 Ruiyan Hou
Ruiyan Hou Lida Wang3†
Lida Wang3† Yi-Jun Wu
Yi-Jun Wu- 1Laboratory of Molecular Toxicology, State Key Laboratory of Integrated Management of Pest Insects and Rodents, Institute of Zoology, Chinese Academy of Sciences, Beijing, China
- 2College of Life Science, University of Chinese Academy of Sciences, Beijing, China
- 3Department of Scientific Research, General Hospital of Heilongjiang Province Land Reclamation Bureau, Harbin, China
ATP-binding cassette (ABC) proteins play important roles in a wide variety of species. These proteins are involved in absorbing nutrients, exporting toxic substances, and regulating potassium channels, and they contribute to drug resistance in cancer cells. Therefore, the identification of ABC transporters is an urgent task. The present study used 188D as the feature extraction method, which is based on sequence information and physicochemical properties. We also visualized the feature extracted by t-Distributed Stochastic Neighbor Embedding (t-SNE). The sample based on the features extracted by 188D may be separated. Further, random forest (RF) is an efficient classifier to identify proteins. Under the 10-fold cross-validation of the model proposed here for a training set, the average accuracy rate of 10 training sets was 89.54%. We obtained values of 0.87 for specificity, 0.92 for sensitivity, and 0.79 for MCC. In the testing set, the accuracy achieved was 89%. These results suggest that the model combining 188D with RF is an optimal tool to identify ABC transporters.
Introduction
The ATP-binding cassette (ABC) transporters are members of the membrane protein superfamily that translocate various molecules across extra- and intra-cellular membranes. ABC transporters are split into eight subfamilies from ABCA to ABCH. In humans, there are only seven subfamilies, designated ABCA through ABCG. Plants do not contain ABCH but instead possess ABCI (Sheps et al., 2004; Ofori et al., 2018). ABC transport proteins bind ATP and consume energy to mediate the movement of a variety of molecules across all cell membranes. As Figure 1 shows, the core architecture is a pair of conversed cytoplasmic domains: the transmembrane domain (TMD) and nucleotide binding domain (NBD) (Maqbool et al., 2015). TMDs are attached transmembrane domains that contain the ligand binding site. In most species, TMDs are composed of five to six α-helical segments (Locher, 2016). NBDs are responsible for ATP binding and hydrolysis (Locher, 2016), and conformational changes in the TMDs. The TMD amino acid sequence and topology are also different in different types of ABC transporters (Beis, 2015). The NBD domains adopt open or closed conformations by forming dimers. When NBD dimers separate, the transporter is inactive. ATP transporters possess ATPase activity when the NBD dimer conformation is closed (Gerber et al., 2008; Kadaba et al., 2008; Ward et al., 2013). During the process of conformational changes, the NBD plays the role of the transporter “engine” (Locher, 2008). Because of the important function of NBDs, the NBD domains are very conserved across different ABC transporter types.
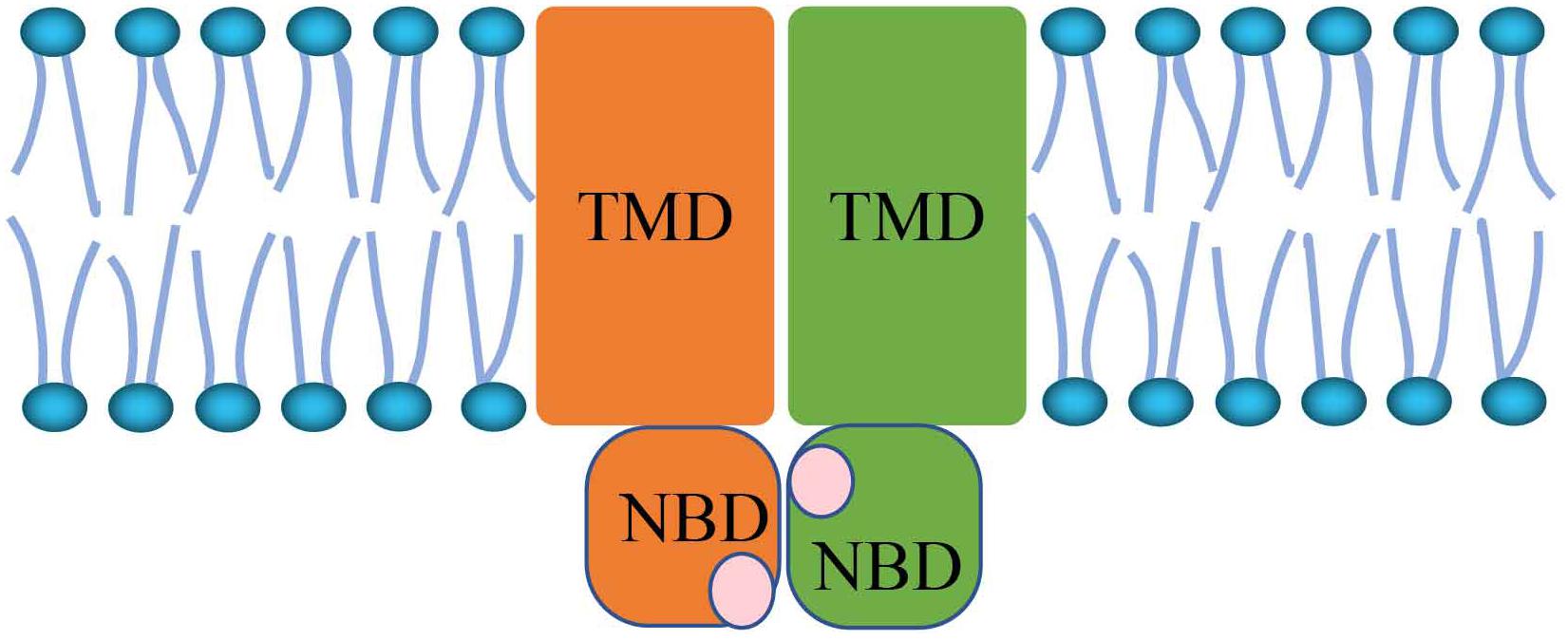
Figure 1. The structure of ATP-binding cassette (ABC) transporters.
ATP-binding cassette transporters play a very important role in many species, from simple bacteria to complex humans. In bacteria, ABC transporters include two types: ABC importers and ABC exporters. ABC importers contribute to the intake of nutrients and micronutrients by importing sugars, metal ions, and vitamins (Davidson et al., 2008; Cui and Davidson, 2011). ABC exporters are predominantly involved in exporting toxic substances and drug resistance (Seeger and van Veen, 2009; Wong et al., 2014). Bacterial ABC exporters also build lipid-linked blocks (Ruiz et al., 2008). Plant ABC transporters are involved in the exchange of secondary metabolites, coating materials, plant hormones and supportive materials. These functions are helpful to the overall development of plants (Hwang et al., 2016). In humans, most of the known functions of ABC transporters involve the transport of antigenic peptides that are relevant to biomedicine and clinical medicine (Leprohon et al., 2011). Mutations of the genes encoding ABC protein contribute to a variety of human disorders, such as cholesterol and bile transport defects, neurological disease, and cystic fibrosis. Mutations also contribute to drug resistance (Dean et al., 2001).
