 John E. Cronan
John E. Cronan
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Genet. , 21 May 2020
Sec. Genetics of Common and Rare Diseases
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.00510
This article is part of the Research Topic Mitochondrial Disorders: Biochemical and Molecular Basis of Disease View all 10 articles
Three human mitochondrial diseases that directly affect lipoic acid metabolism result from heterozygous missense and nonsense mutations in the LIAS, LIPT1, and LIPT2 genes. However, the functions of the proteins encoded by these genes in lipoic acid metabolism remained uncertain due to a lack of biochemical analysis at the enzyme level. An exception was the LIPT1 protein for which a perplexing property had been reported, a ligase lacking the ability to activate its substrate. This led to several models, some contradictory, to accommodate the role of LIPT1 protein activity in explaining the phenotypes of the afflicted neonatal patients. Recent evidence indicates that this LIPT1 protein activity is a misleading evolutionary artifact and that the physiological role of LIPT1 is in transfer of lipoic acid moieties from one protein to another. This and other new biochemical data now define a straightforward pathway that fully explains each of the human disorders specific to the assembly of lipoic acid on its cognate enzyme proteins.
Lipoic acid is an enzyme cofactor that plays a key role in mitochondrial metabolism. All of the mammalian proteins involved in lipoic acid assembly and utilization are located in the mitochondria. The cofactor is covalently attached to subunits of enzymes involved in central pathways of mitochondrial energy and carbon metabolism and thus defects in the synthesis and attachment of lipoic acid have dire effects on human health (Mayr et al., 2014; Tort et al., 2016).
Neonatal patients suffering from mutations in the genes encoding the enzymes of lipoate assembly show defective respiration, accumulation of toxic levels of specific amino acids, and generally die early in life (Mayr et al., 2014; Tort et al., 2016). Note that this review will not discuss the clinical aspects of these diseases but will focus on the biochemistry of lipoic acid assembly. Reviews that discuss the diagnosis and treatment of these disorders are: (Mayr et al., 2014; Tort et al., 2016). Mutations that result in iron–sulfur cluster biosynthesis deficiencies that affect the activity of lipoic acid synthase (LIAS) (Table 1), the enzyme that inserts the lipoate sulfur atoms, will not be discussed because their effects are not specific to lipoic acid synthesis; other key enzymes (e.g., complexes II and III of the respiratory chain) are also affected.
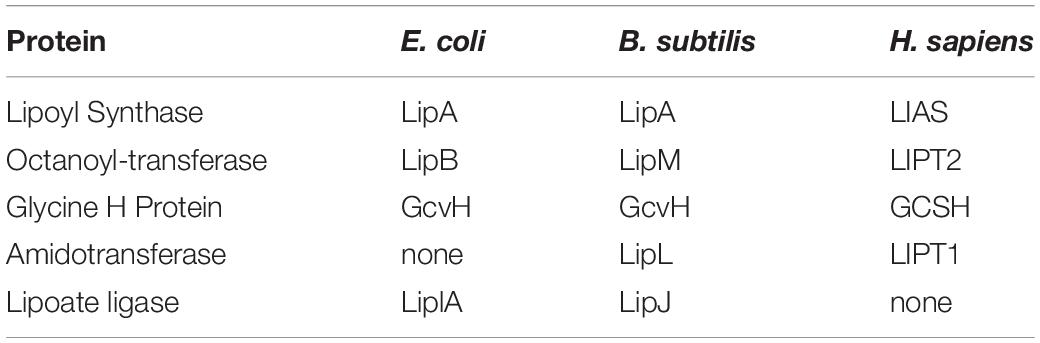
Table 1. The nonenclature differences in E. coli, B. subtilis and humans.
