
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 30 June 2020
Sec. Genomics of Plants and the Phytoecosystem
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.00701
This article is part of the Research Topic Comparative Genomics and Functional Genomics Analyses in Plants View all 37 articles
 Ying Liang1†
Ying Liang1† Shanshan Chen2†
Shanshan Chen2† Kunhua Wei1†
Kunhua Wei1† Zijiang Yang3
Zijiang Yang3 Shengchang Duan4Yuan Du4Peng Qu1Jianhua Miao1*Wei Chen5,6,7*
Shengchang Duan4Yuan Du4Peng Qu1Jianhua Miao1*Wei Chen5,6,7* Yang Dong1,3,5,7*
Yang Dong1,3,5,7*Andrographis paniculata (Chinese name: Chuanxinlian) is an annual dicotyledonous medicinal plant widely grown in China and Southeast Asia. The dried plant has a highly acclaimed usage in the traditional Chinese medicine for its antipyretic, anti-inflammatory, and analgesic effects. In order to help delineate the biosynthetic pathways of various secondary metabolites, we report in this study a high-quality reference genome for A. paniculata. With the help of both PacBio single molecule real time sequencing and Illumina sequencing reads for error correction, the A. paniculata genome was assembled into a total size of 284 Mb with a contig N50 size of 5.14 Mb. The contigs were further assembled into 24 pseudo-chromosomes by the Hi-C technique. We also analyzed the gene families (e.g., KSL, and CYP450) whose protein products are essential for synthesizing bioactive compounds in A. paniculata. In conclusion, the high-quality A. paniculata genome assembly builds the foundation for decoding the biosynthetic pathways of various medicinal compounds.
During the course of human history, all civilizations have tried to explore various plants for medicinal purposes and they formed unique empirical knowledge about how these herbal plants could be used to treat various diseases. Even though much of this knowledge has gradually given way to modern medicine, WHO found that the popularity of herbal medicines increased in almost all parts of the world in recent years (Fitzsimons, 2013). For this reason, herbal plants not only raise the enthusiasm from the general public, but also remain a rich source for discovering novel drug candidates among the researchers.
With the emergence and development in high-throughput sequencing technology, the genome sequences of more than 200 plants have been reported (Lin et al., 2011). Particularly, genome sequencing is a powerful tool for studying various aspects of physiology and genetics in non-model plants, many of which are traditional herbal plants (Zerikly and Challis, 2009; De Luca et al., 2012; Chae et al., 2014). For example, the reference genome of Scutellaria baicalensis (Zerikly and Challis, 2009; De Luca et al., 2012; Chae et al., 2014; Zhao et al., 2019), Panax ginseng (Xu et al., 2017), mint (Vining et al., 2017), and opium poppy (Guo et al., 2018) provided insights into the genes involved in the biosynthesis of unique flavonoids, terpenes, alkaloids and many other secondary metabolites. Additionally, the reference genome of Salvia splendens was very valuable for helping marker-assisted breeding, genome editing, and molecular genetics (Dong et al., 2018). As one of the participants of the Herbal Plant Genomics Initiative, our team has reported the genomes of many Chinese herbal plants in past years, including Salvia miltiorrhiza Bunge (Zhang et al., 2015), Dendrobium officinale (Yan et al., 2015), maca (Zhang et al., 2016), Panax notoginseng (Chen et al., 2017), and fleabane (Yang et al., 2017). The high-quality genome assembly of Andrographis paniculata is presented in this manuscript as a continuum of the bigger research project.
Andrographis paniculata (Figure 1A) is a dicotyledonous medicinal plant widely distributed and used in tropical and subtropical regions of Asia, including India, China, Thailand, and Malaysia (Lim et al., 2012). This annual plant belongs to the family of Acanthaceae in the order of Lamiales. The dried plant has a highly acclaimed usage in the traditional Chinese medicine for its antipyretic, anti-inflammatory, and analgesic effects (Sun et al., 2019). Previous pharmacological research identified andrographolide and neoandrographolide as the main therapeutic constituents in A. paniculata (Srivastava and Akhila, 2010). Andrographolide is a labdane-related diterpenoid and it exhibits anti-cancer (Luo et al., 2014), anti-virus (Chen et al., 2009), antimicrobial and anti-inflammatory activities (Chua, 2014), suggesting potential pharmaceutical values. The leaves of A. paniculata contain major amounts of diterpene lactone compounds, including about 0.1% of deoxyandrographolide, and about 0.2% of neoandrographolide (Srivastava and Akhila, 2010). Even though the biosynthesis of andrographolide and neoandrographolide is achieved by the combination of various isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) (Chen et al., 2011), the complete profile of CYTOCHROME P450 genes (CYPs), COPALYL DIPHOSPHATE SYNTHASE genes (CPSs), and KAURENE SYNTHASE-LIKE PROTEIN genes (KSLs) has not been fully investigated in the A. paniculata genome.
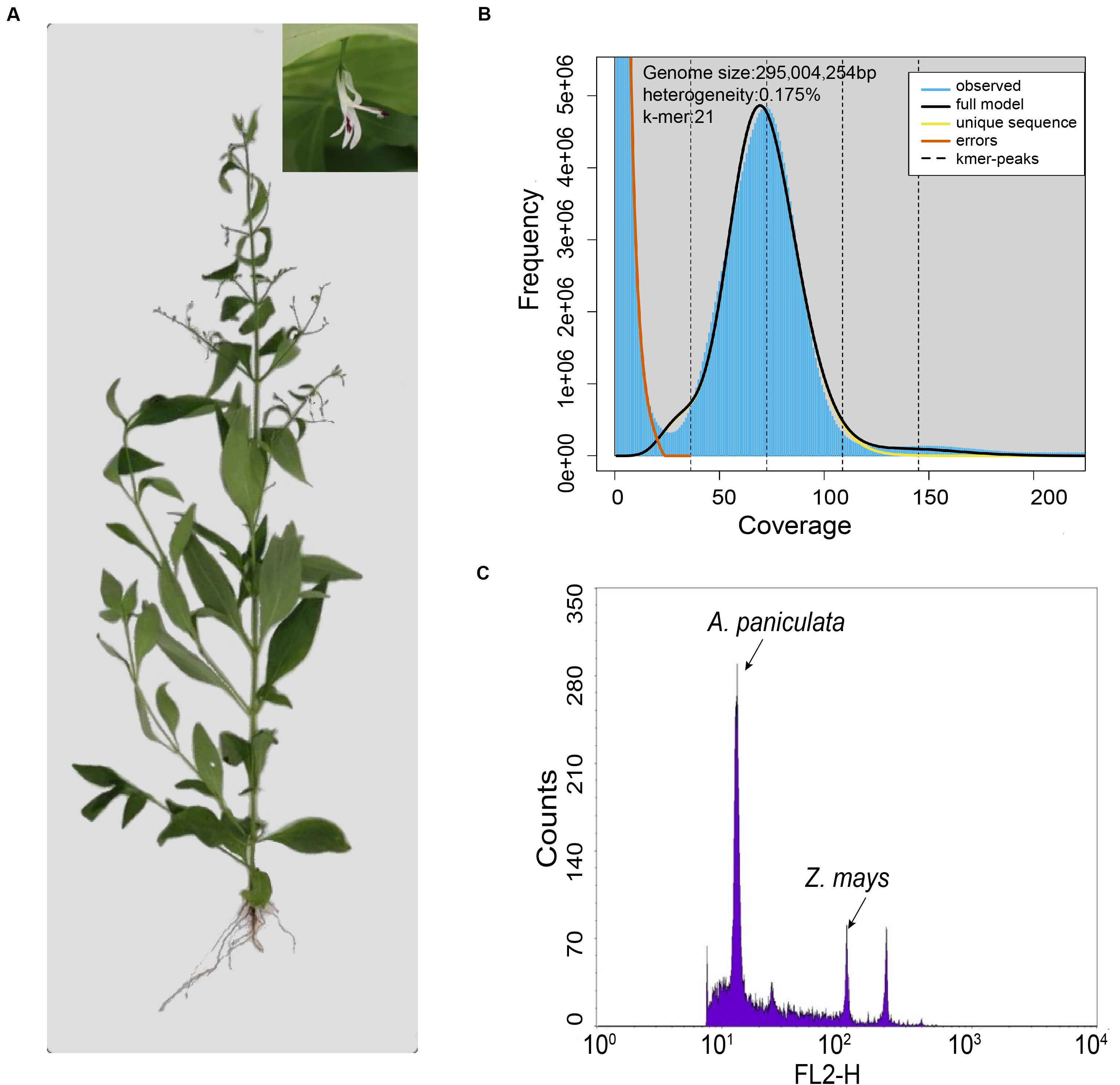
Figure 1. Evaluation of the genome size of A. paniculata. (A) Photo of a single A. paniculata plant with close-up image of the flower. (B) The 21-mer analysis of the A. paniculata genome using GenomeScope. (C) Flow cytometry analysis of A. paniculata genome size comparing with Zea mays.