Many biological laboratories identified ABC transporters by artificial annotation. Pleiotropic drug resistance (PDR) transporters constitute a subfamily that belongs to the ABC superfamily. The Nicotiana tabacum PDR gene NtPDR6 was identified via the Basic Local Alignment Search Tool (BLAST). Previous studies compared the P. hybrida genome sequence to expressed sequence tags of N. tabacum in the National Center for Biotechnology database using BLAST (Xie et al., 2015). They found a sequence that was similar to the P. hybrida PDR gene. Finally, they used the molecular biology to clone this gene and demonstrated its function. Some researchers also identified ABC transporters in monogeneans including Gyrodactylus salaris, Protopolystoma xenopodis, Eudiplozoon nipponicum, and Neobenedenia melleni and identified the transporter subfamily of each species. They identified putative ABC proteins of monogeneans by using BLASTp and screened against the putative proteins in Pfam using the HMMER web server. The server is based on protein structure and discards proteins without the conserved domains (NBD and TMD) (Caña-Bozada et al., 2019). Therefore, this method is based on homology. Putative ABC genes in the Anopheles gambiae genome sequence were detected using various software including GENSCAN and the HMMER package (Bateman et al., 1999) which are ab initio techniques. GENSCAN and HMMER are based on the Hidden Markov Model (HMM) and may be used to predict the location of genes and their exon-intron boundaries (Burge and Karlin, 1997). The accuracy of these methods needs improvement, and the experiments are time-consuming and have a tremendous cost.
With the advent of the era of big data, computational prediction based on machine learning has become a powerful approach for identifying important proteins in biology. This method does not replace biological experiments, but it improves the accuracy of prediction and provides more clues for biological experimentation. There are many examples of the application of machine learning algorithms for protein identification. A web server and software (BinMemPredict) was developed to predict membrane protein types. This approach demonstrated an accuracy of 91.2% for the identification of membrane proteins and an accuracy of 86.1% for selecting membrane protein types (Zou et al., 2013a). Pretata used a novel feature and a dimensionality reduction strategy to predict TATA binding proteins, and it achieved 92.92% prediction accuracy (Zou et al., 2016). Machine learning was also used to combine support vector machine (SVM) and PSSM distance transformation to identify DNA-binding proteins (Xu et al., 2015; Dong et al., 2019; Li Z. L. et al., 2019; Yan et al., 2019). Zou et al., proposed a model using a SVM named AOPs-SVM to identify antioxidant proteins (Jin et al., 2019).
The present study used 188D for feature extraction and employed five classifiers to predict ABC transporters. The method of feature extraction focused on the sequence information and the physical and chemical properties. We also developed the t-SNE algorithm to visualize the features. Finally, we compared five different classifiers and revealed that random forest (RF) was the optimal model to identify ABC transporters. The overall process is shown in Figure 2.
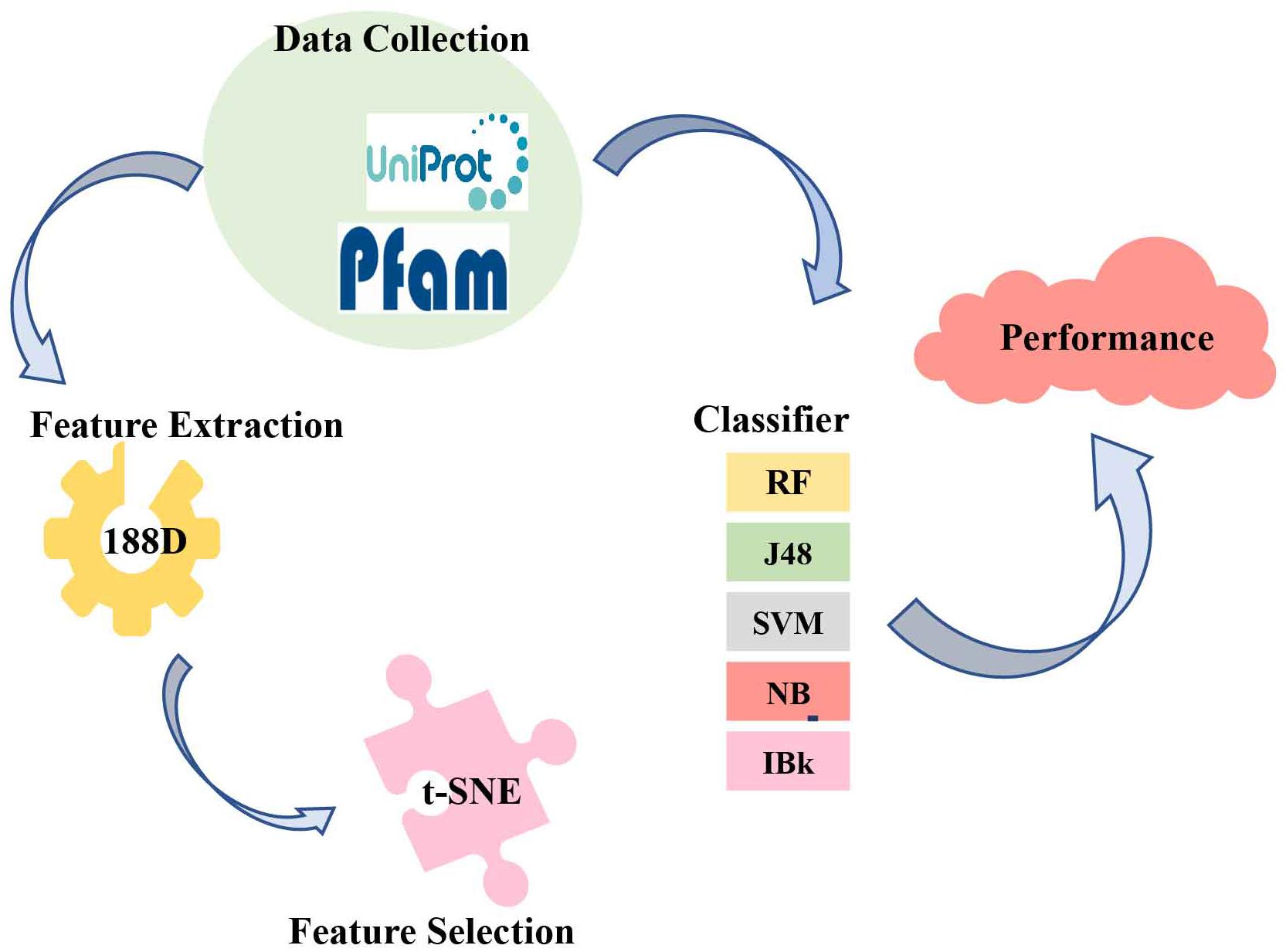
Figure 2. Overall Process of this study.
Materials and Methods
Dataset
The identification of ABC transporters refers to the process of judging whether a protein is an ABC transporter. This classification problem needs two kinds of proteins. The present study used the key word “ABC transporter” to search the sequences in the Uniprot database. This search produced 1509 reviewed sequences that were used as the positive set. After obtaining the positive example set, we constructed a negative set using the following steps. We removed the families, including the above-mentioned positive sequences, from the protein family database PFAM. In these residual families, we extracted the longest protein sequence from each family as a negative sample. A total of 10661 negative example sequences was assigned to the negative data pool.
To ensure reliability of the experimental results, CD-HIT (Fu et al., 2012) was used to eliminate redundant data with a threshold of 0.6. The final dataset contained 875 positive samples and 9736 negative samples.
The positive samples and negative samples were imbalanced. To solve this problem, we randomly selected 875 negative samples from the total 9736 of negative example sequences. Therefore, the numbers of the positive samples and the negative samples were equal. This operation was repeated 10 times and we obtained 10 negative example sets. These 10 negative examples and the same positive example sets formed the 10 training sets, respectively. In every selection process, the remaining 8861 negative samples were assigned to the test set. A total of 10 test sets was obtained.
Feature Extraction
The 188D feature extraction method was used in this study. This method extracts 188 features based on protein sequence information and physical and chemical properties. Previous researchers used the composition-position of proteins and their physical-chemical properties independently to extract protein features (Dubchak et al., 1995; Ding and Dubchak, 2001; Shen et al., 2017, 2019; Wang et al., 2017; Yu et al., 2017; Liu et al., 2018; Qiao et al., 2018; Xiong et al., 2018; Zhang et al., 2018a, b; Zou et al., 2019). In 2003, Cai et al. first combined amino acid sequences with their physicochemical properties to finish feature extraction (Cai et al., 2003). In summary, 188 features are divided into two different categories. One category consists of the amino acid composition that is expressed by 20 features. The other category consists of the physical chemical properties, including hydrophobicity, polarity, polarizability, normalized van der Waals volume, secondary structure, charge, surface tension, and solvent accessibility. The detail about the physicochemical properties is shown in Table 1.
