecular functional category (Figure 3 and Supplementary Table S17). For Qinghai camels and Gansu camels, the most abundant subcategory under biological process was cellular process (GO: 0009987). Under the category of cellular component, the most abundantly expressed genes were cell (GO: 0005623) and cell part (GO: 0044464). Binding (GO: 0005488) represented the main molecular function (Figures 4, 6 and Supplementary Tables S18, S20). For Xinjiang camels, the genes involved in the cellular process (GO: 0009987) were the most abundant subcategory in biological process. Organelle (GO: 0043226) accounted for a high proportion under cellular component. Binding (GO: 0005488) represented the main molecular function (Figure 5 and Supplementary Table S19).
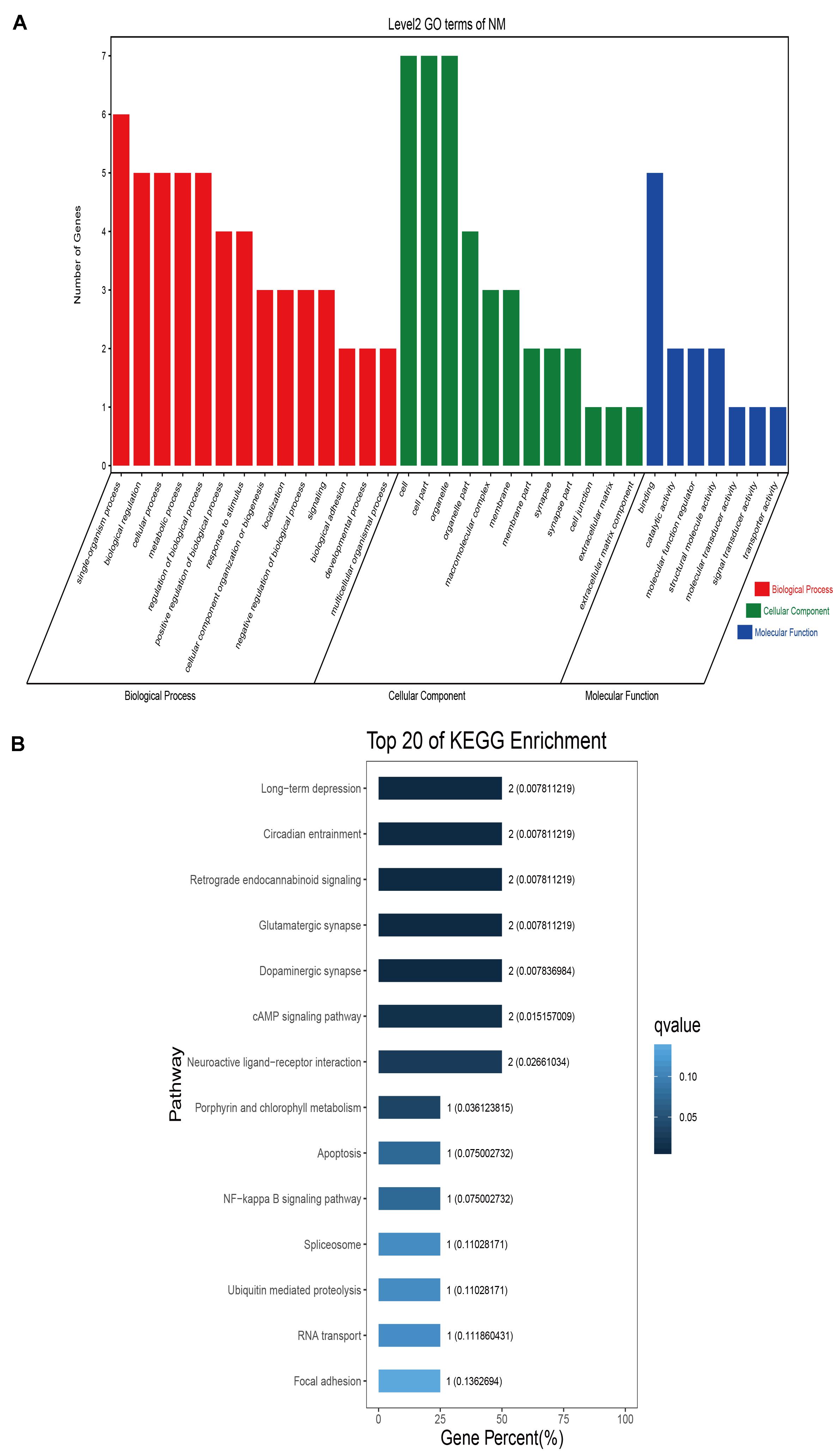
Figure 3. Go classification and KEGG enrichment of the shared selected genes of Inner Mongolia camels. (A) Go classification of the shared selected genes of Inner Mongolia camels. (B) KEGG enrichment of the shared selected genes of Inner Mongolia camels.
