
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 08 June 2020
Sec. Human and Medical Genomics
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.00613
This article is part of the Research TopicCoronavirus Disease (COVID-19): Molecular Mechanisms, Translational Approaches and TherapeuticsView all 118 articles
 Esteban A. Lopera Maya1†
Esteban A. Lopera Maya1† Adriaan van der Graaf1†
Adriaan van der Graaf1† Pauline Lanting1
Pauline Lanting1 Marije van der Geest1
Marije van der Geest1 Jingyuan Fu1,2
Jingyuan Fu1,2 Morris Swertz1
Morris Swertz1 Lude Franke1
Lude Franke1 Cisca Wijmenga1Patrick Deelen1,3
Cisca Wijmenga1Patrick Deelen1,3 Alexandra Zhernakova1
Alexandra Zhernakova1 Serena Sanna1,4* and Lifelines Cohort Study
Serena Sanna1,4* and Lifelines Cohort StudyCoronavirus disease 2019 (COVID-19) shows a wide variation in expression and severity of symptoms, from very mild or no symptoms, to flu-like symptoms, and in more severe cases, to pneumonia, acute respiratory distress syndrome, and even death. Large differences in outcome have also been observed between males and females. The causes for this variability are likely to be multifactorial, and to include genetics. The SARS-CoV-2 virus responsible for the infection depends on two human genes: the human receptor angiotensin converting enzyme 2 (ACE2) for cell invasion, and the serine protease TMPRSS2 for S protein priming. Genetic variation in these two genes may thus modulate an individual's genetic predisposition to infection and virus clearance. While genetic data on COVID-19 patients is being gathered, we carried out a phenome-wide association scan (PheWAS) to investigate the role of these genes in other human phenotypes in the general population. We examined 178 quantitative phenotypes including cytokines and cardio-metabolic biomarkers, as well as usage of 58 medications in 36,339 volunteers from the Lifelines population cohort, in relation to 1,273 genetic variants located in or near ACE2 and TMPRSS2. While none reached our threshold for significance, we observed several interesting suggestive associations. For example, single nucleotide polymorphisms (SNPs) near the TMPRSS2 genes were associated with thrombocytes count (p = 1.8 × 10−5). SNPs within the ACE2 gene were associated with (1) the use of angiotensin II receptor blockers (ARBs) combination therapies (p = 5.7 × 10−4), an association that is significantly stronger in females (pdiff = 0.01), and (2) with the use of non-steroid anti-inflammatory and antirheumatic products (p = 5.5 × 10−4). While these associations need to be confirmed in larger sample sizes, they suggest that these variants could play a role in diseases such as thrombocytopenia, hypertension, and chronic inflammation that are often observed in the more severe COVID-19 cases. Further investigation of these genetic variants in the context of COVID-19 is thus promising for better understanding of disease variability. Full results are available at https://covid19research.nl.
The recent outbreak of the coronavirus disease 2019 (COVID-19) caused by the SARS-CoV-2 virus has quickly become a pandemic and poses a great threat to public health. COVID-19 has a wide range of clinical manifestations: infected people can be asymptomatic, symptomatic with mild respiratory symptoms, or have severe pneumonia (Chen et al., 2020; Huang et al., 2020; Wu and McGoogan, 2020; Xu et al., 2020). Estimates based on reported cases from February 2020 in China indicated that ~20% of patients develop severe respiratory illness requiring hospitalization, and that overall mortality estimates are around 2.3% (Wu and McGoogan, 2020). These estimates are not fixed and are becoming more precise as more cases are reported, screened, and analyzed. Interestingly, there is high variability in these estimates when comparing countries and continents, as well as differences in COVID-19 severity between males and females and between different age groups (Chen et al., 2020; Wu and McGoogan, 2020; Zhou et al., 2020) [WHO Situation Report 70, from March 30, 2020]. Differences in response to SARS-CoV-2 infection between individuals and countries may be explained by diminished immune response in the elderly, comorbidities, or smoking habits (Guan et al., 2020), but severe COVID-19 cases have also been observed in young individuals, seemingly without risk factors. This indicates that most factors explaining COVID-19 severity are still unknown. It is therefore critical to understand the mechanisms behind COVID-19 severity in order to provide appropriate prevention measures and adequate triage strategies, guide the drug discovery process, and ultimately combat the SARS-CoV-2 pandemic.
The large variation in SARS-CoV-2 infection rates and COVID-19 severity could potentially be explained by genetic differences between hosts. While large-scale genetic studies of COVID-19 patients are being assembled, such as those coordinated by the COVID host genetics consortium (The COVID-19 Host Genetics Initiative, 2020; https://www.covid19hg.org/), it is worthwhile to evaluate the effects of genetic variants in genes involved in SARS-CoV-2 infection on human phenotypes, including quantitative traits, taking advantage of already existing cohorts. In fact, while quantitative phenotypes are not always directly associated with a disease, knowledge on the genetic variants that modulate these traits can improve our understanding of disease onset and the variability in symptoms. In one example of how this can work, genetic variants in the BCL11A gene were associated by genome-wide association studies (GWAS) to fetal hemoglobin (HbF) production in the general population (Menzel et al., 2007), and these genetic variants were subsequently found to modulate the severity of beta-thalassemia and sickle cell diseases (Lettre et al., 2008; Uda et al., 2008). This observation explained why certain individuals were naturally predisposed to mild symptoms of these diseases, while others had very severe clinical outcomes and benefitted from HbF increasing drugs. Therefore, understanding the role of genetic variants at genes essential for SARS-CoV-2 infection in human quantitative phenotypes is important to explain the observed variability in infection susceptibility and severity of COVID-19 and this understanding may suggest potential treatments.
Some factors that are necessary for SARS-CoV-2 infection are known (Hoffmann et al., 2020; Yan et al., 2020). Angiotensin converting enzyme 2 (ACE2) is necessary for the invasion of the virus into the host cell through viral spike proteins, and the transmembrane Serine Protease 2 (TMPRSS2) is necessary for the correct maturation of these same viral spike proteins that enter the cell through ACE2 (Yan et al., 2020). According to the GWAS Catalog1, genetic variants in or near TMPRSS2, located on chromosome 21, are associated with susceptibility of prostate cancer and mortality rate in the population, while no associations have been reported for variants in or near ACE2. This can be partly explained by the fact that the ACE2 gene is located on the X chromosome, a part of the genome that is often not analyzed by large scale genome wide association studies (GWAS) due to differences in analysis workflow with the autosomal chromosomes. Potential associations with human phenotypes near ACE2 could have therefore been missed.