The key enzymes that require lipoic acid for activity are pyruvate dehydrogenase, the enzyme required for entry of carbon into the citric acid cycle and α-ketoglutarate dehydrogenase, an enzyme at the midpoint of the citric acid cycle plus the glycine cleavage system. Two other dehydrogenases, the branched chain keto acid dehydrogenase required for degradation of branched-chain amino acids and the α-oxo(keto)adipate dehydrogenase of lysine degradation also require lipoic acid for activity. However, although accumulation of branched-chain amino acids and lysine in body fluids of patients have been reported (Mayr et al., 2011, 2014), enzymological studies have generally focused on the pyruvate and α-ketoglutarate dehydrogenase due to their key roles in energy production. The loss of these two dehydrogenases short-circuits the citric acid cycle, resulting in severe respiratory deficiency and extreme muscle weakness (Mayr et al., 2014; Tort et al., 2016). Lack of glycine cleavage activity additionally results in elevated brain glycine levels which can result in a host of neurological disorders, including neurodegeneration, encephalopathy, and neonatal-onset epilepsy (Mayr et al., 2014; Tort et al., 2016; Stowe et al., 2018). In general, the first indicator of defective lipoic acid metabolism is the presence of very high levels of lactate (resulting from reduction of the pyruvate that accumulates due to loss pyruvate dehydrogenase activity) in urine and other bodily fluids. Subsequent measurement of body fluid glycine levels consigns the patients into two classes (Tort et al., 2016). Normal glycine levels demonstrate that the glycine cleavage system is functional whereas abnormally high glycine levels indicate that glycine cleavage is defective. Tissue fibroblasts (and/or liver biopsy tissue) derived from the patients are then assayed for the cognate enzyme activities plus the levels of lipoylation of the cognate enzyme proteins. Patients that display absent or low lipoylation of all three enzymes have mutations in either of two genes LIAS or LIPT2 whereas patients that retain glycine cleavage activity are mutant in a third gene, LIPT1 (Mayr et al., 2014; Tort et al., 2016; Table 1). The phenotypes of the two classes of patients could be explained by a simple model in which LIAS and LIPT2 are required for lipoic acid synthesis whereas LIPT1 is not required (demonstrated by the normally lipoylated glycine cleavage system H protein (GCSH) of LIPT1 patients). Since it was clear that LIAS was the sulfur insertion enzyme (Mayr et al., 2011), LIPT2 could be assigned a role in providing the substrate of LIAS whereas LIPT1 would function to relay lipoyl groups from GCSH to the pyruvate and α-ketoglutarate dehydrogenases. (Note that the protein products of the genes have the same name as the encoding gene except that protein names lack italics.) However, there was no precedent for such a pathway in nature and the LIPT1 protein had an enzymatic activity that muddled this simple model. It now seems clear that this misleading enzyme activity is an evolutionary artifact (see below).
The background of the lipoic acid assembly and attachment pathway is required to place the products of these genes in context. In bacteria de novo synthesized lipoic acid attached to its cognate enzyme proteins (lipoyl modification) was shown to arise by a markedly unusual pathway: lipoyl groups are assembled on the cognate enzyme proteins (Figure 1). Although this was first uncovered by genetic and biochemical analysis in bacteria (see below), the bacterial and human proteins are members of the same protein family (see below). The “backbone” of lipoic acid is octanoic acid, an eight-carbon fatty acid synthesized by a distinct mitochondrial fatty acid synthesis system. The relevant form of octanoic acid is that attached to a small carrier protein of fatty acid synthesis called acyl carrier protein (ACP). The chemical linkage that couples octanoate to ACP greatly facilitates transfer of the octanoyl group to the glycine cleavage H protein (GCSH) where the covalently bound octanoyl groups are converted to lipoyl groups by insertion of the two sulfur atoms required for function of the cognate enzymes. Following sulfur insertion lipoyl groups are relayed to the E2 subunits of the two dehydrogenases. The human pathway of Figure 1 has been validated by direct enzyme assays of the proteins and their activity in a reconstituted human pathway (Cao et al., 2018). Hence GCSH functions both in glycine cleavage and in assembly of lipoyl moieties. Indeed, bacterial strains that lack GCSH are defective in both glycine cleavage and lipoic acid assembly (Christensen et al., 2011; Cao et al., 2018).