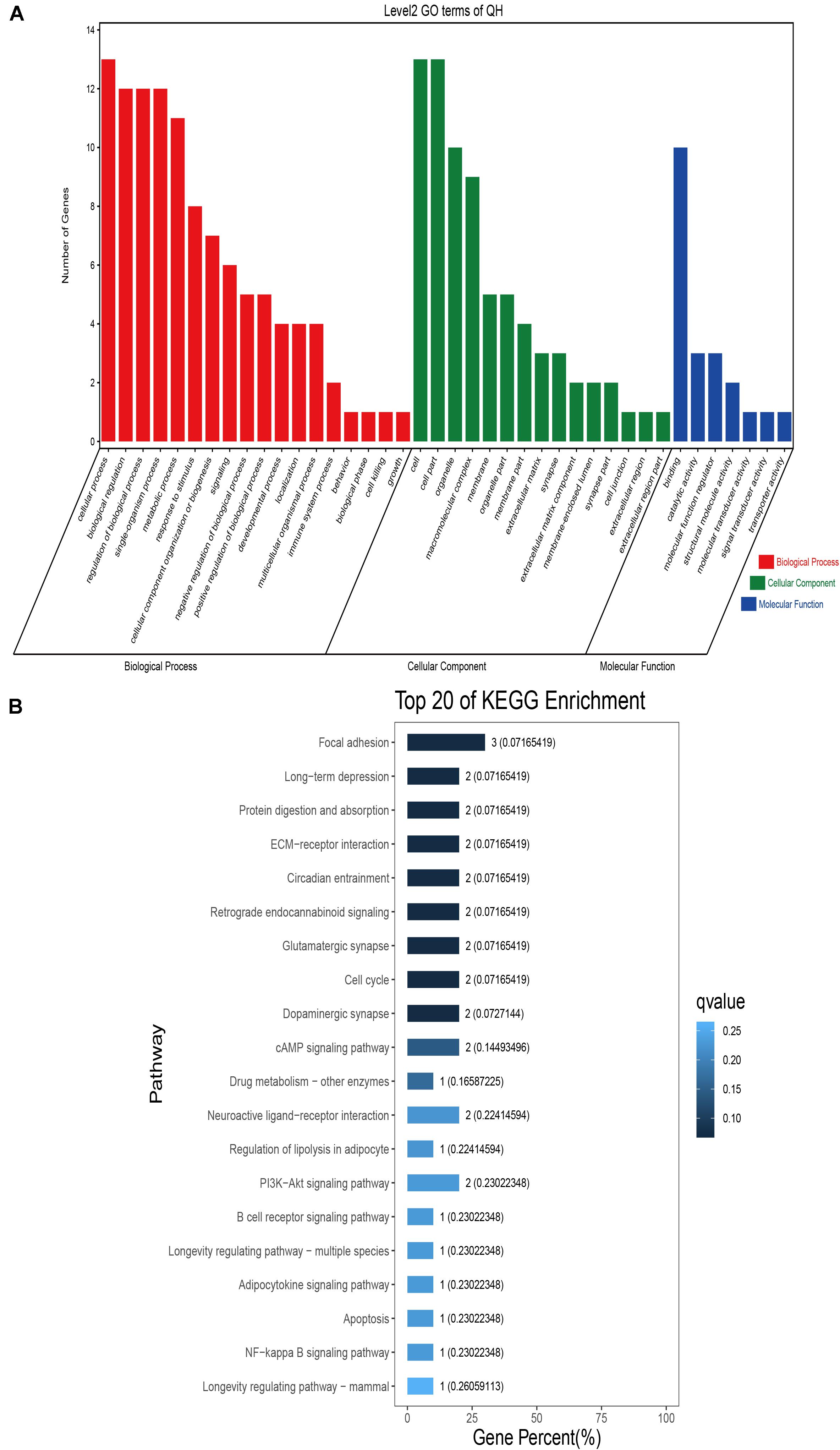
Figure 4. Go classification and KEGG enrichment of the shared selected genes of Qinghai camels. (A) Go classification of the shared selected genes of Qinghai camels. (B) KEGG enrichment of the shared selected genes of Qinghai camels.
