 Chuangye Li1†
Chuangye Li1† Yongjia Yang
Yongjia Yang Yu Zheng
Yu Zheng Yaowang Zhao
Yaowang Zhao- 1Department of Urology, Hunan Children’s Hospital, Changsha, China
- 2The Laboratory of Genetics and Metabolism, Hunan Children’s Research Institute (HCRI), Hunan Children's Hospital, University of South China, Changsha, China
Background: Cystinuria is a rare genetic disorder characterized by defective renal reabsorption of cystine, ornithine, arginine, and lysine. The increased urinary excretion of cystine results in the development of cystine urolithiasis (CU). The mutated SLC3A1 and SLC7A9 genes are the cause of CU, a global disorder. Its frequency and mutation spectrum vary between different populations. In Asia, the data for CU are limited.
Method: Urinary stones were collected from patients of a single center over a five-year period and analyzed via Fourier transform infrared spectroscopy. Genomic DNA was isolated from 13 patients with CU and their parents and from 26 controls affected by calcium oxalate dihydrate stones. The coding regions and the exon–intron boundaries of SLC3A1 and SLC7A9 were subjected to PCR amplification and then sequenced via traditional Sanger sequencing. Genetic variants were functionally annotated using the InterVar, ClinVar, gnom AD, and HGMD databases.
Results: From the 232 samples of urinary stones, we identified 13 patients with CU (10 males and 3 females). The onset age was from 7 months to 9 years. The CU stones varied from 0.26 cm3 to 18.67 cm3. Sanger sequencing detected a total of 14 SLC3A1 (nine were novel) and 10 SLC7A9 (six were novel) rare variants from the 13 CU families. All variants, including 15 novel variants, were pathogenic, disease-causing, or damaging.
Conclusion: All 13 pediatric CU families harbored SLC3A1 or/and SLC7A9 rare variants. A total of 15 novel pathogenic variants in SLC3A1 and SLC7A9 were identified. This study expanded the known mutational spectrum of CU in the Chinese population.
Background
Urolithiasis, also known as urinary calculi disease, develops when a solid piece of material exists in the urinary tract (Miller and Lingeman, 2007). Most kidney stones form because of a combination of genetics and environmental factors (Miller and Lingeman, 2007). Urolithiasis have serious consequences if not treated immediately. Cystine urolithiasis (CU) is a genetic disorder characterized by defective renal reabsorption of cystine, arginine, lysine, and ornithine; the increased urinary excretion of cystine results in the formation of kidney stones (Eggermann et al., 2012; Fattah et al., 2014). Two genes responsible for cystinuria and CU have been identified: SLC3A1, which encodes the heavy subunit rBAT of the renal b0,+ transporter; and SLC7A9, which encodes b0,+AT, the interacting light subunit of the b0,+ transporter (Calonge et al., 1994; Feliubadaló et al., 1999). b0,+ and b0,+AT combine with each other through disulfide linkage, and only the combined b0,+–b0,+AT heterodimer is functional (Chillarón et al., 2010). The inheritance for cystinuria may vary from autosomal recessive (AR) to autosomal dominant (AD) with incomplete penetrance to digenic pattern (Dello Strologo et al., 2002; Sahota et al., 2019). Accordingly, cystinuria is divided into the following subtypes: biallelic SLC3A1 mutations (AA genotype), biallelic SLC7A9 mutations (BB genotype), a single heterozygous SLC3A1 mutation (A genotype), a single heterozygous SLC7A9 mutation (B genotype), and a single heterozygous SLC3A1 mutation combined with a single heterozygous SLC7A9 mutation (AB genotype) (Dello Strologo et al., 2002; Sahota et al., 2019).
Cystinuria is a global disease with a population-specific prevalence (Eggermann et al., 2012). Although the overall frequency is estimated at 1:7,000 in neonates (Segal and Thier, 1995), the frequency varies in different areas. The rate of CU is 1:2,500 in Libyan Jews, 1:15,000 in Americans, and 1:100,000 in Swedes (Weinberger et al., 1974; Segal and Thier, 1995). In Libyan Jews, the mutant gene carrier rate is as high as 1:25 (Weinberger et al., 1974). In Sweden, 0.5% of the general population carries the SLC3A1 p.Met467Thr variant (Harnevik et al., 2001; Harnevik et al., 2003). In Asia, cystinuria is rarely reported and its frequency in the Chinese population is unclear.
In this study, we analyzed the composition of 232 samples of kidney stones and subjected 13 cases of childhood CU to genetic analysis. A total of 24 pathogenic variants in SLC3A1 and SLC7A9 were detected, 15 of which were novel. These data expanded the spectrum of CU variants in the Chinese population.
Methods
Criteria for Pediatric CU and the Controls
Data from pediatric CU patients were collected from 2012 to 2017. A total of 13 CU patients were enrolled in this study (Supplementary Data 1). Patients were diagnosed with pediatric CU if (i) they were aged 0–18 years old, (ii) passed visible urinary stones under a clinical setting, and (iii) passed pure or cystine stones as identified via stone composition analysis. The 26 controls were taken from sporadic cases of patients with calcium oxalate dihydrate stones (Supplementary Data 1).
DNA Isolation and Sanger Sequencing
Peripheral blood samples were obtained from 13 Trios-families and 26 sporadic cases of other kidney stones (calcium oxalate dihydrate stones). DNA was extracted using a genomic DNA isolation kit (D3392-02; Omega Bio-Tek, Inc., Norcross, GA, USA) in accordance with a standard protocol. The coding regions and intron–exon junctions of the SLC3A1 and SLC7A9 genes were amplified via PCR by using primers synthesized by BGI, a local biotech company (Shenzhen, China). Primers were designed with Primer3 software (http://frodo.wi.mit.edu). The primer sequences and PCR conditions are provided in Supplementary Table 1. Sanger sequencing was performed on an Applied Biosystems™ 3500 Dx Genetic Analyzer by using a BigDye 3.1 sequencing kit (Applied Biosystems, CA, USA).
Variant Validation
All Sanger sequencing results were compared with the reference SLC3A1 and SLC7A9 sequences (NM_000341 and NM_014270, GRCh37/hg19, http://genome.ucsc.edu/cgi-bin/hgGateway) by applying SEQMAN software (DNA Star Package, Wisconsin, USA). Genetic variants were functionally annotated using InterVar (version 20180118) and screened for reported mutations by using ClinVar (version 20180603) and the HGMD database (public version). We further customized the clinical classification of each variant in accordance with the ACMG/AMP 2015 guidelines by applying automatically interpreted InterVar results and by integrating individualized information, such as family history and inheritance mode, obtained in this study with the results of previous studies.