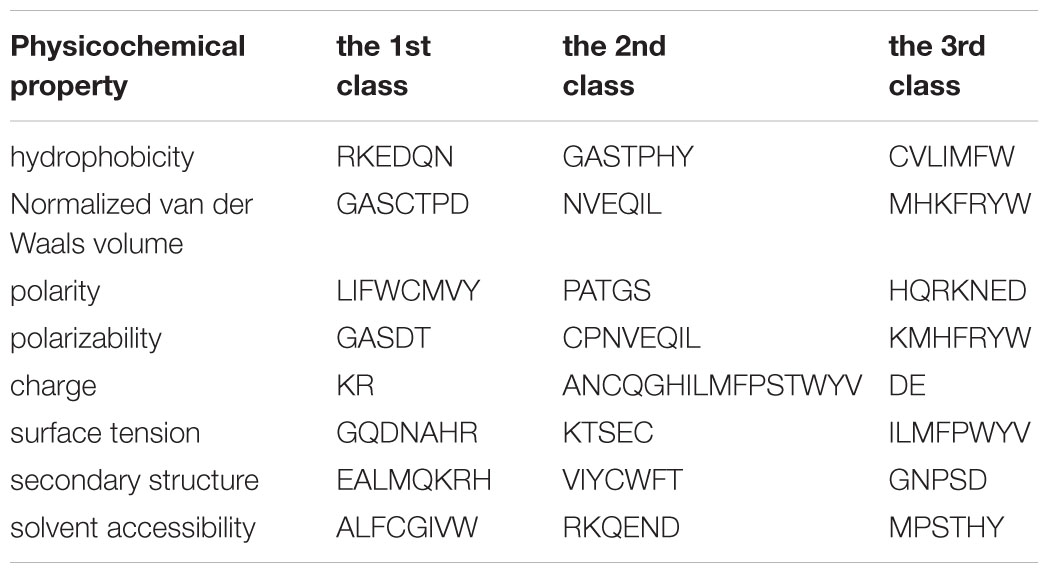
Table 1. Three class divided according to physicochemical property.
There are 20 amino acids. We calculated the respective frequency of the 20 amino acids as n1, n2, n3…n20. The feature can be expressed as:
where F is the feature, and L is the length of sequences.
Next, these 20 amino acids were divided into three types according to their physicochemical properties. The three categories included the content, distribution and the bivalent frequency, which were used to describe the physicochemical properties of proteins. We used surface tension as an example.
First, the 20 amino acids were divided into three groups (Chen et al., 2012), namely, the GQDNAHR group, KTSEC group, and ILMFPWYV group, according to their surface tensions. We calculated the content of the three groups, which are expressed as CS1, CS2, and CS3, respectively. The feature was denoted as:
For the AAs of the GQDNAHR group, the position of the first, the first and 25%, 50%, and 75% of the chain length are represented by DSij where i ranges from 1 to 3 and j ranges from 1 to 5.
The frequencies of occurrence of bigeminal sequences were calculated as (Zou et al., 2013b):
A total of (3 + 3 + 3× 5) = 21 feature vectors were extracted from each property, and we finally extracted all 168 (21 × 8) feature vectors from the eight physicochemical properties. In summary, the 188 (168 + 20) features were used to express the characteristics of ABC transporter protein. The process of feature extraction is illustrated in Figure 3.
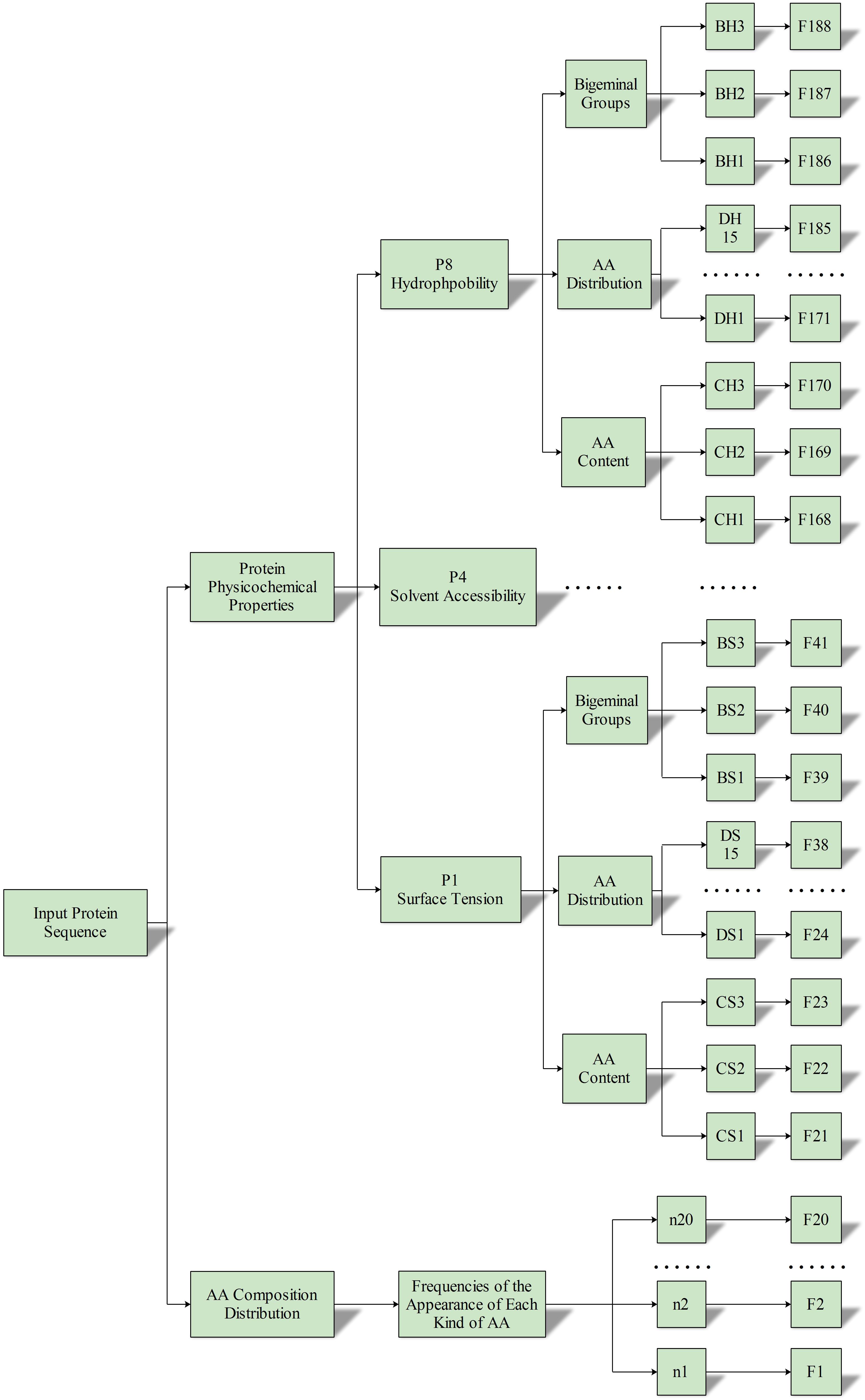
Figure 3. Flowchart of the 188D feature extraction method.