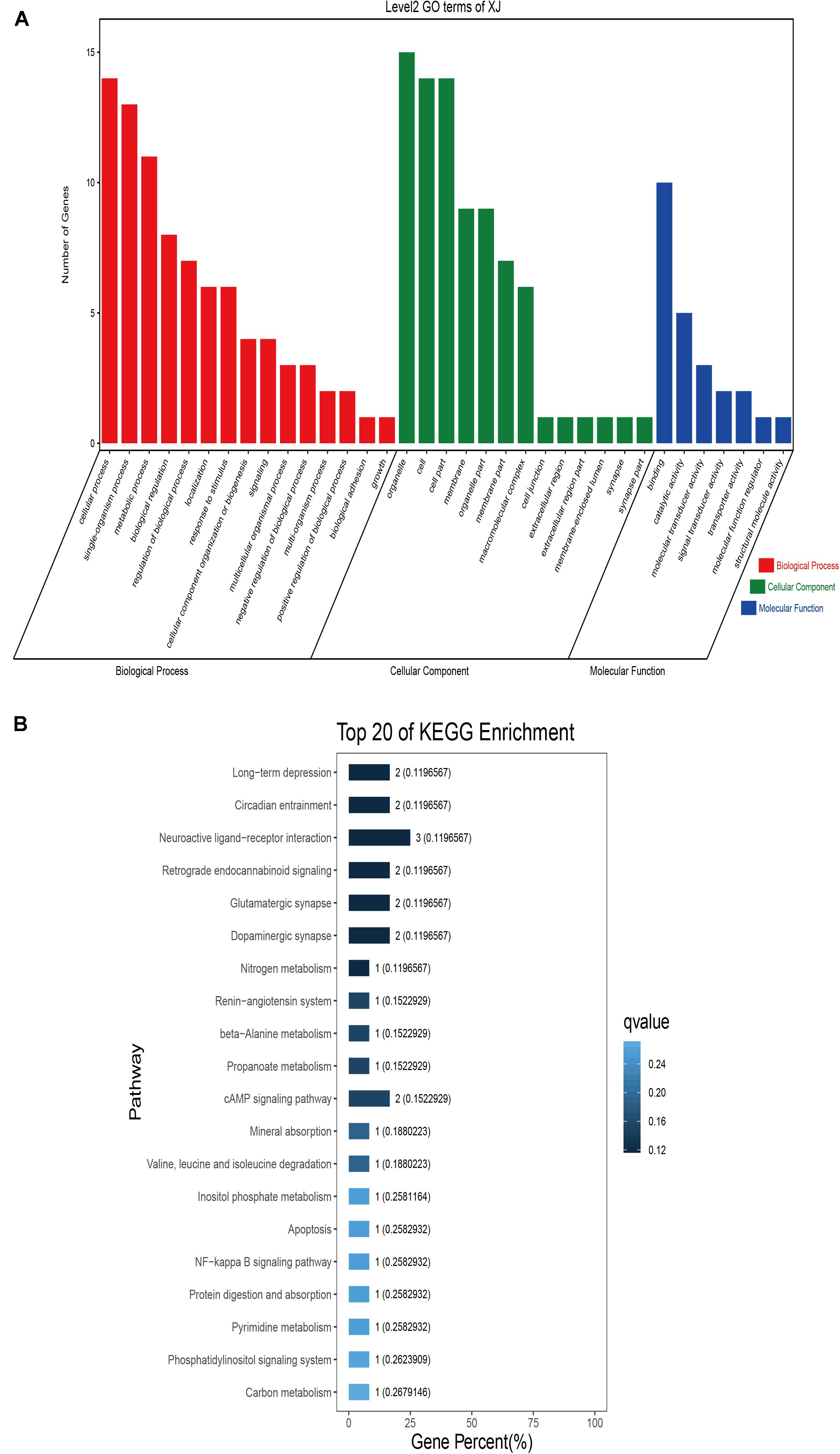
Figure 5. Go classification and KEGG enrichment of the shared selected genes of Xinjiang camels. (A) Go classification of the shared selected genes of Xinjiang camels. (B) KEGG enrichment of the shared selected genes of Xinjiang camels.