The genome of A. paniculata is highly heterozygous and contains many repetitive sequences. These characteristics pose a big challenge in terms of acquiring a high-quality whole-genome reference assembly. A previous research effort reported an A. paniculata genome assembly of ∼269 Mb in total size with a contig N50 of 388 Kb (Sun et al., 2019). These benchmarks suggest the existence of relatively large un-assembled genome and gaps among contigs. Therefore, it is valuable to improve the genome assembly of A. paniculata using better raw data and assembly pipeline. Herein, we reported a reference genome of A. paniculata obtained from the PacBio single-molecule real time sequencing data in the size of 284 Mb. The contig N50 size was improved to 5.14 Mb, which is more than 12-fold longer than before. The resultant contigs were further assembled into 24 pseudo-chromosomes by the Hi-C technology, thereby yielding a high-quality A. paniculata genome assembly.
A single A. paniculata individual was obtained from Guangxi Medicinal Botanical Garden. Plant DNA was extracted from young leaves with the Novel Plant Genomic DNA Rapid Extraction Kit (Genenode Biotech, Beijing, China) according to the product manual.
A total of 15 μg A. paniculata genomic DNA was used to construct eleven PacBio libraries (mean size of 20 kb) with the SMRT Template Prep Kit (Pacific Biosciences, United States). These libraries were sequenced on a PacBio Sequel platform with recommended protocols from the manufacturer (Supplementary Table 1). Sequence reads with a quality score lower than 0.8 were removed.
In order to perform genome survey and genomic base correction, we also obtained Illumina reads for A. paniculata. In brief, 2 μg of genomic DNA was used to construct each library. The genomic DNA was sheared and insert sizes of 241, 313, 424, and 533 bp were selected for the four libraries. All libraries were sequenced on an Illumina HiSeq X Ten platform. The Illumina raw data were processed by fq_filter_V1.5 to remove the low-quality reads, and the parameters were set as follows: -q 33 -t 20 -ta 5 -tb 10 -tc 5 -td 10. We filtered out low-quality reads as specified by the following criteria: (1) filter a read if more than 5% of bases were N or poly-A, (2) filter a read if more than 30 bases were low quality, (3) if the read was contaminated with adaptor sequence, (4) if the size of a read was too small, and (5) if two copies of the paired-end reads had identical sequence (remove both copies). The resultant reads were then corrected by the SOAPec_v2.0.1 package with default settings.
The genome size of A. paniculata was assessed by both flow cytometry and k-mer analysis. For the flow cytometry approach, 20 mg of plant tissue was placed in 1.0 ml ice-cold nuclei isolation buffer in a Petri dish. The plant tissue was minced in the buffer with a new razor blade. The homogenate was filtered through a 42-mm nylon mesh into a labeled sample tube. Propidium iodide was added to a final concentration of 50 mg/ml simultaneously with RNase (50 mg/ml) and the sample was incubated on ice for 20 min before measurement. Zea mays (B73) was used as internal standard, the internal reference maize genome size is 2.3 Gb (Schnable et al., 2009).
For the k-mer analysis, Jellyfish v.2.2.5 (Marcais and Kingsford, 2011) was used to perform k-mer analysis on the Illumina sequencing error-corrected data. First, FastQC software was used to identify the quality of the input sequencing data for the Illumina sequencing data. The first 500,000,000 lines of the lane2 fastq file were extracted, and the extracted reads were used to identify heterozygosity with the ‘–m 21’ option. The graph was generated by GenomeScope (Vurture et al., 2017).
A total of 53.98 Gb of PacBio data were used in the de novo assembly of the A. paniculata genome according to an assembly pipeline named HERA (Du and Liang, 2019). In brief, CANU v1.8 (Koren et al., 2017) was used to correct the raw PacBio data and assemble contigs. The resultant contigs were further improved with HERA. Next, the pipeline used the assembled genome to create an index file using bwa-mem (Li and Durbin, 2010). The processed next-generation sequencing data were aligned to the reference genome. Samtools (Li et al., 2009) was used to sort the resulting bam files. Finally, the bam files were used to polish the contig twice with Pilon (Walker et al., 2014). In this study, a commercial service provider from the Institute of Genetics and Developmental Biology in Beijing was recruited to assemble the A. paniculata genome using a professional version of HERA (Du and Liang, 2019), which is said to run faster and have better performance resolving repetitive sequences.
Three gram of A. paniculata leaves were harvested and crosslinked in a 2% formaldehyde solution for 15min at room temperature. Crosslinking was quenched by adding glycine to a final concentration of 250 mM. The fixed plant tissue was then ground in liquid nitrogen and suspended in extraction buffer for nuclei isolation. After the nuclei were separated, chromatin was solubilized in 0.1% (m/v) SDS at 65°C for 10 min. After SDS was quenched by Triton X-100 (final concentration of 1%), solubilized chromatin was digested by 400 units of DpnII (New England Biolabs, MA, United States) at 37°C overnight. The following steps included biotin labeling of the DNA and blunt-end ligation of DNA fragments. After cross-linking was reversed by the treatment with proteinase K, DNA was purified so that biotin labels could be removed from non-ligated fragment ends. DNA fragments were sonicated into sizes of 400 bp so that paired-end libraries could be obtained. These libraries were sequenced on a NovaSeq 6000 platform (Illumina, United States) to acquire the Hi-C data.
Low-quality Hi-C reads were removed according to the following two criteria: (1) filter a read if more than 10% of bases were N, (2) filter a read if more than 50% bases were low quality (Q ≤ 5). Clean Hi-C reads were mapped to the draft assembly with Juicer (Juicer, juicer_tools.1.7.6_jcuda.0.8.jar) (Durand et al., 2016a). A candidate chromosome-length assembly was generated automatically using the 3d-DNA pipeline to correct misjoins, order, orientation, and then anchor contigs from the draft assembly (Dudchenko et al., 2017). Manual review and refinement of the candidate assembly was performed in Juicebox Assembly Tools (Version 1.9.1) (Durand et al., 2016b) for quality control and interactive correction. And then the genome was finalized using the “run-asm-pipeline-post-review.sh -s finalize –sort-output –bulid-gapped-map” in 3d-DNA with manually adjusted assembly as input (Dudchenko et al., 2017).
The repetitive sequences were identified via sequence alignment and de novo prediction. RepeatMasker (Chen, 2004) was used to compare the assembled genome with the RepBase database (Release16.10)1 using default settings (Bao et al., 2015). Repeatproteinmask searches2 were used for prediction of homologs using default settings. For de novo annotation of repetitive elements, LTR_finder (Xu and Wang, 2007)3, Piler (Edgar and Myers, 2005)4, RepeatScout (Price et al., 2005)5, and RepeatModeler6 were used to construct the de novo library, and annotation was carried out with Repeatmasker (cutf 100, cpu 100, run qsub, -nolow,-no, -norna,-parallel 1). Tandem repeats were identified across the genome with Tandem Repeats Finder (cutf 100, cpu 100, period_size 2000, run qsub Match 2 Mismatch 7 Delta 7 PM 80 PI 10 Minscore 50 MaxPeriod 2000).
According to their characteristics and redundancy, the repeat consensus sequences were first classified using TEsort (Zhang et al., 2019) with REXdb database7. For Copia and Gyspy superfamilies, complete elements were identified based on the presence and order of conserved domains including capsid protein, aspartic proteinase, integrase, reverse transcriptase and RNase H as described in Wicker (Wicker and Keller, 2007). We extracted all reverse transcriptase and multiple sequence alignment of the extracted RT were then conducted by MAFFT (Nakamura et al., 2018) and the phylogenetic tree was constructed with IQTREE (Jana et al., 2016). Itol8 was used for the visualization and edit of the tree. Finally, density of the TE consensus copies according to their lineages were computed along pseudomolecules and visualized using R.
GlimmerHMM (Aggarwal and Ramaswamy, 2002), SNAP (Korf, 2004), GenScan (Aggarwal and Ramaswamy, 2002), and Augustus (Stanke et al., 2006) were used for ab initio prediction of protein-coding genes with default settings. The homology-based prediction utilized reference protein sequence from Arabidopsis thaliana (Michael et al., 2018), Sesamum indicum (Wang et al., 2014), Solanum lycopersicum (Consortium, 2012), and Vitis vinifera (Girollet et al., 2019) according to an established protocol. RNA-seq data sets for A. paniculata leaf and root tissues were obtained from the National Center for Biotechnology Information (NCBI) database (SRX652837, SRX655521), and subsequently used for de novo assembly of the transcriptome. We aligned all RNA reads to the A. paniculata genome using TopHat (Trapnell et al., 2009), assembled the transcripts with Cufflinks (Trapnell et al., 2013) using default parameters, and predicted the open reading frames to obtain reliable transcripts with hidden Markov model (HMM)-based training parameters. Finally, the above 3 gene structure models were compiled by Evidence Modeler tool (Haas et al., 2008) with the following weights: transcripts-set > homology-set > ab initio-set and redundant genes were removed.
The t-RNAscan-SE tool (Chan and Lowe, 2019) was used to predict tRNA in the genome sequence with E-value set to 1e-5. Plant RNA sequences from Rfam database were selected as reference to predict the rRNA by BLASTN with E-value set to 1e-5. The miRNA and snRNA genes were also predicted by BLASTN against the Rfam database with E-value set to e-1.
Gene function was annotated by performing BLASTP (E-value ≤ 1e-5) against the protein databases. SwissProt9, TrEMBL (see footnote 9), KEGG10, and InterPro11 were used for screening the functional domains of the proteins. Gene Ontology (GO) terms for each gene were extracted from the corresponding InterPro entries.