Results
Clinical Data of 13 Patients With Pediatric CU
A total of 13 patients with CU were identified, 10 of whom were males and 3 were females. These patients originated from 232 patients with urinary stones that we treated from 2002 to 2007. The patients had a mean age of 3.82 ± 3.12 years (range: 0.6–9.3) at first stone onset. The mean stone size of the patient group was 4.12 ± 4.96 cm3 (range: 0.26–18.67). Two urethral (3.77%), 2 bladder (3.77%), 14 ureteral (26.42%), 22 renal pelvic or upper/middle calyceal (41.51%), and 13 lower calyceal (46.43%) calculi were noted. Five children had bilateral calculi, whereas two children had staghorn calculi. Seven children had a family history of CU. Twelve patients had normal initial renal function (serum creatinine [CREA] 20–120 μmol/L, blood urea nitrogen [BUN] 1.8–8.2 mmol/L, and serum uric acid [UA] 90–350 μmol/L). The remaining patient with bilateral ureteral calculi exhibited various degrees of renal dysfunction (625.3, 36.82, and 618.8 μmol/L). The mean number of operations was 3.61 ± 2.14 (range: 1–9), including 11 minimally invasive percutaneous nephrolithotomy, 10 retrograde intrarenal surgery, and 26 ureteroscopic lithotripsy. The mean stone-free rate was 46.15% after the final operation. Table 1 lists the clinical features of the 13 patients with CU.
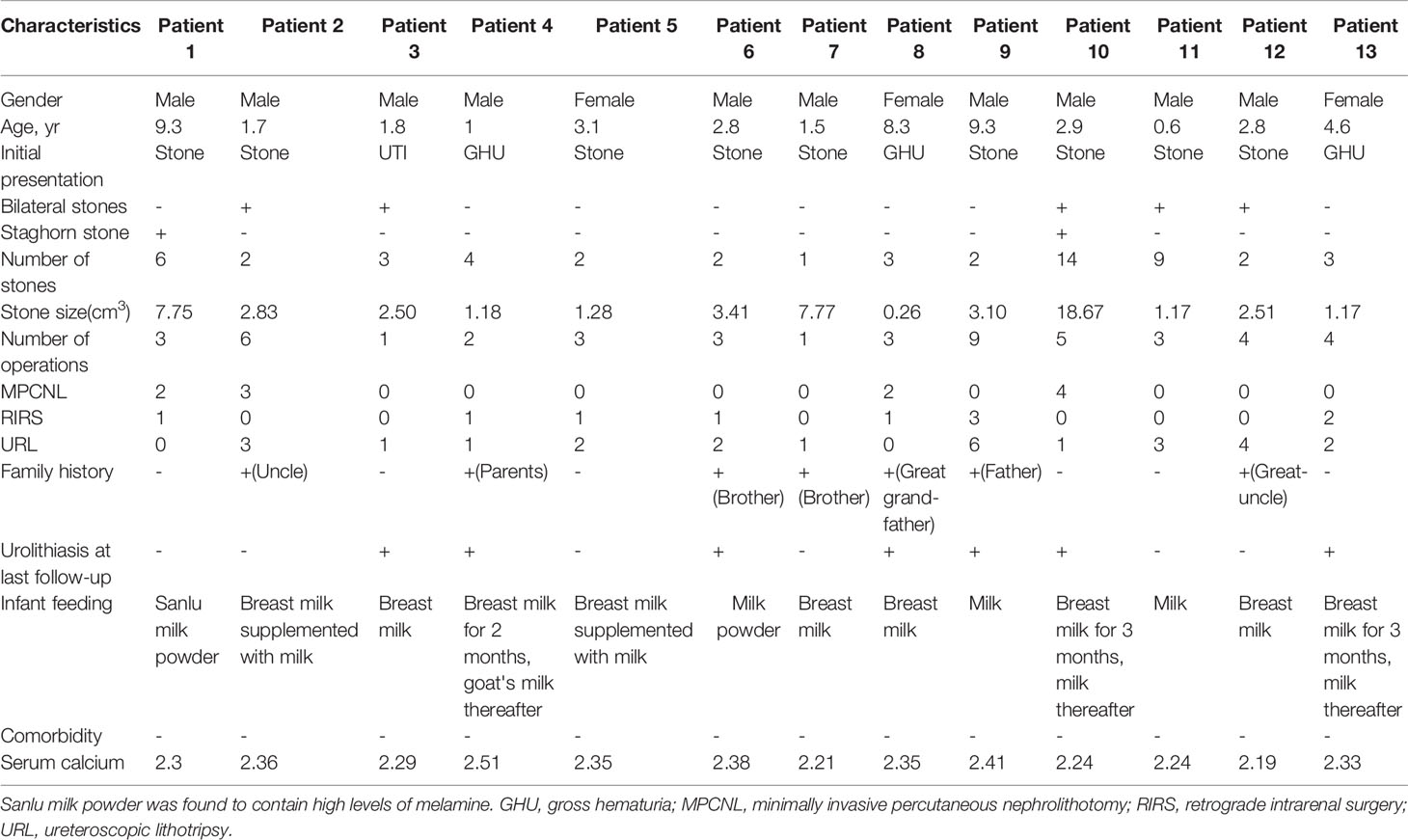
Table 1 Clinical features of 13 patients with cystine urolithiasis.
Identification of SLC3A1 and SLC7A9 Variants on 24 Alleles via Sanger Sequencing
Sanger sequencing was successfully conducted for the 13 patients with pediatric CU and their parents. After filtering out variants with MAF > 0.01 using the gnomAD database and the variants present in 26 controls, we detected 14 SLC3A1 and 8 SLC7A9 variants on 24 alleles from these 13 families (Table 2, Figures 1–3). These variants covered the whole spectrum of mutations and ranged from insertion, deletion, splicing, and missense variants. Six families presented with biallelic SLC3A1 variants (AA genotype) (Figure 1), and four families exhibited biallelic SLC7A9 variants (BB genotype) (Figure 2). The genotypes of the remaining three families included a single heterozygous SLC3A1 variant (A genotype), a single heterozygous SLC7A9 variant (B genotype), and a single heterozygous SLC3A1 variant combined with a single heterozygous SLC7A9 variant (AB genotype) (Figure 3).