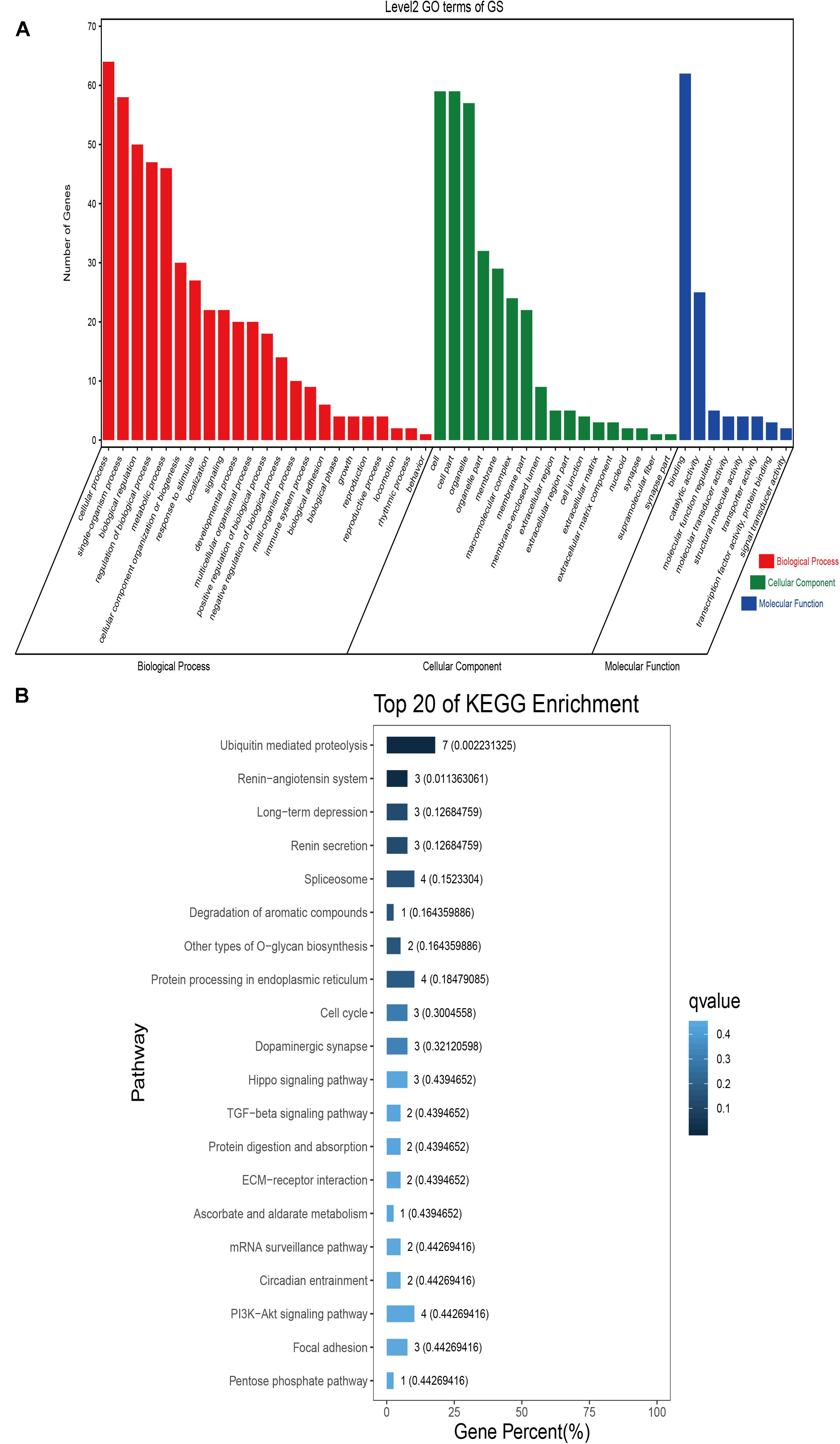
Figure 6. Go classification and KEGG enrichment of the shared selected genes of Gansu camels. (A) Go classification of the shared selected genes of Gansu camels. (B) KEGG enrichment of the shared selected genes of Gansu camels.
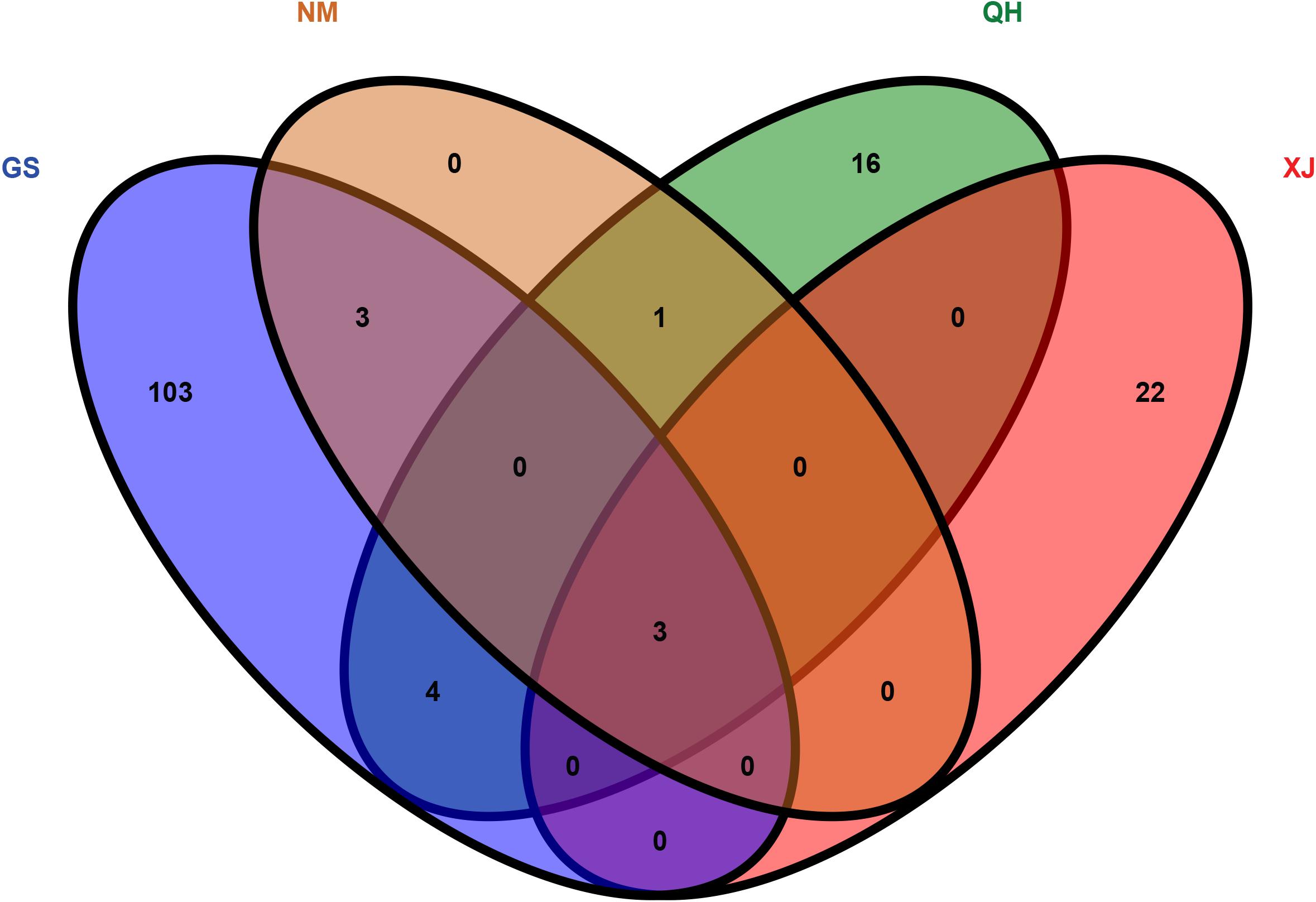
Figure 7. The shared selected genes of the domestic Bactrian camel in China.
KEGG enrichment analysis of the shared selected genes of the domestic Bactrian camel in the four regions was performed. Among the Inner Mongolia camels, the shared selected genes were significantly enriched in eight pathways, including long-term depression and circadian entrainment (Figure 3 and Supplementary Table S21). Among the Qinghai camels, the shared selected genes were mainly enriched in focal adhesion pathway (Figure 4 and Supplementary Table S22). Among Xinjiang camels, the shared selected genes were mainly enriched in neuroactive ligand-receptor interaction pathway (Figure 5 and Supplementary Table S23). Among the Gansu camels, the shared selected genes were significantly enriched in ubiquitin mediated proteolysis and renin-angiotensin system pathway (Figure 6 and Supplementary Table S24).
We reported the identification of 1,568,087 SNP loci in the domestic Bactrian camel in four regions of China, using RAD-seq. When read mapping, we used higher “k”-values, and the parameter settings are within the reasonable range of the software default (Cao et al., 2019; Shibuya and Comin, 2019; Liu et al., 2020). The filtered SNPs were subjected to LD attenuation analysis and nucleotide diversity analysis (π). The results indicated that Xinjiang camels had the largest number of SNPs, the fastest decay of the LD coefficient, the highest nucleotide diversity, and the highest genetic diversity: these features may be attributable to the preservation of the genetic diversity of this group’s ancestors (Abdulla et al., 2009). Population structural analysis, PCA, and Fis results suggested that Qinghai camels were phylogenetically distant from camels in other regions; this