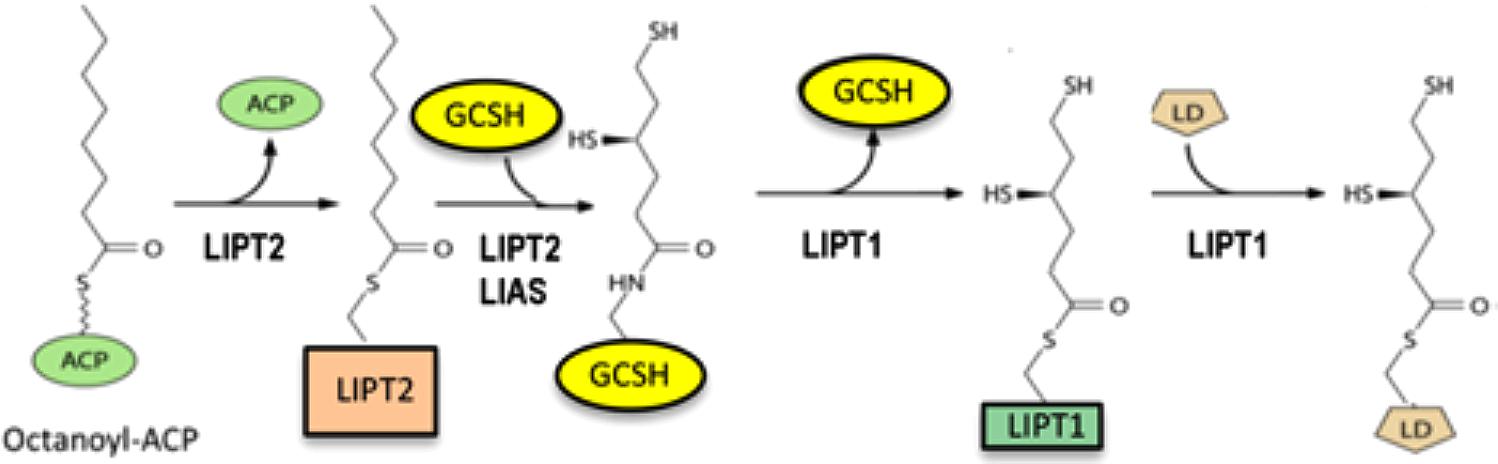
Figure 1. The human pathway of assembly of lipoyl moieties on its cognate proteins. LIPT2 utilizes octanoyl-ACP synthesized by the mitochondrial fatty acid synthesis system to modify GCSH. The reaction proceeds via a LIPT2 acyl enzyme intermediate as shown. The octanoyl-GCSH is then converted to lipoyl-GCSH by LIAS catalyzed sulfur insertion. LIPT1 then transfers the lipoyl moiety from GCSH to form an acyl enzyme intermediate. This in turn is attacked by a specific lysine reside of the lipoyl domain (LD) of the N-termini of the E2 subunits of the dehydrogenases or GCSH to give the active modified enzymes.
Given the pathway (Figure 1) we can now interpret the biochemical phenotypes of the patients. LIPT2 and LIAS proteins are required for lipoic acid assembly because LIPT2 is the enzyme that transfers octanoyl groups from ACP to GCSH to provide the substrate for sulfur insertion by LIAS. Hence patients defective in either of these genes have undetectable or severely decreased levels of all lipoylated proteins and undergo the severe respiratory deficiency and extreme muscle weakness due to citric acid cycle disfunction plus the neurological impairments caused by glycine accumulation in the brain (Tort et al., 2016). In contrast LIPT1 patients generally suffer only the respiratory and muscle weakness problems because GCSH is lipoylated and glycine is cleaved (Tort et al., 2016). The two dehydrogenase proteins lack lipoylation because LIPT1 is the enzyme that transfers lipoyl groups from lipoyl-GCSH to the dehydrogenase proteins. Hence, glycine cleavage is normal and most of these patients escape the neurological problems resulting from glycine accumulation. However, there are reports of neurological involvements in LIPT1 patients (Soreze et al., 2013; Tache et al., 2016; Stowe et al., 2018) which remain unexplained.
Beginning in 1969 Japanese investigators began study of lipoic acid attachment to the glycine cleavage H protein of various vertebrates (Fujiwara et al., 1994, 1996, 1997, 2001). They identified a puzzling enzyme encoded the by the human LIPT1 gene that could transfer the activated intermediate lipoyl-AMP to the H protein. However, the origin of lipoyl-AMP remained a mystery. Since classical ligases in general use ATP to activate the substrate to be ligated as well as perform the transfer of the substrate to the acceptor molecule, the protein seemed a peculiar “half ligase” unable to activate its substrate. This was in contrast to an early report that lipoate was readily attached to pyruvate dehydrogenase in an ATP-requiring reaction that proceeded through a lipoyl-AMP intermediate (Reed et al., 1958) in bacterial extracts. However, since the preparations were only partially purified, the activity could have been due to more than one protein. In 1994 the first homogenous bacterial lipoate ligase was reported (the LplA of E. coli) and unlike the mammalian and avian proteins, it performed the full ligase reaction: incubations containing lipoate and ATP resulted in formation of lipoyl-AMP and the lipoyl moiety was then transferred to the cognate acceptor proteins with release of AMP (Morris et al., 1994). Curiously, this E. coli protein aligned readily with the vertebrate LIPT1 proteins except for the C-terminal 25% of the proteins at which point the sequences diverged greatly (Fujiwara et al., 2001). Subsequent crystal structures of the E. coli LplA and bovine LIPT1 proteins showed that although the N-terminal 75% of the protein structures could be superimposed, the two C-terminal domains were positioned very differently (Fujiwara et al., 2007, 2010). Subsequent studies of other lipoate ligases indicated that the C-terminal domain is required for lipoyl-AMP synthesis consistent with the lack of this activity in the vertebrate proteins (Christensen and Cronan, 2009; Cao and Cronan, 2015). Despite the inability of the vertebrate proteins to perform the overall ligase reaction, several investigators assigned it a role in scavenging dietary lipoate which argued that feeding lipoate to patients should bypass genetic defects in lipoate assembly.