Here we investigated the association of genetic variants within or near (±100 Kb) ACE2 and TMPRSS2 transcripts through a phenome-wide association scan (PheWAS) in 36,339 volunteers from the Lifelines population cohort. We analyzed 72 quantitative phenotypes and the medication usage of 58 different drug categories in the entire cohort, and 92 protein levels in plasma, and 14 cytokines in a subset of ~600 individuals. The quantitative phenotypes selected are anthropometric traits and measurable parameters of lung, hearth, kidney, hematological, immune, and cardio-metabolic functions. Finally, in a sex-stratified anaysis we evaluated whether these variants were sex-specific or differed in their association between males and females to explore potential differences between sexes that could modulate SARS-CoV-2 infection.
The Lifelines cohort (Scholtens et al., 2015) is a multi-disciplinary prospective population-based cohort study, with a unique three generation design, that is examining the health and health-related behaviors of 167,729 individuals living in the North of the Netherlands. It was approved by the medical ethics committee of the University Medical Center Groningen and conducted in accordance with Helsinki Declaration Guidelines. All participants signed an informed consent form prior to enrollment. Lifelines employs a broad range of investigative procedures to assess the impact of biomedical, socio-demographic, behavioral, physical, and psychological factors on multi-morbidity, and complex genetics.
A subset of 38,030 volunteers were genotyped using the Infinium Global Screening Array® (GSA) MultiEthnic Disease Version, according to manufacturer's instructions, at the Rotterdam genotyping center and the Department of Genetics, University Medical Center Groningen. We performed standard quality controls on both samples and markers, including removal of samples and variants with a low genotyping call rate (<99%), variants showing deviation from Hardy-Weinberg equilibrium (p < 1 × 10−6) or excess of Mendelian errors in families (>1% of the parent-offspring pairs), and samples with very high or low heterozygosity. We further checked and removed samples that did not show consistent information between reported sex and genotypes on the X chromosome, between reported familial information and observed identity-by-descent sharing with family members, and between genotypes available from this and previous studies (Francioli et al., 2015; Tigchelaar et al., 2015). A detailed description of the process can be found at the following link: https://covid19research.nl (van der Velde et al., 2019). After quality checks, a total of 36,339 samples and 571,420 autosomal and X-chromosome markers were available for analysis.
The genotyping dataset was then imputed using the Haplotype Reference Consortium (HRC) panel v1.1 at the Sanger imputation server1 (Consortium, 2015), and variants with an imputation quality score higher than 0.4 for variants with a minor allele frequency (MAF) > 0.01 and higher than 0.8 for rare variants (MAF < 0.01) were retained. 58.40% (21,241) of the 36,339 individuals whose genotype passed quality control were female, and the average age at phenotype collection was 39.9 years (±16.3 years).
Quantitative phenotypes were measured as previously described (Scholtens et al., 2015). We removed illegal zero or negative values for the “QRS,” “QT,” “HALB,” “MAP,” “MOP,” “EOP,” “BAP,” “U24HVOL,” “ALT,” “HR,” “EO,” “PQ,” “MO,” and “BA” phenotypes, and removed−999 values from the electrocardiogram phenotypes “P_AXIS,” “T_AXIS,” and “QRS_AXIS” (Table S1). Protein levels in plasma for 92 cardiovascular-related proteins were determined using Olink Proseek Multiplex CVD III panel (OLINK, Uppsala, Sweden), and concentrations of plasma citrulline and cytokines were measured by ProcartaPlex™ multiplex immunoassay (eBioscience, USA) as described previously (Zhernakova et al., 2016, 2018). Medication use was recorded based on drug packaging brought in by the participant's on their first visit to the Lifelines inclusion center. Registration of medication use in this way has been shown to be fairly to highly concordant with health record information (Sediq et al., 2018). After conversion to anatomical therapeutic chemical classification (ATC) codes, the first four letters (level 3) were used to define drug categories for association analyses. ATC codes with less than 100 observations were not considered for analysis, leaving 58 drug categories for analysis (Table S2).
We analyzed quantitative phenotypes using linear-mixed models implemented in SAIGEgds v1.0.0 so as to correct for familial relationships and cryptic population structure (Zheng et al., 2017; Zhou et al., 2018). For the X chromosome, genotypes in males were considered diploid. We tested the additive effect of 1,273 genetic variants within and near (±100 Kb) ACE2 (chrX:15,579,156-15,620,271, GRCh37) and TMPRSS2 (chr21:42,836,478-42,903,043, GRCh37) transcripts. These are all single-nucleotide polymorphisms (SNPs) with minor allele frequency (MAF) > 0.005 that were genotyped or imputed and that passed our quality controls as described above. Analysis through SAIGEgds was carried out for 72 quantitative phenotypes available for all, or a subset of the 36,339 samples (Table S1). Drug categories were analyzed as binary traits (1 = if medication currently in use, 0 otherwise) and restricted only to 1,240 genetic variants with MAF > 0.01. In both analyses age and sex were used as covariates. Inverse-normal transformation was applied to all quantitative traits prior to model fit. We searched for sex-specific effects by analyzing males and females separately (sex-stratified analyses), using only age as covariate and the same transformations as used for the analysis on the entire cohort. We also used the sex-stratified anaysis results to investigated differential genetic effects between sexes at suggestive associations identified in the combined analysis. This approach is typically used in small to moderate studies as an alternative to an analysis with an interaction term (Winkler et al., 2015).
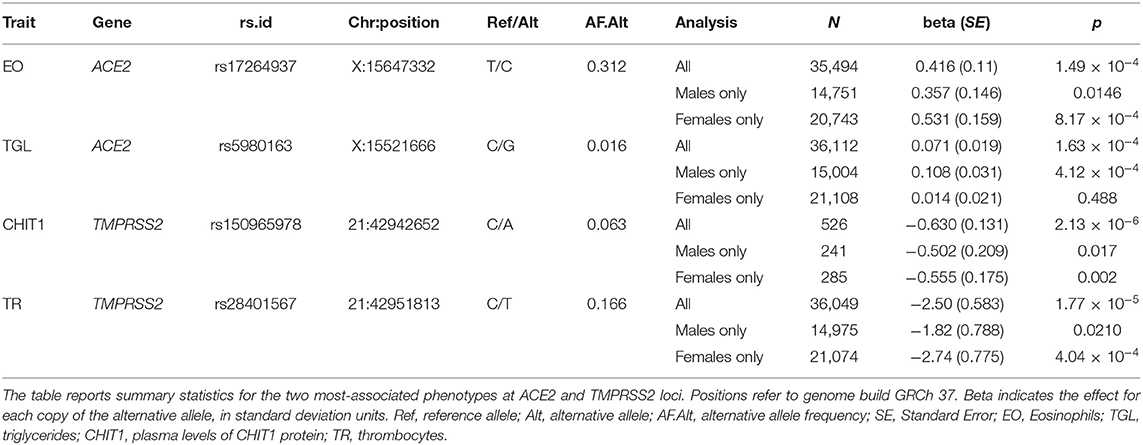
Table 1. Most-significant associations with phenotypes at ACE2 and TMPRSS2 loci.