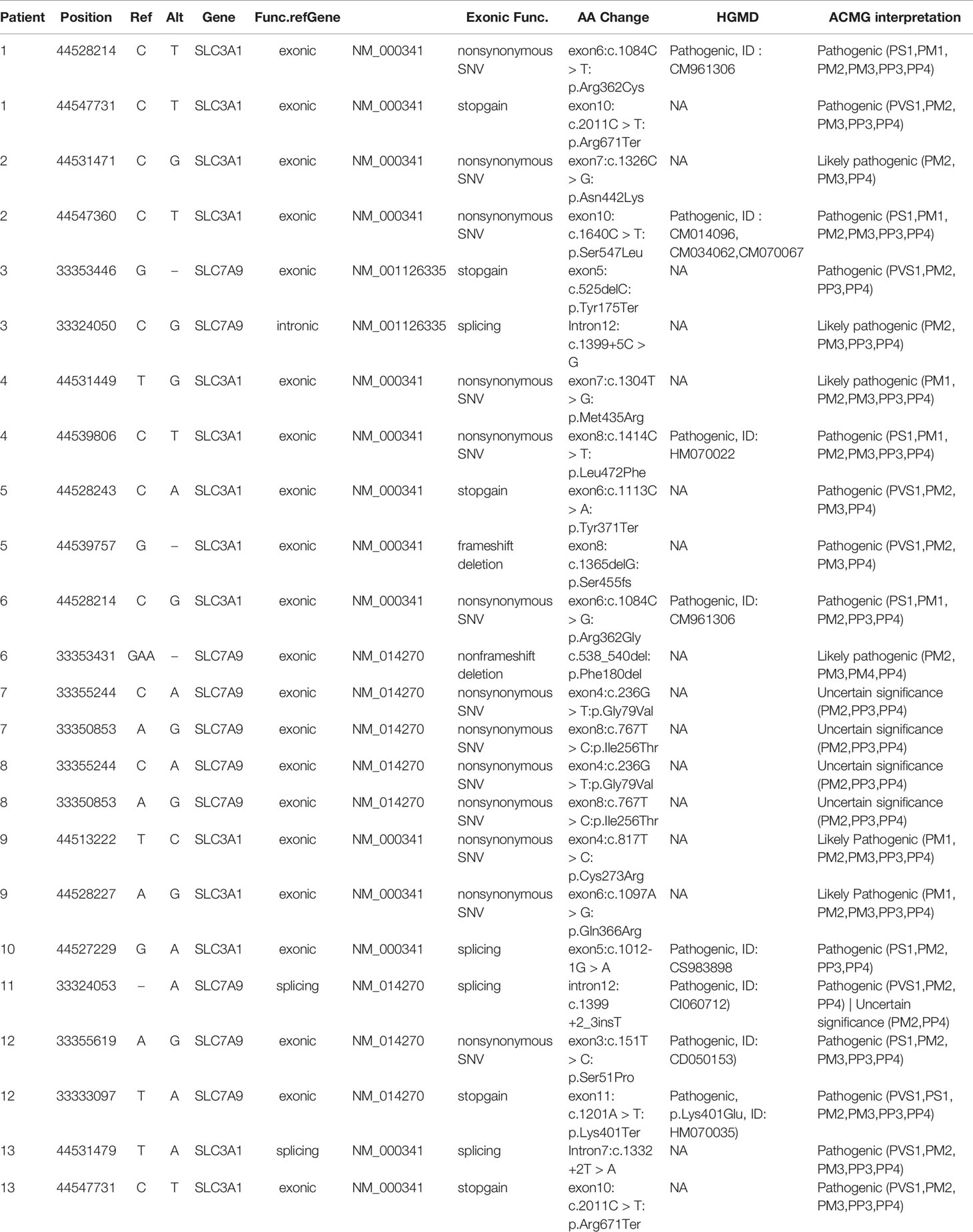
Table 2 Rare variants on SLC3A1 AND SLC7A9 detected in 13 families with cystine urolithiasis.
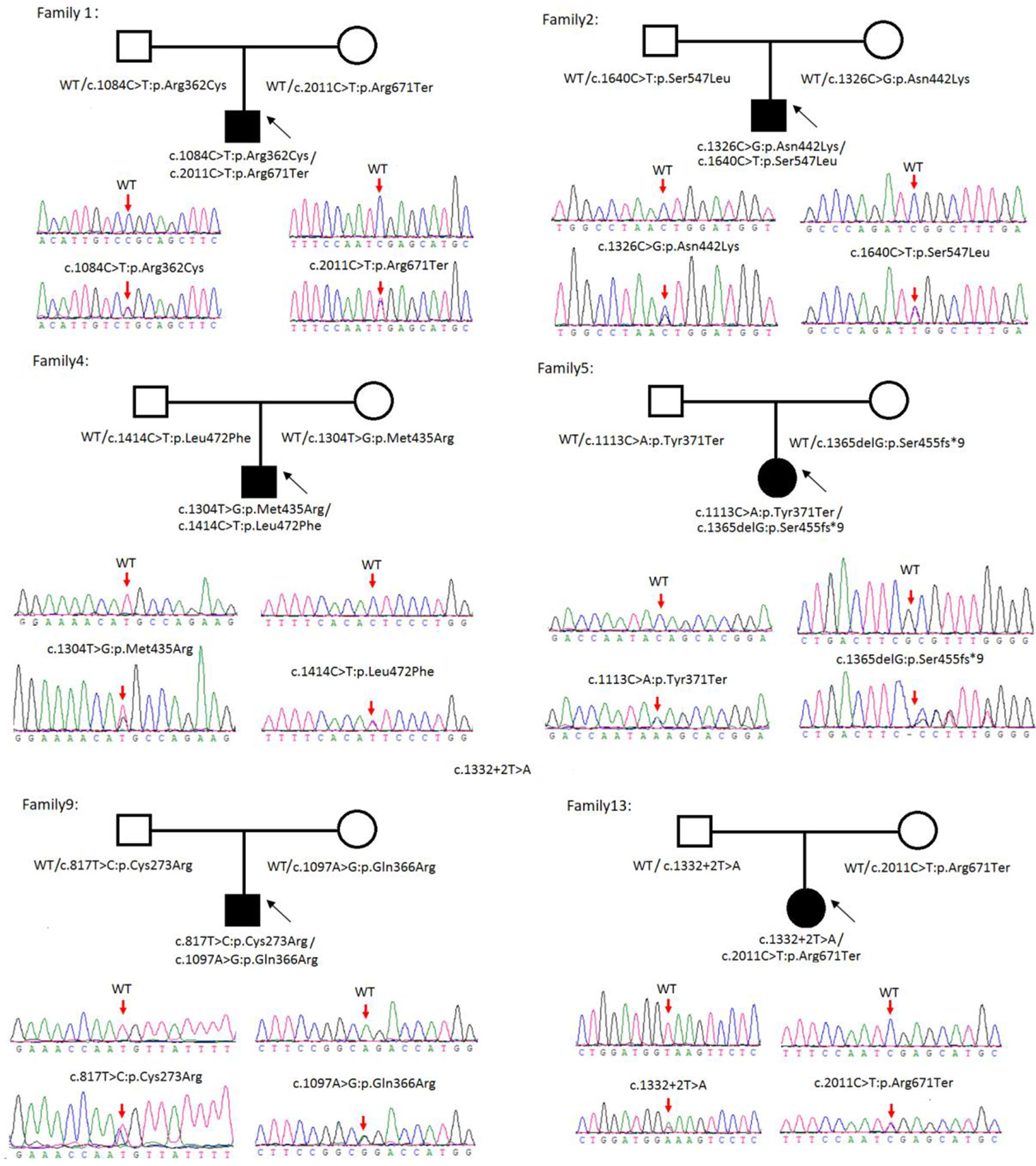
Figure 1 Rare variants of AA genotypes on six cystine urolithiasis (CU) families. Solid squares or circles mean the patients were affected by CU. Open squares or circles denote that the patients were not affected by CU.