The first indication that lipoate supplementation was unable to reverse the genetic defects in lipoic acid metabolism came from studies of LIAS knockout mice (Yi and Maeda, 2005). Despite supplementation of the diets of the heterozygous mothers with lipoic acid during pregnancy, all homozygous LIAS null embryos died in utero and were reabsorbed This was consistent with the findings that lipoic acid supplementation of the diets of human LIAS, LIPT1, and LIPT2 patients or of their fibroblast cultures failed to alleviate the physiological and biochemical effects of the mutant genes (Baker et al., 2014; Tort et al., 2014, 2016; Tsurusaki et al., 2015). Lipoate is known to rapidly enter the bloodstream (Teichert et al., 2003) and thus it should have been available if a fully competent ligase existed. Additional mouse and tissue culture studies also clearly demonstrated the inability of supplemented lipoic acid to become attached to the key lipoic acid modified proteins of central metabolism (Witkowski et al., 2007; Feng et al., 2009; Smith et al., 2012).
Two genetic selections gave E. coli mutants devoid of ligase activity due to disruption of the encoding gene, lplA. These selections were done based on the premise that the ligase played a role in lipoic acid synthesis but this soon found not to be the case. The mutant bacteria had normal levels of lipoylated enzymes and grew normally (Morris et al., 1994). Further work showed that the physiological function of E. coli lipoate ligase (LplA) was to scavenge lipoic acid from the environment, an advantage for an enteric bacterium. These data led to a new hypothesis in which octanoyl-ACP was the substrate for sulfur insertion. Prior studies had isolated mutants that required lipoic acid for growth and these fell into two classes, those lacking the sulfur insertion enzyme (LipA, a homolog of LIAS) and an enzyme (LipB, a homolog of LIPT2) (Table 1) that transferred either lipoic acid or octanoic acid from their ACP thioesters to pyruvate dehydrogenase (Cronan, 2016). Although study of these enzymes led to the first synthesis of lipoic acid in vitro, (Miller et al., 2000) the putative lipoyl-ACP reaction intermediate could not be detected. The reason for this result became clear when LipB mutants were found to grow with octanoic acid supplementation in place of the usual lipoic acid supplementation (Zhao et al., 2003). Moreover, octanoate-dependent growth required LplA ligase activity. Hence, the combination of octanoate supplementation and ligase activity bypassed the lack of LipB transferase activity (Zhao et al., 2003). The most straightforward interpretation of these results was that lipoyl moieties were assembled on the cognate enzyme proteins. That is, octanoyl groups became covalently attached to the dehydrogenase and were then converted to lipoyl moieties by the LipA sulfur insertion enzyme. Hence there was no lipoyl-ACP intermediate to be detected! This “on-site” assembly was a striking departure from the usual pathway of other attached cofactors (e.g., biotin) which are fully assembled and then transferred onto the cognate protein. The pathway of lipoyl-protein assembly was quickly validated by in vivo and in vitro studies in several laboratories (Booker, 2004; Cicchillo et al., 2004; Bryant et al., 2006; Douglas et al., 2006). Hence E. coli lipoyl assembly requires only two enzymes, the LipB octanoyl transferase and the sulfur insertion enzyme, LipA.
Lipoic acid assembly has also been studied in the yeast, Saccharomyces cerevisiae mainly by genetic studies and immunological assays of protein lipoylation (Sulo and Martin, 1993; Schonauer et al., 2008). The picture that emerged is consistent with the human pathway (see below). However, the yeast proteins involved in lipoate assembly form intractable inclusion bodies when expressed in E. coli or in a yeast expression system (Hermes and Cronan, 2013). Hence minimal in vitro enzymology has been done and that was done with proteins refolded from inclusion bodies, a serious caveat (Hermes and Cronan, 2013). Given the lack of direct demonstrations of the enzymatic activities of the proteins, the yeast pathway lacks the biochemical grounding of the human pathway. An unexplained facet is that most of the yeast lipoic acid synthesis mutants were originally isolated as strains defective in processing of mitochondrial RNAs (Sulo and Martin, 1993; Schonauer et al., 2008).