Feature Selection
t-Distributed Stochastic Neighbor Embedding (t-SNE) is a dimensionality reduction tool based on non-linear manners. t-SNE is particularly good at the visualization of high-dimensional datasets (Shao et al., 2018). The present study reduced the 188 features of protein sequences to two-dimensional features by using t-SNE. The t-SNE algorithm uses the joint probabilities to express the similarities between data points. t-SNE endeavors to minimize the Kullback–Leibler discrepancy between the joint probabilities of the low-dimensional and the high-dimensional data. t-SNE was applied to create the two-dimensional embedding in the dataset described above using the R package Rtsne. The settings of Rtsne in the present study were as follows: dims = 2, perplexity = 10, verbose = TRUE, theta = 0, max_iter = 1000, and exaggeration_factor = 8.
Classifier
We used RF, J48, Naïve Bayes, SVM, and IBk as the classifiers. These classifiers were implemented in Weka, which contains a wide variety of machine learning algorithms based on a Java environment (Frank et al., 2004).
Random Forest
Random forest is an ensemble learning method that consists of many classification trees (Breiman, 2001), and it is widely used in bioinformatics research (Ding et al., 2016; Liu, 2017; Liu et al., 2017; Wei et al., 2017a, b, 2019; Zeng et al., 2017a; Yu et al., 2018; Gong et al., 2019; Lv et al., 2019; Ru et al., 2019). The “forest” is built by using bagging and random feature selection methods. The bagging method generally combines various learning models to increase the overall result. RFs are an improvement over bagged decision trees (Breiman, 1996). The details about this algorithm are described below.
First, “M” features were randomly selected from total “n” features, where M <<n. Then, we use the best split point to calculate the node “b” among the “M” features.
Next, by using the best split, the node was split into daughter nodes. These three steps were repeated until “l” number of nodes was reached. Finally, we constructed a “forest” by repeating the above steps for “n” number of times to build “n” number of trees. These are the pseudocode of RF creation.
After the RF model was produced, we made predictions. The test features were taken, and the codes of each randomly produced decision tree were used to predict the results. We calculated the votes for each of the predicted results. Finally, we chose the high voted prediction target as the ultimate prediction (Song et al., 2017).
J48
J48 implements the decision tree algorithm C4.5 (Quinlan, 1986). Ross Quinlan improved ID3 to the C4.5 algorithm in 1993. C4.5 builds decision trees from training data using information entropy. At each step, C4.5 selects an attribute of the data to effectively split into subsets. Examining the standardized information gain or the variation in entropy is the splitting criterion (Radhika and Rao, 2015; Li M. J. et al., 2019). The highest standardized information gain of an attribute is chosen to make the decision. This process recurs on each branch node. When all of the samples included in the branch nodes of the decision belong to the same class, the process is stopped (Jain et al., 2009).
Naïve Bayes
Naïve Bayes is an effective classifier based on the Bayesian Theorem (Cao et al., 2003). The Bayesian Theorem finds the probability of an event occurring when the probability of another event occurring is known. The Bayesian Theorem is primarily based on conditional probability, which is given in the following equation:
where y is a class variable and x is a dependent feature vector. P(y) is called class probability and P(xn| y) is called conditional probability that means the probability of y given x1,……,xn. The formula above may be expressed as:
We can remove the denominator because it remains constant for a given input:
When this formula is applied to a Naïve Bayes, we get the probability of given features for all possible values of the class variable y and select the outcomes with maximum probability. This value may be expressed as:
Support Vector Machine
The SVM is a supervised machine learning algorithm based on the structural risk minimization principle from statistical learning theory. Vapnik first introduced this algorithm in 1995 (Mohammad and Nagarajaram, 2011). In this algorithm, every data point was plotted as a dot in n-dimensional spaces (where n is the number of samples’ features). Then, we find an optimal hyperplane that differentiates the two classes very well. This hyperplane can maximize the margin between the two classes, and support vectors define the hyperplane. SVM has been applied to many tasks in bioinformatics (Wei et al., 2014, 2016, 2018; Ding et al., 2017; He et al., 2018; Zou et al., 2018; Fang et al., 2019; Liu and Li, 2019; Liu et al., 2019; Zeng et al., 2019b, c; Zhang M. et al., 2019; Zhang X. et al., 2019; Zhu et al., 2019).
IBk
The IBk is a machine learning classifier based on the k-nearest-neighbor algorithm. “Feature similarity” is used to predict the values of new data points in the K-nearest-neighbor algorithm. For implementing this algorithm, we choose training and testing data as datasets. Then, we choose an integer as “K.” We use various methods such as Euclidean, Manhattan or Hamming, to calculate the distance between the test data and each line of training data. We sorted each line of training data in increasing order based on the distance value and choose the top K lines from the sorted array. Finally, we assigned the test point to a class based on the most frequent class of these rows.
Prediction System Assessment
The present study used some common evaluation indicators, including the total prediction accuracy (ACC), sensitivity (SN), specificity (SP), and Matthews’ correlation coefficient (MCC) (Matthews, 1975; Yu et al., 2015; Wei et al., 2017c; Zeng et al., 2017b, 2019a; Jia et al., 2018; Hong et al., 2019; Shan et al., 2019; Zhang et al., 2019a, b). These indicators are expressed as follows:
where TP, TN, FP, and FN express the rates of true positives, true negatives, false positives, and false negatives, respectively.
We also used receiver operating characteristic (ROC) curves and the area under the curve (AUC) to judge the performance of each classifier. The ROC curve is used to choose some better classifiers that maximize the true positives and minimize the false positives. Its abscissa is the false positive rate (FPR), and its ordinate is the true positive rate (TPR). We plotted the ROC curves in R. AUC is the area under the ROC curve. Generally, the higher the AUC number, the better the classifier.
Results and Discussion
t-SNE Visualization of the Feature Extracted by 188D
To examine whether the high-level features extracted by 188D had the prediction power and were generalizable, we visualized the features for the training set by applying t-SNE (Figure 4). We visualized 1750 proteins including 875 positive samples and 875 negative samples. All 10 training sets were visualized by t-SNE. Each sample had 188 features. t-SNE mapped the 188 features based on two principal features and minimized the information loss during dimension. As the figure shows, the various protein classes were almost separated clearly. The process suggested that 188D extracted representative features, and the samples were split by using t-SNE. Therefore, the classifier exhibited a sufficient performance based on these features.