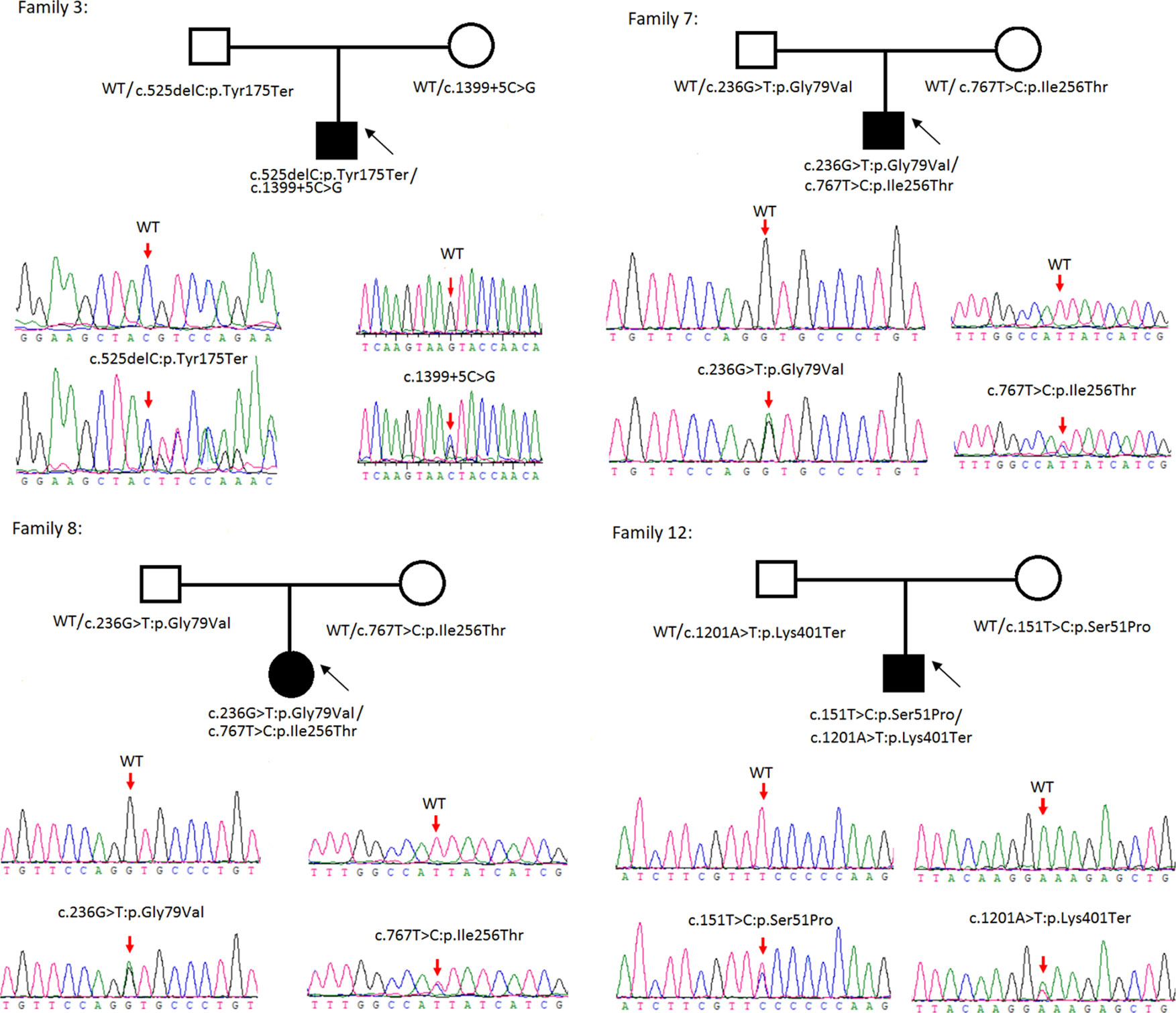
Figure 2 Rare variants of BB genotypes on four cystine urolithiasis (CU) families. Solid squares or circles mean the patients were affected by CU. Open squares or circles denote that the patients were not affected by CU.