The gram-positive bacterium, Bacillus subtilis, was studied because it lacked a gene that encoded a recognizable LipB-like octanoyl transferase although the bacterium had a lipA gene, disruption of which resulted in a growth requirement for lipoic acid (Martin et al., 2009). The ability to use exogenous lipoic acid argued that the bacterium must have a functional lipoate ligase. However, analysis of the B. subtilis genome showed not one but three genes encoding putative lipoate ligases, albeit they had only low sequence identity with E. coli LplA. One of these genes encoded a ligase (LplJ) that functionally replaced LplA in E. coli (Martin et al., 2011) whereas a second putative ligase gene encoded an octanoyl transferase (LipM) that had no ligase activity (Christensen and Cronan, 2010). The protein encoded by third ligase candidate gene, LipL, also lacked ligase activity and seemed an enigma, although its inactivation resulted in a growth requirement for lipoic acid. A series of biochemical experiments with purified proteins presented the perplexing result that the B. subtilis LipM octanoyl transferase was unable to transfer octanoate from octanoyl-ACP to its native pyruvate dehydrogenase E2 subunit, although it was active with a foreign (E. coli) pyruvate dehydrogenase subunit (Christensen et al., 2011). These results argued that a factor or factors was missing. Two were found: GcvH, the GCSH and the enigmatic third ligase candidate, LipL (Christensen et al., 2011). Biochemical analyses showed that the LipM octanoyl transferase attached an octanoyl moiety to GcvH which LipA converted to a lipoyl group. LipL then transferred the lipoyl group from GcvH to the dehydrogenase proteins (Christensen et al., 2011). This was the first biochemical evidence for “lipoyl relay,” the process used in the human and yeast pathways. Indeed, the human pathway reaction scheme given in Figure 1 can describe that of B. subtilis given substitution of LipM for LIPT2, GcvH for GCSH, and LipL for LIPT1.
Why is lipoic acid assembled on its cognate enzymes rather than on octanoyl ACP? A good rationale is economy: the levels of cognate proteins to be modified dictate that the cell can make only the amount of lipoic acid required; no excess is made.
Is there an explanation for why lipoyl moieties are relayed from the glycine cleavage H protein to the dehydrogenase rather than directly assembled on the dehydrogenases? A persuasive argument put forth by Braakman and Smith (2014) is that lipoyl relay seems be a “make-do” device that arose when primitive bacteria having the glycine cleavage H protein as the sole lipoylated protein acquired lipoate-requiring dehydrogenases.
All of the enzymes that transfer octanoate or lipoate are related by their structures but with little sequence conservation and comprise a protein family (Pfam PF03099). Crystal structures show that the lipoate ligases (LplA, LplJ), the octanoyl transferases (LIPT2, LipB, LipM), and the lipoyl relay enzymes (LIPT1 and LipL) are all constructed on the same structural scaffold albeit with only minimal amino acid sequence conservation (generally 15–25%) (Cronan, 2016). Sometimes the scaffold has given rise to the same enzyme activity by seemingly different routes. An example is the two bacterial octanoyl transferases (LipB and LipM) which have their active site cysteine residues lie on different loops of the scaffold (Christensen and Cronan, 2010). The Pfam PF03099 scaffold is also found in the biotin protein ligases that attach biotin to its cognate proteins. The fact that proteins having similar sequences (e.g., LplA and LIPT1) can have different enzyme activities makes assignment of functions based solely on sequence comparisons perilous.
Why has LIPT1 retained a partial lipoate ligase reaction when it clearly functions as an amidotransferase that transfers lipoate from GCSH to the dehydrogenases E2 subunits? The answer seems to lie in a newly appreciated aspect of enzyme evolution called “moonlighting”; a protein can acquire a new enzymatic activity while fully or partially retaining its original activity (Copley, 2012, 2014; Jeffery, 2018, 2019). That is, mutations can enhance the moonlighting function without concomitant elimination of the ancestral function. The original ancestor of LIPT1 seems likely to have been a fully functional lipoate ligase like LplA encoded by the genome of the bacterial endosymbiont that became the mitochondrion. In this scenario the price for retooling the ligase into an amidotransferase was that the carboxyl terminus lost the ability to activate lipoate to lipoyl-AMP and the protein became a “half-ligase” and (temporarily?) retained the partial ligase activity. In other words, the lipoyl-transfer activity is an evolutionary remnant, an aspect often seen in moonlighting proteins (Copley, 2014; Jeffery, 2018). Note that moonlighting as been observed for medically important proteins (Sriram et al., 2005; Houten, 2017) and the possibility that moonlighting could complicate understanding of single gene Mendelian inherited disorders has been raised (Sriram et al., 2005; Franco-Serrano et al., 2018). Indeed, the partial LIPT1 ligase activity markedly hindered our understanding the human pathway of lipoyl-protein assembly.
Although the biochemistry and physiology of the human disorders of lipoic acid metabolism is now understood this knowledge offers few approaches to ameliorate the suffering of these patients (as is the case with other mitochondrial diseases). At present this reviewer can offer only the remote possibility of introducing a bacterial ligase into the mitochondria perhaps by inserting a modified ligase gene into the nuclear genome. The encoded ligase would carry an N-terminal sequence targeting it to the mitochondria. The E. coli lipoate ligase is known to modify human lipoyl enzymes (Quinn, 1997).