Protein sequences of A. paniculata, Sesamum indicum, Salvia miltiorrhiza, Oryza sativa, Catharanthus roseus, Arabidopsis thaliana, Solanum lycopersicum, and Helianthus annuus were downloaded from JGI. A full alignment protein search using BLASTP with the parameter E-value = 1e-5 was performed to verify the gene family clusters in these species and A. paniculata. Ortholog clustering and gene family clustering analyses were performed using OrthoMCL (Li et al., 2003). Venn diagram format was drawn using a web tool12 (Zhang et al., 2016).
An all-against-all BLASTP comparison with a cutoff E-value of 1e-5 was preformed, and the results were clustered into groups of homologous proteins using Markov chain clustering with the default inflation parameter. All 1212 single-copy orthologous genes identified in the gene family cluster analysis from the aforementioned species were used to construct a phylogenetic tree. Multiple sequence alignments were performed for each gene using MUSCLE v.3.713 with default settings (Edgar, 2004).
The MCMCTREE program within the PAML package (Yang, 2007) was used to estimate divergence time of A. paniculata, S. indicum, S. miltiorrhiza, O. sativa, C. s roseus, A. thaliana, S. lycopersicum, and H. annuus. The HKY85 model (model = 4) and independent rates molecular clock (clock = 2) were used for calculation.
CAFE v1.7 (De Bie et al., 2006) is a tool for analyzing the evolution of gene family size based on the stochastic birth and death model. With the calculated phylogeny and the divergence time, this software was applied to identify gene families that had undergone expansion and/or contraction in the aforementioned species with the parameters: -filter -cpu 10 -lrt -simunum 1000.
The fasta and hic.gff files of the published genome assembly (Sun et al., 2019) were downloaded from NCBI. They were combined with the fasta and contig.gff files of our genome assembly by makeblastdb. BLASTP was used to align these sequence, and MCScanX (Wang et al., 2012) was used to perform synteny analysis between two genome assemblies.
We used hmmsearch to perform a preliminary screening of the gene family (CYP450 and terpene sythases, TPSs) in A. paniculata and the gene ID was intercepted with an E-value ≤ 1e-59. The corresponding protein sequence was used as a query for TBLASTN (E = 1e-5) with both versions of the assembled A. paniculata genome sequence. The CYP450 genes from other species were downloaded from the Cytochrome CYP450 homepage14 (Nelson, 2009) and the TPS genes from other species were acquired from a previous publication (Chen et al., 2011). Multiple sequence alignment was carried out with MUSCLE v3.7 (Edgar, 2004) using default parameters. The maximum likelihood (ML) phylogenetic tree was constructed using MEGA7 (Nelson, 2009) with 1,000 bootstraps.
The RNA-Seq data of the roots and leaves of A. paniculata were downloaded from the NCBI database (SRX652837, SRX655521). FPKM value was calculated for each protein-coding gene by Cufflinks (v. 2.1.1) (Trapnell et al., 2013). The heatmap was made with the pheatmap package.
Genome survey with Illumina reads showed that the estimated genome size of A. paniculata with 21-mer analysis was about 295 Mb (Figure 1B). This number was slightly smaller than the estimated genome size of 310 Mb from flow cytometry analysis (Figure 1C), but larger than the previous estimate of 280 Mb (Sun et al., 2019). The difference probably reflects the between-individual variation of the A. paniculata plant. Moreover, the heterozygosity of this sequenced genome was estimated to be 0.175% (Figure 1B).
In order to obtain a high-quality A. paniculata genome assembly, we constructed 11 PacBio SMRT sequencing libraries, which produced 53.96 Gb clean data (Supplementary Table 1). They covered about 183-fold of the estimated genome. We also generated about 238.6 Gb of Illumina sequencing data to polish and correct the error reads that are associated with PacBio sequencing. A combination of these data through a assembly pipeline (see the section on genome assembly methods for details) yielded a draft genome assembly of ∼284.3 Mb with 270 contigs (Table 1). This assembly represented about 91.6 – 96.3% of the estimated genome size. The longest contig was 9.30 Mb in size and the contig N50 was about 5.15 Mb. This benchmark is more than 12-fold longer than that of a previously reported assembly (Sun et al., 2019).
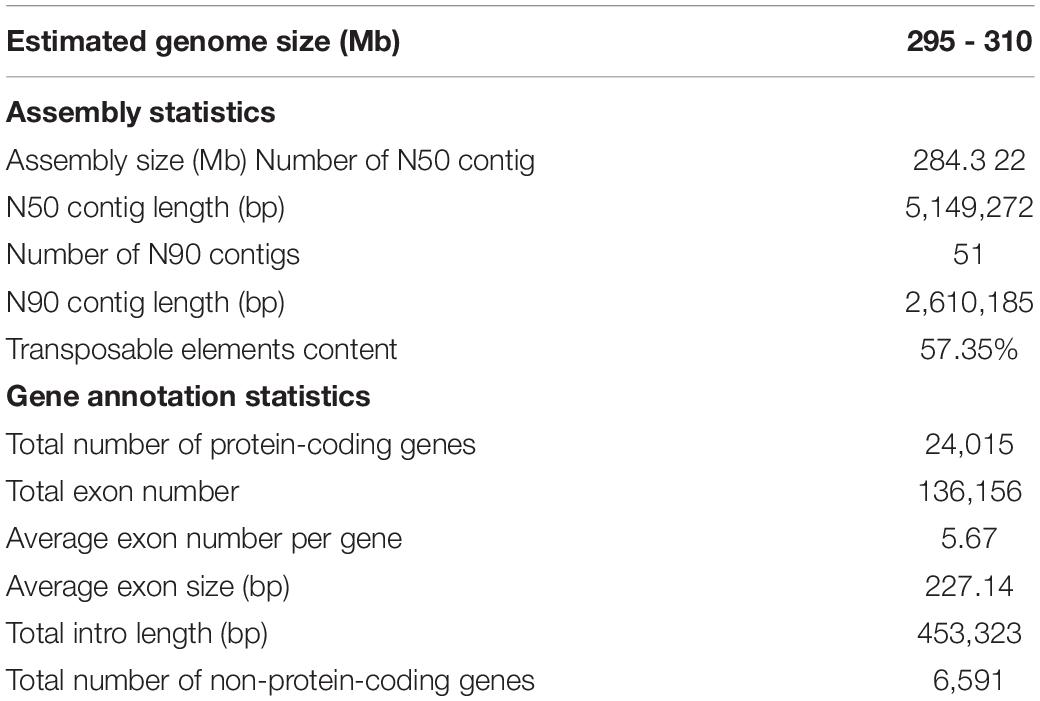
Table 1. The library information and data statistics for the A. paniculata assembly.
We assessed the completeness of the genome assembly of A. paniculata by using the Benchmarking Universal Single-Copy Orthologs (BUSCO) approach (Simao et al., 2015). The result showed that 91.0% of the plant BUSCO genes could be recovered in the genome assembly and 3% of the plant BUSCO genes had partial matches (Supplementary Table 2). We also mapped cleaned Illumina paired reads back to the genome assembly using BWA mem (Li and Durbin, 2010). We found that 97% of the reads could be mapped to the genome assembly and 94% of the reads were found to be properly paired. The high mapping rate indicate high completeness of the assembly. These two benchmarks were also higher than those reported in the previously assembly (Sun et al., 2019). In addition, our contig assembly shared high collinearity with its counterpart from a previous report (Figure 2). These data collectively suggest that the genome assembly of A. paniculata in this study has high quality and could be used for subsequent analyses.