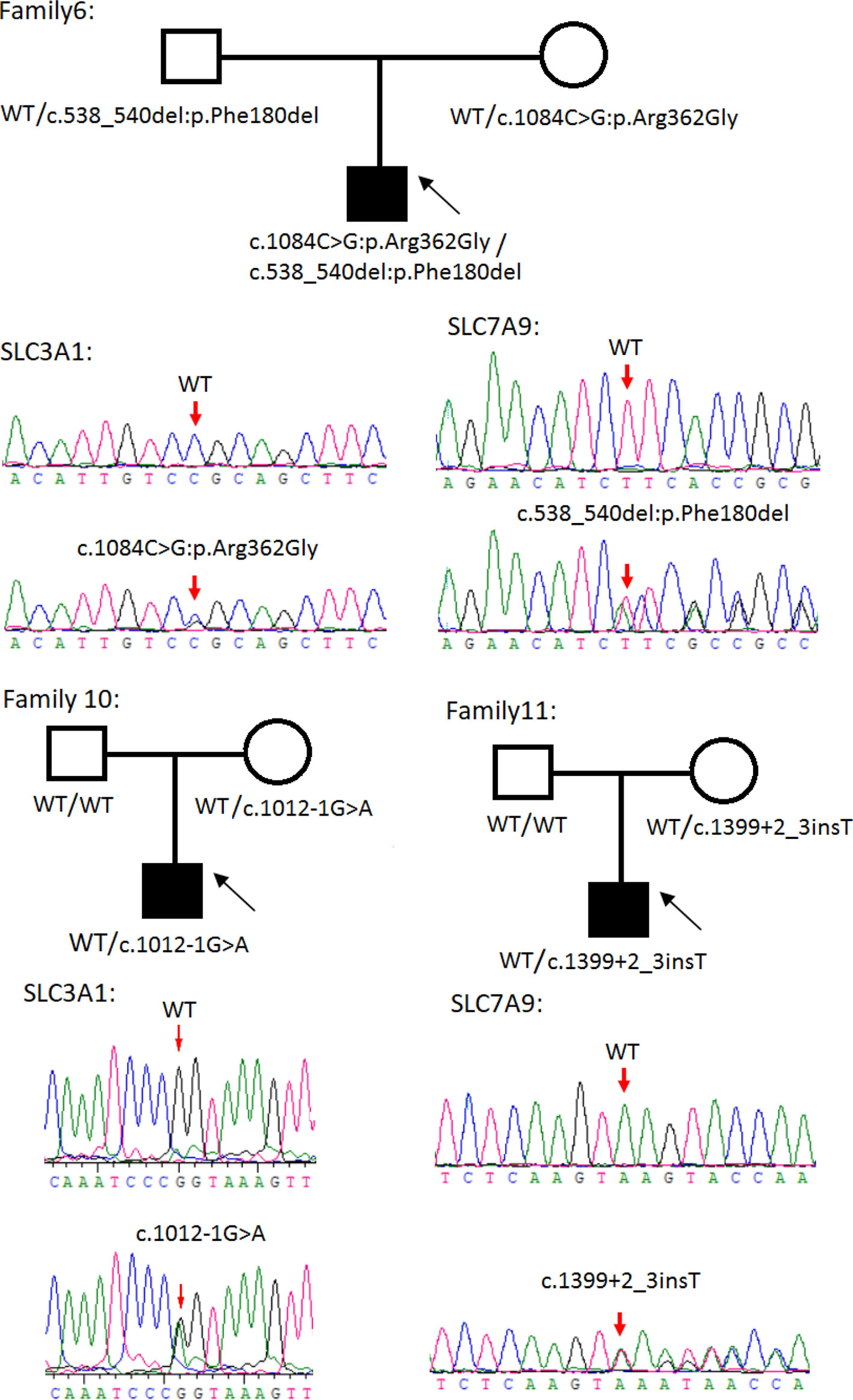
Figure 3 Rare variants of AB, A or B genotypes in three cystine urolithiasis (CU) families. Solid squares or circles mean the patients were affected by CU. Open squares or circles denote that the patients were not affected by CU.
Among these 24 variants, 15 have not been reported elsewhere (9 on SLC3A1, 6 on SLC7A9).
According to the ACMG/AMP 2015 guideline classification (Table 2), five of the nine SLC3A1 variants were pathogenic and four were likely pathogenic; two of the six SLC7A9 variants were pathogenic and another two were likely pathogenic. The classifications of the remaining two variants of SLC7A9 were uncertain. These 15 novel variants included the following: (1) For SLC3A1: c.2011C > T:p.Arg671Ter, c.1326C > G:p.Asn442Lys, c.1304T > G:p.Met435Arg, c.1113C > A:p.Tyr371Ter, c.1365delG:p.Ser455Valfs, c.817T > C:p.Cys273Arg, c.1097A > G:p.Gln366Arg, c.1332+2T > A, and c.2011C > T:p.Arg671Ter. (2)For SLC7A9: c.525delC:p.Tyr175Ter, c.1399+5C > G,c.538_540del:p.180_180del, c.236G > T:p.Gly79Val, c.767T > C:p.Ile256Thr, and exon11:c.1201A > T:p.Lys401Ter.
Further functional effects were tested for all variants via three programs in silico (i.e., REVEL, ClinPred, and MutationTaster). Results showed that 13 of the 15 novel variants were damaging, disease-causing, or pathogenic (Table 3).
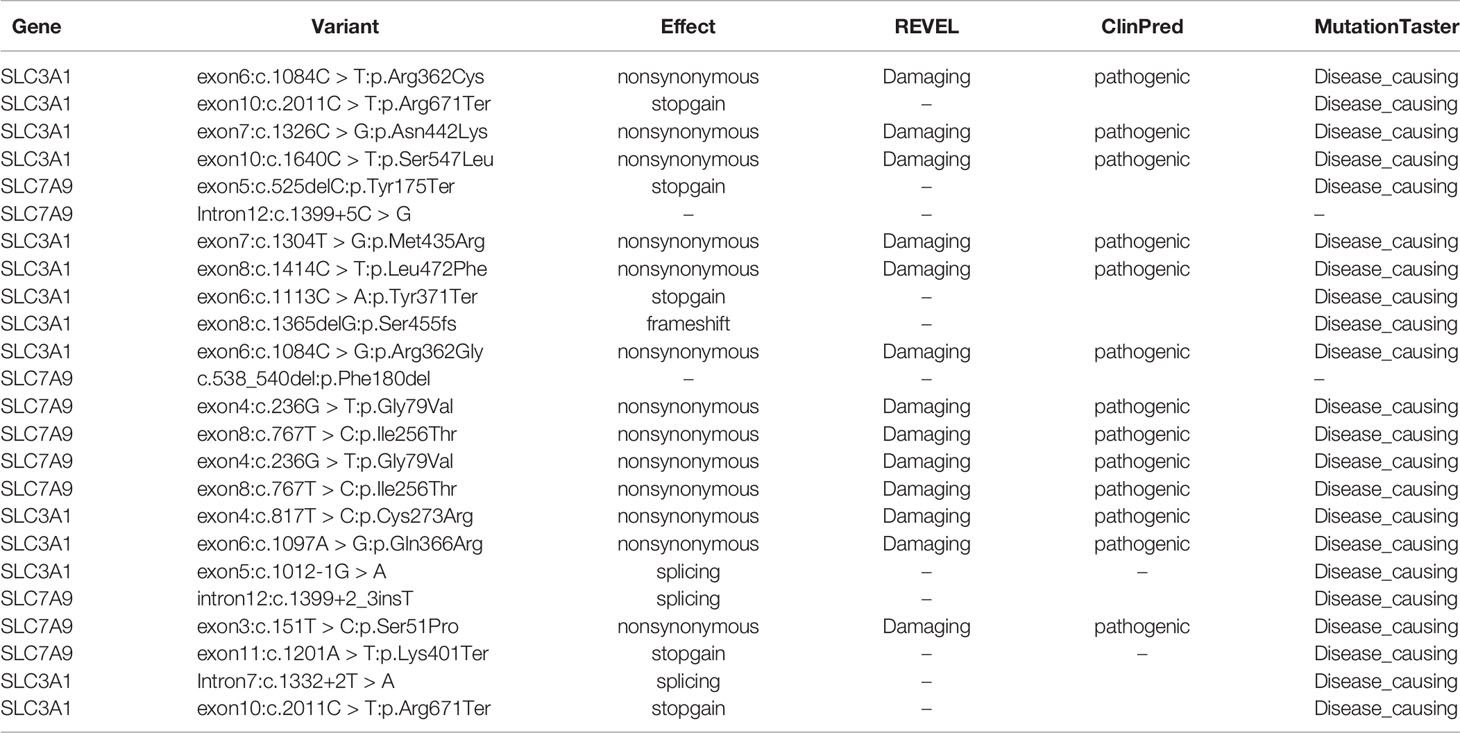
Table 3 Effect predictions of rare variants by three in silico software. REVEL: V0.21.0 (http://revel.github.io/); ClinPred (Alirezaie et al., 2018); MutationTaster (Schwarz et al., 2014).