JC wrote the entire manuscript and takes full responsibility for the contents.
The research from this laboratory discussed herein was supported by Grant AI15650 from the National Institutes of Allergy and Infection Diseases, National Institutes of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Baker, P. R., Friederich, M. W., Swanson, M. A., Shaikh, T., Bhattacharya, K., Scharer, G. H., et al. (2014). Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain 137, 366–379. doi: 10.1093/brain/awt328
Booker, S. J. (2004). Unraveling the pathway of lipoic acid biosynthesis. Chem. Biol. 11, 10–12. doi: 10.1016/j.chembiol.2004.01.002
Braakman, R., and Smith, E. (2014). Metabolic evolution of a deep-branching hyperthermophilic chemoautotrophic bacterium. PLoS One 9:e87950. doi: 10.1371/journal.pone.0087950
Bryant, P., Kriek, M., Wood, R. J., and Roach, P. L. (2006). The activity of a thermostable lipoyl synthase from Sulfolobus solfataricus with a synthetic octanoyl substrate. Anal. Biochem. 351, 44–49. doi: 10.1016/j.ab.2006.01.023
Cao, X., and Cronan, J. E. (2015). The Streptomyces coelicolor lipoate-protein ligase is a circularly permuted version of the Escherichia coli enzyme composed of discrete interacting domains. J. Biol. Chem. 290, 7280–7290. doi: 10.1074/jbc.M114.626879
Cao, X., Hong, Y., Zhu, L., Hu, Y., and Cronan, J. E. (2018). Development and retention of a primordial moonlighting pathway of protein modification in the absence of selection presents a puzzle. Proc. Natl. Acad. Sci. U.S.A. 115, 647–655. doi: 10.1073/pnas.1718653115
Christensen, Q. H., and Cronan, J. E. (2009). The Thermoplasma acidophilum LplA-LplB complex defines a new class of bipartite lipoate-protein ligases. J. Biol. Chem. 284, 21317–21326. doi: 10.1074/jbc.M109.015016
Christensen, Q. H., and Cronan, J. E. (2010). Lipoic acid synthesis: a new family of octanoyltransferases generally annotated as lipoate protein ligases. Biochemistry 49, 10024–10036. doi: 10.1021/bi101215f
Christensen, Q. H., Martin, N., Mansilla, M. C., De Mendoza, D., and Cronan, J. E. (2011). A novel amidotransferase required for lipoic acid cofactor assembly in Bacillus subtilis. Mol. Microbiol. 80, 350–363. doi: 10.1111/j.1365-2958.2011.07598.x
Cicchillo, R. M., Iwig, D. F., Jones, A. D., Nesbitt, N. M., Baleanu-Gogonea, C., Souder, M. G., et al. (2004). Lipoyl synthase requires two equivalents of S-adenosyl-L-methionine to synthesize one equivalent of lipoic acid. Biochemistry 43, 6378–6386. doi: 10.1021/bi049528x
Copley, S. D. (2012). Moonlighting is mainstream: paradigm adjustment required. Bioessays 34, 578–588. doi: 10.1002/bies.201100191
Copley, S. D. (2014). An evolutionary perspective on protein moonlighting. Biochem. Soc. Trans. 42, 1684–1691. doi: 10.1042/BST20140245
Cronan, J. E. (2016). Assembly of lipoic acid on its cognate enzymes: an extraordinary and essential biosynthetic pathway. Microbiol. Mol. Biol. Rev. 80, 429–450. doi: 10.1128/MMBR.00073-15
Douglas, P., Kriek, M., Bryant, P., and Roach, P. L. (2006). Lipoyl synthase inserts sulfur atoms into an octanoyl substrate in a stepwise manner. Angew. Chem. Int. Ed. Engl. 45, 5197–5199. doi: 10.1002/anie.200601910
Feng, D., Witkowski, A., and Smith, S. (2009). Down-regulation of mitochondrial acyl carrier protein in mammalian cells compromises protein lipoylation and respiratory complex I and results in cell death. J. Biol. Chem. 284, 11436–11445. doi: 10.1074/jbc.M806991200
Franco-Serrano, L., Huerta, M., Hernandez, S., Cedano, J., Perez-Pons, J., Pinol, J., et al. (2018). Multifunctional proteins: involvement in human diseases and targets of current drugs. Protein J. 37, 444–453. doi: 10.1007/s10930-018-9790-x
Fujiwara, K., Hosaka, H., Matsuda, M., Okamura-Ikeda, K., Motokawa, Y., Suzuki, M., et al. (2007). Crystal structure of bovine lipoyltransferase in complex with lipoyl-AMP. J. Mol. Biol. 371, 222–234. doi: 10.1016/j.jmb.2007.05.059
Fujiwara, K., Maita, N., Hosaka, H., Okamura-Ikeda, K., Nakagawa, A., and Taniguchi, H. (2010). Global conformational change associated with the two-step reaction catalyzed by Escherichia coli lipoate-protein ligase A. J. Biol. Chem. 285, 9971–9980. doi: 10.1074/jbc.M109.078717
Fujiwara, K., Okamura-Ikeda, K., and Motokawa, Y. (1994). Purification and characterization of lipoyl-AMP:N epsilon-lysine lipoyltransferase from bovine liver mitochondria. J. Biol. Chem. 269, 16605–16609.