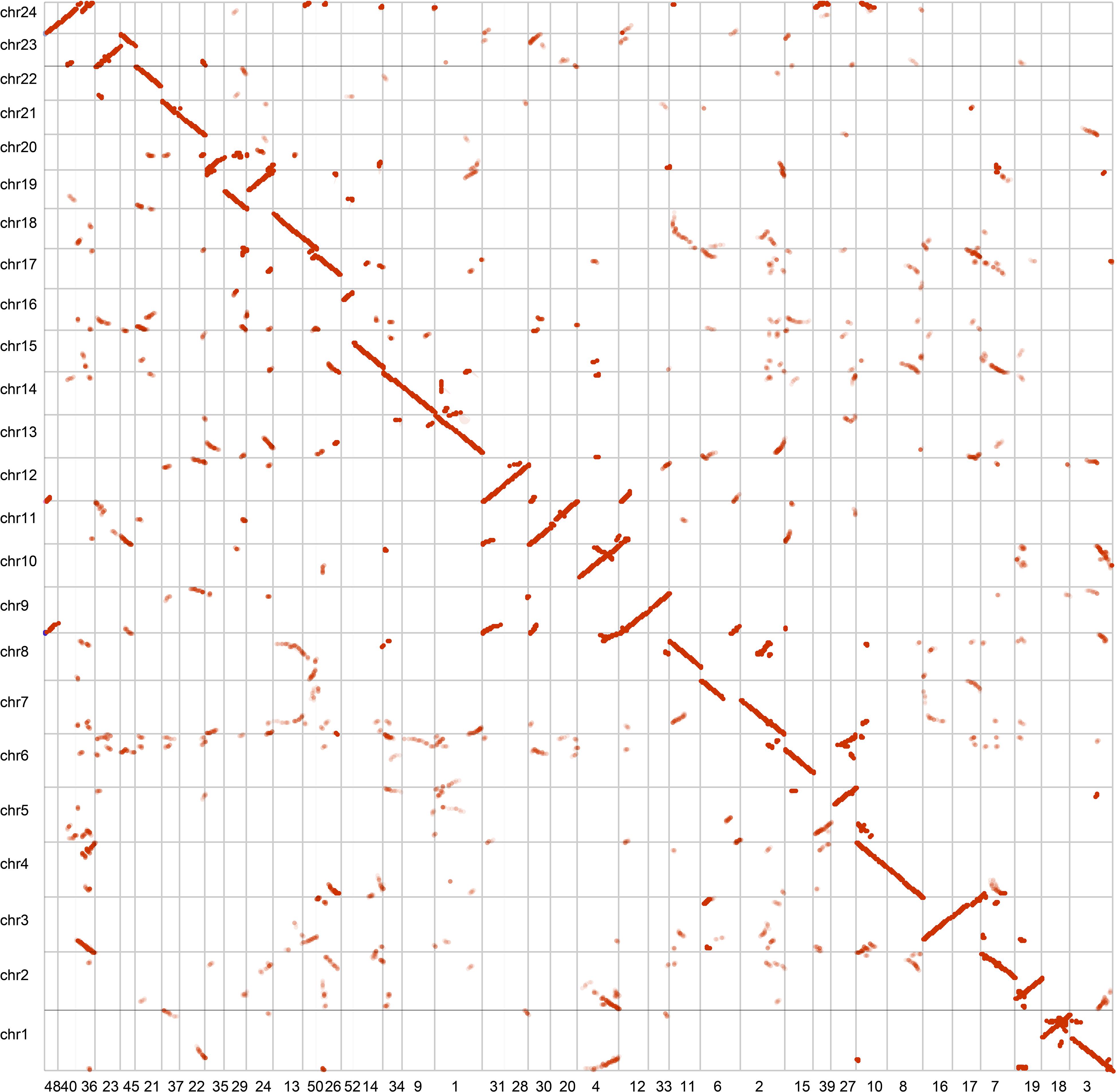
Figure 2. Collinear analysis of the assemblies of A. paniculata genome.
Finally, we obtained about 43.33 Gb (∼152 × coverage) clean Hi-C sequencing data, with which 246,855,874 bp (86.8% of all bases) of the genome assembly were organized into 24 pseudo-chromosomes (Figure 3 and Supplementary Table 3). As expected, the Hi-C interaction decreases as the physical distance between two sequences increases.

Figure 3. Hi-C clustering heatmap for pseudomolecule construction in A. paniculata. In all, 24 pseudomolecules are assembled and indicated by blue boxes.
Transposable elements (TEs) accounted for about 163.1 Mb or 57.35% of the A. paniculata genome (Supplementary Table 4). Breakdown of the TE statistics showed that DNA retrotransposons and long terminal repeat (LTR) retrotransposons were major subtypes in the A. paniculata genome (Supplementary Table 5). TEs constitute an import part in plant genomes. We analyzed the evolutionary history of various Ty3-gypsies and Ty1-copia retroposon elements in the A. paniculata genome and identified unique dynamics of invasion patterns for different TE lineages (Figures 4A,B). For example, Ivana, Ogre, and Ale elements are all relatively young, suggesting an intense and recent burst of insertion or a strong selection against these TE elements. In contrast, Angela elements are the most ancient ones and the bimodal distribution suggests that the burst of insertion occurred twice in the evolutionary history of A. paniculata (Figure 4B). In addition, TE family distribution varied across the genome (Figure 4C). Ogre elements tend to cluster closely, thereby yielding prominent hotspot regions in the genome of A. paniculata.
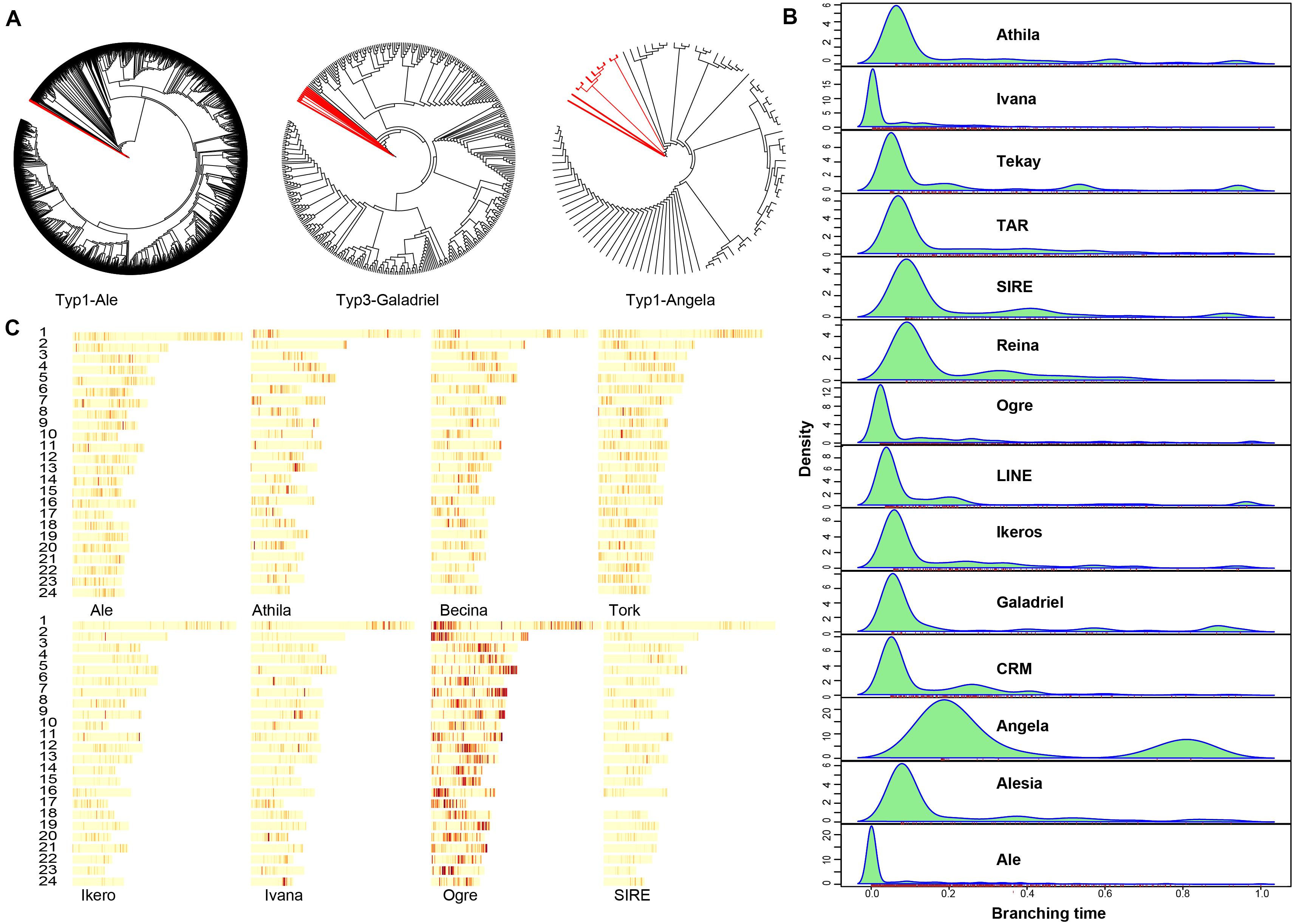
Figure 4. Repeat analysis (A) Neighbor-joining (NJ) trees were built from RT domain sequence similarities among different lineage-specific copies identified in the A. paniculata genome. Deep branching revealed ancient expansion while flat branching is consistent with a recent burst of insertion activity. Red branches correspond to outgroup sequences. (B) The average age of TEs was revealed for the different lineages by the branching distribution in the NJ trees built from RT (light blue). (C) The density of different TE lineages inferred from the detection of their protein-coding domains along pseudomolecules.
A combination of ab initio based, homology based, and RNA-Seq based methods were used to predict 24,015 protein-coding genes in the A. paniculata genome (Supplementary Table 6). The predicted mRNA was on average 3,175 bp in length, containing about 5.67 exons with an average CDS length of 1,287 bp. The number of predicted protein-coding genes was comparable to that of S. indicum, but much smaller than that of O. sativa and H. annuus (Figure 5A). Orthologous clustering analysis showed that the A. paniculata genome contained 4,449 single-copy orthologs, 6,731 multiple-copy orthologs, 1,234 unique paralogs, 7,165 other orthologs, and 4,436 unclustered genes (Supplementary Table 7). In addition, Venn diagram showed that 7,798 gene families were shared by A. paniculata, S. indicum, S. miltiorrhiza, and O. sativa. A total of 525 gene families were unique to A. paniculata. This number was lower than that of the other three plant species (Figure 5B). Among the 24,015 protein-coding genes, a total of 19,824 predicted genes are supported by the RNA-seq expression data (FPKM > 0.05). Functional annotation of predicted protein-coding genes showed that 91.5% could obtain TrEMBL annotation; 62.2% could obtain GO annotation; 77.2% could obtain KEGG annotation; and 81.0% could obtain InterPro annotation (Supplementary Table 8).