may be due to the geographical distribution of the Qinghai camels. The Mohe camel farm is located at an altitude of 3000 meters, which is far more elevated than the location of the domestic Bactrian camels in other regions, resulting in less genetic communication between Qinghai camels and domestic Bactrian camels in other regions. Phylogenetic trees constructed using the NJ method as well as PCA data revealed that the genetic relationship between Xinjiang camels and Inner Mongolia camels was relatively close. According to historical records, Inner Mongolia camels were extensively used to transport military supplies to Xinjiang in the Qing Dynasty, which may have promoted the genetic communication between Inner Mongolia camels and Xinjiang camels (Tong, 2018). The average Fst between Gansu camels and domestic Bactrian camels in other regions was the largest, and were phylogenetically distant from the camels in other regions. This may be related to the geographical location of Gansu camels. Yongchang County is located at the northern foot of the Qilian Mountain, close to the Badain Jaran desert, resulting in less genetic communication between Gansu camels and other domestic Bactrian camels. Analyses of selection signals showed that the number of selected genes shared by Inner Mongolia camels, Qinghai camels, Xinjiang camels and Gansu camels was 7, 24, 25, and 113, respectively. GO classification and KEGG enrichment analysis were performed on the shared selected genes. The results showed that the most abundant subcategory under biological process was cellular process (GO: 0009987). Cell (GO: 0005623), and cell part (GO: 0044464) accounted for a high proportion of the subcategories under cellular component. Binding (GO: 0005488) represented the main molecular function. The shared selected genes of domestic Bactrian camel populations in the four regions were all significantly enriched in the long-term depression pathway. This finding fills the gap in the genome study of the domestic bactrian camel in China (He et al., 2009; Ming et al., 2019).