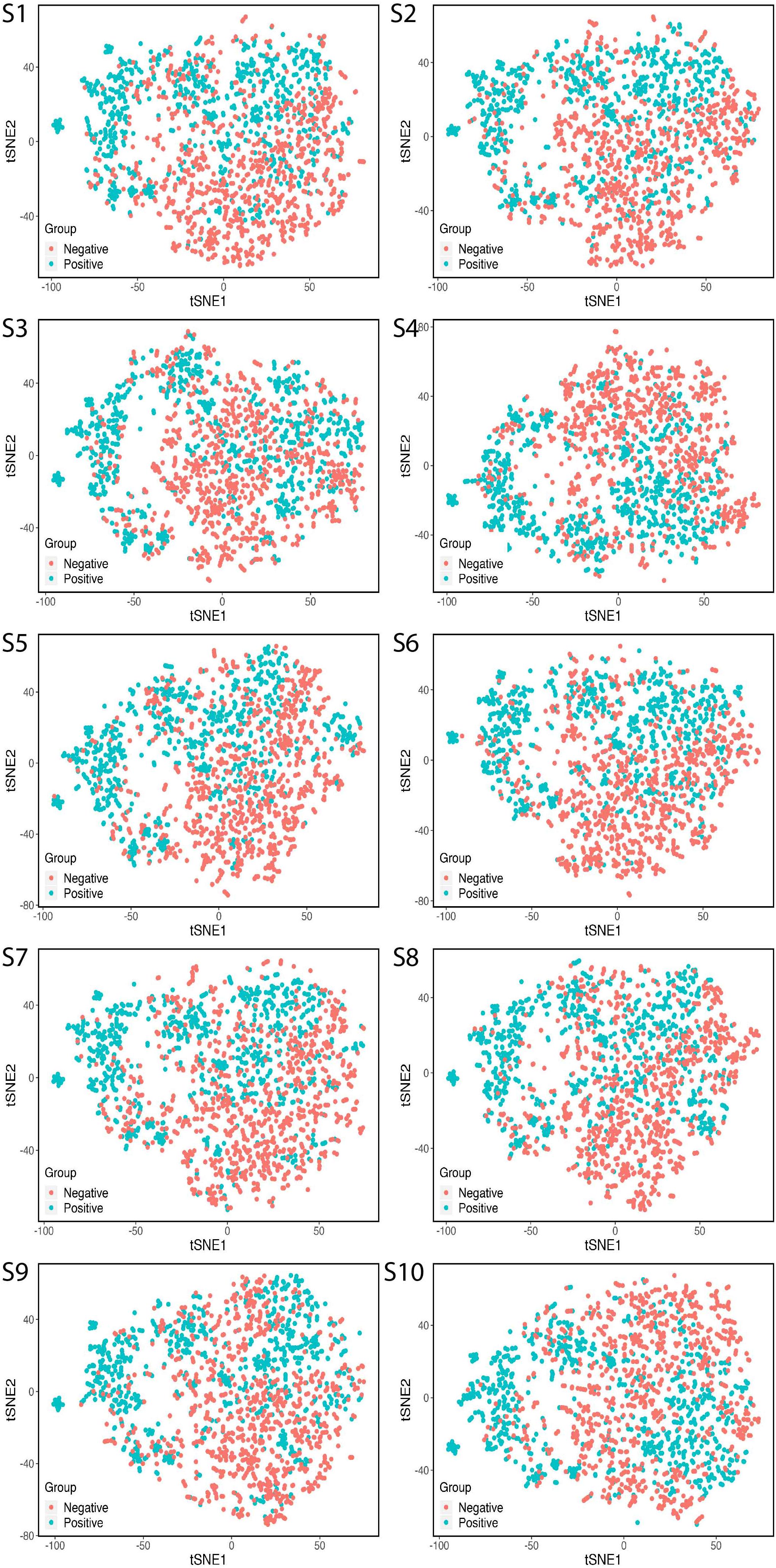
Figure 4. Display of the training features by t-SNE. S is the abbreviation for sample.
Performance of Different Classification Algorithms
To evaluate the performance of different classifiers on our data set, we used 10-fold cross-validation to select the optimal parameters in the training set by implementing WEKA. The excellent parameters in RF were obtained and evaluated on the test set. We repeated the entire process 10 times to ensure the accuracy of the experimental results.
The performances of different classifier models on the training set and testing sets are shown in Figure 5. For the training set, we used the total prediction accuracy, SN, SP, and MCC as the evaluation indicators. The weighted average of these results was used. The accuracy is shown in Figure 5A. The accuracy of RF was 0.8954, and the accuracy of Naïve Bayes was only 0.7397. Surprisingly, for the sensitivity indicator (Figure 5B), Naïve Bayes gave the best performance, and RF was close behind. The highest sensitivity score was 0.9346, and the second sensitivity score was 0.9189. Sensitivity measures the proportion of positives that were correctly identified. This result suggests that Naïve Bayes would recognize the positive samples. However, its specificity score (0.5448) was very poor. This result indicates that SVM occurred at the expense of specificity for higher sensitivity. The specificity scores of Naïve Bayes, IBk, J48, SVM, and RF were 0.5448, 0.7925, 0.8007, 0.8499, and 0.8718, respectively, as shown in Figure 5C. Obviously, the RF classifier showed the best specificity. According to the data shown in Figure 5D, the MCC value of RF is higher than the MCC value of the other algorithm. We achieved an MCC score of 0.7915 using the RF model. However, the lowest MCC value was only 0.521.

Figure 5. Performance of different classifier. (A) Accuracy comparison for the training sets by using all the classification methods. (B) Sensitivity comparison for the training sets using all of the classification methods. (C) Specificity comparison for the training sets by using all of the classification methods. (D) Matthew’s correlation coefficient comparison for training sets by using all the classification methods. (E) Accuracy comparison for the testing sets by using all of the classification methods.
Receiver operating characteristic curves provide a useful approach to compare different classifiers. The performance of all classifiers in ROC plots is shown in Figure 6. The five classification models used in 10 randomly selected training sets performed differently. RF covered the maximum AUC in all training sets followed by Naïve Bayes, SVM, IBk, and J48. All of the AUC values of RF exceeded 95% in 10 training sets.
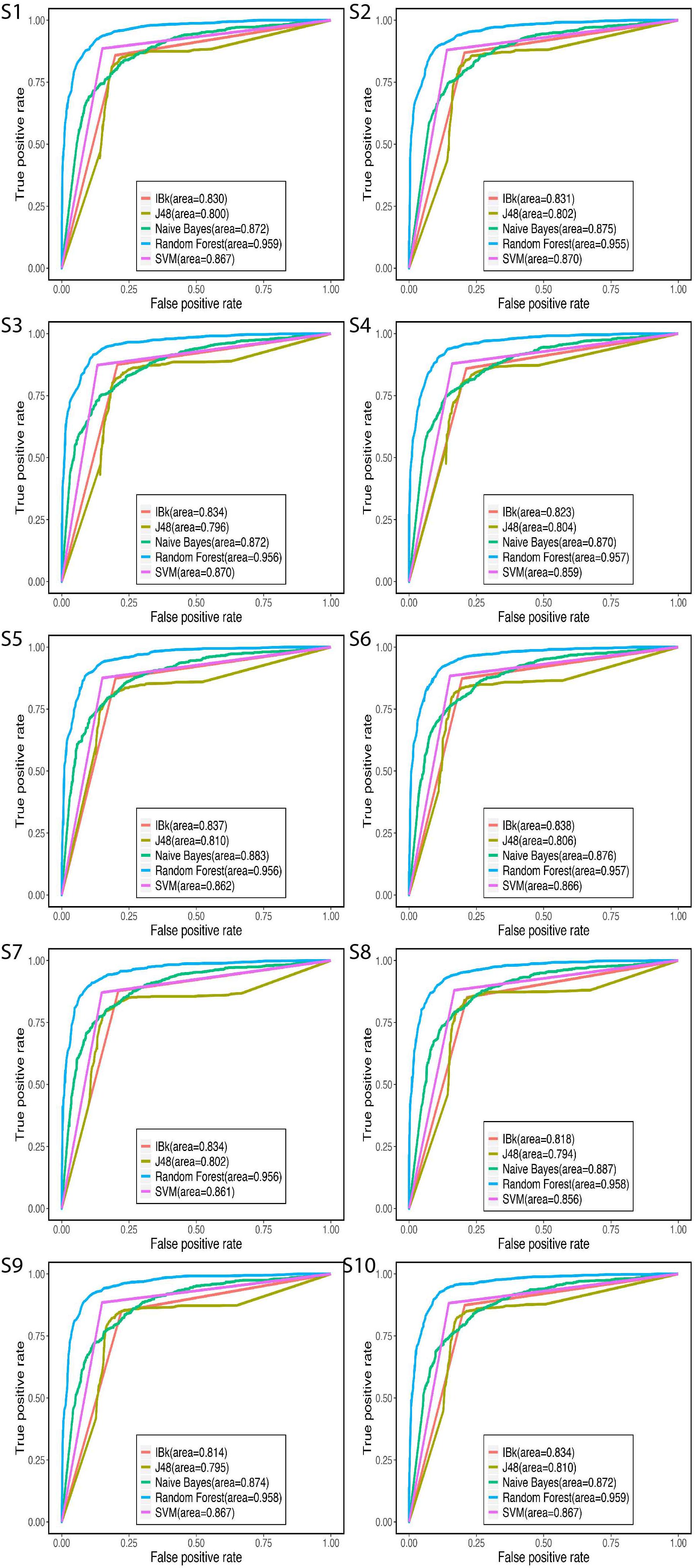
Figure 6. ROC curve to compare difference classifiers. S is the abbreviation for sample.