The 92 circulating plasma proteins and 14 cytokines were measured in a small subset of unrelated individuals and thus did not require correction for familial relationships. These were analyzed using PLINK v2.00a3LM. We performed the association mapping with both sexes jointly, or separately as described above, and using inverse-normal transformation on the traits. We analyzed each variant and trait combination with or without the inclusion of age and sex covariates, as some genetic variants were too highly correlated due to the small sample size, and thus an estimate with covariates included in the model was not possible. To evaluate the statistical power of our study we used the package GeneticsDesign in R (Weilang et al., 2019). For quantitative variable analyses, we used the function GeneticPower.Quantitative.Numeric() and calculated the minimum detectable additive effect (variance explained) with 80% power and at a significance threshold of 5 × 10−8, for an increasing number of samples up to 36,339 (our study size). For binary variables analysis, we used the function GPC.default() and calculated the minimum detectable additive effect size (genotype relative risk) with 80% power and at a significance threshold of 5 × 10−8, for an increasing number of cases in a cohort of 36,339 and for a risk allele frequency varying from 0.05 to 0.5. We set the number of cases up to 4,000 to reflect the maximum number of users for the analyzed drug categories in our study. We also assumed that the causal variant was included in our genotyping data set, therefore we constrained full linkage disequilibrium (Dprime = 1) with the tag marker. Since disease prevalence (pD) could also impact power, we calculated the minimum detectable effect for pD varying from 1 to 20%.
Using a linear-mixed model, we analyzed 1,273 common and low frequency (MAF > 0.005) genetic variants in and near (+/−100Kb) ACE2 and TMPRSS2 transcripts for association with 178 quantitative traits (Table S1). None were found to be significant at the standard genome-wide level (p = 5 × 10−8) or at the Benjamin-Hochberg false discovery rate (FDR < 0.1). The most significant associations found with quantitative traits at the ACE2 locus were with triglycerides (rs5980163, p = 1.6 × 10−4) and with the eosinophil counts (rs17264937, p = 1.5 × 10−4) (Table 1) (Figure 1). The strongest associations at the TMPRSS2 locus were with plasma levels of CHIT1 (rs150965978, p = 2.1 × 10−6) and thrombocytes (rs28401567, p = 1.7 × 10−5) (Table 1) (Figure 2). Only the association at rs5980163 with triglycerides at ACE2 showed a differential effect between males and females (Cochran Q-test pdiff = 0.01), with most of the signal being attributable to males, although the association remains only suggestive (p = 4.12 × 10−4). We did not find any signal that was restricted to either males or females (p > 1 × 10−6 for all associations in the sex-stratified analyses). The SNP-trait associations reported in Table 1 were not replicated in the UK Biobank, based on summary statistics from an analysis that included at least 343,992 samples1 (all p > 0.05). No replication was observed also for the association with CHIT1 plasma levels using results from the INTERVAL study (Sun et al., 2018).
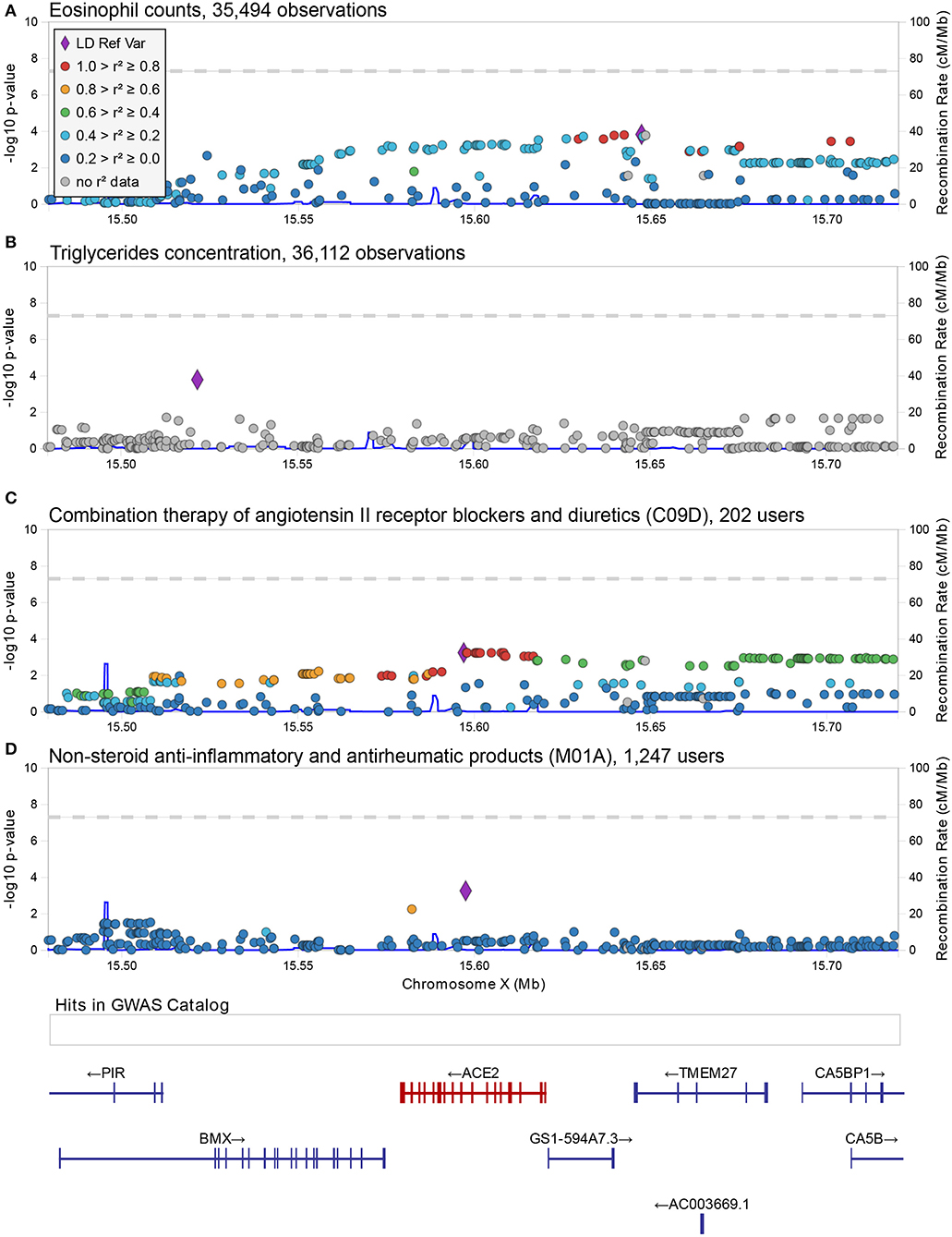
Figure 1. Regional associations plot at the ACE2 locus. Graphical representation of the association results at the ACE2 locus for the SNP-trait associations reported in Table 1 (A,B) and Table 2 (C,D). In each panel, each dot represents a genetic variant, and shown is the association strength (expressed as negative log10 P-values, Y-axis) vs. the genomic position (on the hg19/GRCh37 genomic build, X-axis). The strongest associated variant is depicted with a purple diamond, while other variants are color-coded to reflect their linkage disequilibrium with it (taken from pairwise r2 values calculated from the 1,000 Genomes Europeans). A legend for color-coding is provided in (A). In (D), an additional box shows the location of associations reported in the GWAS catalog (no associations were reported at this locus) and below this box the location of genes is shown with specification of exons and direction of transcription. This figure was drawn using LocusZoom web tool (Pruim et al., 2010).