The shared selected genes of the domestic Bactrian camel between the four regions in China were further analyzed to identify three shared genes (GRIA3, XIAP, THOC2) in the domestic Bactrian camel across all four regions. To our knowledge, this is the first study to identify genes of importance in the evolution of the domestic Bactrian camel in China. GRIA3 (glutamate receptor 3) is the main excitatory neurotransmitter receptor in the mammalian brain, and is activated in many normal neurophysiologic processes. GRIA3 regulates the activity of AMPA glutamate receptor and the NMDA receptor (Kato et al., 2010). Both AMPA receptors and NMDA receptors are classes of vital receptors in learning and memory (Brechet et al., 2017; Wang et al., 2018). According to the report of global human geography, the camel’s memory is second to none in all animals, and the ability of camels to remember routes gives these animals the capacity to navigate journeys accurately during sandstorms. GRIA3 is therefore an interesting target for future research on the genome of the domestic bactrian camel in China. XIAP (E3 ubiquitin-protein ligase XIAP) is a multifunctional protein involved in the cellular response to DNA damage, and can also regulate inflammatory signal transduction and immunity (Deveraux et al., 1997; Damgaard and Gyrd-Hansen, 2011). The harsh environmental conditions of the habitat of the domestic Bactrian camel may promote apoptosis, DNA damage, and inflammatory reactions (Yuan et al., 2017). It can therefore be concluded that XIAP is an interesting target for future research on the genome of the domestic bactrian camel in China. THOC2 (THO complex subunit 2) is a protein-coding gene that is involved in neuronal generation and neuronal development (Straesser et al., 2002). Mutations in this gene can cause hypotonia, gait disturbance, and tremors (Kumar et al., 2015). This gene is also an interesting target for future research on the genome of the domestic bactrian camel in China. A PPI network of the products of the three shared genes, constructed with STRING8, showed that there was interaction between the three encoded proteins (Supplementary Figure S4). Interestingly, the shared selected genes of the domestic Bactrian camel in the four regions were significantly enriched in the long-term depression (LTD) pathway. LTD has previously been assigned an assistant role in signal-to-noise adjustment or “forgetting”. However, LTD also contributes directly to the storage of hippocampal information (Collingridge et al., 2010). Furthermore, LTD plays a dominant role in the processing of precise spatial features. Increasing evidence supports the notion that LTD enables distinct and separate forms of information storage, which together promote the generation of a spatial cognitive map (Kemp and Manahan-Vaughan, 2007).
In brief, we identified the SNPs present in the domestic Bactrian camel genome on a whole-genome basis, and systematically studied the genetic diversity, genetic structure, and genes of importance in the evolution of the domestic Bactrian camel in four regions of China. The results should enable further study of the genetic resources of this mammal, as well as the conservation of these resources. In future studies, we aim to collaborate internationally to collect blood samples from camel populations inhabiting other regions along the Silk Road in order to further explore the available genome resources of this species.
The datasets generated for this study can be found in the NCBI SRA. Bioproject #PRJNA522647 and Biosamples #SAMN10948548–SAMN10948594.
The animal study was reviewed and approved by National Natural Science Foundation of China (31172178). Written informed consent was obtained from the owners for the participation of their animals in this study.
ZR and CL designed the study and wrote the manuscript. HC collected the samples. CZ and XY contributed to the data analysis. All authors read and approved the manuscript.