The remaining two SLC7A9 variants (i.e., c.1399+5C > G and c.538_540del:p.Phe180del) were likely pathogenic. Thus, further confirmatory experiments are needed.
Genotype-Phenotype Correlation for 13 CU Families
The 13 CU families were divided into three groups: classical AR inheritance (six AA and four BB genotypes), AD inheritance (one A and one B genotype), and digenic inheritance (one AB genotype) (Table 4). Several points were observed in this study: (1) SLC3A1 is the main disease gene for childhood CU. SLC3A1 is mutated in 8 out of the 13 patients with CU and the number of stones is 2–9 for AA genotype and 1–3 for BB genotype. (2) Patients with AD inheritance had the highest number of stones. In family number 10 with A genotype, the number of stones is 14. In family number 11 with B genotype, the number of stones is 9. (3) Childhood CU may represent a more aggressive form. By contrast, the pathogenic variants in SLC3A1 or SLC7A9 (Eggermann et al., 2012) cannot all be detected in cystiruia patients. In this study, we detected the pathogenic variants in SLC3A1 or SLC7A9 in all CU patients, suggesting that pediatric CU patients may represent a more aggressive group.
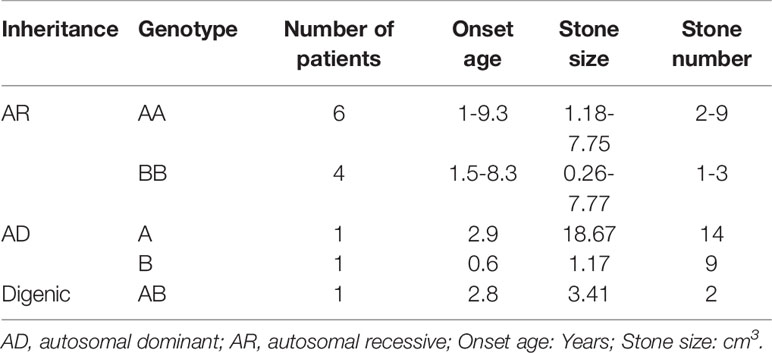
Table 4 In-brief lists of genotype and phenotype data for 13 families with cystine urolithiasis.
Phenotypic Details for Three CU Patients With Heterozygous Variants
Three cases harboring SLC3A1 or (and) SLC7A9 heterozygous variants were identified. For patient number 6 with AB genotype, no clinical symptom of urolithiasis was observed during health examinations. The patient's 24-h urinary calcium was 0.42 mmol/L, 24-h urinary phosphorus was 9.75 mmol/L, serum calcium was 2.38 mmol/L, serum phosphate was 1.61 mmol/L, BUN was 3.39 mmol/L, CREA was 18.32 µmol/L, and UA was 269.31 µmol/L. However, the B ultrasound detected stones in the patient's urinary system.
Patient number 10 with A genotype carried an SLC3A1 spicing variant (c.1012-1G > A) that was previously reported as pathogenic (Saadi et al., 1998). The patient's initial symptom was dysuria but without urinary frequency and urgency. His 24-h urinary calcium was 0.12 mmol/L, 24-h urinary phosphorus was 7.94 mmol/L, serum calcium was 2.24 mmol/L, BUN was 4.42 mmol/L, CREA was 41.65 µmol/L, and serum UA was 182.91 µmol/L.
Patient number 11 with B genotype carried an SLC7A9 spicing variant (1399 + 2_3insT) that was also previously reported as pathogenic (Yuen et al., 2006). The patient had spontaneous discharge of small stones (at 3 months old) according to his parents. The patient's 24-h urinary calcium was 0.59 mmol/L, 24-h urinary phosphorus was 3.92 mmol/L, serum calcium was 2.24 mmol/L, serum phosphate was 2.19 mmol/L, BUN was 3.37 mmol/L, CREA was 32.2 µmol/L, and serum-UA was 217.26 µmol/L.
Discussion
Cystinuria is a metabolic disease caused by defects in the SLC3A1 and SLC7A9 genes. These gene defects result in decreased renal reabsorption of cystine, ornithine, arginine, and lysine. Most patients with cystinuria develop CU, but the age at onset varies (Harnevik et al., 2003). In this study, we subjected 13 patients with pediatric CU to mutation screening and observed typical SLC3A1 and SLC7A9 pathogenic variants consistent with AA, BB, or AB genotypes in 11 of the families with CU and heterozygous SLC3A1 and SLC7A9 rare variants in two of the families with CU (i.e., family numbers 10 and 11). Family numbers 10 and 11 suffer from c.1012-1G > A and 1399 + 2_3insT splicing variants, respectively. These two variants were reported previously as pathogenic (Harnevik et al., 2001; Harnevik et al., 2003). However, no CU phenotypes were observed for both maternal carriers. This situation may be indicative of the non-full penetrance for the heterozygous mutation in females.