The testing set was used to test the models mentioned above. As the Figure 5E shows, except for Naïve Bayes, the accuracy values of the remaining classifiers exceeded 80%, and the accuracy score of RF reached 89%.
All of these indicators demonstrate that RF gives the best performance, and Naïve Bayes is the worst classifier. The RF classifier was considered the optimal classifier for prediction ABC transporter proteins in the dataset.
On the basis of the analysis above, we may draw a conclusion that the optimal strategy of identifying ABC transporter is using 188D as feature extraction method and RF as classifier.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found at Uniprot: https://www.uniprot.org/uniprot/?query=ATP-binding+cassette+transporter&sort=score and https://github.com/Jenny-Jason/Predicting-ABC-transporters.
Author Contributions
Y-JW conceived and designed the project. RH and LW performed the experiments and wrote the manuscript.
Funding
This work was supported in part by the grants from the National Natural Science Foundation of China (No. 31672366).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Bateman, A., Birney, E., Durbin, R., Eddy, S. R., Finn, R. D., and Sonnhammer, E. L. L. (1999). Pfam 3.1: 1313 multiple alignments and profile HMMs match the majority of proteins. Nucleic Acids Res. 27, 260–262. doi: 10.1093/nar/27.1.260
Beis, K. (2015). Structural basis for the mechanism of ABC transporters. Biochem. Soc. Trans. 43, 889–893. doi: 10.1042/BST20150047
Burge, C., and Karlin, S. (1997). Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94. doi: 10.1006/jmbi.1997.0951
Cai, C. Z., Han, L. Y., Ji, Z. L., Chen, X., and Chen, Y. Z. (2003). SVM-Prot: web-based support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Res. 31, 3692–3697. doi: 10.1093/nar/gkg600
Caña-Bozada, V., Morales-Serna, F. N., García-Gasca, A., Llera-Herrera, R., and Fajer-Ávila, E. J. (2019). Genome-wide identification of ABC transporters in monogeneans. Mol. Biochem. Parasitol. 234:111234. doi: 10.1016/j.molbiopara.2019.111234
Cao, J., Panetta, R., Yue, S., Steyaert, A., Young-Bellido, M., and Ahmad, S. (2003). A naive Bayes model to predict coupling between seven transmembrane domain receptors and G-proteins. Bioinformatics 19, 234–240. doi: 10.1093/bioinformatics/19.2.234
Chen, W., Liu, X., Huang, Y., Jiang, Y., Zou, Q., and Lin, C. (2012). Improved method for predicting protein fold patterns with ensemble classifiers. Genet. Mol. Res. 11, 174–181. doi: 10.4238/2012.January.27.4
Cui, J., and Davidson, A. L. (2011). ABC solute importers in bacteria. Essays Biochem. 50, 85–99. doi: 10.1042/bse0500085
Davidson, A. L., Dassa, E., Orelle, C., and Chen, J. (2008). Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol. Mol. Biol. Rev. 72, 317–364. doi: 10.1128/MMBR.00031-07
Dean, M., Hamon, Y., and Chimini, G. (2001). The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 42, 1007–1017. doi: 10.1101/gr.184901
Ding, C. H., and Dubchak, I. (2001). Multi-class protein fold recognition using support vector machines and neural networks. Bioinformatics 17, 349–358. doi: 10.1093/bioinformatics/17.4.349
Ding, Y., Tang, J., and Guo, F. (2016). Predicting protein-protein interactions via multivariate mutual information of protein sequences. BMC Bioinform. 17:398. doi: 10.1186/s12859-016-1253-9
Ding, Y., Tang, J., and Guo, F. (2017). Identification of drug-target interactions via multiple information integration. Inform. Sci. 41, 546–560. doi: 10.1016/j.ins.2017.08.045
Dong, M. H., Wen, S. P., Zeng, Z. G., Yan, Z., and Huang, T. W. (2019). Sparse fully convolutional network for face labeling. Neurocomputing 331, 465–472. doi: 10.1016/j.neucom.2018.11.079
Dubchak, I., Muchnik, I., Holbrook, S. R., and Kim, S.-H. (1995). Prediction of protein folding class using global description of amino acid sequence. Proc. Natl. Acad. Sci. U.S.A. 92, 8700–8704. doi: 10.1073/pnas.92.19.8700
Fang, T., Zhang, Z., Sun, R., Zhu, L., He, J., Huang, B., et al. (2019). RNAm5CPred: prediction of RNA 5-Methylcytosine Sites Based on Three Different Kinds of Nucleotide Composition. Mol. Ther. Nucleic Acids 18, 739–747. doi: 10.1016/j.omtn.2019.10.008
Frank, E., Hall, M., Trigg, L., Holmes, G., and Witten, I. H. (2004). Data mining in bioinformatics using Weka. Bioinformatics 20, 2479–2481. doi: 10.1093/bioinformatics/bth261
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gerber, S., Comellas-Bigler, M., Goetz, B. A., and Locher, K. P. (2008). Structural basis of trans-inhibition in a molybdate/tungstate ABC transporter. Science 321, 246–250. doi: 10.1126/science.1156213
Gong, Y., Niu, Y., Zhang, W., and Li, X. (2019). A network embedding-based multiple information integration method for the MiRNA-disease association prediction. BMC Bioinform. 20:468. doi: 10.1186/s12859-019-3063-3
He, J., Fang, T., Zhang, Z., Huang, B., Zhu, X., and Xiong, Y. (2018). PseUI: pseudouridine sites identification based on RNA sequence information. BMC Bioinform. 19:306. doi: 10.1186/s12859-018-2321-0
Hong, Z., Zeng, X., Wei, L., and Liu, X. (2019). Identifying enhancer–promoter interactions with neural network based on pre-trained DNA vectors and attention mechanism. Bioinformatics 6:btz694. doi: 10.1093/bioinformatics/btz694
Hwang, J.-U., Song, W.-Y., Hong, D., Ko, D., Yamaoka, Y., Jang, S., et al. (2016). Plant ABC transporters enable many unique aspects of a terrestrial plant’s lifestyle. Mol. Plant 9, 338–355. doi: 10.1016/j.molp.2016.02.003
Jain, P., Garibaldi, J. M., and Hirst, J. D. (2009). Supervised machine learning algorithms for protein structure classification. Comput. Biol. Chem. 33, 216–223. doi: 10.1016/j.compbiolchem.2009.04.004
Jia, C., Zuo, Y., and Zou, Q. (2018). O-GlcNAcPRED-II: an integrated classification algorithm for identifying O-GlcNAcylation sites based on fuzzy undersampling and a K-means PCA oversampling technique. Bioinformatics 34, 2029–2036. doi: 10.1093/bioinformatics/bty039
Jin, S., Wang, L., Guo, F., and Zou, Q. (2019). AOPs-SVM: a sequence-based classifier of antioxidant proteins using a support vector machine. Front. Bioeng. Biotechnol. 7:224. doi: 10.3389/fbioe.2019.00224
Kadaba, N. S., Kaiser, J. T., Johnson, E., Lee, A., and Rees, D. C. (2008). The high-affinity E. coli methionine ABC transporter: structure and allosteric regulation. Science 321, 250–253. doi: 10.1126/science.1157987
Leprohon, P., Légaré, D., and Ouellette, M. (2011). ABC transporters involved in drug resistance in human parasites. Essays Biochem. 50, 121–144. doi: 10.1042/bse0500121