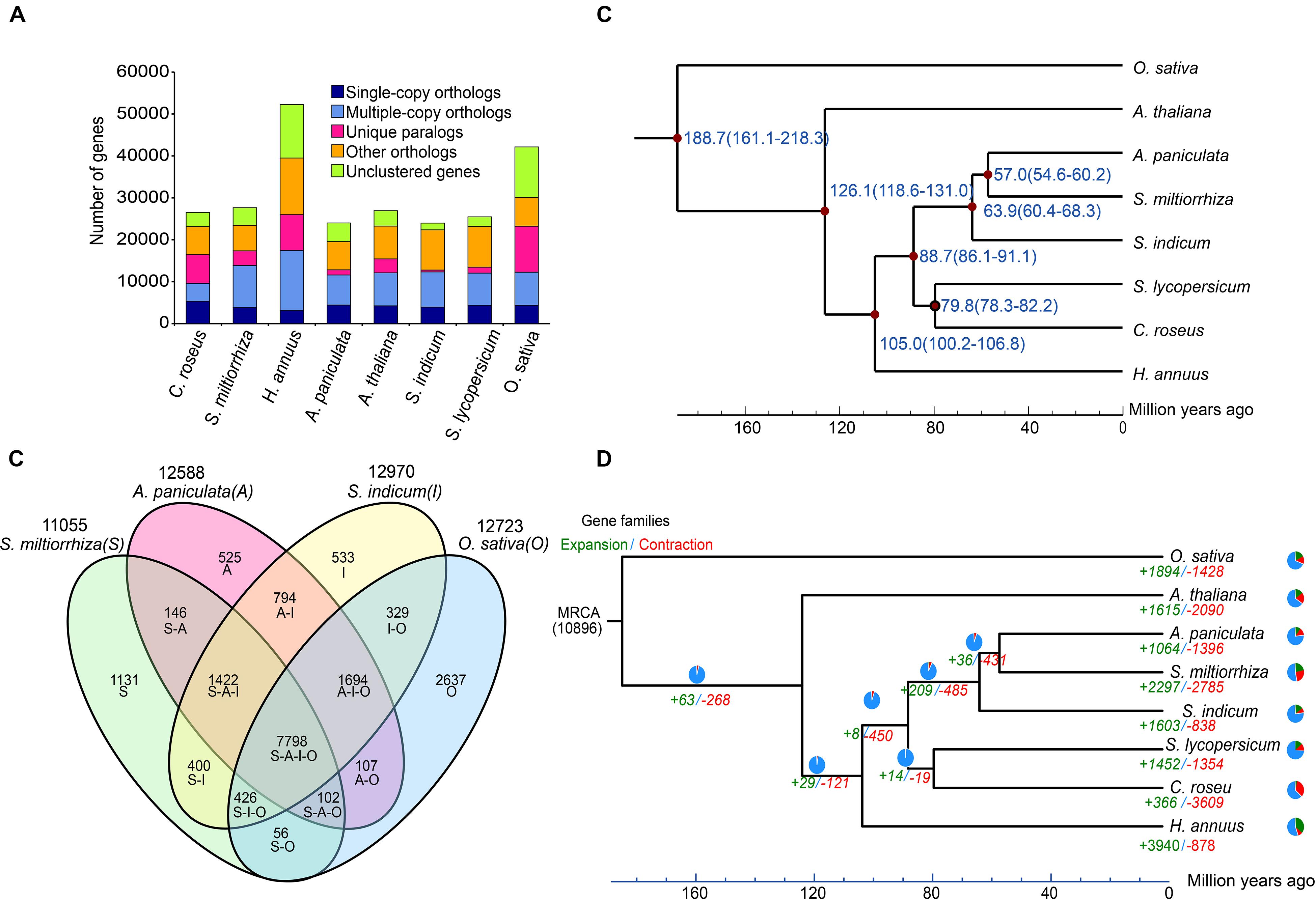
Figure 5. Comparative analyses of the A. paniculata genome. (A) Major groups of orthologous genes in eight plant genomes. (B) Venn diagram of shared orthologous gene families among A. paniculata, S. indicum, S. miltiorrhiza, and O. sativa genomes. (C) Estimation of the time points of divergence (time range shown in parentheses) between A. paniculata and seven other plant species based on orthologous single-copy gene pairs. (D) Expansion and contraction of gene families in eight plant genomes. Pie diagram on each branch and node corresponds to combined change across lineages.
A combination of homolog and ab initio based methods identified a total of 6,591 non-protein-coding genes (Supplementary Table 9). These predicted genes comprised of 93 microRNA genes, 524 transfer RNA (tRNA) genes, 5,785 ribosomal RNA (rRNA) genes, and 189 small nuclear RNA genes (Supplementary Table 9).
Phylogenetic analysis A. paniculata and seven other plant species showed that A. paniculata shared a common ancestor with S. miltiorrhiza approximately 57.0 Myr ago by calculated by r8s (Figure 5C). This estimate corresponds to the report from a previous study (Sun et al., 2019). During the course of evolution, a total of 1,064 and 1,396 gene families in A. paniculata were found to undergo expansion and contraction, respectively (Figure 5D).
Terpenoids are the largest group of plant secondary metabolites that are key targets for pharmaceutical screening and design (Srivastava and Akhila, 2010). Despite the structural diversity, these compounds share a common biosynthetic pathway (Yazaki et al., 2017). Terpenoids are derived from two five-carbon chemicals: IPP and dimethylallyl diphosphate (DMAPP) (Lange et al., 2001). Their biosynthesis involve the classical acetate/mevalonate pathway in the cytosol and the pyruvate/glyceraldehyde-3-phosphate pathway in the plastids (Figure 6A) (Chen et al., 2011). Eventually, the condensation of IPP and DMAPP in various combinations will give rise to the countless terpenoids in plants (Srivastava and Akhila, 2010). In the A. paniculata genome, according to the KEGG annotation results, we identified 41 putative genes that were involved in the terpenoid backbone biosynthesis (Supplementary Table 10). This number is larger than that found in the Panax notoginseng genome (Chen et al., 2017). Additionally, almost all these putative genes exhibited certain levels of expression in the leaf and root tissue of A. paniculata. A previous report showed that the GERANYLGERANYL PYROPHOSPHATE SYNTHASE (GGPPS) group of genes in A. paniculata might be involved in the biosynthesis of andrographolide (Wang et al., 2019). In our genome assembly, a total number of 13 putative GGPPS genes were identified, and 11 out of 13 putative genes were expressed (Supplementary Table 10).
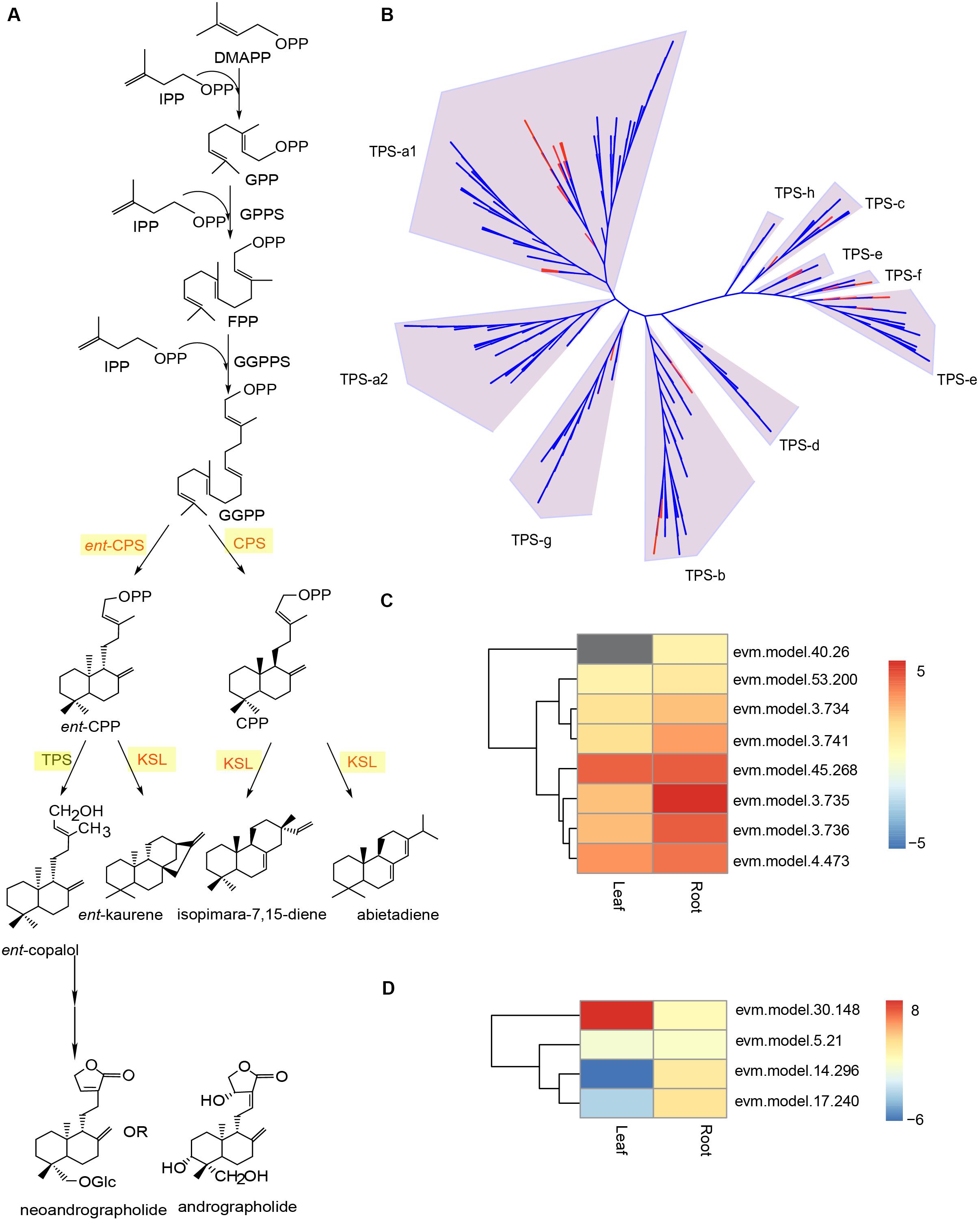
Figure 6. Genomic analysis of putative TPS genes in the A. paniculata genome. (A) Schematic biosynthetic pathway of andrographolide and neoandrographolide. (B) Phylogenetic analysis of TPS genes in A. paniculata and other plants. Red branches indicate putative TPS genes in A. paniculata. (C) Expression levels of putative KS/KSL genes in the leaf and root tissue of A. paniculata. (D) Expression levels of putative CPS genes in the leaf and root tissue of A. paniculata.