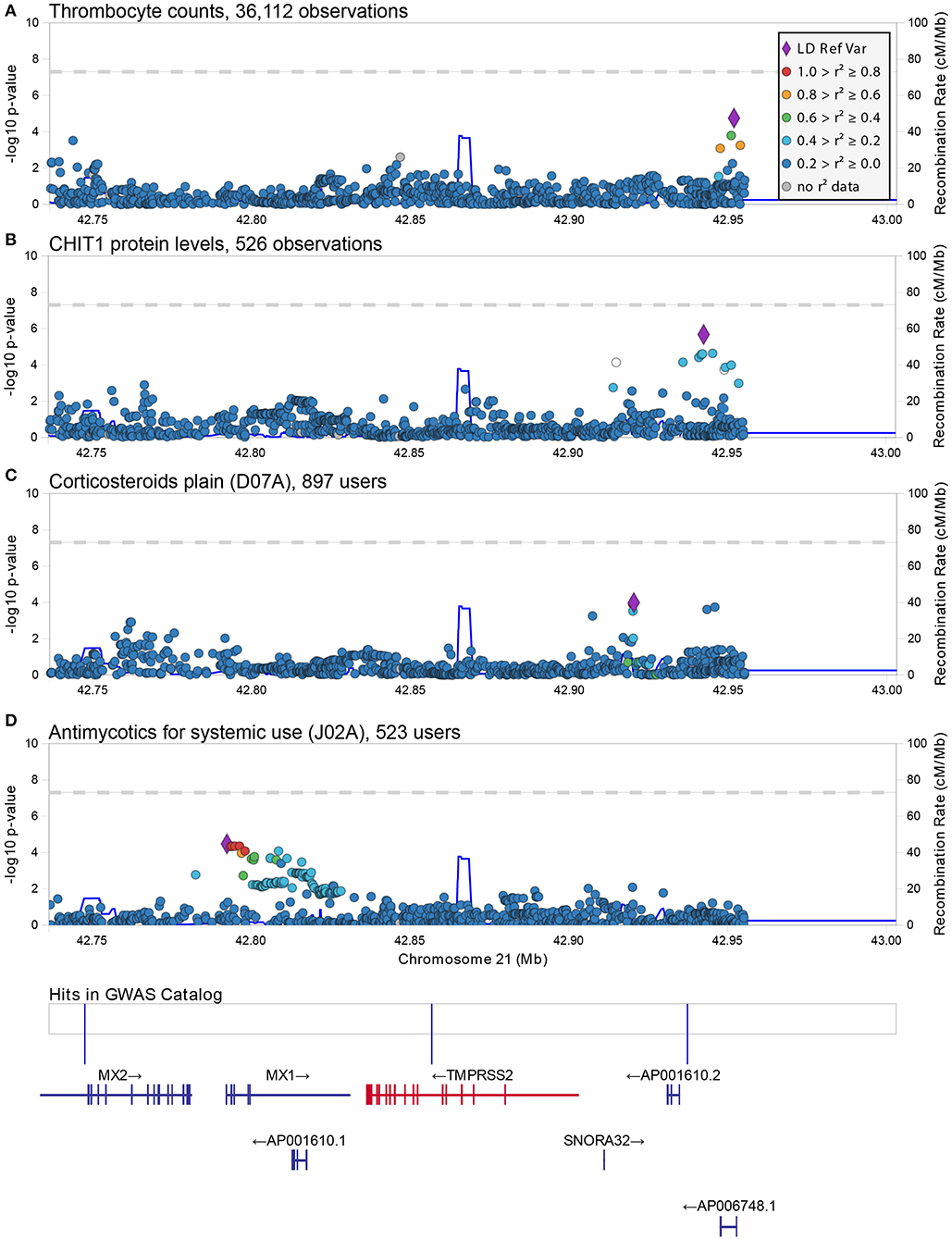
Figure 2. Regional associations plot at the TMPRSS2 locus. Graphical representation of the association results at the TMPRSS2 locus for the SNP-trait associations reported in Table 1 (A,B) and Table 2 (C,D). In each panel, each dot represents a genetic variant and shown is the association strength (expressed as negative log10 P-values, Y-axis) vs. the genomic position (on the hg19/GRCh37 genomic build, X-axis). The strongest associated variant is depicted with a purple diamond, while other variants are color-coded to reflect their linkage disequilibrium with it (taken from pairwise r2 values calculated from the 1,000 Genomes Europeans). A legend for color-coding is provided in (A). In (D), an additional box shows the location of associations reported in the GWAS catalog (associations here reported from left to right are: melanoma, age-related diseases and mortality, and prostate cancer) and below this box the location of genes is shown with specification of exons and direction of transcription This figure was drawn using LocusZoom web tool (Pruim et al., 2010).