Fujiwara, K., Okamura-Ikeda, K., and Motokawa, Y. (1996). Lipoylation of acyltransferase components of alpha-ketoacid dehydrogenase complexes. J. Biol. Chem. 271, 12932–12936. doi: 10.1074/jbc.271.22.12932
Fujiwara, K., Okamura-Ikeda, K., and Motokawa, Y. (1997). Lipoate addition to acyltransferases of alpha-keto acid dehydrogenase complexes and H-protein of glycine cleavage system. Methods Enzymol. 279, 184–193. doi: 10.1016/s0076-6879(97)79022-0
Fujiwara, K., Takeuchi, S., Okamura-Ikeda, K., and Motokawa, Y. (2001). Purification, characterization, and cDNA cloning of lipoate-activating enzyme from bovine liver. J. Biol. Chem. 276, 28819–28823. doi: 10.1074/jbc.M101748200
Hermes, F. A., and Cronan, J. E. (2013). The role of the Saccharomyces cerevisiae lipoate protein ligase homologue, Lip3, in lipoic acid synthesis. Yeast 30, 415–427. doi: 10.1002/yea.2979
Houten, S. M. (2017). Protein moonlighting in inborn errors of metabolism: the case of the mitochondrial acylglycerol kinase. J. Inherit. Metab. Dis. 40, 755–756. doi: 10.1007/s10545-017-0090-y
Jeffery, C. J. (2018). Protein moonlighting: what is it, and why is it important? Philos. Trans. R. Soc. Lond. B Biol. Sci. 373:20160523. doi: 10.1098/rstb.2016.0523
Jeffery, C. J. (2019). Multitalented actors inside and outside the cell: recent discoveries add to the number of moonlighting proteins. Biochem. Soc. Trans. 47, 1941–1948. doi: 10.1042/BST20190798
Martin, N., Christensen, Q. H., Mansilla, M. C., Cronan, J. E., and De Mendoza, D. (2011). A novel two-gene requirement for the octanoyltransfer reaction of Bacillus subtilis lipoic acid biosynthesis. Mol. Microbiol. 80, 335–349. doi: 10.1111/j.1365-2958.2011.07597.x
Martin, N., Lombardia, E., Altabe, S. G., De Mendoza, D., and Mansilla, M. C. (2009). A lipA (yutB) mutant, encoding lipoic acid synthase, provides insight into the interplay between branched-chain and unsaturated fatty acid biosynthesis in Bacillus subtilis. J. Bacteriol. 191, 7447–7455. doi: 10.1128/JB.01160-09
Mayr, J. A., Feichtinger, R. G., Tort, F., Ribes, A., and Sperl, W. (2014). Lipoic acid biosynthesis defects. J. Inherit. Metab. Dis. 37, 553–563.
Mayr, J. A., Zimmermann, F. A., Fauth, C., Bergheim, C., Meierhofer, D., Radmayr, D., et al. (2011). Lipoic acid synthetase deficiency causes neonatal-onset epilepsy, defective mitochondrial energy metabolism, and glycine elevation. Am. J. Hum. Genet. 89, 792–797. doi: 10.1016/j.ajhg.2011.11.011
Miller, J. R., Busby, R. W., Jordan, S. W., Cheek, J., Henshaw, T. F., Ashley, G. W., et al. (2000). Escherichia coli LipA is a lipoyl synthase: in vitro biosynthesis of lipoylated pyruvate dehydrogenase complex from octanoyl-acyl carrier protein. Biochemistry 39, 15166–15178. doi: 10.1021/bi002060n
Morris, T. W., Reed, K. E., and Cronan, J. E. (1994). Identification of the gene encoding lipoate-protein ligase A of Escherichia coli. Molecular cloning and characterization of the lplA gene and gene product. J. Biol. Chem. 269, 16091–16100.
Quinn, J. (1997). Lipoylation of acyltransferase components of 2-oxo acid dehydrogenase complexes. Methods Enzymol. 279, 193–202. doi: 10.1016/s0076-6879(97)79023-2
Reed, L. J., Leach, F. R., and Koike, M. (1958). Studies on a lipoic acid-activating system. J. Biol. Chem. 232, 123–142.