Li, M. J., Xu, H. H., and Deng, Y. (2019). Evidential decision tree based on belief entropy. Entropy 21:14. doi: 10.3390/e21090897
Li, Z. L., Dong, M. H., Wen, S. P., Hu, X., Zhou, P., and Zeng, Z. G. (2019). CLU-CNNs: object detection for medical images. Neurocomputing 350, 53–59. doi: 10.1016/j.neucom.2019.04.028
Liu, B. (2017). BioSeq-Analysis: a platform for DNA, RNA and protein sequence analysis based on machine learning approaches. Brief. Bioinform. 20, 1280–1294. doi: 10.1093/bib/bbx165
Liu, B., Jiang, S., and Zou, Q. (2018). HITS-PR-HHblits: protein remote homology detection by combining PageRank and hyperlink-induced topic search. Brief. Bioinform. 21, 298–308. doi: 10.1093/bib/bby104
Liu, B., Li, C.-C., and Yan, K. (2019). DeepSVM-fold: protein fold recognition by combining support vector machines and pairwise sequence similarity scores generated by deep learning networks. Brief. Bioinform. bbz098. doi: 10.1093/bib/bbz098
Liu, B., and Li, K. (2019). iPromoter-2L2. 0: identifying promoters and their types by combining smoothing cutting window algorithm and sequence-based features. Mol. Ther. Nucleic Acids 18, 80–87. doi: 10.1016/j.omtn.2019.08.008
Liu, B., Yang, F., Huang, D.-S., and Chou, K.-C. (2017). iPromoter-2L: a two-layer predictor for identifying promoters and their types by multi-window-based PseKNC. Bioinformatics 34, 33–40. doi: 10.1093/bioinformatics/btx579
Locher, K. P. (2008). Structure and mechanism of ATP-binding cassette transporters. Philos. Trans. R. Soc. B Biol. Sci. 364, 239–245. doi: 10.1098/rstb.2008.0125
Locher, K. P. (2016). Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat. Struct. Mol. Biol. 23:487. doi: 10.1038/nsmb.3216
Lv, Z., Jin, S., Ding, H., and Zou, Q. (2019). A random forest sub-golgi protein classifier optimized via dipeptide and amino acid composition features. Front. Bioeng. Biotechnol. 7:215. doi: 10.3389/fbioe.2019.00215
Maqbool, A., Horler, R. S., Muller, A., Wilkinson, A. J., Wilson, K. S., and Thomas, G. H. (2015). The substrate-binding protein in bacterial ABC transporters: dissecting roles in the evolution of substrate specificity. Biochem. Soc. Trans. 43, 1011–1017. doi: 10.1042/BST20150135
Matthews, B. W. (1975). Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta Protein Struct. 405, 442–451. doi: 10.1016/0005-2795(75)90109-9
Mohammad, T. A., and Nagarajaram, H. A. (2011). Svm-based method for protein structural class prediction using secondary structural content and structural information of amino acids. J. Bioinform. Comput. Biol. Chem. 9, 489–502. doi: 10.1142/S0219720011005422
Ofori, P. A., Mizuno, A., Suzuki, M., Martinoia, E., Reuscher, S., Aoki, K., et al. (2018). Genome-wide analysis of ATP binding cassette (ABC) transporters in tomato. PLoS One 13:e0200854. doi: 10.1371/journal.pone.0200854
Qiao, Y., Xiong, Y., Gao, H., Zhu, X., and Chen, P. (2018). Protein-protein interface hot spots prediction based on a hybrid feature selection strategy. BMC Bioinformatics 19:14. doi: 10.1186/s12859-018-2009-5
Radhika, V., and Rao, V. S. H. (2015). Computational approaches for the classification of seed storage proteins. J. Food Sci. Technol. 52, 4246–4255. doi: 10.1007/s13197-014-1500-x
Ru, X. Q., Li, L. H., and Zou, Q. (2019). incorporating distance-based Top-n-gram and random forest to identify electron transport proteins. J. Proteom. Res. 18, 2931–2939. doi: 10.1021/acs.jproteome.9b00250
Ruiz, N., Gronenberg, L. S., Kahne, D., and Silhavy, T. J. (2008). Identification of two inner-membrane proteins required for the transport of lipopolysaccharide to the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 105, 5537–5542. doi: 10.1073/pnas.0801196105
Seeger, M. A., and van Veen, H. (2009). Molecular basis of multidrug transport by ABC transporters. Biochim. Biophys. Acta Proteins Proteom. 1794, 725–737. doi: 10.1016/j.bbapap.2008.12.004
Shan, X., Wang, X., Li, C. D., Chu, Y., Zhang, Y., Xiong, Y., et al. (2019). Prediction of CYP450 enzyme-substrate selectivity based on the network-based label space division method. J. Chem. Inf. Model. 59, 4577–4586. doi: 10.1021/acs.jcim.9b00749
Shao, L., Gao, H., Liu, Z., Feng, J., Tang, L., and Lin, H. (2018). Identification of antioxidant proteins with deep learning from sequence information. Front. Pharmacol. 9:1036. doi: 10.3389/fphar.2018.01036
Shen, C., Ding, Y., Tang, J., Song, J., and Guo, F. (2017). Identification of DNA–protein binding sites through multi-scale local average blocks on sequence information. Molecules 22:2079. doi: 10.3390/molecules22122079
Shen, Y., Tang, J., and Guo, F. (2019). Identification of protein subcellular localization via integrating evolutionary and physicochemical information into Chou’s general PseAAC. J. Theor. Biol. 462, 230–239. doi: 10.1016/j.jtbi.2018.11.012
Sheps, J. A., Ralph, S., Zhao, Z., Baillie, D. L., and Ling, V. (2004). The ABC transporter gene family of Caenorhabditis elegans has implications for the evolutionary dynamics of multidrug resistance in eukaryotes. Genom. Biol. 5:R15. doi: 10.1186/gb-2004-5-3-r15
Song, J., Li, C., Zheng, C., Revote, J., Zhang, Z., and Webb, G. I. (2017). MetalExplorer, a bioinformatics tool for the improved prediction of eight types of metal-binding sites using a random forest algorithm with two-step feature selection. Curr. Bioinform. 12, 480–489. doi: 10.2174/2468422806666160618091522
Wang, Y., Ding, Y., Guo, F., Wei, L., and Tang, J. (2017). Improved detection of DNA-binding proteins via compression technology on PSSM information. PLoS One 12:185587. doi: 10.1371/journal.pone.0185587
Ward, A. B., Szewczyk, P., Grimard, V., Lee, C.-W., Martinez, L., Doshi, R., et al. (2013). Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc. Natl. Acad. Sci. U.S.A. 110, 13386–13391. doi: 10.1073/pnas.1309275110
Wei, L., Liao, M., Gao, Y., Ji, R., He, Z., and Zou, Q. (2014). Improved and promising identification of human microRNAs by incorporating a high-quality negative set. IEEE/ACM Trans. Comput. Biol. Bioinform. 11, 192–201. doi: 10.1109/TCBB.2013.146
Wei, L., Xing, P., Shi, G., Ji, Z., and Zou, Q. (2019). Fast prediction of methylation sites using sequence-based feature selection technique. IEEE/ACM Trans. Comput. Biol. Bioinform. 16, 1264–1273. doi: 10.1109/TCBB.2017.2670558
Wei, L., Xing, P., Su, R., Shi, G., Ma, Z., and Zou, Q. (2017a). CPPred-RF: a sequence-based predictor for identifying cell-penetrating peptides and their uptake efficiency. J. Proteome Res. 16, 2044–2053. doi: 10.1021/acs.jproteome.7b00019
Wei, L., Xing, P., Tang, J., and Zou, Q. (2017b). PhosPred-RF: a novel sequence-based predictor for phosphorylation sites using sequential information only. IEEE Trans. Nanobiosci. 16, 240–247. doi: 10.1109/TNB.2017.2661756