For this analysis, we focused on 1,240 variants with MAF > 0.01. As with the quantitative phenotypes, none of the genetic variants showed genome-wide significant association with medication use (Table S2). The strongest associations at the ACE2 locus were observed for the group of drugs that contains non-steroid anti-inflammatory and antirheumatic products (NSAIDs) (ATC = M01A) [odds ratio (OR) = 1.34, 95% C.I. = 1.14–1.58, p = 5.5 × 10−4 for the G allele of rs4646190] (Table 2) (Figure 1), and for the group that contains angiotensin II receptor blockers (ARBs) in combination with other antihypertensive drugs (ATC = C09D) (OR = 1.35, 95% C.I. = 1.14–1.62 p = 5.7 × 10−4 for the T allele of rs4646156) (Table 2) (Figure 1). These SNPs are both located in intron eight of the ACE2 transcript and only 525 bp apart, but they are not in linkage disequilibrium (r2 = 0.05 in 1,000 Genomes Europeans).
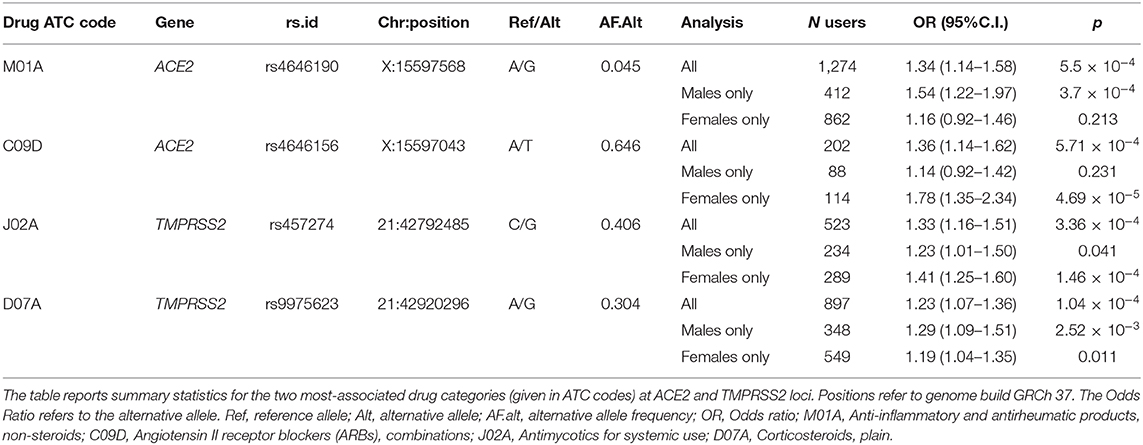
Table 2. Most-significant associations with medications use at ACE2 and TMPRSS2 loci.
NSAIDs are used for treating pain, fever and inflammation, and include ibuprofen. The significance of rs4646190 was stronger in males (p = 3.7 × 10−4) than in females (p = 0.08), but the effect sizes were not statistically different (pdiff = 0.054).
The second group of drugs encodes for a combined therapy used to treat hypertension. Combination therapy of ARBs with other hypertensive drugs is usually initiated as a second option when the antihypertensive effect of an ARB alone is not sufficient (Ram, 2004; Flack, 2007). Our results indicate that individuals carrying at least one T allele at the rs4646156 polymorphism were more likely to take this combined therapy compared to individuals with the other allele. The effect of this SNP was also not significant when considering only ARBs intake (ATC = C09C, p = 0.66). Thus, the association with ARB combination therapies could indicate that individuals in whom it is difficult to manage hypertension may be genetically predisposed to this state by rs4646156. Interestingly, when analyzing males and females separately, we found that the signal of rs4646156 on ARB combination therapy was mostly attributable to females, even when accounting for differences in number of users (OR = 1.78, 95% C.I. = 1.35–2.34, p = 4.7 × 10−5 in females vs. OR = 1.14, 95% C.I. = 0.92–1.42, p = 0.23 in males, pdiff = 0.01).
We reiterate that none of these associations (in the combined and in the sex-specific analyses) meet either the genome-wide or FDR thresholds for significance. To confirm these findings larger sample sizes are necessary.
The strongest associations at the TMPRSS2 locus were observed for the group of drugs containing antimycotics prescriptions (ATC = J02A) (p = 3.65 × 10−5) and for corticosteroids (ATC = D07A) (p = 1.0 × 10−4) (Table 2) (Figure 2). No significant difference in effect size between sexes was observed for these two associations (pdiff > 0.2). These SNPs were independent from each other and from the top associations with quantitative traits described in Table 1.
We attempted to validate our findings on medication use using again the UK Biobank public GWAS summary statistics1, although their data refers to the use of individual medications rather than drug categories. When considering the medications most commonly used (>1,000 users in the UK Biobank cohort) in the categories of interest (M01A, C09D, J02A, and D07A), we found nominal association with the same direction of effects only for glucosamine use (ATC = M01A, p = 0.002 in the combined analysis and p = 0.008 in males only) and with candesartan cilexetin (ATC = C09D, p = 0.008 in females only) (Table S3). A similar detailed analysis in our cohort was underpowered to detect an association signal for single medications of C09D and M01A categories (Table S4). This lack of replication could be attributable to differences in medication usage reporting between studies. While both are based on self-reported information, in the Lifelines study records are confirmed by medication packaging collected by a nurse during the recruitment.
Recent studies have demonstrated that SARS-CoV-2 uses ACE2 as the key receptor to invade cells (Yan et al., 2020) and that ACE2-mediated cell invasion is enhanced by TMPRSS2 expression (Hoffmann et al., 2020). Genetic variations in these two genes that interfere with the gene function may thus be involved in the observed variability of SARS-CoV-2 susceptibility and COVID-19 severity. The association of these genetic variants with human phenotypes in the general population may suggest potential treatments and help to better identify at-risk individuals. Here we used a cohort of 36,339 individuals from the Lifelines general population cohort to investigate the impact of variants near and within these two genes on 178 quantitative traits including measurable parameters of lung, hearth, kidney, hematological, immune, and cardio-metabolic functions.
We found no significant evidence that common and low frequency variants in these loci were associated with the measured quantitative traits in the general population. We did observe suggestive signals for phenotypes (triglycerides and thrombocytes) that are involved in cardiovascular diseases, which are considered risk factors for COVID-19 diseases (Wu and McGoogan, 2020), but none of the genetic variants reached statistical significance despite our large sample size. Nevertheless, we cannot exclude a role of these variants in the regulation of COVID-19 severity through other relevant phenotypes such as specific immune cell types or cytokine levels that were not measured in our cohort.