According to the ACMG/AMP 2015 guidelines or the predictions by the software in silico, all 24 rare variants are pathogenic, likely pathogenic, disease-causing, or damaging (Tables 2 and 3). This result is similar to that obtained by another study (Shen et al., 2017). Shen et al. (2017) examined 13 pediatric patients with CU and found that all patients harbored pathogenic SLC3A1 or SLC7A9 variants. These results raise the question of whether newborns with mutations in the SLAC3A1 or SLC7A9 genes should be afforded certain precautions, especially as next-generation sequencing has become widely available.
As a global disorder, the prevalence of cystinuria varies in different populations, and hotspot mutations have been described previously. The typical mutation hotspot site is SLC3A1 p.Met467Thr (Weinberger et al., 1974; Harnevik et al., 2001; Botzenhart et al., 2002; Eggermann et al., 2012). A study reported that 0.5% of the Swedish population carries the hotspot variant (Harnevik et al., 2001). China is the most populated country in the world. However, to the best of our knowledge, only four studies (including the present study) have been conducted in China concerning this topic. These studies involved 40 patients in total (Yuen et al., 2006; Shen et al., 2017; Ma et al., 2018). A total of 26 SLC3A1 and 21 SLC7A9 pathogenic variants have been reported in the Chinese population. As shown in Figure 4, the Chinese variants were evenly distributed through the SLC3A1 or SLC7A9 genes, and only five SLC3A1 and six SLC7A9 variants were reported twice. No SLC3A1-p.Met467Thr was detected in the Chinese patients (Figure 4). Therefore, the SLC3A1 and SLC7A9 variants in the patients likely represent mutation spectra that differ from those in other populations.
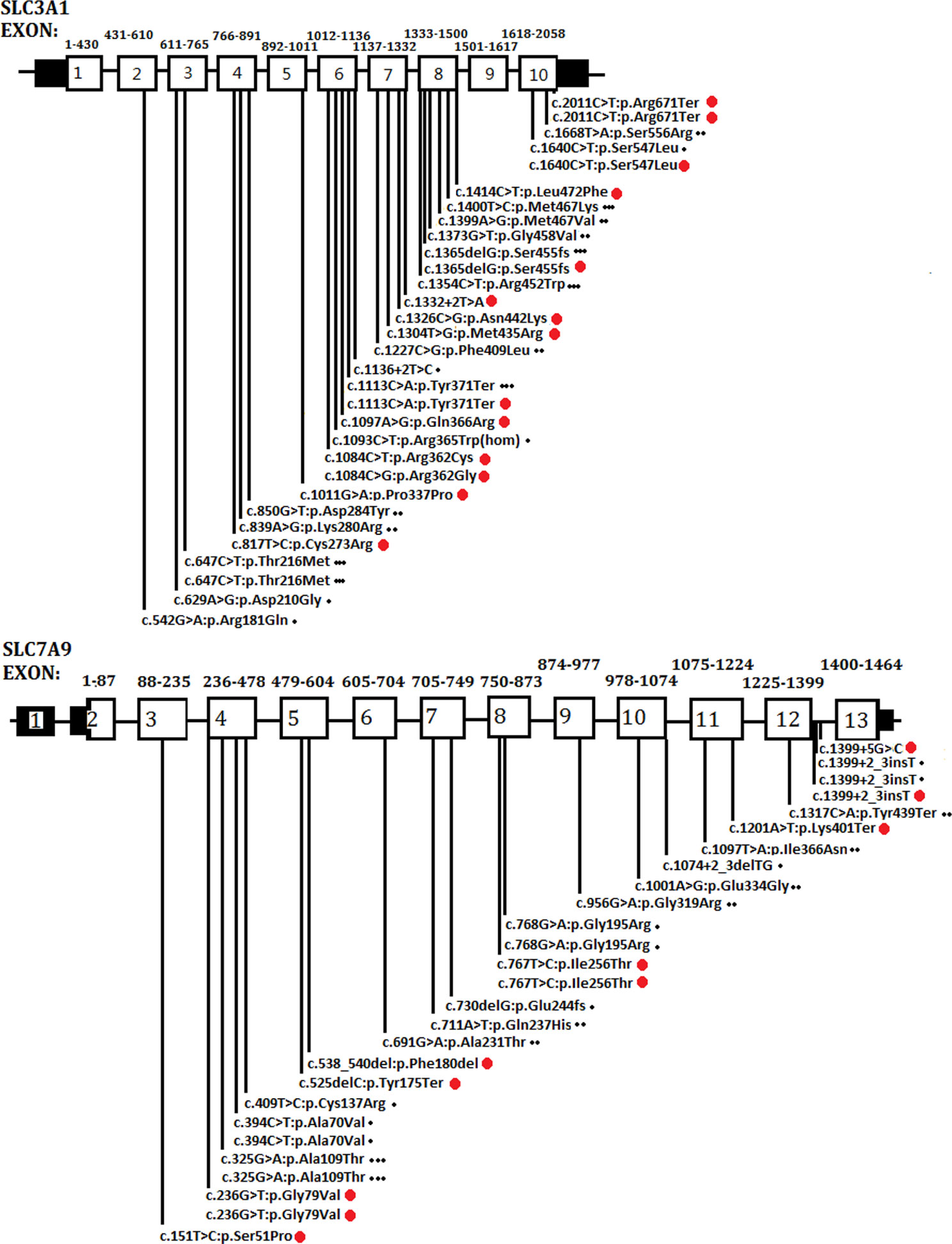
Figure 4 Schematic presentation of SLC3A1 or SLC7A9 pathogenic variants in the Chinese population that have been reported thus far. One dot (at the end of the variants) represents the variants reported by Yuen et al. (2006). Two dots denote the variants reported by Shen et al. (2017). Three dots mean the variants reported by Ma et al. (2018). Red dots are the variants identified in the present study.
Conclusions
We detected a total of 24 rare variants in the SCL3A1 and SLC7A9 genes from 13 families with CU. Among these variants, 15 were novel. Functional evaluation analysis confirmed that all these novel variants are pathogenic, damaging, or disease-causing. These data expanded the spectrum of CU variants in the Chinese population.
Data Availability Statement
The SLC3A1 and SLC7A9 genetic polymorphism data can be found in ClinVar, accession number SUB6699202.