Wei, L., Xing, P., Zeng, J., Chen, J., Su, R., and Guo, F. (2017c). Improved prediction of protein–protein interactions using novel negative samples, features, and an ensemble classifier. Artif. Intell. Med. 83, 67–74. doi: 10.1016/j.artmed.2017.03.001
Wei, L., Zhou, C., Chen, H., Song, J., and Su, R. (2018). ACPred-FL: a sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics 34, 4007–4016. doi: 10.1093/bioinformatics/bty451
Wei, L., Zou, Q., Liao, M., Lu, H., and Zhao, Y. (2016). A novel machine learning method for cytokine-receptor interaction prediction. Comb. Chem. High Throughput Screen. 19, 144–152. doi: 10.2174/1386207319666151110122621
Wong, K., Ma, J., Rothnie, A., Biggin, P. C., and Kerr, I. D. (2014). Towards understanding promiscuity in multidrug efflux pumps. Trends Biochem. Sci. 39, 8–16. doi: 10.1016/j.tibs.2013.11.002
Xie, X., Wang, G., Yang, L., Cheng, T., Gao, J., Wu, Y., et al. (2015). Cloning and characterization of a novel Nicotiana tabacum ABC transporter involved in shoot branching. Physiol. Plant. 153, 299–306. doi: 10.1111/ppl.12267
Xiong, Y., Wang, Q., Yang, J., Zhu, X., and Wei, D. Q. (2018). PredT4SE-stack: prediction of bacterial type iv secreted effectors from protein sequences using a stacked ensemble method. Front. Microbiol. 9:2571. doi: 10.3389/fmicb.2018.02571
Xu, R., Zhou, J., Wang, H., He, Y., Wang, X., and Liu, B. (2015). Identifying DNA-binding proteins by combining support vector machine and PSSM distance transformation. BMC Syst. Biol. 9(Suppl. 1):S10. doi: 10.1186/1752-0509-9-S1-S10
Yan, Z., Liu, W. W., Wen, S. P., and Yang, Y. (2019). Multi-label image classification by feature attention network. IEEE Access. 7, 98005–98013. doi: 10.1109/access.2019.2929512
Yu, L., Huang, J. B., Ma, Z. X., Zhang, J., Zou, Y. P., and Gao, L. (2015). Inferring drug-disease associations based on known protein complexes. BMC Med. Genom. 8:13. doi: 10.1186/1755-8794-8-s2-s2
Yu, L., Zhao, J., and Gao, L. (2017). Drug repositioning based on triangularly balanced structure for tissue-specific diseases in incomplete interactome. Artif. Intell. Med. 77, 53–63. doi: 10.1016/j.artmed.2017.03.009
Yu, L., Zhao, J., and Gao, L. (2018). Predicting potential drugs for breast cancer based on miRNA and tissue specificity. Int. J. Biol. Sci. 14, 971–980. doi: 10.7150/ijbs.23350
Zeng, X., Liao, Y., Liu, Y., and Zou, Q. (2017a). Prediction and validation of disease genes using hetesim scores. IEEE/ACM Trans. Comput. Biol. Bioinform. 14, 687–695. doi: 10.1109/tcbb.2016.2520947
Zeng, X., Lin, W., Guo, M., and Zou, Q. (2017b). A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput. Biol. 13:e1005420. doi: 10.1371/journal.pcbi.1005420
Zeng, X., Lin, Y., He, Y., Lv, L., Min, X., and Rodríguez-Paton, A. (2019a). Deep collaborative filtering for prediction of disease genes. IEEE/ACM Trans. Comput. Biol. Bioinform. doi: 10.1109/TCBB.2019.2907536 [Epub ahead of print].
Zeng, X., Zhong, Y., Lin, W., and Zou, Q. (2019b). Predicting disease-associated circular RNAs using deep forests combined with positive-unlabeled learning methods. Brief. Bioinform. bbz080. doi: 10.1093/bib/bbz080
Zeng, X., Zhu, S., Liu, X., Zhou, Y., Nussinov, R., and Cheng, F. (2019c). deepDR: a network-based deep learning approach to in silico drug repositioning. Bioinformatics 35, 5191–5198. doi: 10.1093/bioinformatics/btz418
Zhang, M., Li, F., Marquez-Lago, T. T., Leier, A., Fan, C., Kwoh, C. K., et al. (2019). MULTiPly: a novel multi-layer predictor for discovering general and specific types of promoters. Bioinformatics 35, 2957–2965. doi: 10.1093/bioinformatics/btz016
Zhang, W., Jing, K., Huang, F., Chen, Y., Li, B., Li, J., et al. (2019a). SFLLN: a sparse feature learning ensemble method with linear neighborhood regularization for predicting drug–drug interactions. Inform. Sci. 497, 189–201. doi: 10.1016/j.ins.2019.05.017
Zhang, W., Li, Z., Guo, W., Yang, W., and Huang, F. (2019b). A fast linear neighborhood similarity-based network link inference method to predict microRNA-disease associations. IEEE/ACM Trans. Comput. Biol. Bioinform. 2931546. doi: 10.1109/TCBB.2019.2931546
Zhang, X., Zou, Q., Rodriguez-Paton, A., and Zeng, X. (2019). Meta-path methods for prioritizing candidate disease miRNAs. IEEE/ACM Trans. Comput. Biol. Bioinform. 16, 283–291. doi: 10.1109/TCBB.2017.2776280
Zhang, W., Qu, Q., Zhang, Y., and Wang, W. (2018a). The linear neighborhood propagation method for predicting long non-coding RNA–protein interactions. Neurocomputing 273, 526–534. doi: 10.1016/j.neucom.2017.07.065
Zhang, W., Yue, X., Tang, G., Wu, W., Huang, F., and Zhang, X. (2018b). SFPEL-LPI: sequence-based feature projection ensemble learning for predicting LncRNA-protein interactions. PLoS Comput. Biol. 14:e1006616. doi: 10.1371/journal.pcbi.1006616
Zhu, X., He, J., Zhao, S., Tao, W., Xiong, Y., and Bi, S. (2019). A comprehensive comparison and analysis of computational predictors for RNA N6-methyladenosine sites of Saccharomyces cerevisiae. Brief. Funct. Genom. 18, 367–376. doi: 10.1093/bfgp/elz018
Zou, Q., Li, X., Jiang, Y., Zhao, Y., and Wang, G. (2013a). BinMemPredict: a web server and software for predicting membrane protein types. Curr. Proteom. 10, 2–9. doi: 10.2174/15701646112098880001
Zou, Q., Lin, G., Jiang, X., Liu, X., and Zeng, X. (2018). Sequence clustering in bioinformatics: an empirical study. Brief. Bioinform. 21, 1–10. doi: 10.1093/bib/bby090
Zou, Q., Wang, Z., Guan, X., Liu, B., Wu, Y., and Lin, Z. (2013b). An approach for identifying cytokines based on a novel ensemble classifier. Biomed. Res. Int. 2013:686090. doi: 10.1155/2013/686090
Zou, Q., Wan, S., Ju, Y., Tang, J., and Zeng, X. (2016). Pretata: predicting TATA binding proteins with novel features and dimensionality reduction strategy. BMC Syst. Biol. 10:114. doi: 10.1186/s12918-016-0353-5
Keywords: ABC transporters, random forest, classify, 188D, t-SNE
Citation: Hou R, Wang L and Wu Y-J (2020) Predicting ATP-Binding Cassette Transporters Using the Random Forest Method. Front. Genet. 11:156. doi: 10.3389/fgene.2020.00156
Received: 26 December 2019; Accepted: 11 February 2020;
Published: 25 March 2020.
Edited by:
Li Guo, Nanjing University of Posts and Telecommunications, ChinaReviewed by:
Fei Guo, Tianjin University, ChinaXiucai Ye, University of Tsukuba, Japan
Hao Lin, University of Electronic Science and Technology of China, China
Copyright © 2020 Hou, Wang and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi-Jun Wu, d3V5akBpb3ouYWMuY24=
†These authors have contributed equally to this work