To evaluate the effect of genetic variation in clinically relevant phenotypes, we investigated the association of genetics with medication use. We observed a marginal association of variants within ACE2 with use of ARBs combination therapy (ATC = C09D, rs4646156) and with use of non-steroidal anti-inflammatory and antirheumatic drugs (NSAIDs, rs4646190). Interestingly, a marginal association with ARBs (C09C category) was also observed at the TMPRSS2 locus (rs75833467, p = 3.5 × 10−4). These results are intriguing considering the current debate about whether the use of ARBs and NSAIDs could worsen COVID-19 severity (Kuster et al., 2020; Little, 2020; Russell et al., 2020), and their potential effect on increasing ACE2 expression. No significant associations were found for these variants with blood pressure measurements or inflammatory markers in our cohort (p < 0.05), not even when the use of such drugs were added as covariates (data not shown). Association with diastolic and systolic blood pressure was also not observed in the large UK Biobank cohort. Thus, these variants are likely to be associated only with clinical conditions such as hypertension and chronic inflammation or with a better drug response. It has to be noted that our sample size allowed sufficient statistical power to detect genetic variants with small effects (down to 0.001 of variance explained), and thus we are confident in claiming lack of association at TMPRS22 and ACE2 loci with the quantitative phenotypes assessed. For analyses on medication usage we were instead sufficiently powered to find small effects (genotype relative risk ~1.1) only for very common SNPs (frequency >0.3), but we are underpowered for smaller effects and, in general, at less common variants (Figure S1). Therefore, our suggestive results for medication usage could indicate a real effect for which we were underpowered to find a genome-wide significance evidence. Further exploration of these associations is needed.
ARBs are the preferred alternative for patients who experience ACE-inhibitor induced coughing. However, as rs4646156 is not associated with this adverse drug reaction (ADR), our results are likely independent of the switch to ARBs due to ACE-inhibitor induced coughing (Mas et al., 2011). Interestingly, the association of this SNP with ARBs was specific to ARBs combination therapy, thus pointing to individuals with difficult-to-manage hypertension. The major allele (T) of rs4646156 has different frequencies across populations: 0.653 in Europeans, 0.997 in East Asians and 0.797 South Asians, according to 1,000 Genomes1. Likewise, the G allele at the rs4646190 SNP, associated with a higher probability of NSAIDs use, shows substantial different frequencies among populations. It is mostly absent in Asians but not in Europeans: 0.03 in Europeans, 0 in East Asians and 0.003 in South Asians, according to 1,000 Genomes1.
The suggestive genetics associations we find for NSAIDs and ARBs combination therapy indicate that, depending on their genotype, certain individuals are predisposed to take these drugs, and thus to suffer from hypertension and chronic inflammation, diseases often described among COVID-19 comorbidities. This, together with the observed different allele frequencies across continents and the sex-related differential effects could explain the observed variation in COVID-19 severity between countries and sexes. Unfortunately, we could only speculate around this hypothesis as this study is not suited to prove that these genetic associations are directly related to SARS-CoV-2 susceptibility or COVID-19 severity, nor we can determine if ARBs or NSAIDs improve or worsen COVID-19 severity. A role of ARBs in worsing severity seems however unlikely (Gill et al., 2020; Mancia et al., 2020; Mehra et al., 2020).
We acknowledge the following limitations in our study. First, only age and sex were used as covariates in our analyses, which may not be sufficient to correct for confounders for all traits, such as drug usage or diseases, although the effect of these confounders should be mitigated by our sample size. Secondly, our analyses on medication use are underpowered given the limited number of individuals in the general population who use the medications that we tested, and thus none of the associations found here met the multiple-testing adjusted significance. Third, our results for medication use did not include low frequency and none of the analysis include rare variants (MAF < 0.005) which could still be relevant. Fourth, while we can speculate about potential connections of our results with current knowledge of COVID-19, longitudinal and well-characterized data on patients is needed to further explore our hypothesis.
In conclusion we carried out an extensive screening of potential genetic associations at common and low frequency variants in the ACE2 and TMPRSS2 genes, and found a lack of substantial effect in human quantitative phenotype variation in the general population. Genetic analyses in more phenotypes are needed to evaluate their functional role in other physiological processes.
Finally, since genetic variation in other genes, for example those involved in regulating the immune system, could also be important in determining SARS-CoV-2 susceptibility and disease severity, large scale genetic initiatives like the COVID-19 host genetics consortium (The COVID-19 Host Genetics Initiative, 2020; https://www.covid19hg.com/) that directly involve patients with COVID-19 and deeply characterization of genomes and phenotypes are urgently needed.
The data analyzed in this study was obtained from the Lifelines biobank, under project application number OV18_0463. Requests to access this dataset should be directed to Lifelines Research Office (cmVzZWFyY2hAbGlmZWxpbmVzLm5s). Full summary statistics of the results are available at https://covid19research.nl.
The studies involving human participants were reviewed and approved by the medical ethics committee of the University Medical Center Groningen and conducted in accordance with Helsinki Declaration Guidelines. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
EL and AG performed statistical analyses. EL, AG, PL, PD, AZ, and SS interpreted results. MG and MS provided computing infrastructure and web portal. Lifelines Cohort Study, LF, CW, JF, and AZ provided access to the data. EL, AG, and SS wrote the manuscript draft with critical input from PL, LF, CW, PD, JF, and AZ. All authors read and approved the manuscript.
Raul Aguirre-Gamboa (1), PD (1), LF (1), Jan A Kuivenhoven (2), EL (1), Ilja M Nolte (3), SS (1), Harold Snieder (3), MS (1), Judith M Vonk (3), CW(1)
(1) Department of Genetics, University of Groningen, University Medical Center Groningen, Netherlands.
(2) Department of Pediatrics, University of Groningen, University Medical Center Groningen, Netherlands.
(3) Department of Epidemiology, University of Groningen, University Medical Center Groningen, Netherlands.