Besides GGPPS genes, previous studies suggest that COPALYL DIPHOSPHATE SYNTHASE (CPS) genes are implicated in the biosynthetic pathway of andrographolide and neoandrographolide (Garg et al., 2015; Shen et al., 2016a, b). The CPS enzymes are very similar in protein sequence to kaurene synthase (KS) and kaurene synthase-like (KSL) proteins (Zi et al., 2014). Despite their distinct catalytic activity, CPSs and KSLs belong to the c-type and e-type subfamilies of the terpene synthase (TPS) enzymes, respectively (Chen et al., 2011). In the A. paniculata genome, we identified a total of 53 putative TPS genes (Supplementary Table 11). Phylogenetic analysis of the TPS protein sequences from A. paniculata and eight other species showed that there were 24 putative TPS-a1 genes, 10 putative TPS-b genes, 4 putative TPS-c genes, 8 putative TPS-e genes, 5 putative TPS-f genes, and 2 putative TPS-g genes (Figure 6B). This suggest that the A. paniculata genome may have four CPS genes and eight KS/KSL genes, all of which show certain levels of expression in the leaf and root tissues of the plant (Figures 6C,D).
Various biosynthetic pathways in plants rely on members of the CYTOCHROME P450 (CYP450) gene family to accomplish chemical modification (Schuler, 1996; Mizutani and Ohta, 1998; Morant et al., 2003). For this reason, we investigated the putative CYP450 genes in A. paniculata, and found a total of 205 candidates (Supplementary Table 12). The phylogenetic tree of the putative CYP450 protein sequences exhibited nine major clans (Figure 7A). The majority of candidate genes belonged to the clan 71 (104 genes), clan 72 (33 genes), clan 85 (33 genes), and clan 86 (25 genes), respectively. We also found that some CYP450 genes appeared in a cluster in the contigs of the assembly (Figure 7B). This result agrees with the findings that it is fairly common to have gene clusters for specific biosynthetic pathways in plant genomes (Nutzmann and Osbourn, 2014). Among the 205 candidate genes, we identified 111 putative CYP450 genes that were differentially expressed in the root and leaf of A. paniculata (Figure 7C). Additionally, the expression level of evm.49.509 in the CYP450 family was the highest in the root of A. paniculata, followed by evm.model.49.208, evm.model.7.226, and evm.model.54.219. In comparison, evm.model.19.136 was the highest expressed gene in the leaves of A. paniculata, followed by evm.model.54.197 and evm.model.11.4.
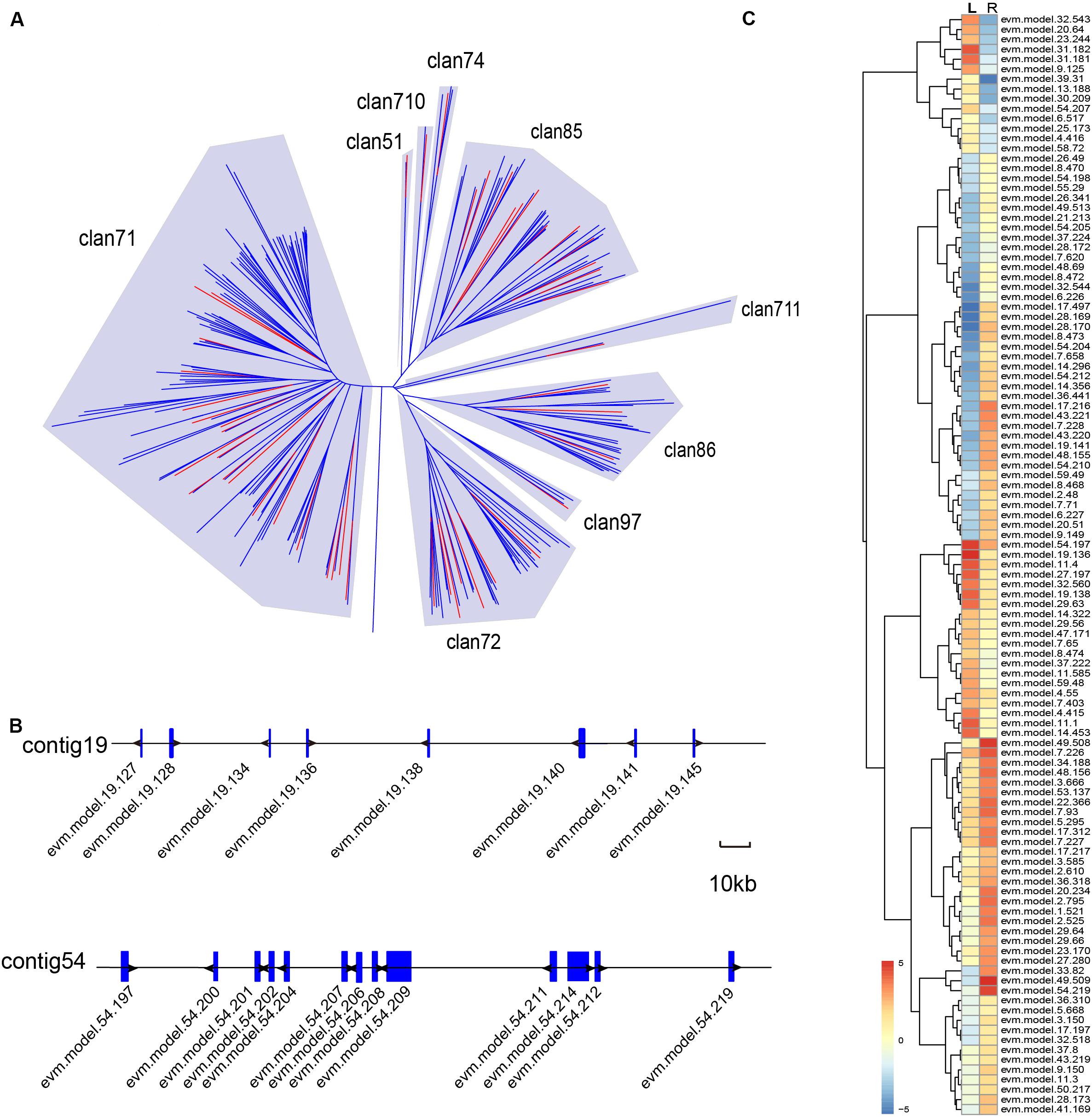
Figure 7. Genomic analysis of putative CYP450 genes in the A. paniculata genome. (A) Phylogenetic analysis of CYP450 genes in A. paniculata and other plants. Red branches indicate putative CYP450 genes in A. paniculata. (B) The CYP450 gene clusters on the contigs. (C) Differentially expressed putative CYP450 genes in A. paniculata.
With the advancement and cost reduction of genome sequencing technologies, more and more plant species have revealed their near-complete genetic composition at a single-base resolution. Because most medicinal plant have highly repetitive and/or heterozygous genomes, a high-quality draft assembly is usually difficult and costly to secure. Particularly, lower contiguity of the genome assembly at the contig level will impede genomic analysis of functional genes, delineation of biosynthetic pathways, and the development of novel pharmaceutical candidate.
We reported a chromosome-level reference genome for A. paniculata with improved benchmark values. Our updated A. paniculata genome was 284 Mb with a contig N50 size of 5.14 M. This number was more than 12-fold longer than that of the previous report (Sun et al., 2019). The BUSCO value reached 91.7%, which demonstrated the high completeness of the genome assembly.