This study was supported by the National Natural Science Foundation of China, 31172178.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00797/full#supplementary-material
Abdulla, M. A., Ahmed, I., Assawamakin, A., Bhak, J., Brahmachari, S. K., Calacal, G. C., et al. (2009). Mapping human genetic diversity in Asia. Science 326, 1541–1545. doi: 10.1126/science.1177074
Baird, N. A., Etter, P. D., Atwood, T. S., Currey, M. C., Shiver, A. L., Lewis, Z. A., et al. (2008). Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 3:e3376. doi: 10.1371/journal.pone.0003376
Brechet, A., Buchert, R., Schwenk, J., Boudkkazi, S., and Fakler, B. (2017). AMPA-receptor specific biogenesis complexes control synaptic transmission and intellectual ability. Nat. Commun. 8, 1–14. doi: 10.1038/ncomms15910
Cao, D. N., Wang, M., Ge, Y., and Gong, S. P. (2019). Draft genome of the big-headed turtle Platysternon megacephalum. Sci. Data 6:60. doi: 10.1038/s41597-019-0067-9
Chen, H. L., Ren, Z. J., Zhao, J. P., Zhang, C. D., and Yang, X. J. (2018). Y-chromosome polymorphisms of the domestic bactrian camel in China. J. Genet. 97, 3–10. doi: 10.1007/s12041-017-0852-1
Collingridge, G. L., Peineau, S., Howland, J. G., and Wang, Y. T. (2010). Long-term depression in the CNS. Nat. Rev. Neurosci. 11, 459–473. doi: 10.1038/nrn2867
Damgaard, R. B., and Gyrd-Hansen, M. (2011). Inhibitor of apoptosis (IAP) proteins in regulation of inflammation and innate immunity. Discov. Med. 11, 221–231.
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Davey, J. W., Hohenlohe, P. A., Etter, P. D., Boone, J. Q., Blaxter, M. L., and Catchen, J. M. (2011). Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 12, 499–510. doi: 10.1038/nrg3012
DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. doi: 10.1038/ng.806
Deveraux, Q. L., Takahashi, R., Salvesen, G. S., and Reed, J. C. (1997). X-linked IAP is a direct inhibitor of cell-death proteases. Nature 388, 300–304. doi: 10.1038/40901
Díaz-Arce, N., Arrizabalaga, H., Murua, H., Irigoien, X., and Rodríguez-Ezpeleta, N. (2016). RAD-seq derived genome-wide nuclear markers resolve the phylogeny of tunas. Mol. Phylogenet. Evol. 102, 202–207. doi: 10.1016/j.ympev.2016.06.002
Guo, C. Y. (2012). Association Analysis Of Genetic Diversity Of Jianghuai Soybean Breeding Germplasm and QTL of Breeding Target Traits. Master’s thesis, Nanjing Agriculture University, Nanjing, IL.
He, X. H., Han, X. L., and Ma, Y. H. (2009). Progress in the study of genetic diversity of bactrian camel. Acta Ecol. Anim. Domast. 30, 9–13.
Ji, R., Cui, P., Ding, F., Geng, J., and Meng, H. (2009). Monophyletic origin of domestic bactrian camel (Camelus bactrianus) and its evolutionary relationship with the extant wild camel (Camelus bactrianus ferus). Anim. Genet. 40, 377–382. doi: 10.1111/j.1365-2052.2008.01848.x
Kato, A. S., Gill, M. B., Ho, M. T., Yu, H., and Bredt, D. S. (2010). Hippocampal AMPA receptor gating controlled by both TARP and cornichon proteins. Neuron 68, 1082–1096. doi: 10.1016/j.neuron.2010.11.026
Kemp, A., and Manahan-Vaughan, D. (2007). Hippocampal long-term depression: master or minion in declarative memory processes? Trends Neurosci. 30, 111–118. doi: 10.1016/j.tins.2007.01.002
Kumar, R., Corbett, M. A., van Bon, B. W. M., Woenig, J. A., Douglas, E., Friend, K. L., et al. (2015). THOC2 mutations implicate mRNA-export pathway in X-linked intellectual disability. Am. J. Hum. Genet. 97, 302–310. doi: 10.1016/j.ajhg.2015.05.021
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Liu, C. M., Chen, H. L., Ren, Z. J., Zhang, C. D., and Yang, X. J. (2019). Population genetic analysis of the domestic bactrian camel in China by RAD-seq. Ecol. Evol. 9, 11232–11242. doi: 10.1002/ece3.5624
Liu, X., Li, C., Chen, M., Liu, B., Yan, X. J., Ning, J. H., et al. (2020). Draft genomes of two Atlantic bay scallop subspecies Argopecten irradians irradians and A. i. concentricus. Sci. Data 7:99. doi: 10.1038/s41597-020-0441-7
Mao, X., Cai, T., Olyarchuk, J. G., and Wei, L. (2005). Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21, 3787–3793. doi: 10.1093/bioinformatics/bti430
Meadow, R. H., and Zeder, M. A. (1978). Approaches to faunal analysis in the Middle East. Peabody Museum Bull. 2, 91–103.