This work was supported by the Netherlands Organization for Scientific Research (NWO): NWO Spinoza Prize SPI 92-266 (to CW). The Lifelines Biobank initiative has been made possible by funding from the Dutch Ministry of Health, Welfare and Sport; the Dutch Ministry of Economic Affairs; the University Medical Center Groningen (UMCG the Netherlands); the University of Groningen and the Northern Provinces of the Netherlands. The generation and management of GWAS genotype data for the Lifelines Cohort Study was supported by the UMCG Genetics Lifelines Initiative (UGLI). JF was supported by NWO Gravitation Netherlands Organ-on-Chip Initiative (024.003.001) and the Netherlands Heart Foundation CVON grant 2018-27. MS was supported by EOSC-Life, funded by European Union's Horizon 2020 programme under grant agreement 824087 and NWO-VIDI grant VIDI 917.164.455. AZ was supported by ERC Starting Grant 715772, NWO-VIDI grant 016.178.056, the Netherlands Heart Foundation CVON grant 2018-27, and NWO Gravitation grant ExposomeNL (024.004.017).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer JK declared a shared affiliation with no collaboration with one of the authors PD to the handling editor at time of review.
The authors wish to acknowledge the services of the Lifelines Cohort Study, the contributing research centers delivering data to Lifelines, and all the study participants. We also thank K. McIntyre for editorial assistance, and the UMCG Genomics Coordination center, the UG Center for Information Technology and their sponsors BBMRI-NL & TarGet for storage and computational infrastructure. This manuscript has been released as a pre-print at https://www.medrxiv.org/ (Lopera et al., 2020).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00613/full#supplementary-material
Figure S1. Statistical power estimates. (A–D) show statistical power estimates for binary trait analyses, and (E) for quantitative trait analyses. In (A–D), each line shows the minimum detectable additive effect size (genotype relative risk, Y-axis) with 80% power and at a significance threshold of 5 × 10−8, for an increasing number of cases in a cohort of 36,339 (X-axis). The different lines depict these values when risk allele frequency varies from 0.05 to 0.5. The label of the X-axis indicates the disease prevalence (pD) considered in each panel. In (E), the line shows the minimum detectable additive effect (variance explained, Y-axis) with 80% power and at a significance threshold of 5 × 10−8, for an increasing number of samples up to 36,339 (X-axis).
1. ^1000 Genomes study: https://www.internationalgenome.org/; Sanger imputation server: https://imputation.sanger.ac.uk; GWAS catalogue: https://www.ebi.ac.uk/gwas/home (accessed on April 6, 2020); UK biobank all phenotype associations: http://www.nealelab.is/uk-biobank.
Chen, N., Zhou, M., Dong, X., Qu, J., Gong, F., Han, Y., et al. (2020). Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 395, 507–513. doi: 10.1016/S0140-6736(20)30211-7
Consortium (2015). The 1,000 genomes project. A global reference for human genetic variation. Nature 526, 68–74. doi: 10.1038/nature15393
Flack, J. M. (2007). Maximising antihypertensive effects of angiotensin II receptor blockers with thiazide diuretic combination therapy: focus on irbesartan/hydrochlorothiazide. Int. J. Clin. Practice 61, 2093–2102. doi: 10.1111/j.1742-1241.2007.01577.x
Francioli, L. C., Paz, P. P., Koren, A., Menelaou, A., Chun, S., Renkens, I., et al. (2015). Genome-wide patterns and properties of de novo mutations in humans. Nat. Genet. 47, 822–826. doi: 10.1038/ng.3292
Gill, D., Arvanitis, M., Carter, P., Hernandez Cordero, A. I., Jo, B., Karhunen, V., et al. (2020). ACE inhibition and cardiometabolic risk factors, lung ACE2 and TMPRSS2 gene expression, and plasma ACE2 levels: a mendelian randomization study. MedRxiv. doi: 10.1101/2020.04.10.20059121
Guan, W.-j., Liang, W.-h., Zhao, Y., Liang, H.-r., Chen, Z.-s, Li, Y.-m., et al. (2020). Comorbidity and its impact on 1,590 patients with Covid-19 in China: a nationwide analysis. Eur. Respiratory J. 55:2000547. doi: 10.1183/13993003.00547-2020
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Krüger, N., Herrler, T., Erichsen, S., et al. (2020). SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8. doi: 10.1016/j.cell.2020.02.052
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., et al. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506. doi: 10.1016/S0140-6736(20)30183-5
Kuster, G. M., Pfister, O., Burkard, T., Zhou, Q., Twerenbold, R., Haaf, P., et al. (2020). SARS-CoV2: should inhibitors of the renin–angiotensin system be withdrawn in patients with COVID-19? Eur. Heart J. 41, 1801–1803. doi: 10.1093/eurheartj/ehaa235
Lettre, G., Sankaran, V. G., Bezerra, M. A. C., Araújo, A. S., Uda, M., Sanna, S., et al. (2008). DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc. Natl. Acad. Sci. U. S. A. 105, 11869–11874. doi: 10.1073/pnas.0804799105
Little, P. (2020). Non-steroidal anti-inflammatory drugs and covid-19. BMJ 368:1185. doi: 10.1136/bmj.m1185
Lopera, E., van der Graaf, A., Lanting, P., van der Geest, M., Fu, J., Swertz, M., et al. (2020). Lack of Association between Genetic Variants at ACE2 and TMPRSS2 genes Involved in SARS-CoV-2 Infection and Human Quantitative Phenotypes. MedRxiv. 2:2020.04.22.20074963. doi: 10.1101/2020.04.22.20074963
Mancia, G., Rea, F., Ludergnani, M., Apolone, G., and Corrao, G. (2020). Renin–angiotensin–aldosterone system blockers and the risk of covid-19. N. Engl. J. Med. doi: 10.1056/NEJMoa2006923. [Epub ahead of print].
Mas, S., Gass,ò, P., Alvarez, S., Ortiz, J., Sotoca, J. M., Francino, A., et al. (2011). Pharmacogenetic predictors of angiotensin-converting enzyme inhibitor-induced cough: the role of ACE, ABO, and BDKRB2 genes. Pharmacogenet. Genom. 21, 531–538. doi: 10.1097/FPC.0b013e328348c6db
Mehra, M. R., Desai, S. S., Kuy, S., Henry, T. D., and Patel, A. N. (2020). Cardiovascular disease, drug therapy, and mortality in covid-19. N. Engl. J. Med. doi: 10.1056/NEJMoa2007621. [Epub ahead of print].