Schonauer, M. S., Kastaniotis, A. J., Hiltunen, J. K., and Dieckmann, C. L. (2008). Intersection of RNA processing and the type II fatty acid synthesis pathway in yeast mitochondria. Mol. Cell. Biol. 28, 6646–6657. doi: 10.1128/MCB.01162-08
Smith, S., Witkowski, A., Moghul, A., Yoshinaga, Y., Nefedov, M., De Jong, P., et al. (2012). Compromised mitochondrial fatty acid synthesis in transgenic mice results in defective protein lipoylation and energy disequilibrium. PLoS One 7:e47196. doi: 10.1371/journal.pone.0047196
Soreze, Y., Boutron, A., Habarou, F., Barnerias, C., Nonnenmacher, L., Delpech, H., et al. (2013). Mutations in human lipoyltransferase gene LIPT1 cause a Leigh disease with secondary deficiency for pyruvate and alpha-ketoglutarate dehydrogenase. Orphanet. J. Rare Dis. 8:192. doi: 10.1186/1750-1172-8-192
Sriram, G., Martinez, J. A., Mccabe, E. R., Liao, J. C., and Dipple, K. M. (2005). Single-gene disorders: what role could moonlighting enzymes play? Am. J. Hum. Genet. 76, 911–924. doi: 10.1086/430799
Stowe, R. C., Sun, Q., Elsea, S. H., and Scaglia, F. (2018). LIPT1 deficiency presenting as early infantile epileptic encephalopathy, Leigh disease, and secondary pyruvate dehydrogenase complex deficiency. Am. J. Med. Genet A 176, 1184–1189. doi: 10.1002/ajmg.a.38654
Sulo, P., and Martin, N. C. (1993). Isolation and characterization of LIP5. A lipoate biosynthetic locus of Saccharomyces cerevisiae. J. Biol. Chem. 268, 17634–17639.
Tache, V., Bivina, L., White, S., Gregg, J., Deignan, J., Boyadjievd, S. A., et al. (2016). Lipoyltransferase 1 gene defect resulting in fatal lactic acidosis in two siblings. Case Rep. Obstet. Gynecol. 2016:6520148. doi: 10.1155/2016/6520148
Teichert, J., Hermann, R., Ruus, P., and Preiss, R. (2003). Plasma kinetics, metabolism, and urinary excretion of alpha-lipoic acid following oral administration in healthy volunteers. J. Clin. Pharmacol. 43, 1257–1267. doi: 10.1177/0091270003258654
Tort, F., Ferrer-Cortes, X., and Ribes, A. (2016). Differential diagnosis of lipoic acid synthesis defects. J. Inherit. Metab. Dis. 39, 781–793. doi: 10.1007/s10545-016-9975-4
Tort, F., Ferrer-Cortes, X., Thio, M., Navarro-Sastre, A., Matalonga, L., Quintana, E., et al. (2014). Mutations in the lipoyltransferase LIPT1 gene cause a fatal disease associated with a specific lipoylation defect of the 2-ketoacid dehydrogenase complexes. Hum. Mol. Genet. 23, 1907–1915. doi: 10.1093/hmg/ddt585
Tsurusaki, Y., Tanaka, R., Shimada, S., Shimojima, K., Shiina, M., Nakashima, M., et al. (2015). Novel compound heterozygous LIAS mutations cause glycine encephalopathy. J. Hum. Genet. 60, 631–635. doi: 10.1038/jhg.2015.72
Witkowski, A., Joshi, A. K., and Smith, S. (2007). Coupling of the de novo fatty acid biosynthesis and lipoylation pathways in mammalian mitochondria. J. Biol. Chem. 282, 14178–14185. doi: 10.1074/jbc.M701486200
Yi, X., and Maeda, N. (2005). Endogenous production of lipoic acid is essential for mouse development. Mol. Cell. Biol. 25, 8387–8392. doi: 10.1128/MCB.25.18.8387-8392.2005
Keywords: lipoic acid, lipoate assembly, pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, glycine cleavage system, LIPT1, LIPT2, LIAS
Citation: Cronan JE (2020) Progress in the Enzymology of the Mitochondrial Diseases of Lipoic Acid Requiring Enzymes. Front. Genet. 11:510. doi: 10.3389/fgene.2020.00510
Received: 06 March 2020; Accepted: 27 April 2020;
Published: 21 May 2020.
Edited by:
Al-Walid Mohsen, University of Pittsburgh, United StatesReviewed by:
Sandro Ghisla, University of Konstanz, GermanyCopyright © 2020 Cronan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John E. Cronan, amVjcm9uYW5AaWxsaW5vaXMuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.