Repetitive elements account for a large percentage of plant genomes. For example, the genome of lettuce contains about 74.2% of the sequence as repeats and the genome of sunflower includes more than 75% LTRs (Badouin et al., 2017; Reyes-Chin-Wo et al., 2017). We analyzed the repetitive element sequences in the A. paniculata genome and found that the repeats of the A. paniculata genome is as high as 57%, among which long terminal repeat retrotransposons (LTR-RTs) are predominant. Phylogenetic analyses of various copia and gypsies RT subclasses showed that the burst of invasion of many RT were recent events except Angela elements. This is in contrast to the pea genome where Angela elements drives the invasion fairly recently (Kreplak et al., 2019). Given these results, it is worth noting that TE annotation remains a challenging task for plant genomes. For instance, about 21.8% of de novo identified TEs in the A. paniculata genome could not be assigned to a particular category. Moreover, TEs tend to insert into the structures of existing TE elements, creating nested TEs in the genome. With our data, it would be interesting to use newly developed pipelines (e.g., Extensitve de novo TE Annotator) (Ou et al., 2019) to deconvolute nested TEs in the genome of A. paniculata.
In the present study, we analyzed the Cyp450 gene family in A. paniculata, and identified 205 putative CYP450 genes with conserved motifs. The results showed that all major classes of Cyp450 reported by the Nutzmann and Osbourn (2014) could be found in the A. paniculata genome. The number of Cyp450 genes with high expressions is larger in roots than in leaf. Furthermore, terpene synthesis (TPSs) is one of the main drivers of terpene diversification. The terpene synthase (TPS) family genes are generally categorized into seven clades (Shen et al., 2018). In the A. paniculata genome, we identified a total of 53 putative TPS genes, most of which belong to the TPS-a and TPS-b subfamilies. This is in line with the fact that TPS-a and TPS-b subfamilies represent the angiosperm-specific genes that have diverged from the other TPS genes (Shen et al., 2018).
The high-quality genome of A. paniculata will not only lay out the foundation for investigating the genetic basis for secondary metabolite biosynthesis, but also serve as an important resource for the study of other plant species in the Andrographis genus.
All raw sequence reads and the genome assembly have been deposited at NCBI under the BioProject accession number PRJNA549104.
YL, KW, and PQ collected the samples and performed the experiments. SC, ZY, YDu, and SD completed the data analysis. YDo, WC, and JM edited and modified the manuscript. All authors read and approved the manuscript.
This study was supported by the National Key R&D Program of China (2019YFC1711100), Yunnan Provincial Key Programs of Yunnan Eco-friendly Food International Cooperation Research Center Project (2019ZG00908), the Guangxi Innovation-Driven Development Project (GuiKe AA18242040), National Natural Science Foundation of China (81460582 and 81473309), China Agriculture Research System (CARS-21), and Guangxi Science and Technology Project (GuiKe AD17129044).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00701/full#supplementary-material
BUSCO, Benchmarking Universal Single-Copy Orthologs; CPS, copalyl diphosphate synthase; CYP, cytochrome P450; DMAPP, dimethylallyl diphosphate; GGPPS, geranylgeranyl pyrophosphate synthase; GO, gene ontology; IPP, isopentenyl diphosphate; KSL, kaurene synthase-like protein; TE, transposable element.
Aggarwal, G., and Ramaswamy, R. (2002). Ab initio gene identification: prokaryote genome annotation with GeneScan and GLIMMER. J. Biosci. 27, 7–14. doi: 10.1007/BF02703679
Badouin, H., Gouzy, J., Grassa, C. J., Murat, F., Staton, S. E., Cottret, L., et al. (2017). The sunflower genome provides insights into oil metabolism, flowering and asterid evolution. Nature 546, 148–152. doi: 10.1038/nature22380
Bao, W., Kojima, K. K., and Kohany, O. (2015). Repbase update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6:11. doi: 10.1186/s13100-015-0041-9
Chae, L., Kim, T., Nilo-Poyanco, R., and Rhee, S. Y. (2014). Genomic signatures of specialized metabolism in plants. Science 344, 510–513. doi: 10.1126/science.1252076
Chan, P. P., and Lowe, T. M. (2019). TRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol. Biol. 1962, 1–14. doi: 10.1007/978-1-4939-9173-0_1
Chen, F., Tholl, D., Bohlmann, J., and Pichersky, E. (2011). The family of terpene synthases in plants: a mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 66, 212–229. doi: 10.1111/j.1365-313X.2011.04520.x
Chen, J. X., Xue, H. J., Ye, W. C., Fang, B. H., Liu, Y. H., Yuan, S.-H., et al. (2009). Activity of andrographolide and its derivatives against influenza virus in vivo and in vitro. Biol. Pharm. Bull. 32, 1385–1391. doi: 10.1248/bpb.32.1385
Chen, N. (2004). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 25, 4.10.1–4.10.14. doi: 10.1002/0471250953.bi0410s05
Chen, W., Kui, L., Zhang, G., Zhu, S., Zhang, J., Wang, X., et al. (2017). Whole-genome sequencing and analysis of the Chinese herbal plant Panax notoginseng. Mol. Plant 10, 899–902. doi: 10.1016/j.molp.2017.02.010
Chua, L. S. (2014). Review on liver inflammation and antiinflammatory activity of Andrographis paniculata for hepatoprotection. Phytother. Res. 28, 1589–1598. doi: 10.1002/ptr.5193
Consortium, T. G. (2012). The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641. doi: 10.1038/nature11119
De Bie, T., Cristianini, N., Demuth, J. P., and Hahn, M. W. (2006). CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271. doi: 10.1093/bioinformatics/btl097
De Luca, V., Salim, V., Atsumi, S. M., and Yu, F. (2012). Mining the biodiversity of plants: a revolution in the making. Science 336, 1658–1661. doi: 10.1126/science.1217410
Dong, A. X., Xin, H. B., Li, Z. J., Liu, H., Sun, Y. Q., Nie, S., et al. (2018). High-quality assembly of the reference genome for scarlet sage, Salvia splendens, an economically important ornamental plant. Gigascience 7:giy068. doi: 10.1093/gigascience/giy068
Du, H., and Liang, C. (2019). Assembly of chromosome-scale contigs by efficiently resolving repetitive sequences with long reads. Nat. Commun. 10:5360. doi: 10.1038/s41467-019-13355-3
Dudchenko, O., Batra, S. S., Omer, A. D., Nyquist, S. K., Hoeger, M., Durand, N. C., et al. (2017). De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95. doi: 10.1126/science.aal3327
Durand, N. C., Robinson, J. T., Shamim, M. S., Machol, I., Mesirov, J. P., Lander, E. S., et al. (2016a). Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101. doi: 10.1016/j.cels.2015.07.012
Durand, N. C., Shamim, M. S., Machol, I., Rao, S. S., Huntley, M. H., Lander, E. S., et al. (2016b). Juicer provides a one-click system for analyzing loop-resolution hi-C experiments. Cell Syst. 3, 95–98. doi: 10.1016/j.cels.2016.07.002
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Edgar, R. C., and Myers, E. W. (2005). PILER: identification and classification of genomic repeats. Bioinformatics 21, (Suppl. 1), i152–i158. doi: 10.1093/bioinformatics/bti1003
Garg, A., Agrawal, L., Misra, R. C., Sharma, S., and Ghosh, S. (2015). Andrographis paniculata transcriptome provides molecular insights into tissue-specific accumulation of medicinal diterpenes. BMC Genomics 16:659. doi: 10.1186/s12864-015-1864-y
Girollet, N., Rubio, B., and Bert, P. F. (2019). De novo phased assembly of the Vitis riparia grape genome. Sci. Data 6:127. doi: 10.1038/s41597-019-0133-3
Guo, L., Winzer, T., Yang, X., Li, Y., Ning, Z., He, Z., et al. (2018). The opium poppy genome and morphinan production. Science 362, 343–347. doi: 10.1126/science.aat4096
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9:R7. doi: 10.1186/gb-2008-9-1-r7
Jana, T., Lam-Tung, N., Arndt, V. H., and Quang, M. B. (2016). W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235. doi: 10.1093/nar/gkw256
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., Phillippy, A. M., et al. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Korf, I. (2004). Gene finding in novel genomes. BMC Bioinformatics 5:59. doi: 10.1186/1471-2105-5-59
Kreplak, J., Madoui, M., Cápal, P., Novák, P., and Burstin, J. (2019). A reference genome for pea provides insight into legume genome evolution. Nat. Genet. 51, 1411–1422. doi: 10.1038/s41588-019-0480-1
Lange, B. M., Ketchum, R. E., and Croteau, R. B. (2001). Isoprenoid biosynthesis. Metabolite profiling of peppermint oil gland secretory cells and application to herbicide target analysis. Plant Physiol. 127, 305–314. doi: 10.1104/pp.127.1.305
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Lim, J. C., Chan, T. K., Ng, D. S., Sagineedu, S. R., Stanslas, J., Fred Wong, W. S., et al. (2012). Andrographolide and its analogues: versatile bioactive molecules for combating inflammation and cancer. Clin. Exp. Pharmacol. Physiol. 39, 300–310. doi: 10.1111/j.1440-1681.2011.05633.x
Lin, Y., Li, J., Shen, H., Zhang, L., Papasian, C. J., Deng, H. W., et al. (2011). Comparative studies of de novo assembly tools for next-generation sequencing technologies. Bioinformatics 27, 2031–2037. doi: 10.1093/bioinformatics/btr319
Luo, X., Luo, W., Lin, C., Zhang, L., and Li, Y. (2014). Andrographolide inhibits proliferation of human lung cancer cells and the related mechanisms. Int. J. Clin. Exp. Med. 7, 4220–4225.