Ethics Statement
The studies involving human participants were reviewed and approved by Academic Committee of Hunan Children's Hospital (Approval number: HCHLL58, Changsha City, Hunan Province, China). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
Writing of the first draft: CL, YY, YZ, and YwZ. Review and Critique: YY and YwZ. Conception: CL, YY, YwZ. Organization: YY, YZ. Execution: YY, CL, YwZ, YZ. Clinical support: FS, LL, YL, LpL. Material support: YY, YwZ, CL, YZ, YL and LL.
Funding
This work was supported by the Natural Science Foundation of Hunan Province, China (2017JJ2139) and Hunan Health Commission Research Fund (B2019019).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the subjects for their participation in this research study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00074/full#supplementary-material
References
Alirezaie, N., Kernohan, K. D., Hartley, T., Majewski, J., Hocking, T. D. (2018). ClinPred: prediction tool to identify disease-relevant nonsynonymous single-nucleotide variants. Am. J. Hum. Genet. 103 (4), 474–483. doi: 10.1016/j.ajhg.2018.08.005
Botzenhart, E., Vester, U., Schmidt, C., Hesse, A., Halber, M., Wagner, C., et al. (2002). Cystinuria in children: distribution and frequencies of mutations in the SLC3A1 and SLC7A9 genes. Kidney Int. 62, 1136–1142. doi: 10.1111/j.1523-1755.2002.kid552.x
Calonge, M. J., Gaparini, P., Chillaron, J., Chillon, M., Gallucci, M., Rousaud, F., et al. (1994). Cystinuria caused by mutations in rBAT, a gene involved in the transport of cystine. Nat. Genet. 6, 420–425. doi: 10.1038/ng0494-420
Chillarón, J., Font-Llitjós, M., Fort, J., Zorzano, A., Goldfarb, D. S., Nunes, V., et al. (2010). Pathophysiology and treatment of cystinuria. Nat. Rev. Nephrol. 6, 424–434. doi: 10.1038/nrneph.2010.69
Dello Strologo, L., Pras, E., Pontesilli, C., Beccia, E., Ricci-Barbini, V., de Sanctis, L., et al. (2002). Comparison between SLC3A1 and SLC7A9 cystinuria patients and carriers: a need for a new classifcation. J. Am. Soc. Nephrol. 13, 2547–2553. doi: 10.1097/01.ASN.0000029586.17680.E5
Eggermann, T., Venghaus, A., Zerres, K. (2012). Cystinuria: an inborn cause of urolithiasis. Orphanet J. Rare Dis. 7, 19. doi: 10.1186/1750-1172-7-19
Fattah, H., Hambaroush, Y., Goldfarb, D. S. (2014). Cystine nephrolithiasis. Transl. Androl. Urol. 3 (3), 228–233. doi: 10.3978/j.issn.2223-4683.2014.07.04
Feliubadaló, L., Font, M., Purroy, J., Rousaud, F., Estivill, X., Nunes, V., et al. (1999). International cystinuria consortium. Non-type I cystinuria caused by mutations in SLC7A9, encoding a subunit (bo,+AT) of rBAT. Nat. Genet. 23, 52–57. doi: 10.1038/12652
Harnevik, L., Fjellstedt, E., Molbaek, A., Tiselius, H. G., Denneberg, T., Söderkvist, P. (2001). Identification of 12 novel mutations in the SLC3A1 gene in Swedish cystinuria patients. Hum. Mutat. 18, 516–525. doi: 10.1002/humu.1228
Harnevik, L., Fjellstedt, E., Molbaek, A., Denneberg, T., Söderkvist, P. (2003). Mutation analysis of SLC7A9 in cystinuria patients in Sweden. Genet. Test. 7, 13–20. doi: 10.1089/109065703321560886
Ma, Y. Y., Liu, Y. P., Li, D., Li, X. Y., Song, J. Q., Yang, YL. (2018). Clinical, Biochemical, and genetic findings of Cystinuria in Chinese children. Clin. Lab. 64, 1145–1151. doi: 10.7754/Clin.Lab.2018.180110
Miller, N. L., Lingeman, J. E. (2007). Management of kidney stones. BMJ 334 (7591), 468–472. doi: 10.1136/bmj.39113.480185.80
Saadi, I., Chen, X. Z., Hediger, M., Ong, P., Pereira, P., Goodyer, P., et al. (1998). Molecular genetics of cystinuria: mutation analysis of SLC3A1 and evidence for another gene in type I (silent) phenotype. Kidney Int. 54, 48–55. doi: 10.1046/j.1523-1755.1998.00956.x
Sahota, A., Tischfield, J. A., Goldfarb, D. S., Ward, M. D., Hu, L. (2019). Cystinuria: genetic aspects, mouse models, and a new approach to therapy. Urolithiasis 47, 57–66. doi: 10.1007/s00240-018-1101-7
Schwarz, J. M., Cooper, D. N., Schuelke, M., Seelow, D. (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11 (4), 361–362. doi: 10.1038/nmeth.2890
Segal, S., Thier, S. (1995). The Metabolic and molecular basis of inherited disease. Eds. Scriver, C. H., Beaudet, A. L., Sly, W. S., Valle, D. (New York: McGraw-Hill;: Cystinuria;), 3581–3601.
Shen, L., Cong, X., Zhang, X., Wang, N., Zhou, P., Xu, Y., et al. (2017). Clinical and genetic characterization of Chinese pediatric cystine stone patients. J. Pediatr. Urol. 13, 629.e1–629.e5. doi: 10.1016/j.jpurol.2017.05.021
Weinberger, A., Sperling, O., Rabinovtz, M., Brosh, S., Adam, A., De Vries, A. (1974). High frequency of cystinuria among Jews of Libyan origin. Hum. Hered. 24, 568–572. doi: 10.1159/000152696
Keywords: cystine urolithiasis, SLC3A1, SLC7A9, pathogenic variants, Chinese population
Citation: Li C, Yang Y, Zheng Y, Shen F, Liu L, Li Y, Li L and Zhao Y (2020) Genetic and Clinical Analyses of 13 Chinese Families With Cystine Urolithiasis and Identification of 15 Novel Pathogenic Variants in SLC3A1 and SLC7A9. Front. Genet. 11:74. doi: 10.3389/fgene.2020.00074
Received: 09 October 2019; Accepted: 22 January 2020;
Published: 18 February 2020.
Edited by:
Enrico Baruffini, University of Parma, ItalyReviewed by:
Fazil YM Marickar, Mount Zion Medical College, IndiaMark Nicholas Wass, University of Kent, United Kingdom
Copyright © 2020 Li, Yang, Zheng, Shen, Liu, Li, Li and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaowang Zhao, eXc1MDhAc2luYS5jb20=
†These authors have contributed equally to this work