Menzel, S., Garner, C., Gut, I., Matsuda, F., Yamaguchi, M., Heath, S., et al. (2007). A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat. Genet. 39, 1197–1199. doi: 10.1038/ng2108
Pruim, R. J., Welch, R. P., Sanna, S., Teslovich, T. M., Chines, P. S., Gliedt, T. P., et al. (2010). LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337. doi: 10.1093/bioinformatics/btq419
Ram, C. V. S. (2004). Angiotensin receptor blockers and diuretics as combination therapy: clinical implications. Am. J. Hypertens. 17, 277–280. doi: 10.1016/j.amjhyper.2003.09.002
Russell, B., Moss, C., Rigg, A., and Van Hemelrijck, M. (2020). COVID-19 and treatment with NSAIDs and corticosteroids: should we be limiting their use in the clinical setting? Ecancermedicalscience 14:1023. doi: 10.3332/ecancer.2020.1023
Scholtens, S., Smidt, N., Swertz, M. A., Bakker, S. J. L., Dotinga, A., Vonk, J. M., et al. (2015). Cohort profile: lifelines, a three-generation cohort study and biobank. Int. J. Epidemiol. 44, 1172–1180. doi: 10.1093/ije/dyu229
Sediq, R., van der Schans, J., Dotinga, A., Alingh, R.A., Wilffert, B., Bos, J. H. J., et al. (2018). Concordance assessment of self-reported medication use in the netherlands three-generation lifelines cohort study with the pharmacy database IaDB.Nl: the pharmLines initiative. Clin. Epidemiol. 10, 981–989. doi: 10.2147/CLEP.S163037
Sun, B. B., Maranville, J. C., Peters, J. E., Stacey, D., Staley, J. R., Blackshaw, J., et al. (2018). Genomic atlas of the human plasma proteome. Nature 558, 73–79. doi: 10.1038/s41586-018-0175-2
The COVID-19 Host Genetics Initiative (2020). The COVID-19 host genetics initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur. J. Hum. Genet. 28, 715–718. doi: 10.1038/s41431-020-0636-6
Tigchelaar, E. F., Zhernakova, A., Dekens, J. A. M., Hermes, G., Baranska, A., Mujagic, Z., et al. (2015). Cohort profile: lifelines DEEP, a prospective, general population cohort study in the Northern Netherlands: study design and baseline characteristics. BMJ Open 5:e006772. doi: 10.1136/bmjopen-2014-006772
Uda, M., Galanello, R., Sanna, S., Lettre, G., Sankaran, V. G., Chen, W., et al. (2008). Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of β-thalassemia. Proc. Natl. Acad. Sci. U. S. A. 105, 1620–1625. doi: 10.1073/pnas.0711566105
van der Velde, K. J., Imhann, F., Charbon, B., Pang, C., van Enckevort, D., Slofstra, M., et al. (2019). MOLGENIS research: advanced bioinformatics data software for non-bioinformaticians. Bioinformatics 35, 1076–1078. doi: 10.1093/bioinformatics/bty742
Weilang, Q., Lazarus, R., Warnes, G., Duffy, D., and Man, M. (2019). Power Calculation for Testing If Disease Is Associated with Marker in a Case-Control Study Using the GeneticsDesign Package (version 1.55.0). R. Available online at: https://www.bioconductor.org/packages/release/bioc/html/GeneticsDesign.html (accessed May 13, 2020).
Winkler, T. W., Justice, A. E., Graff, M., Barata, L., Feitosa, M. F., Chu, S., et al. (2015). The influence of age and sex on genetic associations with adult body size and shape: a large-scale genome-wide interaction study. PLOS Genetics 11:e1005378. doi: 10.1371/journal.pgen.1005378
Wu, Z., and McGoogan, J. M. (2020). Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72 314 cases from the chinese center for disease control and prevention. JAMA. 1239–1242. doi: 10.1001/jama.2020.2648
Xu, X.-W., Xiao-Xin, W., Xian-Gao, J., Kai-Jin, X., Ling-Jun, Y., Chun-Lian, M., et al. (2020). Clinical findings in a group of patients infected with the 2019 novel coronavirus (SARS-Cov-2) outside of Wuhan, China: retrospective case series. BMJ 368:606. doi: 10.1136/bmj.m606
Yan, R., Zhang, Y., Li, Y., Xia, L., Guo, Y., and Zhou, Q. (2020). Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367, 1444–1448. doi: 10.1126/science.abb2762
Zheng, X., Gogarten, S. M., Lawrence, M., Stilp, A., Conomos, M. P., Weir, B. S., et al. (2017). SeqArray-a storage-efficient high-performance data format for WGS variant calls. Bioinformatics 33, 2251–2257. doi: 10.1093/bioinformatics/btx145
Zhernakova, A., Kurilshikov, A., Bonder, M. J., Tigchelaar, E. F., Schirmer, M., Vatanen, T., et al. (2016). Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science. 352, 565–569. doi: 10.1126/science.aad3369
Zhernakova, D. V., Le, T. H., Kurilshikov, A., Atanasovska, B., Bonder, M. J., Sanna, S., et al. (2018). Individual variations in cardiovascular-disease-related protein levels are driven by genetics and gut microbiome. Nat. Genet. 50, 1524–1132. doi: 10.1038/s41588-018-0224-7
Zhou, F., Yu, T., Du, R., Fan, G., Liu, Y., Liu, Z., et al. (2020). Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395, 1054–1062. doi: 10.1016/S0140-6736(20)30566-3
Keywords: PheWAS, ACE2, TMPRSS2, NSAIDs (non-steroidal anti-inflammatory drugs), ARBs (angiotensin II receptor blockers), COVID-19, SARS-CoV-2
Citation: Lopera Maya EA, van der Graaf A, Lanting P, van der Geest M, Fu J, Swertz M, Franke L, Wijmenga C, Deelen P, Zhernakova A, Sanna S and Lifelines Cohort Study (2020) Lack of Association Between Genetic Variants at ACE2 and TMPRSS2 Genes Involved in SARS-CoV-2 Infection and Human Quantitative Phenotypes. Front. Genet. 11:613. doi: 10.3389/fgene.2020.00613
Received: 17 April 2020; Accepted: 19 May 2020;
Published: 08 June 2020.
Edited by:
Marika Kaakinen, University of Surrey, United KingdomReviewed by:
Markus H. Hoffmann, University of Erlangen Nuremberg, GermanyCopyright © 2020 Lopera Maya, van der Graaf, Lanting, van der Geest, Fu, Swertz, Franke, Wijmenga, Deelen, Zhernakova, Sanna and Lifelines Cohort Study. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Serena Sanna, c2VyZW5hLnNhbm5hQGlyZ2IuY25yLml0
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.