Marcais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Michael, T. P., Jupe, F., Bemm, F., Motley, S. T., Sandoval, J. P., Lanz, C., et al. (2018). High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nat. Commun. 9:541. doi: 10.1038/s41467-018-03016-2
Mizutani, M., and Ohta, D. (1998). Two isoforms of NADPH:cytochrome P450 reductase in Arabidopsis thaliana. Gene structure, heterologous expression in insect cells, and differential regulation. Plant Physiol. 116, 357–367. doi: 10.1104/pp.116.1.357
Morant, M., Bak, S., Moller, B. L., and Werck-Reichhart, D. (2003). Plant cytochromes P450: tools for pharmacology, plant protection and phytoremediation. Curr. Opin. Biotechnol. 14, 151–162. doi: 10.1016/s0958-1669(03)00024-7
Nakamura, T., Yamada, K. D., Tomii, K., Katoh, K., and Hancock, J. (2018). Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 34, 2490–2492. doi: 10.1093/bioinformatics/bty121
Nutzmann, H. W., and Osbourn, A. (2014). Gene clustering in plant specialized metabolism. Curr. Opin. Biotechnol. 26, 91–99. doi: 10.1016/j.copbio.2013.10.009
Ou, S., Su, W., Liao, Y., Chougule, K., Agda, J., Hellinga, A. J., et al. (2019). Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20:275. doi: 10.1186/s13059-019-1905-y
Price, A. L., Jones, N. C., and Pevzner, P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics 21, (Suppl. 1), i351–i358. doi: 10.1093/bioinformatics/bti1018
Reyes-Chin-Wo, S., Wang, Z., Yang, X., Kozik, A., Arikit, S., Song, C., et al. (2017). Genome assembly with in vitro proximity ligation data and whole-genome triplication in lettuce. Nat. Commun. 8:14953. doi: 10.1038/ncomms14953
Schnable, P. S., Ware, D., Fulton, R. S., Stein, J. C., Wei, F., Pasternak, S., et al. (2009). The B73 maize genome: complexity, diversity, and dynamics. Science 326, 1112–1115. doi: 10.1126/science.1178534
Schuler, M. A. (1996). The role of cytochrome P450 monooxygenases in plant-insect interactions. Plant Physiol. 112, 1411–1419. doi: 10.1104/pp.112.4.1411
Shen, Q., Li, L., Jiang, Y., and Wang, Q. (2016a). Functional characterization of ent-copalyl diphosphate synthase from Andrographis paniculata with putative involvement in andrographolides biosynthesis. Biotechnol. Lett. 38, 131–137. doi: 10.1007/s10529-015-1961-7
Shen, Q., Liu, Q., Congcong, L. I., Yuping, F. U., and Wang, Q. (2016b). Functional characterization of ApCPS involved in andrographolides biosynthesis by virus-induced gene silencing. Acta Bot. Boreali 36, 17–22.
Shen, Q., Zhang, L., Liao, Z., Wang, S., Yan, T., Shi, P., et al. (2018). The genome of Artemisia annua provides insight into the evolution of asteraceae family and artemisinin biosynthesis. Mol. Plant 11, 776–788. doi: 10.1016/j.molp.2018.03.015
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Srivastava, N., and Akhila, A. (2010). Biosynthesis of andrographolide in Andrographis paniculata. Phytochemistry 71, 1298–1304. doi: 10.1016/j.phytochem.2010.05.022
Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S., Morgenstern, B., et al. (2006). AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439. doi: 10.1093/nar/gkl200
Sun, W., Leng, L., Yin, Q., Xu, M., Huang, M., Xu, Z., et al. (2019). The genome of the medicinal plant Andrographis paniculata provides insight into the biosynthesis of the bioactive diterpenoid neoandrographolide. Plant J. 97, 841–857. doi: 10.1111/tpj.14162
Trapnell, C., Hendrickson, D. G., Sauvageau, M., Goff, L., Rinn, J. L., Pachter, L., et al. (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 31, 46–53. doi: 10.1038/nbt.2450
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. doi: 10.1093/bioinformatics/btp120
Vining, K. J., Johnson, S. R., Ahkami, A., Lange, I., Parrish, A. N., Trapp, S. C., et al. (2017). Draft genome sequence of Mentha longifolia and development of resources for mint cultivar improvement. Mol. Plant 10, 323–339. doi: 10.1016/j.molp.2016.10.018
Vurture, G. W., Sedlazeck, F. J., Nattestad, M., Underwood, C. J., Fang, H., Gurtowski, J., et al. (2017). GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204. doi: 10.1093/bioinformatics/btx153
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, J., Lin, H. X., Su, P., Chen, T., Guo, J., Gao, W., et al. (2019). Molecular cloning and functional characterization of multiple geranylgeranyl pyrophosphate synthases (ApGGPPS) from Andrographis paniculata. Plant Cell Rep. 38, 117–128. doi: 10.1007/s00299-018-2353-y
Wang, L., Yu, S., Tong, C., Zhao, Y., Liu, Y., Song, C., et al. (2014). Genome sequencing of the high oil crop sesame provides insight into oil biosynthesis. Genome Biol. 15:R39. doi: 10.1186/gb-2014-15-2-r39
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Wicker, T., and Keller, B. (2007). Genome-wide comparative analysis of Copia retrotransposons in Triticeae, rice, and Arabidopsis reveals conserved ancient evolutionary lineages and distinct dynamics of individual copia families. Genome Res. 17, 1072–1081. doi: 10.1101/gr.6214107
Xu, J., Chu, Y., Liao, B., Xiao, S., Yin, Q., Bai, R., et al. (2017). Panax ginseng genome examination for ginsenoside biosynthesis. Gigascience 6, 1–15. doi: 10.1093/gigascience/gix093
Xu, Z., and Wang, H. (2007). LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268. doi: 10.1093/nar/gkm286
Yan, L., Wang, X., Liu, H., Tian, Y., Lian, J., Yang, R., et al. (2015). The genome of Dendrobium officinale illuminates the biology of the important traditional Chinese orchid herb. Mol. Plant 8, 922–934. doi: 10.1016/j.molp.2014.12.011
Yang, J., Zhang, G., Zhang, J., Liu, H., Chen, W., Wang, X., et al. (2017). Hybrid de novo genome assembly of the Chinese herbal fleabane Erigeron breviscapus. Gigascience 6, 1–7. doi: 10.1093/gigascience/gix028
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yazaki, K., Arimura, G. I., and Ohnishi, T. (2017). ‘Hidden’ terpenoids in plants: their biosynthesis, localization and ecological roles. Plant Cell Physiol. 58, 1615–1621. doi: 10.1093/pcp/pcx123
Zerikly, M., and Challis, G. L. (2009). Strategies for the discovery of new natural products by genome mining. Chembiochem 10, 625–633. doi: 10.1002/cbic.200800389
Zhang, G., Tian, Y., Zhang, J., Shu, L., Yang, S., Wang, W., et al. (2015). Hybrid de novo genome assembly of the Chinese herbal plant danshen (Salvia miltiorrhiza Bunge). Gigascience 4:62. doi: 10.1186/s13742-015-0104-3
Zhang, J., Tian, Y., Yan, L., Zhang, G., Wang, X., Zeng, Y., et al. (2016). Genome of plant maca (Lepidium meyenii) illuminates genomic basis for High-Altitude adaptation in the central andes. Mol. Plant 9, 1066–1077. doi: 10.1016/j.molp.2016.04.016
Zhang, R., Wang, Z., Ou, S., and Li, G. (2019). TEsorter: lineage-level classification of transposable elements using conserved protein domains. bioRxiv [Preprint] doi: 10.1101/800177
Zhao, Q., Yang, J., Cui, M. Y., Liu, J., Fang, Y., et al. (2019). The reference genome sequence of Scutellaria baicalensis provides insights into the evolution of wogonin biosynthesis. Mol. Plant 12, 935–950. doi: 10.1016/j.molp.2019.04.002
Keywords: PacBio sequencing, Hi-C, genome assembly, medicinal plant, Andrographis paniculata
Citation: Liang Y, Chen S, Wei K, Yang Z, Duan S, Du Y, Qu P, Miao J, Chen W and Dong Y (2020) Chromosome Level Genome Assembly of Andrographis paniculata. Front. Genet. 11:701. doi: 10.3389/fgene.2020.00701
Received: 20 February 2020; Accepted: 09 June 2020;
Published: 30 June 2020.
Edited by:
Xiaoming Song, North China University of Science and Technology, ChinaReviewed by:
Marcio Resende, University of Florida, United StatesCopyright © 2020 Liang, Chen, Wei, Yang, Duan, Du, Qu, Miao, Chen and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianhua Miao, bWpoMTk2MkB2aXAuMTYzLmNvbQ==; Wei Chen, d2NoZW5udHJAZ21haWwuY29t; Yang Dong, bG95YWx5YW5nQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.