Miller, M. R., Dunham, J. P., Amores, A., Cresko, W. A., and Johnson, E. A. (2007). Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 17, 240–248. doi: 10.1101/gr.5681207
Ming, L., Yi, L., He, J., Hai, L., and Ji, R. M. T. (2019). Identification of microsatellites and parentage testing development of bactrian camel (Camelus bactrianus). J. Camel. Pract. Res. 26, 133–142.
Ming, L., Yi, L., Sa, R., Wang, Z. X., Wang, Z., and Ji, R. M. T. (2017). Genetic diversity and phylogeographic structure of bactrian camels shown by mitochondrial sequence variations. Anim. Genet. 48, 217–220. doi: 10.1111/age.12511
Nei, M., and Li, W. H. (1979). Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. U.S.A. 76, 5269–5273. doi: 10.1073/pnas.76.10.5269
Pfeifer, B., Wittelsbürger, U., Ramos-Onsins, S. E., and Lercher, M. J. (2014). PopGenome: an efficient Swiss army knife for population genomic analyses in R. Mol. Biol. Evol. 31, 1929–1936. doi: 10.1093/molbev/msu136
Ren, A. Y., Du, K., Jia, X. B., Yang, R., Wang, J., Chen, S. Y., et al. (2019). Genetic diversity and population structure of four Chinese rabbit breeds. PLoS One 14:e0222503. doi: 10.1371/journal.pone.0222503
Sambrook, J., and Russell, D. W. (2002). Translated by Huang, PT 444 Molecular Cloning A Laboratory Manual. Beijing: Science Press.
Schraiber, J. G., and Akey, J. M. (2015). Methods and models for unravelling human evolutionary history. Nat. Rev. Genet. 16, 727–740. doi: 10.1038/nrg4005
Shibuya, Y., and Comin, M. (2019). Better quality score compression through sequence-based quality smoothing. BMC Bioinform. 20:302. doi: 10.1186/s12859-019-2883-5
Straesser, K., Masuda, S., Mason, P., Pfannstiel, J., Oppizzi, M., Rodriguez-Navarro, S., et al. (2002). TREX is a conserved complex coupling transcription with messenger RNA export. Nature 417, 304–308. doi: 10.1038/nature746
Tong, B. (2018). Study on camel transportation of the inner Mongolian Hui people on “silk tea camel road” in grassland. Tea in Fujian. 11:451.
Wang, J., Lv, X., Wu, Y., Xu, T., and Qiu, S. (2018). Postsynaptic RIM1 modulates synaptic function by facilitating membrane delivery of recycling NMDARs in hippocampal neurons. Nat. Commun. 9, 1–15. doi: 10.1038/s41467-018-04672-0
Yang, J., Lee, S. H., Goddard, M. E., and Visscher, P. M. (2011). GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82. doi: 10.1016/j.ajhg.2010.11.011
Ye, J., Fang, L., Zheng, H. K., Zhang, Y., Chen, J., Zhang, Z. J., et al. (2006). WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 34, 293–297. doi: 10.1093/nar/gkl031
Yuan, S. P., Wen, J., Zheng, R. C., and Qin, S. (2017). Progress in research on squamous cell carcinoma of the skin caused by ultraviolet radiation. J. Diagn. Ther. Dermato Venereol. 24, 423–426.
Zhang, B. D., Xue, D. X., Wang, J., Li, Y. L., Liu, B. J., and Liu, J. X. (2016). Development and preliminary evaluation of a genomewide single nucleotide polymorphisms resource generated by RAD-seq for the small yellow croaker (Larimichthys polyactis). Mol. Ecol. Resour. 16, 755–768. doi: 10.1111/1755-0998.12476
Keywords: RAD sequencing, Bactrian camel, single nucleotide polymorphisms, genomic resource, genetic analysis
Citation: Liu C, Chen H, Ren Z, Yang X and Zhang C (2020) Development of Genomic Resources and Identification of Genetic Diversity and Genetic Structure of the Domestic Bactrian Camel in China by RAD Sequencing. Front. Genet. 11:797. doi: 10.3389/fgene.2020.00797
Received: 23 December 2019; Accepted: 03 July 2020;
Published: 30 July 2020.
Edited by:
Guilherme J. M. Rosa, University of Wisconsin-Madison, United StatesReviewed by:
Derek M. Bickhart, United States Department of Agriculture (USDA), United StatesCopyright © 2020 Liu, Chen, Ren, Yang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhanjun Ren, UmVuemhhbmp1bkBud3N1YWYuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.